

Abstract
Long non-coding RNA HOX Transcript Antisense RNA (HOTAIR) is overexpressed in multiple cancers with diverse genetic profiles. Importantly, since HOTAIR heavily contributes to cancer progression by promoting tumor growth and metastasis, HOTAIR becomes a potential target for cancer therapy. However, the underlying mechanism leading to HOTAIR deregulation is largely unexplored. Here, we performed a pan-cancer analysis using more than 4,200 samples and found that intragenic exon CpG island (Ex-CGI) was hypermethylated and was positively correlated to HOTAIR expression. Also, we revealed that Ex-CGI methylation promotes HOTAIR expression through enhancing the transcription elongation process. Furthermore, we linked up the aberrant intragenic tri-methylation on H3 at lysine 4 (H3K4me3) and Ex-CGI DNA methylation in promoting transcription elongation of HOTAIR. Targeting the oncogenic CDK7-CDK9-H3K4me3 axis downregulated HOTAIR expression and inhibited cell growth in many cancers. To our knowledge, this is the first time that a positive feedback loop that involved CDK9-mediated phosphorylation of RNA Polymerase II Serine 2 (RNA PolII Ser2), H3K4me3, and intragenic DNA methylation, which induced robust transcriptional elongation and heavily contributed to the upregulation of oncogenic lncRNA in cancer has been demonstrated. Targeting the oncogenic CDK7-CDK9-H3K4me3 axis could be a novel therapy in many cancers through inhibiting the HOTAIR expression.
Keywords: HOTAIR, cancer, DNA methylation, transcription elongation
Graphical abstract
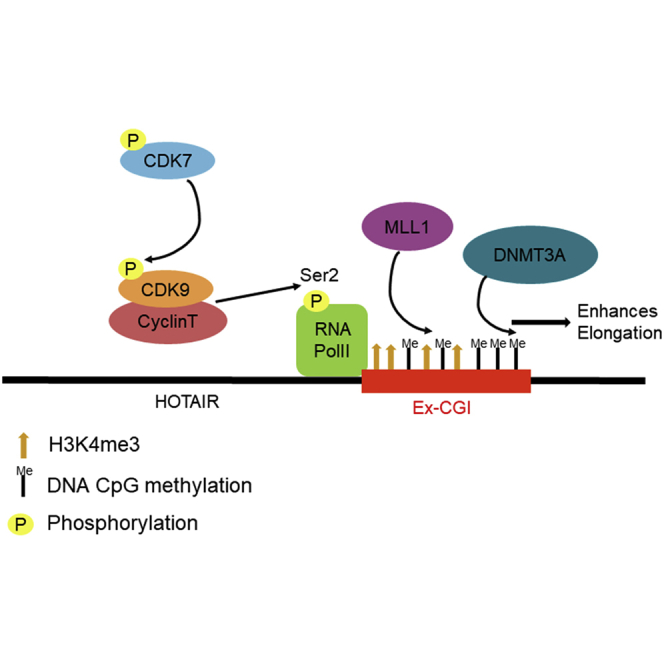
We unravel a novel molecular pathway in upregulating the oncogenic lncRNA in cancer, which links up intragenic histone methylation, DNA methylation, and RNA transcription elongation. We reveal that the oncogenic CDK7-CDK9-H3K4me3 axis regulates Ex-CGI DNA methylation, and then leads to subsequent HOTAIR transcription elongation and expression in cancers.
Introduction
HOX Transcript Antisense RNA (HOTAIR), as one of the well-studied long non-coding RNAs (lncRNAs), is significantly overexpressed across different cancer types with diverse genetic profiles.1 HOTAIR drives important cancer phenotypes via inducing deregulations on critical genes by PRC2-mediated H3K27 trimethylation, LSD1/CoREST/REST-mediated H3K4 demethylation, and interacting with microRNAs (miRNAs) as competitive RNA.2, 3, 4 However, the mechanism in regulating HOTAIR expression in cancer remains unexplored.
DNA methylation at cytosine of CpG dinucleotides has been reported to regulate gene expression.5 A previous report hypothesized that methylation in downstream CpG island (DS-CGI) of HOTAIR gene facilitates the transcription of HOTAIR in breast cancer.6 The DS-CGI, which is located between the HOTAIR gene and HOXC12 gene, may control the expression of HOTAIR. When DS-CGI is methylated, transcription of HOXC12 may terminate at DS-CGI and HOTAIR may be transcribed normally. However, when DS-CGI is unmethylated, transcription of HOXC12 may continue and disrupt HOTAIR transcription. In this study, we attempted to validate this hypothesis and delineated the underlying mechanism leading to the deregulation of HOTAIR in cancer.
Here, we found that the methylation of DS-CGI may not be involved in the regulation of HOTAIR expression. Importantly, we demonstrated a general mechanism that contributed to HOTAIR upregulation in cancer. Methylation of exon CpG island in the HOTAIR gene promotes HOTAIR expression by facilitating the transcription elongation process. In addition, the methylation of exon CpG island is regulated by CDK9-mediated phosphorylation of RNA PolII Ser2 and MLL1-mediated H3K4me3.
Results
Methylation of downstream CpG island may not be involved in regulating HOTAIR expression
HOTAIR reportedly plays important roles in diverse types of cancer.1 Consistently, we found that HOTAIR was frequently upregulated in cancers (Figures S1A–S1G). Also, we found that HOTAIR overexpression in non-tumor human pancreatic ductal epithelial (HPDE) cells promoted cell growth (Figures S1H and S1I). On the other hand, HOTAIR knockdown inhibited cell growth in pancreatic ductal adenocarcinoma (PDAC) PANC-1 and SW1990 cells (Figures S1J and S1K). These suggested that HOTAIR played important roles in cancer progression. However, the mechanism in regulating HOTAIR expression in cancer remains unexplored.
The previous report hypothesized that the DNA methylation of CpG island, which is downstream of the HOTAIR gene, i.e., DS-CGI, is required for the transcription of full-length HOTAIR transcript (Figure S2A).6 Therefore, we attempted to validate this hypothesis. First, we profiled the HOTAIR expression and the DNA methylation status of DS-CGI in cancer cells. We failed to observe the correlation between HOTAIR expression and DS-CGI DNA methylation (Figures S2B and S2C). Furthermore, both the downstream 5.8-kb and the upstream 4-kb transcript of the DS-CGI could be detected in all breast cancer cells regardless of the DNA methylation status (Figure S2C). Regarding the situation in PDAC, both the 5.8-kb and the 4-kb transcript were not detected in non-tumor HPDE cells with a low HOTAIR level, while both the 5.8-kb and the 4-kb transcript could be detected in PDAC cells with methylated DS-CGI (Figure S2D). Chromatin immunoprecipitation (ChIP) analysis also demonstrated the binding of RNA Polymerase II to the 4-kb transcript, suggesting active transcription of the 4-kb transcript in SW1990 cells with high HOTAIR level (Figure S2E). These findings contradicted the previous theory that DNA methylation of DS-CGI hindered the transcription of the downstream 4-kb transcript. Therefore, we here demonstrated that DNA methylation of DS-CGI may not be involved in regulating HOTAIR expression.
DNA methylation in exon CpG island contributed to HOTAIR expression in cancer
Then we tried to investigate how HOTAIR was regulated in cancer. DNA methylation at the cytosine residues has been frequently reported to be an important regulator of gene expression. To study the association between DNA methylation and HOTAIR expression, we located all CGIs in the HOTAIR gene region. Apart from DS-CGI, which is not involved in regulating HOTAIR expression, we observed another two CGIs in the HOTAIR gene (Figure 1A). We failed to locate any CGI in the promoter region (Figure S3A). Instead, we found that one CGI was in the intron 2 (Intron-CGI); and the remaining CGI overlapped with exon 4 (Ex-CGI). Studies found that more than a half of the DNA methylation occurred at the intragenic regions and played important roles in the regulation of transcription process and gene expression.7, 8, 9, 10, 11 Also, a recent study revealed the importance of an evolutionarily conserved region on the HOTAIR gene, which covers Ex-CGI, in regulating HoxC and HoxD cluster genes.12 Hence, we hypothesized that the DNA methylation of the evolutionarily conserved Ex-CGI of the HOTAIR gene promoted the transcription process of HOTAIR. To investigate the association between Ex-CGI DNA methylation and HOTAIR expression, quantitative methylation-specific PCR (qMSP) was first conducted. We found that Ex-CGI DNA was frequently methylated in most PDAC cell lines with high HOTAIR expression, but not in the non-tumor HPDE cells and PDAC CFPAC-1 cells with low HOTAIR expression (Figure 1B). In addition, most hepatocellular carcinoma (HCC), colorectal cancer (CRC), and breast cancer cells had high expression of HOTAIR, while their Ex-CGI DNA was also hypermethylated (Figures 1C–1E, S1E, S1F, and 2B). In contrast, low Ex-CGI DNA methylation level was observed in MIHA, DLD-1, and MCF10A cells, which expressed relatively low HOTAIR level (Figures 1C–1E, S1E, S1F, and 2B). This suggested that Ex-CGI DNA methylation is positively correlated with HOTAIR expression in cancers.
Figure 1.

DNA methylation of Ex-CGI was positively associated with HOTAIR expression in cancer cells
(A) Schematic of CpG island (CGI) distribution in HOTAIR gene. Two CGIs were found in the intragenic region of HOTAIR. One CGI was in the intron 2 (Intron-CGI); and the remaining one CGI overlapped with exon 4 (Ex-CGI). Eighteen CpG sites were found in Ex-CGI. (B–E) Ex-CGI was frequently hypermethylated in (B) PDAC, (C) HCC, (D) CRC, and (E) breast cancer cells. Data are from at least three independent experiments and plotted as means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Figure 2.

Pan-cancer analysis revealed the positive correlation between DNA methylation of Ex-CGI and HOTAIR expression in cancers
Ex-CGI was frequently hypermethylated in tumor and was positively correlated with HOTAIR expression in (A) PAAD, (B) LIHC, (C) COAD, (D) STAD, (E) ESCA, (F) BLCA, (G) HNSC, (H) KIRC, (I) LUAD, and (J) BRCA, n = 4,271 subjects.
To investigate the clinical importance of Ex-CGI DNA methylation in cancer, qMSP at the HOTAIR Ex-CGI was performed in human PDAC primary tumors. We found that Ex-CGI DNA of human PDAC primary tumors was significantly hypermethylated (Figure S3B). Subsequently, pyrosequencing had confirmed that several CpG sites at Ex-CGI were frequently hypermethylated and were positively associated with HOTAIR expression level in PDAC primary tumors (Figures S3C and S3D). Importantly, we performed pan-cancer analysis of Ex-CGI DNA methylation in gastrointestinal (GI) cancers, including pancreatic adenocarcinoma (PAAD), liver hepatocellular carcinoma (LIHC), colorectal adenocarcinoma (COAD), stomach adenocarcinoma (STAD), esophageal cancer (ESCA), and bladder cancer (BLCA) (1,873 subjects in total), using The Cancer Genome Atlas (TCGA) datasets. We found that Ex-CGI DNA was hypermethylated and was positively correlated to HOTAIR expression in GI cancers (Figures 2A–2F). In addition, pan-cancer analysis in non-GI cancers with high incidence and mortality rate, including breast carcinoma (BRCA), lung adenocarcinoma (LUAD), head and neck squamous cell carcinoma (HNSC), and kidney renal clear cell carcinoma (KIRC), also revealed a positive correlation between Ex-CGI DNA methylation and HOTAIR expression (2,398 subjects in total) (Figures 2G–2J). Collectively, our results suggested that DNA methylation in Ex-CGI plays an important role in regulating HOTAIR expression in cancers.
DNA methylation in exon CpG island promoted transcription elongation of HOTAIR gene
We next investigated the mechanism of Ex-CGI DNA methylation on HOTAIR expression. Previous studies suggested that intragenic DNA methylation altered chromatin structure to promote transcription process.13 Therefore, we hypothesized that Ex-CGI DNA methylation promoted the HOTAIR transcription through stabilizing protein factors on the intragenic regions. Since de novo exonic DNA methylation pattern is established by DNA methyltransferase DNMT3A and DNMT3B,14 we first profiled DNMT occupancies along the HOTAIR gene region. Upregulation of DNMT3A occupancy, but not DNMT3B nor DNMT1, was observed at Ex-CGI in cancer cells (Figures S4A–S4C). In addition, we found that DNA demethylation of Ex-CGI using 5-Aza in PDAC, HCC, and CRC cells resulted in the decrease in HOTAIR expression (Figures 3A and S4D). Consistently, only the knockdown of DNMT3A, but not DNMT3B nor DNMT1, inhibited HOTAIR transcription (Figures 3B and S4E–S4H). Also, we found that knockdown of DNMT3A inhibited cancer cell growth (Figure S4I). Conversely, in vitro DNA methylation of the HOTAIR gene by SssI DNA methyltransferase induced the expression of HOTAIR in HPDE cells and Cos7 cells where endogenous human HOTAIR was not expressed (Figure 3C). Importantly, CRISPR-based dCas9-SunTag DNMT3A system, which enabled the highly specific DNA methylation at the Ex-CGI, also revealed the upregulation of the HOTAIR expression (Figure 3D). These suggested that DNMT3A-mediated Ex-CGI DNA methylation promotes HOTAIR expression in cancer.
Figure 3.

DNA methylation of Ex-CGI promotes HOTAIR expression through facilitating its transcription elongation
(A and B) (A) 5-Aza treatment or (B) knockdown of DNMT3A inhibited HOTAIR expression in PDAC, HCC, and CRC cells. (C) Transfecting pEGFP-N1 with methylated HOTAIR gene in HPDE cells and Cos7 cells, where endogenous human HOTAIR was not expressed, promoted HOTAIR expression. (D) CRISPR-based dCas9-SunTag DNMT3A system with sgRNA targeting Ex-CGI promoted HOTAIR expression in HPDE cells. (E) Expression of 3′ end of HOTAIR was greatly hindered in HPDE and CFPAC-1 cells with a low Ex-CGI DNA methylation level. (F) Transcription elongation efficiency was significantly decreased in HPDE and CFPAC-1 cells, as revealed by the nuclear run-on assay, followed by the northern dot plot. (G) RNA PolII Ser2 phosphorylation on Ex-CGI was significantly upregulated in PDAC, HCC, and CRC cells. (H) Open chromatin structure was observed in PDAC, HCC, and CRC cells with high Ex-CGI DNA methylation and HOTAIR expression, as revealed by the MspI chromatin accessibility assay. Restriction enzyme MspI mimicked the binding of RNA PolII to the HOTAIR gene. (I) RNA PolII Ser2 phosphorylation on Ex-CGI was reduced after 5-Aza treatment in PANC-1 and SW1990 cells. (J) Nuclear accessibility to Ex-CGI was decreased after inhibition of DNMT in PDAC, HCC, and CRC cells. (K) Transcription elongation efficiency to Ex-CGI was significantly decreased after inhibition of DNMT in PDAC and HCC cells. Data are from at least three independent experiments and plotted as means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
We further investigated the detailed mechanism of how Ex-CGI DNA methylation promoted HOTAIR expression in cancer. We first profiled the expression of different exons in PDAC cells. We found that although transcription of exon 2 occurred in both non-tumor and cancer cells, transcription after Ex-CGI was greatly hindered in HPDE and CFPAC-1 cells with low Ex-CGI DNA methylation level (Figures 3E and 3F). ChIP assay also revealed that Ser2-phosphorylated RNA PolII, which is involved in transcription elongation,15 was significantly enriched at Ex-CGI in PDAC, HCC, and CRC cells with high Ex-CGI DNA methylation level and HOTAIR expression (Figures 3G, S5A, and S5B). Furthermore, ChIP sequencing (ChIP-seq) showed an increase in Ser2-phosphorylated RNA PolII binding at Ex-CGI in cancer cells, as compared with non-tumor cells (Figure S5C). Also, open chromatin structure was observed in PDAC, HCC, and CRC cells with high Ex-CGI DNA methylation and HOTAIR expression (Figure 3H). In contrast, DNA demethylation at Ex-CGI significantly reduced the chromatin accessibility of Ex-CGI, the occupancy of Ser2-phosphorylated RNA PolII, and transcription efficiency downstream Ex-CGI (Figures 3I–3K). These results suggested that Ex-CGI DNA methylation plays an important role in stabilizing the HOTAIR transcription process. In tumor cells with methylated DNA at Ex-CGI, chromatin is maintained in an open state, which facilitates the transcription of HOTAIR, and in turn allows the complete transcription of HOTAIR RNA.
MLL1-mediated H3K4me3 promoted the establishment of exon CpG island DNA methylation
DNMT3A and DNMT3B recognize intragenic histone modifications, which were the marks of active transcription elongation process,16,17 to promote de novo intragenic methylation.18,19 To investigate histone modification-mediated Ex-CGI DNA methylation, we performed ChIP assay to profile the histone modification status along the HOTAIR gene region. We found that H3K4me3, but not the total H3, and its methyltransferase MLL1 were significantly enriched at Ex-CGI (Figures 4A, 4B, and S5D). ChIP-seq also showed the increase in H3K4me3 at Ex-CGI in cancer cells, as compared with non-tumor cells (Figure S5E). Moreover, knockdown of MLL1 inhibited H3K4me3 occupancy and DNA methylation at Ex-CGI in PDAC, HCC, and CRC cells (Figures 4C, 4D, and S5F). This in turn hindered the transcription elongation process by RNA PolII and inhibited HOTAIR expression (Figures 4D and 4E). Collectively, our results suggested that H3K4me3 promotes DNMT3A-mediated Ex-CGI DNA methylation and HOTAIR expression.
Figure 4.

MLL1-H3K4me3 promotes Ex-CGI DNA methylation and HOTAIR expression
(A and B) ChIP analysis revealed that the occupancies of (A) H3K4me3 and (B) MLL1 on Ex-CGI were significantly increased in PDAC, HCC, and CRC cells. Enrichment levels of H3K4me3 and MLL1 in cancer cells were compared with corresponding non-tumor cells. (C–E) (C) The DNA methylation levels of Ex-CGI, (D) MLL1 occupancies, H3K4me3 and RNA PolII Ser2 phosphorylation on the HOTAIR Ex-CGI, and (E) HOTAIR expression were decreased after knockdown of MLL1 in PDAC, HCC, and CRC cells. Data are from at least three independent experiments and plotted as means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001
CDK7-CDK9 axis promoted HOTAIR expression by induction of Ex-CGI H3K4 trimethylation and DNA methylation
We further explored the regulatory mechanism of MLL1-H3K4me3-Ex-CGI DNA methylation in regulating HOTAIR expression. Preceding works suggested the role of positive transcription elongation factor b (p-TEFb) on promoting transcription elongation through indirectly mediating epigenetic changes in the coding region.20,21 We hypothesized that p-TEFb promoted the H3K4me3-Ex-CGI-DNA-methylation to facilitate transcription of HOTAIR in PDAC, HCC, and CRC cells. First, the pharmacological suppression of p-TEFb by DRB inhibited HOTAIR expression (Figure S6A). Also, inhibition of p-TEFb regulator Brd4,22 which promotes PDAC progression,23 by PFI-2 inhibited HOTAIR expression (Figure S6B). Small interfering RNA-mediated inhibition and pharmacological inhibition of CDK9, which is the key component of p-TEFb, by LDC-06724 in cancer cells resulted in a reduction of Ex-CGI DNA methylation and HOTAIR expression (Figures 5A and S6C–S6F). The lack of CDK9 activity also impaired the transcription elongation process, resulting in the failure in producing full-length functional HOTAIR transcript (Figures 5B–5E and S6G). Since CDK9 can be regulated by multiple pathways, which target different phosphorylation sites on CDK9,25 we next investigated which CDK9 regulator was involved in HOTAIR expression. Knockdown of CDK7 (but not other CDK9 regulators CDK2 and PP1) inhibited Ex-CGI DNA methylation and HOTAIR expression (Figure S7A–S7D). Also, pharmacological inhibition of CDK7 by THZ126 in cancer cells inhibited Ex-CGI methylation and HOTAIR expression, with Protein Kinase D1 (PRKD1), MYB, and Baculoviral IAP Repeat Containing 3 (BIRC3) as the positive controls (Figures 6A, S7E, and S7F). Furthermore, Ser2-phosphorylated RNA PolII binding along the HOTAIR gene after CDK7 inhibition in cancer was analyzed by ChIP-seq. We found that inhibition of CDK7 decreased the RNA PolII Ser-2 phosphorylation along the HOTAIR gene (Figure S7G), which suggested that CDK7-CDK9 promotes HOTAIR expression through Ex-CGI H3K4me3 and DNA methylation.
Figure 5.

CDK9 promotes transcription of HOTAIR in cancers
(A) HOTAIR expression was significantly inhibited after inhibiting CDK9 by LDC-067 in PDAC, HCC, and CRC cells. (B and C) ChIP analysis revealed that the occupancies of (B) H3K4me3 and (C) RNA PolII Ser2 phosphorylation on Ex-CGI were greatly decreased after inhibiting CDK9. (D) The nuclear accessibility to the 3′ end of HOTAIR gene was inhibited after inhibition of CDK9 in PDAC, HCC, and CRC cells. MspI mimicked the binding of RNA PolII to the HOTAIR gene. (E) Nuclear run-on qPCR analysis revealed the transcription elongation efficiency of HOTAIR after inhibition of CDK9 in PDAC and HCC cells. The expression of exon 6 was compared with the expression of exon 2. Transcription efficiency of exon 6 was decreased after inhibition of CDK9. Data are from at least three independent experiments and plotted as means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Figure 6.

The upregulated of CDK7-CDK9 axis promotes HOTAIR expression
(A) HOTAIR expression was inhibited after inhibiting CDK7 by THZ1 in PDAC, HCC, and CRC cells. (B–E) (B) CDK7, (C) CDK7 (T170) phosphorylation (p-CDK7), (D) CDK9, and (E) CDK9 (T170) phosphorylation (p-CDK9) were upregulated in PDAC tumors (n = 42). CDK7, p-CDK7, and p-CDK9 were positively correlated with HOTAIR expression level. Scale bar, 200 μm. (F) CDK7 expression was positively correlated with HOTAIR expression in PAAD, LIHC, BRCA, KIRC, OV, and STAD; TCGA datasets, n = 2,482 subjects. (F) p-CDK9 on Ex-CGI was significantly reduced in PDAC and CRC cells after 5-Aza treatment. Data are from at least three independent experiments and plotted as means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To study the clinical significance of CDK7-CDK9-HOTAIR axis, we examined the phosphorylation level of CDK7 (CDK7 phosphor T170, p-CDK7) and CDK9 (CDK9 phosphor T186, p-CDK9) in PDAC cells and primary tumors. We found that CDK7 and CDK9 were robustly phosphorylated in PDAC cells and primary tumors and were positively associated with HOTAIR expression level (Figures 6B–6E and S8A). Pan-cancer analysis of TCGA datasets also revealed the positive correlation between CDK7 and HOTAIR expression levels in PAAD, LIHC, BRCA, KIRC, ovarian cancer (OV), and STAD (2,482 samples in total) (Figure 6F). Also, we found that knockdown of CDK7 and CDK9 inhibited the growth of cancer cells (Figures S8B and S8C). More importantly, we demonstrated that DNA methylation played a role in recruiting CDK9 to the HOTAIR gene. DNA demethylation induced by 5-Aza could reduce the binding of p-CDK9 at the Ex-CGI of HOTAIR (Figure 6G). As such, it suggested that intragenic methylation at the exon of HOTAIR could promote the binding of CDK9, and enhanced the elongation process via RNA PolII phosphorylation. The positive feedback loop involving the methylation of H3K4 and exon CpG island and the recruitment of CDK9 would then be established, which allowed the persistent expression of HOTAIR. Collectively, these results demonstrated the CDK7-CDK9 axis contributes to the establishment of a positive feedback loop that corporates Ser2 phosphorylation of RNA PolII, induction of Ex-CGI H3K4 and DNA methylation, and recruitment of more CDK9, and in turn promotes persistent HOTAIR overexpression (Figure 7).
Figure 7.

Schematic diagram describing the role of CDK7-CDK9-mediated intragenic H3K4me3 and DNA methylation in promoting HOTAIR transcription in cancer
Phosphorylation of oncogenic CDK7 (T170) activates CDK9, which is the key component of p-TEFb, by phosphorylating T186, which in turn promoted the elongation process of HOTAIR by activating RNA Polymerase II Ser2. Ex-CGI DNA methylation in the HOTAIR gene was regulated by MLL1-mediated H3K4 trimethylation during the transcription elongation process. The DNA methylation of Ex-CGI enhanced the elongation process, resulting in the upregulation of HOTAIR in cancers.
Discussion
HOTAIR, one of the well-characterized lncRNAs, is frequently upregulated in cancers. Studies have demonstrated that HOTAIR plays important roles in promoting cancer progression by regulating gene expression.1 HOTAIR guilds the EZH2/PRC2 complex to tumor suppressor for triggering gene silencing H3K27me3.2 Also, HOTAIR interacts with H3K4 demethylase LSD1 and induces gene silencing.4 However, the mechanism contributing to the deregulation of HOTAIR in cancers is still largely unknown. A previous report hypothesized that methylation of DS-CGI, which is located between the HOTAIR gene and HOXC12 gene, facilitated the transcription of HOTAIR in breast cancer.6 Here, instead of DS-CGI, DNA methylation of Ex-CGI, which overlaps with exon 4 of the HOTAIR gene, promoted HOTAIR expression by facilitating the transcription of full-length transcript. Importantly, we demonstrated the intragenic DNA methylation was regulated by CDK7-CDK9-RNA PolII-mediated H3K4me3.
DNA methylation at the cytosine residues has been reported to be an important regulator of gene expression. DNA methylation at the promoter frequently links to gene silencing. However, only 10% to 20% of DNA methylation occurs at the promoter, while up to 60% is at the intragenic regions.7,8 Intragenic or specifically exon DNA methylation may result in either gene activation or gene silencing. Genome-wide and pan-cancer analysis reveal both positive and negative correlations between intragenic DNA methylation and gene expression.27 DNA methylation at the first exon inhibits gene expression by blocking transcription initiation.28 Genes with hypomethylated intragenic DNA are maintained in an open chromatin.9 Also, intragenic regions with DNA methylation are depleted of RNA PolII binding and histone markers of active transcription elongation.11 On the other hand, another study found that oncogenes were frequently hypermethylated in intragenic DNA and 5′UTR DNA.29 This intragenic DNA hypermethylation promotes gene expression by maintaining open chromatin structure.30,31 Furthermore, several reports found that the blockage of spurious RNA PolII entry to the intragenic cryptic transcript start sites (TSS) by intragenic DNA methylation could prevent cryptic transcription initiation, and in turn facilitated transcription of canonical transcription.32,33 Therefore, further studies are required to explore the detailed roles of intragenic methylation on gene expression.
Here, we identified two intragenic CGIs (Intron-CGI and Ex-CGI) in the HOTAIR gene region. Recent study revealed that HOTAIR ancient sequence, which overlaps with Ex-CGI, played important roles in regulating the expressions of HoxC and HoxD cluster genes.12 We observed the DNA hypermethylation of Ex-CGI and its positive correlation with HOTAIR expression in cancers. Although many cancers, particularly BRCA, LUAD, HNSC, and ESCA, show a moderate correlation between hypermethylation of Ex-CGI and HOTAIR expression, a few cancer types have a relatively week correlation. There may be some CpG sites at the Ex-CGI that may also heavily contribute to HOTAIR expression that are not covered by the DNA methylation microarray used in the TCGA datasets. A more comprehensive measurement of CpG methylation in TCGA tumors can help to further confirm the correlation between Ex-CGI DNA methylation and HOTAIR expression in cancers. Notably, depletion of Ex-CGI DNA methylation by 5-Aza or knockdown of DNMT3A resulted in the formation of heterochromatin, and in turn, impaired RNA PolII transcription elongation process and decreased in HOTAIR expression. On the other hand, we found that in vitro DNA methylation of Ex-CGI promoted HOTAIR expression. Although HOTAIR is expressed in both normal cells and cancer cells, intragenic methylation of HOTAIR had no effect on its expression in normal cells.34 These results suggested that the cancer-specific DNA methylation of Ex-CGI promotes HOTAIR expression by facilitating the transcription elongation process.
Although many studies suggested that intragenic DNA methylation is involved in gene regulation, how it is regulated is largely unexplored. Several studies demonstrated the link between DNA methylation and histone modifications in regulating gene expression.35, 36, 37 For instance, DNA methylation and H3K4me3 have an opposite transcriptional activity on gene promoter.36 DNA methylation and H3K9 methylation often co-exist on same promoter, resulting in gene silencing.37 Also, in the intragenic region, PWWP domain of DNMT3A and DNMT3B recognizes H3K36me3, which is a hallmark of the active transcription elongation process, to promote de novo intragenic DNA methylation.11,18,21 Mutation in H3K36 methyltransferase SETD2 resulted in DNA hypomethylation and heterochromatin formation.38,39 In addition, the establishment of intragenic DNA methylation further induces the histone modification for the transcription process.40 Here, we observed the increased H3K4me3 level, which is also a hallmark histone modification of active transcription elongation,41, 42, 43 at Ex-CGI in cancer cells with high HOTAIR expression and Ex-CGI DNA methylation. Also, knockdown of H3K4me3 methyltransferase MLL1 reduced Ex-CGI DNA methylation level and HOTAIR expression. These suggested that DNMT3A recognizes H3K4me3 to promote Ex-CGI DNA methylation and HOTAIR expression.
CDK9, a key component of p-TEFb, and its regulator CDK7 are fundamentally important for the formation of transcription elongation complex, by releasing the RNA PolII from the promoter and triggering transcription elongation processes. CDK9 phosphorylates the Ser2 position of the highly conserved heptad repeat of RNA PolII subunit Rpb1 C-terminal domain, which is the regulatory unit of RNA PolII, for the recruiting of other members of the transcription elongation complex, including WAC and PAF.21 Inhibition of either CDK7 or CDK9 decreased transcription elongation rate by pausing RNA PolII.44,45 Also, studies also demonstrated the link between CDK7-CDK9 and intragenic histone modification markers in promoting the transcription elongation process.43 Through CDK7-CDK9-mediated RNA PolII Ser2 phosphorylation, histone modification enzymes such as SETD1 and SETD2 are recruited to the gene body for the establishment of elongation-promoting H3K4 and H3K36 methylation.21,44 Upon inhibition of CDK7 and CDK9, the spreading of H3K4me3 and H3K36me3 downstream of the TSS were greatly reduced.44 In this study, we found that the increase in H3K4me3 at Ex-CGI, which contributed to Ex-CGI DNA methylation and HOTAIR expression, was regulated by CDK7-CDK9 axis. Inhibition of CDK9 decreased the H3K4me3 and DNA methylation level at Ex-CGI, and in turn, inhibited HOTAIR expression. These suggested the importance of CDK7-CDK9-dependent RNA PolII Ser2 phosphorylation and intragenic histone modifications in the transcription elongation process.
Furthermore, several reports in many cancer types revealed the upregulation of CDK7 and CDK9 in promoting the expression of oncogenes.26,45, 46, 47, 48 Inhibition of either CDK7 or CDK9 may serve as novel therapeutic targets for multiple cancers.4,49, 50, 51, 52 In this study, we found that CDK7 and its phosphorylated form p-CDK7, and its downstream target CDK9 and the phosphorylated form p-CDK9 were frequently upregulated in PDAC. Pan-cancer analysis revealed the positive correlation between CDK7 and HOTAIR expression. Further pan-cancer analysis on the protein level of CDK7 and its phosphorylated form will confirm the role of CDK7 on regulating Ex-CGI DNA methylation and HOTAIR expression in cancers. Also, knockdown of either CDK7 or CDK9 inhibited cancer growth. Importantly, inhibition of these transcription elongation regulators CDK7 and CDK9 inhibited Ex-CGI DNA methylation and HOTAIR expression. Importantly, depletion of Ex-CGI DNA methylation reduced the enrichment levels of p-CDK9 at the gene body of HOTAIR. A study in leukemia also reported that 5-Aza treatment inhibited transcription elongation-related RNA PolII Ser2 phosphorylation.53 These suggested the presence of a feedback loop that intragenic DNA methylation recruits CDK9 for the persistent expression of HOTAIR.
Conclusions
In summary, we unraveled a novel molecular pathway in upregulating oncogenic lncRNA in cancer, which links up intragenic histone methylation, DNA methylation, and RNA transcription elongation. We revealed for the first time that the oncogenic CDK7-CDK9-H3K4me3 axis regulates Ex-CGI DNA methylation, and then leads to subsequent HOTAIR expression in cancers. The identification of this pathway can provide a novel therapeutic potential in targeting oncogenic lncRNAs, which are ubiquitously overexpressed in multiple cancer types with diverse genetic profiles.
Materials and methods
Cell culture and drug treatment
PDAC cell lines PANC-1, SW1990, CAPAN-2, CFPAC-1, PANC0403, and BxPC-3; HCC cell lines HepG2, Hep3B, and PLC/PRF/5 (PLC); CRC cell lines DLD-1, SW1116, SW480, HCT116, HT29, and LS180; breast cancer cell lines MDA-MB-231, MCF7, MDA-MB-415, and MDA-MB-453; mammary non-tumorigenic epithelial cell line MCF10A; HEK293, and HEK293T were purchased from American Type Culture Collection. The HPDE cell line was gifted from Dr. Ming-Sound Tsao (University Health Network, Ontario Cancer Institute and Princess Margaret Hospital Site, Toronto).54 The non-tumorigenic human hepatocyte cell line MIHA was kindly provided by Dr. J.R. Chowdhury's laboratory at Albert Einstein College of Medicine.55 The human HCC cell line Huh7 was obtained from Dr. H. Nakabayashi, Hokkaido University School of Medicine, Sapporo, Japan,56 and Bel-7404 was obtained from Cell Bank of the Chinese Academy of Science.57 HPDE cells were cultured in keratinocyte serum free medium supplemented with 50 μg/mL bovine pituitary extract, 0.2 ng/mL human epithelial growth factor (Invitrogen, Waltham, MA, USA) and 3% antibiotic and antimycotic (ThermoFisher Scientific, Waltham, MA, USA). BxPC-3 cells were maintained in RPMI-1640 supplemented with 10% fetal bovine serum (FBS) and 100 units/mL penicillin and 100 μg/mL streptomycin (ThermoFisher Scientific). The remaining cell lines were maintained in DMEM containing 10% FBS, 100 units/mL penicillin, and 100 μg/mL streptomycin. For drug treatment, cells were treated with 5-azacytidine (5-Aza) (Sigma, St. Louis, MO, USA), THZ1 (MedChem Express, Monmouth Junction, NJ, USA), LDC-067 (MedChem Express), PFI-2 (ApexBio, Houston, TX, USA), and DRB (ApexBio) for 72 h before the experiment as described.
Clinical sample and histology
Sixty pairs of PDAC tumors and adjacent non-tumor tissues were obtained from patients who underwent pancreatic resection at the Prince of Wales Hospital, Hong Kong.58 The study was carried out according to the ethical guidelines and with the approval of the Joint CUHK-NTEC Clinical Research Ethics Committee in accordance with Declaration of Helsinki. Written informed consent was obtained from all patients recruited. PDAC tissue specimens were fixed in 4% buffered formalin for 24 h and stored in 70% ethanol until paraffin embedding; 5-μm sections were stained with H&E to locate tumors and adjacent non-tumor tissues.
Constructs and cell transduction
The lentiviral vector with full length of HOTAIR was purchased from abm (Richmond, Canada). To achieve stable ectopic expression, lentivirus was produced by co-transfecting HEK293T cells with HOTAIR-containing lentiviral vector and three packaging vectors: pMDLg/pRRE, pRSV-REV, and pCMV-VSVG, as previously described.59,60 Then HPDE cells were transduced by lentivirus with polybrene, followed by antibiotic selection. The efficiency of HOTAIR overexpression was validated by qRT-PCR.
Quantitative methylation-specific PCR
DNA was isolated from cell lines by NK Lysis Buffer.61 DNA from formalin-fixed paraffin-embedded (FFPE) samples was isolated by QIAamp DNA FFPE Tissue Kit (Qiagen) according to the manufacturer's protocol. Bisulfite conversion was performed using the EZ DNA Methylation-Lightning Kit (Zymo Research, Irvine, CA, USA). The methylation state of HOTTIP CpG island was analyzed by both PCR and qPCR using methylation-specific primers and unmethylation-specific primers.
Pyrosequencing
Each pyrosequencing primer set consisted of one unlabeled forward primer, one biotinylated reverse primer, and one sequencing primer. Four sequencing primers specific to bisulfite-treated exon CpG island were used to analyze the methylation status of each CpG site. Pyrosequencing was performed on the PSQ 96MA system (Qiagen) and the resulting methylation percentage of cytosine in each CpG dinucleotide was calculated by Pyro Q-CpG software.
In vitro DNA methylation assay
HOTAIR gene was cloned into expression vector pEGFP-N1. In vitro DNA methylation was performed by CpG Methyltransferase (M.SssI) (New England Biolabs, Ipswich, MA, USA) in the presence of 320 μM S-adenosylmethionine (New England Biolabs). pEGFP-N1 with HOTAIR being methylated was transfected in Cos7 cells and HPDE cells using Lipofectamine 3000 Reagent (Thermo Fisher Scientific) and P3000 Reagent (Thermo Fisher Scientific). RNA was extracted by TRIzol Reagent (ThermoFisher) and analyzed by qRT-PCR.
Targeted DNA methylation of the Ex-CGI
Targeted Ex-CGI DNA methylation was performed using the CRISPR-based dCas9-SunTag DNMT3A system, which was a gift from Ryan Lister.62 HPDE cells were transfected with pEF1a-NLS-scFvGCN4-DNMT3a (Addgene plasmid # 100941; http://n2t.net/addgene:100941; RRID:Addgene_100941), pGK-dCas9-SunTag-BFP (Addgene plasmid # 100957; http://n2t.net/addgene:100957; RRID:Addgene_100957), and Ex-CGI-specific single guide RNA (sgRNA) for the targeted DNA methylation at the Ex-CGI. The sgRNA target sequences are listed in Table S1.
Nuclear run-on assay
Nuclear run-on assay was performed according to reported protocols with modifications.63 Details are provided in the supplemental information.
Chromatin accessibility assay
The accessibility of the HOTAIR gene was assayed with MspI digestion, which mimicked the binding of RNA Polymerase II, according to the previous report with minor modifications.64 Details are provided in the supplemental information.
Analysis of publicly available datasets
Publicly available patient datasets (PAAD, LIHC, COAD, STAD, ESCA, BLCA, BRCA, LUAD, HNSC, KIRC, and OV) were obtained from TCGA. Processed data were used in the analysis of CDK7 and HOTAIR expression. Processed data in beta value were used in the analysis of Ex-CGI DNA methylation level. For the association analysis between HOTAIR expression and Ex-CGI DNA methylation level, and between HOTAIR expression and CDK7 expression, linear regression was used.
Publicly available ChIP-seq data of RNA Polymerase II (RNA PolII) and RNA Polymerase II Serine 2 (RNA PolII Ser2) phosphorylation were obtained from Gene Expression Omnibus (GEO) GSE33281, GSE20040, GSE100040, GSE97589, GSE132233, GSE104545, and Encyclopedia of DNA Elements (ENCODE).38,65, 66, 67, 68, 69, 70 ChIP-seq data of H3K4me3 and total H3 were obtained from ENCODE, GEO GSE142579, GSE113336, GSE78158, GSE76344, GSE91401, GSE103728, GSE117306, and GSE103734.70, 71, 72, 73, 74, 75, 76, 77, 78 The processed data were visualized by The Integrative Genomics Viewer.
Statistical analysis
GraphPad Prism 7 (GraphPad Software) was used for statistical analysis. Two-tailed Student's t test was used to compare the differences between two groups, unless noted otherwise. Data are represented as mean ± SD. A p value less than 0.05 was considered statistically significant. For association study between CGI methylation and HOTAIR expression level in PDAC tissues, we first calculated the beta value of each CpG site in each sample and compared with the beta value of their corresponding adjacent non-tumor tissue.79 Then mean beta value in each pair of PDAC tissues was used for correlation analysis with their corresponding relative HOTAIR expression.
Detailed materials and methods can be found in the supplemental information.
Data availability
Microarray data are available at ArrayExpress E-MTAB-7305.
Acknowledgments
This work was supported by General Research Fund, Research Grants Council of Hong Kong [CUHK462713, CUHK14102714, CUHK14171217, CUHK14120618, and CUHK14120419 to Y.C.]; National Natural Science Foundation of China [81672323 to Y.C.]; and Direct Grant from CUHK to Y.C.
Author contributions
C.H.W. and C.H.L. designed and performed the experiments and drafted the manuscript. Q.H., U.K.L., Z.L., and J.W. assisted in obtaining data. J.H.M.T. and K.F.T. provided some research materials. D.Z. assisted in preparation of the experiments. Y.C. was the PI of the grant, oversaw the whole progress, and revised the manuscript.
Declaration of interests
The authors declare that they have no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.01.038.
Supplemental information
References
- 1.Tang Q., Hann S.S. HOTAIR: an oncogenic long non-coding RNA in human cancer. Cell Physiol. Biochem. 2018;47:893–913. doi: 10.1159/000490131. [DOI] [PubMed] [Google Scholar]
- 2.Li C.H., Xiao Z., Tong J.H., To K.F., Fang X., Cheng A.S., Chen Y. EZH2 coupled with HOTAIR to silence microRNA-34a by the induction of heterochromatin formation in human pancreatic ductal adenocarcinoma. Int. J. Cancer. 2017;140:120–129. doi: 10.1002/ijc.30414. [DOI] [PubMed] [Google Scholar]
- 3.Liu X.H., Liu Z.L., Sun M., Liu J., Wang Z.X., De W. The long non-coding RNA HOTAIR indicates a poor prognosis and promotes metastasis in non-small cell lung cancer. BMC Cancer. 2013;13:464. doi: 10.1186/1471-2407-13-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang H., Diab A., Fan H., Mani S.K., Hullinger R., Merle P., Andrisani O. PLK1 and HOTAIR accelerate proteasomal degradation of SUZ12 and ZNF198 during hepatitis B virus-induced liver carcinogenesis. Cancer Res. 2015;75:2363–2374. doi: 10.1158/0008-5472.CAN-14-2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antequera F. Structure, function and evolution of CpG island promoters. Cell Mol. Life Sci. 2003;60:1647–1658. doi: 10.1007/s00018-003-3088-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu L., Zhu G., Zhang C., Deng Q., Katsaros D., Mayne S.T., Risch H.A., Mu L., Canuto E.M., Gregori G., et al. Association of large noncoding RNA HOTAIR expression and its downstream intergenic CpG island methylation with survival in breast cancer. Breast Cancer Res. Treat. 2012;136:875–883. doi: 10.1007/s10549-012-2314-z. [DOI] [PubMed] [Google Scholar]
- 7.Nones K., Waddell N., Song S., Patch A.M., Miller D., Johns A., Wu J., Kassahn K.S., Wood D., Bailey P., et al. Genome-wide DNA methylation patterns in pancreatic ductal adenocarcinoma reveal epigenetic deregulation of SLIT-ROBO, ITGA2 and MET signaling. Int. J. Cancer. 2014;135:1110–1118. doi: 10.1002/ijc.28765. [DOI] [PubMed] [Google Scholar]
- 8.Dong L., Ma L., Ma G.H., Ren H. Genome-wide analysis reveals DNA methylation alterations in obesity associated with high risk of colorectal cancer. Sci. Rep. 2019;9:5100. doi: 10.1038/s41598-019-41616-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aran D., Toperoff G., Rosenberg M., Hellman A. Replication timing-related and gene body-specific methylation of active human genes. Hum. Mol. Genet. 2011;20:670–680. doi: 10.1093/hmg/ddq513. [DOI] [PubMed] [Google Scholar]
- 10.Maunakea A.K., Nagarajan R.P., Bilenky M., Ballinger T.J., D'Souza C., Fouse S.D., Johnson B.E., Hong C., Nielsen C., Zhao Y., et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–257. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singer M., Kosti I., Pachter L., Mandel-Gutfreund Y. A diverse epigenetic landscape at human exons with implication for expression. Nucleic Acids Res. 2015;43:3498–3508. doi: 10.1093/nar/gkv153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nepal C., Taranta A., Hadzhiev Y., Pundhir S., Mydel P., Lenhard B., Müller F., Andersen J.B. Ancestrally duplicated conserved noncoding element suggests dual regulatory roles of HOTAIR in cis and trans. iScience. 2020;23:101008. doi: 10.1016/j.isci.2020.101008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lorincz M.C., Dickerson D.R., Schmitt M., Groudine M. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat. Struct. Mol. Biol. 2004;11:1068–1075. doi: 10.1038/nsmb840. [DOI] [PubMed] [Google Scholar]
- 14.Okano M., Bell D.W., Haber D.A., Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 15.Komarnitsky P., Cho E.J., Buratowski S. Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 2000;14:2452–2460. doi: 10.1101/gad.824700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morselli M., Pastor W.A., Montanini B., Nee K., Ferrari R., Fu K., Bonora G., Rubbi L., Clark A.T., Ottonello S., et al. In vivo targeting of de novo DNA methylation by histone modifications in yeast and mouse. Elife. 2015;4:e06205. doi: 10.7554/eLife.06205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wen H., Li Y., Xi Y., Jiang S., Stratton S., Peng D., Tanaka K., Ren Y., Xia Z., Wu J., et al. ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature. 2014;508:263–268. doi: 10.1038/nature13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhayalan A., Rajavelu A., Rathert P., Tamas R., Jurkowska R.Z., Ragozin S., Jeltsch A. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem. 2010;285:26114–26120. doi: 10.1074/jbc.M109.089433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hahn M.A., Wu X., Li A.X., Hahn T., Pfeifer G.P. Relationship between gene body DNA methylation and intragenic H3K9me3 and H3K36me3 chromatin marks. PLoS One. 2011;19:e18844. doi: 10.1371/journal.pone.0018844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eissenberg J.C., Shilatifard A. Leaving a mark: the many footprints of the elongating RNA polymerase II. Curr. Opin. Genet. Dev. 2006;16:184–190. doi: 10.1016/j.gde.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 21.Tanny J.C. Chromatin modification by the RNA Polymerase II elongation complex. Transcription. 2004;5:e988093. doi: 10.4161/21541264.2014.988093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Itzen F., Greifenberg A.K., Bösken C.A., Geyer M. Brd4 activates P-TEFb for RNA polymerase II CTD phosphorylation. Nucleic Acids Res. 2014;42:7577–7590. doi: 10.1093/nar/gku449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y.H., Sui Y.N., Yan K., Wang L.S., Wang F., Zhou J.H. BRD4 promotes pancreatic ductal adenocarcinoma cell proliferation and enhances gemcitabine resistance. Oncol. Rep. 2015;33:1699–1706. doi: 10.3892/or.2015.3774. [DOI] [PubMed] [Google Scholar]
- 24.Albert T.K., Rigault C., Eickhoff J., Baumgart K., Antrecht C., Klebl B., Mittler G., Meisterernst M. Characterization of molecular and cellular functions of the cyclin-dependent kinase CDK9 using a novel specific inhibitor. Br. J. Pharmacol. 2014;171:55–68. doi: 10.1111/bph.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nekhai S., Petukhov M., Breuer D. Regulation of CDK9 activity by phosphorylation and dephosphorylation. Biomed. Res. Int. 2014;2014:964964. doi: 10.1155/2014/964964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwiatkowski N., Zhang T., Rahl P.B., Abraham B.J., Reddy J., Ficarro S.B., Dastur A., Amzallag A., Ramaswamy S., Tesar B., et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511:616–620. doi: 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spainhour J.C., Lim H.S., Yi S.V., Qiu P. Correlation patterns between DNA methylation and gene expression in the cancer genome atlas. Cancer Inform. 2019;18 doi: 10.1177/1176935119828776. 1176935119828776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brenet F., Moh M., Funk P., Feierstein E., Viale A.J., Socci N.D., Scandura J.M. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS One. 2011;6:e14524. doi: 10.1371/journal.pone.0014524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arechederra M., Daian F., Yim A., Bazai S.K., Richelme S., Dono R., Saurin A.J., Habermann B.H., Maina F. Hypermethylation of gene body CpG islands predicts high dosage of functional oncogenes in liver cancer. Nat. Commun. 2018;9:3164. doi: 10.1038/s41467-018-05550-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang X., Han H., De Carvalho D.D., Lay F.D., Jones P.A., Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26:577–590. doi: 10.1016/j.ccr.2014.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu H., Coskun V., Tao J., Xie W., Ge W., Yoshikawa K., Li E., Zhang Y., Sun Y.E. Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science. 2010;329:444–448. doi: 10.1126/science.1190485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jjingo D., Conley A.B., Yi S.V., Lunyak V.V., Jordan I.K. On the presence and role of human gene-body DNA methylation. Oncotarget. 2012;3:462–474. doi: 10.18632/oncotarget.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neri F., Rapelli S., Krepelova A., Incarnato D., Parlato C., Basile G., Maldotti M., Anselmi F., Oliviero S. Intragenic DNA methylation prevents spurious transcription initiation. Nature. 2017;543:72–77. doi: 10.1038/nature21373. [DOI] [PubMed] [Google Scholar]
- 34.Kalwa M., Hänzelmann S., Otto S., Kuo C.C., Franzen J., Joussen S., Fernandez-Rebollo E., Rath B., Koch C., Hofmann A., et al. The lncRNA HOTAIR impacts on mesenchymal stem cells via triple helix formation. Nucleic Acids Res. 2016;44:10631–10643. doi: 10.1093/nar/gkw802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kondo Y. Epigenetic cross-talk between DNA methylation and histone modifications in human cancers. Yonsei Med. J. 2009;50:455–463. doi: 10.3349/ymj.2009.50.4.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X., Wang X., He K., Ma Y., Su N., He H., Stolc V., Tongprasit W., Jin W., Jiang J., et al. High-resolution mapping of epigenetic modifications of the rice genome uncovers interplay between DNA methylation, histone methylation, and gene expression. Plant Cell. 2008;20:259–276. doi: 10.1105/tpc.107.056879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baylin S.B., Jones P.A. Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 2016;8:a019505. doi: 10.1101/cshperspect.a019505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ebmeier C.C., Erickson B., Allen B.L., Allen M.A., Kim H., Fong N., Jacobsen J.R., Liang K., Shilatifard A., Dowell R.D., et al. Human TFIIH kinase CDK7 regulates transcription-associated chromatin modifications. Cell Rep. 2017;20:1173–1186. doi: 10.1016/j.celrep.2017.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simon J.M., Hacker K.E., Singh D., Brannon A.R., Parker J.S., Weiser M., Ho T.H., Kuan P.F., Jonasch E., Furey T.S., et al. Variation in chromatin accessibility in human kidney cancer links H3K36 methyltransferase loss with widespread RNA processing defects. Genome Res. 2014;24:241–250. doi: 10.1101/gr.158253.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bewick A.J., Ji L., Niederhuth C.E., Willing E.M., Hofmeister B.T., Shi X., Wang L., Lu Z., Rohr N.A., Hartwig B., et al. On the origin and evolutionary consequences of gene body DNA methylation. Proc. Natl. Acad. Sci. U S A. 2016;113:9111–9116. doi: 10.1073/pnas.1604666113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen K., Chen Z., Wu D., Zhang L., Lin X., Su J., Rodriguez B., Xi Y., Xia Z., Chen X., et al. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat. Genet. 2015;47:1149–1157. doi: 10.1038/ng.3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ding Y., Ndamukong I., Xu Z., Lapko H., Fromm M., Avramova Z. ATX1-generated H3K4me3 is required for efficient elongation of transcription, not initiation, at ATX1-regulated genes. PLoS Genet. 2012;8:e1003111. doi: 10.1371/journal.pgen.1003111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okitsu C.Y., Hsieh J.C., Hsieh C.L. Transcriptional activity affects the H3K4me3 level and distribution in the coding region. Mol. Cell. Biol. 2010;30:2933–2946. doi: 10.1128/MCB.01478-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eissenberg J.C., Shilatifard A., Dorokhov N., Michener D.E. Cdk9 is an essential kinase in Drosophila that is required for heat shock gene expression, histone methylation and elongation factor recruitment. Mol. Genet. Genomics. 2007;277:101–114. doi: 10.1007/s00438-006-0164-2. [DOI] [PubMed] [Google Scholar]
- 45.Greenall S.A., Lim Y.C., Mitchell C.B., Ensbey K.S., Stringer B.W., Wilding A.L., O'Neill G.M., McDonald K.L., Gough D.J., Day B.W., et al. Cyclin-dependent kinase 7 is a therapeutic target in high-grade glioma. Oncogenesis. 2017;15:e336. doi: 10.1038/oncsis.2017.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalan S., Amat R., Schachter M.M., Kwiatkowski N., Abraham B.J., Liang Y., Zhan T., Olson C.M., Larochelle S., Young R.A., et al. Activation of the p53 transcriptional program sensitizes cancer cells to CDK7 inhibitors. Cell Rep. 2017;21:467–481. doi: 10.1016/j.celrep.2017.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y., Zhang T., Kwiatkowski N., Abraham B.J., Lee T.I., Xie S., Yuzugullu H., Von T., Li H., Lin Z., et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell. 2015;163:174–186. doi: 10.1016/j.cell.2015.08.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Booth G.T., Parua P.K., Sansó M., Fisher R.P., Lis J.T. Cdk9 regulates a promoter-proximal checkpoint to modulate RNA polymerase II elongation rate in fission yeast. Nat. Commun. 2018;9:543. doi: 10.1038/s41467-018-03006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chow P.M., Liu S.H., Chang Y.W., Kuo K.L., Lin W.C., Huang K.H. The covalent CDK7 inhibitor THZ1 enhances temsirolimus-induced cytotoxicity via autophagy suppression in human renal cell carcinoma. Cancer Lett. 2020;471:27–37. doi: 10.1016/j.canlet.2019.12.005. [DOI] [PubMed] [Google Scholar]
- 50.Czudor Z., Balogh M., Bánhegyi P., Boros S., Breza N., Dobos J., Fábián M., Horváth Z., Illyés E., Markó P., et al. Novel compounds with potent CDK9 inhibitory activity for the treatment of myeloma. Bioorg. Med. Chem. Lett. 2018;28:769–773. doi: 10.1016/j.bmcl.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 51.Wang L., Hu C., Wang A., Chen C., Wu J., Jiang Z., Zou F., Yu K., Wu H., Liu J., et al. Discovery of a novel and highly selective CDK9 kinase inhibitor (JSH-009) with potent antitumor efficacy in preclinical acute myeloid leukemia models. Invest. New Drugs. 2020;38:1272–1281. doi: 10.1007/s10637-019-00868-3. [DOI] [PubMed] [Google Scholar]
- 52.Zhang H., Pandey S., Travers M., Sun H., Morton G., Madzo J., Chung W., Khowsathit J., Perez-Leal O., Barrero C.A., et al. Targeting CDK9 reactivates epigenetically silenced genes in cancer. Cell. 2018;175:1244–1258. doi: 10.1016/j.cell.2018.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheng J.X., Chen L., Li Y., Cloe A., Yue M., Wei J., Watanabe K.A., Shammo J.M., Anastasi J., Shen Q.J., et al. RNA cytosine methylation and methyltransferases mediate chromatin organization and 5-azacytidine response and resistance in leukaemia. Nat. Commun. 2018;9:1163. doi: 10.1038/s41467-018-03513-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ouyang H., Mou L.J., Luk C., Liu N., Karaskova J., Squire J., Tsao M.S. Immortal human pancreatic duct epithelial cell lines with near normal genotype and phenotype. Am. J. Pathol. 2000;157:1623–1631. doi: 10.1016/S0002-9440(10)64800-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakabayashi H., Taketa K., Miyano K., Yamane T., Sato J. Growth of human hepatoma cell lines with differentiated functions in chemically defined medium. Cancer Res. 1982;42:3858–3863. [PubMed] [Google Scholar]
- 56.Brown J.J., Parashar B., Moshage H., Tanaka K.E., Engelhardt D., Rabbani E., Roy-Chowdhury N., Roy-Chowdhury J. A long-term hepatitis B viremia model generated by transplanting nontumorigenic immortalized human hepatocytes in Rag-2-deficient mice. Hepatology. 2000;31:173–181. doi: 10.1002/hep.510310126. [DOI] [PubMed] [Google Scholar]
- 57.Chen R., Zhu D., Ye X., Shen D., Lu R. Establishment of three human liver carcinoma cell lines and some of their biological characteristics in vitro. Sci. Sin. 1980;23:236–247. [PubMed] [Google Scholar]
- 58.Wong C.H., Li C.H., He Q., Chan S.L., Tong J.H., To K.F., Lin L.Z., Chen Y. Ectopic HOTTIP expression induces noncanonical transactivation pathways to promote growth and invasiveness in pancreatic ductal adenocarcinoma. Cancer Lett. 2020;477:1–9. doi: 10.1016/j.canlet.2020.02.038. [DOI] [PubMed] [Google Scholar]
- 59.Xu F., Li C.H., Wong C.H., Chen G.G., Lai P.B.S., Shao S., Chan S.L., Chen Y. Genome-wide screening and functional analysis identifies tumor suppressor long noncoding RNAs epigenetically silenced in hepatocellular carcinoma. Cancer Res. 2019;79:1305–1317. doi: 10.1158/0008-5472.CAN-18-1659. [DOI] [PubMed] [Google Scholar]
- 60.Wong C.H., Lou U.K., Li Y., Chan S.L., Tong J.H., To K.F., Chen Y. CircFOXK2 promotes growth and metastasis of pancreatic ductal adenocarcinoma by complexing with RNA-binding proteins and sponging MiR-942. Cancer Res. 2020;80:2138–2149. doi: 10.1158/0008-5472.CAN-19-3268. [DOI] [PubMed] [Google Scholar]
- 61.Chen S., Sanjana N.E., Zheng K., Shalem O., Lee K., Shi X., Scott D.A., Song J., Pan J.Q., Weissleder R., et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell. 2015;160:1246–1260. doi: 10.1016/j.cell.2015.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pflueger C., Tan D., Swain T., Nguyen T.V., Pflueger J., Nefzger C., Polo J.M., Ford E., Lister R. A modular dCas9-SunTag DNMT3A epigenome editing system overcomes pervasive off-target activity of direct fusion dCas9-DNMT3A constructs. Genome Res. 2018;28:1193–1206. doi: 10.1101/gr.233049.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roberts T.C., Hart J.R., Kaikkonen M.U., Weinberg M.S., Vogt P.K., Morris K.V. Quantification of nascent transcription by bromouridine immunocapture nuclear run-on RT-qPCR. Nat. Protoc. 2015;10:1198–1211. doi: 10.1038/nprot.2015.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nguyen C.T., Gonzales F.A., Jones P.A. Altered chromatin structure associated with methylation-induced gene silencing in cancer cells: correlation of accessibility, methylation, MeCP2 binding and acetylation. Nucleic Acids Res. 2001;29:4598–4606. doi: 10.1093/nar/29.22.4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang W., Prakash C., Sum C., Gong Y., Li Y., Kwok J.J., Thiessen N., Pettersson S., Jones S.J., Knapp S., et al. Bromodomain-containing protein 4 (BRD4) regulates RNA polymerase II serine 2 phosphorylation in human CD4+ T cells. J. Biol. Chem. 2012;287:43137–43155. doi: 10.1074/jbc.M112.413047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Barski A., Chepelev I., Liko D., Cuddapah S., Fleming A.B., Birch J., Cui K., White R.J., Zhao K. Pol II and its associated epigenetic marks are present at Pol III-transcribed noncoding RNA genes. Nat. Struct. Mol. Biol. 2010;17:629–634. doi: 10.1038/nsmb.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bayles I., Krajewska M., Pontius W.D., Saiakhova A., Morrow J.J., Bartels C., Lu J., Faber Z.J., Fedorov Y., Hong E.S., et al. Ex vivo screen identifies CDK12 as a metastatic vulnerability in osteosarcoma. J. Clin. Invest. 2019;129:4377–4392. doi: 10.1172/JCI127718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Iannelli F., Galbiati A., Capozzo I., Nguyen Q., Magnuson B., Michelini F., D'Alessandro G., Cabrini M., Roncador M., Francia S., et al. A damaged genome's transcriptional landscape through multilayered expression profiling around in situ-mapped DNA double-strand breaks. Nat. Commun. 2017;8:15656. doi: 10.1038/ncomms15656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Trizzino M., Barbieri E., Petracovici A., Wu S., Welsh S.A., Owens T.A., Licciulli S., Zhang R., Gardini A. The tumor suppressor ARID1A controls global transcription via pausing of RNA polymerase II. Cell Rep. 2018;23:3933–3945. doi: 10.1016/j.celrep.2018.05.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Davis C.A., Hitz B.C., Sloan C.A., Chan E.T., Davidson J.M., Gabdank I., Hilton J.A., Jain K., Baymuradov U.K., Narayanan A.K., et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018;46:D794–D801. doi: 10.1093/nar/gkx1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mallm J.P., Iskar M., Ishaque N., Klett L.C., Kugler S.J., Muino J.M., Teif V.B., Poos A.M., Großmann S., Erdel F., et al. Linking aberrant chromatin features in chronic lymphocytic leukemia to transcription factor networks. Mol. Syst. Biol. 2019;15:e8339. doi: 10.15252/msb.20188339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Choi S.H., Gearhart M.D., Cui Z., Bosnakovski D., Kim M., Schennum N., Kyba M. DUX4 recruits p300/CBP through its C-terminus and induces global H3K27 acetylation changes. Nucleic Acids Res. 2016;44:5161–5173. doi: 10.1093/nar/gkw141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bosnakovski D., Gearhart M.D., Toso E.A., Ener E.T., Choi S.H., Kyba M. Low level DUX4 expression disrupts myogenesis through deregulation of myogenic gene expression. Sci. Rep. 2018;8:16957. doi: 10.1038/s41598-018-35150-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Berger A., Brady N.J., Bareja R., Robinson B., Conteduca V., Augello M.A., Puca L., Ahmed A., Dardenne E., Lu X., et al. N-Myc-mediated epigenetic reprogramming drives lineage plasticity in advanced prostate cancer. J. Clin. Invest. 2019;129:3924–3940. doi: 10.1172/JCI127961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Perez S., Gevor M., Davidovich A., Kaspi A., Yamin K., Domovich T., Meirson T., Matityahu A., Brody Y., Stemmer S.M., et al. Dysregulation of the cohesin subunit RAD21 by Hepatitis C virus mediates host-virus interactions. Nucleic Acids Res. 2019;47:2455–2471. doi: 10.1093/nar/gkz052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhao D., Lu X., Wang G., Lan Z., Liao W., Li J., Liang X., Chen J.R., Shah S., Shang X., et al. Synthetic essentiality of chromatin remodelling factor CHD1 in PTEN-deficient cancer. Nature. 2017;542:484–488. doi: 10.1038/nature21357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zheng D., Trynda J., Sun Z., Li Z. NUCLIZE for quantifying epigenome: generating histone modification data at single-nucleosome resolution using genuine nucleosome positions. BMC Genomics. 2019;20:541. doi: 10.1186/s12864-019-5932-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Org T., Hensen K., Kreevan R., Mark E., Sarv O., Andreson R., Jaakma Ü., Salumets A., Kurg A. Genome-wide histone modification profiling of inner cell mass and trophectoderm of bovine blastocysts by RAT-ChIP. PLoS One. 2019;14:e0225801. doi: 10.1371/journal.pone.0225801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Du P., Zhang X., Huang C.C., Jafari N., Kibbe W.A., Hou L., Lin S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Microarray data are available at ArrayExpress E-MTAB-7305.