

Abstract
A 34‐year‐old man visited our Department of Gastroenterology and Metabolism, Nagoya City University Graduate School of Medical Sciences, Nagoya, Japan, because of dry mouth and weight loss. His plasma glucose level was 32.8 mmol/L and serum levels of ketone bodies were increased, but with metabolic alkalemia. He was also suffering from renal tubular hypomagnesemia and hypokalemia. Abdominal computed tomography showed bilateral renal cysts. These findings were suggestive of maturity‐onset diabetes of the young type 5. Genetic testing showed heterozygous hepatocyte nuclear factor 1 beta gene deletion. In the present case, it seemed reasonable to view hepatocyte nuclear factor 1 beta gene deletion as the common cause of maturity‐onset diabetes of the young type 5‐associated diabetic ketoacidosis and tubular malfunction‐induced hypokalemic alkalosis. This case exemplifies the importance of hepatocyte nuclear factor 1 beta gene abnormality as a potential cause of diabetic ketoacidosis with alkalemia.
Keywords: Diabetic ketoacidosis with alkalemia, HNF1B, Maturity‐onset diabetes of the young type 5
Genetic mutations of hepatocyte nuclear factor 1 beta cause multiple organ disorders, as well as maturity‐onset diabetes of the young type 5, collectively known as the hepatocyte nuclear factor 1 beta syndrome. It can lead to the development of diabetic ketoacidosis with alkalemia.

INTRODUCTION
Maturity‐onset diabetes of the young (MODY) is a group of monogenic forms of diabetes that are inherited in an autosomal dominant manner. Although the prevalence of MODY syndromes is estimated to be between 0.6 and 2% of all diabetes 1 , hepatocyte nuclear factor 1 beta (HNF1B)‐MODY type 5 (MODY5) is exceedingly rare, comprising less than 5% of all MODY subtypes 2 . More importantly, MODY5 is uniquely associated with a broad clinical spectrum from renal phenotypes to pancreatic β‐cell dysfunction 3 . Clinical symptoms, such as polyuria and/or weight loss, are present in 47% of patients, but ketoacidosis is rare at the time of diabetes diagnosis in patients with MODY5 4 .
Here, we report a patient newly diagnosed as MODY5, who presented with diabetic ketoacidosis with renal tubular alkalemia, and later was diagnosed genetically as HNF1B syndrome.
CASE REPORT
A 34‐year‐old man visited Department of Gastroenterology and Metabolism, Nagoya City University Graduate School of Medical Sciences, Nagoya, Japan, because of dry mouth and weight loss of 10 kg within 1 year. He had no medical history, except for hyperglycemia identified in a health examination 1 year before that was left untreated. He had no family history of diabetes or kidney disease. His height was 170 cm and bodyweight was 47.0 kg. Laboratory examination showed that the casual plasma glucose level was 32.8 mmol/L, with the hemoglobin A1c level was 189 mmol/mol (Table 1). Although serum levels of ketone bodies were increased, blood gas analysis showed a pH of 7.48 and HCO3 − of 36.7 mmol/L, suggesting an existence of metabolic alkalosis. Therefore, he was diagnosed with diabetic ketoacidosis with alkalemia and treated with linagliptin as a tentative pharmacotherapy. He was admitted to our hospital 1 week later for further examination and treatment.
Table 1.
Postprandial laboratory data on patient's first visit
| Hematology | Glycometabolism tests | ||
| White blood cells (×109/L) | 9.3 | Glucose (mmol/L) | 32.8 |
| Hemoglobin (g/L) | 170 | HbA1c (mmol/mol) | 189 |
| Platelets (×109/L) | 236 | Insulin (pmol/mL) | 12.5 |
| C‐peptide (nmol/L) | 0.1 | ||
| Biochemistry | Anti‐GAD antibodies (IU/mL) | <5.0 | |
| Total protein (g/L) | 78 | Anti‐IA2 antibodies (IU/mL) | <0.4 |
| Albumin (mmol/L) | 0.6 | Anti‐insulin antibodies (IU/mL) | <0.4 |
| AST (µmol/s L) | 0.47 | Anti‐ZnT8 antibodies (IU/mL) | <10.0 |
| ALT (µmol/s L) | 0.62 | ||
| LDH (µmol/s L) | 4.22 | Endocrinological tests | |
| γGTP (µmol/s L) | 1.17 | Plasma renin activity (μg/L/h) | 22.0 |
| ALP (µmol/s L) | 6.8 | Aldosterone (pmol/L) | 1,144 |
| Creatine kinase (µmol/s L) | 2.85 | ACTH (pmol/L) | 4.17 |
| Amylase (µmol/s L) | 1.08 | Cortisol (nmol/L) | 303.5 |
| Uric acid (µmol/L) | 244 | TSH (mIU/L) | 3.68 |
| Creatinine (µmol/L) | 91.1 | Free T4 (pmol/L) | 19 |
| BUN (mmol/L) | 8.39 | Free T3 (pmol/L) | 3.01 |
| eGFR (mL/min/1.73 m2) | 68.3 | ||
| Sodium (mmol/L) | 133 | Venous blood gas analysis | |
| Potassium (mmol/L) | 3.7 | pH | 7.48 |
| Chloride (mmol/L) | 79 | PaCO2 (kPa) | 6.66 |
| Calcium (mmol/L) | 2.6 | PaO2 (kPa) | 8.53 |
| Magnesium (mmol/L) | 0.5 | HCO3 − (mmol/L) | 36.7 |
| Phosphate (mmol/L) | 1.1 | BE (mmol/L) | 11.3 |
| Triglyceride (mmol/L) | 2.6 | ||
| HDL cholesterol (mmol/L) | 1.8 | Urinalysis | |
| LDL cholesterol (mmol/L) | 2.1 | Specific gravity | 1.031 |
| pH | 5.0 | ||
| Ketone body fractions | Glucose | (4+) | |
| Total ketone bodies (mmol/L) | 1.016 | Protein | (±) |
| Acetoacetic acid (mmol/L) | 0.395 | Ketone body | (−) |
| β‐hydroxybutyric acid (mmol/L) | 0.621 |
γGTP, gamma‐glutamyl transpeptidase; ACTH, adrenocorticotropic hormone; ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BE, base excess; BUN, blood urea nitrogen; eGFR, estimated glomerular filtration rate; GAD, glutamate decarboxylase; HDL, high‐density lipoprotein; IA2, islet antigen 2; LDH, lactate dehydrogenase; LDL, low‐density lipoprotein; TSH, thyroid‐stimulating hormone; ZnT8, zinc transporter 8.
After admission, linagliptin was stopped and multiple daily injections of insulin were initiated. Urinalysis showed a rather low level of urinary C‐peptide secretion (9.8 nmol/day), suggesting a certain level of β‐cell dysfunction. Anti‐islet antibodies in the serum were negative. The patient’s hyperglycemia improved a few days later. During the course, however, he developed profound hypomagnesemia and hypokalemia. Additional urine electrolyte analysis showed an inappropriately high fractional excretion of magnesium (FeMg: 22.7%) and increased urinary potassium excretion (38 mmol/day), suggesting renal magnesium and potassium wasting. A challenge with hydrochlorothiazide showed a blunted response. We then initially suspected that along with diabetes, the patient was complicated with Gitelman syndrome.
Abdominal computed tomography showed bilateral multiple renal cysts and pyelectasis (Figure 1). Based on these clinical observations, MODY5 was suspected. Furthermore, the HNF1B score, a pivotal tool for rational genetic testing 5 , was 19, confirming a high level of clinical suspicion for HNF1B‐related disease. We carried out multiplex ligation probe amplification using patient‐derived lymphocyte, and identified heterozygous entire deletion of the HNF1B gene (Figure 2).
Figure 1.
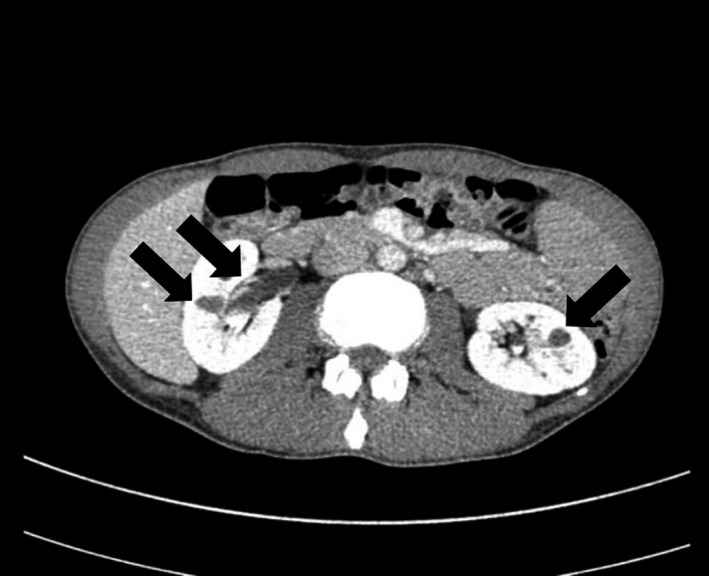
Contrast agent‐enhanced computed tomography scan of the abdominal plane. Bilateral multiple renal cysts and pyelectasis were visualized. Pancreatic and genital tract were spared.
Figure 2.
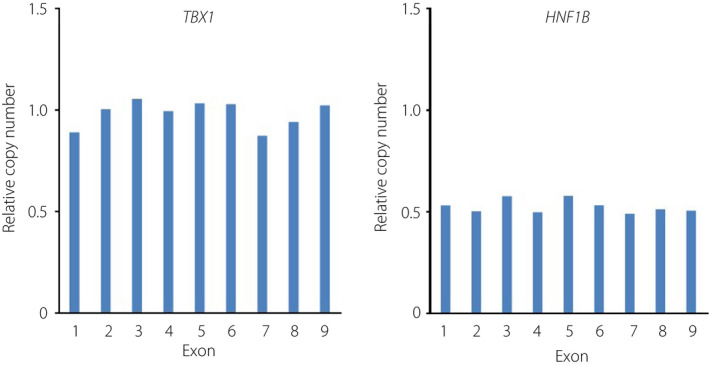
Multiplex ligation probe amplification analysis. Briefly, genomic deoxyribonucleic acid was denatured and hybridized with the probe to detect the deletion of HNF1B. Ligation and polymerase chain reaction amplification were carried out with the SALSA P357 MPLA kit and analyzed by capillary electrophoresis using Gene Mapper v.3.7 (Thermo Fisher, Waltham, MA, USA). Heterozygous deletion in the HNF1B gene was identified, whereas no copy number alterations in the TBX1 gene, located in chromosome 22q11.2 region, was detected.
After the diagnosis, we continued multiple daily injections of insulin, taking into consideration a decline of insulin secretion in the future. We also started supplementation of potassium and magnesium with high confidence in the prevention of muscle weakness, convulsion or arrhythmia.
DISCUSSION
We herein report a case of MODY5 presenting diabetic ketoacidosis with alkalemia as an initial manifestation, possibly due to the coexistence of HNF1B‐associated renal tubular dysfunction.
Genetic mutations of HNF1B, located on chromosome 17q12, cause multiple organ disorders, collectively known as the HNF1B syndrome. Among 33 Japanese patients with HNF1B‐related disorders, including the present case, whole‐gene deletion and heterozygous variants account for up to 14 and 19 cases, respectively 6 . Although the genotype–phenotype relationship has not yet been conclusive, a report suggests that patients with HNF1B deletion less often show end‐stage chronic kidney disease than those with point mutations 4 , consistent with an absence of kidney dysfunction in the present case.
It is well known that manifestations of HNF1B syndrome vary among patients 3 . For example, diabetes or renal morphological abnormalities, such as renal cystic disease, was present in 38.7 and 77.4% of patients, respectively 6 . The co‐existence of renal cysts in diabetes‐complicating cases is known as renal cysts and diabetes syndrome, as found in the current case. Clinical features observed in the present case are summarized in Figure 3.
Figure 3.
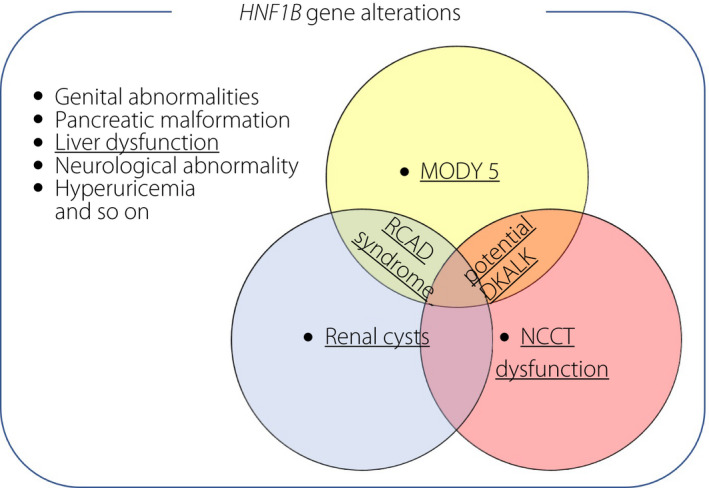
Schematic diagram of the clinical manifestations in the present case within the broad spectrum of HNF1B‐related multi‐organ disorders. Underlines indicate disorders found in the patient. When maturity‐onset diabetes of the young type 5 (MODY5) and Na‐Cl cotransporter (NCCT) dysfunction overlaps in a single patient, potential occurrence of diabetic ketoacidosis with alkalemia (DKALK) is suggested, as in the present case. RCAD, renal cysts and diabetes.
It has been reported that HNF1B mutations can also affect Na‐Cl cotransporter function in the distal convoluted tube, leading to hypokalemia, hypomagnesemia and metabolic alkalosis 7 , 8 . In the present case, hypokalemic alkalosis potentially caused by Na‐Cl cotransporter dysfunction seems to predominate over a mild diabetic ketoacidosis caused by insulin deficiency in MODY5 (Figure 3).
Diabetic ketosis typically manifests with acidemia due to an accumulation of acidic ketone bodies. However, it can present as alkalemia under certain, but limited, conditions, including vomiting and the use of diuretics, for which it was not relevant in the present case.
The absence of clinical manifestation in the patient’s parents shows that the deletion of the gene might be a spontaneous de novo mutation, although we could not obtain agreement from his parents for their genetic analysis. De novo mutations reportedly occur relatively frequently, as seen in 50–60% of patients with HNF1B gene abnormality 9 . This is believed to be the case, because chromosome 17q12 contains a sequence with high homogeny, and meiotic cross‐over might sometimes lead to mistakes during gene replication 10 .
Collectively, when a young diabetes patient presents with diabetic ketoacidosis with alkalemia or electrolyte abnormalities, genetic testing for MODY5 is recommended. Conversely, when a patient is genetically diagnosed as MODY5, clinical screening tests for HNF1B mutation‐related multi‐organ complications need to be carried out.
COMPLIANCE WITH ETHICAL STANDARDS
Genetic testing was carried out in accordance with the ethical standards of Nagoya City University and Kobe University, related laws, and the Declaration of Helsinki, and under the permission of the institutional review board of Kobe University (no. 301). Patient's written informed consent was obtained.
DISCLOSURE
The authors declare no conflict of interest.
Approval of the research protocol: Research protocol was approved by the institutional review board of Kobe University.
Informed consent: Written informed consent was obtained from the patient.
Approval date of registry and the registration no. of study/trial: Approval date of the registry was 25 January 2021. The registration number of the study is 301.
Animal studies: N/A.
J Diabetes Investig. 2022; 13: 923–926
REFERENCES
- 1. McDonald TJ, Ellard S. Maturity onset diabetes of the young: identification and diagnosis. Ann Clin Biochem 2013; 50: 403–415. [DOI] [PubMed] [Google Scholar]
- 2. Mateus JC, Rivera C, O’Meara M, et al. Maturity‐onset diabetes of the young type 5 a MULTISYSTEMIC disease: a CASE report of a novel mutation in the HNF1B gene and literature review. Clin Diabetes Endocrinol 2020; 6: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Horikawa Y. Maturity‐onset diabetes of the young (MODY) as a model for elucidating the multifactorial origin of type 2 diabetes mellitus. J Diabetes Investig 2018; 9: 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dubois‐Laforgue D, Cornu E, Saint‐Martin C, et al. Diabetes, associated clinical spectrum, long‐term prognosis, and genotype/phenotype correlations in 201 adult patients with hepatocyte nuclear factor 1B (HNF1B) molecular defects. Diabetes Care 2017; 40: 1436–1443. [DOI] [PubMed] [Google Scholar]
- 5. Faguer S, Chassaing N, Bandin F, et al. The HNF1B score is a simple tool to select patients for HNF1B gene analysis. Kidney Int 2014; 86: 1007–1015. [DOI] [PubMed] [Google Scholar]
- 6. Nagano C, Morisada N, Nozu K, et al. Clinical characteristics of HNF1B‐related disorders in a Japanese population. Clin Exp Nephrol 2019; 23: 1119–1129. [DOI] [PubMed] [Google Scholar]
- 7. Bech AP, Wetzels JF, Bongers EM, et al. Thiazide responsiveness testing in patients with renal magnesium wasting and correlation with genetic analysis: a diagnostic test study. Am J Kidney Dis 2016; 68: 168–170. [DOI] [PubMed] [Google Scholar]
- 8. Ito S, Uchimaru R, Watanabe M, et al. A case of MODY 5 exhibiting acute onset of diabetic ketoacidosis in combination with metabolic alkalosis, hypokalemia and hypomagnesemia. J Jpn Diabetes Soc 2013; 56: 93–101 (Japanese). [Google Scholar]
- 9. Ulinski T, Lescure S, Beaufil S, et al. Renal phenotype related to hepatocyte nuclear factor‐1 beta (TCF2) mutations in a pediatric cohort. J Am Soc Nephrol 2006; 17: 497–503. [DOI] [PubMed] [Google Scholar]
- 10. Shchelochkov OA, Cheung SW, Lupski JR. Genomic and clinical characteristics of microduplications in chromosome 17. Am J Med Genet A 2010; 152A: 1101–1110. [DOI] [PubMed] [Google Scholar]