

Abstract
Mucopolysaccharidosis type IIIA (MPS-IIIA) is an autosomal recessive disorder caused by mutations in SGSH involved in the degradation of heparan sulfate. MPS-IIIA presents severe neurological symptoms such as progressive developmental delay and cognitive decline, for which there is currently no treatment. Brain targeting represents the main challenge for therapeutics to treat MPS-IIIA, and the development of small-molecule-based treatments able to reach the CNS could be a relevant advance for therapy. Using cell-based high content imaging to survey clinically approved drugs in MPS-IIIA cells, we identified fluoxetine, a selective serotonin reuptake inhibitor. Fluoxetine increases lysosomal and autophagic functions via TFEB activation through a RagC-dependent mechanism. Mechanistically, fluoxetine increases lysosomal exocytosis in mouse embryonic fibroblasts from MPS-IIIA mice, suggesting that this process may be responsible for heparan sulfate clearance. In vivo, fluoxetine ameliorates somatic and brain pathology in a mouse model of MPS-IIIA by decreasing the accumulation of glycosaminoglycans and aggregated autophagic substrates, reducing inflammation, and slowing down cognitive deterioration. We repurposed fluoxetine for potential therapeutics to treat human MPS-IIIA disease.
Key words: MPS-IIIA, fluoxetine, lysosomal exocytosis, autophagy, TFEB, high content imaging, drug repurposing, lysosomal storage disorders
Graphical abstract
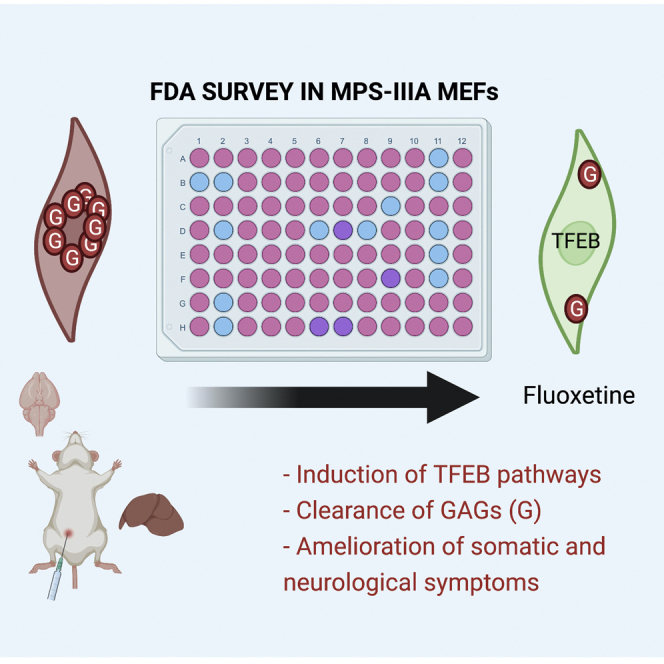
Medina and colleagues combined HCI screening and FDA repurposing to identify correctors of lysosomal dysfunction in MPS-IIIA disease. They identified fluoxetine, an antidepressant that boosts lysosomal function and autophagy and promotes GAG clearance via lysosomal exocytosis in a TFEB-dependent manner. Thus, fluoxetine may be a suitable drug to treat human MPS-IIIA.
Introduction
Mucopolysaccharidosis type IIIA (MPS-IIIA) is a rare inherited neurodegenerative disorder caused by deficiency of the lysosomal enzyme sulfamidase (SGSH), which is responsible for the breakdown of heparan sulfate (HS). Pathological hallmarks of MPS-IIIA are progressive neurodegeneration with subsequent mental decline and a shortened lifespan. Other clinical symptoms include hyperactivity, aggressive behavior, and sleep disturbances.1 The pathogenic cascades recognized in MPS-IIIA cellular and mouse models consist primarily of the accumulation of HS within the lysosomal compartment, lysosomal aggregation in the cellular perinuclear area, and a partial block of autophagy due to defects in autophagosome (AV)-lysosome fusion.2 Due to an inefficient degradation process, the accumulation of aggregate-prone proteins, such as α-synuclein and poly-ubiquitin, has been observed in the MPS-IIIA mouse brain.2,3 Cumulative evidence indicates that targeting lysosomal and autophagic pathways by activating transcription factor EB (TFEB), a positive regulator of these pathways, may represent a novel approach to treat lysosomal storage diseases (LSDs) and more common neurodegenerative diseases.4, 5, 6, 7, 8, 9, 10.
Here, we developed a cell-based high-content imaging (HCI) screening assay for the repurposing of US Food and Drug Administration (FDA)-approved compounds boosting the lysosomal degradation of exogenously added bovine serum albumin (BSA) derivative (DQ-BSA) in MPS-IIIA murine embryonic fibroblasts (MEFs). Upon screening, we identified the antidepressant fluoxetine (FLX) and found that its activation of lysosomal degradative capacity requires the induction of TFEB, a master gene of lysosomal function and autophagy. In vivo, FLX treatment ameliorates somatic and CNS pathology in a spontaneous mouse model of MPS-IIIA by promoting lysosomal exocytosis and reducing glycosaminoglycan (GAG) accumulation. Also, FLX reduces the accumulation of polyubiquitinated proteins and autophagic substrates such as p62 and aggregated α-synuclein. Moreover, FLX decreases neuroinflammation and slows down the cognitive impairment of MPS-IIIA-treated mice. Thus, we identified the CNS-approved drug FLX as a candidate compound to treat MPS disorders by activating lysosomal and autophagic functions via TFEB.
Results
DQ-BSA screening identifies fluoxetine as a candidate drug for MPS-IIIA treatment
While developing a cell-based HCI assay to evaluate the functional capacity of the lysosome to degrade cargo by using a self-quenched BSA (DQ-BSA), we observed that MPS-IIIA MEFs experience a significant impairment in their ability to degrade DQ-BSA when compared with wild-type (WT) counterpart (Figure S1A). Thus, to identify compounds that can promote lysosomal activity in MPS-IIIA cells, we screened an FDA collection of 1,280 approved drugs using a cell-based HCI DQ-BSA assay. The analysis of the Pearson coefficient confirms a good correlation between well-plate replicates (Figure 1A). Compound hits were selected when the mean of “DQ-BSA spots per cell” was higher than the mean of DQ-BSA spots in untreated MPS-IIIA cells plus three times the standard deviation (SD) (Figures S1B and S1C) and p ≤ 0.01 (∗∗) (Figure 1B). Also, we excluded compound hits reducing cell viability to more than 50% of untreated cells. By applying these constraints, we identified 36 compounds inducing a significant elevation of DQ-BSA fluorescence in MPS-IIIA cells (Figure 1C). We found that more than half of the compound hits (58%) are known to cross the blood-brain barrier (BBB), and therefore can reach the CNS, the major MPS-IIIA disease targeted tissue. Interestingly, the 25% of compound hits were selective serotonin reuptake inhibitors (SSRIs), including FLX. FLX is included in the World Health Organization Model List of Essential Medicines and is considered one of the most widely prescribed medications, including for pediatric use.11, 12, 13, 14 Also, some reports have described the neuroprotective and neuroplastic effects of FLX in different physio-pathological conditions.15, 16, 17, 18, 19 Thus, we focused on FLX for further validation by performing a dose-response analysis of its potency in the DQ-BSA assay, which resulted in a half-maximal effective concentration (EC50) of ∼3.0 μM (Figure 1D). Fluoxetine activity on the DQ-BSA assay was well separated from potential toxicity (Figure 1D). Also, we excluded that the FLX-mediated induction of DQ-BSA puncta was due to an elevation of its uptake by endocytosis. We found that the levels of endocytosed green BSA were similar in vehicle- and FLX-treated cells (Figure S2A). We also observed an induction of lysosomal cathepsin B (CTSB) activity (Figure S2B), confirming the activity of FLX boosting the lysosomal degradative capacity. Interestingly, the mRNA expression levels of genes encoding for the lysosomal enzymes cathepsin B and D were upregulated upon FLX treatment, suggesting that this drug may induce the transcription of these lysosomal proteases (Figure S2C).
Figure 1.

Cell-based DQ-BSA HCI assay identifies fluoxetine (FLX) as a compound hit in MPS-IIIA MEFs
(A) Plot representing the Pearson correlation coefficient of the 3 replicas of the 4 Prestwick sub-libraries screened. (B) Plot representing the results of the screening. The y axis shows the DQ-BSA spots per cell values ±SEMs of the hit compounds obtained from the screening, compared to the untreated cells value (DMSO, median of the 4 sub-libraries), and the positive control value (Torin-1, median of the 4 sub-libraries). ANOVA test, ∗∗p < 0.01 versus DMSO; ∗∗∗p < 0.001 versus DMSO. (C) Hit compounds classification. The pie chart shows the drug class distribution of the hit compounds in percentage. (D) FLX re-testing in dose-response format. Dose-response curves of DQ-BSA spots per cell and viability (nuclei count) ± SEMs with relative EC50 and IC50 values, derived from the treatment of MPS-IIIA MEFs with FLX at scalar doses (from 30 to 0, 1 μM) of n > 100 cells from 2 independent experiments. The HCI images of the DQ-BSA spots are representative of the treatment with FLX at 3 μM compared to the untreated cells.
FLX induces autophagy in vitro
We asked whether the induction of lysosomal degradation involves the activation of autophagy in MPS-IIIA MEFs. We observed that FLX increases the co-localization of DQ-BSA+ spots with the AV marker LC3, indicating the formation of autolysosomes (Figure S2D).20 Also, FLX induces the formation of WD repeat domain, phosphoinositide interacting 2-positive (WIPI2+) puncta, a marker of early AV 21 (Figure 2A), further increases the lipidation of LC3 protein in the presence of bafilomycin A1, and reduces the accumulation of the autophagic substrate SQSTM1/p62 (Figure 2B) in MPS-IIIA MEFs. Similarly, the analysis of the co-staining of LC3 with the lysosomal marker LAMP-1 resulted in an elevation in their co-localization upon FLX treatment, suggesting that FLX induces AV-lysosome fusion in MPS-IIIA MEFs (Figure S2E). A similar elevation in autolysosome number was obtained using human WT ARPE-19 cells overexpressing the autophagic reporter red fluorescent protein (RFP)-enhanced green fluorescent protein (EGFP)-LC322 (Figure 2C).
Figure 2.

FLX induces autophagy
(A) Representative confocal images of endogenous WIPI2+ puncta in MPS-IIIA MEFs untreated and treated with Torin-1 or FLX for 3 h. The plot shows the means of WIPI2 spots per cell ±SEMs of n = 102 cells from 3 independent experiments. ANOVA test, ∗∗p < 0.01 versus untreated; ∗∗∗p < 0.001 versus untreated. (B) Representative image of immunoblot analysis of endogenous p62 and LC3I/LC3II in MPS-IIIA MEFs upon 24 h of FLX treatment alone or in the presence of BafA1 for the last 3 h. The plot shows the densitometry of p62 (at left) and the LC3-II band (on right) normalized to actin as mean values ±SEMs of n = 6 lysates per condition pooled from 2 independent experiments. ANOVA test, ∗∗∗p < 0.001 versus untreated; ⋅⋅p < 0.01 versus FLX; ⋅⋅⋅p < 0.001 versus FLX. (C) Representative images from the high-content assay of ARPE-19 cells stably overexpressing mRFP-EGFP-LC3 plasmid starved with HBSS or untreated and treated with bafilomycin (BafA1) or FLX. The plot shows the quantification of autophagosomes (EGFP+ + mRFP+ spots per cell) compared to the autolysosomes (RFP+/GFP− spots per cell). Values are means ± SEMs of n > 1,000 cells pooled from 3 independent experiments. ANOVA test, ∗∗∗p < 0.001 versus untreated/autolysosomes; ⋅⋅⋅p < 0.001 versus untreated/autophagosomes.
Then, we asked whether FLX induces autophagy in vivo. We treated GFP-LC3 transgenic mice23 with 10 mg/kg FLX for 48 h. We observed an elevation in LC3-GFP puncta in liver sections of mice treated with FLX compared with their vehicle-treated counterparts (Figure S3A). Most important, immunoblot analysis of p62 liver extracts from the same transgenic mice clearly shows a reduction in p62 protein levels (Figure S3B). In agreement with previous reports,16, 17, 18, 19 our experimental evidence suggests that FLX induces autophagy in vitro and in vivo.
FLX reduces the accumulation of heparan sulfate in MPS-IIIA MEFs by inducing lysosomal exocytosis
Next, we evaluated whether FLX reduces the primary HS storage in MPS-IIIA MEFs. As expected, double immunofluorescence (IF) using anti-HS antibodies and sequential permeabilization of cells showed that endogenous HS mostly localizes at the plasma membrane (PM) in WT MEFs (Figure 3A). In agreement with the inability to degrade HS, MPS-IIIA MEFs showed an additional pool of intracellular HS that was accumulated in a vesicular compartment (Figure 3A).24 Conversely, FLX-treated MPS-IIIA cells presented a significant reduction in the lysosomal HS accumulation, detected by co-localization with LAMP-1 marker (Figure 3B). FLX treatment did not alter the PM pool of HS in WT cells (Figure 3B). Most important, we found a normalization of HS localization to the PM25 in FLX-treated MPS-IIIA cells. The relocation of HS to the PM upon FLX treatment of MPS-IIIA MEFs may indicate the activation of lysosomal exocytosis, a process that we showed can promote the clearance of pathological substrates in various LSDs, including MPSs.5 Lysosomal exocytosis is a two-step process involving the pre-docking of lysosomes underneath the PM and the subsequent fusion of lysosomes with the cell surface.5 We found that FLX induces a redistribution of LAMP-1+ lysosomes from the perinuclear area toward the PM in MPS-IIIA MEFs (Figure 3C). Similar redistribution was obtained by measuring the positioning of lysosomes by electron microscopy (Figure 3D). Finally, lysosomal fusion and exocytosis were confirmed by the evidence that FLX induces the release of the mature form of lysosomal cathepsin D in the culture media of treated MPS-IIIA MEFs (Figure 3E).26 The absence of the cytosolic enzyme lactate dehydrogenase (LDH) excluded the potential deleterious effects of FLX during this treatment, whereas the membrane permeabilizing compound digitonin releases both cathepsin D and LDH (Figure 3E). This experimental evidence indicates that FLX induces lysosomal exocytosis and the clearance of HS in MPS-IIIA MEFs.
Figure 3.

FLX reduces intracellular accumulation of HS by inducing lysosomal exocytosis
(A) Representative confocal images of endogenous HS in WT and MPS-IIIA MEFs before (green) and after (red) cell permeabilization showing the HS intracellular aggregation in MPS-IIIA MEFs. (B) Representative confocal images of HS and LAMP-1 in WT and MPS-IIIA MEFs untreated and treated for 24 h with FLX. The plot shows the percentage of HS colocalization in LAMP-1 ± SEMs of n = 100 cells from 3 independent experiments. ANOVA test, ∗∗p < 0.01 versus WT untreated; ••p < 0.01 versus MPS-IIIA untreated. (C) Representative confocal images of lysosome distribution marked by LAMP-1 in MPS-IIIA MEFs untreated and treated with FLX overnight; the plot shows the perinuclear index of LAMP-1 ± SEMs of n = 100 cells from 3 independent experiments. t test, ∗∗∗ p < 0.001 versus untreated. (D) Representative immunoelectron microscopy (EM) images of untreated and FLX-treated MPS-IIIA MEFs, labeled with an antibody against LAMP-1. The plot shows the distance of lysosomes to the plasma membrane (PM) (nm) of n ≥ 50 counts. t test, ∗∗∗p < 0.001 versus untreated. The black arrows indicate the vesicle structures; the white arrowheads indicate the PM. (E) Representative images of immunoblot analysis of active cathepsin-D (CTSD) and lactate dehydrogenase (LDH) from the protein precipitated from the growth media (at left) and from the cell lysate (at right) of MPS-IIIA MEFs untreated and treated with FLX (24 h) or digitonin (1 h). The plot shows the normalization of CTSD in the medium on CTSD in the cell lysate. ANOVA test, ∗p < 0.05 versus untreated.
FLX promotes lysosomal clearance through the activation of TFEB
Our observations indicate that FLX improves the lysosomal-autophagic pathways and promotes the clearance of HS through lysosomal exocytosis in MPS-IIIA MEFs (Figures 1, 2, 3, and S2). Interestingly, we previously showed that the overexpression of the transcription factor TFEB, a master gene controlling lysosomal and autophagic functions,4, 5, 6, 7, 8, 9, 10 can induce GAG clearance via lysosomal exocytosis in various in vitro and in vivo models of lysosomal storage disorders, including MPS-IIIA and multiple sulfatase deficiency (MSD).5 Thus, we asked whether FLX could exert its functions through the activation of TFEB. We found that FLX induces TFEB nuclear translocation in HeLa cells stably expressing TFEB fused to the GFP (HeLa TFEB-GFP) (Figure 4A). Immunoblot analysis of FLX-treated MPS-IIIA MEFs further confirmed the activation of endogenous TFEB by looking at the increase in the fast-migrating band corresponding to its activating dephosphorylation (Figure 4B) and to the upregulation of various TFEB target genes by quantitative PCR (qPCR) analysis (Figure 4C). FLX-mediated dephosphorylation of TFEB was further confirmed by immunoblot detection of two critical serine residues involved in the subcellular localization of TFEB localization (S142 and S211) (Figure S4A).27 Interestingly, a time course analysis of FLX-mediated TFEB nuclear translocation showed a very fast induction and a progressive elevation of nuclear TFEB localization at longer time points (Figure S4B), suggesting that TFEB activation is a very early event upon FLX treatment. Most important, the depletion of TFEB/TFE3 by gene editing fully blocks FLX-mediated induction of DQ-BSA degradation in HeLa cells (HeLa TFEB/TFE3 knockout [KO]) (Figure 4D) and inhibits FLX-mediated clearance of the autophagic substrate p62 (Figure S4C). These results strongly indicate that FLX activity ameliorating the lysosomal phenotype in MPS-IIIA cells requires TFEB.
Figure 4.

FLX activates TFEB in vitro
(A) Representative HCI images of HeLa stably expressing TFEB-GFP plasmid and untreated or treated with FLX for 3 h. (B) Representative image of immunoblot analysis of endogenous TFEB in MPS-IIIA MEFs untreated or treated with FLX, showing the shift from phosphorylated (pTFEB) to the dephosphorylated form of TFEB. As a positive control, cells are starved with HBSS. The plot shows the ratio between the 2 bands (TFEB/pTFEB). ANOVA test, ∗∗p < 0.01 versus untreated. (C) Graph of quantitative RT-PCR (qRT-PCR) showing the mRNA levels of a subset of TFEB target genes in MPS-IIIA MEFs upon 24 h of FLX treatment. The data in the graphs are mean values ±SEMs; n = 4 samples per condition. t test, ∗∗p < 0.01 versus untreated; ∗∗∗p < 0.001 versus untreated. (D) Representative HCI images of DQ-BSA assay in HeLa WT and TFEB/TFE3 KO untreated and treated with BafA1 or FLX. The plot shows the number of DQ-BSA spots per cell as means ± SEMs of n > 1,000 cells. ANOVA test, ∗∗∗p < 0.001 versus untreated. (E) Representative confocal images of HeLa stably expressing TFEB-GFP transfected with HA-GST-RagCS75L plasmid and treated with FLX for 3 h. The plot shows the nuclear:cytosol TFEB ratio as means ± SEMs of n > 50 cells from 3 independent experiments, in the 2 distinct populations, not transfected (HA−) and transfected (HA+, indicated with an asterisk). t test, ∗∗∗p < 0.001 versus HA−. (F) Representative HCI images of MPS-IIIA MEFs transfected with HA-GST-RagCS75L plasmid, pre-treated with FLX for 3 h and incubated overnight with DQ-BSA. The plot shows the DQ-BSA spots per cell as means ± SEMs of n > 1,000 cells from 3 independent experiments, in the 3 distinct populations, not transfected (HA−) and transfected (HA+, indicated with an asterisk). t test, ∗∗p < 0.01 versus HA−.
FLX induces TFEB through the inhibition of mammalian target of rapamycin complex 1 (mTORC1) via a Rag-dependent mechanism
TFEB is negatively regulated by the mTOR kinase-mediated phosphorylation,27 whereas the calcium pathway involving TRPML1-calcineurin regulates TFEB dephosphorylation.28 Thus, we asked whether FLX induces TFEB nuclear translocation by modulating these pathways. Depletion of the TRPML1-calcineurin axis by silencing either TRPML1 or calcineurin was not effective at reducing FLX-mediated induction of TFEB nuclear translocation in HeLa cells (Figure S4D). Similarly, pharmacological chelation of calcium using 1,2-bis-(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetra(acetoxymethyl) ester (BAPTA-AM) did not affect the efficacy of FLX in inducing TFEB nuclear translocation in HeLa cells, as well as the increased degradation of DQ-BSA in MPS-IIIA MEFs (Figure S4E). Then, we investigated whether FLX inhibits mTORC1, a negative regulator of TFEB activation.29 Interestingly, FLX did not affect mTOR kinase activity toward its canonical substrates S6K and 4EPB, although the levels of major mTOR-dependent phosphorylation sites on TFEB (Ser142 and Ser211) were reduced (Figure S4A). Recent work has shown that mTORC1-mediated phosphorylation of TFEB is strictly dependent on the activation of RagC and RagD GTPases.30 The overexpression of a constitutive active form of RagC (hemagglutinin-glutathione S-transferase-RagCS75L [HA-GST-RagCS75L]) was able to block FLX-mediated TFEB nuclear translocation in HeLa TFEB-GFP cells and to significantly reduce FLX induction of DQ-BSA degradation in MPS-IIIA MEFs (Figures 4E and 4F). Thus, our results indicate that FLX induces TFEB through the inhibition of mTORC1 via a Rag-dependent mechanism.
FLX ameliorates the liver phenotype of MPS-IIIA mice
Next, we evaluated the in vivo efficacy of FLX in the naturally occurring mouse model of MPS-IIIA.31, 32, 33 These mice recapitulate human MPS-IIIA disease, representing an excellent model for evaluating therapeutic strategies 1,33, 34, 35 Although in MPS-IIIA mice, GAGs begin to accumulate into the lysosomes postnatally, the autophagy-lysosomal pathway becomes defective at ∼6 months of age, when neurological defects, including neuroinflammation, synaptic dysfunction, and cognitive deficits also appeared and then worsened progressively as the mice aged.31,36 Thus, we selected 6 months to start the treatment. Six-month-old male mice were intraperitoneally (i.p.) injected with 10 mg/kg FLX every other day until 8 months of age, which represents a late stage of the pathology.37 Compared to WT mice, IF evaluation of liver sections from MPS-IIIA mice showed a dramatic vacuolization that was positive to the lysosomal marker LAMP-1 (Figure 5A). Surprisingly, the treatment with FLX showed a significant reduction in LAMP-1+ vacuolization in the livers of MPS-IIIA-treated mice; similar to what was observed in vitro, LAMP-1 seems to be distributed more to the periphery (Figure 5A). More in-depth analyses of both vacuolization and vesicle redistribution were obtained using the electron microscopy (EM) approach (Figures 5B and 5C); the reduction in LAMP-1 area in liver tissues was accompanied by an elevation in small vesicles, which may indicate the induction of lysosomal biogenesis (Figure 5B). Moreover, FLX-treated mutant mice revealed a decrement in the distance of the vesicles to the PM, suggesting the activation of lysosomal exocytosis (Figure 5C). Then, quantitative analysis of GAG accumulation in MPS-IIIA compared with WT age-matched mice38 resulted in a significant reduction in GAG storage in FLX-treated mutant mice compared to vehicle-treated MPS-IIIA mice (Figure 6A). Similarly, histological analysis of GAGs by Alcian blue staining confirmed these results (Figure 6B). Immunohistochemical (IHC) staining using TFEB antibodies (Figure 6C) and RT-PCR of TFEB target genes (Figure 6D) in mutant mice treated with FLX confirmed the induction of TFEB in vivo. These observations indicate that FLX reduces lysosomal vacuolization, promotes the clearance of primary GAG storage, and increases nuclear TFEB in MPS-IIIA mice.
Figure 5.

FLX reduces lysosomal vacuolization and induces exocytosis in MPS-IIIA liver tissues
(A) Representative confocal images of LAMP-1 in liver sections from WT (n = 4 per groups) and MPS-IIIA (n = 5 per groups) mice that were FLX-treated and compared with the vehicle-treated groups. The plot shows the mean intensity of LAMP-1 per area ±SEMs. ANOVA test, ∗∗∗p < 0.001 versus WT; ⋅⋅⋅p < 0.001 versus MPS-IIIA vehicle. (B) Representative EM images of liver tissues from WT and MPS-IIIA mice vehicle- or FLX-treated. The plot shows the diameter (nm) of n > 50 lysosomal-like structures derived from 2 different animals per group. ANOVA test, ∗∗∗p < 0.001 versus WT; ⋅⋅⋅p < 0.001 versus MPS-IIIA. The 2 histograms analyze the size-frequency distribution. The black arrows indicate the vesicle structures. (C) Representative EM images of liver tissues from vehicle- and FLX-treated MPS-IIIA mice showing the distance of the vesicles to the PM. The plot shows the number of vesicles close to the PM as ±SEMs of n > 50 lysosomal-like structures derived from 2 different animals per group. ANOVA test, ∗∗∗p < 0.001 versus vehicle. The black arrows indicate the vesicle structures; the white arrowheads indicate the PM. BC, bile canaliculus.
Figure 6.

FLX ameliorates the hallmarks of liver MPS-IIIA pathology
(A) Plot showing the results of the colorimetric quantification of GAGs (μg/μg DNA) ± SEMs in liver extracts from WT, MPS-IIIA-treated with vehicle, and MPS-IIIA-treated with FLX mice (n = 5 per group). The results are expressed as the fold change on WT. ANOVA test, ∗∗p < 0.01 versus WT; ⋅p < 0.05 versus MPS-IIIA. (B) Representative IHC images from AxioScan of colorimetric GAGs staining in liver sections from WT, MPS-IIIA-treated with vehicle, and FLX-treated MPS-IIIA mice; the plot shows the percentage of blue areas in MPS-IIIA untreated and treated (n = 5 per groups) with FLX. t test, ∗∗∗p < 0.001 versus untreated. (C) Representative confocal images of TFEB staining in liver sections of WT and MPS-IIIA mice vehicle- or FLX-treated. (D) Graph of qRT-PCR showing the mRNA levels of a subset of TFEB target genes in liver samples from vehicle- and FLX-treated WT and MPS-IIIA mice (n = 5). The data in the graphs are means ±SEMs. t test, ∗p < 0.05 versus WT vehicle; ⋅⋅p < 0.01 versus MPS-IIIA vehicle; ⋅p < 0.05 versus MPS-IIIA vehicle. (E) Representative image of immunoblot analysis of p62 in liver protein extracts from vehicle- and FLX-treated MPS-IIIA mice. The plot shows the densitometry of p62 band normalized to vinculin (n = 5 per group) as mean values ±SEMs. t test ∗∗p < 0.01 versus vehicle. (F) Representative confocal images of CD68 in liver sections from vehicle- and FLX-treated WT and MPS-IIIA mice. The plot shows the percentage of CD68+ cells per area ±SEMs (n = 5 per group). ANOVA test, ∗∗∗p < 0.001 versus WT vehicle; ⋅⋅p < 0.01 versus MPS-IIIA vehicle.
MPS-IIIA mice present impairment in the autophagy pathway due to the lysosomal storage and a consequent block of fusion between lysosomes and autophagosomes,2,39 as confirmed by immunoblot of p62 (Figure S5A). In agreement with our results in WT LC3-GFP mice showing FLX-mediated induction of autophagy (Figures S3A and S3B), FLX reduces the accumulation of the autophagic receptor p62 in the livers of MPS-IIIA mice compared to vehicle-treated animals (Figure 6E).
We then investigated whether FLX ameliorates inflammation in MPS-IIIA-treated mice compared with untreated mutant age-matched mice. IF analysis of the CD68 marker showed a reduction in macrophage infiltration in liver samples of MPS-IIIA-treated mice compared to vehicle-treated MPS-IIIA animals (Figure 6F). FLX shows in vivo efficacy ameliorating somatic pathology in the livers of MPS-IIIA mice.
FLX ameliorates the neuropathology in MPS-IIIA mice
To evaluate the efficacy of FLX in the treatment of lysosomal CNS pathology, we performed an IHC analysis of LAMP-1. Like the effect on the liver tissue samples, FLX treatment reduces vacuolization in the cerebral cortex of MPS-IIIA-treated mice compared to the age-matched vehicle-treated MPS-IIIA mice (Figure 7A). A similar reduction in vacuolization, coupled to the induction of small-vesicle biogenesis, was observed by EM analysis of cortical tissues from MPS-IIIA-treated mice (Figure 7B). Moreover, quantitative analysis of GAGs by the Blyscan assay showed a significant reduction in brain tissues from FLX-treated mice, whereas untreated mutant mice accumulate GAGs compared to age-matched WT (Figure 7C).34 In agreement with our in vitro data, FLX treatment increases the nuclear localization of TFEB in the parietal cortex and hippocampus of WT and MPS-IIIA-treated mice (Figure S6A) and upregulates TFEB target genes (Figure S6B).
Figure 7.

FLX ameliorates the storage pathology in the brain of MPS-IIIA-treated mice
(A) Representative immunostaining using antibodies against the LAMP-1 protein in parietal cortex and hippocampus (CA3) sections from WT and MPS-IIIA mice that were FLX treated compared with vehicle. Nuclei were stained with hematoxylin II (blue). The data are expressed as the percentage of the LAMP-1 area. The graphs showed means ± SEMs (n = 4 per group). ANOVA test, ∗∗∗p < 0.001 versus WT: ⋅p < 0.05 versus MPS-IIIA. (B) Representative EM images of parietal cortex tissues from WT and MPS-IIIA mice that were vehicle- or FLX-treated. The plots show the diameter (nm) of n > 25 lysosomal-like structures derived from 2 different animals per group. ANOVA test, ∗∗∗p < 0.001 versus WT; ⋅⋅⋅p < 0.001 versus MPS-IIIA. The 2 histograms analyze the size-frequency distribution. The black arrows indicate the vesicle structures. (C) Plot showing the results of the colorimetric quantification of GAGs (μg/μg DNA ±SEMs) in brain extracts from WT, vehicle, and treated MPS-IIIA mice (n = 5 per group). Top, the fold change on WT. ANOVA test, ∗∗p < 0.01 versus WT; ⋅p < 0.05 versus MPS-IIIA.
As expected, FLX regimen reduces the accumulation of the autophagic receptor p62 (Figure 8A), which also accumulated in MPS-IIIA brains compared to those of age-matched WT mice (Figure S5B). To further confirm the amelioration of the autophagic pathway in MPS-IIIA brains, we investigated whether α-synuclein, another autophagic substrate and pathological hallmark of neurodegenerative diseases, including MPS-IIIA,3,36 may be reduced upon FLX treatment. IHC analysis shows that the aggregated form of α-synuclein (PS129-α-syn) accumulates in the cortical region of vehicle-treated MPS-IIIA mice compared to the vehicle-treated WT mice, and its levels were significantly reduced by FLX treatment (Figure 8B).
Figure 8.

FLX ameliorates the neurological hallmarks of MPS-IIIA mice
(A) Representative images of immunoblot analysis of p62 in brain protein extracts from vehicle and FLX-treated MPS-IIIA mice. The plot shows the densitometry of the p62 band normalized to actin as mean values ±SEMs (n = 5 per group). t test, ∗∗p < 0.01 versus MPS-IIIA. (B) Representative immunostaining of phopsho-α-synuclein marker (pS129 α-syn) in brain parietal cortex sections from WT and MPS-IIIA mice that were FLX treated compared with the vehicle group. Nuclei were stained with hematoxylin II (blue). The plot shows the mean percentage of p-129-α-synuclein area on the total area ±SEMs (n = 4 per group). ANOVA test, ∗∗p < 0.01 versus WT vehicle; ⋅⋅p < 0.01 versus MPS-IIIA vehicle. (C) Representative images of the immunoblot analysis of ubiquitin in brain protein extracts from WT and MPS-IIIA vehicle and FLX-treated mice. The plot shows the densitometry of polyubiquitinated proteins normalized to actin as mean values ±SEMs (n = 6 per group). ANOVA test, ∗∗p < 0.01 versus WT vehicle; ⋅p < 0.05 versus MPS-IIIA vehicle. (D) FLX-treated WT (n = 9) and MPS-IIIA (n = 9) and relative control vehicle-treated WT (n = 7) and MPS-IIIA (n = 9) were tested at 8 months of age in the fear contextual conditioning test. Unequal post hoc analysis, ∗p < 0.05 versus train; ⋅p < 0.05 versus MPS-IIIA.
The ubiquitin-proteasome system (UPS) maintains intracellular proteostasis by the selective degradation of abnormal or redundant proteins. Cumulative evidence suggests the disruption of UPS underlying the accumulation of aggregate-prone proteins in common neurodegenerative disorders such as Alzheimer’s disease.40 Polyubiquitinated proteins accumulate in MPS-IIIA brains.2 Most important, we found that FLX also reduces the accumulation of polyubiquitinated proteins in MPS-IIIA brain extracts measured by immunoblotting of ubiquitin (Figure 8C), suggesting that FLX may protect proteostatic pathways by diminishing cellular proteotoxic load in the brain.
To evaluate whether the FLX treatment ameliorates progressive cognitive deterioration in MPSIIIA mice, we tested its effect in the fear contextual conditioning test. This test evaluates conditional response to a negative stimulus (mild foot shock), which is altered in MPS-IIIA mice.41,42 As shown in Figure 8D, vehicle-treated MPS-IIIA mice are less responsive than the age-matched WT mice to the foot shock. FLX treatment significantly improved the response by increasing the freezing time of the treated mice during the test phase compared to the vehicle-treated MPS-IIIA mice, slowing down the progressive memory deficit (Figure 8D). Our results strongly indicate that FLX ameliorates neurological hallmarks in MPS-IIIA mice.
Discussion
Here, we developed a cell-based HCI assay (DQ-BSA assay) to identify FDA compounds that can boost impaired lysosomal degradative capacity in MPS-IIIA cells. Thus, we identified FLX, a SSRI used as an antidepressant in adults and children. We found that FLX increases the lysosomal degradation of DQ-BSA in part through the induction of autophagy, as DQ-BSA partially co-localizes with the autophagosome marker LC3. In addition, we observed that FLX promotes the clearance of HS, the primary lysosomal storage cargo accumulated in MPS-IIIA disease, and induces lysosomal exocytosis. Cumulative evidence indicates that the activation of the transcription factor TFEB, a master gene of lysosomal function, can promote the clearance of pathological lysosomal storage in different LSDs, such as MSD and MPS-IIIA,5 Pompe disease,43 Batten disease,44,45 Gaucher and Tay Sachs disease,46 and cystinosis.47 This effect is most likely the consequence of the ability of TFEB to induce lysosomal exocytosis, autophagy, and lysosome biogenesis. Surprisingly, we found that FLX, which also activates the latter biological processes, induces TFEB nuclear translocation in WT and MPS-IIIA models. Also, FLX induces TFEB target genes in cells and tissues of mutant mice, strongly suggesting the activation of the TFEB pathway. Hence, we hypothesize that FLX may promote lysosomal clearance via TFEB. Mechanistically, FLX induces TFEB nuclear translocation through the inhibition of mTOR in a Rag-dependent manner, while mTOR activity toward canonical substrates is not affected. Thus, FLX has the advantage of promoting TFEB activation without the potential adverse effects that may represent the complete inhibition of mTOR kinase, a master regulator of cell growth and proliferation. Thus, FLX may represent a better alternative to rapamycin, an allosteric inhibitor of mTORC1 frequently proposed as a therapeutic approach for neurodegenerative diseases, which may push cargo toward lysosomal-mediated catabolism in neurodegenerative models with already poorly functioning lysosomes48 and without the ability to induce TFEB29 to boost lysosomal function. By contrast, FLX can induce proteostasis, pushing cargo toward lysosomes and in parallel alleviating lysosomal engulfment by promoting TFEB-mediated lysosomal biogenesis and exocytosis.
In vivo, FLX treatment induces autophagy, reduces autophagic cargoes such as p62 and p-129-α-synuclein, and reduces lysosomal vacuolization, promoting GAG clearance. Recent reports have shown the potential beneficial effects of FLX on in vivo models of neurological disorders, including ischemic stroke and brain injury through autophagy activation.16, 17, 18, 19,49,50 In agreement with these observations, we demonstrated a significant reduction in neuroinflammation with a slowing of memory deficits in MPS-IIIA mice treated with FLX. We hypothesize that FLX activity requires the activation of TFEB to ameliorate global lysosomal phenotypes and autophagy and promote pathological clearance. However, further studies are needed to clarify whether the slowing down of neurological and cognitive deterioration observed is due to TFEB or involves the synergy with other FLX targets as the serotoninergic pathway. Intriguingly, recent works suggest that antidepressant drugs bind the neurotrophic receptor TRKB (tropomyosin receptor kinase B), in a cholesterol-sensitive manner, facilitating brain-derived neurotrophic factor (BDNF) signaling and antidepressant action.51 Since MPS-IIIA models accumulate cholesterol,39 it could be interesting to investigate whether the FLX-mediated attenuation of cognitive deficits in MPS-IIIA mice is linked to this pathway via the stabilization of TRKB signaling.
In line with other in vivo studies,16,17,50 because of the shorter half-life of FLX in rodents relative to humans,52,53 our proof of principle in vivo has used high drug doses (10 mg/kg) compared to those shown to be clinically effective for humans,52,53 and therefore more preclinical studies are required to determine the human oral dose for pediatric use in MPS patients.
In summary, by combining cell-based HCI screening and repurposing of FDA drugs, we identified a new indication for FLX, one of the most prescribed CNS drugs in adults and children. This may represent a promising therapeutic strategy, alone or in combination with enzyme replacement therapy (ERT) or gene therapy, to treat MPS-IIIA and perhaps other neurodegenerative diseases.
Materials and methods
Cellular models, drug treatments, plasmids, and small interfering RNAs (siRNAs) transfection
Immortalized mouse embryonic fibroblasts were isolated from embryonic day 14 (E14) WT and MPS-IIIA mouse embryos, as previously described.2 HeLa TFEB-GFP cells were described in Settembre et al.29 Human ARPE-19 LC3-Tandem cells were generated by the stable expression of ARPE-19 cells with a vector encoding the mRFP-EGFP-LC3 construct. The HeLa TFEB/TFE3 KO cell line was generated by Dr. Richard Youle of the National Institutes of Health. All of the cell lines were grown in DMEM supplemented with 10% FBS and penicillin/streptomycin. The compounds used were Torin-1 (1 μM for 3 h; Tocris Bioscience), bafilomycin A1 (200 nM for 3 h; Sigma-Aldrich), FLX hydrochloride (European Pharmacopoeia, Reference Standard, 5 μM during 5 min–24 h; Sigma-Aldrich), BAPTA-AM (10 μM pre-treatment for 30 min and then used in co-treatment with FLX for 3 h; Thermo Fisher Scientific), digitonin (100 μM 1 h, Sigma-Aldrich). The starvation medium was prepared using Hank’s balanced salt solution (HBSS) (Thermo Fisher Scientific) plus 10 mM HEPES (Sigma-Aldrich). HeLa TFEB-GFP cells were silenced using 40 nM of siRNAs against TRPML1 (sequences 5′–3′: #1 CCUUCGCCGUCGUCUCAAA; #2 AUCCGAUGGUGGUUACUGA; #3 GAUCACGUUUGACAACAAA), calcineurin, catalytic subunit PPP3CB (Ambion, 4390824), and regulatory subunit PPP3R1 (Invitrogen, HSS108410-3, HSS183046-3, HSS108411-3) for 72 h using the Lipofectamine RNAimax (Thermo Fisher Scientific) reagent according to the protocol provided by the manufacturers. The efficiency of silencing is reported in the corresponding figures. HeLa TFEB-GFP cells and MPS-IIIA MEFs were transfected with pRK5-HA GST RagC-S75L (Addgene plasmids) for 48 h using Lipofectamine Transfection Reagent (Thermo Fisher Scientific) for HeLa cells and Lipofectamine Stem Transfection Reagent (Thermo Fisher Scientific) for MEFs.
RNA extraction and qPCR
Total RNA was extracted from cells or tissues using the RNeasy Plus Mini Kit (Qiagen). Reverse transcription was performed using QuantiTect Rev Transcription Kit (Qiagen). Real-time qRT-PCR was performed using the LightCycler System 2.0 (Roche Applied Science). Hypoxanthine phosphoribosyltransferase (HPRT) was used for qRT-PCR as the reference gene. qRT-PCR amplification was performed according to Roche’s recommendations. The following primers were used in this study. Mouse primers: DPP7: fw: CGCCAGCAATACTGTCTGGATAC, rev: AAATGATGTTGCTGGCTGCTTTA; PGC1alpha: fw: GAATCAAGCCACTACAGACACCG, rev: CATCCCTCTTGAGCCTTTCGTG; RAGD: fw: GCTGGAGAATCTGCTGAACA; rev: CACAGCAGAGCTCGTACGTC; ATP6V1D: fw: AGGAGCACAGACTGGTCGAAAC, rev: CTCAGCCAATGAGAAGGCAGCT; ATP6V0D1: fw: GCATCTCAGAGCAGGACCTTGA, rev: GGATAGGACACATGGCATCAGC; ATP6V1H: fw: GTTGCTGCTCACGATGTTGGAG, rev: TGTAGCGAACCTGCTGGTCTTC; TFEB: fw: GCGAGAGCTAACAGATGCTGA, rev: CCGGTCATTGATGTTGAA-CC; TRPML1: fw: ATGTGGACCCAGCCAATGATACCTT, rev: TGTCTTCAGCTGGAAGTGGATTGGT; and HPRT: fw: CAAGCTTGCTGGTGAAAAGG, rev: GTCAAGGGCATATCCAACAAC. Human primers: TRPML1: fw: GAGTGGGTGCGACAAGTTTC, rev: TGTTCTCTTCCCGGAATGTC; HPRT: fw: TGGCGTCGTGATTAGTGATG, rev: AACACCCTTTCCAAATCCTCA; PP3CB: fw: CAACCATGAATGCAGACACC, rev: AAGTGCAGCAAGAGGCAAAC; and PP3R1: fw: GAGGGCGTCTCTCAGTTCAG rev: GATTGTTCCCCACCATCATC.
DQ-BSA assay and screening
For the lysosomal degradation assay, DQ Red BSA (Thermo Fisher Scientific) was used. After its internalization by endocytosis, proteolysis of DQ-BSA in lysosomes or autolysosomes (derived from the fusion of amphysomes with lysosomes) results in dequenching and release of brightly fluorescent fragments, allowing the visualization of both lysosomal degradative capacity and the completion of the autophagic pathway.20 Torin-1, a selective inhibitor of the mTOR kinase, and bafilomycin A1 (BafA-1), an inhibitor of lysosomal activity, was used as positive and negative controls, respectively54 (Figure S1A). For the screening, MPS-IIIA MEFs were seeded at a concentration of 3.2 × 103 cells/well in 384-well plates. After 24 h of growth, the cells were treated with 1 μM of the positive control Torin-1 and 10 μM of the drugs from the Prestwick Chemical Library for 3 h; the 1,280 FDA-approved compounds were divided into 4 sublibraries, and each of them was tested in triplicate. After drug pre-treatment, the cells were incubated overnight with 10 μg/mL of DQ Red BSA added in a normal medium. Cells were fixed with 4% paraformaldehyde (PFA) and washed twice with PBS. Nuclei were counterstained with Hoechst (Invitrogen). Images (at least 20 images per well) were acquired on random fields from every well using the Opera imager (PerkinElmer) with a 40X objective. Image analysis was performed using Columbus Image Data Storage and Analysis System (PerkinElmer), and the DQ-BSA spots per cell value was obtained by dividing the brightest spots—selected by a threshold of intensity—for the number of nuclei. Quality control parameters such as plate distribution, plate correlation (Pearson coefficient), and z factor were used to determine the robustness of the screening. ANOVA test for multiple comparisons followed by Bonferroni’s test using GraphPad Prism 5 was performed on the parameter called DQ-BSA spots per cell for each compound-treated cell compared with the DMSO-treated cells. Thus, compound hits were selected based on two quantitative criteria: (1) the mean of DQ-BSA spots per cell of treated cells > the mean of DQ-BSA spots per cell in control cells plus three times SD, and (2) p ≤ 0.01 (∗∗). Similar protocols and analyses were performed for the DQ-BSA assay in 96-well formats, by plating the cells at a concentration of 1.0 × 104 cells/well. For DQ-BSA in dose-response format, the cells were treated with 6 scalar concentrations (1:3) of FLX (from 30 to 0.1 μM) in quadruplicate. EC50 and half-maximal inhibitory concentration (IC50) curves and relative values were obtained by nonlinear regression (curve fit) analysis on GraphPad Prism using the equation log (agonist/inhibitor) versus response variable slop. As the control of endocytosed BSA rate, cells were incubated with 10 μg/mL green BSA (Invitrogen) in complete medium overnight.
IF
The following antibodies were used for IF: WIPI2 (anti-mouse 2A2, ab105459, 1:1,000), LC3 (anti-rabbit MBL PM036, 1:600), LAMP-1 (anti-rat 1D4b, sc-19992, 1:800), HS (mouse anti-human HS mAb 10 × 104, 1/50, US Biological/Amsbio), and HA tag (mouse anti-HA.11 clone 16B12 Biolegend 901501 1:500). For WIPI2 staining, cells were fixed with ice-cold methanol for 5 min on ice and permeabilized in 0.1% (w/v) saponin, 0.5% (w/v) BSA, and 50 mM NH4Cl in PBS (blocking buffer saponin). For LAMP-1, LC3, HS, and HA tag, cells were fixed in PFA 4% for 10 min and permeabilized with blocking buffer saponin (LC3, LAMP-1, HA tag) or with Triton buffer (HS). Cells were incubated with the indicated primary antibodies for 1 h and subsequently incubated with secondary antibodies for 45 min. For the double HS staining, the cells were fixed with PFA (methanol-free grade) and, without permeabilization, incubated with the primary antibody diluted in PBS for 3 h 1:100, 4°C, and with the secondary antibody (Alexa Fluor 488 1:400 room temperature [RT]) for 1 h; then, the cells were fixed again, permeabilized with Triton buffer for 20 min, and incubated with the same primary antibody for 3 h and then with the secondary antibody (Alexa Fluor 568 1:400 RT). To test the specificity of the antibody, the cells were treated overnight with the heparanase III enzyme from Flavobacterium heparinum (H889110UN, Sigma-Aldrich). For confocal imaging, the samples were examined under a Zeiss LSM 700 confocal microscope. Optical sections were obtained under 63× or 40× immersion objectives at a definition of 1,024 × 1,024 pixels, adjusting the pinhole diameter to 1 Airy unit for each emission channel to have all of the intensity values between 1 and 254 (linear range). ImageJ software (National Institutes of Health) was used for the image analysis.
Cathepsin B activity assay
The cathepsin B activity was assessed using the Magic Red kit (ImmunoChemistry Technologies), which acts as an enzyme substrate, emitting fluorescence upon cleavage. Briefly, MPS-IIIA MEFs were plated on 96-well plates at a concentration of 3.5 × 103 per well. After 24 h of growth, cells were treated overnight with 5 μM FLX. Then, Magic Red was added to the culture media (1:250) together with the NucBlue Live ReadyProbes (Thermo Fisher Scientific), for 20 min at 37°C. The plate was acquired by OPERA in live mode; cathepsin B activity was quantified by measuring the number of spots per cell by Columbus software (PerkinElmer).
LC3 tandem assay
ARPE-19-LC3 Tandem cells were plated in 96-well plates at a concentration of 1.0 × 104 cells/well. The next day, cells were starved for 2 h or treated with bafilomicin (BafA1) and FLX for 3 h. Then, the cells were fixed with PFA 4% and stained for nuclei (Hoechst, Invitrogen) and cytoplasm (CellMask, Thermo Fisher Scientific). HC- Images (at least 10 images per well) were acquired using the OPERA system (PerkinElmer). Image analysis was performed using Columbus software. The population of spots (mRFP+ spots/EGFP+ spots) was selected by an intensity threshold. Then, mRFP+ spots that were also positive to EGFP (the autophagosomes) were quantified by selecting the EGFP intensity threshold in the mRFP population. The autolysosomes instead were selected as a spot population positive only for mRFP.
TFEB nuclear translocation assay
HeLa TFEB-GFP cells were seeded in 96- (1.0 × 104 cells/well) or 384- (4.0 × 103 cells/well) well plates, incubated for 24 h, and then starved or treated with FLX for the indicated time points. Then, the cells were fixed with 4% PFA and stained for nuclei (Hoechst) and cytoplasm (CellMask). At least 10 images were acquired per well by using the OPERA system. A dedicated script was developed to quantify the ratio derived from the mean intensity of GFP signal in a fixed ring region around the perinuclear area and the nucleus.
Perinuclear index
For the quantification of lysosome distribution, confocal images of LAMP-1 staining in MPS-IIIA MEFs were analyzed by Columbus software; cytoplasm was segmented into two fixed regions (perinuclear and peripheral), and the mean intensity was measured in both areas. The perinuclear index was obtained by the ratio of the two mean intensities (perinuclear/peripheral).
Mice
GFP-LC3 transgenic mice were generated by Dr. N. Mizushima23 and purchased from RIKEN (Japan). Homozygous mutant (B6Cg-Sgsh[mps3a/PstJ] mouse [Sgsh−/−] from The Jackson Laboratory, phenotypically MPS-IIIA affected) and healthy (Sgsh+/+) C57BL/6 mice were used for the in vivo efficacy study.31 Experiments were conducted following the guidelines of the Animal Care and Use Committee of Cardarelli Hospital in Naples and authorized by the Italian Ministry of Health.
Treatments and tissue collection
For GFP-LC3 mice, acute FLX treatment (single dose, i.p.) was performed at 2 months of age and mice were sacrificed 48 h after the injection. MPS-IIIA mice and age-matched WT mice were i.p. injected with 10 mg/kg compound dissolved in 100 μL of water every other day for 2 months (from 6 to 8 months of age). Animal body weight was monitored every month. Mice were euthanized at 8 months by injection with ketamine and xylazine before blood collection. For tissue collection, mice were intracardially perfused with PBS (pH 7.4), and brain and liver samples were removed. The brain was divided into two halves for, respectively, biochemical and IHC analysis. Three different lobes from the liver were collected and used for biochemical and IHC analysis.
Western blotting
Total cell lysates were prepared by the solubilization of samples in Tris HCl 10 mM pH 8.0 and 0.2% SDS supplemented with 1× protease inhibitor cocktail (Roche) and 1× phosphatase inhibitor cocktail (Sigma-Aldrich). For tissues, brain and liver samples were disrupted and homogenized using the TissueLyser (Qiagen) and RIPA buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 1× protease inhibitor cocktail [Roche] and 1× phosphatase inhibitor cocktail [Sigma-Aldrich]). Approximately 30 μg of proteins (determined by the Bradford method) for each sample were separated by SDS-PAGE gels and electrotransferred to the polyvinylidene fluoride (PVDF) membranes. After immunoblotting, the protein was incubated with the different antibodies and visualized by chemiluminescence methods (Luminata Crescendo Western HRP substrate, Millipore) using peroxidase-conjugated anti-rabbit or anti-mouse secondary antibodies (Millipore). Membranes were developed using a Chemidoc UVP imaging system (Ultra-Violet Products). Densitometric quantification was performed on low-exposure images from different blots using ImageJ. The antibodies used were β-actin (Santa Cruz SC 47778, 1:5,000), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (6C5) (sc-32233, 1:15,000), vinculin (SIGMA 9264, 1:5,000), LC3 (Novus NB100-2220, 1:1,000), TFEB (against mouse: Bethyl Laboratories, A303-673A, 1:250; against human: Cell Signaling Technology 37785S; 1:250), TFEB-pSer142 (EMD-Millipore ABE1971, 1:15,000), TFEB-pS211 (custom generated in collaboration with Bethyl Laboratories, 1:1,000), p62 (BD 610833, 1:1,000), P-4EBP (S65, Cell Signaling Technology 9456S 1:1,000), 4EBP (Cell Signaling Technology, 9644S 1:1,000), phospho-p70 S6 kinase/Thr389 (Cell Signaling Technology 9205 1:1,000) p70 S6 kinase (Cell Signaling Technology 2708, 1:1,000), Ubiquitin (Novus Biological, NBP-300/130, 1:1,000), cathepsin D (rabbit anti-cathepsin D[H75] Santa Cruz sc-10725, 1:500).
Protein concentration from growth medium and western blot analysis
For cathepsin D secretion experiments, MPS-IIIA MEFs were plated into 6-well plate at a concentration of 5 × 105. After 24 h, medium was replaced with fresh DMEM supplemented with 1% BSA alone or with FLX at a 5-μM concentration for 24 h; digitonin was added at a concentration of 100 μM for the last 1 h of growth. Then, the medium was collected and centrifuged at maximum velocity for 10 min to remove cell debris. In parallel, protein lysate from cells was obtained as previously described. Proteins in the medium were concentrated using TCA/DOC buffer (50% trichloroacetic acid, 2 mg/mL sodium deoxycholate); the pellet was washed with cold acetone and then resuspended in 50 mM TRIS HCL loading buffer, pH 8. Cathepsin-D levels in the medium were determined by western blot analysis, performed as previously described.
Quantitative and qualitative analysis of GAG
Quantification of GAG was performed using the Blyscan kit (Biocolor). Briefly, liver and brain tissues were digested in papain extraction reagent for 3 h at 65°C and then centrifuged. Aliquots of supernatant, normalized on DNA content, were processed with the Blyscan dye reagent to precipitate GAGs. The absorbance of the resuspended samples was measured at 656 nm and compared with a standard curve. For the qualitative detection of GAGs in liver samples, tissues were fixed in methacarn buffer solution (methanol 60%, chloroform 30%, and acetic acid 10%) for 24 h at 4°C and the next day embedded in paraffin after their dehydration with a 70%–100% ethanol gradient. Serial 4-μm sections were obtained by cutting with a microtome (Leica). After deparaffinization and rehydration, the tissue slides were stained with 1% Alcian blue in hydrochloric acid, counterstained with nuclear-fast red reagent, and processed by ZEISS Axio Scan.Z1 at 40×. Alcian blue was quantified by measuring RGB intensity on four different regions randomly selected for each histological section, using ImageJ software. Positive Alcian blue areas were selected by a common threshold of RGB intensity, presented as a percentage on the total analyzed region, and then averaged for each animal and each group of animals.
IF staining on liver tissues
Liver tissues (left lobes) were fixed with 4% PFA and subjected to a sucrose gradient (from 10% to 30%) before optimal cutting temperature compound (OCT) embedding. Cryosections of 8 μm were collected on slides (Superfrost Plus; Thermo Fisher Scientific) and incubated overnight with the primary antibodies (LAMP-1, D4b sc-19992, 1:800; CD68, AbD Serotec, 1:250; TFEB Bethyl A303-673A 1:600); then, slides were incubated with secondary antibodies Alexa Fluor 488 anti-rat or anti-mouse (1:1,000) and DAPI (1:500, Vector Laboratories H-1200) for 1 h at RT, and finally mounted with PBS/glycerol (Eukitt Mounting Medium; EMS). Confocal images were acquired by the LSM700 Zeiss Confocal Microscopy system using 40× immersion objective at a definition of 1,024 × 1,024 pixels, adjusting the pinhole diameter to 1 Airy unit for each emission channel to make all of the intensity values between 1 and 254 (linear range). Mean spot intensity was quantified on four different regions randomly selected for each histological section, using ImageJ software and presented as LAMP-1 mean intensity per cell or as a percentage of CD68+ cells.
Automated IHC
IHC assays were performed in 5-μm paraffin sections with the VENTANA BenchMark Ultra automated staining instrument (Ventana Medical Systems, Roche), using VENTANA reagents except as noted, according to the manufacturer’s instructions. Slides were deparaffinized using EZ Prep solution (950-102) for 16 min at 72°C. Epitope retrieval was accomplished with CC1 solution (950-224) at a high temperature (95°C) for a period that is suitable for a specific tissue type. Antibodies were titered with a blocking solution into user-fillable dispensers for use on the automated stainer. For bright-field detection, slides were developed using the VENTANA ultra-view Universal DAB detection kit (760-500) according to the manufacturer’s instructions. Slides were then counterstained with hematoxylin II (790-2208) for 8 min, followed by bluing reagent (760-2037) for 4 min. For fluorescent detection, slides were developed using the DISCOVERY FAM Kit (760-243) for 8 min. Slides were then counterstained with DISCOVERY QD DAPI (760-4196) for 8 min. Bright-field sections were scanned with ZEISS Axio Scan.Z1. The whole digital slides were viewed by ZEISS’s Zen Blue software. The positive pixel count algorithm was selected and adjusted to cover each positive staining of each marker analysis. The LAMP-1 and pS129-α-synuclein data were presented as positivity, which was obtained from the following formula: Positivity (%) = positive area (μm2)/total analyzed area (μm2) × 100%. The antibodies used were LAMP-1 (Santa Cruz Laboratories, sc-19992), anti-pS129 α-Syn (Abcam, ab51253), and TFEB (Bethyl A303-673A).
EM
MEF cells were fixed with a mixture of 4% PFA and 0.05% GA prepared in 0.2 M HEPES for 10 min and then fixed with 4% PFA for 30 min. Then, the cells were permeabilized and labeled with a primary antibody against LAMP-1 and subsequently incubated with a secondary antibody conjugated to 1.4-nm gold particles. Gold enhancement, post-fixation, and embedding procedures were performed as described in Polishchuk et al.55 The distances between PM and LAMP-1+ structures were measured using ITEM software. A specific brain region (cortex) and liver tissues were fixed with a mixture of 2% PFA and 1% glutaraldehyde prepared in 0.2 M HEPES (pH 7.3). The specimens were then post-fixed, dehydrated, embedded in epoxy resin, and polymerized as described in Polishchuk et al.55 Thin 60-nm sections were cut at the Leica EM UC7 microtome. EM images were acquired from thin sections using an FEI Tecnai-12 electron microscope equipped with a VELETTA CCD digital camera (FEI). Morphometric analysis on the size and number of lysosome-like structures, as well as the measure of the distance to PM, were performed on 10 fields of view per sample, using ITEM software).
Statistical analysis
For all of the experiments, the data were expressed as means ± SEMs. The significance of difference among the experimental groups was calculated by two-tailed Student’s t test, one-way or two-way ANOVA, followed by Bonferroni post hoc test. A p < 0.05 was considered statistically significant. Before applying the ANOVA test, data distribution normality was tested with the Kolmogorov-Smirnov test.
Contextual fear conditioning test
The contextual fear conditioning test was carried out in a behavioral testing room maintained under constant light, temperature, and humidity. The number of subjects per group was determined according to the limitation of our internal and national animal welfare committees. We were authorized to set power analysis parameters to effect size 0.4; α: 0.05 (1 − β): 0.9, which are in line with international guidelines.56 The mice were tested between 9 a.m. and 6 p.m. by an experimenter who was blinded to the genotype and the treatment. Before testing, animals were habituated to the testing room for at least 30 min. The test was performed as previously described.41,42 Briefly, each mouse was trained in a conditioning chamber (30 cm × 24 cm × 21 cm; Ugo Basile s.r.l.) that had a removable grid floor through which the shock was delivered. The shock intensity was 0.5 mA, with a duration of 2 s (unconditioned stimulus), and was presented 3 times; the conditioning chamber had striped walls (conditioned stimulus). Mice were tested 24 h after the training, without the foot shock within the same context, and expected to show freezing in response to the conditioned stimulus. Freezing behavior was defined as a complete lack of movement, except for respiration, and scored using a video-tracking system (ANY-maze, Stoelting). Data were expressed as freezing time in the training and testing phases. Data from the behavioral tests were expressed as means ± SEMs. The statistical significance was assessed using two-way ANOVA for repeated measures (two levels: training and test; factors: genotype, two levels: WT and MPS-IIIA; treatment, two levels: vehicle and FLX), followed by unequal post hoc test. Before applying the ANOVA test, the data distribution normality was tested with the Kolmogorov-Smirnov test.
Acknowledgments
We would like to thank TIGEM’s facilities: the High Content Screening Facility for the drug screening; the Advanced Histopathology Facility for embedding, IHC processing, and imaging the tissue samples; and the Advanced Microscopy and Imaging Core, the Behavioral Core, and the Bioinformatic Core for their support in the statistical data analysis. We acknowledge financial support from the Cure Sanfilippo Foundation (D.L.M. and A.C.) and Orphan Disease Center (D.L.M. and A.C.). The graphical abstract was generated using Biorender.com.
Author contributions
D.L.M. conceived the project, designed the experiments, and wrote the manuscript. N.C.S. designed the in vivo study and supervised the research; she contributed to the analysis of the in vivo data and the writing of the manuscript. A.C. designed, performed, and analyzed the in vitro and in vivo experiments. S.M., V.C., and M.S. contributed to the performance and analysis of the in vivo experiments. E.N. contributed to the performance of the in vivo experiments. E.D.L. designed the behavioral tests; M.T.P. and M.D.R. contributed to the performance and analysis of the behavioral tests. G.N. and A.E. designed and performed the in vitro experiments involving the mTOR pathway. E.P. designed and performed the EM experiments.
Declaration of interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.01.037.
Contributor Information
Nicolina Cristina Sorrentino, Email: sorrentino@tigem.it.
Diego Luis Medina, Email: medina@tigem.it.
Supplemental information
References
- 1.Hemsley K.M., Hopwood J.J. Development of motor deficits in a murine model of mucopolysaccharidosis type IIIA (MPS-IIIA) Behav. Brain Res. 2005;158:191–199. doi: 10.1016/j.bbr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 2.Settembre C., Fraldi A., Jahreiss L., Spampanato C., Venturi C., Medina D., de Pablo R., Tacchetti C., Rubinsztein D.C., Ballabio A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008;17:119–129. doi: 10.1093/hmg/ddm289. [DOI] [PubMed] [Google Scholar]
- 3.Monaco A., Maffia V., Sorrentino N.C., Sambri I., Ezhova Y., Giuliano T., Cacace V., Nusco E., De Risi M., De Leonibus E., et al. The amyloid inhibitor CLR01 relieves autophagy and ameliorates neuropathology in a severe lysosomal storage disease. Mol. Ther. 2020;28:1167–1176. doi: 10.1016/j.ymthe.2020.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sardiello M., Palmieri M., di Ronza A., Medina D.L., Valenza M., Gennarino V.A., Di Malta C., Donaudy F., Embrione V., Polishchuk R.S., et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 5.Medina D.L., Fraldi A., Bouche V., Annunziata F., Mansueto G., Spampanato C., Puri C., Pignata A., Martina J.A., Sardiello M., et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell. 2011;21:421–430. doi: 10.1016/j.devcel.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Settembre C., Ballabio A. TFEB regulates autophagy: an integrated coordination of cellular degradation and recycling processes. Autophagy. 2011;7:1379–1381. doi: 10.4161/auto.7.11.17166. [DOI] [PubMed] [Google Scholar]
- 7.Settembre C., Di Malta C., Polito V.A., Garcia Arencibia M., Vetrini F., Erdin S., Erdin S.U., Huynh T., Medina D., Colella P., et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Medina D.L., Ballabio A. Lysosomal calcium regulates autophagy. Autophagy. 2015;11:970–971. doi: 10.1080/15548627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moskot M., Montefusco S., Jakóbkiewicz-Banecka J., Mozolewski P., Węgrzyn A., Di Bernardo D., Węgrzyn G., Medina D.L., Ballabio A., Gabig-Cimińska M. The phytoestrogen genistein modulates lysosomal metabolism and transcription factor EB (TFEB) activation. J. Biol. Chem. 2014;289:17054–17069. doi: 10.1074/jbc.M114.555300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Contreras P.S., Tapia P.J., González-Hódar L., Peluso I., Soldati C., Napolitano G., Matarese M., Heras M.L., Valls C., Martinez A., et al. c-Abl inhibition activates TFEB and promotes cellular clearance in a lysosomal disorder. iScience. 2020;23:101691. doi: 10.1016/j.isci.2020.101691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mouti A., Reddihough D., Marraffa C., Hazell P., Wray J., Lee K., Kohn M. Fluoxetine for Autistic Behaviors (FAB trial): study protocol for a randomized controlled trial in children and adolescents with autism. Trials. 2014;15:230. doi: 10.1186/1745-6215-15-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perez-Caballero L., Torres-Sanchez S., Bravo L., Mico J.A., Berrocoso E. Fluoxetine: a case history of its discovery and preclinical development. Expert Opin. Drug Discov. 2014;9:567–578. doi: 10.1517/17460441.2014.907790. [DOI] [PubMed] [Google Scholar]
- 13.Selph S.S., McDonagh M.S. Depression in children and adolescents: evaluation and treatment. Am. Fam. Physician. 2019;100:609–617. [PubMed] [Google Scholar]
- 14.Kumar B., Prakash A., Sewal R.K., Medhi B., Modi M. Drug therapy in autism: a present and future perspective. Pharmacol. Rep. 2012;64:1291–1304. doi: 10.1016/s1734-1140(12)70927-1. [DOI] [PubMed] [Google Scholar]
- 15.Rein T. Is autophagy involved in the diverse effects of antidepressants? Cells. 2019;8:44. doi: 10.3390/cells8010044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou Y., Tao X., Wang Z., Feng L., Wang L., Liu X., Pan R., Liao Y., Chang Q. Hippocampus metabolic disturbance and autophagy deficiency in olfactory bulbectomized rats and the modulatory effect of fluoxetine. Int. J. Mol. Sci. 2019;20:4282. doi: 10.3390/ijms20174282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shu X., Sun Y., Sun X., Zhou Y., Bian Y., Shu Z., Ding J., Lu M., Hu G. The effect of fluoxetine on astrocyte autophagy flux and injured mitochondria clearance in a mouse model of depression. Cell Death Dis. 2019;10:577. doi: 10.1038/s41419-019-1813-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J.R., Xu H.Z., Nie S., Peng Y.C., Fan L.F., Wang Z.J., Wu C., Yan F., Chen J.Y., Gu C., et al. Fluoxetine-enhanced autophagy ameliorates early brain injury via inhibition of NLRP3 inflammasome activation following subrachnoid hemorrhage in rats. J. Neuroinflammation. 2017;14:186. doi: 10.1186/s12974-017-0959-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang F., Zhou H., Wilson B.C., Shi J.S., Hong J.S., Gao H.M. Fluoxetine protects neurons against microglial activation-mediated neurotoxicity. Parkinsonism Relat. Disord. 2012;18:S213–S217. doi: 10.1016/S1353-8020(11)70066-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vázquez C.L., Colombo M.I. Assays to assess autophagy induction and fusion of autophagic vacuoles with a degradative compartment, using monodansylcadaverine (MDC) and DQ-BSA. Methods Enzymol. 2009;452:85–95. doi: 10.1016/S0076-6879(08)03606-9. [DOI] [PubMed] [Google Scholar]
- 21.Scotto Rosato A., Montefusco S., Soldati C., Di Paola S., Capuozzo A., Monfregola J., Polishchuk E., Amabile A., Grimm C., Lombardo A., et al. TRPML1 links lysosomal calcium to autophagosome biogenesis through the activation of the CaMKKβ/VPS34 pathway. Nat. Commun. 2019;10:5630. doi: 10.1038/s41467-019-13572-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klionsky D.J., Abeliovich H., Agostinis P., Agrawal D.K., Aliev G., Askew D.S., Baba M., Baehrecke E.H., Bahr B.A., Ballabio A., et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mizushima N., Yamamoto A., Matsui M., Yoshimori T., Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell. 2004;15:1101–1111. doi: 10.1091/mbc.e03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ziolkowski A.F., Popp S.K., Freeman C., Parish C.R., Simeonovic C.J. Heparan sulfate and heparanase play key roles in mouse β cell survival and autoimmune diabetes. J. Clin. Invest. 2012;122:132–141. doi: 10.1172/JCI46177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J.P., Kusche-Gullberg M. Heparan sulfate: biosynthesis, structure, and function. Int. Rev. Cell Mol. Biol. 2016;325:215–273. doi: 10.1016/bs.ircmb.2016.02.009. [DOI] [PubMed] [Google Scholar]
- 26.Zhitomirsky B., Assaraf Y.G. Lysosomal accumulation of anticancer drugs triggers lysosomal exocytosis. Oncotarget. 2017;8:45117–45132. doi: 10.18632/oncotarget.15155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puertollano R., Ferguson S.M., Brugarolas J., Ballabio A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018;37:e98804. doi: 10.15252/embj.201798804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Medina D.L., Di Paola S., Peluso I., Armani A., De Stefani D., Venditti R., Montefusco S., Scotto-Rosato A., Prezioso C., Forrester A., et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015;17:288–299. doi: 10.1038/ncb3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Settembre C., Zoncu R., Medina D.L., Vetrini F., Erdin S., Erdin S., Huynh T., Ferron M., Karsenty G., Vellard M.C., et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–1108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Napolitano G., Di Malta C., Esposito A., de Araujo M.E.G., Pece S., Bertalot G., Matarese M., Benedetti V., Zampelli A., Stasyk T., et al. A substrate-specific mTORC1 pathway underlies Birt-Hogg-Dubé syndrome. Nature. 2020;585:597–602. doi: 10.1038/s41586-020-2444-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhaumik M., Muller V.J., Rozaklis T., Johnson L., Dobrenis K., Bhattacharyya R., Wurzelmann S., Finamore P., Hopwood J.J., Walkley S.U., Stanley P. A mouse model for mucopolysaccharidosis type III A (Sanfilippo syndrome) Glycobiology. 1999;9:1389–139610. doi: 10.1093/glycob/9.12.1389. [DOI] [PubMed] [Google Scholar]
- 32.Bhattacharyya R., Gliddon B., Beccari T., Hopwood J.J., Stanley P. A novel missense mutation in lysosomal sulfamidase is the basis of MPS III A in a spontaneous mouse mutant. Glycobiology. 2001;11:99–103. doi: 10.1093/glycob/11.1.99. [DOI] [PubMed] [Google Scholar]
- 33.Crawley A.C., Gliddon B.L., Auclair D., Brodie S.L., Hirte C., King B.M., Fuller M., Hemsley K.M., Hopwood J.J. Characterization of a C57BL/6 congenic mouse strain of mucopolysaccharidosis type IIIA. Brain Res. 2006;1104:1–17. doi: 10.1016/j.brainres.2006.05.079. [DOI] [PubMed] [Google Scholar]
- 34.Sorrentino N.C., Fraldi A. Brain targeting in MPS-IIIA. Pediatr. Endocrinol. Rev. 2016;13:630–638. [PubMed] [Google Scholar]
- 35.Fraldi A., Biffi A., Lombardi A., Visigalli I., Pepe S., Settembre C., Nusco E., Auricchio A., Naldini L., Ballabio A., Cosma M.P. SUMF1 enhances sulfatase activities in vivo in five sulfatase deficiencies. Biochem. J. 2007;403:305–312. doi: 10.1042/BJ20061783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sambri I., D'Alessio R., Ezhova Y., Giuliano T., Sorrentino N.C., Cacace V., De Risi M., Cataldi M., Annunziato L., De Leonibus E., Fraldi A. Lysosomal dysfunction disrupts presynaptic maintenance and restoration of presynaptic function prevents neurodegeneration in lysosomal storage diseases. EMBO Mol. Med. 2017;9:112–132. doi: 10.15252/emmm.201606965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sorrentino N.C., D'Orsi L., Sambri I., Nusco E., Monaco C., Spampanato C., Polishchuk E., Saccone P., De Leonibus E., Ballabio A., Fraldi A. A highly secreted sulphamidase engineered to cross the blood-brain barrier corrects brain lesions of mice with mucopolysaccharidoses type IIIA. EMBO Mol. Med. 2013;5:675–690. doi: 10.1002/emmm.201202083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Langford-Smith A., Wilkinson F.L., Langford-Smith K.J., Holley R.J., Sergijenko A., Howe S.J., Bennett W.R., Jones S.A., Wraith J., Merry C.L., et al. Hematopoietic stem cell and gene therapy corrects primary neuropathology and behavior in mucopolysaccharidosis IIIA mice. Mol. Ther. 2012;20:1610–1621. doi: 10.1038/mt.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fraldi A., Annunziata F., Lombardi A., Kaiser H.J., Medina D.L., Spampanato C., Fedele A.O., Polishchuk R., Sorrentino N.C., Simons K., Ballabio A. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. EMBO J. 2010;29:3607–3620. doi: 10.1038/emboj.2010.237. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Weng F.L., He L. Disrupted ubiquitin proteasome system underlying tau accumulation in Alzheimer's disease. Neurobiol. Aging. 2021;99:79–85. doi: 10.1016/j.neurobiolaging.2020.11.015. [DOI] [PubMed] [Google Scholar]
- 41.Sorrentino N.C., Cacace V., De Risi M., Maffia V., Strollo S., Tedesco N., Nusco E., Romagnoli N., Ventrella D., Huang Y., et al. Enhancing the therapeutic potential of sulfamidase for the treatment of mucopolysaccharidosis IIIA. Mol. Ther. Methods Clin. Dev. 2019;15:333–342. doi: 10.1016/j.omtm.2019.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Risi M., Tufano M., Alvino F.G., Ferraro M.G., Torromino G., Gigante Y., Monfregola J., Marrocco E., Pulcrano S., Tunisi L., et al. Altered heparan sulfate metabolism during development triggers dopamine-dependent autistic-behaviours in models of lysosomal storage disorders. Nat. Commun. 2021;12:3495. doi: 10.1038/s41467-021-23903-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spampanato C., Feeney E., Li L., Cardone M., Lim J.A., Annunziata F., Zare H., Polishchuk R., Puertollano R., Parenti G., et al. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol. Med. 2013;5:691–706. doi: 10.1002/emmm.201202176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palmieri M., Pal R., Nelvagal H.R., Lotfi P., Stinnett G.R., Seymour M.L., Chaudhury A., Bajaj L., Bondar V.V., Bremner L., et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat. Commun. 2017;8:14338. doi: 10.1038/ncomms14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soldati C., Lopez-Fabuel I., Wanderlingh L.G., Garcia-Macia M., Monfregola J., Esposito A., Napolitano G., Guevara-Ferrer M., Scotto Rosato A., Krogsaeter E.K., et al. Repurposing of tamoxifen ameliorates CLN3 and CLN7 disease phenotype. EMBO Mol. Med. 2021;13:e13742. doi: 10.15252/emmm.202013742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song W., Wang F., Savini M., Ake A., di Ronza A., Sardiello M., Segatori L. TFEB regulates lysosomal proteostasis. Hum. Mol. Genet. 2013;22:1994–2009. doi: 10.1093/hmg/ddt052. [DOI] [PubMed] [Google Scholar]
- 47.Rega L.R., Polishchuk E., Montefusco S., Napolitano G., Tozzi G., Zhang J., Bellomo F., Taranta A., Pastore A., Polishchuk R., et al. Activation of the transcription factor EB rescues lysosomal abnormalities in cystinotic kidney cells. Kidney Int. 2016;89:862–873. doi: 10.1016/j.kint.2015.12.045. [DOI] [PubMed] [Google Scholar]
- 48.Carosi J.M., Sargeant T.J. Rapamycin and Alzheimer disease: a double-edged sword? Autophagy. 2019;15:1460–1462. doi: 10.1080/15548627.2019.1615823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gulbins A., Schumacher F., Becker K.A., Wilker B., Soddemann M., Boldrin F., Müller C.P., Edwards M.J., Goodman M., Caldwell C.C., et al. Antidepressants regulate autophagy by targeting acid sphingomyelinase. Mol. Psychiatr. 2018;23:2251. doi: 10.1038/s41380-018-0319-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu F.Y., Cai J., Wang C., Ruan W., Guan G.P., Pan H.Z., Li J.R., Qian C., Chen J.S., Wang L., Chen G. Fluoxetine attenuates neuroinflammation in early brain injury after subarachnoid hemorrhage: a possible role for the regulation of TLR4/MyD88/NF-κB signaling pathway. J. Neuroinflammation. 2018;15:347. doi: 10.1186/s12974-018-1388-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Casarotto P.C., Girych M., Fred S.M., Kovaleva V., Moliner R., Enkavi G., Biojone C., Cannarozzo C., Sahu M.P., Kaurinkoski K., et al. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell. 2021;184:1299–1313.e19. doi: 10.1016/j.cell.2021.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perrone J.A., Chabla J.M., Hallas B.H., Horowitz J.M., Torres G. Weight loss dynamics during combined fluoxetine and olanzapine treatment. BMC Pharmacol. 2004;4:27. doi: 10.1186/1471-2210-4-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alvarez J.C., Bothua D., Collignon I., Advenier C., Spreux-Varoquaux O. Determination of fluoxetine and its metabolite norfluoxetine in serum and brain areas using high-performance liquid chromatography with ultraviolet detection. J. Chromatogr. B Biomed. Sci. Appl. 1998;707:175–180. doi: 10.1016/s0378-4347(97)00588-. [DOI] [PubMed] [Google Scholar]
- 54.Palm W., Park Y., Wright K., Pavlova N.N., Tuveson D.A., Thompson C.B. The utilization of extracellular proteins as nutrients is suppressed by mTORC1. Cell. 2015;162:259–270. doi: 10.1016/j.cell.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Polishchuk E.V., Merolla A., Lichtmannegger J., Romano A., Indrieri A., Ilyechova E.Y., Concilli M., De Cegli R., Crispino R., Mariniello M., et al. Activation of autophagy, observed in liver tissues from patients with wilson disease and from ATP7B-deficient animals, protects hepatocytes from copper-induced apoptosis. Gastroenterology. 2019;156:1173–1189.e5. doi: 10.1053/j.gastro.2018.11.032. [DOI] [PubMed] [Google Scholar]
- 56.Sonzogni M., Wallaard I., Santos S.S., Kingma J., du Mee D., van Woerden G.M., Elgersma Y. A behavioral test battery for mouse models of Angelman syndrome: a powerful tool for testing drugs and novel Ube3a mutants. Mol. Autism. 2018;9:47. doi: 10.1186/s13229-018-0231-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.