

Abstract
The androgen receptor (AR) plays a pivotal role in driving prostate cancer (PCa) development. However, when stimulated by high levels of androgens, AR can also function as a tumor suppressor in PCa cells. While the high-dose testosterone (high-T) treatment is currently being tested in clinical trials of castration-resistant prostate cancer (CRPC), there is still a pressing need to fully understand the underlying mechanism and thus develop treatment strategies to exploit this tumor-suppressive activity of AR. In this study, we demonstrate that retinoblastoma (Rb) family proteins play a central role in maintaining the global chromatin binding and transcriptional repression program of AR and that Rb inactivation desensitizes CRPC to the high-dose testosterone treatment in vitro and in vivo. Using a series of patient-derived xenograft (PDX) CRPC models, we further show that the efficacy of high-T treatment can be fully exploited by a CDK4/6 inhibitor, which strengthens the chromatin binding of the Rb-E2F repressor complex by blocking the hyperphosphorylation of Rb proteins. Overall, our study provides strong mechanistic and preclinical evidence on further developing clinical trials to combine high-T with CDK4/6 inhibitors in treating CRPC.
Key words: prostate cancer, androgen receptor, androgen deprivation therapy, transcriptional repression, high-dose androgen, DNA replication, retinoblastoma protein, Rb, E2F1, CDK4/6 inhibitor
Graphical abstract
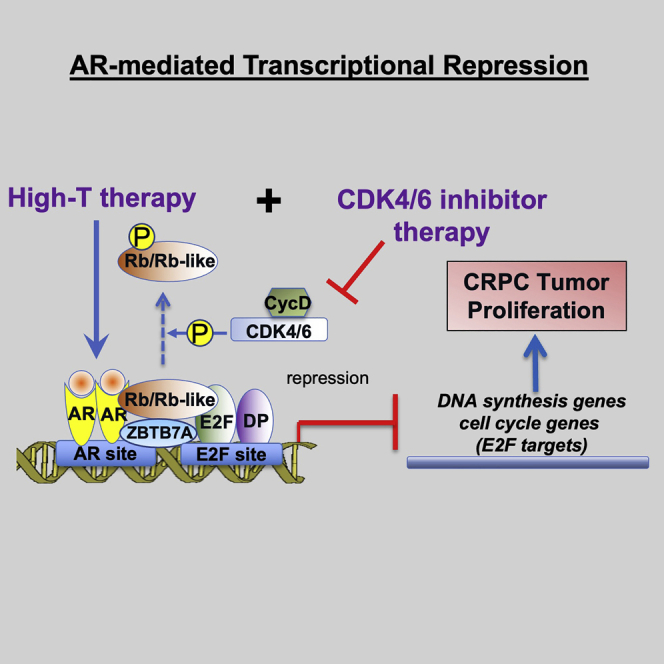
We show that Rb family proteins play a central role in maintaining the global chromatin binding and transcriptional repression program of AR in prostate cancer cells and that the high-dose-androgen-induced transcriptional repression activity can be fully exploited by CDK4/6 inhibitor treatment, which blocks the hyperphosphorylation of Rb proteins.
Introduction
Androgens exert their actions by binding to the androgen receptor (AR) and thus induce the transcriptional activity of AR.1 While AR is well known for its transcriptional activation function in normal prostate and prostate cancer (PCa) cells, it can also function as a transcriptional repressor by recruiting repressive cofactors (such as LSD1 and EZH2) to suppress the expression of a subset of genes, including AR and its splice variants (AR-Vs), androgen synthetic genes, and genes mediating DNA replication and repair.2, 3, 4, 5 The transcriptional repression of AR and AR-Vs and androgen synthetic genes (such as AKR1C3 and HSD17B6) functions as a negative-feedback loop and plays a critical role to restore AR signaling during tumor progression to the castration-resistant stage of PCa (CRPC).1 More importantly, the repression activity of AR on DNA synthesis and cell-cycle pathways provides a molecular mechanism for the high-dose testosterone (high-T) therapy in PCa.5,6 Previous studies have shown that high-T treatment alone or in combination with DNA-damaging reagents can suppress the growth of CRPC or CRPC that is resistant to enzalutamide in preclinical studies using patient-derived xenograft (PDX) models.7,8 Clinical studies of the bipolar androgen therapies (BATs) that periodically treat the CRPC patients with rapid cycling of high-T and AR-signaling-inhibition agents to disrupt the adaptive regulation of AR signaling in CRPC patients also show encouraging results in recent phase II clinical trials.9, 10, 11 Several additional mechanisms have been suggested, including androgen-stimulated DNA-damaging effect, licensing factor function of AR on DNA replication, androgen-induced expression of tumor suppressor genes (such as ZBTB16), and androgen-repressed expression of oncogenes (such as MYC) and anti-apoptotic proteins.7,12, 13, 14, 15, 16, 17, 18, 19 Nonetheless, there is an urgent need to fully understand the tumor suppressor activity of AR and thus identify treatment strategies that can further exploit the anti-tumor activity of high-dose androgens.
Retinoblastoma protein (Rb) (encoded by RB1 gene) is a well-established tumor-suppressor protein by forming a repressor complex with E2F transcription factors.20 The Rb-E2F complex plays an important role in cells to repress the transcription of genes mediating DNA synthesis and cell cycle progression. Hyperphosphorylation of Rb by cyclin D-CDK4/6 and cyclin E-CDK2 disrupts the interaction of Rb with E2Fs and thus activates E2F transcriptional activities.21 Recent sequencing studies in CRPC tumor samples have revealed a high frequency of RB1 deletion in ∼10%–15% of tumors, and the loss of RB1 is associated with worse patient outcomes,22,23 indicating a critical role of Rb in preventing the progression of CRPC. RB1 loss leads to an expansion of E2F1 cistrome, increased redox metabolism, and lineage plasticity, and is associated with the resistance to AR-signaling-inhibition therapies.24, 25, 26, 27, 28 Our previous studies have shown that AR can recruit hypophosphorylated Rb to DNA replication gene loci and strengthens the activity of Rb-E2F suppressor complex.5 However, it is still unclear how Rb globally mediates this transcriptional repressor activity of AR in CRPC and whether enhancing Rb-E2F transcriptional repression activity can exploit the tumor-suppressive function of high-T in vivo.
In this study, using integrated transcriptomic and cistromic analyses, we found that Rb depletion in CRPC cells broadly compromised the AR-mediated direct transcriptional repression on E2F-regulated genes, but not the indirect repression on Myc-regulated genes. We then demonstrated that the Rb-like pocket protein p130 can similarly mediate the transcriptional repression activity of AR and partially compensate for the function of Rb. Moreover, we also found that Rb depletion can reprogram the transcriptional repression activity of AR by altering AR chromatin binding. Consistent with these mechanistic studies, the Rb-proficient CRPC cells were more sensitive to the high-T treatment in vitro and in vivo in comparison with Rb-deficient cells. Since Rb/p130 activities are highly dependent on CDK4/6, we next determined whether high-T treatment can be combined with CDK4/6 inhibitors. Significantly, we found that a clinically approved CDK4/6 inhibitor, palbociclib, can enhance the efficacy of high-T treatment in a set of CRPC cell line and PDX models. Overall, this study demonstrates a critical role of Rb family proteins in mediating the tumor-suppressive activity of high-T and suggests a therapeutic strategy to enhance the efficacy of high-T treatment with combined treatment of CDK4/6 inhibitors.
Results
High-dose androgen treatment enhances global Rb binding to suppress E2F signaling
Our previous study has shown that AR can transcriptionally repress genes mediating DNA synthesis through direct chromatin binding and the interaction with hypophosphorylated Rb.5 Therefore, we hypothesize that the AR-promoted Rb-E2F repressor complex activity is a major mechanism for the tumor-suppressive function of high-T in PCa. To further study the role of Rb in this process, we generated a stable cell line expressing doxycycline-inducible lentiviral short hairpin RNA (shRNA) against RB1 in LNCaP-derived C4-2 CRPC cells (C4-2-tet-shRB).29 Consistent with the previous studies in VCaP model,2,30 while 0.1 nM dihydrotestosterone (DHT) treatment can sufficiently activate classic androgen-induced genes, higher concentrations of DHT (>1 nM) were required to induce AR-mediated transcription repression on previously identified DNA synthesis and cell cycle genes in C4-2 model (Figures S1A and S1B). We then globally characterized the transcriptional repression activity of AR in Rb-positive CRPC cells by comparing the high-dose-androgen (10 nM DHT, comparable to the androgen levels in pre-castrated men)-induced AR transcriptome in uninduced C4-2-tet-shRB cells, VCaP cells, and VCaP-CR cells (derived from castration-resistant VCaP xenograft).2,31 While androgen-induced genes were enriched primarily in lipid biosynthesis pathways (Figure S2A), consistent with previous studies,32,33 androgen-repressed genes were highly enriched in E2F and Myc transcriptional target gene sets (Figures 1A and 1B and S2B). Both pathways are known to mediate DNA synthesis and cell cycle progression. Interestingly, androgen-repressed genes were also enriched for a previously identified gene set (NE-up) that is upregulated in the neuroendocrine (NE) subtype of CRPC (NEPC),34 suggesting that AR may transcriptionally repress NE differentiation, a consistent finding with previous studies.16,35, 36, 37 Moreover, combining the chromatin immunoprecipitation sequencing (ChIP-seq) analyses in these models with the transcriptome data, we found that those AR-repressed genes associated with nearby AR bindings (potentially AR directly repressed genes) were similarly enriched for E2F targets and NE-up genes but less enriched for Myc targets (Figure 1C), suggesting that E2F target genes, as well as NE genes, are the major direct targets of AR-mediated transcriptional repression. However, the androgen-repressed expression of Myc target genes is likely an indirect effect that is possibly mediated through repression of the MYC gene15,18 or other Myc cofactors. Consistent with previous findings, AR directly activated genes were enriched for lipid biosynthesis pathways (Figure S2C).2,33
Figure 1.

High-dose androgen treatment enhances global Rb binding to suppress E2F signaling
(A) C4-2 cells were stably infected by doxycycline-inducible lentiviral shRNA against non-target control (shNTC) or RB1 (shRB, C4-2-tet-shRB). RNA-seq was done in C4-2-tet-shRB cells (no doxycycline treatment) stimulated by ethanol or 10 nM DHT for 24 h (hours). The gene profiling data for VCaP and VCaP-CR cells stimulated by ethanol and DHT (10 nM, 24 h) were obtained from a previous study.2 Gene set enrichment analysis (GSEA) was done to compare the androgen-repressed genes in C4-2-tet-shRB versus VCaP/VCaP-CR cells. NES, normalized enrichment score. (B) Enrichments of HALLMARK_E2F_TARGETS and HALLMARK_MYC_TARGET_V1 gene sets were plotted. (C) GSEA was done to compare directly androgen-repressed genes (AR-repressed genes with nearby AR binding) in these cells. (D–G) ChIP-seq analyses were performed in C4-2 cells treated with ethanol or DHT (10 nM, 4 h). Venn diagrams (D and E) or heatmap view (F and G) for Rb or c-Myc binding peaks were shown. (H) Binding and expression target analysis (BETA) for the association of Rb or c-Myc binding sites (DHT stimulated) with the expression of AR-repressed genes identified from RNA-seq (A). n.s., not significant.
Next, we determined whether androgen treatment may globally affect Rb or c-Myc chromatin binding. ChIP-seq analyses of Rb and c-Myc were conducted in C4-2 cells in the absence or presence of 10 nM DHT. As shown in Figures 1D and 1E, the number and intensity of Rb binding peaks were noticeably increased by the androgen treatment, consistent with our previous findings that AR can recruit Rb to strengthen the Rb-E2F repressor complex. However, although the number of c-Myc chromatin binding peaks was increased, the intensity of those binding peaks did not appear to be significantly affected (Figures 1F and 1G). Furthermore, using binding and expression target analysis (BETA),38,39 we found that Rb chromatin binding was highly associated with the transcriptional repression function of AR, while c-Myc binding was not significantly associated with AR-regulated genes (Figure 1H). Together, these data indicate that AR can directly repress E2F signaling and indirectly repress Myc signaling and that the direct regulation of Rb-E2F activity may be one primary mechanism of AR-mediated transcriptional repression function in CRPC cells.
Rb mediates AR repression of E2F signaling and cell cycle progression
The protein expression of Rb was efficiently depleted by the doxycycline treatment (within 3 days) in C4-2-tet-shRB cells (Figures 2A and S3A), and this Rb ablation impaired the androgen-repressed cell cycle progression (Figure 2B). We then performed RNA-seq analyses in these stable cells with both short-term (3 days) and long-term (∼30 days) treatments of doxycycline. As shown in Figures 2C and S3B, while Rb depletion did not appear to broadly affect DHT-induced gene expression, it clearly impaired the repression activity of DHT on a large subset of genes (clusters 1 and 3). We then compared the effect of Rb depletion on the E2F pathway versus the Myc pathway genes. As shown in Figures 2D (upper panel) and S3C–S3E and Table S1, E2F target genes were broadly repressed by DHT, but this repression effect was significantly attenuated by Rb silencing. Many E2F family genes, particularly activator E2Fs (E2F1–3), are known as direct targets of Rb-E2F repressor complex,40 and their expression was also AR repressed and mediated by Rb (Figure 2E). One major activity of Rb-E2F complex is to suppress DNA synthesis via transcriptional repression of MCM helicase and other DNA replication factors.41 A group of these genes (77 genes) was previously identified as AR-repressed genes,5 and the full repression activity of DHT on these genes was also dependent on Rb expression (Figure S3F). On the contrary, MYC, PCAT-1 (function to stabilize c-Myc protein),42 and many Myc target genes were repressed by androgen treatment, but this repression effect was not significantly affected by Rb depletion (Figures 2D, lower panel, and 2F).
Figure 2.

Rb depletion compromises global AR repression activity
(A) Immunoblotting for Rb in C4-2-tet-shRB cells treated with doxycycline (0.05 μg/mL) at 0, 3, or 30 d (days). (B) The flow cytometry cell cycle analysis for C4-2-tet-shRB cells treated with or without doxycycline and with or without 10 nM DHT for 24 h. (C) RNA-seq analyses were done in C4-2-tet-shRB cells treated with doxycycline (0.05 μg/mL at 0, 3, or 30 days) and stimulated with or without DHT (10 nM, 24 h). Heatmap view for DHT-repressed genes was shown. (D) Box plots for the change of expression (Log2(fold-change)) for E2F target genes or Myc target genes in these samples are shown. (E) Heatmap view for E2Fs (upper panel) and a panel of E2F target genes (lower panel) is shown. (F) Heatmap view for MYC and its coregulators (upper panel) and a panel of Myc target genes (lower panel) is shown. (G) Immunoblotting for Rb, AR, and FOXA1 in control C4-2 cells versus selected clones of C4-2 with RB1 knockout (KO1–4) using CRISPR-Cas9 approach is shown. (H) The proliferation assay for control C4-2 cells and two RB-KO lines treated with 0–20 μM of enzalutamide for 6 d is shown. (I) The flow cytometry cell cycle analysis for the control C4-2 and two RB-KO clones is shown. (J) Quantitative real-time PCR for several E2F-regulated genes in control lines versus RB-KO lines is shown. Data in bar graphs represent the mean ± SD. n.s., not significant. ∗p < 0.05.
Since increased E2F expression is a result of Rb depletion, we next examined whether the impairment of AR-mediated repression is directly due to the elevated expression of E2Fs. To test this hypothesis, we stably expressed a doxycycline-inducible E2F1 in C4-2 cells (Figure S4A). Interestingly, overexpression of E2F1 modestly enhanced AR binding to the activation sites and the DHT-induced expression of AR-activated genes (Figures S4B and S4C). However, the AR binding to the suppression sites and DHT-induced repression of DNA-replication genes were not affected (Figures S4D and S4E), suggesting that overexpression of E2F is not sufficient to impair the transcriptional repression activity of AR on DNA synthesis and cell cycle genes. The epigenetic modifier EZH2 was also shown to be a critical target of Rb-E2F, and RB1 loss can induce EZH2 activity in neuroendocrine PCa.24,25,43 Since EZH2 was reported to involve in the transcriptional repression activity of AR,3 we next examined whether EZH2 inhibition can impair the AR-mediated repression on DNA synthesis and cell cycle genes. As shown in Figure S5, treating C4-2 cells with an EZH2-specific inhibitor (GSK126) modestly decreased the basal expression of DNA synthesis genes but did not appear to significantly alter the DHT-induced transcriptional repression on these genes, indicating that EZH2 is not involved in this specific activity of AR.
To further confirm the Rb dependence in androgen-induced transcriptional repression of DNA synthesis and cell cycle, we also generated RB1 knockout (RB-KO) stable clones using the CRISPR-Cas9 approach (Figure 2G). These RB-KO clones developed resistance to enzalutamide treatment (Figure 2H), consistent with previous findings.25,27 Importantly, the androgen-induced cell cycle repression was significantly impaired in RB-KO cells (Figure 2I). A similar effect was also observed when examining the androgen-induced repression of several DNA synthesis genes (although clonal variation was observed; Figure 2J), confirming the critical role of Rb in mediating the transcriptional repression activity of AR. Overall, these in vitro studies clearly demonstrated a critical role of Rb in mediating the transcriptional repression and tumor-suppressive activities of high-dose androgens in CRPC.
p130 compensates for the absence of Rb in mediating the transcriptional repression activity of AR in Rb-depleted cells
Since Rb depletion cannot completely prevent the androgen-mediated transcriptional repression of E2F signaling (see Figure 2D), it suggests that additional factors may be involved in mediating E2F activity when Rb expression level is low or absent. One possible factor is ZBTB7A, which can act as a corepressor of AR and mediate AR transcriptional repression of E2F signaling.6 Other factors may include Rb-like pocket proteins, p107 and p130 (encoded by RBL1 and RBL2, respectively), which were known to form complex with E2Fs (such as DREAM complex)44 and play an important role in regulating E2F signaling. It is also known that these pocket proteins (Rb/p107/p130) have partly redundant activities and can often compensate for each other if one is inactivated or absent. In addition, p130 has been shown to regulate the AR-mediated repression of EZH2 in PCa cells.14,45 While RBL1 expression was generally low in CRPC patient samples (SU2C metastatic CRPC dataset),23 RBL2 expression was much higher in CRPC. Moreover, RBL2 deep deletions were detected in ∼3.5% of CRPC samples (SU2C, analyzed through cBioPortal)46,47 and appear to be mutually exclusive to RB1 deletion (Figure S6A), suggesting RBL2 may play a similar tumor suppressor function in CRPC progression. Therefore, we next focused on examining whether RBL2/p130 is involved in mediating the transcriptional repression activity of AR.
We first examined the chromatin binding of p130 in C4-2-tet-shRB cells using ChIP-seq analyses. In the untreated cells, the majority of Rb-binding sites (∼80%) and E2F1-binding sites28 (∼60%) were overlapped with p130 binding (∼25% and ∼70% of its total binding sites, respectively; Figures 3A and 3B), suggesting that p130 may function alternatively to Rb to form complexes with E2Fs. Examining several previously identified Rb-binding sites, we confirmed the occupancy of p130, which can be similarly enhanced by the androgen treatment (Figure S6B). Interestingly, the total number of p130 binding peaks was decreased when Rb expression was silenced, but the remaining p130 peaks showed increased overlapping with E2F1 binding (∼80% of p130 peaks) and were strongly associated with Rb-E2F-repressed genes (Figures 3C, 3D, and S6C). Moreover, the interaction between p130 and E2F1 was markedly increased by Rb depletion and can be further strengthened by a CDK4/6 inhibitor, palbociclib, which blocked the phosphorylation of p130 (Figures 3E–3G), suggesting that p130 may be similarly regulated by CDKs and function to compensate for the loss of Rb to repress the activity of E2Fs.
Figure 3.

p130 compensates for the absence of Rb in mediating the transcriptional repression activity of AR
(A and B) ChIP-seq analysis of p130 was performed in uninduced C4-2-tet-shRB cells (no doxycycline). The Venn diagram for p130 and Rb (A) or p130 and E2F1 (B) overlapping binding peaks was shown. (C) ChIP-seq analysis of p130 was performed in C4-2-tet-shRB cells treated with vehicle or doxycycline (3 d). The Venn diagram for p130 binding peaks in both conditions was shown. (D) BETA for the association of p130 binding peaks (doxycycline treated) with the expression of Rb-repressed genes identified from RNA-seq of C4-2-tet-shRB (doxycycline versus vehicle) is shown. (E) Immunoprecipitation (IP) of E2F1 in C4-2-tet-shRB cells (doxycycline for 0, 3, or 30 d), followed by immunoblotting for p130 or E2F1, is shown. (F and G) IP of p130 (F) or E2F1 (G) in C4-2-tet-shRB cells cultured with or without doxycycline and treated with vehicle or palbociclib (1μM, 24 h), followed by immunoblotting for total p130, p-p130 (S672 phosphorylated), or E2F1 is shown. (H) BETA for the association of p130 binding peaks (doxycycline treated) with the expression of AR-repressed genes identified from RNA-seq of C4-2-tet-shRB (doxycycline treated) is shown. (I–K) C4-2-tet-shRB cells stably infected by lentiviral shRNAs against NTC or RBL2 were treated with vehicle or doxycycline (6 d), followed by immunoblotting for Rb, p130, and E2F1 (I), quantitative real-time PCR for E2F-regulated genes (J), and the flow cytometry cell cycle analysis (K). Data in bar graphs represent the mean ± SD. n.s., not significant. ∗p < 0.05.
Next, we determined whether p130 contributes to the transcriptional repression activity of AR in Rb-depleted cells. The RBL2 mRNA expression was slightly decreased by Rb ablation (Figure S6D), but its protein expression was barely affected (see Figure 3E). Notably, the chromatin binding of p130 was significantly associated with AR repression function when Rb expression was silenced (Figure 3H). Interestingly, knocking down RBL2 also decreased Rb protein and mRNA expression (Figures 3I and S6E), indicating an additional function of RBL2 to support RB1 expression in C4-2 cells. Nonetheless, co-silencing both RB1 and RBL2 further blocked the repression activity of DHT on E2F-regulated DNA replication genes and the cell cycle progression (Figures 3J and 3K). Similar results were also obtained from co-silencing RB1 and RBL2 in VCaP model (Figures S7A and S7B). Together, these data indicate that p130 participates in the androgen-induced transcriptional repression of E2F signaling and may partially compensate for the absence of Rb.
Rb depletion alters AR chromatin binding
While our data indicated that Rb depletion may broadly compromise the androgen-induced repression of genes, it is unclear whether Rb is important to maintain AR chromatin binding. To determine this, we carried out AR ChIP-seq in C4-2-tet-shRB cells treated with or without doxycycline and stimulated by high-dose androgens (10 nM DHT) to identify AR binding sites. As shown in Figures 4A and 4B, DHT treatment markedly increased AR chromatin binding in both doxycycline-treated (3 d) and untreated cells. However, the DHT-induced AR binding sites were significantly altered in Rb-depleted cells versus the control cells (Figure 4C), indicating a rapid redistribution of AR chromatin binding by Rb inactivation. We then performed ChIP-seq of FOXA1, a critical pioneer factor of AR,48,49 to determine whether Rb depletion can affect FOXA1 chromatin binding prior to AR recruitment. To minimize the feedback effect of AR on FOXA1 chromatin binding,50 we carried out this experiment in the absence of DHT. Interestingly, unlike the massive redistribution of AR binding, the FOXA1 binding sites were largely conserved in Rb-depleted cells (Figure 4D), indicating that Rb depletion did not alter FOXA1 binding. Significantly, ∼70% of AR binding sites (13,256) were lost by Rb depletion, and these “lost” sites had weaker AR binding than the “conserved” sites but were strongly associated with AR and FOXA1 binding motifs (Figures 4E and 4F). Moreover, ∼30% (223/806) of AR-repressed genes contained at least one of these “lost” sites and the repression of these genes was impaired by Rb depletion (Figures 4G and 4H), suggesting that Rb depletion may disrupt AR chromatin binding at a subset of suppressive androgen response elements (AREs). On the contrary, AR-activated genes containing lost sites did not seem to be affected by Rb depletion (Figures S8A and S8B).
Figure 4.

Rb depletion reprograms the transcriptional repression activity of AR
(A–C) ChIP-seq analyses of AR were performed in C4-2-tet-shRB cells cultured with vehicle or doxycycline (3 days) and then stimulated with ethanol or DHT (10 nM, 4 h). Venn diagrams for AR binding peaks in cells treated with DHT versus ethanol in the absence of doxycycline (A), DHT versus ethanol in the presence of doxycycline (B), and doxycycline versus vehicle in the presence of DHT (C) were shown. (D) ChIP-seq analyses of FOXA1 were performed in C4-2-tet-shRB cells cultured with vehicle or doxycycline. The Venn diagram for FOXA1 binding peaks in both conditions was shown. (E) Heatmap view for ChIP-seq signal intensity of AR and FOXA1 at clustered AR-binding sites (lost, conserved, and gained) is shown. (F) Motif enrichment analysis for these AR binding sites is shown. (G) The Venn diagram for AR-repressed genes and the gene annotation of the “lost” AR sites is shown. (H) Box plots for the change of expression of AR-repressed genes containing at least one lost AR site in C4-2-tet-shRB cells are shown. (I) Heatmap view for RB1-silencing-induced AR-reprogramming targets determined by gained AR binding and regulation (fold-change > 1.5) is shown. (J) Gene ontology analyses for the reprogrammed AR-repressed genes are shown. (K) Heatmap view for a panel of reprogrammed AR-repressed genes is shown.
Rb depletion also resulted in 13,189 “gained” AR sites, but these sites were not occupied by FOXA1 or enriched for AR/FOXA1 binding motifs. Indeed, the gained AR binding sites appeared to contain binding motifs for a distinct set of transcription factors, including zinc finger and BTB-domain-containing proteins (ZBTB3), nuclear receptor family members (HNF4A and VDR), glial cells missing transcription factors (GCM1), nescient helix loop helix 1 (NHLH1), and E2Fs. We next conducted a combined analysis using these ChIP-seq data and RNA-seq data from C4-2-tet-shRB cells to identify genes that were associated with gained AR chromatin binding and AR regulation upon Rb depletion. This analysis resulted in the identification of 41 new androgen-repressed genes but only 24 new androgen-induced genes (Figure 4I). While we did not find any pathway enrichment in the androgen-induced genes, these new androgen-repressed genes were enriched in the pathways of maintaining cell polarity (Figure 4J), which may contribute to cancer progression and regulation of cell lineage plasticity. These genes include FRMD4B, MARK1, and FGF13 (Figure 4K), and the expression of cytoplasmic FGF13 protein has been previously reported to correlate with increased risk of biochemical recurrence of PCa.51 Overall, these results indicate that Rb expression is critical for maintaining AR chromatin binding and that Rb depletion can disrupt canonical AR binding at a subset of AR-repressed gene loci and may also promote noncanonical AR binding to repress a set of new targets.
Rb depletion desensitizes CRPC responses to high-dose androgen treatment
Deletion of RB1 is commonly found in CRPC and contributes to its progression.23 While the above results clearly indicated a central role of Rb family proteins in mediating the global repression activity of AR, it is not clear whether CRPC tumors with RB1 loss may completely turn into non-responders to high-T treatment. Therefore, we next sought to determine whether Rb activity affects the tumor response to high-T treatment in vivo. Xenograft tumors derived from C4-2-tet-shRB cells were established in castrated male severe combined immunodeficiency (SCID) mice, and RB1 silencing was induced by switching the regular diet to a doxycycline-supplemented diet (Figure S9A). These mice were then treated with two doses of testosterone treatments, both of which were well tolerated by the animals (Figures S9B and S9C). The lower dose of testosterone treatment (40 mg/kg) that was used in the experiment is comparable with the dose used in the previous preclinical studies and clinical trials.8, 9, 10 The growth of Rb-proficient C4-2 tumors was markedly repressed by both testosterone treatments (Figure 5A). However, while the higher dose of testosterone (160 mg/kg) can still suppress the growth of Rb-deficient tumors, the lower dose of testosterone treatment showed a reduced efficacy in treating these tumors (Figure 5B). To further confirm this finding, we also selected an RB-KO clone (Figure 5C) to determine the effect of RB1 loss on the tumor-suppressive effect of DHT. While 10, 100, or 1,000 nM DHT treatment can similarly repress C4-2 cell proliferation, RB1 loss appeared to have a stronger effect on the lower doses of DHT versus higher doses of DHT (Figure 5D). We then generated xenograft tumors using this RB-KO line and similarly treated mice with two doses of testosterone. As shown in Figure 5E, the RB-KO tumor did not respond to the lower dose of testosterone but modestly responded to the higher dose. This result is consistent with the observation in the C4-2-tet-shRB model, but the tumor-suppressive effect of high-T appeared to be more significantly impaired by the complete silencing of RB1.
Figure 5.

Rb depletion desensitizes CRPC tumor response to high-T
(A and B) Xenograft tumors derived from C4-2-tet-shRB cells were established in castrated male SCID mice. Tumor-bearing mice fed with a regular diet (A) or a doxycycline-supplemented diet (B) were daily injected with vehicle or testosterone (40 mg/kg or 160 mg/kg). Tumor volume was measured manually by caliber (n = 6). Note: the two xenograft experiments were done independently. (C) Cell proliferation assay for control C4-2 cell line and two RB-KO cell lines treated with or without DHT (10 nM, 0–6 d) is shown. (D) Cell proliferation assay for control C4-2 cell line and RB-KO-3 cell line treated with or without DHT (0–1,000 nM, 0–5 d) is shown. (E) Xenograft tumors derived from RB-KO-3 line were established in castrated male SCID mice. Tumor-bearing mice were then daily injected with vehicle or testosterone (40 mg/kg or 160 mg/kg). Tumor volume was measured manually by caliber (n = 4). Data in growth curves represent the mean ± SE. n.s., not significant.
We next performed an RNA-seq analysis in the tumor samples from C4-2-tet-shRB xenografts to determine the impact of high-T (higher dose) on tumor cell transcriptomes (Figures 6A and S10A). The androgen-repressed genes were significantly enriched for E2F signaling, cell cycle pathways (G2M checkpoint and mitotic spindle), and NE-up genes in Rb-proficient tumors but were noticeably less enriched for these pathways in Rb-depleted tumors, despite that the tumor growth was still repressed by this dose of high-T. In contrast, the enrichment of Myc signaling (MYC_TARGET_V1) and DNA repair pathway were not significantly affected by Rb silencing, indicating a persistent repression activity on these pathways. Interestingly, we have also observed the enrichment of the oxidative phosphorylation pathway in both conditions, which was not seen in the cell line studies. Moreover, the high-T treatment specifically repressed p53 and protein secretion pathways in Rb-silenced cells. The androgen-mediated repression of the AR gene itself was not affected by Rb depletion (Figure S10B). As expected, high-T upregulated genes were enriched for the androgen response pathway and the enrichment was not significantly affected by Rb depletion (see Figure S10A).
Figure 6.

Rb depletion impairs high-T-induced transcriptional repression on E2F signaling
(A) RNA-seq analyses were performed using tumor samples (treated with 160 mg/kg testosterone) at the end of the experiments. GSEA was done to compare the androgen-repressed genes in Rb-proficient tumors versus Rb-deficient tumors. (B) Box plots for the change of expression (Log2(fold-change)) for E2F target genes or Myc target genes in these samples are shown. (C) Heatmap view for E2Fs (upper panel) and a panel of E2F target genes (lower panel) is shown. (D) Heatmap view for MYC and its coregulator genes (upper panel) and a panel of Myc target genes (lower panel) is shown. (E) Heatmap view for a panel of noncanonical AR-repressed genes is shown.
Next, we compared the androgen-repressed expression of E2F signaling targets versus Myc signaling targets in these tumor samples. As shown in Figures 6B–6D and S10C–S10E, the E2F signaling was strongly repressed by the high-T treatment, but this repression effect was significantly compromised by Rb depletion. On the contrary, the repression on Myc signaling was much weaker than E2F signaling, and it was clearly less affected by Rb depletion, suggesting that sustained repression on Myc signaling may contribute to the effectiveness of high-T treatment (with increased dose) on Rb-deficient tumors. We next examined whether Rb-depletion-induced reprogramming of AR could contribute to the tumor-suppressive effect of high-T. Indeed, many noncanonical AR repression targets, including the identified cell polarity genes, were repressed in Rb-deficient tumors, but not in Rb-proficient tumors (Figure 6E). Together, these results suggest that RB1-loss CRPC is less sensitive to the high-T treatment but may still respond to higher doses of testosterone treatments (super high-T), possibly through mediating multiple Rb-independent mechanisms.
CDK4/6 inhibition enhances the efficacy of high-T treatment in CRPC models
CDK4/6 inhibitor treatment has been approved in treating ER-positive and HER2-negative breast cancer52 but appears to be less effective in the clinical trials of prostate cancer.53 One major downstream effect of CDK4/6 inhibition is to block the hyperphosphorylation of Rb, which disrupts the Rb-E2F repressor complex. In addition, Rb-like proteins were also previously demonstrated as the direct targets of CDK4/6. Therefore, we first examined whether palbociclib, a US Food and Drug Administration (FDA)-approved CDK4/6 inhibitor,54,55 can prevent the hyperphosphorylation of Rb and p130 in CRPC cells. In C4-2-tet-shRB cells, palbociclib treatment markedly repressed the phosphorylation of Rb and p130 in the absence or presence of androgen treatments (Figure 7A), indicating that CDK4/6 inhibition can effectively target Rb/p130-E2F complex. We next determined whether CDK4/6 inhibition can be used to improve high-T treatment in CRPC cell lines. As seen in Figures 7B and 7C, palbociclib treatment clearly enhanced the transcriptional repression effect of DHT on DNA synthesis genes and the suppression effect on cell cycle progression.
Figure 7.

CDK4/6 inhibition enhances the growth-suppressive activity of high-T in CRPC
(A) Immunoblotting for total Rb, p-Rb (S780 phosphorylated), total p130, p-p130 (S672 phosphorylated), and E2F1 in C4-2-tet-shRB cells treated with vehicle, doxycycline (3 d), palbociclib (1 μM), or DHT (10 nM) for 24 h. (B and C) Quantitative real-time PCR for the expression of E2F-regulated genes (B) or flow cytometry cell cycle analysis (C) in C4-2 cells treated with DHT (10 nM, 24 h), palbociclib (1 μM, 24 h), or the combination is shown. (D) mRNA expression (from RNA-seq) for AR, RB1, and RBL2 in tumor samples from LuCaP PDXs is shown. (E) Signature scores of Rb-target genes (directly repressed) in tumor samples from LuCaP PDXs are shown. (F–I) Castrated SCID male mice bearing LuCaP35CR (F), 77CR (G), 96CR (H), and 70CR (I) xenograft tumors were daily treated with vehicle, testosterone (40 mg/kg), palbociclib (150 mg/kg), or the combination via intraperitoneal injection (for high-T) or oral gavage (for palbociclib; n = 6). Tumor volume was measured by caliper. (J) Signature scores of Rb-target genes (directly repressed) in SU2C mCRPC dataset are shown. Data in bar graphs represent the mean ± SD. Data in growth curves represent the mean ± SE. n.s., not significant. ∗,p < 0.05.
Since Rb depletion altered AR chromatin binding (see Figure 4), we next determined whether Rb enhancement by CDK4/6 inhibition can also redistribute AR binding in C4-2 cells. Using ChIP-seq analysis of AR in C4-2 cells treated with DHT and palbociclib, we identified 9,974 high-confidence AR-binding peaks. This distinct AR-binding program had very limited overlapping sites with AR bindings in Rb-proficient C4-2 cells (1,707 sites) or Rb-depleted cells (1,528 sites; Figure S11A). We then performed a motif enrichment analysis for the lost, conserved, and gained AR sites in comparison with the regular AR-binding program (Figure S11B). While the lost and the conserved AR-binding sites contained classic AREs, the gained AR-binding sites have no enrichment of known AR-binding sequences. Instead, AR appeared to bind to sites that contain E2F or altered E2F-binding nucleotide sequences (Figure S11C). Moreover, examining a panel of gene loci with gained AR binding, we also found that the gained AR-binding sites appeared to be also co-occupied by Rb (Figure S11D). These data strongly suggest that CDK4/6 inhibition may redistribute AR binding to the sites that are occupied by Rb-E2F repressor complex.
To test the efficacy of the combination treatment in vivo, we passaged a series of LuCaP CRPC PDX models in castrated SCID mice, including 35CR, 70CR, 77CR, and 96CR. While all four models have AR gene amplification, 35CR is RB1+/+, 70CR and 77CR are RB1+/−, and 96CR is RB1−/−.56 Using the reported RNA-seq data generated from these models,8 we showed that the expression levels of RB1 and RBL2 were higher in 35CR and lower in LuCaP96CR (Figure 7D). These results were also validated by ChIP-qPCR using tumor RNA samples (Figure S12A). Since RB1 copy-number change and mRNA expression may not fully reflect the deficiency of Rb, we next assessed the Rb activity in these models using a recently developed gene signature of Rb directly repressed targets.57 Surprisingly, the score of Rb targets in 70CR tumors appeared to be much higher than other models (Figure 7E), suggesting that Rb may be inactivated in this model due to other mechanisms. Interestingly, we also consistently detected Rb mRNA expression in the 96CR model (comparable to the expression level in C4-2) despite that it was previously reported as an RB1-loss model.56 Indeed, the phosphorylated Rb proteins were detected in 96CR tumor samples (Figure S12B), suggesting that 96CR tumors may be heterogeneous and still contain a significant portion of Rb-positive cells. All four models expressed comparable or higher levels of AR in comparison with C4-2 CRPC xenograft tumors (Figures S12C and S12D).
We next treated all these PDX models with the combination treatment. As shown in Figures 7F–7H, the high-T treatment alone significantly repressed the tumor growth in Rb-proficient models, including 35CR, 77CR, and 96CR, consistent with previous findings.8 While palbociclib treatment alone repressed tumor growth in 35CR and 77CR, but not 96CR, the enhanced tumor-suppression effect in combination with high-T was observed in all these three responders. On the contrary, the possible Rb-deficient 70CR tumors showed no response to high-T or the combination treatment (Figure 7I). These data suggest that the repressor activity of Rb may be a critical molecular determinant for the efficacy of high-T and the combination treatment in CRPC patients. Applying the Rb-target signature in the public mCRPC patient dataset (SU2C mCRPC),23 we can then separate tumors with lower Rb activity (higher scores) versus higher Rb activity (lower scores) and predict that the tumors with high Rb activity may respond better to the high-T and the combination treatments (Figure 7J).
We next conducted an RNA-seq analysis using tumor samples from the LuCaP35CR study. The inhibition of Rb phosphorylation by palbociclib was confirmed by immunohistochemistry staining (Figure S13A). While 1,220 genes or 1,403 genes were downregulated by high-T or palbociclib treatment, respectively, significantly more genes (2,750) were repressed by the combination treatment (Figure 8A). The combination treatment also showed a much stronger repression effect on these genes than any single-agent treatment (Figure 8B). Using gene set enrichment analysis (GSEA), we found that the downregulated genes by the single or combination treatment were enriched for E2F signaling, Myc signaling, cell cycle regulation, and DNA damage repair pathway, while the upregulated genes were enriched for androgen response and apoptosis (Figures 8C and S13B–S13D). We next examined the effects of those treatments on E2F and Myc signaling genes. As shown in Figure 8D, both pathways were repressed by either high-T or palbociclib treatment, and the repression effect was further enhanced by the combination treatment. However, the repression effect on E2F pathway genes appeared to be stronger than the effect on Myc pathway genes, consistent with the in vitro study that showed higher repression activity of AR on E2F target genes. This enhancing effect was also seen from a panel of E2F or Myc target genes (Figures 8E and 8F). On the contrary, the AR gene was repressed only by high-T, but not palbociclib, and no additive effect was observed (Figure S13E). Furthermore, we also examined how redistributed AR binding induced by CDK4/6 inhibition may affect gene expression regulated by the combination treatment. By comparing the RNA-seq data from combination treatment versus high-T alone and using AR ChIP-seq in C4-2 model treated with DHT and palbociclib, we found 1,159 upregulated genes and 832 downregulated that were associated with nearby AR-binding sites (Figure S14A). Importantly, while the upregulated genes were only enriched for tumor necrosis factor alpha (TNFα) pathway, the downregulated genes were highly enriched for E2F targets, NE_up genes, and cell cycle genes, but not Myc targets (Figure S14B), suggesting that the CDK4/6-inhibition-induced alteration of AR binding may contribute to the overall combinatory effect of palbociclib and testosterone. Together, these data highly suggest that the high-T treatment can be combined with CDK4/6 inhibitors to treat patients with AR-positive and Rb/p130-proficient CRPC.
Figure 8.

Gene profiling in LuCaP35CR tumors receiving the combination treatment
(A) RNA-seq analyses were performed using tumor samples from the LuCaP35CR study (at the end of the experiment). The Venn diagram for high-T-repressed genes, palbociclib-repressed genes, and the combination treatment-repressed genes is shown. (B) Heatmap view for the combination treatment-repressed genes in all tumor samples is shown. (C) GSEA for the downregulated genes by single treatments versus the combination treatment is shown. (D) Box plots for the change of expression of E2F target genes or Myc target genes in these samples are shown. (E and F) Heatmap view for E2Fs and E2F target genes (E) or MYC and its coregulator genes and a panel of Myc target genes (F) is shown.
Discussion
Rb is a transcriptional repression partner of E2Fs, and Rb-E2F repressor complex is critical for cells to repress DNA synthesis and other events prior to cell cycle transition from G1 to S phase.20 Hyperphosphorylation of Rb by cyclin D-CDK4/6 and cyclin E-CDK2 disrupts the Rb-E2F complex and leads to the transcriptional activation of E2Fs.58 A previous study has identified an androgen-induced AR-Rb interaction at chromatin that promotes the activity of Rb-E2F repressor complex. Therefore, in this study, we attempted to determine whether this activity of AR is a primary mechanism for the anti-tumor activity of high-dose androgens. We performed a series of global studies and demonstrated that E2F signaling is the major direct downstream target of the transcriptional repression function of AR induced by high-dose androgen treatment, which is mediated through increased global chromatin binding of the Rb-E2F complex (see Figure 1). Furthermore, we determined that Rb activity is important for the CRPC sensitivity to high-dose androgen treatments in vitro and in vivo. Mechanistically, we also demonstrated that an Rb-like pocket protein, p130, was similarly involved in this activity by interacting with E2Fs particularly when Rb expression is low. While we did not examine the requirement of p130 for the high-T treatment in vivo, we predict that depleting p130 expression would also desensitize CRPC tumor to high-T. Future studies are clearly needed to demonstrate the in vivo role of p130 in the presence or absence of Rb using CRPC xenograft models. Importantly, our finding is also consistent with a recent study using a series of castration-resistant LuCaP PDX models, which demonstrated that the most robust molecular phenotype for high-T treatment is the suppression of E2F transcriptional output.8
RB1 loss is common in CRPC, and this event causes profound genomic consequences. In this study, we show that RB1 loss may also reshape AR cistrome by broadly disrupting AR binding from canonical AREs and redistributing AR chromatin binding to low-affinity noncanonical sites lacking classic FOXA1- and AR-binding motifs (see Figure 4). These new AR-binding sites appeared to be enriched for the binding motif of ZBTB family proteins, and our recent study indicates that ZBTB7A can function as a corepressor of AR to directly mediate its transcriptional repression activity.6 Therefore, the reprogrammed sites may favor the interaction of AR and ZBTB7A to repress a distinct subset of genes. Indeed, this redistribution of AR appeared to have a stronger impact on reprogramming AR transcriptional repression activity than activation activity, and we have identified a large set of canonical AR-repressed genes with reduced AR-repression activity and a small set of genes that may gain AR-mediated repression in Rb-depleted cells. Interestingly, these noncanonical targets enriched for pathways of maintaining cell polarity and deregulation of cell polarity proteins have been known to be crucial for cancer progression, including lineage plasticity.59,60 One of the new AR-repressed genes is a member of fibroblast growth factor (FGF) homologous factors, FGF13, which has been shown to associate with poor patient outcomes in PCa51 and promotes the metastasis of triple-negative breast cancer.61 Interestingly, FGF signaling has been also shown to drive PCa progression in double-negative (AR-negative and NE-null) CRPC.62 Therefore, possibly suppressing FGF signaling by high-T may represent a novel tumor-suppressor activity of AR to prevent lineage plasticity and cancer progression in RB1-loss CRPC. Nonetheless, the biological significance of this noncanonical AR repression activity in Rb-deficient cells remains to be determined in future studies. Interestingly, enhancing Rb-E2F complex with CDK4/6 inhibition can also significantly redistribute AR-binding sites. These new sites appeared to be enriched for E2F binding sequences, which may further strengthen AR interaction with Rb-E2F complex. Future analyses are required to comprehensively characterize this AR reprogramming induced by CDK4/6 inhibitor treatment.
Another target pathway consistently repressed by high-T is Myc signaling, which also regulates DNA synthesis and cell cycle progression.63,64 However, the androgen-induced repression effect on Myc signaling appeared to be indirect and much weaker than the effect on E2F signaling. Unlike AR-induced Rb binding, c-Myc chromatin binding was not strongly altered by high-T and was not associated with AR-repressed genes (see Figure 1). Therefore, the indirect suppression effect of high-T on Myc targets is possibly mediated by the transcriptional repression on MYC gene18 and activation on MXD1 gene (encodes Mad protein, a negative regulator of Myc; see Figure 2F), which promotes Myc/Mad repressor complex.65 Importantly, our data revealed persistent repression of Myc signaling by the high-T treatment (super high dose) in Rb-depleted CRPC tumors, suggesting that the repression on Myc signaling may play a role in mediating the tumor-suppressive function of high-T in RB1-loss CRPC. Therefore, combining high-T with therapies targeting Myc signaling may be a potential combination strategy for treating RB1-loss CRPC.
While the high-T treatment in the setting of BAT appeared to be effective in the recent two phase II clinical trials,10,11 CDK4/6 inhibitor treatment was not promising in treating hormone-sensitive metastatic PCa when it was combined with AR-signaling-inhibition treatment.53 Therefore, our study provides an important therapeutic strategy to combine CDK4/6 inhibition with high-T treatment in treating Rb-proficient CRPC, and our data from the preclinical studies using PDX models support this strategy (see Figure 7). However, most of the PDX models that we tested appeared to be sensitive to the high-T treatment, and therefore, only a modest enhancing effect was observed with the combination treatment. Future experiments should be done using lower doses of high-T in order to observe a stronger synergetic effect. Interestingly, CDK4/6 inhibitor and high-T cotarget many pathways in addition to E2F signaling and cell cycle regulations (see Figure 8). These pathways include Myc signaling, DNA repair, and oxidative phosphorylation. The suppression effects on Myc signaling and Myc-regulated homologous recombination pathways by CDK4/6 inhibition have been previously reported in breast and ovarian cancers.66,67 Therefore, the overall synergetic effect of the combination therapy is likely contributed by the suppression of multiple cancer-promoting pathways. However, biomarkers that can predict the tumor response to high-T or the combination treatment have not been identified. In this study, we suggest that Rb repressor activity, determined by a recently developed Rb-target gene signature,57 could be used as a prognosis marker to predict the treatment response (see Figures 7E and 7J). Future clinical studies are needed to further validate the use of such biomarkers in the clinical trials of high-T treatment.
In summary, we have determined a central role of Rb/p130-E2F complex in mediating the tumor-suppressive effect of high-T in treating CRPC and demonstrated that the efficacy of high-T treatment depends on Rb activity and that the effect of high-T can be enhanced by CDK4/6 inhibitor treatment in preclinical CRPC models. This study provides a strong rationale for the further development of such combination treatments that can exploit the tumor-suppressor activity of AR in CRPC in clinical trials.
Materials and methods
Cell culture and establishment of stable cell lines are as follows: C4-2 cell lines were obtained from ATCC and were examined for mycoplasma contamination using MycoAlert Mycoplasma Detection Kit (Lonza). Tetracycline-inducible shRNA constructs (pTRIPZ) against non-target control or RB1 (tet-shRB) were obtained from Dharmacon. Lentifect Purified lentiviral particles of shRNA against RBL2 were obtained from GeneCopoeia. To establish the stable cell line, C4-2 cells were stably infected with lentivirus-expressing tet-shRB and further selected by puromycin. For establishing RB-KO cell lines, parental C4-2 cells were infected with lentiviral TLCV2-RB1, which expresses doxycycline-inducible Cas9 and single-guide RNA (sgRNA) against RB, followed by further screening and selection for control and knockout clones (sg-RB1: GCTCTGGGTCCTCCTCAGGA). TLCV2-RB1 plasmid was acquired from Addgene (no. 87836) as a gift from Dr. Adam Karpf. All C4-2 and its derived cell lines were maintained in RPMI 1640 medium with reduced steroid hormone (8% charcoal-stripped fetal bovine serum [FBS] [CSS] plus 2% regular FBS). All stable cell lines were maintained using tetracycline-free FBS.
Cell cycle and proliferation assays are as follows: for cell cycle analysis, cells were collected by trypsinization, fixed by 70% ice-cold ethanol for over 3 h, and then stained with Muse Cell Cycle Kit. The stained cells were counted by Guava Muse Cell Analyzer. For proliferation analysis, cells were collected by trypsinization and then examined for cell number and viability by using Muse Count & Viability Kit. The fluorescence signal of the stained cells was measured using Guava Muse Cell Analyzer.
Immunoprecipitation and immunoblotting are as follows: for immunoprecipitation (IP), cells were lysed in Triton lysis buffer supplemented with protein inhibitor cocktails (Thermo Fisher Scientific), followed by brief sonication. Cell lysates were then immunoprecipitated with anti-E2F1 (Abcam) or anti-p130 (Cell Signaling Technology). Proteins were eluted by boiling in Laemmli buffer with 5% beta-mercaptoethanol. For immunoblotting, cells were washed with PBS and then lysed in 2% SDS with boiling. The primary antibodies that were used are anti-Rb (Cell Signaling Technology), anti-p-Rb (Ser780) (Cell Signaling Technology), anti-p130 (Cell Signaling Technology), anti-p-p130 (Ser672) (Abcam), anti-E2F1 (Cell Signaling Technology), and anti-GAPDH (Abcam).
ChIP and ChIP-seq analysis
For conducting ChIP experiments with DHT treatments, cells were grown in hormone-depleted medium (5% CSS) for 3 days and then treated with DHT for 4 h. For the preparation of ChIP, cells were fixed with 1% formaldehyde and lysed by the ChIP lysis buffer (1% SDS, 5 mM EDTA, and 50 mM Tris-HCl pH 8.1). Chromatin was then sheared to ∼300-bp fragments using Bioruptor Sonicator (Diagenode). Immunoprecipitation was carried out using anti-Rb antibody (Cell Signaling Technology and BD Pharmingen), anti-Myc (Cell Signaling Technology), anti-AR antibody (Abcam), anti-FOXA1 (EMD Millipore), and anti-p130 antibody (Cell Signaling Technology). Precipitated protein-DNA complexes were then reverse-crosslinked at 65°C, followed by DNA purification. ChIP-seq libraries were constructed using the SMARTer ThruPLEX DNA-Seq Prep Kit (Takara Bio USA). Next-generation sequencing (51 nt, single-end) was performed using Illumina HiSeq2500. ChIP-seq reads were mapped to the hg19 human genome using bwa (v.0.7.9a) with aln and samse sub-commands. Samtools (v.1.2) was used to convert sam files to bam format. The significance of enriched ChIP regions was evaluated by using MACS2 (v.2.1.0).68 The R package IRanges (v.2.18.3) was used to analyze peak intervals and determine the overlapped regions. Venn diagrams were generated using VennDiagram (v.1.6.20) R package.69 The signals associated with genomic regions were visualized by using compueMatrix and plotHeatmap tools from deepTools (v.3.3.0).70 computeMatrix with reference-point mode was used to calculate scores for each genomic region, and plotHeatmap was used to create a heatmap for scores associated with genomic regions. Motif enrichment analysis was performed by using SeqPos with the default setting in Galaxy/Cistrome.38 Binding and expression target analysis (BETA) was performed by BETA software package (v.1.0.7)39 with default parameters to integrate ChIP-seq and with differential gene expression to predict direct targets. Peak interval files from MACS2 and differential expression results from limma were used as inputs.
Quantitative real-time PCR and RNA-seq
RNA from the cell lines was extracted with TRIzol reagent (Invitrogen). RNA from tumor tissue samples was extracted by using TissueLyser LT (QIAGEN) and RNeasy Kit (QIAGEN). Quantitative real-time PCR was performed using Fast 1-step Mix (Thermo Fisher Scientific) at QuantStudio 3 PCR machine. All quantitative real-time PCR results were normalized with GAPDH. All Taqman primers and probes were predesigned by and obtained from Thermo Fisher Scientific.
For RNA-seq, the library preparation was done using TruSeq Stranded mRNA (Illumina). Next-generation sequencing (51 nt, single-end) was performed using Illumina HiSeq2500. Transcriptome sequencing reads were aligned to the human reference genome (hg19) using STAR (v.2.7.2) followed by counting with featureCounts (v.1.6.4) from GRCh37 Ensembl reference. All gene counts were processed with R package limma (v.3.40.6)71 to evaluate the differential expression using the Benjamini-Hochberg false discovery rate (FDR)-adjusted p value. The expression values were centered and scaled across samples and then displayed using the ComplexHeatmap (v.2.0.0) R package. GSEA was done by using R package fgsea (v.1.10.1). The assession number for ChIP-seq and RNA-seq data generated in this study is GEO: GSE179688.
Xenograft study
All animal experiments were approved by the University of Massachusetts, Boston Institutional Animal Care and Use Committee and were conducted following institutional and national (USA) guidelines. C4-2-tet-shRB cells were resuspended in serum-free RPMI 1640 medium and mixed in 1:1 ratio with Matrigel (BD Biosciences) prior to subcutaneous implantation (2 × 106 cells per injection) on flanks of castrated SCID mice (∼4 to 6 weeks old; Taconic). Xenograft tumors, including PDXs, were further passaged in castrated male SCID mice. To silence Rb, mice bearing tumors derived from C4-2-tet-shRB were fed with doxycycline-supplemented food and drinking water. Tumor length (L) and width (W) were measured by caliper at the indicated time, and tumor volumes were calculated (L × W2/2).
Statistical analysis
Data in bar graphs represent the mean ± SD of at least three biological repeats. Data in the tumor growth curves represent the mean ± SE of at least 4 independent tumor samples. For most studies, statistical analyses were performed using Student's t test by comparing treatment versus vehicle or otherwise as indicated. p < 0.05 (∗) was considered to be statistically significant. The results for immunoblotting are representative of at least three experiments. Boxplots of signature scores and gene expression were compared using the Wilcoxon test for comparison between two groups of samples. The difference in tumor growth was determined using two-way ANOVA. All other statistical analyses and visualization were performed with R (v.3.6.0) unless otherwise specified.
Acknowledgments
This work is supported by grants from the NIH (R00 CA166507 and R01 CA211350 to C.C., U54 CA156734 to J.A.M., and P01 CA163227 and P50 CA090381 to S.P.B.), DOD (W81XWH-16-1-0445, W81XWH-19-1-0361, and W81XWH-21-1-0267 to C.C. and W81XWH-19-1-0777 to S.G.), CIHR (142246, 152863, 152864, and 159567 to H.H.H.), and Terry Fox Frontiers Program Project Grants (1090 P3 to H.H.H.). The establishment, characterization, and maintenance of the LuCaP PDXs are funded by the NIH (P50 CA097186 and P01 CA163227). M. Liu was supported by the graduate fellowship from Integrative Biosciences Program at University of Massachusetts Boston. W.H., Z.W., and A.B. were supported by CSM (College of Science and Mathematics) Dean's Doctoral Research Fellowship from University of Massachusetts Boston. C.C. is supported by Proposal Development Grant Program from University of Massachusetts Boston. H.H.H. holds the Joey and Toby Tanenbaum Brazilian Ball Chair in Prostate Cancer. H.-M.L. and E.C. are supported by the Institute of Prostate Cancer Research (IPCR).
Author contributions
C.C., S.G., S.P.B., E.C., and W.H. designed the study. W.H., M. Liu, D.H., M. Li, A.A.T., Z.W., A.B., S.G., and H.-M.L. performed experiments and analyzed the results. W.H., D.H., M. Liu, S.P., and J.A.M. performed deep sequencing analyses. C.C. and W.H. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Declaration of interests
No potential conflicts of interest were disclosed.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.01.039.
Supplemental information
Note: DHT-downregulated genes (fold-change < 1.5) are highlighted in red.
References
- 1.Yuan X., Cai C., Chen S., Chen S., Yu Z., Balk S.P. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene. 2014;33:2815–2825. doi: 10.1038/onc.2013.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cai C., He H.H., Chen S., Coleman I., Wang H., Fang Z., Nelson P.S., Liu X.S., Brown M., Balk S.P. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell. 2011;20:457–471. doi: 10.1016/j.ccr.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao J.C., Yu J., Runkle C., Wu L., Hu M., Wu D., Liu J.S., Wang Q., Qin Z.S., Yu J. Cooperation between Polycomb and androgen receptor during oncogenic transformation. Genome Res. 2012;22:322–331. doi: 10.1101/gr.131508.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu Z., Chen S., Sowalsky A.G., Voznesensky O.S., Mostaghel E.A., Nelson P.S., Cai C., Balk S.P. Rapid induction of androgen receptor splice variants by androgen deprivation in prostate cancer. Clin. Cancer Res. 2014;20:1590–1600. doi: 10.1158/1078-0432.CCR-13-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao S., Gao Y., He H.H., Han D., Han W., Avery A., Macoska J.A., Liu X., Chen S., Ma F., et al. Androgen receptor tumor suppressor function is mediated by recruitment of retinoblastoma protein. Cell Rep. 2016;17:966–976. doi: 10.1016/j.celrep.2016.09.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han D., Chen S., Han W., Gao S., Owiredu J.N., Li M., Balk S.P., He H.H., Cai C. ZBTB7A mediates the transcriptional repression activity of the androgen receptor in prostate cancer. Cancer Res. 2019;79:5260–5271. doi: 10.1158/0008-5472.CAN-19-0815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chatterjee P., Schweizer M.T., Lucas J.M., Coleman I., Nyquist M.D., Frank S.B., Tharakan R., Mostaghel E., Luo J., Pritchard C.C., et al. Supraphysiological androgens suppress prostate cancer growth through androgen receptor-mediated DNA damage. J. Clin. Invest. 2019;129:4245–4260. doi: 10.1172/JCI127613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lam H.M., Nguyen H.M., Labrecque M.P., Brown L.G., Coleman I.M., Gulati R., Lakely B., Sondheim D., Chatterjee P., Marck B.T., et al. Durable response of enzalutamide-resistant prostate cancer to supraphysiological testosterone is associated with a multifaceted growth suppression and impaired DNA damage response transcriptomic program in patient-derived xenografts. Eur. Urol. 2019 doi: 10.1016/j.eururo.2019.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schweizer M.T., Antonarakis E.S., Wang H., Ajiboye A.S., Spitz A., Cao H., Luo J., Haffner M.C., Yegnasubramanian S., Carducci M.A., et al. Effect of bipolar androgen therapy for asymptomatic men with castration-resistant prostate cancer: results from a pilot clinical study. Sci. translational Med. 2015;7:269ra2. doi: 10.1126/scitranslmed.3010563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Teply B.A., Wang H., Luber B., Sullivan R., Rifkind I., Bruns A., Spitz A., DeCarli M., Sinibaldi V., Pratz C.F., et al. Bipolar androgen therapy in men with metastatic castration-resistant prostate cancer after progression on enzalutamide: an open-label, phase 2, multicohort study. Lancet Oncol. 2018;19:76–86. doi: 10.1016/S1470-2045(17)30906-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denmeade S.R., Wang H., Agarwal N., Smith D.C., Schweizer M.T., Stein M.N., Assikis V., Twardowski P.W., Flaig T.W., Szmulewitz R.Z., et al. TRANSFORMER: a randomized phase II study comparing bipolar androgen therapy versus enzalutamide in asymptomatic men with castration-resistant metastatic prostate cancer. J. Clin. Oncol. 2021;39:1371–1382. doi: 10.1200/JCO.20.02759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang H., Zegarra-Moro O.L., Benson D., Tindall D.J. Androgens repress Bcl-2 expression via activation of the retinoblastoma (RB) protein in prostate cancer cells. Oncogene. 2004;23:2161–2176. doi: 10.1038/sj.onc.1207326. [DOI] [PubMed] [Google Scholar]
- 13.D'Antonio J.M., Vander Griend D.J., Isaacs J.T. DNA licensing as a novel androgen receptor mediated therapeutic target for prostate cancer. Endocr. Relat. Cancer. 2009;16:325–332. doi: 10.1677/ERC-08-0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bohrer L.R., Chen S., Hallstrom T.C., Huang H. Androgens suppress EZH2 expression via retinoblastoma (RB) and p130-dependent pathways: a potential mechanism of androgen-refractory progression of prostate cancer. Endocrinology. 2010;151:5136–5145. doi: 10.1210/en.2010-0436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chuu C.P., Kokontis J.M., Hiipakka R.A., Fukuchi J., Lin H.P., Lin C.Y., Huo C., Su L.C., Liao S. Androgen suppresses proliferation of castration-resistant LNCaP 104-R2 prostate cancer cells through androgen receptor, Skp2, and c-Myc. Cancer Sci. 2011;102:2022–2028. doi: 10.1111/j.1349-7006.2011.02043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kregel S., Kiriluk K.J., Rosen A.M., Cai Y., Reyes E.E., Otto K.B., Tom W., Paner G.P., Szmulewitz R.Z., Vander Griend D.J. Sox2 is an androgen receptor-repressed gene that promotes castration-resistant prostate cancer. PLoS One. 2013;8:e53701. doi: 10.1371/journal.pone.0053701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsieh C.L., Botta G., Gao S., Li T., Van Allen E.M., Treacy D.J., Cai C., He H.H., Sweeney C.J., Brown M., et al. PLZF, a tumor suppressor genetically lost in metastatic castration-resistant prostate cancer, is a mediator of resistance to androgen deprivation therapy. Cancer Res. 2015;75:1944–1948. doi: 10.1158/0008-5472.CAN-14-3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo H., Wu Y., Nouri M., Spisak S., Russo J.W., Sowalsky A.G., Pomerantz M.M., Wei Z., Korthauer K., Seo J.H., et al. Androgen receptor and MYC equilibration centralizes on developmental super-enhancer. Nat. Commun. 2021;12:7308. doi: 10.1038/s41467-021-27077-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiang Z., Sun Y., You B., Zhang M., Huang C., Yu J., You X., Wu D., Chang C. Suppressing BCL-XL increased the high dose androgens therapeutic effect to better induce the Enzalutamide-resistant prostate cancer autophagic cell death. Cell Death Dis. 2021;12:68. doi: 10.1038/s41419-020-03321-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dick F.A., Rubin S.M. Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cel. Biol. 2013;14:297–306. doi: 10.1038/nrm3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giacinti C., Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220–5227. doi: 10.1038/sj.onc.1209615. [DOI] [PubMed] [Google Scholar]
- 22.Robinson D., Van Allen E.M., Wu Y.M., Schultz N., Lonigro R.J., Mosquera J.M., Montgomery B., Taplin M.E., Pritchard C.C., Attard G., et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abida W., Cyrta J., Heller G., Prandi D., Armenia J., Coleman I., Cieslik M., Benelli M., Robinson D., Van Allen E.M., et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. U S A. 2019;116:11428–11436. doi: 10.1073/pnas.1902651116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ku S.Y., Rosario S., Wang Y., Mu P., Seshadri M., Goodrich Z.W., Goodrich M.M., Labbe D.P., Gomez E.C., Wang J., et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science. 2017;355:78–83. doi: 10.1126/science.aah4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mu P., Zhang Z., Benelli M., Karthaus W.R., Hoover E., Chen C.C., Wongvipat J., Ku S.Y., Gao D., Cao Z., et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science. 2017;355:84–88. doi: 10.1126/science.aah4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McNair C., Xu K., Mandigo A.C., Benelli M., Leiby B., Rodrigues D., Lindberg J., Gronberg H., Crespo M., De Laere B., et al. Differential impact of RB status on E2F1 reprogramming in human cancer. J. Clin. Invest. 2018;128:341–358. doi: 10.1172/JCI93566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nyquist M.D., Corella A., Coleman I., De Sarkar N., Kaipainen A., Ha G., Gulati R., Ang L., Chatterjee P., Lucas J., et al. Combined TP53 and RB1 loss promotes prostate cancer resistance to a spectrum of therapeutics and confers vulnerability to replication stress. Cell Rep. 2020;31:107669. doi: 10.1016/j.celrep.2020.107669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mandigo A.C., Yuan W., Xu K., Gallagher P., Pang A., Guan Y.F., Shafi A.A., Thangavel C., Sheehan B., Bogdan D., et al. RB/E2F1 as a master regulator of cancer cell metabolism in advanced disease. Cancer Discov. 2021 doi: 10.1158/2159-8290.CD-20-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pfitzenmaier J., Quinn J.E., Odman A.M., Zhang J., Keller E.T., Vessella R.L., Corey E. Characterization of C4-2 prostate cancer bone metastases and their response to castration. J. Bone Miner Res. 2003;18:1882–1888. doi: 10.1359/jbmr.2003.18.10.1882. [DOI] [PubMed] [Google Scholar]
- 30.Korenchuk S., Lehr J.E., Clean L.M., Lee Y.G., Whitney S., Vessella R., Lin D.L., Pienta K.J. VCaP, a cell-based model system of human prostate cancer. In Vivo. 2001;15:163–168. [PubMed] [Google Scholar]
- 31.Cai C., Wang H., Xu Y., Chen S., Balk S.P. Reactivation of androgen receptor-regulated TMPRSS2:ERG gene expression in castration-resistant prostate cancer. Cancer Res. 2009;69:6027–6032. doi: 10.1158/0008-5472.CAN-09-0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cai C., Chen S., Ng P., Bubley G.J., Nelson P.S., Mostaghel E.A., Marck B., Matsumoto A.M., Simon N.I., Wang H., et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011;71:6503–6513. doi: 10.1158/0008-5472.CAN-11-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han W., Gao S., Barrett D., Ahmed M., Han D., Macoska J.A., He H.H., Cai C. Reactivation of androgen receptor-regulated lipid biosynthesis drives the progression of castration-resistant prostate cancer. Oncogene. 2018;37:710–721. doi: 10.1038/onc.2017.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beltran H., Rickman D.S., Park K., Chae S.S., Sboner A., MacDonald T.Y., Wang Y., Sheikh K.L., Terry S., Tagawa S.T., et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011;1:487–495. doi: 10.1158/2159-8290.CD-11-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Svensson C., Ceder J., Iglesias-Gato D., Chuan Y.C., Pang S.T., Bjartell A., Martinez R.M., Bott L., Helczynski L., Ulmert D., et al. REST mediates androgen receptor actions on gene repression and predicts early recurrence of prostate cancer. Nucleic Acids Res. 2014;42:999–1015. doi: 10.1093/nar/gkt921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Srinivasan D., Senbanjo L., Majumdar S., Franklin R.B., Chellaiah M.A. Androgen receptor expression reduces stemness characteristics of prostate cancer cells (PC3) by repression of CD44 and SOX2. J. Cell Biochem. 2018 doi: 10.1002/jcb.27573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim D.H., Sun D., Storck W.K., Welker Leng K., Jenkins C., Coleman D.J., Sampson D., Guan X., Kumaraswamy A., Rodansky E.S., et al. BET bromodomain inhibition blocks an AR-repressed, E2F1-activated treatment-emergent neuroendocrine prostate cancer lineage plasticity program. Clin. Cancer Res. 2021 doi: 10.1158/1078-0432.CCR-20-4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu T., Ortiz J.A., Taing L., Meyer C.A., Lee B., Zhang Y., Shin H., Wong S.S., Ma J., Lei Y., et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 2011;12:R83. doi: 10.1186/gb-2011-12-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang S., Sun H., Ma J., Zang C., Wang C., Wang J., Tang Q., Meyer C.A., Zhang Y., Liu X.S. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat. Protoc. 2013;8:2502–2515. doi: 10.1038/nprot.2013.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Araki K., Nakajima Y., Eto K., Ikeda M.A. Distinct recruitment of E2F family members to specific E2F-binding sites mediates activation and repression of the E2F1 promoter. Oncogene. 2003;22:7632–7641. doi: 10.1038/sj.onc.1206840. [DOI] [PubMed] [Google Scholar]
- 41.Yoshida K., Inoue I. Regulation of Geminin and Cdt1 expression by E2F transcription factors. Oncogene. 2004;23:3802–3812. doi: 10.1038/sj.onc.1207488. [DOI] [PubMed] [Google Scholar]
- 42.Prensner J.R., Chen W., Han S., Iyer M.K., Cao Q., Kothari V., Evans J.R., Knudsen K.E., Paulsen M.T., Ljungman M., et al. The long non-coding RNA PCAT-1 promotes prostate cancer cell proliferation through cMyc. Neoplasia. 2014;16:900–908. doi: 10.1016/j.neo.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bracken A.P., Pasini D., Capra M., Prosperini E., Colli E., Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323–5335. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sadasivam S., DeCaprio J.A. The DREAM complex: master coordinator of cell cycle-dependent gene expression. Nat. Rev. Cancer. 2013;13:585–595. doi: 10.1038/nrc3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang J., Pan Y., Regan K.M., Wu C., Zhang X., Tindall D.J., Huang H. Androgens repress expression of the F-box protein Skp2 via p107 dependent and independent mechanisms in LNCaP prostate cancer cells. Prostate. 2012;72:225–232. doi: 10.1002/pros.21430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E., et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E., et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao N., Zhang J., Rao M.A., Case T.C., Mirosevich J., Wang Y., Jin R., Gupta A., Rennie P.S., Matusik R.J. The role of hepatocyte nuclear factor-3 alpha (Forkhead Box A1) and androgen receptor in transcriptional regulation of prostatic genes. Mol. Endocrinol. 2003;17:1484–1507. doi: 10.1210/me.2003-0020. [DOI] [PubMed] [Google Scholar]
- 49.Lupien M., Eeckhoute J., Meyer C.A., Wang Q., Zhang Y., Li W., Carroll J.S., Liu X.S., Brown M. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008;132:958–970. doi: 10.1016/j.cell.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Swinstead E.E., Miranda T.B., Paakinaho V., Baek S., Goldstein I., Hawkins M., Karpova T.S., Ball D., Mazza D., Lavis L.D., et al. Steroid receptors reprogram FoxA1 occupancy through dynamic chromatin transitions. Cell. 2016;165:593–605. doi: 10.1016/j.cell.2016.02.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu L., Toriseva M., Tuomala M., Seikkula H., Elo T., Tuomela J., Kallajoki M., Mirtti T., Taimen P., Bostrom P.J., et al. Increased expression of fibroblast growth factor 13 in prostate cancer is associated with shortened time to biochemical recurrence after radical prostatectomy. Int. J. Cancer. 2016;139:140–152. doi: 10.1002/ijc.30048. [DOI] [PubMed] [Google Scholar]
- 52.Finn R.S., Crown J.P., Lang I., Boer K., Bondarenko I.M., Kulyk S.O., Ettl J., Patel R., Pinter T., Schmidt M., et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16:25–35. doi: 10.1016/S1470-2045(14)71159-3. [DOI] [PubMed] [Google Scholar]
- 53.Palmbos P.L., Daignault-Newton S., Tomlins S.A., Agarwal N., Twardowski P., Morgans A.K., Kelly W.K., Arora V.K., Antonarakis E.S., Siddiqui J., et al. A randomized phase II study of androgen deprivation therapy with or without palbociclib in RB-positive metastatic hormone-sensitive prostate cancer. Clin. Cancer Res. 2021 doi: 10.1158/1078-0432.CCR-21-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Finn R.S., Martin M., Rugo H.S., Jones S., Im S.A., Gelmon K., Harbeck N., Lipatov O.N., Walshe J.M., Moulder S., et al. Palbociclib and letrozole in advanced breast cancer. N. Engl. J. Med. 2016;375:1925–1936. doi: 10.1056/NEJMoa1607303. [DOI] [PubMed] [Google Scholar]
- 55.Turner N.C., Slamon D.J., Ro J., Bondarenko I., Im S.A., Masuda N., Colleoni M., DeMichele A., Loi S., Verma S., et al. Overall survival with palbociclib and fulvestrant in advanced breast cancer. N. Engl. J. Med. 2018;379:1926–1936. doi: 10.1056/NEJMoa1810527. [DOI] [PubMed] [Google Scholar]
- 56.Nguyen H.M., Vessella R.L., Morrissey C., Brown L.G., Coleman I.M., Higano C.S., Mostaghel E.A., Zhang X., True L.D., Lam H.M., et al. LuCaP prostate cancer patient-derived xenografts reflect the molecular heterogeneity of advanced disease an--d serve as models for evaluating cancer therapeutics. Prostate. 2017;77:654–671. doi: 10.1002/pros.23313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Han W., Liu M., Han D., Li M., Toure A.A., Wang Z., Besschetnova A., Patalano S., Macoska J.A., Gao S., et al. RB1 loss in castration-resistant prostate cancer confers vulnerability to LSD1 inhibition. Oncogene. 2022 doi: 10.1038/s41388-021-02135-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McCartney A., Migliaccio I., Bonechi M., Biagioni C., Romagnoli D., De Luca F., Galardi F., Risi E., De Santo I., Benelli M., et al. Mechanisms of resistance to CDK4/6 inhibitors: potential implications and biomarkers for clinical practice. Front Oncol. 2019;9:666. doi: 10.3389/fonc.2019.00666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martin-Belmonte F., Perez-Moreno M. Epithelial cell polarity, stem cells and cancer. Nat. Rev. Cancer. 2011;12:23–38. doi: 10.1038/nrc3169. [DOI] [PubMed] [Google Scholar]
- 60.Ellenbroek S.I., Iden S., Collard J.G. Cell polarity proteins and cancer. Semin. Cancer Biol. 2012;22:208–215. doi: 10.1016/j.semcancer.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 61.Johnstone C.N., Pattison A.D., Harrison P.F., Powell D.R., Lock P., Ernst M., Anderson R.L., Beilharz T.H. FGF13 promotes metastasis of triple-negative breast cancer. Int. J. Cancer. 2020;147:230–243. doi: 10.1002/ijc.32874. [DOI] [PubMed] [Google Scholar]
- 62.Bluemn E.G., Coleman I.M., Lucas J.M., Coleman R.T., Hernandez-Lopez S., Tharakan R., Bianchi-Frias D., Dumpit R.F., Kaipainen A., Corella A.N., et al. Androgen receptor pathway-independent prostate cancer is sustained through FGF signaling. Cancer Cell. 2017;32:474–489 e476. doi: 10.1016/j.ccell.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dominguez-Sola D., Gautier J. MYC and the control of DNA replication. Cold Spring Harb Perspect. Med. 2014;4 doi: 10.1101/cshperspect.a014423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bretones G., Delgado M.D., Leon J. Myc and cell cycle control. Biochim. Biophys. Acta. 2015;1849:506–516. doi: 10.1016/j.bbagrm.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 65.Grandori C., Cowley S.M., James L.P., Eisenman R.N. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev Biol. 2000;16:653–699. doi: 10.1146/annurev.cellbio.16.1.653. [DOI] [PubMed] [Google Scholar]
- 66.Yi J., Liu C., Tao Z., Wang M., Jia Y., Sang X., Shen L., Xue Y., Jiang K., Luo F., et al. MYC status as a determinant of synergistic response to Olaparib and Palbociclib in ovarian cancer. EBioMedicine. 2019;43:225–237. doi: 10.1016/j.ebiom.2019.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhu X., Chen L., Huang B., Li X., Yang L., Hu X., Jiang Y., Shao Z., Wang Z. Efficacy and mechanism of the combination of PARP and CDK4/6 inhibitors in the treatment of triple-negative breast cancer. J. Exp. Clin. Cancer Res. 2021;40:122. doi: 10.1186/s13046-021-01930-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen H., Boutros P.C. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics. 2011;12:35. doi: 10.1186/1471-2105-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ramirez F., Dundar F., Diehl S., Gruning B.A., Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014;42:W187–W191. doi: 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: DHT-downregulated genes (fold-change < 1.5) are highlighted in red.