

Abstract
Exercise and its regulated molecules have myocardial protective effects against cardiac ischemia/reperfusion (I/R) injury. The muscle-enriched miR-486 was previously identified to be upregulated in the exercised heart, which prompted us to investigate the functional roles of miR-486 in cardiac I/R injury and to further explore its potential in contributing to exercise-induced protection against I/R injury. Our data showed that miR-486 was significantly downregulated in the heart upon cardiac I/R injury. Both preventive and therapeutic interventions of adeno-associated virus 9 (AAV9)-mediated miR-486 overexpression could reduce cardiac I/R injury. Using AAV9 expressing miR-486 with a cTnT promoter, we further demonstrated that cardiac muscle cell-targeted miR-486 overexpression was also sufficient to protect against cardiac I/R injury. Consistently, miR-486 was downregulated in oxygen-glucose deprivation/reperfusion (OGDR)-stressed cardiomyocytes, while upregulating miR-486 inhibited cardiomyocyte apoptosis through PTEN and FoxO1 inhibition and AKT/mTOR activation. Finally, we observed that miR-486 was necessary for exercise-induced protection against cardiac I/R injury. In conclusion, miR-486 is protective against cardiac I/R injury and myocardial apoptosis through targeting of PTEN and FoxO1 and activation of the AKT/mTOR pathway, and mediates the beneficial effect of exercise for myocardial protection. Increasing miR-486 might be a promising therapeutic strategy for myocardial protection.
Keywords: miR-486, cardiac ischemia/reperfusion injury, cardiomyocyte, apoptosis, exercise
Graphical abstract
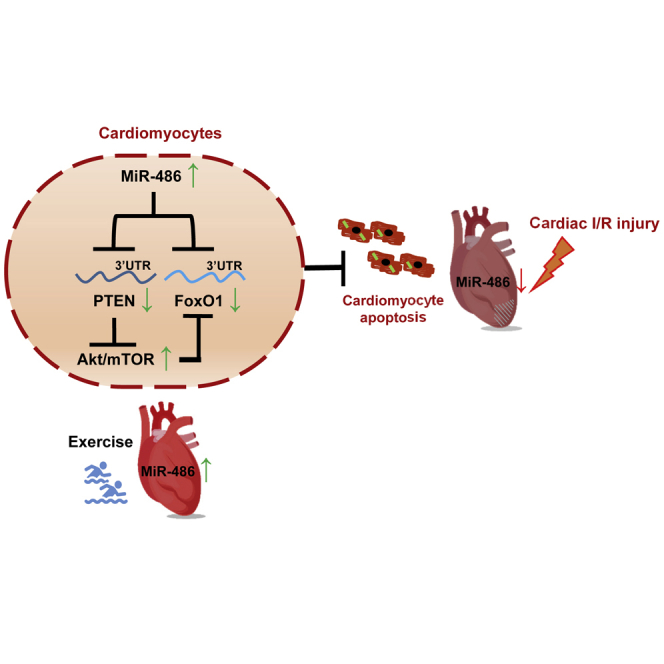
MiR-486 attenuates cardiomyocyte apoptosis and cardiac ischemia/reperfusion (I/R) injury through targeting PTEN and FoxO1 and activating Akt/mTOR pathway, and mediates the beneficial effect of exercise for myocardial protection.
miR-486 is protective against cardiac ischemia/reperfusion (I/R) injury and myocardial apoptosis through targeting of PTEN and FoxO1 and activation of the AKT/mTOR pathway, and mediates the beneficial effect of exercise for myocardial protection. Increasing miR-486 might be a promising therapeutic strategy for myocardial protection.
Introduction
Timely and effective cardiac reperfusion is the key strategy to limit infarct size after acute myocardial infarction (AMI).1 The mortality from AMI has decreased during past decades where interventional reperfusion is available; however, reperfusion per se will inevitably cause further injury to the myocardium, which may develop cardiac remodeling and eventually heart failure (HF).2,3 Effective strategies to reduce cardiac ischemia/reperfusion (I/R) injury are still lacking.4
MicroRNAs (miRNAs, miRs) are a large class of small non-coding RNAs regulating target genes at the post-transcriptional level, and they play important roles in cardiovascular physiology and disease.5 miRNA-486-5p (miR-486) is a skeletal and cardiac muscle-enriched miRNA encoded by the muscle-specific gene Ankyrin, which was found to be decreased in atrophied skeletal muscle but increased during postnatal cardiac growth.6 Increasing evidence indicates that cardiac-enriched miRNAs, such as miR-1, miR-133a, miR-208a/b, and miR-499, are critically involved in cardiogenesis and cardiac function.7 In the cardiovascular system, miR-486 was previously shown to prevent hydrogen peroxide (H2O2)-induced and coronary microembolization-induced cardiomyocyte apoptosis,8,9 and to mediate the protective effect of circulating extracellular vesicles against cardiomyocyte apoptosis.10 However, the functional roles and regulatory mechanisms of miR-486 in cardiac I/R injury remain to be elucidated.
Exercise is an effective and economical way to prevent and treat cardiovascular diseases.11,12 In experimental cardiac I/R injury models, chronic exercise training was proved to be effective at reducing infarct size.13 Interestingly, exercise-regulated molecules also have the potential to exert myocardial protective effects.14, 15, 16, 17 miR-486 was previously found to be upregulated in the heart after exercise training.18 However, the relationship between exercise and miR-486, and whether miR-486 could contribute to exercise-induced protection against cardiac I/R injury, was largely unclear.
In the present study, we investigated the functional roles of miR-486 in murine models of cardiac I/R injury and an oxygen-glucose deprivation/reperfusion (OGDR)-induced apoptosis model of cardiomyocytes. We further identified target genes and molecular mechanisms underlying the myocardial protective effect of miR-486. Also, we explored the potential regulatory effect of exercise on miR-486 and the role of miR-486 in exercise-induced protection against cardiac I/R injury.
Results
miR-486 overexpression prevents cardiac I/R injury and cardiac dysfunction
To study the functional roles of miR-486 in regulating cardiac I/R injury, we first examined the change in miR-486 expression after cardiac I/R injury. A marked downregulation of miR-486 was observed in heart tissues from a murine model of cardiac I/R injury in vivo (Figure 1A). To further investigate the functional roles of miR-486 in vivo, mice injected with adeno-associated virus 9 (AAV9) expressing miR-486 (miR-486-AAV9) were subjected to acute cardiac I/R injury (Figure S1A). miR-486 was significantly upregulated in mouse heart tissues after miR-486-AAV9 injections (Figure S1B). At 24 h after cardiac I/R injury, mice with AAV9-mediated miR-486 overexpression in the heart had significantly reduced infarct size compared with those injected with AAV9 controls (CTL-AAV9) (Figure 1B). Moreover, miR-486 significantly reduced TUNEL-positive cardiomyocytes (Figure 1C), decreased Bax/Bcl-2 ratio and caspase-3 cleavage (Figure S1C), and downregulated CTGF, Col1a1, Col3a1, and BNP mRNA levels (Figure S1D) in mouse hearts upon acute I/R injury. These data demonstrate markedly protective effects of miR-486 against acute cardiac I/R injury in vivo.
Figure 1.

AAV9-mediated miR-486 overexpression prevents cardiac ischemia/reperfusion injury
(A) RT-PCR for miR-486 expression in heart tissues from mice at 24 h after cardiac ischemia/reperfusion (I/R) injury compared with sham controls (n = 5–6). (B) The 2,3,5-triphenyltetrazolium chloride (TTC) staining for the infarct size at 24 h after cardiac I/R injury as determined by the infarct-size/area-at-risk (INF/AAR) ratio. The area-at-risk/left-ventricle-weight (AAR/LV) ratio represents the homogeneity of surgery (n = 7–8). (C) TUNEL staining for myocardial apoptosis in α-actinin-labeled cardiomyocytes (n = 6). Scale bar, 100 μm. (D) Echocardiography for left-ventricular ejection fraction (EF, %) and fractional shortening (FS, %) in mice at 3 weeks after I/R injury (n = 9–10). (E) Masson trichrome staining for cardiac fibrosis in mouse heart tissues (n = 5 for sham groups, n = 8–9 for I/R injury [IRI] groups). Scale bar, 100 μm. (F) RT-PCR for CTGF, Col1a1, Col3a1, and BNP expression in mouse heart tissues (n = 9–10). Data between two groups were compared by unpaired two-tailed Student's t test. Data among four groups were compared by two-way ANOVA followed by Tukey’s post hoc test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To further investigate whether miR-486 overexpression could protect the heart in the long term, we injected mice with miR-486-AAV9 and then subjected them to cardiac I/R injury for 3 weeks (Figures S2A and S2B). At 3 weeks post I/R injury, mice injected with miR-486-AAV9 had well-preserved cardiac function compared with the CTL-AAV9-injected I/R group (Figure 1D). Meanwhile, miR-486 overexpression reduced the Bax/Bcl-2 ratio and caspase-3 cleavage (Figure S2C), and also reduced cardiac fibrotic areas (Figure 1E) and downregulated CTGF, Col1a1, Col3a1, and BNP expression (Figure 1F) in mouse hearts after cardiac I/R injury. During a 6-week long-term functional experiment of miR-486 in vivo, we observed that only three mice died post I/R injury in the CTL-AAV9 + I/R injury (IRI) group, while no mice died in the sham groups or in the I/R group injected with miR-486-AAV9, indicating a low mortality due to the I/R surgery in our study. Although no difference was found in the survival rate among different groups (Figure S3A), the beneficial effect of miR-486 was still obvious in attenuated cardiac remodeling and cardiac dysfunction at 6 weeks after I/R injury (Figures S3B–S3E).
In contrast, we constructed an miR-486 sponge AAV9 to examine the functional role of inhibiting miR-486 in cardiac I/R injury. We first observed that inhibiting miR-486 did not further increase the infarct size at 24 h after I/R injury (Figure S4A). Next, miR-486 sponge AAV9 was injected via tail vein at 1 week before cardiac I/R injury, and heart tissues were harvested at 3 weeks post I/R injury (Figure S4B). We demonstrated that miR-486 sponge AAV9 significantly reduced miR-486 expression levels in heart tissues (Figure S4C). Meanwhile, luciferase reporter assay verified that miR-486 sponge was able to target and bind miR-486 directly, but had no binding activity with other miRNAs such as miR-210 (Figure S4D). However, inhibiting miR-486 was not able to further aggravate cardiac I/R injury and cardiac dysfunction at 3 weeks post I/R injury (Figures S4E–S4H).
Taken together, these data indicate that miR-486 overexpression is sufficient to prevent myocardial apoptosis, cardiac remodeling, and cardiac dysfunction after I/R injury
Therapeutic delivery of miR-486-AAV9 attenuates cardiac I/R injury and cardiac dysfunction
To investigate further whether delivery of miR-486-AAV9 shortly after myocardial reperfusion still had a beneficial effect, we treated mice with miR-486-AAV9 through tail vein injection within 30 min of cardiac I/R injury (Figure 2A). At 3 weeks post I/R injury, an increase in miR-486 expression was confirmed in heart tissues from mice treated with miR-486-AAV9 (Figure 2B). Moreover, therapeutic delivery of miR-486-AAV9 had beneficial effects in attenuating cardiac dysfunction, cardiac fibrosis, and apoptosis in mice with cardiac I/R injury (Figures 2C–2F). These data demonstrate that therapeutic delivery of miR-486-AAV9 shortly after myocardial reperfusion still has protective effects against cardiac I/R injury and cardiac dysfunction.
Figure 2.

Therapeutic delivery of miR-486-AAV9 attenuates cardiac ischemia/reperfusion injury
(A) Schematic diagram showing that miR-486-AAV9 or CTL-AAV9 was injected via tail vein within 30 min of myocardial reperfusion, and echocardiography and tissue harvest were performed at 3 weeks post cardiac I/R injury. (B) RT-PCR for miR-486 expression in mouse heart tissues at 3 weeks post cardiac I/R injury (n = 6). (C) Echocardiography for left-ventricular ejection fraction (EF, %) and fractional shortening (FS, %) in mice at 3 weeks after I/R injury (n = 8–10). (D) Masson trichrome staining for cardiac fibrosis in mouse heart tissues (n = 7–9). Scale bar, 100 μm. (E) Western blot for Bax/Bcl-2 ratio and cleaved-caspase-3/caspase-3 ratio in mouse heart tissues (n = 6). (F) RT-PCR for CTGF, Col1a1, Col3a1, and BNP expression in mouse heart tissues (n = 6). Data were compared by two-way ANOVA followed by Tukey’s post hoc test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Increasing miR-486 inhibits cardiomyocyte apoptosis
Next, we isolated neonatal rat cardiomyocytes (NRCMs) and determined the functional role of miR-486 in cardiomyocytes in vitro. Consistent with reduced miR-486 expression in cardiac I/R injury, miR-486 was also found to be downregulated in an OGDR-induced apoptosis model of NRCMs in vitro (Figure 3A). After confirming that transfection of miR-486 mimic or inhibitor could efficiently regulate miR-486 expression in NRCMs (Figure S5A), we further examined OGDR-induced cardiomyocyte apoptosis using TUNEL staining and western blot for apoptosis-associated proteins. Our data showed that miR-486 mimic significantly reduced TUNEL-positive cardiomyocytes, while miR-486 inhibitor increased them (Figure 3B). Meanwhile, OGDR treatment caused elevated Bax/Bcl-2 ratio and cleaved-caspase-3/caspase-3 ratio at the protein level, which could be significantly reversed by miR-486 mimic (Figure 3C). On the other hand, miR-486 inhibitor transfection further increased Bax/Bcl-2 ratio and caspase-3 cleavage in OGDR-treated NRCMs (Figure 3D). These data provide direct evidence that increasing miR-486 exerts anti-apoptotic effects in cardiomyocytes.
Figure 3.

Overexpression of miR-486 inhibits cardiomyocyte apoptosis
(A) RT-PCR for miR-486 expression in primary cultured neonatal rat cardiomyocytes (NRCMs) treated with oxygen-glucose deprivation/reperfusion (OGDR) (n = 4). (B) TUNEL staining for α-actinin-labeled NRCMs transfected with miR-486 mimic, inhibitor, or negative control (NC) under OGDR stress (n = 4). Scale bar, 100 μm. (C and D) Western blot for Bax/Bcl-2 ratio and cleaved-caspase-3/caspase-3 ratio in OGDR-treated NRCMs transfected with miR-486 mimic (C), inhibitor (D), or negative controls (n = 6). (E) RT-PCR for miR-486 expression in human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) treated with OGDR (n = 3). (F) TUNEL staining for cTnT-labeled human cardiomyocytes treated with OGDR (n = 3 technique repeats). Scale bar, 100 μm. Data between two groups were compared by unpaired two-tailed Student's t test. Data among three groups were compared by one-way ANOVA. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To further test the potential role of miR-486 in human cardiomyocytes, we performed OGDR stress on human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs). The purity of the hiPSC-CMs was confirmed by cardiac troponin T (cTnT) immunofluorescence staining (Figure S5B). In line with downregulated expression in OGDR-treated NRCMs, miR-486 was also decreased in hiPSC-CMs with OGDR stress compared with controls (Figure 3E). Further, we efficiently transfected an miR-486 mimic into hiPSC-CMs (Figure S5C), and explored whether miR-486 overexpression could inhibit OGDR-induced apoptosis in human cardiomyocytes. Although the hiPSC-CM numbers were not significantly changed after OGDR stress (Figure S5D), OGDR increased the ratio of apoptotic hiPSC-CMs as demonstrated by TUNEL assay (Figure 3F). Interestingly, hiPSC-CMs overexpressing miR-486 showed clearly lower levels of apoptosis under OGDR stress compared with negative controls (Figure 3F), suggesting that myocardial protection by miR-486 is also conserved in human cardiomyocytes.
miR-486 targets PTEN and FoxO1 in cardiomyocytes
To identify the target genes of miR-486, we applied bioinformatic analysis using miRDB, Target Scan, and miRTarBase algorithms, and further subjected the predicted target genes of miR-486 in the Enrichr database. Among the common predicted target genes, PTEN and FoxO1 were critically involved in the pathways sorted by KEGG (Figures 4A and 4B). Although PTEN and FoxO1 were previously reported as target genes of miR-486 in various cancer cells and also in the heart,6,19 whether they mediate the roles of miR-486 in myocardial apoptosis and I/R injury remained unclear. To confirm whether miR-486 directly targets PTEN and FoxO1, we constructed luciferase reporter plasmids containing 3′ untranslated region (3′ UTR) binding sequences or mutated sequences of PTEN and FoxO1. Co-transfection with miR-486 mimic and plasmids containing PTEN or FoxO1 3′ UTR binding sequences significantly decreased luciferase activity in 293T cells, while the reduction of luciferase activity was not present when the 3′ UTR binding site was mutated, indicating that miR-486 could directly target the 3′ UTR of PTEN or FoxO1 (Figures 4C and 4D). In NRCMs, we further observed that miR-486 mimic was effective in downregulating PTEN and FoxO1 at both mRNA and protein levels, while miR-486 inhibitor upregulated PTEN and FoxO1 expression (Figures 4E–4H). Meanwhile, we observed a trend toward downregulation of PTEN and a statistically significant downregulation of FoxO1 at the mRNA level in hiPSC-CMs transfected with miR-486 mimic (Figure S6). These results prove that miR-486 directly targets and negatively regulates PTEN and FoxO1 in cardiomyocytes.
Figure 4.

miR-486 targets PTEN and FoxO1 in cardiomyocytes
(A and B) Bioinformatic analysis using miRDB, Target Scan, and miRTarBase algorithms (A) and KEGG pathway analysis (B). (C and D) Luciferase reporter assays performed in 293T cells transfected with miR-486 mimic (or negative control, NC mimic) and luciferase reporter plasmids containing the binding sitein the 3′ UTR of PTEN (C, PTEN 3'UTR) or FoxO1 (D, FoxO1 3'UTR) or the mutated binding site in the 3'UTR of PTEN (C, PTEN 3'UTR mut) or FoxO1 (D, FoxO1 3'UTR mut) (n = 6). (E and F) RT-PCR for PTEN and FoxO1 expression in primary cultured neonatal rat cardiomyocytes (NRCMs) transfected with miR-486 mimic (E), inhibitor (F), or NC (n = 6). (G and H) Western blot for PTEN and FoxO1 expression in NRCMs transfected with miR-486 mimic (G), inhibitor (H), or NC (n = 6). Data between two groups were compared by unpaired two-tailed Student's t test. Data among four groups were compared by two-way ANOVA followed by Tukey’s post hoc test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
miR-486 inhibits cardiomyocyte apoptosis by targeting PTEN and FoxO1
To further determine whether PTEN and FoxO1 mediate the functional role of miR-486 in cardiomyocytes, we performed co-transfection of miR-486 inhibitor with small interfering RNA (siRNA) targeting PTEN or FoxO1. We first confirmed that PTEN or FoxO1 siRNA transfection could significantly downregulate their expression in NRCMs (Figure S7). In NRCMs co-transfected with miR-486 inhibitor and PTEN siRNA, we observed that silencing PTEN was able to abolish the pro-apoptotic effect of miR-486 inhibitor in OGDR-treated cardiomyocytes as determined by TUNEL staining (Figure 5A) and western blot (Figure S8). Similarly, siRNA targeting FoxO1 also attenuated the deleterious effect of miR-486 inhibitor on cardiomyocyte apoptosis (Figures 5B and S9). Overexpressing PTEN or FoxO1 abolished the beneficial effect of miR-486 mimic in reducing cardiomyocyte apoptosis (Figure S10). Interestingly, both preventive and therapeutic delivery of miR-486-AAV9 were able to reduce PTEN and FoxO1 expression in mouse hearts both in the sham and in the I/R groups (Figures S11A, S11B, 5C, and 5D). These experiments indicate that PTEN and FoxO1 are critical target genes of miR-486 in regulating cardiomyocyte apoptosis, and provide in vivo evidence that miR-486 could modulate PTEN and FoxO1 in heart tissues under both normal and I/R injury conditions.
Figure 5.

miR-486 inhibits cardiomyocyte apoptosis via targeting of PTEN and FoxO1
(A and B) TUNEL staining for α-actinin-labeled neonatal rat cardiomyocytes (NRCMs) transfected with miR-486 inhibitor and siRNA targeting PTEN (A) or FoxO1 (B) in the condition of oxygen glucose deprivation/reperfusion (OGDR) treatment (n = 4). Scale bar, 100 μm. (C and D) RT-PCR (C) and western blot (D) for PTEN and FoxO1 expression in heart tissues from mice treated with miR-486-AAV9 within 30 min of myocardial reperfusion. Heart tissues were harvested at 3 weeks post cardiac ischemia/reperfusion (I/R) injury (n = 6). Data were compared by two-way ANOVA followed by Tukey’s post hoc test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Anti-apoptotic effect of miR-486 is associated with AKT/mTOR activation
PTEN is well known for its inactivation effect on AKT signaling, which regulates cell growth and survival.20 As PTEN functions as a target gene of miR-486 in cardiomyocytes, we further determined whether miR-486 could regulate AKT phosphorylation and subsequent mTOR activation. In NRCMs transfected with miR-486 mimic or inhibitor, we observed that miR-486 overexpression significantly increased AKT phosphorylation and induced mTOR and P70S6K activation (Figure 6A), while miR-486 inhibitor dampened the phosphorylation levels of the AKT/mTOR/P70S6K pathway (Figure 6B). Next, we co-treated NRCMs with miR-486 mimic and AKT inhibitor (MK2206) or mTOR inhibitor (rapamycin) in the condition of OGDR. As shown by TUNEL staining and western blot, AKT or mTOR inhibition was sufficient to attenuate the protective effect of miR-486 mimic against cardiomyocyte apoptosis (Figures 6C, 6D, and S12). Moreover, AAV9-mediated miR-486 overexpression was also sufficient to increase Akt phosphorylation levels in heart tissues suffering from I/R injury (Figures 6E and S11C). Collectively, these results highly suggest that miR-486 may target PTEN and subsequently activate AKT/mTOR pathways to protect against myocardial apoptosis.
Figure 6.

miR-486 inhibits cardiomyocyte apoptosis by activating the AKT/mTOR pathway
(A and B) Western blot for phosphorylation levels of the AKT/mTOR/P70S6K pathway in NRCMs transfected with miR-486 mimic (A), inhibitor (B), or negative controls (NC) (n = 6). (C and D) TUNEL staining for α-actinin-labeled NRCMs treated with miR-486 mimic and AKT inhibitor MK2206 (C) (n = 4) or mTOR inhibitor rapamycin (D) (n = 4). Scale bar, 100 μm. (E) Western blot for AKT phosphorylation levels in heart tissues from mice treated with miR-486-AAV9 within 30 min of myocardial reperfusion. Heart tissues were harvested at 3 weeks post cardiac ischemia/reperfusion (I/R) injury (n = 6). Data between two groups were compared by unpaired two-tailed Student's t test. Data among four groups were compared by two-way ANOVA followed by Tukey’s post hoc test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Cardiomyocyte-specific overexpression of miR-486 alleviates cardiac I/R injury
miR-486 was previously reported to protect against tissue fibrosis, including myocardial interstitial fibrosis, through targeting of Smad1 and/or Smad2.21 Interestingly, using isolated neonatal rat cardiac fibroblasts (NRCFs), we found that miR-486 mimic also inhibited NRCF proliferation and differentiation into myofibroblasts under stimulation of transforming growth factor β (TGF-β), while miR-486 inhibitor enhanced both NRCF proliferation and their differentiation into myofibroblasts regardless of TGF-β stimulation (Figure S13). Meanwhile, we demonstrated that miR-486 negatively regulated Smad1 and Smad2 at both mRNA and protein levels in NRCFs (Figure S14). These data indicate that miR-486 also has inhibitory effects on cardiac fibroblast proliferation and activation, which may contribute to the beneficial effect of AAV9-mediated miR-486 overexpression in heart tissues against cardiac I/R injury.
Under these circumstances, we further constructed AAV9 expressing miR-486 with a cardiac muscle cell-specific promoter, cTnT (cTnT-miR-486 AAV9), to investigate whether overexpression of miR-486 in cardiomyocytes could be sufficient to protect against cardiac I/R injury. The therapeutic delivery of cTnT-miR-486 AAV9 was performed within 30 min of cardiac I/R injury (Figure 7A), and miR-486 was significantly increased in heart tissues from mice treated with cTnT-miR-486 AAV9 (Figure 7B). Meanwhile, immunofluorescence imaging showed an obvious co-localization of ZsGreen (indicative of AAV9) and α-actinin staining in heart tissues from mice injected with cTnT-miR-486 AAV9 or cTnT-control vectors (Figure S15A), supporting an efficient delivery of AAV9 in cardiomyocytes in vivo. We further demonstrated that cTnT-miR-486 AAV9 was able to attenuate cardiac dysfunction and cardiac fibrosis, and downregulated apoptosis- and fibrosis-associated proteins or gene markers at 3 weeks post I/R injury (Figures 7C–7F). Moreover, cTnT-miR-486 AAV9 significantly downregulated PTEN and FoxO1 expression (Figures S15B and S15C) and activated AKT phosphorylation (Figure S15D) in heart tissues. These data suggest that in addition to miR-486 overexpression in the whole heart, cardiomyocyte-targeted miR-486 overexpression is also effective at alleviating cardiac I/R injury in association with its myocardial protection effect through targeting of PTEN and FoxO1 and activation of AKT.
Figure 7.

Cardiomyocyte-specific overexpression of miR-486 is a therapeutic to alleviate cardiac ischemia/reperfusion injury
(A) Schematic diagram showing that AAV9 expressing miR-486 with a cardiac muscle cell-specific promoter, cTnT (cTnT-miR-486 AAV9), or cTnT-CTL AAV9 was injected via the tail vein within 30 min of myocardial reperfusion, and echocardiography and tissue harvest were performed at 3 weeks post cardiac I/R injury. (B) RT-PCR for miR-486 expression in mouse heart tissues at 3 weeks post cardiac I/R injury (n = 7–10). (C) Echocardiography for left-ventricular ejection fraction (EF, %) and fractional shortening (FS, %) in mice at 3 weeks after I/R injury (n = 7–10). (D) Masson trichrome staining for cardiac fibrosis in mouse heart tissues (n = 5). Scale bar, 100 μm. (E) Western blot for Bax/Bcl-2 ratio and cleaved-caspase-3/caspase-3 ratio in mouse heart tissues (n = 6). (F) RT-PCR for CTGF, Col1a1, Col3a1, and BNP expression in mouse heart tissues (n = 7–10). Data were compared by two-way ANOVA followed by Tukey’s post hoc test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
miR-486 is necessary for exercise-induced myocardial protection
miR-486 was previously reported to be induced in heart tissues after exercise training.18 In the present study, we further examined the regulatory effect of exercise on miR-486 in a mouse model of regular swimming exercise. Here we observed that swimming exercise significantly induced miR-486 expression (Figure 8A), and also regulated known cardioprotective factors responsive to exercise, such as CITED4, miR-222, and lncRNA CPhar, in mouse heart tissues (Figure S16A). Specifically, miR-486 was increased in mouse isolated cardiomyocytes, but not in cardiac fibroblasts from swimming mouse hearts (Figure 8A). Meanwhile, we observed that miR-486 was upregulated in the serum samples as well as in the gastrocnemius and anterior tibialis from swimming mice (Figures S16B–S16D). These data suggest that a regular exercise training such as swimming is effective at inducing miR-486 expression, which prompted us to further investigate whether miR-486 is necessary to mediate the beneficial effect of exercise against cardiac I/R injury.
Figure 8.

miR-486 is necessary for exercise-induced myocardial protection
(A) RT-PCR for miR-486 expression in the mouse heart and isolated mouse cardiomyocytes (CMs) or cardiac fibroblasts (CFs) from swimming or sedentary mice (n = 5–6). (B) Schematic diagram showing that mice were injected with miR-486 sponge AAV9 and then subjected to 3 weeks of swimming exercise before cardiac ischemia/reperfusion (I/R) injury. (C) The 2,3,5-triphenyltetrazolium chloride (TTC) staining for infarct size at 24 h after cardiac I/R injury as determined by the infarct-size/area-at-risk (INF/AAR) ratio. The area-at-risk/left-ventricle-weight (AAR/LV) ratio represents the homogeneity of surgery (n = 6 for sedentary mice, n = 10 for swimming mice). (D) RT-PCR for miR-486 expression in mouse heart tissues at 1 week post cardiac I/R injury (n = 6 for sedentary mice, n = 11–12 for swimming mice). (E) Echocardiography for left-ventricular ejection fraction (EF, %) and fractional shortening (FS, %) in mice at 1 week after I/R injury (n = 6 for sedentary mice, n = 11–12 for swimming mice). (F) TUNEL staining for myocardial apoptosis in α-actinin-labeled cardiomyocytes (n = 6). Scale bar, 100 μm. (G) Western blot for Bax/Bcl-2 ratio, cleaved-caspase-3/caspase-3 ratio, PTEN, and FoxO1 in mouse heart tissues (n = 6). Data between two groups were compared by unpaired two-tailed Student's t test. Data among four groups were compared by two-way ANOVA followed by Tukey’s post hoc test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Mice were injected with miR-486 sponge AAV9 or CTL-AAV9 and then subjected to swimming exercise before cardiac I/R injury (Figure 8B). Our data showed a protective effect of exercise in reducing the infarct size upon acute I/R injury, which was, however, attenuated by miR-486 inhibition (Figure 8C). At 1 week after I/R injury, we confirmed that miR-486 sponge AAV9 significantly reduced exercise-induced miR-486 level in I/R hearts (Figure 8D). The protective effects of exercise against cardiac dysfunction, myocardial apoptosis, and cardiac remodeling were also abolished by miR-486 inhibition (Figures 8E–8G and S17A). Moreover, exercise training downregulated PTEN and FoxO1 in the heart, while miR-486 sponge AAV9 abolished these modulations (Figures 8G and S17B). Similarly, miR-486 knockout (KO) mice and wild-type (WT) littermates were subjected to swimming exercise before I/R injury (Figure S18A). We also observed that miR-486 KO swimming mice had significantly increased infarct size compared with WT swimming mice (Figure S18B). Collectively, these data suggest that miR-486 is necessary for exercise-induced myocardial protection upon I/R injury.
Discussion
Exercise and its regulated molecules have myocardial protective effects and can protect against cardiac I/R injury.22 miR-486, enriched in muscle, was previously identified to be upregulated in the heart after exercise training,18 which prompted us to investigate the functional roles of miR-486 in cardiac I/R injury and to explore further the potential of miR-486 contributing to exercise-induced protection against I/R injury.
In the present study, a consistent downregulation of miR-486 was observed in both cardiac I/R injury in vivo and OGDR-treated cardiomyocytes in vitro. Functionally, AAV9-mediated miR-486 overexpression significantly prevented cardiac I/R injury in vivo, as evidenced by reduced infarct size in the acute-phase and preserved cardiac function at long term. Noteworthy, therapeutic delivery of miR-486-AAV9 shortly after myocardial reperfusion still exerted obvious protective effects against cardiac I/R injury and cardiac dysfunction, suggesting a practicable therapeutic intervention by increasing miR-486 in the treatment of cardiac I/R injury. Moreover, to specifically elucidate the functional role of miR-486 in myocardial protection, we treated mice with AAV9 expressing miR-486 with a cardiac muscle cell-specific promoter, cTnT, and found that cardiac muscle cell-targeted overexpression of miR-486 was still sufficient to protect against cardiac I/R injury in vivo. Collectively, these data provide strong evidence indicating that therapeutic miR-486 overexpression has myocardial protective effects against cardiac I/R injury.
Cardiomyocyte apoptosis is a critical pathological feature of I/R injury, which may further contribute to the development of cardiac remodeling and heart failure.23 Our data further demonstrated that both preventive and therapeutic interventions of miR-486 overexpression in vivo could reduce myocardial apoptosis in I/R mice, and miR-486 also inhibited OGDR-induced apoptosis in primary cultured NRCMs in vitro. Previous studies reported that miR-486 reduced H2O2-induced apoptosis in H9C2 cells,8 and prevented microembolization-induced myocardial apoptosis in rats.9 miR-486 was also shown to mediate the protective effect of bone marrow stromal cell-derived exosomes and circulating extracellular vesicles against cardiac I/R injury.10,24 Here we provide direct evidence that miR-486 overexpression is an efficient way to reduce cardiac I/R injury and cardiomyocyte apoptosis, as well as improving cardiac function at long term. Furthermore, we demonstrate that miR-486 also exerts anti-apoptotic effects in human cardiomyocytes, highlighting its potential effect in human myocardial protection.
miRNAs are known to function by binding to the 3′ UTR of target genes, which leads to mRNA degradation and/or translation inhibition.25 Here we performed bioinformatic algorithms and identified that miR-486 directly interacted with the 3′ UTR of PTEN and FoxO1. PTEN was previously reported to be regulated by miR-486 in various cancer cell lines and also in the heart,9,19 and increasing evidence has indicated that PTEN inactivation, which is associated with reduced apoptosis, prevents cardiac I/R injury.26 However, whether PTEN mediates the role of miR-486 in myocardial apoptosis and I/R injury remained unclear. In addition, FoxO1 was identified as another target gene of miR-486,6,27 although little was known about the functional relationship between miR-486 and FoxO1 in the heart. In the present study, we first observed that miR-486 negatively regulated PTEN and FoxO1 at both mRNA and protein levels in cardiomyocytes, and miR-486 overexpression led to downregulated PTEN and FoxO1 in the heart. We further showed that silencing PTEN or FoxO1 was able to rescue the pro-apoptotic effect of miR-486 inhibitor in cardiomyocytes, while overexpressing downstream targets abolished the effect of miR-486 in reducing cardiomyocyte apoptosis. These data indicate that PTEN and FoxO1 are direct targets of miR-486 in the regulation of myocardial apoptosis and I/R injury.
AKT/mTOR signaling is essentially involved in the regulation of cell growth and survival.28 PTEN inactivation and subsequent activation of the AKT/mTOR pathway is a desirable means to promote cell survival while inhibiting apoptosis.29,30 In line with the downregulation of PTEN, our data demonstrated that miR-486 overexpression led to an activation of the AKT/mTOR/P70S6K pathway in cardiomyocytes. On the other hand, the AKT/mTOR/P70S6K phosphorylation levels were significantly reduced in cardiomyocytes with miR-486 inhibition. Function rescue experiments further indicated that AKT and mTOR activation is necessary to mediate the protective effect of miR-486 against cardiomyocyte apoptosis. Interestingly, AKT activation in turn can lead to repression of FoxO1 activity, which is an important approach to prevent apoptosis.31,32 In our study, we also observed that miR-486 overexpression was able to increase AKT phosphorylation levels in heart tissues from I/R mice, indicative of an in vivo activation of AKT by increasing miR-486 during cardiac I/R injury.
Mounting evidence has shown that exercise per se and exercise-regulated molecules can have myocardial protective effects against I/R injury and cardiac remodeling,16,22 and miR-486 was previously identified to be upregulated in the heart after exercise training.18 Swimming exercise has been previously demonstrated to be beneficial for myocardial protection.17 In the present study, we demonstrated that miR-486 was significantly upregulated in the heart tissues and serum samples after swimming exercise. Next, we injected mice with miR-486 sponge AAV9 and then subjected mice to swimming exercise before cardiac I/R injury. Interestingly, we observed that miR-486 inhibition significantly attenuated the beneficial effect of exercise against cardiac I/R injury, myocardial apoptosis, and cardiac dysfunction. Swimming exercise led to downregulated PTEN and FoxO1 in the I/R hearts, which was, however, reversed by miR-486 inhibition. We further demonstrated that swimming-exercised mice with miR-486 KO had significantly increased infarct size after I/R injury compared with WT exercised mice. These data consistently indicate that miR-486 is necessary for the myocardial protective effect of exercise against cardiac I/R injury.
In conclusion, miR-486 is protective against cardiac I/R injury and myocardial apoptosis through the targeting of PTEN and FoxO1 and activation of the AKT/mTOR pathway, and mediates the beneficial effect of exercise for myocardial protection (see the graphical abstract). With the encouraging development of non-coding RNA-based therapeutics, increasing miR-486 might be a promising therapeutic strategy for myocardial protection.
Materials and methods
Animals and cardiac I/R injury model
Male C57BL/6J mice ages 8–10 weeks were purchased from Cavens Lab Animal (Changzhou, China) and maintained in the specific-pathogen-free (SPF) laboratory animal facility of Shanghai University (Shanghai, China). miR-486 KO mice were generated using CRISPR-Cas9-mediated genome engineering by VIEWSOLID BIOTECH (Beijing, China). Briefly, guide RNA (gRNA) targeting vectors were constructed based on the mature sequence of mmu-miR-486 on mouse chromosome 8. The Cas9 mRNA and validated gRNA generated by in vitro transcription were co-injected into fertilized eggs for KO mouse production. The pups were genotyped by PCR followed by sequence analysis, which showed a deletion of 254 bp containing the mature sequence of miR-486. All animal experiments were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals for biomedical research published by the National Institutes of Health (No. 85-23, revised 1996) and approved by the Committee for the Ethics of Animal Experiments of Shanghai University. To induce acute cardiac I/R injury, mice were anesthetized with 4% chloral hydrate (intraperitoneally [i.p.], 10 μL/g body weight) followed by endotracheal ventilation, and the left anterior descending (LAD) coronary artery was ligated for 30 min and then reperfused for 24 h as previously reported.18,23 Sham mice were subjected to the same surgery procedures, but without LAD ligation.
To study the preventive effect of miR-486 in vivo, AAV9 expressing miR-486 (miR-486-AAV9) was injected via the tail vein at a dose of 1011 virus genomes (v.g.) per mouse. miR-486-AAV9 and control vectors (CTL-AAV9) were purchased from Hanbio Biotechnology (Shanghai, China). Briefly, the mmu-miR-486a-5p was inserted between the BamHI and the EcoRI restriction enzyme sites of the pHBAAV9-CMV-MCS-WPRE vector (Hanbio Biotechnology). AAVs were generated using packaging plasmids pAAV-rep/cap vector and pHelper vector together with AAV9-CMV-mmu-miR-486a-5p constructs. Three weeks after AAV9 injections, acute cardiac I/R injury was induced as described above. At 24 h after I/R injury, 2,3,5-triphenyltetrazolium chloride (TTC) staining and heart tissue harvest were performed. In another set of animal experiments, we constructed miR-486 sponge AAV9 and injected mice with miR-486 sponge AAV9 or control vectors via the tail vein at a dose of 1011 v.g. per mouse to investigate whether miR-486 inhibition could further aggravate cardiac I/R injury. The infarct size was measured using TTC staining at 24 h after I/R injury, and heart tissues were harvested for further examination. To study the effect of miR-486 on cardiac I/R injury in the long term, mice were injected with miR-486-AAV9 as described above, and 1 week later subjected to cardiac I/R injury. At 3 and 6 weeks after injury, echocardiography and heart tissue harvest were performed.
To study the potential therapeutic effect of miR-486 overexpression in cardiac I/R injury, mice were injected with miR-486-AAV9, AAV9 expressing miR-486 with the cardiac muscle cell-specific promoter cTnT (cTnT-miR-486 AAV9) (Hanbio Biotechnology), or control vectors at a dose of 1011 v.g. per mouse, within 30 min of cardiac I/R injury. Echocardiography and heart tissue harvest were performed after 3 weeks of I/R injury.
Swimming exercise training
To establish the mouse model of swimming exercise, mice were adapted to 5 min swimming in the morning and in the afternoon before formal exercise training. At the first day of swimming training, mice swam for 10 min twice a day, and the swimming time increased by 10 min each day until 90 min twice a day as previously reported.17,18 After 4 weeks of swimming exercise, miR-486 expression levels were determined in mouse heart tissues, gastrocnemius and anterior tibialis muscles, and serum samples, as well as in the isolated cardiomyocytes and cardiac fibroblasts from adult swimming mice or sedentary mice as previously described.16 To investigate the role of miR-486 in exercise-induced protection against cardiac I/R injury, mice were injected with miR-486 sponge AAV9 or CTL-AAV9 at 1 week before swimming, and then subjected to swimming exercise. After 3 weeks of swimming exercise, cardiac I/R injury was induced by LAD ligation and reperfusion as described above. Finally, TTC staining was performed to evaluate the infarct size at 24 h after I/R injury, and heart tissue harvest was performed at 1 week post I/R injury. In addition, 8- to 10-week-old male miR-486 KO mice and WT littermates received swimming exercise for 3 weeks and then were subjected to cardiac I/R injury for determination of the infarct size using TTC staining at 24 h after I/R injury.
TTC staining
At 24 h after cardiac I/R injury, mice were anesthetized with 4% chloral hydrate (i.p., 10 μL/g body weight) before the left ventricle was injected with 1 mL of 1% Evans blue. The heart was then sliced into 1-mm-thick tissue sections and stained with 1% TTC. The area-at-risk to left-ventricle-weight ratio (AAR/LV) was used to confirm homogeneity of surgery. The infarct-size to area-at-risk ratio (INF/AAR) was used to evaluate cardiac I/R injury.
Echocardiography for cardiac function
Mice were anesthetized with inhaled isoflurane and evaluated for cardiac function using echocardiography (FUJIFILM Visual Sonics). Left-ventricular ejection fraction (EF, %) and fractional shortening (FS, %) were measured and calculated through three consecutive cardiac cycles based on M-mode long-axis view of the left ventricle at the papillary muscle level.
Primary culture and treatment of neonatal rat cardiomyocytes and cardiac fibroblasts
NRCMs and NRCFs were isolated from 1- to 3-day-old Sprague-Dawley rats using collagenase II (Gibco, 17101015)/pancreatin from porcine pancreas (Sigma, P3292) digestion and Percoll (GE Healthcare, 17-0891-01) centrifugation protocol.17,18 NRCMs were maintained in Dulbecco's modified Eagle's medium (DMEM, Corning) containing 4.5 g/L glucose supplemented with 10% horse serum (Gibco) and 5% fetal bovine serum (BioInd, Israel). To mimic cardiac I/R injury in vitro, NRCMs were treated with oxygen-glucose deprivation for 8 h followed by reperfusion for 12 h as previously described.23 To study the functional role of miR-486 in OGDR-induced cardiomyocyte apoptosis, NRCMs were transfected with miR-486 mimic (50 nM), inhibitor (100 nM), or negative controls (RiboBio, Guangzhou, China) for 48 h using Lipofectamine 2000 (Invitrogen). Transfection of siRNAs targeting PTEN or FoxO1 (75 nM, RiboBio, Guangzhou, China), or plasmids expressing PTEN or FoxO1, was conducted in OGDR-treated NRCMs for 48 h to investigate the target genes of miR-486 in regulating cardiomyocyte apoptosis. Moreover, NRCMs were treated with the AKT inhibitor MK2206 (10 nM, Selleck) or mTOR inhibitor rapamycin (100 nM, Sigma) for 24 h under OGDR stress to elucidate whether AKT/mTOR activation was involved in the functional role of miR-486 in cardiomyocytes. NRCFs were maintained in DMEM (Corning) containing 4.5 g/L glucose supplemented with 10% fetal bovine serum. The functional roles of miR-486 were determined in NRCFs transfected with miR-486 mimic (50 nM) or inhibitor (100 nM) under stimulation of recombinant human TGF-β (Peprotech, Rocky Hill, NJ, USA) at 20 ng/mL for 48 h.
hiPSC-derived cardiomyocytes and treatment
The hiPSCs (hHSC_Iso4_ADCF_SeV-iPS2; alternative name, MHHi001) were reprogrammed from CD34-positive human cord blood hematopoietic stem cells of a healthy female donor as previously published.33 Informed consent was obtained, in accordance with local regulations and Institutional Review Board requirements. The hiPSCs were maintained under feeder-free culture conditions using Geltrex (Thermo Scientific)-coated polystyrene plates (Greiner CELLSTAR) and StemMACS full medium with supplements (Milteny).33 Directed differentiation of hiPSCs toward mesodermal lineage was performed as described previously,34 followed by a metabolic selection process to obtain a purified population of hiPSC-CMs.35 hiPSC-CMs were maintained as described previously,36 and transfected with 100 nM pre-miR hsa-miR-486 (AM17100) for 48 h and further put under OGDR stress. For OGDR stress, hiPSC-CMs were placed in a 1% O2 incubator for 8 h with no glucose medium (supplemented with lactate). Then, the cells recovered in a 21% O2 incubator for 12 h in normal maintenance medium. hiPSC-CMs transfected with pre-miR negative control 2 (Ambion, AM17111) served as controls. At the endpoint, TUNEL assays were performed to measure cell apoptosis.
Immunofluorescence staining
Cardiomyocyte apoptosis was examined using a TUNEL FITC apoptosis detection kit (Vazyme) according to the manufacturer's manuals. Briefly, frozen heart tissue sections or cultured cardiomyocytes were fixed with 4% paraformaldehyde (PFA) and permeabilized with 0.5% Triton X-100 before being blocked with 5% bovine serum albumin (BSA) for 1 h. Heart tissue sections or NRCMs were incubated with a 1:200 dilution of mouse anti-α-actinin (Sigma, A7811) at 4°C overnight, and then incubated with Cy3-AffiniPure goat anti-mouse IgG (H + L) (The Jackson Laboratory) for 1 h at room temperature. For hiPSC-CMs, cardiomyocytes were immunofluorescently labeled with cTnT (Thermo, MS-295-P0). Finally, heart tissue sections or cardiomyocytes were stained with the TUNEL FITC apoptosis detection kit (Vazyme) according to the manufacturer's instructions, and nuclei were counterstained with DAPI or Hoechst. A total of at least 10 fields per tissue or per well were photographed and analyzed using a confocal microscope (Carl Zeiss). The percentage of TUNEL-positive cardiomyocytes was determined to evaluate cardiomyocyte apoptosis upon cardiac I/R injury or OGDR stress. For immunofluorescence staining of α-SMA and EdU, NRCFs were pre-incubated with EdU working solution for 24 h and incubated with α-SMA-Cy3 antibody (Sigma, C6198) overnight at 4°C. EdU labeling was performed using the Click-iT Plus EdU Alexa Fluor 488 Imaging Kit (Invitrogen) according to the manufacturer's instructions, and nuclei were counterstained with Hoechst. A total of at least 10 fields per well were viewed under a confocal microscope (Carl Zeiss). The percentage of EdU-positive NRCFs and the immunofluorescence intensity of α-SMA were determined to evaluate cardiac fibroblast proliferation and differentiation to myofibroblasts.
Masson's trichrome staining
Heart tissues were embedded in paraffin, and 5-μm-thick sections were stained with Masson's trichrome (Solarbio, G1340) according to the manufacturer’s manual. The percentage of fibrotic area to total area was evaluated for cardiac fibrosis. Images were analyzed using ImageJ software 1.50i (NIH, USA).
Luciferase reporter assay
To study the direct interaction between miR-486 and the 3′ UTR of PTEN and FoxO1, we constructed the luciferase reporter vector pGL3-Basic (Promega) containing the binding site (or mutated) with miR-486 in the 3′ UTR of PTEN (719–726) or FoxO1 (182–189). 293T cells were transfected with WT or mutated (mut) luciferase reporter plasmid with miR-486 mimic or negative control. The dual-luciferase reporter assay (Promega) was performed according to the manufacturer's instructions to study the direct binding of miR-486 in the 3′ UTR of PTEN or FoxO1 as determined by firefly/Renilla luciferase activity. To verify that miR-486 sponge was sufficient to target and bind miR-486, we constructed luciferase reporter vector pGL3-Basic (Promega) containing multiple tandem binding sites for miR-486. 293T cells were co-transfected with the miR-486 binding site-carrying luciferase reporter plasmids and the negative control (NC) mimic, miR-486 mimic, or miR-210 mimic. The group co-transfected with miR-210 mimic was designed to confirm that miR-486 had no binding activity with other miRNAs. Dual-luciferase reporter assay (Promega) was performed according to the manufacturer's instructions.
Real-time PCR
Total RNA was isolated from tissues or cultured cells using Trizol RNAiso Plus (TaKaRa). RNA concentration and quality were measured by NanoDrop (Thermo Scientific). A total of 100–400 ng RNA was then reverse transcribed to cDNA using a RevertAid First Strand cDNA synthesis kit (Thermo Scientific K1622). Real-time PCR was conducted using TaKaRa SYBR Premix Ex Taq (Tli RNaseH Plus, Japan) on a Roche LightCycler 480 PCR system. The primers for genes (forward and reverse 5′-3′) are listed in Table S1. For miRNA quantification in murine tissues and/or cells, real-time PCR was performed using a Bulge-Loop miRNA qRT-PCR primer set for miRNA (RiboBio, Guangzhou, China). 18s (or GAPDH) and 5s were used as internal controls for normal gene and miRNA, respectively. For quantification of miR-486 in mouse serum samples, total RNA was isolated using the MirVana miRNA isolation kit (Thermo Scientific, AM1561). Caenorhabditis elegans miR-39 (cel-miR-39) was added as the spike-in control. TaqMan assay (Thermo Fisher) was applied to measure miR-486-5p (assay 001278) expression in hiPSC-CMs, and U6 (assay 001973) was used as an internal control.
Western blotting
Total protein was lysed from heart tissues or cultured cells with RIPA lysis buffer (KeyGEN BioTECH, China) complemented with 1% phenylmethylsulfonyl fluoride (PMSF) and Pierce protease and phosphatase inhibitor (Thermo Scientific, 88668). Equal quantities of total protein were loaded on SDS-PAGE gels and transferred to PVDF membranes. Protein blots were blocked with 5% milk and incubated with primary antibodies for Bax (Abclonal, A0207), Bcl-2 (Abclonal, A2845), caspase-3 (Cell Signaling, 9662), PTEN (Cell Signaling, 9552), FoxO1 (Proteintech, 18592-1-AP), p-AKT 473 (Cell Signaling, 4051), p-AKT 308 (Cell Signaling, 2965), p-mTOR (Cell Signaling, 2971), p-P70S6K (Cell Signaling, 9205), Smad1 (Abclonal, A1101), and Smad2 (Abclonal, A19114) at 4°C overnight. Corresponding secondary antibodies were incubated on the next day before detection for protein bands using an enhanced chemiluminescence (ECL) kit. For phosphorylated proteins, protein blots were stripped and reblotted with specific antibodies for corresponding total protein, including AKT (Proteintech, 10176-2-AP), mTOR (Cell Signaling, 2972), and P70S6K (Cell Signaling, 9202). GAPDH or β-actin was used as an internal control. Finally, protein band intensity was analyzed using ImageJ software.
Statistical analysis
All data were analyzed using SPSS software version 20.0 or GraphPad Prism 8. Experimental data were presented as the mean ± SD using GraphPad Prism 8. Data between two groups were compared by unpaired two-tailed Student's t test. Data among three groups were compared by one-way ANOVA. For more than three groups, data were compared by two-way ANOVA followed by Tukey’s post hoc test. A p < 0.05 was considered statistically significant.
Acknowledgments
This work was supported by grants from the National Key Research and Development Project (2018YFE0113500 to J.J.X.), National Natural Science Foundation of China (82020108002 and 81911540486 to J.J.X., 81770401 and 81970335 to Y.H.B.), Innovation Program of Shanghai Municipal Education Commission (2017-01-07-00-09-E00042 to J.J.X.), Science and Technology Commission of Shanghai Municipality (21XD1421300 and 20DZ2255400 to J.J.X.), “Dawn” Program of Shanghai Education Commission (19SG34 to J.J.X.), Shanghai Rising-Star Program (19QA1403900 to Y.H.B.), and Shanghai Committee of Science and Technology (21SQBS00100 to Y.H.B.). C.B. and T.T. received funding from the German Research Foundation, DFG (SFB/Transregio TRR267).
Author contributions
J.J.X. and T.T. designed the study, supervised all experiments, and drafted the manuscript. Y.H.B., D.C.L., and C.B. performed the experiments and analyzed the data. Y.J.Z., Z.Z.H., and M.Y.H. analyzed miR-486 in the murine model of swimming exercise. M.W., P.J.Y., S.Y.L., and K.W. established murine models of myocardial injury. S.C., A.C., I.R., and F.C.M. provided hiPSC-CMs and technical assistance. All authors read and approved the final manuscript.
Declaration of interests
T.T. is a founder and shareholder of Cardior Pharmaceuticals GmbH. T.T. has filed and licensed patents on non-coding RNAs (outside of this work). The other authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.01.031.
Contributor Information
Thomas Thum, Email: thum.thomas@mh-hannover.de.
Junjie Xiao, Email: junjiexiao@shu.edu.cn.
Supplemental information
References
- 1.Ibanez B., Heusch G., Ovize M., Van de Werf F. Evolving therapies for myocardial ischemia/reperfusion injury. J. Am. Coll. Cardiol. 2015;65:1454–1471. doi: 10.1016/j.jacc.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 2.Hausenloy D.J., Yellon D.M. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J. Clin. Invest. 2013;123:92–100. doi: 10.1172/JCI62874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heusch G., Libby P., Gersh B., Yellon D., Bohm M., Lopaschuk G., Opie L. Cardiovascular remodelling in coronary artery disease and heart failure. Lancet. 2014;383:1933–1943. doi: 10.1016/S0140-6736(14)60107-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heusch G., Gersh B.J. The pathophysiology of acute myocardial infarction and strategies of protection beyond reperfusion: a continual challenge. Eur. Heart J. 2017;38:774–784. doi: 10.1093/eurheartj/ehw224. [DOI] [PubMed] [Google Scholar]
- 5.Beermann J., Piccoli M.T., Viereck J., Thum T. Non-coding RNAs in development and disease: background, mechanisms, and therapeutic approaches. Physiol. Rev. 2016;96:1297–1325. doi: 10.1152/physrev.00041.2015. [DOI] [PubMed] [Google Scholar]
- 6.Small E.M., O'Rourke J.R., Moresi V., Sutherland L.B., McAnally J., Gerard R.D., Richardson J.A., Olson E.N. Regulation of PI3-kinase/Akt signaling by muscle-enriched microRNA-486. Proc. Natl. Acad. Sci. U S A. 2010;107:4218–4223. doi: 10.1073/pnas.1000300107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chistiakov D.A., Orekhov A.N., Bobryshev Y.V. Cardiac-specific miRNA in cardiogenesis, heart function, and cardiac pathology (with focus on myocardial infarction) J. Mol. Cell Cardiol. 2016;94:107–121. doi: 10.1016/j.yjmcc.2016.03.015. [DOI] [PubMed] [Google Scholar]
- 8.Sun Y., Su Q., Li L., Wang X., Lu Y., Liang J. MiR-486 regulates cardiomyocyte apoptosis by p53-mediated BCL-2 associated mitochondrial apoptotic pathway. BMC Cardiovasc. Disord. 2017;17:119. doi: 10.1186/s12872-017-0549-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu H.H., Wang X.T., Sun Y.H., He W.K., Liang J.B., Mo B.H., Li L. MicroRNA-486-5p targeting PTEN protects against coronary microembolization-induced cardiomyocyte apoptosis in rats by activating the PI3K/AKT pathway. Eur. J. Pharmacol. 2019;855:244–251. doi: 10.1016/j.ejphar.2019.03.045. [DOI] [PubMed] [Google Scholar]
- 10.Wang H., Maimaitiaili R., Yao J., Xie Y., Qiang S., Hu F., Li X., Shi C., Jia P., Yang H., et al. Percutaneous intracoronary delivery of plasma extracellular vesicles protects the myocardium against ischemia-reperfusion injury in Canis. Hypertension. 2021;78:1541–1554. doi: 10.1161/HYPERTENSIONAHA.121.17574. [DOI] [PubMed] [Google Scholar]
- 11.McMullen J.R., Amirahmadi F., Woodcock E.A., Schinke-Braun M., Bouwman R.D., Hewitt K.A., Mollica J.P., Zhang L., Zhang Y., Shioi T., et al. Protective effects of exercise and phosphoinositide 3-kinase(p110alpha) signaling in dilated and hypertrophic cardiomyopathy. Proc. Natl. Acad. Sci. U S A. 2007;104:612–617. doi: 10.1073/pnas.0606663104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bei Y., Zhou Q., Sun Q., Xiao J. Exercise as a platform for pharmacotherapy development in cardiac diseases. Curr. Pharm. Des. 2015;21:4409–4416. doi: 10.2174/1381612821666150803150008. [DOI] [PubMed] [Google Scholar]
- 13.Bei Y., Fu S., Chen X., Chen M., Zhou Q., Yu P., Yao J., Wang H., Che L., Xu J., et al. Cardiac cell proliferation is not necessary for exercise-induced cardiac growth but required for its protection against ischaemia/reperfusion injury. J. Cell Mol. Med. 2017;21:1648–1655. doi: 10.1111/jcmm.13078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bostrom P., Mann N., Wu J., Quintero P.A., Plovie E.R., Panakova D., Gupta R.K., Xiao C., MacRae C.A., Rosenzweig A., et al. C/EBPbeta controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell. 2010;143:1072–1083. doi: 10.1016/j.cell.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bezzerides V.J., Platt C., Lerchenmuller C., Paruchuri K., Oh N.L., Xiao C., Cao Y., Mann N., Spiegelman B.M., Rosenzweig A. CITED4 induces physiologic hypertrophy and promotes functional recovery after ischemic injury. JCI Insight. 2016;1:e85904. doi: 10.1172/jci.insight.85904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao R., Wang L., Bei Y., Wu X., Wang J., Zhou Q., Tao L., Das S., Li X., Xiao J. Long noncoding RNA cardiac physiological hypertrophy-associated regulator induces cardiac physiological hypertrophy and promotes functional recovery after myocardial ischemia-reperfusion injury. Circulation. 2021;144:303–317. doi: 10.1161/CIRCULATIONAHA.120.050446. [DOI] [PubMed] [Google Scholar]
- 17.Shi J., Bei Y., Kong X., Liu X., Lei Z., Xu T., Wang H., Xuan Q., Chen P., Xu J., et al. miR-17-3p contributes to exercise-induced cardiac growth and protects against myocardial ischemia-reperfusion injury. Theranostics. 2017;7:664–676. doi: 10.7150/thno.15162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X., Xiao J., Zhu H., Wei X., Platt C., Damilano F., Xiao C., Bezzerides V., Bostrom P., Che L., et al. miR-222 is necessary for exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell Metab. 2015;21:584–595. doi: 10.1016/j.cmet.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lopez-Bertoni H., Kotchetkov I.S., Mihelson N., Lal B., Rui Y., Ames H., Lugo-Fagundo M., Guerrero-Cazares H., Quinones-Hinojosa A., Green J.J., et al. A sox2:miR-486-5p Axis regulates survival of GBM cells by inhibiting tumor suppressor networks. Cancer Res. 2020;80:1644–1655. doi: 10.1158/0008-5472.CAN-19-1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ola R., Kunzel S.H., Zhang F., Genet G., Chakraborty R., Pibouin-Fragner L., Martin K., Sessa W., Dubrac A., Eichmann A. SMAD4 prevents flow induced arteriovenous malformations by inhibiting casein kinase 2. Circulation. 2018;138:2379–2394. doi: 10.1161/CIRCULATIONAHA.118.033842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao H., Yang H., Geng C., Chen Y., Tang Y., Li Z., Pang J., Shu T., Nie Y., Liu Y., et al. Elevated IgE promotes cardiac fibrosis by suppressing miR-486a-5p. Theranostics. 2021;11:7600–7615. doi: 10.7150/thno.47845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hill J.A. Braking bad hypertrophy. N. Engl. J. Med. 2015;372:2160–2162. doi: 10.1056/NEJMcibr1504187. [DOI] [PubMed] [Google Scholar]
- 23.Bei Y., Pan L.L., Zhou Q., Zhao C., Xie Y., Wu C., Meng X., Gu H., Xu J., Zhou L., et al. Cathelicidin-related antimicrobial peptide protects against myocardial ischemia/reperfusion injury. BMC Med. 2019;17:42. doi: 10.1186/s12916-019-1268-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun X.H., Wang X., Zhang Y., Hui J. Exosomes of bone-marrow stromal cells inhibit cardiomyocyte apoptosis under ischemic and hypoxic conditions via miR-486-5p targeting the PTEN/PI3K/AKT signaling pathway. Thromb. Res. 2019;177:23–32. doi: 10.1016/j.thromres.2019.02.002. [DOI] [PubMed] [Google Scholar]
- 25.Jae N., Dimmeler S. Noncoding RNAs in vascular diseases. Circ. Res. 2020;126:1127–1145. doi: 10.1161/CIRCRESAHA.119.315938. [DOI] [PubMed] [Google Scholar]
- 26.Ruan H., Li J., Ren S., Gao J., Li G., Kim R., Wu H., Wang Y. Inducible and cardiac specific PTEN inactivation protects ischemia/reperfusion injury. J. Mol. Cell Cardiol. 2009;46:193–200. doi: 10.1016/j.yjmcc.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 27.Lange S., Banerjee I., Carrion K., Serrano R., Habich L., Kameny R., Lengenfelder L., Dalton N., Meili R., Borgeson E., et al. miR-486 is modulated by stretch and increases ventricular growth. JCI Insight. 2019;4:e125507. doi: 10.1172/jci.insight.125507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wendel H.G., De Stanchina E., Fridman J.S., Malina A., Ray S., Kogan S., Cordon-Cardo C., Pelletier J., Lowe S.W. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428:332–337. doi: 10.1038/nature02369. [DOI] [PubMed] [Google Scholar]
- 29.Yue H.W., Liu J., Liu P.P., Li W.J., Chang F., Miao J.Y., Zhao J. Sphingosylphosphorylcholine protects cardiomyocytes against ischemic apoptosis via lipid raft/PTEN/Akt1/mTOR mediated autophagy. Biochim. Biophys. Acta. 2015;1851:1186–1193. doi: 10.1016/j.bbalip.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 30.Lu C., Wang X., Ha T., Hu Y., Liu L., Zhang X., Yu H., Miao J., Kao R., Kalbfleisch J., et al. Attenuation of cardiac dysfunction and remodeling of myocardial infarction by microRNA-130a are mediated by suppression of PTEN and activation of PI3K dependent signaling. J. Mol. Cell Cardiol. 2015;89:87–97. doi: 10.1016/j.yjmcc.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X., Tang N., Hadden T.J., Rishi A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta. 2011;1813:1978–1986. doi: 10.1016/j.bbamcr.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 32.Puthanveetil P., Wan A., Rodrigues B. FoxO1 is crucial for sustaining cardiomyocyte metabolism and cell survival. Cardiovasc. Res. 2013;97:393–403. doi: 10.1093/cvr/cvs426. [DOI] [PubMed] [Google Scholar]
- 33.Haase A., Gohring G., Martin U. Generation of non-transgenic iPS cells from human cord blood CD34(+) cells under animal component-free conditions. Stem Cell Res. 2017;21:71–73. doi: 10.1016/j.scr.2017.03.022. [DOI] [PubMed] [Google Scholar]
- 34.Zangi L., Oliveira M.S., Ye L.Y., Ma Q., Sultana N., Hadas Y., Chepurko E., Spater D., Zhou B., Chew W.L., et al. Insulin-like growth factor 1 receptor-dependent pathway drives epicardial adipose tissue formation after myocardial injury. Circulation. 2017;135:59–72. doi: 10.1161/CIRCULATIONAHA.116.022064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tohyama S., Hattori F., Sano M., Hishiki T., Nagahata Y., Matsuura T., Hashimoto H., Suzuki T., Yamashita H., Satoh Y., et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell. 2013;12:127–137. doi: 10.1016/j.stem.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 36.Foinquinos A., Batkai S., Genschel C., Viereck J., Rump S., Gyongyosi M., Traxler D., Riesenhuber M., Spannbauer A., Lukovic D., et al. Preclinical development of a miR-132 inhibitor for heart failure treatment. Nat. Commun. 2020;11:633. doi: 10.1038/s41467-020-14349-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.