

ABSTRACT
Aims/Introduction
It is not unclear whether the complement system is involved in the pathogenesis of diabetic nephropathy (DN). We explored the role of the complement system in glomeruli from patients with DN using integrated transcriptomic bioinformatics analysis and renal histopathology.
Materials and Methods
Four datasets (GSE30528, GSE104948, GSE96804 and GSE99339) from the Gene Expression Omnibus database were integrated. We used a protein–protein interaction network and the Molecular Complex Detection App to obtain hub genes. Gene ontology and the Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses were carried out to identify significant pathways. We also investigated the associations of C1q and C3 deposition on renal histopathology with clinical data, pathological parameters and renal survival in DN patients.
Results
We identified 47 up‐ and 48 downregulated genes associated with DN. C3, C1QB and C1QA were found to be complement‐related hub genes. The gene ontology and Kyoto Encyclopedia of Genes and Genomes analyses identified complement activation and humoral immune response as the significant oncology terms, with C1QB and C3 positioned at the center of the pathway. Regarding renal histopathology, patients with both C1q and C3 deposition had more severe glomerular classes. Multivariate Cox proportional hazards regression showed that the deposition of glomerular C1q and C3 was an independent risk factor for kidney failure. Patients with high C1q, C3 or C4d expression in glomeruli were more likely to progress to kidney failure, whereas glomerular mannose‐binding lectin was rare.
Conclusions
Complement activation is involved in the development of DN, and activation of the classical complement pathway in glomeruli might accelerate disease progression.
Keywords: Complement system, Diabetic nephropathy, Glomeruli
Local complement activation is involved in the development of diabetic nephropathy and deposition of complement C1q and C3 in glomeruli may accelerate the progression of diabetic nephropathy. New approaches to modulate the complement system might lead to the development of new agents to prevent or slow the progression of diabetic nephropathy.

INTRODUCTION
Diabetic nephropathy (DN) is the leading cause of end‐stage renal disease (ESRD) worldwide 1 . The estimated global prevalence of diabetes in 2019 was 9.3% (463 million), and is projected to increase to 10.9% (700 million) by 2045 2 . Nearly 30–40% of diabetes patients develop DN, and most patients with DN die of cardiovascular complications or infections before they progress to ESRD. Although various drugs are available, the existing drugs have limited therapeutic effect on DN. Therefore, it is necessary to explore its etiopathogenesis 3 . Pathophysiological studies have shown that the progression of DN is closely related to metabolic abnormalities, oxidative stress, hemodynamic changes and innate immunity 4 , 5 . As an important part of the innate immune system, the complement system has attracted much attention for its role in the development of DN 6 , 7 , 8 , 9 .
The complement system is the first barrier to accelerated inflammation, and it ameliorates the clearance of impaired cells and pathogenic microorganisms from the body through phagocytes and antibodies 10 . However, it remains unclear whether the complement system is involved in the pathogenesis of DN.
Omics analyses, especially transcriptomics and metabolomics, have been widely used to detect genes associated with the pathogenesis of DN 11 . Transcriptomic profiling using microarray chips provides important information about the molecular and cellular mechanisms of disease pathogenesis and progression 12 . However, independent microarray analyses might lead to a high false positive rate. The R software package, RobustRankAggreg (The R Foundation For Statistical Computing, Vienna, Austria), is helpful for ranking differentially expressed genes (DEGs) obtained from different platforms using a robust rank aggregation (RRA) approach 13 . Therefore, we integrated four transcriptomic microarray datasets from kidney tissue using RRA to determine whether complement‐related genes are hub genes involved in DN. Then, we investigated the clinical and pathological significance of complement deposition in the renal histopathology of patients with DN to explore the potential role of the complement system in DN, which should provide new insights into potential targets for DN therapeutics.
MATERIALS AND METHODS
Collection of gene expression datasets and platform records
All datasets, derived from glomerular transcriptional profiles from DN patients, were downloaded from the Gene Expression Omnibus Database (https://www.ncbi.nlm.nih.gov/geo/). The gene expression profiling microarray data were eligible if they provided transcriptome comparison of glomeruli from kidneys with DN and control glomeruli tissues obtained from healthy living donors, and had more than three samples in each group. Finally, four datasets (GSE30528 14 , GSE104948 15 , GSE96804 16 and GSE99339 17 ) were included in the present study (Table 1).
Table 1.
Four datasets from transcriptional profiles of human glomeruli included in the analysis
| Reference | GEO Dataset | Platform | Control glomeruli | DN glomeruli |
|---|---|---|---|---|
| Woroniecka 2011 14 | GSE30528 | GPL571 | 13 | 9 |
| Grayson 2018 15 | GSE104948 | GPL22945 | 18 | 7 |
| Pan 2018 16 | GSE96804 | GPL17586 | 20 | 41 |
| Shved 2017 17 | GSE99339 | GPL19184 | 8 | 7 |
DN, diabetic nephropathy; GEO, Gene Expression Omnibus.
Datasets pre‐processing and identification of DEGs
We first used the “limma” package, a Bioconductor package of R (version 4.0.5), to normalize and select DEGs from the messenger ribonucleic acid expression microarray data. Then, we used the “RobustRankAggreg” package in R to integrate these four datasets with the RRA algorithm. Based on the criteria (|logFC (fold change)| >1 and adjusted P‐value <0.05), the DEG frame results were finally established.
Functional enrichment analyses of DEGs
To explore the potential functions of the DEGs, we carried out gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses using the “clusterProfiler” package in R. The adjusted P‐value <0.05 was considered significant. Using the “ggplot2” package in R, we visualized all data frame results.
Identification of hub genes
We used protein–protein interaction (PPI) 18 analysis to further explore the hub genes that were embedded in gene regulatory networks. First, the Search Tool for the Retrieval of Interacting Genes (STRING; http://string‐db.org) database was used to search for the PPI network of DEGs with the highest confidence‐score (>0.9). Then, the PPI network data, which were exported from STRING, were imported into Cytoscape 3.7.2 (Institute for Systems Biology, Seattle, WA, USA) for their visualization. Subsequently, we used the Molecular Complex Detection (MCODE), a clustering algorithm of Cytoscape, to carry out network analysis to obtain hub genes (the parameter criteria were as follows: degree cut‐off = 2, k‐score = 2, node score cut‐off = 0.2 and max depth = 100). Consequently, the GO and KEGG pathway analysis of hub genes were carried out with the R package.
Clinical validation of DN patients
We retrospectively collected patients with type 1 or type 2 diabetes and nephropathy who underwent renal biopsy at the China‐Japan Friendship Hospital, Beijing, China, between January 2013 and December 2020. All participants met the diagnostic criteria for diabetes proposed by the American Diabetes Association in 2021 19 and the criteria of DN followed by the Renal Pathology Society in 2010 20 . We recorded the classic histopathological characteristics, such as expansion of the mesangial matrix, glomerular hypertrophy, thickening of the glomerular basement membrane and nodular glomerulosclerosis. The exclusion criteria were as follows: (i) coincident non‐diabetic renal disease, estimated glomerular filtration rate (eGFR) <15 mL/min/1.73 m2 at baseline, inadequate renal tissue (<5 glomeruli) and follow‐up interval <6 months or no follow‐up information.
Clinical characteristics of DN patients
The baseline data of included patients were collected from the Electronic Medical Record System, including age, sex, duration of diabetes, 24‐h urinary protein excretion, serum levels of albumin, creatinine, complement C1q (C1q), complement 3 (C3) and complement 4 (C4). The eGFR was calculated using the creatinine‐based Chronic Kidney Disease Epidemiology Collaboration equation 21 . The follow‐up interval was assessed from the time of renal biopsy to one of four end‐points: ESRD, death, loss to follow up or study termination (October 2021). ESRD was defined as a need for maintenance dialysis or kidney transplantation. The primary end‐point in the present study was the composite of a doubling of the baseline serum creatinine concentration or ESRD.
Renal histopathology
Renal biopsy specimens were evaluated using light microscopy, immunofluorescence staining and electron microscopy. Classification of DN and histological scoring were carried out according to the criteria of Tervaert et al 20 . The glomerular classification was as follows. Class I, isolated glomerular basement membrane thickening, and only mild, non‐specific light microscopy changes that do not meet the criteria of class II through IV. Class II, mesangial expansion, mild (IIa) or severe (IIb): glomeruli classified as mild or severe mesangial expansion, but not meeting the criteria for class III or IV. Class III, nodular sclerosis (Kimmelstiel–Wilson lesions): at least one glomerulus with Kimmelstiel–Wilson nodules and <50% global glomerulosclerosis. Class IV, advanced diabetic glomerulosclerosis: >50% global glomerulosclerosis. The interstitial fibrosis and tubular atrophy (IFTA) scores were classified as follows: 0, absent; 1, <25%; 2, 25–50%; and 3, >50% of the total area. The interstitial inflammation was scored as follows: 0, absent; 1, infiltration only in areas related to IFTA; and 2, infiltration in areas without IFTA. Scores of vascular lesions were evaluated based on the presence of large‐vessel arteriosclerosis and arteriolar hyalinosis. The intensity of immunofluorescence staining of the complement (C1q and C3) deposits in glomeruli was graded using a semiquantitative method on a scale of 0 ~ ++++. When the presence of glomerular complement (C1q or C3) staining was detected at 1+ or a higher grade, it was taken as positive.
Immunohistochemistry of C1q, C3, mannose‐binding lectin and C4d
The renal tissues embedded in paraffin were sectioned at 2‐µm thickness. After dewaxing and hydration steps, the high‐pressure steam repair method was used to promote antigen repair. Endogenous peroxidase for samples was blocked with 3% peroxide‐methanol and incubated in goat serum working fluid for 30 min at room temperature. Then the following primary antibodies (all from Abcam, Cambridge, UK) were separately used: (i) anti‐C1q antibody (ab268120) diluted 1:400; (ii) anti‐C3 antibody (ab97462) diluted 1:300; (iii) anti‐C4d antibody (ab183311) diluted 1:100; and (iv) anti‐mannose‐binding lectin (MBL) antibody (ab23457) diluted 1:25. After an overnight incubation at 4°C, the reactions were visualized by staining with horseradish peroxidase and diaminobenzidine.
Immunohistochemical images were obtained from sections observed microscopically at ×200 magnification using an integrated digital camera system (Nikon, Tokyo, Japan). An average optical density was obtained by analysis of 10 different fields, and quantification was carried out semiquantitatively in a blinded manner. The Image‐Pro Plus software 6.0 (Media Cybernetics, Bethesda, MD, USA) was used to analyze the average optical density of the stained areas.
Statistical analysis
We used SPSS 21.0 (SPSS Inc.; www.spss.com) to analyze clinical and pathological data; graphs were created using GraphPad Prism 8.0 (GraphPad Software Inc.; www.graphpad.com). Continuous variables were represented as mean ± standard deviation. Categorical variables were summarized as numbers (percentages). Student's t‐test or one‐way anova was used for continuous variables. The χ2‐test or Fisher's exact test was used for categorical variables. We used the linear regression analysis to calculate the slopes of the rates of change in eGFR. The results were also analyzed by calculating the difference between the baseline and the final eGFR and dividing by time. Cox proportional hazard models were carried out to assess the hazard ratio (HR) and 95% confidence interval (CI) for parameters associated with the definitive renal outcome. We first used univariate log‐rank tests to estimate the association of baseline parameters with the definitive renal outcome. The variables that had P‐values <0.1 by univariate analysis or were clinically significant were subsequently entered into a final multivariate Cox regression model. Differences with a two‐sided P‐value <0.05 was considered statistically significant.
RESULTS
Construction of DEGs by integrated analysis
The flowchart of the present study is shown in Figure 1. By integrating four microarray datasets (GSE30528, GSE104948, GSE96804 and GSE99339), we identified 95 DEGs, including 47 upregulated genes and 48 downregulated genes, which were the most significant genes that were associated with DN (Table S1). The top 20 significantly upregulated and downregulated genes ranked according to the fold change value were shown in Figure 2a. GO analysis showed that upregulated genes were mainly enriched in humoral immune response (9.69E−10), extracellular matrix organization (1.58E−9), extracellular structure organization (1.62E−9) and regulation of complement activation (9.74E−9), whereas downregulated genes were mainly enriched in striated muscle tissue development (3.25E−8), muscle organ development (4.97E−8), muscle tissue development (5.20E−8) and carbohydrate biosynthetic process (9.08E−7; Figures S1 and S2). We then carried out KEGG pathway enrichment analysis of upregulated genes, and found that the top three enriched pathways were complement and coagulation cascades (3.47E−07), pertussis (5.25E−06) and extracellular matrix‐receptor interaction (1.08E−05; Figure 2b). In contrast, the top three pathways that were related to downregulated genes, were mainly focused in HIF‐1 signaling pathway (0.000567), growth hormone synthesis, secretion and action (0.000788), and FoxO signaling pathway (0.00113; Figure 2c).
Figure 1.
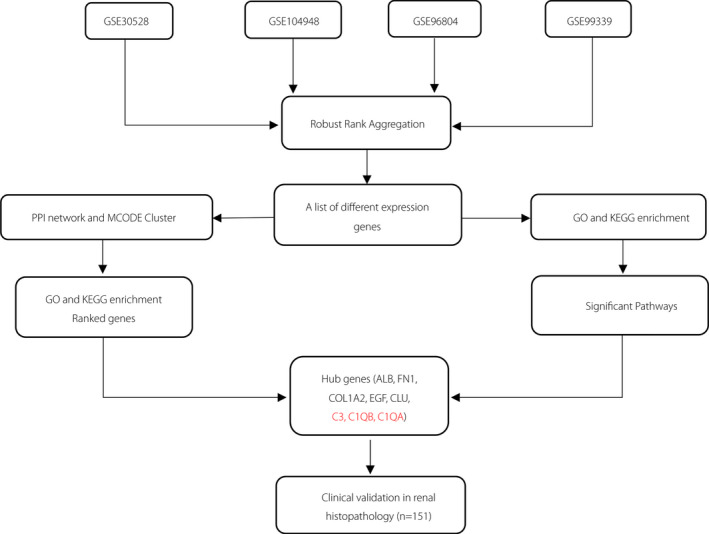
Study flowchart. GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; MCODE, Molecular Complex Detection; PPI, protein–protein interaction.
Figure 2.
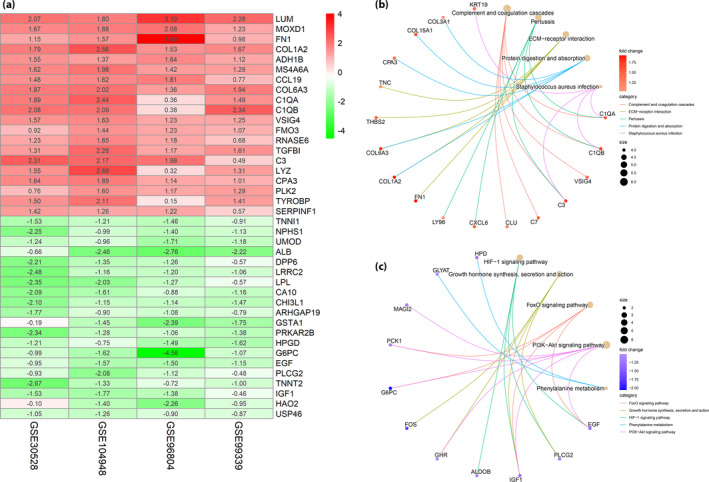
Collection of differentially expressed genes (DEGs) and Kyoto Encyclopedia of Genes and Genomes pathway analysis in diabetic nephropathy. (a) Heat map of DEGs. Each column represents one dataset and each row represents one gene. Green represents a lower level of gene expression, red represents a higher expression level. (b) The upregulated DEGs enriched pathway analysis. (c) The downregulated DEGs enriched pathway analysis. PPI, protein–protein interaction. ECM, extracellular matrix.
PPI analysis and identification of hub genes
The PPI network of the aforementioned 95 DEGs was built according to the STRING database and was then ranked with topological analysis on nodes. The exported data was visualized by Cytoscape software (Figure S3). Four discrete subclusters were obtained through MCODE plug‐in by Cytoscape software, and the top 24 hub genes ranked based on the degree score was shown in Table S2.
GO and KEGG enrichment analysis of hub genes
GO analysis of hub genes were found to be associated with 96 biological process terms. Figure 3a shows the top 10 terms of GO analysis. We found that the hub genes were mainly enriched in extracellular organization (2.76E−10), complement activation (1.87E−07) and humoral immune response (4.01E−07). To further explore the pathogenesis of DN, we again carried out the KEGG pathway analysis. Figure 3b presents the results of the top five terms of the KEGG enrichment analyses, and most of the hub genes were linked to complement and coagulation cascades (6.29E−07), extracellular matrix‐receptor interaction (7.49E−07), focal adhesion (2.12E−06) and PI3K–protein kinase B signaling pathway (3.79E−06). Based the above GO and KEGG analyses results, we could find that some hub genes (C1QA, C1QB and C3), which were involved in the complement activation process, did play a vital role in the pathogenesis of DN.
Figure 3.
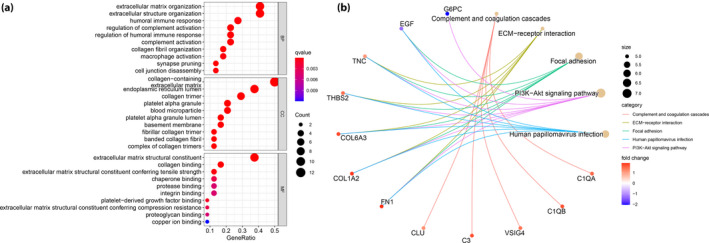
(a) Gene Ontology annotation and (b) Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis. ECM, extracellular matrix.
Comparison of the clinical and pathological characteristics of DN patients
This study enrolled 151 patients with pathologically diagnosed DN (Figure S4). C3 or C1q deposits were found in glomerular capillary walls, the mesangium, Bowman’s capsule and the tubular basement membrane. The grouping of C3 or C1q deposits in the present study was based on the presence of deposition in the glomeruli. On direct immunofluorescence microscopy, C1q and C3 co‐deposits (C1q+ C3+), only C1q or C3 deposits (C1q+ C3−/C1q− C3+), and no complement deposits (C1q− C3−) were found in 24 of 151 (15.89%), 55 of 151 (36.42%) and 72 of 151 (47.68%) patients, respectively. Patients with C1q+ C3+ deposition in glomeruli had significantly lower serum albumin (P < 0.01) and higher urinary protein excretion (P < 0.05) levels compared with those with only one or no complement deposition in glomeruli. Furthermore, patients with C1q+ C3+ deposition in glomeruli had significantly more severe glomerular classes (P < 0.05; Table 2).
Table 2.
Characteristics of patients with diabetic nephropathy according to C3 and C1q deposition
| C1q+ C3+ (n = 24) | C1q+ C3−/C1q− C3+ (n = 55) | C1q− C3− (n = 72) | P | |
|---|---|---|---|---|
| Clinical characteristics | ||||
| Males (%) | 18 (75%) | 42 (76.4%) | 58 (80.6%) | 0.83 |
| Age (years) | 53.08 ± 13.45 | 51.47 ± 9.49 | 54.11 ± 10.60 | 0.39 |
| HbA1c (%) | 7.54 ± 1.77 | 7.38 ± 1.25 | 7.29 ± 1.67 | 0.78 |
| Duration of diabetes (years) | 14.04 ± 5.75 | 12.09 ± 7.61 | 11.54 ± 6.65 | 0.31 |
| Serum albumin (g/L) † | 31.00 ± 4.67 | 33.80 ± 6.94 | 35.86 ± 6.76 | <0.01 |
| Urinary protein (g/24 h) ‡ | 6.83 ± 3.46 | 5.91 ± 3.86 | 4.71 ± 3.64 | 0.03 |
| Serum C1q (mg/dL) | 188.88 ± 47.27 | 190.76 ± 43.40 | 195.14 ± 47.37 | 0.79 |
| Serum C3 (mg/dL) | 85.92 ± 15.11 | 92.25 ± 23.10 | 93.11 ± 19.45 | 0.31 |
| Serum C4 (mg/dL) | 25.54 ± 6.98 | 27.38 ± 7.02 | 26.01 ± 7.14 | 0.45 |
| Serum creatinine (μmol/L) | 146.83 ± 61.14 | 153.96 ± 74.36 | 145.57 ± 59.12 | 0.76 |
| eGFR (mL/min/1.73 m2) | 52.50 ± 25.26 | 53.35 ± 25.55 | 53.65 ± 23.84 | 0.98 |
| CKD stages (1/2/3a/3b/4) | 2/5/5/7/5 | 4/17/13/12/9 | 6/21/14/14/17 | 0.95 |
| Pathological characteristics | ||||
| Glomerular class (I/IIa/IIb/III/IV) | 0/0/6/16/2 | 0/0/22/30/3 | 2/10/28/29/3 | <0.05 |
| IFTA score (0/1/2/3) | 0/4/13/7 | 0/11/30/14 | 0/20/37/15 | 0.75 |
| Interstitial inflammation (0/1/2) | 0/11/13 | 2/28/25 | 5/36/31 | 0.75 |
| Vascular lesion (0/1/2) | 4/8/12 | 8/28/19 | 17/37/18 | 0.19 |
| Global sclerosis (%) | 21.78 ± 18.73 | 26.53 ± 14.06 | 23.62 ± 14.22 | 0.36 |
| Using ACEI or ARB | 16 (66.7%) | 39 (70.9%) | 53 (73.6%) | 0.80 |
| Hypoglycemic agents (OHA therapy/insulin therapy) | 16/8 | 27/28 | 40/32 | 0.3 |
ACEI, angiotensin‐converting enzyme inhibitor; ARB, angiotensin II type I receptor blocker; CKD, chronic kidney disease; eGFR, estimated glomerular filtration rate; HbA1c, glycated hemoglobin; IFTA, interstitial fibrosis and tubular atrophy; insulin therapy, treatment with insulin including basal supported oral hypoglycemic agent; OHA, oral hypoglycemic agent.
C1q+ C3+ vs C1q− C3−, P = 0.006.
C1q+ C3+ vs C1q− C3−, P = 0.048.
Progression of kidney failure
During follow up, the eGFR values at baseline and before dialysis were tracked in 123 patients. Table 3 shows the mean rate of decline in kidney function for each group according to C3 and C1q deposition, as determined by linear regression analysis of eGFR. The rate of decline in eGFR was significantly slower in patients without C3 and C1q (C1q− C3−) deposition (0.4 ± 1.13 mL/min/1.73 m2/month) than in those with C1q+ C3+ deposition (1.16 ± 2.1 mL/min/1.73 m2/month) or C1q+ C3−/C1q− C3+ deposition (1.11 ± 1.35 mL/min/1.73 m2/month). When the difference between the initial and final eGFR over time was used to calculate the rate of decline, the decline was 0.79 ± 0.78 mL/min/1.73 m2/month in patients without C3 and C1q (C1q− C3−) deposition, and 1.33 ± 1.04 mL/min/1.73 m2/month in patients with C1q+ C3+ deposition (P < 0.05).
Table 3.
Two assessments of the decline in estimated glomerular filtration rate over time in patients with diabetic nephropathy according to C3 and C1q deposition
| C1q+ C3+ (n = 19) | C1q+ C3−/C1q− C3+ (n = 48) | C1q− C3− (n = 56) | P | |
|---|---|---|---|---|
| Mean follow up (months) | 17.9 ± 8.38 | 20.84 ± 13.0 | 24.65 ± 13.61 | – |
| Assessment by linear regression analysis † | ||||
| Change in eGFR (mL/min/1.73 m2/month) | 1.16 ± 2.1 | 1.11 ± 1.35 | 0.40 ± 1.13 ‡ , § | <0.05 |
| Assessment by calculation of differences between initial and final values | ||||
| Change in eGFR (mL/min/1.73 m2/month) | 1.33 ± 1.04 | 1.13 ± 1.06 | 0.79 ± 0.78# | 0.05 |
eGFR, estimated glomerular filtration rate.
Values shown are the slopes of the regression line for each group.
Compared with the C1q+ C3+ group, P < 0.05.
Compared with the C1q+ C3−/C1q− C3+ group, P < 0.01.
Survival analysis according to C1q and C3 deposition in DN patients
Of the 151 DN patients, ESRD or doubling of the baseline serum creatinine level was noted in 90 cases. Patients who died before they reached the composite renal outcome were excluded from the present study. The mean follow up of this cohort was 23 ± 19 months. As shown in Figure 4a, patients with C1q+ C3+ deposition in glomeruli had worse renal survival compared with those with C1q− C3− or with C1q+ C3−/C1q− C3+ (P < 0.05). The event‐free survival probability was significantly lower in DN patients with than in those without C3 deposition in glomeruli (P < 0.05 Figure 4b), as well as in those with than in those without C1q deposition in glomeruli (P < 0.001, Figure 4c). These results showed that glomerular C1q and C3 deposition were associated with poorer renal survival.
Figure 4.
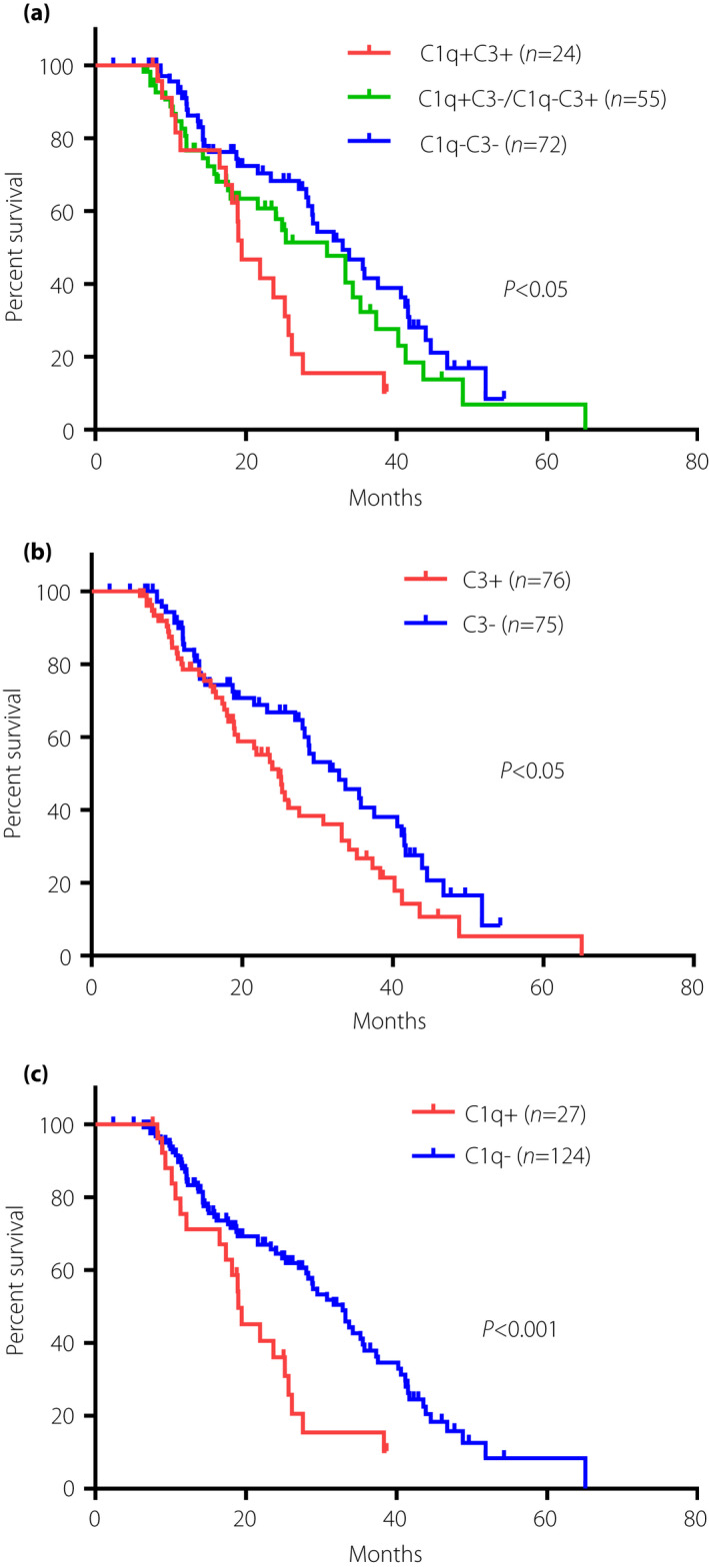
The event‐free survival probability of the Kaplan–Meier analysis. Kaplan–Meier curves comparison when dividing patients in strata of (a) glomerular C1q and C3 deposition pattern (with either or neither of C1q and C3 deposition; P = 0.0077); (b) C3 deposition versus without C3 deposition (P = 0.0345); and (c) C1q deposition versus without C1q deposition (P = 0.0018).
Predictors of composite renal outcome in DN
As shown in Figure 5a, univariate Cox regression analysis showed that glomerular C1q deposition, glomerular C3 deposition, and combined glomerular C1q and C3 deposition all predicted renal survival in DN patients (HR 2.13, 95% CI 1.27–3.56, P = 0.004; HR 1.53, 95% CI 1.0–2.33, P = 0.048; HR 2.33, 95% CI 1.31–4.14, P = 0.01; respectively). Regarding other pathological characteristics, the glomerular class (HR 1.65, 95% CI 1.19–2.28, P = 0.003), IFTA (HR 2.6, 95% CI 1.8–3.75, P < 0.001) and interstitial inflammation (HR 2.26, 95% CI 1.51–3.4, P < 0.001) were also associated with renal survival of DN. A higher eGFR predicted better renal survival (HR 0.96, 95% CI 0.95–0.97, P < 0.001), whereas the 24‐h urinary protein excretion predicted unfavorable renal survival (HR 1.15, 95% CI 1.09–1.22, P < 0.001).
Figure 5.
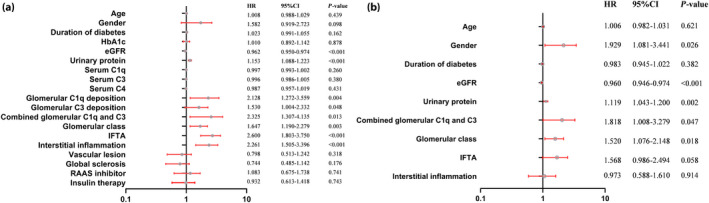
Risk factors for the composite renal outcome determined by (a) univariate or (b) multivariate Cox hazard analysis in diabetic nephropathy. CI, confidence interval; eGFR, estimated glomerular filtration rate; HbA1c, glycated hemoglobin; HR, hazard ratio; IFTA, interstitial fibrosis and tubular atrophy; RAAS, renin–angiotensin–aldosterone system.
Multivariate Cox regression analysis showed that combined glomerular C1q and C3 deposition (HR 1.82, 95% CI 1.01–3.28, P = 0.047) and the glomerular class (HR 1.52, 95% CI 1.08–2.15, P = 0.02) were independent predictors of renal survival (Figure 5b), as were sex (HR 1.93, 95% CI 1.08–3.44, P = 0.03), eGFR (HR 0.96, 95% CI 0.95–0.97, P < 0.001) and 24‐h urinary protein excretion (HR 1.12, 95% CI 1.04–1.2, P = 0.002).
Immunohistochemical analyses of C3, C1q, C4d and MBL in kidney tissues
C3 deposition in the kidney indicates activation of the common complement pathway, so further research is required on the complement pathways involved in DN. We carried out a preliminarily immunohistochemical analysis of complement components in kidney tissues from 54 DN patients who were followed for 1 and 2 years. Of the 54 patients, seven were followed up for only 1 year. Figure 6 shows immunohistochemical staining for C1q, C3, C4d and MBL in glomeruli (Figure 6a–d), and the relationship between the expression of the four complement components at baseline and the progression of kidney failure after 1 (Figure 6e) or 2 (Figure 6f) years. Baseline C1q, C3 and C4d expression in glomeruli was correlated with the composite renal outcome. By contrast, the glomerular MBL expression was very low at baseline regardless of whether the patient had an end‐point. As such, it provides two important observations: (i) the expression of MBL in glomeruli was very low; and (ii) activation of the classical complement pathway in glomeruli might play a role in the progression of DN.
Figure 6.
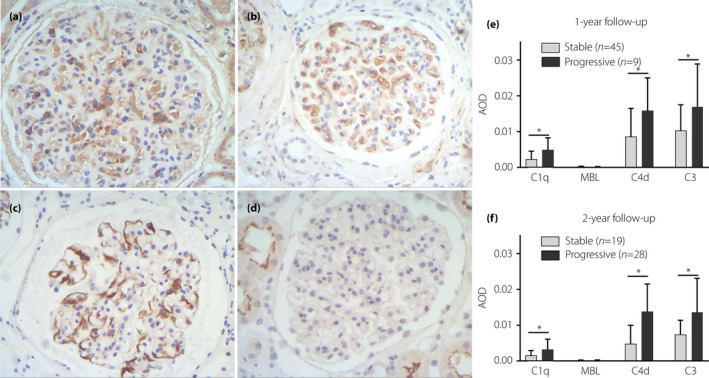
Immunohistochemical staining for (a) C1q, (b) C3, (c) C4d and (d) mannose‐binding lectin (MBL) expression in the glomeruli (original magnification: ×200), and the expression levels of C1q, C3, C4d and MBL in the stable and progressive groups. The progressive group was defined as those with the occurrence of the composite renal outcome; on the contrary, the stable group referred to those who did not enter the endpoints. *P < 0.05 versus stable group. AOD, average optical density.
Transcriptional levels of C1QA and C3 in different types of kidney diseases
We then further explored the gene expression levels of complement components in DN when compared with other kidney diseases. The transcriptional data were downloaded from Nephroseq v5 online platform (https://www.nephroseq.org/). The levels of gene expression of C1QA and C3 in DN glomeruli were comparable to those in glomeruli from immunoglobulin A nephropathy or lupus nephritis, but even significantly higher than those from membranous nephropathy and minimal change disease (Figure 7).
Figure 7.
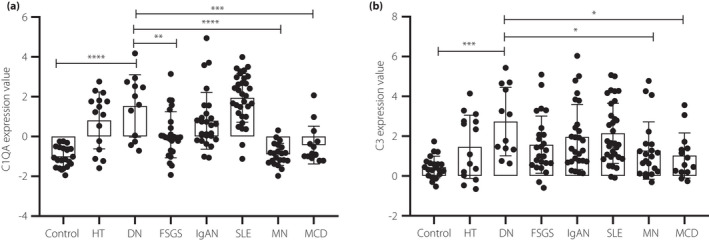
The gene expression levels of C1QA (a) and C3 (b) in glomeruli from patients with diabetic nephropathy when compared with those with other kidney diseases. *P < 0.05; **P < 0.01; ***P < 0.001. FSGS, focal segmental glomerulosclerosis; HT, hypertension; IgAN, IgA nephropathy; MCD, minimal change disease; MN, membranous nephropathy; SLE, systematic lupus erythematosus.
DISCUSSION
In the present study, we identified DEGs associated with DN from four independent datasets using RRA, which overcomes noise in different individual studies. A total of 95 overlapping DEGs, including 47 up‐ and 48 downregulated genes, were identified. The top 24 hub genes ranked based on the degree score by MCODE were selected for GO and KEGG enrichment analyses to investigate their molecular mechanisms. In the GO analysis, most hub genes were enriched in extracellular matrix organization (2.76E−10), complement activation (1.87E−07) and humoral immune response (4.01E−07). The C1QA, C1QB, VSIG4, C3 and CLU hub genes were found to be involved in both complement activation and humoral immune response, which is consistent with previous studies 22 , 23 , 24 . In KEGG pathway enrichment analysis, we found that most of the top 10 hub genes were linked to the complement and coagulation cascades, supporting the potential role of the complement system in the development of DN.
Five of the top 24 hub genes were involved in complement system function. The hub gene, CLU (the abbreviation of clusterin), a kind of complement regulatory protein, combines with C5b‐7 and hinders the production of the membrane attack complex 25 . Clusterin was upregulated in both the urine and glomeruli of DN patients 23 , 26 , and it could attenuate the development of renal fibrosis 27 . The B7 family‐related protein, VSIG4, acts as a complement receptor for C3 28 , but it remains unclear how VSIG4 interacts with the complement system to affect the course of DN. C3 not only plays a pivotal role in the classical/lectin complement pathway, but also in the alternative complement pathway as the central component in the complement cascade 29 .
Next, we investigated the renal histopathology of 151 patients with DN. C1q and C3 staining was routinely applied by direct immunofluorescence. We compared the clinical and histological characteristics according to whether there was C3 and C1q deposition in the glomeruli of patients with DN, and found that patients with both glomerular C3 and C1q deposition had lower serum albumin levels, higher 24‐h urinary protein excretion, and more severe renal pathologic lesions compared with those without glomerular C3 and C1q deposition. Sun et al. 7 found an association between C1q and C3c complement deposition on renal histopathology and more severe kidney damage in DN patients. These results emphasize the potential role of local complement activation in DN. Several studies have also shown the vital role of complement in the pathogenesis of DN 30 , 31 , 32 , 33 . For instance, Pelletier et al. 31 found that renal C1q was strongly associated with inflammation, fibrosis, proteinuria and the rate of renal function decline in patients with DN, and renal C1q deposition mainly in glomeruli hili and arterioles was correlated with the occurrence of DN 30 . Angeletti et al. 32 suggested that loss of C3 convertase regulator prompted podocyte injury and glomerulosclerosis. BTBR ob/ob mice, which show renal C3 deposition and increased expression of the C3a and C3a receptor, developed glomerular injury, podocyte loss and albuminuria 33 . Consistently, a wide range of inflammatory processes was confirmed in glomeruli from DN mice by single‐cell sequencing 34 . Complement activation is a primary pathogenic mechanism in numerous inflammatory disorders 35 .
Here, we found that the rate of kidney function decline was significantly higher in patients with both C3 and C1q deposition than in patients without either C3 or C1q deposition in the glomeruli, and that glomerular C1q+ C3+ deposition was an independent risk factor for renal survival. This suggests that complement activation plays a role in DN progression. The deposition of C3 in glomeruli indicates activation of the common complement pathway, and it is necessary to determine which complement pathway is activated in DN. Therefore, we examined the immunohistochemical expression of C1q (classical pathway), MBL (lectin pathway), C4d and C3 in glomeruli. C4d is a C4 cleavage product that binds covalently to the target tissue, and can arise from both the classical and MBL pathways. Those with high baseline C1q, C3 or C4d expression in glomeruli were more likely to progress to ESRD, whereas glomerular MBL was rare in the present cohort. These findings suggest that local classical complement pathway in glomeruli is involved in the progression of DN. Therefore, suppression of the complement system might be a novel therapy for DN 36 . Further study of the role of the complement system in DN might help to initiate new therapeutic approaches 37 .
The present study had several limitations. First, how the complement system plays a role in DN and its mechanism requires further research. Second, complement deposition in renal tubules and vessels was not evaluated, but it might also be involved in the development of DN. Further research is required to verify the present results and elucidate the precise mechanism of complement in the development and progression of DN.
In conclusion, local complement system activation in glomeruli is involved in DN, and C1q and C3 deposition in glomeruli might be associated with worse renal survival in patients with DN. A new method of regulating the complement system might lead to the development of new drugs to prevent or slow the progression of DN.
DISCLOSUE
The authors declare no conflict of interest.
Approval of the research protocol: The protocol for this study was approved by the ethics committee of the China‐Japan Friendship Hospital (approval number: 2018‐43‐K32).
Informed consent: Informed consent from patients for their medical data to be used in the clinical study were obtained.
Approval date of registry and the registration no. of the study/trial: N/A.
Animal studies: N/A.
Supporting information
Table S1 | The 47 upregulated differential expressed genes and 48 downregulated differentially expressed genes identified through integrating four transcriptomics microarray datasets.
Table S2 | Top 24 hub genes identified in protein–protein interaction network for differentially expressed genes
Figure S1 | Gene Ontology of upregulated differentially expressed genes (DEGs).
Figure S2 | Gene Ontology of down‐regulated differentially expressed genes (DEGs).
Figure S3 | PPI network of DEGs. DEG, differentially expressed genes; PPI, protein–protein interaction.
Figure S4 | Flowchart of screening clinical data for diabetic nephropathy.
ACKNOWLEDGMENTS
This work was supported by grants from Beijing Municipal Natural Science Foundation (7202179), National Natural Science Foundation of China (81870495) and National Key R&D Program of China (2016YFC1305500).
J Diabetes Investig. 2022; 13: 839–849
Contributor Information
Shimin Jiang, Email: minjiang101@163.com.
Wenge Li, Email: wenge_lee2002@126.com.
REFERENCES
- 1. GBD Chronic Kidney Disease Collaboration . Global, regional, and national burden of chronic kidney disease, 1990‐2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020; 395: 709–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sinclair A, Saeedi P, Kaundal A, et al. Diabetes and global ageing among 65‐99‐year‐old adults: findings from the International Diabetes Federation Diabetes Atlas, 9 edition. Diabetes Res Clin Pract 2020; 162: 108078. [DOI] [PubMed] [Google Scholar]
- 3. Jonatan B‐C, Frédéric J. Pathophysiologic mechanisms in diabetic kidney disease: a focus on current and future therapeutic targets. Diabetes Obes Metab 2020; 22: 16–31. [DOI] [PubMed] [Google Scholar]
- 4. Thomas MC, Brownlee M, Susztak K, et al. Diabetic kidney disease. Nat Rev Dis Primers 2015; 1: 15018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tang SCW, Yiu WH. Innate immunity in diabetic kidney disease. Nat Rev Nephrol 2020; 16: 206–222. [DOI] [PubMed] [Google Scholar]
- 6. Li L, Chen L, Zang J, et al. C3a and C5a receptor antagonists ameliorate endothelial‐myofibroblast transition via the Wnt/β‐catenin signaling pathway in diabetic kidney disease. Metabolism 2015; 64: 597–610. [DOI] [PubMed] [Google Scholar]
- 7. Sun Z‐J, Li X‐Q, Chang D‐Y, et al. Complement deposition on renal histopathology of patients with diabetic nephropathy. Diabetes Metab 2019; 45: 363–368. [DOI] [PubMed] [Google Scholar]
- 8. Vaisar T, Durbin‐Johnson B, Whitlock K, et al. Urine complement proteins and the risk of kidney disease progression and mortality in type 2 diabetes. Diabetes Care 2018; 41: 2361–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sircar M, Rosales IA, Selig MK, et al. Complement 7 is up‐regulated in human early diabetic kidney disease. Am J Pathol 2018; 188: 2147–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Flyvbjerg A. The role of the complement system in diabetic nephropathy. Nat Rev Nephrol 2017; 13: 311–318. [DOI] [PubMed] [Google Scholar]
- 11. Haring R, Wallaschofski H. Diving through the "‐omics": the case for deep phenotyping and systems epidemiology. OMICS 2012; 16: 231–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eddy S, Mariani LH, Kretzler M. Integrated multi‐omics approaches to improve classification of chronic kidney disease. Nat Rev Nephrol 2020; 16: 657‐668. [DOI] [PubMed] [Google Scholar]
- 13. Kolde R, Laur S, Adler P, et al. Robust rank aggregation for gene list integration and meta‐analysis. Bioinformatics 2012; 28: 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Woroniecka KI, Park ASD, Mohtat D, et al. Transcriptome analysis of human diabetic kidney disease. Diabetes 2011; 60: 2354–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grayson Peter C, Sean E, Taroni Jaclyn N, et al. Metabolic pathways and immunometabolism in rare kidney diseases. Ann Rheum Dis 2018; 77: 1226–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pan Y, Jiang S, Hou Q, et al. Dissection of glomerular transcriptional profile in patients with diabetic nephropathy: SRGAP2a protects podocyte structure and function. Diabetes 2018; 67: 717–730. [DOI] [PubMed] [Google Scholar]
- 17. Shved N, Warsow G, Eichinger F, et al. Transcriptome‐based network analysis reveals renal cell type‐specific dysregulation of hypoxia‐associated transcripts. Sci Rep 2017; 7: 8576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Franceschini A, Szklarczyk D, Frankild S, et al. STRING v9.1: protein‐protein interaction networks, with increased coverage and integration. Nucleic Acids Res 2013; 41: D808–D815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. American Diabetes Association . Standards of medical care in diabetes‐20212. Classification and diagnosis of diabetes. Diabetes Care 2021; 44: S15–S33. [DOI] [PubMed] [Google Scholar]
- 20. Thijs WCT, Mooyaart AL, Amann K, et al. Pathologic classification of diabetic nephropathy. J Am Soc Nephrol 2010; 21: 556–563. [DOI] [PubMed] [Google Scholar]
- 21. Inker LA, Schmid CH, Tighiouart H, et al. Estimating glomerular filtration rate from serum creatinine and cystatin C. N Engl J Med 2012; 367: 20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li X‐Q, Chang D‐Y, Chen M, et al. Complement activation in patients with diabetic nephropathy. Diabetes Metab 2019; 45: 248–253. [DOI] [PubMed] [Google Scholar]
- 23. Junling HE, Dijkstra Kyra L, Kim B, et al. Glomerular clusterin expression is increased in diabetic nephropathy and protects against oxidative stress‐induced apoptosis in podocytes. Sci Rep 2020; 10: 14888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhao L, Zhang Y, Liu F, et al. Urinary complement proteins and risk of end‐stage renal disease: quantitative urinary proteomics in patients with type 2 diabetes and biopsy‐proven diabetic nephropathy. J Endocrinol Investig 2021; 44: 2709–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yamada K, Hori Y, Hanafusa N, et al. Clusterin is up‐regulated in glomerular mesangial cells in complement‐mediated injury. Kidney Int 2001; 59: 137–146. [DOI] [PubMed] [Google Scholar]
- 26. Kim SS, Song SH, Kim JH, et al. Urine clusterin/apolipoprotein J is linked to tubular damage and renal outcomes in patients with type 2 diabetes mellitus. Clin Endocrinol 2017; 87: 156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jung G‐S, Kim M‐K, Jung Y‐A, et al. Clusterin attenuates the development of renal fibrosis. J Am Soc Nephrol 2012; 23: 73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vogt L, Schmitz N, Kurrer MO, et al. VSIG4, a B7 family‐related protein, is a negative regulator of T cell activation. J Clin Investig 2006; 116: 2817–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. West EE, Kolev M, Kemper C. Complement and the regulation of T cell responses. Annu Rev Immunol 2018; 36: 309–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bus P, Chua JS, Klessens CQF, et al. Complement activation in patients with diabetic nephropathy. Kidney Int Rep 2017; 3: 302–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pelletier K, Bonnefoy A, Chapdelaine H, et al. Clinical value of complement activation biomarkers in overt diabetic nephropathy. Kidney Int Rep 2019; 4: 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Angeletti A, Cantarelli C, Petrosyan A, et al. Loss of decay‐accelerating factor triggers podocyte injury and glomerulosclerosis. J Exp Med 2020; 217: e20191699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Morigi M, Perico L, Corna D, et al. C3a receptor blockade protects podocytes from injury in diabetic nephropathy. JCI Insight 2020; 5: e131849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fu J, Akat KM, Sun Z, et al. Single‐cell RNA profiling of glomerular cells shows dynamic changes in experimental diabetic kidney disease. J Am Soc Nephrol 2019; 30: 533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mastellos DC, Ricklin D, Lambris JD. Clinical promise of next‐generation complement therapeutics. Nat Rev Drug Discov 2019; 18: 707–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Komeno M, Pang X, Shimizu A, et al. Cardio‐ and reno‐protective effects of dipeptidyl peptidase III in diabetic mice. J Biol Chem 2021; 296: 100761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Budge K, Dellepiane S, Yu S‐W, et al. Complement, a therapeutic target in diabetic kidney disease. Front Med 2021; 7: 599236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 | The 47 upregulated differential expressed genes and 48 downregulated differentially expressed genes identified through integrating four transcriptomics microarray datasets.
Table S2 | Top 24 hub genes identified in protein–protein interaction network for differentially expressed genes
Figure S1 | Gene Ontology of upregulated differentially expressed genes (DEGs).
Figure S2 | Gene Ontology of down‐regulated differentially expressed genes (DEGs).
Figure S3 | PPI network of DEGs. DEG, differentially expressed genes; PPI, protein–protein interaction.
Figure S4 | Flowchart of screening clinical data for diabetic nephropathy.