

Abstract
Brain pericytes regulate cerebral blood flow, maintain the integrity of the blood-brain barrier (BBB), and facilitate the removal of amyloid β (Aβ), which is critical to healthy brain activity. Pericyte loss has been observed in brains from patients with Alzheimer's disease (AD) and animal models. Our previous data demonstrated that friend leukemia virus integration 1 (Fli-1), an erythroblast transformation-specific (ETS) transcription factor, governs pericyte viability in murine sepsis; however, the role of Fli-1 and its impact on pericyte loss in AD remain unknown. Here, we demonstrated that Fli-1 expression was up-regulated in postmortem brains from a cohort of human AD donors and in 5xFAD mice, which corresponded with a decreased pericyte number, elevated inflammatory mediators, and increased Aβ accumulation compared with cognitively normal individuals and wild-type (WT) mice. Antisense oligonucleotide Fli-1 Gapmer administered via intrahippocampal injection decelerated pericyte loss, decreased inflammatory response, ameliorated cognitive deficits, improved BBB dysfunction, and reduced Aβ deposition in 5xFAD mice. Fli-1 Gapmer-mediated inhibition of Fli-1 protected against Aβ accumulation-induced human brain pericyte apoptosis in vitro. Overall, these studies indicate that Fli-1 contributes to pericyte loss, inflammatory response, Aβ deposition, vascular dysfunction, and cognitive decline, and suggest that inhibition of Fli-1 may represent novel therapeutic strategies for AD.
Keywords: Fli-1, pericytes, Alzheimer's disease, cognitive deficit, antisense oligonucleotide, Gapmer, 5xFAD, blood-brain barrier
Graphical abstract
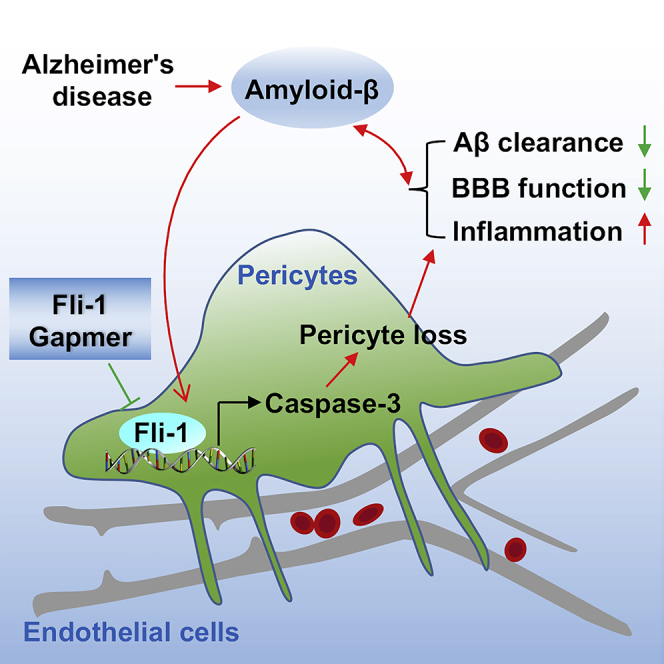
Fli-1 expression was significantly up-regulated in postmortem brains from patients with AD and in 5xFAD mice. Antisense oligonucleotide Fli-1 Gapmer decelerated pericyte loss, suppressed inflammatory response, ameliorated cognitive deficits, improved BBB function, and reduced Aβ deposition in 5xFAD mice. Thus, inhibition of Fli-1 may represent a novel therapeutic strategy for AD.
Introduction
Alzheimer's disease (AD) is a complex disease associated with cognitive impairment, accumulation of amyloid β-peptide (Aβ), vascular dysfunction, and neuroinflammation.1, 2, 3 Cerebral vascular dysfunction, such as decreases in cerebral blood flow (CBF) and disruption of the blood-brain barrier (BBB), has been recognized as an early and pivotal contributor to AD pathogenesis and a reliable predictor of cognitive decline.4,5 BBB damage in patients with AD and in animal models is associated with degeneration of brain pericytes, which are vascular mural cells that regulate CBF, maintain BBB integrity, mediate neuroinflammation, and exhibit phagocytic activity to remove toxic endogenous proteins, including Aβ.6, 7, 8, 9, 10 A deficiency of brain pericytes in the murine central nervous system leads to BBB breakdown.11,12 Furthermore, a reduced pericyte number was observed and correlated with BBB breakdown and Aβ deposition in the hippocampus and retina from patients with AD and in animal models.10,11,13,14 Moreover, pericyte deficiency in APP/PS1 mice was reported to accelerate BBB breakdown and increase Aβ accumulation in the brain.15 However, the processes that govern pericyte viability and their role in AD development have not been fully elucidated.
Our previous studies have demonstrated that friend leukemia virus integration 1 (Fli-1), an erythroblast transformation-specific (ETS) transcription factor, governs lung pericyte dysfunction and viability via the mediation of pericyte pyroptosis.16 Specifically, Fli-1 binds to the promoter regions of caspase-1/3 and regulates caspase-1/3 expression.17,18 In addition, Fli-1 is involved in a wide spectrum of biological processes, including cancer development, fibrosis, vasculopathy, and inflammation,19, 20, 21, 22 and it regulates the expression of important cytokines and matrix metalloproteases (MMPs) such as IL-6 and MMP3, which are implicated in AD.22, 23, 24, 25 However, the role of Fli-1 and its impact on pericyte degeneration in AD have not been previously investigated.
Here, we examined potential mechanisms by which Fli-1 may modulate pericyte stability within the BBB and contribute to AD pathogenesis. We found that Fli-1 expression is increased in the brain, including within pericytes from patients with AD and from 5xFAD mice. We further showed that inhibition of Fli-1 by an antisense oligonucleotide Gapmer, retards pericyte loss, ameliorates cognitive deficits, reduces Aβ deposition, and decelerates BBB breakdown in 5xFAD mice. Knockdown of Fli-1 prevented Aβ accumulation-induced human brain pericyte apoptosis in vitro. Thus, Fli-1 contributes to pericyte degeneration and cognitive impairment in AD, and may represent a novel therapeutic target to modify AD progression.
Results
Increased Fli-1 corresponded with pericyte loss in the hippocampus from postmortem brains of AD patients
To investigate the possible role of Fli-1 in AD, we first detected Fli-1 expression levels in patients with AD. The demographics for the human subjects in the study are shown in Table 1. Fli-1 mRNA levels determined by real-time PCR were significantly increased in the hippocampus and superior temporal gyrus of AD patients (n = 8–11, Figure 1A; p < 0.05). These data are from a subcohort of human subjects with AD diagnosis (age mean ± SD 83.7 ± 6.7 years, 7 females/4 males; postmortem interval [PMI] mean ± SD 5.6 ± 1.7 h) and are compared with those of cognitively normal controls (age mean ± SD 73.2 ± 19.8 years, 4 females/4 males; PMI mean ± SD 9.2 ± 3.3 h). Similarly, the Fli-1 levels determined by immunostaining in the hippocampus were also significantly increased in AD patients (n = 10) compared with cognitively normal controls (n = 10, Figure 1B; p < 0.05). The immunostaining was performed on postmortem brains isolated from another subcohort of human subjects with AD diagnosis (age mean ± SD 77.2 ± 2.7 years, 4 females/6 males; PMI mean ± SD 14.4 ± 6.3 h) and compared with those of cognitively normal controls (age mean ± SD 76.2 ± 11.5 years, 5 females/5 males; PMI mean ± SD 15.2 ± 9.1 h). Expression of TNFα and MMP3 but not IL-6 was increased in the hippocampus; however, increased TNFα expression was observed only in the superior temporal gyrus of AD patients (Figure S1; same cohort as in Figure 1A). In addition, Fli-1 levels were significantly (p < 0.05) up-regulated within pericytes in the hippocampus of AD patients (n = 10, Figure 1C; same cohort as in Figure 1B) as evidenced by increased Fli-1+ pericyte number. However, the absolute number of pericytes (CD13 positive) in the hippocampus was significantly (p < 0.05) decreased by 34% in AD patients compared with cognitively normal controls (n = 10, Figure 1D; same cohort as in Figure 1B). Quantification of active caspase-3+ pericyte number confirmed pericyte apoptosis in the hippocampus, which was significantly (p < 0.05) higher in AD patients (n = 10, Figure 1E; same cohort as in Figure 1B).
Table 1.
Demographic data for all human brain donors
| N = 38 | Cognitively normal controls | Alzheimer's disease |
|---|---|---|
| No. of subjects | 17 | 21 |
| Females (%) | 9 (52.9%) | 11 (52.4%) |
| Age ± SD (years) | 73.9 ± 15.2 | 80.6 ± 6.1 |
| Race (%) | 15 W (88.2%) | 19 W (90.5%) |
| 1 B (5.9%) | 2 N/A (9.5%) | |
| 1 A (5.9%) | ||
| PMI ± SD (h) | 12.2 ± 7.7 | 9.8 ± 6.3 |
SD, standard deviation; W, White; B, Black; A, Asian; N/A, not available; PMI, postmortem interval. Values are presented as mean ± SD.
Figure 1.

Fli-1 levels are increased and pericyte number is decreased in the hippocampus from postmortem brains of AD patients
(A) RNAs were isolated from 100 mg brain hippocampal or superior temporal tissue from human donors with AD (n = 11) and cognitively normal controls (n = 8), and Fli-1 mRNA levels were determined by RT-PCR. (B) Representative fluorescence images of brain hippocampus stained for Fli-1 (green) and nuclei (DAPI, blue) in human donors with AD (n = 10) and cognitively normal controls (n = 10). Scale bar: 60 μm. Fli-1 levels were analyzed. (C) Representative fluorescence images of brain hippocampus stained for Fli-1 (green), pericytes (CD13, red), and nuclei (DAPI, blue) in human donors with AD (n = 10) and cognitively normal controls (n = 10). Scale bar: 60 μm. Representative Fli-1+ pericytes are indicated by arrows, and Fli-1+ pericyte numbers were analyzed. (D) Representative fluorescence images of brain hippocampus stained for pericytes (CD13, red) and nuclei (DAPI, blue) in human donors with AD (n = 10) and cognitively normal controls (n = 10). Scale bar: 60 μm. CD13+ pericyte numbers were analyzed. (E) Representative fluorescence images of brain hippocampus stained for active caspase-3 (green), pericytes (CD13, red), and nuclei (DAPI, blue) in human donors with AD (n = 10) and cognitively normal controls (n = 10). Scale bar: 60 μm. Representative active caspase-3+ pericytes are indicated by arrows, and active caspase-3+ pericyte numbers were analyzed. Data are expressed as mean ± standard error of the mean. ∗p < 0.05 compared with CON group. CON, control; AD, Alzheimer's disease.
Increased Fli-1 and pericyte loss in the hippocampus of 5xFAD mice
Fli-1 levels and pericyte loss in the hippocampus were further investigated in a transgenic AD mouse model (5xFAD) compared with age- and sex-matched wild-type (WT) littermates (6.5 months of age). We observed a significant increase in Fli-1 mRNA and protein levels in the hippocampus of 5xFAD mice compared with the WT mice (Figures 2A–2C; p < 0.05). Fli-1 mRNA levels were also significantly increased in the cortex of AD mice (Figure S2A; p < 0.05). Increased expression of inflammatory mediators and MMPs, including TNFα, IL-6, IL-10, and MMP3, which are implicated in AD development,23,24,26 was observed in the hippocampus and/or cortex of 5xFAD mice (Figures 2D, S2B–S2D; p < 0.05). Increased Fli-1 within pericytes and decreased pericyte number by 29% in the hippocampus of 5xFAD mice were confirmed by immunostaining (Figures 2E–2G; p < 0.05), analogous to observations in human brain tissues.
Figure 2.

Up-regulated Fli-1 levels are associated with increased pericyte loss in the hippocampus of 5xFAD mice
The hippocampus was isolated from WT and 5xFAD mice at 6.5 months of age. (A) Fli-1 mRNA and (B) Fli-1 protein levels were determined by RT-PCR and western blot; n = 5 mice/group. (C) Representative fluorescence images of brain hippocampus stained for Fli-1 (green) and nuclei (DAPI, blue). Scale bar: 50 μm. Quantification analysis of Fli-1 fluorescence intensity was performed; n = 18 random fields from three mice/group. (D) Expression levels of TNFα, IL-6, IL-10, and MMP3 were determined by RT-PCR; n = 5 mice/group. (E) Representative fluorescence images of brain hippocampus stained for Fli-1 (green), pericytes (CD13, red), and nuclei (DAPI, blue). Scale bars: 150 and 50 μm. Representative Fli-1+ pericytes are indicated by arrows. (F and G) Fli-1+ pericyte numbers (F) and CD13+ pericyte numbers (G) were analyzed; n = 18 random fields from three mice/group. Data are expressed as mean ± standard error of the mean. ∗p < 0.05 compared with WT group.
Fli-1 Gapmer treatment ameliorates cognitive deficits in 5xFAD mice
To further investigate the possible association between Fli-1 and cognitive deficits in AD, we knocked down Fli-1 by intrahippocampal injection of Fli-1 antisense oligonucleotide Gapmers into 5xFAD mice at ages 3 and 4.5 months (Figure S3). Decreased Fli-1 mRNA and protein levels in the hippocampus but not in the cortex of 6.5-month-old 5xFAD mice were confirmed in the Fli-1 Gapmer group (Figures 3A and S4A). Significant impairment in working memory and spatial memory has been observed in 5xFAD mice.27,28 To investigate the impairment in working memory, we performed the novel object recognition (NOR) test in 6-month-old 5xFAD mice. We demonstrated that 6-month-old 5xFAD mice had significantly fewer interactions with the novel object (frequency recognition index) and spent significantly less time exploring the novel object (time recognition index) than WT mice (Figure 3B; p < 0.05), indicating the impairment in working memory of 5xFAD mice. However, Fli-1 Gapmer administration significantly improved the recognition index of frequency and time in 5xFAD mice compared with the control Gapmer group (Figure 3C; p < 0.05). The Morris water maze (MWM) test was used to explore spatial memory limitations in 6-month-old 5xFAD mice. After training trials on day 1, the escape latency time on day 2 of the 5xFAD mice was significantly longer than that of the WT mice (Figure 3D; p < 0.05), indicating the impairment in spatial memory of 5xFAD mice. However, the escape latency of the 5xFAD mice on day 2 of the Fli-1 Gapmer group was significantly reduced compared with that of the control Gapmer group (Figure 3E; p < 0.05).
Figure 3.

Inhibition of Fli-1 by Fli-1 Gapmer via intrahippocampal injection attenuates cognitive deficits in 5xFAD mice
Fli-1 or control Gapmers were injected into both sides of the hippocampus of 5xFAD mice at 3 and 4.5 months of age. (A) The expression of Fli-1 in the hippocampus was determined by RT-PCR and western blot in 5xFAD mice treated with control or Fli-1 Gapmers at 6.5 months of age. n = 3–5 mice/per group. NOR and MWM tests were performed on wild-type mice (n = 8), 5xFAD mice (n = 8), and 5xFAD mice injected with control (n = 6) or Fli-1 Gapmers (n = 7–8) at 6 months of age. The frequency and time of recognition of novel object index (frequency to visit and time spent with novel object versus both objects) for WT and 5xFAD mice (B), and 5xFAD mice treated with control or Fli-1 Gapmers (C) were recorded and analyzed. The average time for WT and 5xFAD mice (D), and 5xFAD mice treated with control or Fli-1 Gapmers (E) to find the hidden platform was recorded and analyzed. Data are expressed as mean ± standard error of the mean. ∗p < 0.05 compared with WT group or 5xFAD + Con Gapmer group. #p < 0.05 compared with 5xFAD + Con Gapmer group.
Fli-1 Gapmer treatment reduces pericyte loss and improves BBB dysfunction in the hippocampus of 5xFAD mice
To further investigate the possible role of Fli-1 in AD pathology, inflammatory mediators, MMPs, pericyte loss, and BBB dysfunction were assessed in 5xFAD mice treated with Fli-1 Gapmer or control Gapmer at age 6.5 months. As revealed in Figure 4A, TNFα, IL-6, and MMP3, but not IL-10, expression in the hippocampus of 5xFAD mice was significantly (p < 0.05) decreased in the Fli-1 Gapmer group. As intrahippocampal injection of Fli-1 Gapmer did not affect the Fli-1 levels in the cortex, we consistently observed unchanged TNFα, IL-6, and MMP3 expression in the cortex of the Fli-1 Gapmer group (Figures S4B–S4D). Fli-1 expression decreased within pericytes, although the overall pericyte number increased by 22% in the hippocampus of 5xFAD mice treated with Fli-1 Gapmer compared with the control Gapmer group (Figures 4B–4D; p < 0.05). In addition, inhibition of Fli-1 significantly attenuated the vascular leakage of IgG in the hippocampus of 5xFAD mice, suggesting that Fli-1 Gapmer treatment improved BBB dysfunction (Figures 4E and 4F; p < 0.05).
Figure 4.

Inhibition of Fli-1 by Fli-1 Gapmer via intrahippocampal injection ameliorates pericyte loss and BBB dysfunction in the hippocampus of 5xFAD mice
Fli-1 or control Gapmers were injected into both sides of the hippocampus of 5xFAD mice at 3 and 4.5 months of age. The hippocampus was isolated from WT and 5xFAD mice at 6.5 months of age. (A) TNFα, IL-6, IL-10, and MMP3 levels were determined by RT-PCR; n = 5 mice/group. (B) Representative fluorescence images of brain hippocampus stained for Fli-1 (green), pericytes (CD13, red), and nuclei (DAPI, blue). Scale bars: 150 and 50 μm. Representative Fli-1+ pericytes are indicated by arrows. (C) and (D) Fli-1+ pericyte numbers (C) and CD13+ pericyte numbers (D) were analyzed; n = 18 random fields from three mice/group. (E) Representative fluorescence images of brain hippocampus stained for IgG (red) and CD31 (green). Scale bar: 25 μm. (F) Quantitative analysis of IgG fluorescence intensity; n = 18 random fields from three mice/group. Data are expressed as mean ± standard error of the mean. ∗p < 0.05 compared with 5xFAD + Con Gapmer group. #p < 0.05 compared with WT group.
Fli-1 Gapmer treatment attenuates Aβ accumulation in the hippocampus of 5xFAD mice
Pericyte loss has been associated with increased BBB breakdown and Aβ deposition in the hippocampus from AD patients and animal models.10,11 Thus, we further determined the effect of Fli-1 Gapmers via intrahippocampal injection on Aβ deposition in 5xFAD mice. By 6.5 months, significant Aβ plaques were observed by thioflavin-S staining (Figure 5A) in the hippocampus of 5xFAD mice but were barely found in their WT littermates. Fli-1 Gapmer treatment significantly reduced both the number and the area of amyloid plaque deposition in the hippocampus (Figures 5B–5D; p < 0.05). Moreover, Aβ load as assessed by human Aβ antibody staining (Figure 5E) was observed in the hippocampus of 5xFAD mice but was barely found in their WT littermates. The 5xFAD mice treated with Fli-1 Gapmer had less Aβ load in the hippocampus compared with the control Gapmer group (Figures 5F and 5G; p < 0.05). To strengthen these results, we further determined soluble human Aβ levels in the hippocampus by ELISA. Consistently, we found that soluble Aβ40 and Aβ42 levels in the hippocampus were significantly increased in 5xFAD mice but reduced in those injected with Fli-1 Gapmer (Figures 5H and 5I; p < 0.05). However, soluble Aβ40 or Aβ42 levels in the cortex were not affected by intrahippocampal injection of Fli-1 Gapmers due to the undisrupted Fli-1 levels (Figures S4E and S4F).
Figure 5.

Inhibition of Fli-1 by Fli-1 Gapmer via intrahippocampal injection reduces Aβ accumulation in the hippocampus of 5xFAD mice
(A) Representative fluorescence images of brain hippocampus from WT and 5xFAD mice at 6.5 months of age stained for Aβ plaque (thioflavin-S, green). Scale bar: 100 μm. Fli-1 or control Gapmers were injected into both sides of the hippocampus of 5xFAD mice at 3 and 4.5 months of age. The hippocampus was isolated from 5xFAD mice treated with control or Fli-1 Gapmers at 6.5 months of age. (B) Representative fluorescence images of brain hippocampus stained for Aβ plaque (thioflavin-S, green). Scale bar: 50 μm. (C) and (D) Aβ plaque number (C) and immunoreactive area (D) were analyzed; n = 19–24 random fields from three mice/group. (E) Representative fluorescence images of brain hippocampus from WT and 5xFAD mice at 6.5 months of age stained for human Aβ (82E1, red) and nuclei (DAPI, blue). Scale bar: 50 μm. (F) Representative fluorescence images of brain hippocampus from 5xFAD mice treated with control or Fli-1 Gapmers stained for human Aβ (82E1, red) and nuclei (DAPI, blue). Scale bar: 50 μm. (G) Human Aβ immunoreactive area was analyzed; n = 19 random fields from three mice/group. (H) Soluble human Aβ40 levels in the hippocampus were detected by ELISA; n = 5 mice/group. (I) Soluble human Aβ42 levels in the hippocampus were detected by ELISA; n = 5 mice/group. Data are expressed as mean ± standard error of the mean. ∗p < 0.05 compared with 5xFAD + Con Gapmer group. #p < 0.05 compared with WT group.
Fli-1 Gapmer treatment prevents Aβ accumulation-induced pericyte apoptosis
To investigate the role of Fli-1 in pericyte apoptosis, we cultured human brain pericytes in vitro. Aβ40 or TNFα treatment significantly increased Fli-1 mRNA levels in human brain pericytes (Figure 6A; p < 0.05). Prolonged accumulation of Aβ40 has been reported to cause cell apoptosis of brain pericytes.10,15 Consistently, cell viability assays showed that 7 days of continuous treatment with freshly aggregated Aβ40 treatment significantly decreased pericyte cell viability, and transfection with Fli-1 Gapmer treatment significantly decreased Fli-1 levels (Figure 6B; p < 0.05) and increased cell viability in brain pericytes (Figure 6C; p < 0.05). Knockdown of Fli-1 significantly reduced Aβ40-induced caspase-3 mRNA levels (Figure 6D; p < 0.05). To assess whether Fli-1 Gapmer-prevented pericyte death is attributed to inhibited apoptosis, we next performed terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and active caspase-3 staining, two typical markers of apoptosis induction. Indeed, the Aβ40-induced increase in fluorescence intensity of TUNEL and active caspase-3 expression in human brain pericytes was significantly suppressed in the Fli-1 Gapmer group compared with the control Gapmer group (Figures 6E–6G; p < 0.05).
Figure 6.

Inhibition of Fli-1 by Fli-1 Gapmer reduces Aβ accumulation-induced pericyte apoptosis and caspase-3 expression in human brain pericytes
Human brain pericytes were stimulated with freshly aggregated Aβ (1–40, 10 μM) or human TNFα (10 ng/mL) for 12 h. (A) Fli-1 levels were determined by RT-PCR; n = 3 independent experiments. Human brain pericytes were transfected with Fli-1 or Con Gapmers for 48 h and treated with freshly aggregated Aβ (1–40, 10 μM) for 5 or 7 consecutive days. (B) Fli-1 levels were determined by RT-PCR; n = 3 independent experiments. (C) Cell viability was evaluated by PrestoBlue cell viability reagent. Results are expressed as percentage of Con Gapmer alone group; n = 12 replicates from four independent experiments. (D) Caspase-3 mRNA levels were measured by RT-PCR; n = 3 independent experiments. (E) Representative fluorescence images of brain pericytes stained for TUNEL (green) or active caspase-3 (red). Scale bar: 100 μm. Quantification analysis of (F) TUNEL fluorescence intensity and (G) active caspase-3 expression is shown; n = 10 random fields from three independent experiments. Data are expressed as mean ± standard error of the mean. ∗p < 0.05 compared with control or Con Gapmer group. #p < 0.05 compared with Aβ40 + Con Gapmer group. Con, control.
Discussion
AD is the most common neurodegenerative disorder and a leading cause of dementia among elderly people. It accounted for approximately 70% of the 50 million people suffering from dementia worldwide in 2018, and this figure is anticipated to triple by 2050, with associated costs approaching $4 trillion.5 Pericyte loss in AD and its central role in regulating BBB integrity, Aβ accumulation, and cognitive function have been recognized recently and hold promise for therapeutic interventions.6, 7, 8, 9, 10 In the present study, we demonstrated that the antisense oligonucleotide Gapmer-mediated inhibition of Fli-1 alleviates AD progression via its protective effects against pericyte loss. This notion can be supported by our several novel discoveries. First, Fli-1 expression level is increased with pericyte loss in postmortem brains from AD patients and in 5xFAD mice. Second, knockdown of Fli-1 via intrahippocampal injection of antisense Gapmers ameliorates cognitive deficits, pericyte loss, BBB dysfunction, and Aβ deposition in 5xFAD mice. Last, Fli-1 Gapmer treatment inhibits Aβ accumulation-induced pericyte apoptosis in cultured human brain pericytes.
Inflammatory response is a key factor in the development of AD.23 As a critical regulator of inflammation, Fli-1 belongs to the ETS transcription factor family and is expressed in macrophages, B cells, T cells, endothelial cells, and pericytes.16,19,21,22,29 Abnormal Fli-1 expression has been observed in several diseases, including Ewing sarcoma, systemic lupus erythematosus (SLE), systemic sclerosis, and sepsis, and plays a critical role in the pathological development of these diseases.16,30, 31, 32, 33, 34 Our previous study demonstrated that Fli-1 expression is significantly increased in lung pericytes and contributes to the inflammatory response and vascular leak in a murine model of sepsis.16 In this study, we provide evidence that pericyte Fli-1 levels were elevated in the hippocampus of AD patients and in 5xFAD mice along with increased inflammatory mediators TNFα and IL-6 (Figures 1, 2, and S1). It is likely that the increased Fli-1 expression in brain pericytes is associated with inflammation and Aβ accumulation in AD, as its expression can be up-regulated in human brain pericytes upon stimulation with TNFα or Aβ40 (Figure 6). We further noticed that inhibition of Fli-1 via intrahippocampal injection of antisense Gapmers significantly decreased the expression of Fli-1 (Figure 3). Interestingly, this phenomenon was observed at 2 months after the injection of Fli-1 Gapmers. There are two possible mechanisms behind this finding: (1) the long-lasting effect of Fli-1 Gapmer in vivo and (2) Fli-1 Gapmer injection decreased Fli-1 levels leading to reduced TNFα levels and Aβ deposition in the hippocampus, which in turn decelerated the increase in Fli-1 levels during AD. However, the dose-dependent and time-dependent effects of Fli-1 Gapmer treatment on in vivo Fli-1 levels and AD development need further study. In addition, Fli-1 Gapmer treatment significantly decreased TNFα and IL-6 levels in the hippocampus of 5xFAD mice (Figure 4); however, unaffected TNFα and IL-6 expression levels were observed when Fli-1 expression was not disrupted in the cortex (Figure S4). TNFα and IL-6 are proinflammatory cytokines produced in the brain during AD and contribute to Aβ deposition and BBB dysfunction.23 The TNFα blocking agents showed improvement in cognitive deficits of AD animal models and reduced risks of AD in human patients with rheumatoid arthritis and psoriasis.35,36 Neutralization of IL-6 in the brains alleviated memory impairment in APP/PS1 mice.37 As an important regulator of inflammation, Fli-1 regulates expression of several important cytokines, including IL-6, by directly binding to the promoter, and inhibition of Fli-1 exerts beneficial effects in inflammatory diseases, including sepsis and SLE.16,22,38 The collective evidence together with our present data indicate that Fli-1 is an important regulator of inflammation during AD, and the beneficial effects of Fli-1 knockdown in AD may be partly attributed to the attenuated inflammatory response.
In addition to inflammation, the contribution of pericyte loss to AD progression has gained increasing attention.15 Aβ can cause death of cerebrovascular cells, including pericytes.39 Previous studies demonstrated that pericyte number and coverage in the hippocampus of patients with AD are reduced by up to 60% compared with cognitively normal controls.11 Decreased pericyte number in the hippocampus of APP/PS1 mice was also observed.10 In addition, increased pericyte loss via apoptosis in the retina from patients with AD was identified.14 Consistent with these findings, we confirmed pericyte loss in the hippocampus from patients with AD and 5xFAD mice (Figures 1 and 2). A recent study suggests that Fli-1 deficiency promotes proliferation and cell survival of endothelial cells.40 Our previous data demonstrated that Fli-1 plays a critical role in pericyte dysfunction and viability in sepsis by mediating pericyte pyroptosis.16 In addition, Fli-1 directly binds to the promoter region of caspase-1/3 and thereby mediates caspase-1/3 expression.17,18 Thus, Fli-1 may regulate brain pericyte loss in AD. Indeed, we further demonstrated that knockdown of Fli-1 attenuated pericyte loss in the hippocampus of 5xFAD mice and reduced Aβ-induced apoptosis and caspase-3 expression in cultured human brain pericytes (Figures 4 and 6). Accelerated BBB breakdown and microvascular reductions have been documented in pericyte-deficient AD mice.15 A recent publication demonstrated that soluble PDGFR-β is a cerebrospinal fluid biomarker of pericyte injury during early cognitive impairment and correlates with BBB disruption independent of Aβ and tau.41 We previously reported that knockout of Fli-1 improved vascular dysfunction in the lung and kidney from septic mice.16 Therefore, Fli-1 may also regulate BBB dysfunction in AD. Here, we provide evidence that inhibition of Fli-1 via intrahippocampal injection of antisense Gapmers significantly improved BBB breakdown as indicated by reduced IgG extravasation in the hippocampus (Figure 4). These observations indicate that Fli-1 may contribute to pericyte loss via regulation of caspase-3, leading to BBB dysfunction in AD.
In the brain, Aβ is produced by neurons and other cells, and plays an indispensable role in the development of AD.23,42 Dysfunction of Aβ clearance, rather than Aβ overproduction, is chiefly responsible for the accumulation of Aβ in the brain.43 Pericytes also show potent phagocytic activity and play a critical role in the removal of Aβ.6,7,15,44 Other recent studies using pericyte-deficient transgenic mouse models demonstrated that pericyte deficiency elevates brain Aβ40 and Aβ42 levels and accelerates amyloid angiopathy by diminishing clearance of soluble Aβ.15 From one end, loss of pericytes disrupts BBB integrity, which may in turn accelerate Aβ plaque buildup.15,45 From the other end, pericyte loss reduces early clearance of soluble Aβ, leading to the accumulation of Aβ in the brain, which in turn self-amplifies Aβ-induced pericyte loss.15 In this study, our data showed that antisense Gapmer-mediated inhibition of Fli-1 significantly ameliorated Aβ plaque deposition and soluble Aβ levels in the hippocampus of 5xFAD mice (Figure 5), which may be partly attributed to the protection against pericyte loss. Thus, Fli-1 is involved in a wide spectrum of activities in the brain by manipulating inflammation, pericyte loss, BBB dysfunction, and Aβ accumulation, which all contribute to the cognitive decline in AD.4,5,10,23 Therefore, Fli-1 may contribute to the cognitive deficits in AD. Indeed, we further demonstrated that inhibition of Fli-1 in the hippocampus rescues spatial learning and memory deficits in the AD developing 5xFAD mice (Figure 3). These findings indicate that antagonizing pericyte loss via Fli-1 Gapmer can decelerate vascular damage, Aβ accumulation, and cognitive deficits in AD.
Fli-1 plays an important role in vascular development, and its deficiency results in embryonic lethality.46 Mice with a conditional deletion of Fli-1 from endothelial cells also showed markedly increased vessel permeability.47 These findings indicated that extremely low levels of Fli-1 impair vascular homeostasis and cause vascular injury. However, high levels of Fli-1 were associated with increased inflammation and vascular leak in sepsis.16 Furthermore, Fli-1 transgenic mice that overexpress Fli-1 developed a high incidence of a progressive immunological renal disease associated with increased renal inflammation, and ultimately died of renal failure caused by tubulointerstitial nephritis and immune-complex glomerulonephritis.48 Therefore, extremely low or high levels of Fli-1 perturb the physiological homeostasis and result in vascular and/or organ injury. Our study demonstrated that Fli-1 levels were markedly increased by 4.4-fold in the hippocampus in 5xFAD mice compared with WT mice, which is associated with increased inflammation, pericyte loss, and Aβ deposition, while Fli-1 Gapmer treatment reduced Fli-1 levels by 4.7-fold in the hippocampus of 5xFAD mice and led to attenuated inflammation, pericyte loss, and Aβ deposition. Thus, Fli-1 Gapmer treatment did not completely knock out Fli-1 levels but decreased the Fli-1 levels back to the baseline, which is helpful in maintaining homeostasis in the brain during AD. The intrahippocampal injection of Fli-1 Gapmer we used in this study is a proof of concept that guarantees future efforts to treat AD with systemic Fli-1 Gapmers, which could be a viable way to translate these results into a therapeutic solution.
There are several limitations to our study. Increased Fli-1 expression levels were confirmed in patients with AD and in 5xFAD mice at 6.5 months of age. It would be of clinical significance to investigate further whether Fli-1 levels are associated with AD severity in patients from a larger cohort or in 5xFAD mice via long-term observations. We demonstrated the beneficial effect of Fli-1 Gapmer in the hippocampus of 5xFAD mice; however, the role of Fli-1 in the cortex in AD needs further investigation using systemic injection of Fli-1 Gapmers or Fli-1 knockout mice. The contribution of Fli-1 to AD progression in 5xFAD mice may be partly attributed to the mediation of pericyte loss. However, whether Fli-1 has an impact on other cell types in AD, such as astrocytes, microglia, neurons, and endothelial cells, remains unknown and could be an interesting topic for future study.
In summary, our studies identify Fli-1 as a novel regulator of pericyte dysfunction in AD in association with protective effects of Fli-1 Gapmer against BBB dysfunction, Aβ accumulation, and cognitive decline. Thus, we provide evidence that signaling pathways reducing Fli-1 expression or its downstream mediators may represent novel therapeutic strategies for preventing or mitigating AD-induced cognitive dysfunction.
Materials and methods
Human brain donors
Brain samples were obtained from the Carroll A. Campbell, Jr., Neuropathology Laboratory (brain bank) at the Department of Pathology and Laboratory Medicine at the Medical University of South Carolina. These studies were in accordance with the 1964 Helsinki declaration and its later amendments or comparable ethical standards and approved by the Institutional Review Board at the Medical University of South Carolina. Brain tissues were collected from 21 clinically and neuropathologically confirmed AD patients and 17 cognitively normal individuals (females and males showing neither clinical cognitive impairment/dementia nor brain pathology). The entire human cohort demographics are listed in Table 1. The groups had no significant differences in age, sex, or PMI hours. All samples were previously anonymized.
Animals
The male and female 5xFAD transgenic mice on the C57BL/6J background were purchased from The Jackson Laboratory and bred in the animal facility at the Medical University of South Carolina. All animals were allowed free access to food and water and maintained under a facility with a 12 h light/dark cycle and constant temperature. All procedures complied with the standards for care and use of animal subjects as stated in the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Resources, National Academy of Sciences, Bethesda, MD). The protocol for all animal studies was approved by the Institutional Animal Care and Use Committee at the Medical University of South Carolina. The in vivo experimental design can be found in Figure S3.
Intrahippocampal injection of control or Fli-1 Gapmers
Control or Fli-1 antisense oligonucleotide Gapmers (Qiagen, Germantown, MD) were injected into the hippocampus of 5xFAD transgenic mice as previously described.49 Each mouse received two injections at 3 and 4.5 months of age (Figure S3). Briefly, mice were anesthetized with vaporized isoflurane and fixed in a stereotaxic apparatus. Then, a 2 μL volume of control or Fli-1 Gapmers (25 μM) was injected bilaterally into the hippocampus (anterior-posterior [AP], −2.1 mm; medial-lateral [ML], ±1.6 mm; dorsal-ventral [DV], −1.4 mm) using a Hamilton 5 μL syringe and a 27 G needle at 0.2 μL/min for a 10 min duration.
Novel object recognition
NOR was performed as described previously50,51 in 6-month-old mice. The NOR test Plexiglas box was located in an isolated and illuminated animal testing room. Mice were first given a 10 min habituation trial with no objects in the test box, and the NOR test was performed 24 h later. In the first trial of NOR, two identical objects were placed in diagonally opposite corners of the test box and the mice were allowed to explore them for 6 min. After 30 min, one of the objects was replaced with a novel object and the mice were again placed in the test box for a second 6 min trial. Mouse behavior/activity and time spent near each object were recorded by live video tracking and EthoVision XT v.13 software (Noldus Information Technology, Leesburg, VA). Both the objects and the box were cleaned with alcohol and dried after each trial to remove olfactory cues. Recognition index (RI) of time was calculated based on the total time (second trial) spent exploring the novel object (TN) and the familiar (TF) object: RItime = TN/(TN + TF). RI of frequency was calculated based on the number of visits (second trial) to the novel object (FN) and the familiar (FF) object: RIfrequency = FN/(FN + FF).
Morris water maze
MWM test was performed as described previously52 in 6-month-old mice. The MWM consists of a raised tank filled halfway with water, which was warmed to a temperature of 25°C and made opaque with the addition of non-toxic, white tempera paint. A Plexiglas escape platform on a raised base was placed in one of four quadrants of the maze, to be slightly submerged (1 cm) under water. Visual cues were placed on the surrounding walls of the maze (N, S, E, W) and remained in place for each day of testing. In brief, each mouse was trained four times at 2 min intervals on day 1. In each trial, the mouse was placed in water at one of four randomly selected starting positions and given 90 s to find the platform; the elapsed time for finding the platform was defined as the escape latency for each trial. Latency to reach the location of the platform was monitored and recorded using a high-speed camera and EthoVision XT v.13 software (Noldus Information Technology, Leesburg, VA). If the mouse did not locate the platform within 90 s, the mouse was guided gently by hand to the platform and allowed to rest there for 10 s; the escape latency was recorded as 90 s. Following the trial, each mouse was placed in a dry container and allowed to rest for 2 min. At day 2, one trial was performed for each mouse to test spatial learning and memory ability, and the mice were allowed to swim for 90 s at the starting position opposite the platform, and the escape latency was recorded. The test was performed blindly. Mean swim latency for all the trials on each day in each group was calculated.
Human brain pericyte culture and stimulation
Primary human brain pericytes were purchased from ScienCell Research Laboratories and cultured in human pericyte medium (#1201, ScienCell Research Laboratories, Carlsbad, CA). Pericytes were transfected with control or Fli-1 antisense Gapmers (Qiagen, Germantown, MD) for 48 h and cultured with or without freshly aggregated Aβ40 (10 μM, rPeptide, Watkinsville, GA) for 5 consecutive days. Total RNA was collected for further analysis. In another set of experiments, pericytes were transfected with control or Fli-1 antisense Gapmers (Qiagen, Germantown, MD) for 48 h and cultured with or without freshly aggregated Aβ40 (10 μM, rPeptide, Watkinsville, GA) for 7 consecutive days. Immunostaining and cell viability assays were performed. Fresh medium with or without 10 μM Aβ40 was replaced every 2 days until the end of the experiment.
Cell viability
Cell viability was detected by PrestoBlue cell viability reagent (Invitrogen, Waltham, MA) according to the instructions.
Immunocytochemistry
After deparaffinization, postmortem paraffin-embedded human hippocampus sections (4 μm) were treated with antigen retrieval solution at 98°C for 20 min and washed in PBS. Frozen mouse brain tissues were cut into 8 μm sections, fixed with 4% paraformaldehyde (PFA) for 15 min at room temperature, and then washed with PBS. The sections from human or mouse were then incubated in blocking buffer followed by primary antibody overnight at 4°C with the following combinations: Fli-1 (1:400; Proteintech, Rosemont, IL), CD13 (1:2,000; Cell Signaling, Danvers, MA), Aβ (82E1, 1:2,000; IBL America), Fli-1 (1:400; Proteintech, Rosemont, IL)/CD13 (1:2,000; Cell Signaling, Danvers, MA), and cleaved caspase-3 (1:400; Cell Signaling, Danvers, MA)/CD13 (1:2,000; Cell Signaling, Danvers, MA). Sections were washed three times with PBS and incubated with Alexa Fluor 488 goat anti-rabbit IgG (H + L) secondary antibody (Invitrogen, Waltham, MA) or Alexa Fluor 594 goat anti-mouse IgG (H + L) secondary antibody (Invitrogen, Waltham, MA) diluted 1:200 in PBS for 1 h at room temperature. For thioflavin-S staining, brain sections were incubated with 0.2% thioflavin-S (T1892, Sigma-Aldrich, St. Louis, MO) diluted in PBS for 10 min. After a wash with PBS, images were acquired using a Keyence BZ-X800 microscope and processed using the Keyence software package.
Human brain pericytes grown on 12 well plates coated with 0.2% gelatin were fixed with 4% PFA for 15 min at room temperature and then washed with DPBS. After being blocked with 1% BSA for 1 h at room temperature, the cells were incubated overnight at 4°C with cleaved caspase-3 antibody (1:400; Cell Signaling, Danvers, MA). Cells were washed with DPBS and incubated for 1 h at room temperature with Alexa Fluor 594 goat anti-rabbit IgG (H + L) secondary antibody (Invitrogen, Waltham, MA) diluted 1:200 in DPBS. After a wash with DPBS, images were acquired using a Keyence BZ-X800 microscope and processed using the Keyence software package.
TUNEL staining
TUNEL staining was performed using a TUNEL kit according to the manufacturer's instructions (Roche, Indianapolis, IN).
Immunostaining quantification
For each biomarker, the fluorescence of specific signals was captured using the same setting and exposure time. For Figure 1 of postmortem paraffin-embedded human hippocampus sections, quantification was performed from 10 AD donors and 10 age- and sex-matched cognitive normal controls. For Figure 1B, four to six images at 20× objective were taken randomly from each subject. The fluorescence intensity of acquired images was analyzed in the NIH ImageJ software, averaging the value per subject. For Figure 1C, four to six random fields at 20× objective per subject were analyzed for Fli-1+ pericyte number, averaging the number per square millimeter per subject. For Figure 1D, three images at 20× objective were taken randomly from each subject, and CD13+ pericyte number in each acquired image was analyzed, averaging the number per square millimeter per subject. For Figure 1E, five or six random fields at 20× objective per subject were analyzed for active caspase-3+ pericyte number, averaging the number per square millimeter per subject. For Figure 2 of frozen brain tissues, quantification was performed from WT and 5xFAD mice (three mice per group). For Figure 2C, six images at 20× objective were taken randomly from the hippocampus area per mouse, and the fluorescence intensity in each acquired image was analyzed in the NIH ImageJ software. For Figures 2F and 2G, six random images in the hippocampus area per mouse at 20× objective were taken, and the Fli-1+ pericyte number and CD13+ pericyte number in each acquired image were analyzed. For Figure 4 of frozen brain tissues, quantification was performed from WT mice and 5xFAD mice treated with control or Fli-1 Gapmers (three mice per group). For Figures 4C and 4D, six random images in the hippocampus area per mouse at 20× objective were taken, and Fli-1+ pericyte number and CD13+ pericyte number in each acquired image were analyzed. For Figure 4F, six random images in the hippocampus area per mouse at 20× objective were taken, and the fluorescence intensity of acquired images was analyzed in the NIH ImageJ software. For Figure 5 of frozen brain tissues, quantification was performed from 5xFAD mice treated with control or Fli-1 Gapmers (three mice per group). For Figures 5C and 5D, six to nine images at 20× objective were taken randomly from the hippocampus area per mouse. The plaque number in each image was counted, and the plaque area in each image was analyzed in the NIH ImageJ software. For Figure 5G, six or seven images at 20× objective were taken randomly from the hippocampus area per mouse. The Aβ area in each image was analyzed in the NIH ImageJ software. For Figure 6 of human brain pericyte immunostaining, quantification was performed from three independent experiments. For Figures 6F and 6G, 10 random images per group at 10× objective were taken, and the fluorescence intensity in each acquired image was analyzed in the NIH ImageJ software.
ELISA
The concentration of soluble human Aβ40 and Aβ42 in the hippocampus or cortex from WT mice and 5xFAD mice treated with control or Fli-1 Gapmers at age 6.5 months was detected. In brief, mice were anesthetized, and hippocampal and cortex tissues were removed and homogenized in RIPA buffer (Cell Signaling, Danvers, MA) on ice for 30 min. Samples were then centrifuged for 10 min at 4°C at 12,000 × g to obtain the supernatant. Protein concentration was determined by BCA assay and equal amounts of proteins were used to detect the concentration of soluble Aβ40 or Aβ42 using an Aβ40 or Aβ42 Human ELISA Kit (Invitrogen, Waltham, MA) according to the manufacturer's instructions. Results are presented as picograms of Aβ40 or Aβ42 per microgram of total protein.
Real-time reverse transcription-polymerase chain reaction
Total RNA was extracted from cultured human brain pericytes, human brain samples, or mouse brain tissues using the RNeasy Plus Mini Kit (Qiagen, Germantown, MD). cDNA was synthesized with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Waltham, MA). Quantitative real-time PCR was performed using the SYBR Green PCR Kit (Qiagen, Germantown, MD) and CFX96 Real-Time PCR system (Bio-Rad, Hercules, CA). Primers used in this study were purchased from Qiagen (Germantown, MD). Data were analyzed with 2−ΔΔCt value calculation using GAPDH for normalization.
Western blot analysis
Mouse brain tissues were lysed with ice-cold RIPA lysis buffer (Cell Signaling, Danvers, MA). All lysed samples were kept on ice for 30 min and centrifuged for 10 min at 4°C at 12,000 × g. Cell lysates were subjected to 12% SDS-PAGE and transferred onto a polyvinylidene difluoride membrane. The membranes were blocked with 7% milk in TBST (20 mM Tris, 500 mM NaCl, and 0.1% Tween 20) for 1 h. After being washed with TBST twice, membranes were incubated with primary antibody overnight at 4°C. Primary antibodies to β-actin were obtained from Cell Signaling (Danvers, MA). Fli-1 primary antibody was obtained from Proteintech (Rosemont, IL). The membranes were washed twice with TBST and incubated with horseradish peroxidase (HRP)-conjugated secondary antibody in blocking buffer for 1 h. After being washed three times with TBST, immunoreactive bands were visualized by incubation with ECL Plus detection reagents (GE Healthcare, Chicago, IL). Images were acquired by a ChemiDoc Touch Imaging System (Bio-Rad, Hercules, CA), and the densitometry of bands was quantified with ImageJ2 software.
Data analysis
Statistical significance was determined by analysis of variance (ANOVA) or Student’s t test using GraphPad Prism software. A value of p < 0.05 was considered statistically significant.
Acknowledgments
This work was supported in part by National Institutes of Health grants (1R01GM113995 [H.F.], 1R01GM130653 [H.F.], 3R01GM130653-03S1 [H.F.], 1R41AI157378 [H.F.], TL1TR001451 [P.V.H.], and ULTR001450 [P.V.H., A.G.]).
Author contributions
P.L. and Y.W. performed experiments, data collection, and analysis and wrote and edited the manuscript. E.H. performed experiments and edited the manuscript. A.G. and P.H. helped with study design and manuscript editing. S.C. and M.L. assisted with data interpretation and manuscript editing. H.F. was responsible for study conception and design, data analysis and interpretation, study supervision, and manuscript writing and editing. All authors have read and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.01.023.
Supplemental information
References
- 1.Jack C.R., Jr., Bennett D.A., Blennow K., Carrillo M.C., Dunn B., Haeberlein S.B., Holtzman D.M., Jagust W., Jessen F., Karlawish J., et al. NIA-AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535–562. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Medrano-Jimenez E., Jimenez-Ferrer Carrillo I., Pedraza-Escalona M., Ramirez-Serrano C.E., Alvarez-Arellano L., Cortes-Mendoza J., Herrera-Ruiz M., Jimenez-Ferrer E., Zamilpa A., Tortoriello J., et al. Malva parviflora extract ameliorates the deleterious effects of a high fat diet on the cognitive deficit in a mouse model of Alzheimer's disease by restoring microglial function via a PPAR-gamma-dependent mechanism. J. Neuroinflammation. 2019;16:143. doi: 10.1186/s12974-019-1515-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sweeney M.D., Sagare A.P., Zlokovic B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018;14:133–150. doi: 10.1038/nrneurol.2017.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kisler K., Nelson A.R., Montagne A., Zlokovic B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017;18:419–434. doi: 10.1038/nrn.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iadecola C., Duering M., Hachinski V., Joutel A., Pendlebury S.T., Schneider J.A., Dichgans M. Vascular cognitive impairment and dementia: JACC scientific expert panel. J. Am. Coll. Cardiol. 2019;73:3326–3344. doi: 10.1016/j.jacc.2019.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halliday M.R., Rege S.V., Ma Q., Zhao Z., Miller C.A., Winkler E.A., Zlokovic B.V. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer's disease. J. Cereb. Blood Flow Metab. 2016;36:216–227. doi: 10.1038/jcbfm.2015.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma Q., Zhao Z., Sagare A.P., Wu Y., Wang M., Owens N.C., Verghese P.B., Herz J., Holtzman D.M., Zlokovic B.V. Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-beta42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol. Neurodegener. 2018;13:57. doi: 10.1186/s13024-018-0286-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tachibana M., Yamazaki Y., Liu C.C., Bu G., Kanekiyo T. Pericyte implantation in the brain enhances cerebral blood flow and reduces amyloid-beta pathology in amyloid model mice. Exp. Neurol. 2018;300:13–21. doi: 10.1016/j.expneurol.2017.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown L.S., Foster C.G., Courtney J.M., King N.E., Howells D.W., Sutherland B.A. Pericytes and neurovascular function in the healthy and diseased brain. Front. Cell Neurosci. 2019;13:282. doi: 10.3389/fncel.2019.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu Q., Yuan X., Bai J., Han R., Li Z., Zhang H., Xiu R. MicroRNA-181a protects against pericyte apoptosis via directly targeting FOXO1: implication for ameliorated cognitive deficits in APP/PS1 mice. Aging (Albany NY) 2019;11:6120–6133. doi: 10.18632/aging.102171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sengillo J.D., Winkler E.A., Walker C.T., Sullivan J.S., Johnson M., Zlokovic B.V. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer's disease. Brain Pathol. 2013;23:303–310. doi: 10.1111/bpa.12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Armulik A., Genove G., Mae M., Nisancioglu M.H., Wallgard E., Niaudet C., He L., Norlin J., Lindblom P., Strittmatter K., et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- 13.Shi H., Koronyo Y., Fuchs D.T., Sheyn J., Wawrowsky K., Lahiri S., Black K.L., Koronyo-Hamaoui M. Retinal capillary degeneration and blood-retinal barrier disruption in murine models of Alzheimer's disease. Acta Neuropathol. Commun. 2020;8:202. doi: 10.1186/s40478-020-01076-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi H., Koronyo Y., Rentsendorj A., Regis G.C., Sheyn J., Fuchs D.T., Kramerov A.A., Ljubimov A.V., Dumitrascu O.M., Rodriguez A.R., et al. Identification of early pericyte loss and vascular amyloidosis in Alzheimer's disease retina. Acta Neuropathol. 2020;139:813–836. doi: 10.1007/s00401-020-02134-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sagare A.P., Bell R.D., Zhao Z., Ma Q., Winkler E.A., Ramanathan A., Zlokovic B.V. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 2013;4:2932. doi: 10.1038/ncomms3932. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Li P., Zhou Y., Goodwin A.J., Cook J.A., Halushka P.V., Zhang X.K., Wilson C.L., Schnapp L.M., Zingarelli B., Fan H. Fli-1 governs pericyte dysfunction in a murine model of sepsis. J. Infect. Dis. 2018;218:1995–2005. doi: 10.1093/infdis/jiy451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sohn E.J., Li H., Reidy K., Beers L.F., Christensen B.L., Lee S.B. EWS/FLI1 oncogene activates caspase 3 transcription and triggers apoptosis in vivo. Cancer Res. 2010;70:1154–1163. doi: 10.1158/0008-5472.CAN-09-1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li P., Goodwin A.J., Cook J.A., Halushka P.V., Zhang X.K., Fan H. Fli-1 transcription factor regulates the expression of caspase-1 in lung pericytes. Mol. Immunol. 2019;108:1–7. doi: 10.1016/j.molimm.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takahashi T., Asano Y., Sugawara K., Yamashita T., Nakamura K., Saigusa R., Ichimura Y., Toyama T., Taniguchi T., Akamata K., et al. Epithelial Fli1 deficiency drives systemic autoimmunity and fibrosis: possible roles in scleroderma. J. Exp. Med. 2017;214:1129–1151. doi: 10.1084/jem.20160247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akamata K., Asano Y., Yamashita T., Noda S., Taniguchi T., Takahashi T., Ichimura Y., Toyama T., Trojanowska M., Sato S. Endothelin receptor blockade ameliorates vascular fragility in endothelial cell-specific Fli-1-knockout mice by increasing Fli-1 DNA binding ability. Arthritis Rheumatol. 2015;67:1335–1344. doi: 10.1002/art.39062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Theisen E.R., Pishas K.I., Saund R.S., Lessnick S.L. Therapeutic opportunities in Ewing sarcoma: EWS-FLI inhibition via LSD1 targeting. Oncotarget. 2016;7:17616–17630. doi: 10.18632/oncotarget.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sato S., Lennard Richard M., Brandon D., Jones Buie J.N., Oates J.C., Gilkeson G.S., Zhang X.K. A critical role of the transcription factor fli-1 in murine lupus development by regulation of interleukin-6 expression. Arthritis Rheumatol. 2014;66:3436–3444. doi: 10.1002/art.38818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cai Z., Hussain M.D., Yan L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer's disease. Int. J. Neurosci. 2014;124:307–321. doi: 10.3109/00207454.2013.833510. [DOI] [PubMed] [Google Scholar]
- 24.Pillai J.A., Maxwell S., Bena J., Bekris L.M., Rao S.M., Chance M., Lamb B.T., Leverenz J.B., Alzheimer's Disease Neuroimaging Initiative Key inflammatory pathway activations in the MCI stage of Alzheimer's disease. Ann. Clin. Transl. Neurol. 2019;6:1248–1262. doi: 10.1002/acn3.50827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ho H.H., Ivashkiv L.B. Downregulation of Friend leukemia virus integration 1 as a feedback mechanism that restrains lipopolysaccharide induction of matrix metalloproteases and interleukin-10 in human macrophages. J. Interferon Cytokine Res. 2010;30:893–900. doi: 10.1089/jir.2010.0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kantarci A., Aytan N., Palaska I., Stephens D., Crabtree L., Benincasa C., Jenkins B.G., Carreras I., Dedeoglu A. Combined administration of resolvin E1 and lipoxin A4 resolves inflammation in a murine model of Alzheimer's disease. Exp. Neurol. 2018;300:111–120. doi: 10.1016/j.expneurol.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 27.Maiti P., Bowers Z., Bourcier-Schultz A., Morse J., Dunbar G.L. Preservation of dendritic spine morphology and postsynaptic signaling markers after treatment with solid lipid curcumin particles in the 5xFAD mouse model of Alzheimer's amyloidosis. Alzheimers Res. Ther. 2021;13:37. doi: 10.1186/s13195-021-00769-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Jawhar S., Trawicka A., Jenneckens C., Bayer T.A., Wirths O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Abeta aggregation in the 5XFAD mouse model of Alzheimer's disease. Neurobiol. Aging. 2012;33:196 e9–140. doi: 10.1016/j.neurobiolaging.2010.05.027. [DOI] [PubMed] [Google Scholar]
- 29.Lou N., Lennard Richard M.L., Yu J., Kindy M., Zhang X.K. The Fli-1 transcription factor is a critical regulator for controlling the expression of chemokine C-X-C motif ligand 2 (CXCL2) Mol. Immunol. 2017;81:59–66. doi: 10.1016/j.molimm.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 30.Taniguchi T., Asano Y., Akamata K., Noda S., Takahashi T., Ichimura Y., Toyama T., Trojanowska M., Sato S. Fibrosis, vascular activation, and immune abnormalities resembling systemic sclerosis in bleomycin-treated Fli-1-haploinsufficient mice. Arthritis Rheumatol. 2015;67:517–526. doi: 10.1002/art.38948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kubo M., Czuwara-Ladykowska J., Moussa O., Markiewicz M., Smith E., Silver R.M., Jablonska S., Blaszczyk M., Watson D.K., Trojanowska M. Persistent down-regulation of Fli1, a suppressor of collagen transcription, in fibrotic scleroderma skin. Am. J. Pathol. 2003;163:571–581. doi: 10.1016/S0002-9440(10)63685-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zampeli E., Klinman D.M., Gershwin M.E., Moutsopoulos H.M. A comprehensive evaluation for the treatment of lupus nephritis. J. Autoimmun. 2017;78:1–10. doi: 10.1016/j.jaut.2016.12.011. [DOI] [PubMed] [Google Scholar]
- 33.Parikh S.V., Almaani S., Brodsky S., Rovin B.H. Update on lupus nephritis: core curriculum 2020. Am. J. Kidney Dis. 2020;76:265–281. doi: 10.1053/j.ajkd.2019.10.017. [DOI] [PubMed] [Google Scholar]
- 34.Elzi D.J., Song M., Houghton P.J., Chen Y., Shiio Y. The role of FLI-1-EWS, a fusion gene reciprocal to EWS-FLI-1, in Ewing sarcoma. Genes Cancer. 2015;6:452–461. doi: 10.18632/genesandcancer.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou M., Xu R., Kaelber D.C., Gurney M.E. Tumor Necrosis Factor (TNF) blocking agents are associated with lower risk for Alzheimer's disease in patients with rheumatoid arthritis and psoriasis. PLoS One. 2020;15:e0229819. doi: 10.1371/journal.pone.0229819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Decourt B., Lahiri D.K., Sabbagh M.N. Targeting tumor necrosis factor Alpha for alzheimer's disease. Curr. Alzheimer Res. 2017;14:412–425. doi: 10.2174/1567205013666160930110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lyra E.S.N.M., Goncalves R.A., Pascoal T.A., Lima-Filho R.A.S., Resende E.P.F., Vieira E.L.M., Teixeira A.L., de Souza L.C., Peny J.A., Fortuna J.T.S., et al. Pro-inflammatory interleukin-6 signaling links cognitive impairments and peripheral metabolic alterations in Alzheimer's disease. Transl. Psychiatry. 2021;11:251. doi: 10.1038/s41398-021-01349-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X., Oates J.C., Helke K.L., Gilkeson G.S., Zhang X.K. Camptothecin and topotecan, inhibitors of transcription factor fli-1 and topoisomerase, markedly ameliorate lupus nephritis in (NZB x NZW)F1 mice and reduce the production of inflammatory mediators in human renal cells. Arthritis Rheumatol. 2021;73:1478–1488. doi: 10.1002/art.41685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilhelmus M.M., Otte-Holler I., van Triel J.J., Veerhuis R., Maat-Schieman M.L., Bu G., de Waal R.M., Verbeek M.M. Lipoprotein receptor-related protein-1 mediates amyloid-beta-mediated cell death of cerebrovascular cells. Am. J. Pathol. 2007;171:1989–1999. doi: 10.2353/ajpath.2007.070050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toyama T., Asano Y., Miyagawa T., Nakamura K., Hirabayashi M., Yamashita T., Saigusa R., Miura S., Ichimura Y., Takahashi T., et al. The impact of transcription factor Fli1 deficiency on the regulation of angiogenesis. Exp. Dermatol. 2017;26:912–918. doi: 10.1111/exd.13341. [DOI] [PubMed] [Google Scholar]
- 41.Nation D.A., Sweeney M.D., Montagne A., Sagare A.P., D'Orazio L.M., Pachicano M., Sepehrband F., Nelson A.R., Buennagel D.P., Harrington M.G., et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019;25:270–276. doi: 10.1038/s41591-018-0297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leissring M.A. Abeta-degrading proteases: therapeutic potential in alzheimer disease. CNS Drugs. 2016;30:667–675. doi: 10.1007/s40263-016-0364-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mawuenyega K.G., Sigurdson W., Ovod V., Munsell L., Kasten T., Morris J.C., Yarasheski K.E., Bateman R.J. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sweeney M.D., Ayyadurai S., Zlokovic B.V. Pericytes of the neurovascular unit: key functions and signaling pathways. Nat. Neurosci. 2016;19:771–783. doi: 10.1038/nn.4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winkler E.A., Sagare A.P., Zlokovic B.V. The pericyte: a forgotten cell type with important implications for Alzheimer's disease? Brain Pathol. 2014;24:371–386. doi: 10.1111/bpa.12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hart A., Melet F., Grossfeld P., Chien K., Jones C., Tunnacliffe A., Favier R., Bernstein A. Fli-1 is required for murine vascular and megakaryocytic development and is hemizygously deleted in patients with thrombocytopenia. Immunity. 2000;13:167–177. doi: 10.1016/s1074-7613(00)00017-0. [DOI] [PubMed] [Google Scholar]
- 47.Asano Y., Stawski L., Hant F., Highland K., Silver R., Szalai G., Watson D.K., Trojanowska M. Endothelial Fli1 deficiency impairs vascular homeostasis: a role in scleroderma vasculopathy. Am. J. Pathol. 2010;176:1983–1998. doi: 10.2353/ajpath.2010.090593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang L., Eddy A., Teng Y.T., Fritzler M., Kluppel M., Melet F., Bernstein A. An immunological renal disease in transgenic mice that overexpress Fli-1, a member of the ets family of transcription factor genes. Mol. Cell Biol. 1995;15:6961–6970. doi: 10.1128/mcb.15.12.6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Butler A.A., Johnston D.R., Kaur S., Lubin F.D. Long noncoding RNA NEAT1 mediates neuronal histone methylation and age-related memory impairment. Sci. Signal. 2019;12:eaaw9277. doi: 10.1126/scisignal.aaw9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watson L.S., Stone T.D., Williams D., Williams A.S., Sims-Robinson C. High-Fat diet impairs tactile discrimination memory in the mouse. Behav. Brain Res. 2020;382:112454. doi: 10.1016/j.bbr.2019.112454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stover K.R., Campbell M.A., Van Winssen C.M., Brown R.E. Early detection of cognitive deficits in the 3xTg-AD mouse model of Alzheimer's disease. Behav. Brain Res. 2015;289:29–38. doi: 10.1016/j.bbr.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 52.Min L.J., Kobayashi Y., Mogi M., Tsukuda K., Yamada A., Yamauchi K., Abe F., Iwanami J., Xiao J.Z., Horiuchi M. Administration of bovine casein-derived peptide prevents cognitive decline in Alzheimer disease model mice. PLoS One. 2017;12:e0171515. doi: 10.1371/journal.pone.0171515. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.