

CONSPECTUS:
At the heart of synthetic chemistry is the holy grail of predictable catalyst design. In particular, researchers involved in reaction development in asymmetric catalysis have pursued a variety of strategies toward this goal. This is driven by both the pragmatic need to achieve high selectivities and the inability to readily identify why a certain catalyst is effective for a given reaction. While empiricism and intuition have dominated the field of asymmetric catalysis since its inception, enantioselectivity offers a mechanistically rich platform to interrogate catalyst-structure response patterns that explain the performance of a particular catalyst or substrate.
In the early stages of an asymmetric reaction development campaign, the overarching mechanism of the reaction, catalyst speciation, the turnover limiting step, and many other details are unknown or posited based on related reactions. Considering the unclear details leading to a successful reaction, initial enantioselectivity data are often used to intuitively guide the ultimate direction of optimization. However, if the conditions of the Curtin–Hammett principle are satisfied, then measured enantioselectivity can be directly connected to the ensemble of diastereomeric transition states (TSs) that lead to the enantiomeric products, and the associated free energy difference between competing TSs (ΔΔG‡ = −RT ln[(S)/(R)], where (S) and (R) represent the concentrations of the enantiomeric products). We, and others, speculated that this important piece of information can be leveraged to guide reaction optimization in a quantitative way.
Although traditional linear free energy relationships (LFERs), such as Hammett plots, have been used to illuminate important mechanistic features, we sought to develop data science derived tools to expand the power of LFERs in order to describe complex reactions frequently encountered in modern asymmetric catalysis. Specifically, we investigated whether enantioselectivity data from a reaction can be quantitatively connected to the attributes of reaction components, such as catalyst and substrate structural features, to harness data for asymmetric catalyst design.
In this context, we developed a workflow to relate computationally derived features of reaction components to enantioselectivity using data science tools. The mathematical representation of molecules can incorporate many aspects of a transformation, such as molecular features from substrate, product, catalyst, and proposed transition states. Statistical models relating these features to reaction outputs can be used for various tasks, such as performance prediction of untested molecules. Perhaps most importantly, statistical models can guide the generation of mechanistic hypotheses that are embedded within complex patterns of reaction responses. Overall, merging traditional physical organic experiments with statistical modeling techniques creates a feedback loop that enables both evaluation of multiple mechanistic hypotheses and future catalyst design. In this Account, we highlight the evolution and application of this approach in the context of a collaborative program based on chiral phosphoric acid catalysts (CPAs) in asymmetric catalysis.
Graphical Abstract

INTRODUCTION
Before entering our long-term collaboration, our teams (Sigman and Toste) had similar interests in conceptual aspects of asymmetric catalyst design and often applied physical organic tools in investigating mechanistic hypotheses. However, it was a serendipitous alignment of events that lead to our collaboration described in this Account. During discussions about early work in the area of chiral catalyst parametrization, Dr. Andrew Neel (a graduate student in the Toste group at the time) described his interest in optimizing and understanding a phase transfer chiral anion catalysis system under investigation. These systems are not readily studied by traditional physical organic tools, as the phase transfer events are often rate limiting and the nature of asymmetric induction is “kinetically silent”. The coalescence of shared interest provided a platform to investigate the combination of new data science tools (being developed by Dr. Anat Milo, a postdoc at the time in the Sigman lab) with traditional physical organic experiments to facilitate reaction development.1 At the outset of this collaboration, we sought to address the following questions: (i) Can one simultaneously optimize a reaction while gaining rapid insight into the mechanism? (ii) If so, how can one identify “mechanistic breaks” wherein a change in speciation or enantioselectivity determining events occurs as a response to a change in reaction conditions (substrate, additive, or catalyst)? (iii) Asymmetric induction often arises from differential noncovalent interactions (NCIs) in diastereomeric TSs. Can statistical and computational tools reveal the specific nature of attractive and repulsive NCIs in enantioselective catalysis?
To answer these questions, we investigated the use of a number of data science tools, including computational featurization of reaction components, linear regression modeling, statistical classifications, and data set design.4–6 Our approach emphasizes the mantra that all data are useful data because poor performing reactions are just as information-rich as those with excellent performance metrics.5,6 Importantly, these data science tools are related to (and supplemented with) the results of traditional physical organic experiments such as the study of nonlinear effects (NLEs)7,8 and kinetic isotope effects (KIEs).9 This integrated approach has furthered our understanding of asymmetric catalytic processes, thereby providing a platform for predictable catalyst design. In this Account, we aim to showcase the tools we developed and exemplify how our programs have traditional physical organic chemistry at their heart.
MECHANISTIC BREAKS
A change in mechanism, such as a difference in speciation or rate-determining step, can result from modest alterations to a reaction component. A classical experimental result that points to such a scenario is a break in a LFER, exemplified by two different regions of a Hammett plot (V-shaped).10–15 In contrast to the relatively trivial observation in a Hammett correlation, it is quite challenging to identify such a mechanistic break in multivariable linear regression (MLR), as poor correlations could result from insufficient data or descriptor space.16 To address this issue, our groups explored a data visualization strategy for the identification of unique responses from certain combinations of reaction components and found that statistical modeling of identified classes within a data set led to more precise and interpretable models.1 The following case studies demonstrate how this technique can be used.
In 2016, our groups reported the palladium-catalyzed enantioselective 1,1-diarylation of benzyl acrylates via chiral anion phase transfer catalysis (CAPT) using chiral phosphoric acid (CPA) derivatives (Figure 1A).17–21 In this reaction, the insoluble aryldiazonium salt undergoes salt metathesis with a chiral phosphate anion (PA) to form a soluble chiral ion pair. The aryldiazonium undergoes oxidative addition to a palladium catalyst, wherein the PA counterion remains associated with the metal and dictates the enantioselectivity of the reaction. During the study, 12 PAs and 18 acrylate substrates were evaluated to provide 145 data points for subsequent analysis.2 Graphical analysis of a subset of results revealed that the anthracenyl-substituted PA does not follow the same pattern as the remainder of the catalysts. Specifically, the observation of unique results with this PA and the 3,5-disubstituted aryl substrates indicated that the reaction appeared to be influenced by the substrate in this case, while the other reactions seemed to be mainly under catalyst control. Hence, the anthracenyl-substituted PA was evaluated separately and investigated by transition state calculations. The results indicated that this PA engages in a unique π-stacking interaction with the aryl group of the acrylate, thus clarifying its distinct response in the visualization of the data set.
Figure 1.

Data visualization highlights mechanistic breaks in (A) Pd-catalyzed enantioselective 1,1-diarylation of benzyl acrylates and (B) enantioselective fluorination of homoallylic alcohols. Continuous lines in the graphs are purely for visualization purposes.
In another example, a data set was designed for the study of the enantioselective fluorination of homoallylic alcohols via CAPT catalysis that included systematic variation of both the BINOL-derived PA catalyst and boronic acid (BA) directing group (Figure 1B).21 Similar reactivities were observed for the three PAs tested with the ortho- and para-substituted BAs. However, in the case of meta-substituted BAs, the isopropyl-substituted PA afforded the opposite enantiomer of the product compared to the other catalysts. This result was interpreted as a change in mechanism for these specific substrate/catalyst pairings; however, further mechanistic interrogation was required to identify the underlying cause of this break. One possibility is a change in the number of chiral catalysts involved in the enantio-determining step that is often ascertained using a traditional test of nonlinear effects (NLEs).8
NONLINEAR EFFECTS
Identifying NLEs within asymmetric catalysis, wherein the observed enantioselectivity of product does not directly correlate to the enantiopurity of catalyst, can be a powerful technique to increase mechanistic understanding (Figure 2).7,8 The observation of NLEs can be consistent with an autocatalytic process being at play23,24 or off-cycle reservoirs of homo- or heterochiral catalysts25 or, more commonly, more than one catalyst molecule being involved in a mechanistic step that influences enantioselection.8,26 In the course of a mechanistic study of the aforementioned diarylation of benzyl acrylates, it was found that while the reaction is zeroth-order in CPA, suggesting that the CPA is likely not involved in the rate-determining step of the reaction,2,17 increasing its concentration nevertheless afforded increasing amounts of product 2 relative to the Heck byproduct 3 (Figure 2A). This observation suggests that the formation of 3 results from a more complex kinetic scenario in competition with the transmetalation step. Experiments employing CPA catalysts with various ee’s revealed that an NLE was observed only for the product/Heck product ratio (2/3) and not for the product enantioselectivity. There are two reasonable proposals that explain this result: (a) the PA activates the boronic acid toward transmetalation to form 2 or (b) the PA deprotonates palladium hydride to form 3. The NLE experiments alone cannot distinguish between these two pathways, but an experiment was designed wherein the ratio between the PA and the Pd(0) catalyst was varied, which should have an effect on the 2/3 ratio. For the deprotonation pathway, increasing the amount of PA (decreasing the Pd/PA ratio) should afford greater amounts of 3 (lower 2/3 ratio). In the transmetalation pathway, increasing the amount of PA (higher 2/3 ratio) should correspond to greater 2 formation as the presence of PA would convert the boronic acid to a boronic ester for efficient transmetalation. A direct relationship between the 2/3 ratio and Pd/PA ratio was observed; that is, decreasing amounts of PA (high Pd/PA product ratio) lead to greater amounts of 2 (high 2/3 ratio) whereas increasing the concentration of PA (low Pd/PA product ratio) favored the formation of the 3 (low 2/3 ratio). This normal relationship between the ratios suggests that the deprotonation pathway is dominant. In this case, multiple catalysts are involved in determining the product distribution but not in the enantio-determining step.
Figure 2.
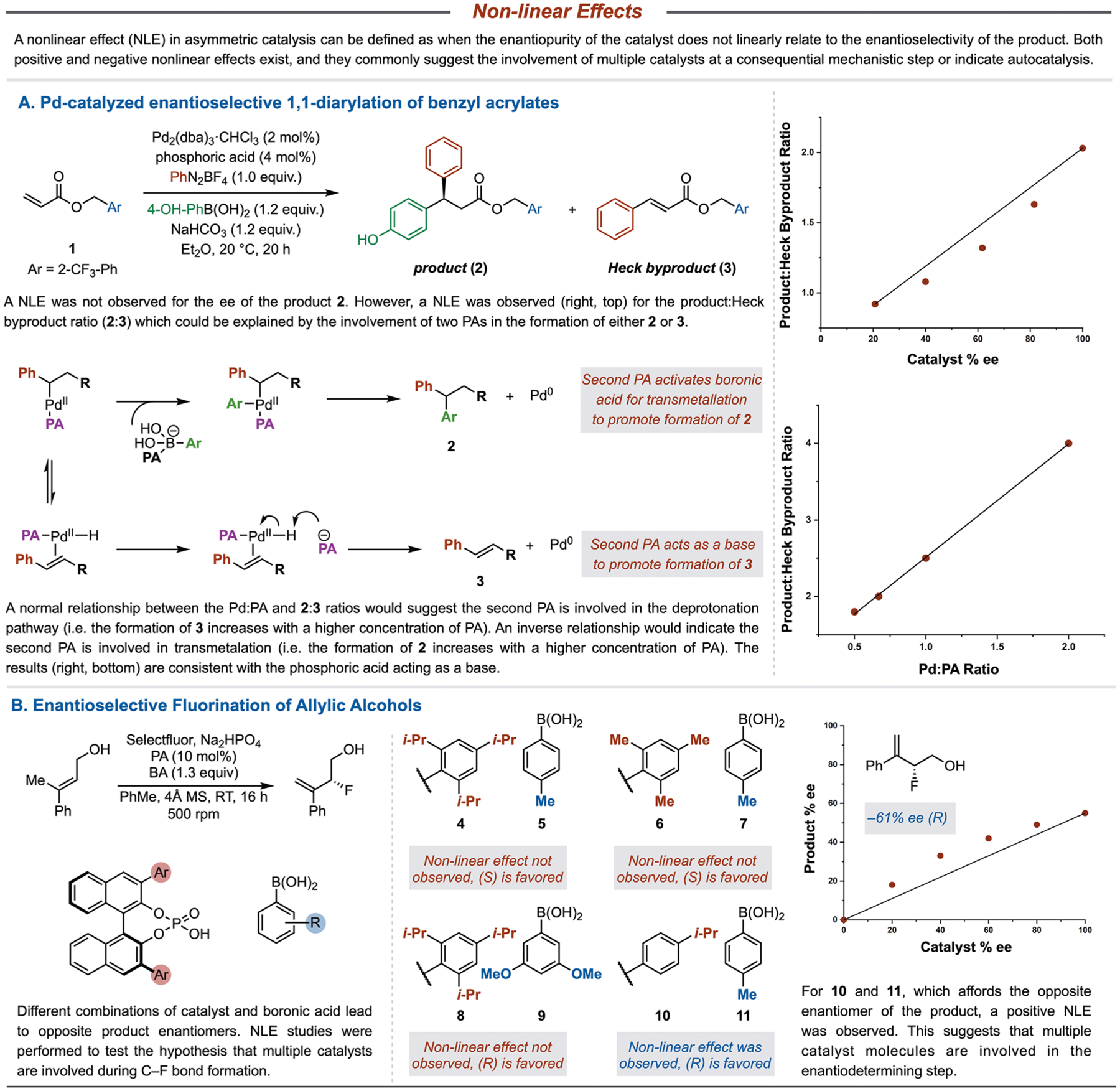
Nonlinear effect experiments, especially in conjunction with statistical modeling, provide important insight into the general structure of key transition states.
In the course of exploring an enantioselective fluorination of allylic alcohols through CAPT catalysis,22 divergent selectivity patterns were observed wherein different combinations of substrates, boronic acids, and catalysts led to opposite enantiomers (Figure 2B). Because of the doubly cationic nature of Selectfluor, a NLE study was initiated to investigate whether the involvement of multiple catalysts led to the divergent enantioselectivity response.7,8 For three different combinations (4 and 5, 6 and 7, 8 and 9), a NLE was not observed, suggesting that a single catalyst molecule is involved in the enantio-determining step.22 However, a NLE was observed with the combination of 10 and 11 that afforded the opposite enantiomer, likely because the lack of the ortho substituents allows for multiple catalysts to be in close proximity to one another. Intriguingly, although the combination of 8 and 9 also afforded the opposite enantiomer to the other combinations, a NLE was not observed, suggesting that there is a different underlying mechanistic reason for a large change in the geometry of the enantio-determining transition state. Hence, isotopic labeling experiments were employed to investigate the specific role of key hydrogen atoms in the enantio-divergent process.
KINETIC ISOTOPE EFFECTS
A KIE is a change in reaction rate due to the incorporation of an isotope, most commonly deuterium, into a reactant.9,27–29 In enantioselective reactions, an enantiomeric excess (ee) value provides a direct readout of the relative rates of the reactions leading to the two enantiomers. The study of KIEs can identify which atoms are involved in the rate- or product-determining step of a reaction, and variations in KIEs can signify changes in mechanism.30–32 During the development of the enantioselective fluorination of allylic alcohols (Figure 3), opposite selectivity was observed with the 3,5-(OMe)2-BA directing group (9, −77% ee) compared to BAs substituted in the 4-position (e.g., 65% ee with 4-Me-BA 11).22 As described above, the absence of a NLE suggested that the divergent selectivity was not due to a change in catalyst molecularity. The investigation of KIEs was employed to gain greater insight into the nature of the enantio-determining step (EDS). A deuterated analogue of the starting material, 12-d, was prepared and submitted to the standard reaction conditions. A significant KIE was observed with 9 (−77% eeH vs −90% eeD, 0.5 kcal/mol difference) but not with 11 (65% eeH vs 63% eeD). These results suggest a change in mechanism, with the C–H bond of the substrate only involved in the EDS with 9. This was ultimately rationalized through a concerted process with 9 compared to a stepwise process with 11.
Figure 3.

Kinetic isotope effects provide insight into key bond breaking steps in the enantioselective fluorination of allylic alcohols.
EARLY INVESTIGATION OF NONCOVALENT INTERACTIONS
In 2015, our teams investigated a chiral triazole-phosphoric acid catalyzed intramolecular dehydrogenative C–N coupling, a reaction in which little was known about the origin of selectivity (Figure 4).1,33 The aim of the investigation was to utilize a data-driven approach, in combination with classical physical organic techniques, for mechanistic elucidation. Data-driven analysis necessitated the evaluation of a diverse library of substrates and catalysts. Hence, each reaction component was strategically modified at positions hypothesized to influence enantioselectivity: the benzyl and the distal aryl rings for the substrates and the aryl ring attached to the triazole for the catalyst. The specific choice of substituents was determined by synthetic accessibility and a consideration of electronic (Hammet σpara) and steric (Sterimol B1) descriptor values. The combinations of the resulting 11 catalysts and 12 substrates were evaluated in the reaction, providing a wide range of enantioselectivity values. The data were plotted to enable visualization of selectivity trends, wherein three distinct regions appeared based on the substituents of the catalyst (Figure 4A). Linear regression was applied to the three individual subsets, and the resulting correlations with steric and electronic parameters were interpreted to suggest a π-interaction between the catalyst and substrate. At this point, KIE experiments were performed to establish the EDS (Figure 4B). The observation of a KIE with the enantiomeric catalysts and a stereo-defined deuterated substrate 14-d indicated that the chiral phosphoric acid is involved in the rate-determining oxidation. Although the enantioselectivity of the product 15 is formally set during the cyclization, it is conceivable that selectivity could arise from preorganization of a catalyst–substrate intermediate during oxidation. If this were the case, then different enantioselectivities would be expected from the reaction of stereo-defined substrate 14-d with (S)- and (R)-catalysts. However, the observation of opposite but equal product ee’s obtained with opposite enantiomer of CPA catalysts is consistent with an enantio-determining cyclization.
Figure 4.
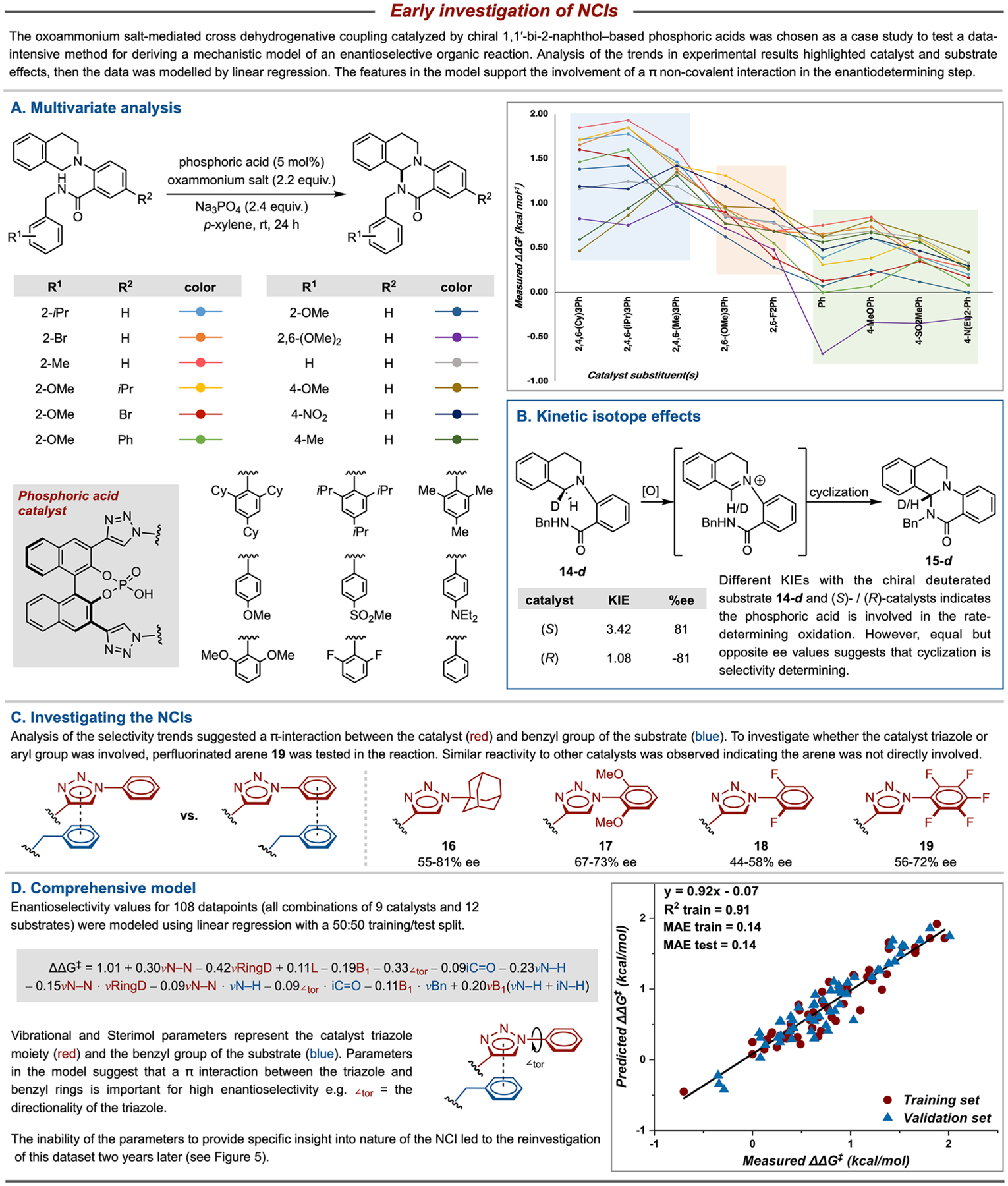
Early work seeking to understand the role of NCIs within CAPT catalysis.
Following the KIE studies, a series of catalysts were tested in the reaction to investigate whether the triazole or the aryl substituent of the catalyst was involved in the π-interaction with the substrate (Figure 4C). NCIs are often strongly affected by the charge distribution of an arene, so the observation of relatively similar results with the perfluorinated arene 19 compared to the other catalysts (16–18) was interpreted to suggest that it was the triazole that was involved in the NCI.34 Energy stabilization gained from a π-interaction is also affected by the distance and geometry of the rings. This was reflected in a comprehensive model of the results that included a term for the torsion angle of the catalyst arene (which influences the geometry of the triazole ring), along with a variety of steric and electronic terms (Figure 4D). Several new catalysts were synthesized based on model predictions, resulting in extrapolation to the highest overall selectivities for the reaction. However, the model lacked simplicity due to the large number of variables, thereby precluding the development of more detailed mechanistic hypotheses.35 Overall, the true nature of the putative NCIs at the heart of asymmetric induction remained unclear, and this unsatisfactory understanding prompted the development of superior molecular features for NCIs.
DEVELOPMENT OF DESCRIPTORS FOR NONCOVALENT INTERACTIONS
It is challenging to identify and quantify the role of NCIs through purely empirical means, as the individual contribution of each of these stabilizing interactions is quite small (<2 kcal/mol) and NCIs can be highly dynamic.36 As shown above (Figure 4C), the presence of NCIs can be probed through modification of catalyst structure, wherein the introduction of functional groups should modulate the strength of the hypothesized NCI.37,38 However, the representation of NCIs within the context of statistical modeling remained unclear.
Inspired by the work of Wheeler and Houk that correlates interaction energies of stacked π systems to Hammett values,39 we investigated the application of interaction energies (Eπ) and distances (Dπ) as mechanistically driven molecular descriptors for NCIs (Figure 5A).2 The incorporation of these descriptors in a statistically validated MLR model would suggest the presence of a NCI in the reaction(s) under investigation. A NCI between substrate arenes and catalyst triazolyl substituents was hypothesized to be a key stereo-controlling element an oxidative amination.40 Therefore, the reaction data were reanalyzed using Eπ and Dπ parameters to gain further insight into this reaction. There are three potential rings (A, B, and C, Figure 5B) in the oxidized substrate intermediate 21 that could engage in a NCI with the CPA triazole motif. Computational analysis of each of these possible interactions was undertaken. Descriptors relevant to the substrate were extracted from the corresponding uncatalyzed transition states (TSs), which were hypothesized to resemble their catalyzed variants. This approach provided a platform in which highly relevant descriptors could be used. Two major conformations exist for the uncatalyzed TSs, which primarily differ through the orientation of the benzyl substituent (TS A and TS B, Figure 5B). As NCIs are highly distance and orientation dependent, the ability of the substrate to access these conformations is likely critical. Therefore, a parameter describing the difference in energy between these two conformers of the uncatalyzed TS was computed for each substrate. Again, considering the key possible orientations of the catalyst and substrate, electrostatic potential maps (ESPs) can be used to match areas of low and high electron density.41–43 These ESPs highlight that the triazole region of the catalyst is an area of high electron density, which, based on measured distances, would match well with the electron-deficient iminium if the nucleophilic amide N is engaged in hydrogen bonding with the CPA.40
Figure 5.
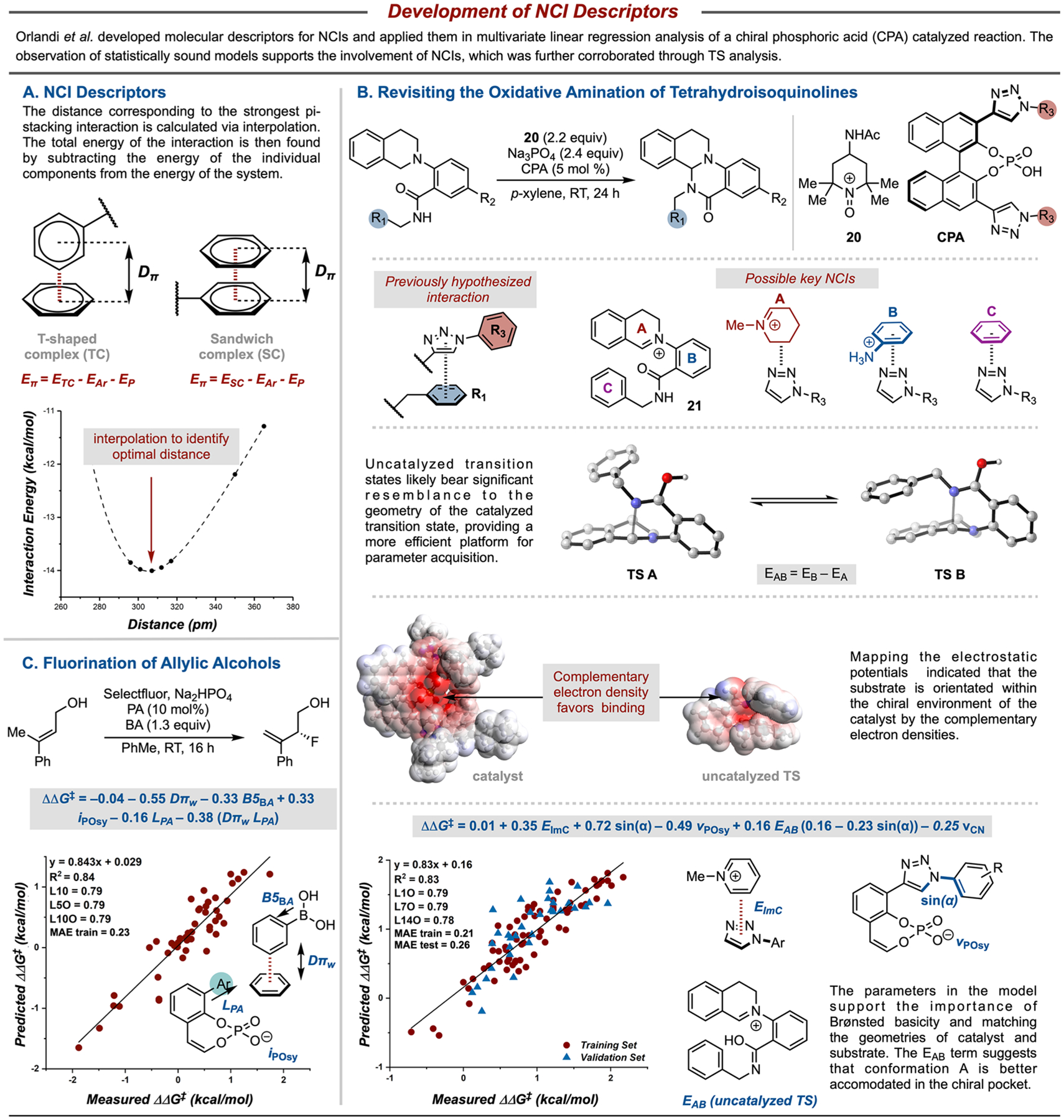
Development of specific NCI descriptors enabled the evaluation of numerous mechanistic hypotheses.
Finally, a 6-parameter global model describing 103 catalyst and substrate combinations was developed (Figure 5B, bottom), a significant improvement to the previously developed 13-term model.44,45 The model highlights the roles of Brønsted basicity (vPOSy) and NCIs (EImC), and the importance of substrate conformation (EAB). The remaining terms suggest the geometrical importance of the orientation of the aryl rings (sin(α)) and the substrate nucleophilicity (vCN).
Further analysis of the previously reported allylic fluorination reaction incorporating these developed NCI descriptors also emphasizes the role of attractive NCIs in enantio-determining transition states and the effectiveness of these descriptors at highlighting these weak, additive interactions (Figure 5C).2,21 These models illustrate how statistical modeling can be enhanced by incorporating mechanistic hypotheses into descriptor development, thereby providing greater insight into the transformation. Moreover, the combination of transition state analysis, statistical modeling, and traditional physical organic tools can maximize mechanistic insight.
THE COMBINATION OF STATISTICAL MODELING AND TRANSITION STATE ANALYSIS
The advancement of density functional theory (DFT) in recent years has led to the general feasibility of studying catalytic reactions computationally.46–49 This approach has provided mechanistic insights into a wide variety of reactions. However, the computational cost of TS analysis generally prohibits its application to the full scope of a reaction, so a model catalyst/substrate is often used. Hence, this approach is complementary to statistical modeling, which can easily take advantage of the entire reaction data set by utilizing ground state structures for descriptor acquisition.1,4,6
In 2020, our groups reported the first highly enantioselective allenoate-Claisen rearrangement using doubly axially chiral phosphate (DAP) sodium salts as catalysts (Figure 6A).3 A chiral Lewis acid phosphate counterion coordinates to the allenoate creating a large adaptable chiral pocket in which NCIs were hypothesized to play a key role. However, the multitude of weak interactions and flexibility of the system made this reaction difficult to study computationally. Hence, computational analysis of the uncatalyzed and sodium cation-catalyzed reactions was first performed to identify the EDS, which was found to be the [3,3]-sigmatropic rearrangement. This step was then studied computationally using the full phosphate catalyst (Figure 6B). The Boltzmann-weighted average of 14 TSs for the rearrangement resulted in a computed ee of 77%, comparable to the experimental value of 66% ee. The sodium cation assembled the TSs leading to the major and minor enantiomers of the product through several NCIs. Furthermore, in the major TS two edge-to-face interactions were observed between the catalyst arene (Ar2, green), substrate arene (Ar1, red), and catalyst naphthyl (yellow). In contrast, a staggered sandwich arene–arene interaction was observed the between catalyst arene and substrate arene in the minor TS. While the TS analysis was performed using a single catalyst–substrate combination, the allyl amine component was varied significantly to afford a range of β-amino γ,δ-disubstituted esters (Figure 6C). MLR models were developed using the previously described descriptors (Figure 5) in order to test the hypothesis that changes in the structure of the substrate and catalyst dictate the enantioselectivity through modulation of NCIs. Based on the TS analysis, interaction energies and distances were calculated for the edge-to-face and sandwich complexes found in the major and minor TSs, respectively (see molecular renderings, Figure 6C). MLR of the selectivities from 24 reactions afforded a model with reasonable statistics (R2 = 0.77, test R2 = 0.54), supporting the NCI-driven mechanistic hypothesis. The introduction of further descriptors that capture other elements of the catalyst variation resulted in improved MLR model (R2 = 0.87, test R2 = 0.76). Overall, this computationally driven analysis highlights the role of NCIs for these flexible DAP catalysts.
Figure 6.
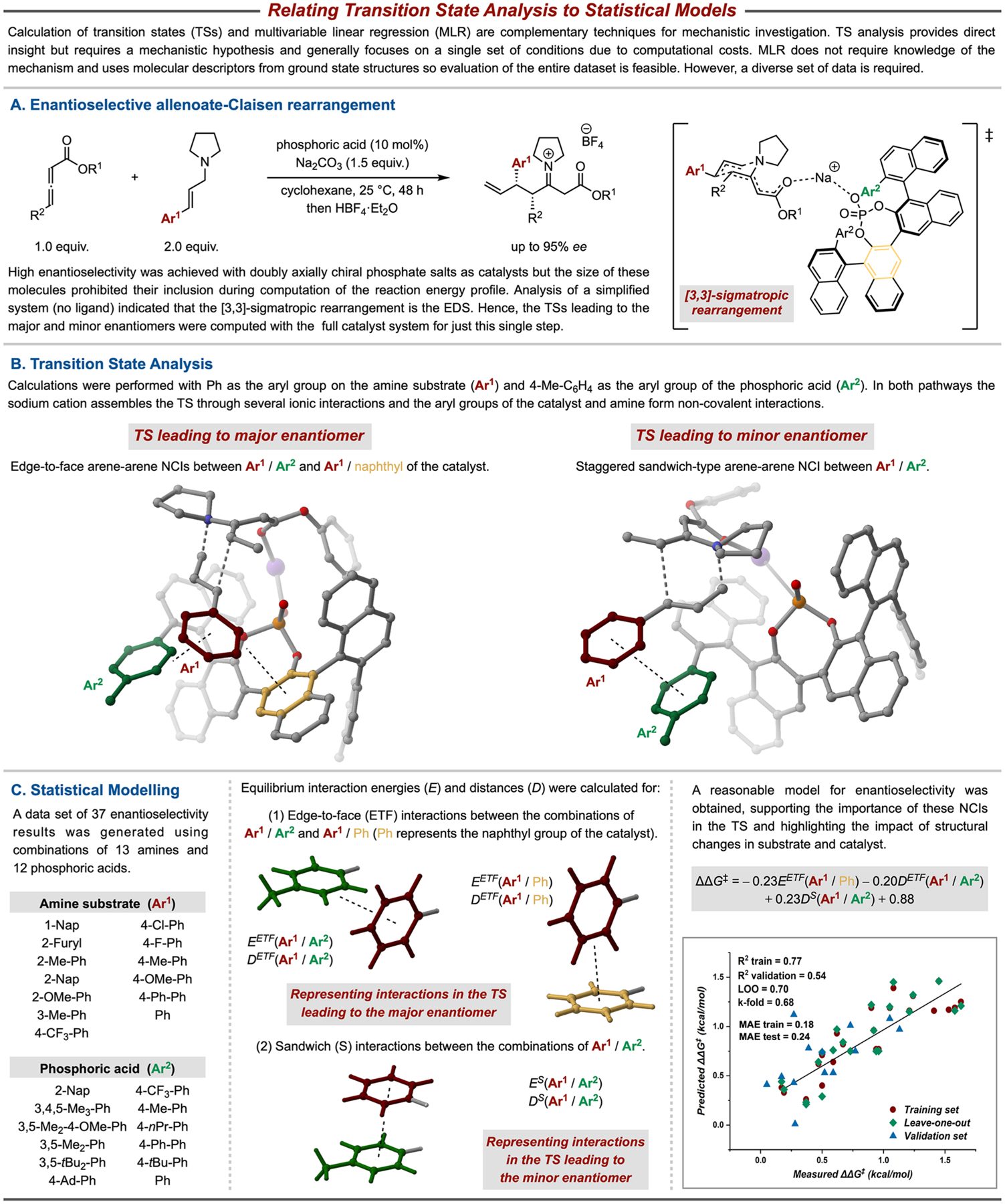
Statistical modeling and transition state analysis are complementary techniques for mechanistic interrogation.
CONFORMATIONAL DYNAMICS IN CATALYSIS
Although rigidifying elements are generally incorporated within the small molecule catalysts that are used for enantioselective methods in order to increase selectivity, flexible catalysts, like the DAP catalysts described above, may provide unique opportunities to maximize stabilizing NCIs throughout the catalytic cycle and perhaps provide greater substrate generality.50 For example, tetrapeptidic catalysts 22, pioneered by the Miller group, can adopt multiple conformations and have been shown to be highly effective for a variety of transformations, including those traditionally catalyzed by BINOL-derived CPAs 23.51–55 Various mechanistic tools, driven by data science, were employed to directly compare these two disparate catalyst scaffolds in the study of an atroposelective cyclodehydration (Figure 7A).56 It was hoped that further insight into the key features of privileged catalysts could be gained through this comparison, which may enable predictable catalyst design.57
Figure 7.
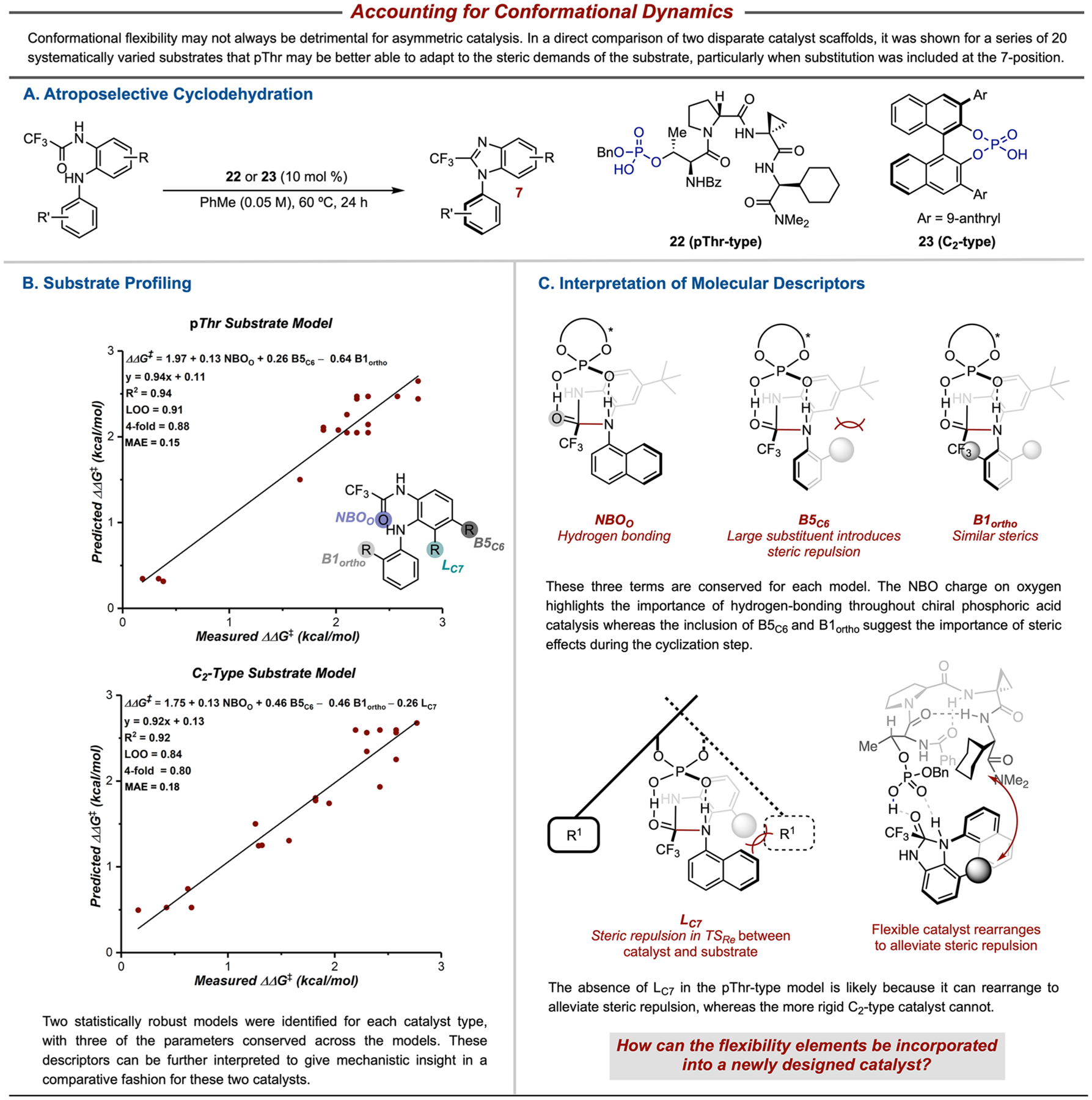
Direct comparison of two disparate catalyst scaffolds, one rigid and one flexible, through a substrate profiling technique alludes to how flexibility may impart generality. Adapted with permission from ref 56. Copyright 2019 American Chemical Society.
One particularly powerful tool that enables direct comparison is using a substrate profiling technique, wherein each substrate is tested with the optimal catalyst from each catalyst type. In this case, 20 diverse substrates were tested with the two catalysts, and the results were used for the development of comparative MLR models. Although both catalysts performed similarly for many of the substrates tested, there is a notable difference between the two when incorporating large, bulky substituents at the 7-position. The peptidic catalyst 22 leads to high enantiomeric excesses for this series whereas the more rigid BINOL-derived catalyst 23 results in more moderate enantioselectivity.
The terms of the two MLR models were analyzed in order to gain greater insight into these results (Figure 7B). Interpretation and comparison of the descriptors used in the models can give insight into key catalyst–substrate interactions, providing a consistent method to simultaneously evaluate a flexible and a rigid catalyst. Importantly, three terms were conserved across both catalyst classes: the NBO charge of the carbonyl oxygen (NBOO), the B5 value of the substituent at the 6-position (B5C6), and the B1 value of the ortho substituents of the bottom aryl ring (B1ortho). These descriptors highlight the general importance of hydrogen bonding in CPA catalysis in addition to implicating steric effects during the enantio-determining cyclization step. The only nonconserved term is the length of the substituent at the 7-position (LC7). This suggests that the more flexible peptide catalyst 22 may be able to rearrange to adapt to the steric demands of the substrate whereas the more rigid BINOL scaffold 23 cannot.
The high atroposelectivity observed with both disparate catalyst scaffolds supports the hypothesis that flexibility may not be inherently detrimental within asymmetric catalysis. However, further investigation into the extent to which flexibility is beneficial is required. Hence, our teams are exploring the incorporation of flexibility as a design element such that multiple stabilizing NCIs can be accessed that adapt to a variety of intermediates and transition states throughout a catalytic cycle.
CONCLUSION AND OUTLOOK
Through our collaborative efforts, we have demonstrated how a strategy relying on the intersection between data science and traditional physical organic chemistry has enabled optimization of particular reactions while simultaneously providing mechanistic insights. In considering the future of this overarching strategy, we are enthusiastic that only the surface of this field has been investigated. There are a number of exciting questions one can consider, especially in the area of asymmetric catalysis. First, considering the nature and widespread use of privileged catalysts, can modern physical organic tools be used to generate a holistic understanding of underlying structural features that enable selective asymmetric catalysis? We have begun to address these questions in the context of chiral phosphoric acid catalysis,58 hydrogen bond donating catalysts,59 and bisoxazoline ligands.60 However, a diverse range of privileged catalyst scaffolds remain unexplored.
Second, how will the incorporation of data science reshape a chemist’s approach to the development of new synthetic methods? For instance, a change in perspective is required wherein no data are wasted and “negative” results are viewed as just as valuable as “positive” ones. This mindset enables the identification of subtle trends within the data, even when a particular result may be unexpected from a chemical intuition standpoint, which can guide further screening, hypothesis development, and future optimization campaigns. The expansion and distribution of databases of physical organic features will help to increase the accessibility of the data science workflow to chemists in a variety of fields.61,62 It should be noted that the incorporation of data science principles to project design goes hand-in-hand with modern advances in automation that streamline the data collection process.63,64
Finally, what might the future of our laboratories look like if we fully embrace this philosophy? Through the integration of data science and physical organic, synthetic, and computational chemistry, each experiment becomes a physical organic experiment. All data can be analyzed and contribute to a greater understanding of the reactions under investigation. More importantly, this understanding can be transferred and compared in the development of new processes. Thus, we embrace this strategy and pedagogical restructuring required to integrate the computer science and chemistry disciplines. This multidisciplinary approach is integral to identifying and understanding the key connections and patterns hidden within the data and accelerating our fundamental understanding of chemical reactions and reactivity.
ACKNOWLEDGMENTS
M.S.S. and F.D.T. have had the opportunity to work with many inspirational, talented, and motivated co-workers during this collaborative effort. We especially thank the two individuals, Anat Milo and Andrew Neel, who initiated the program and established an integrated and stimulating blueprint for collaborative work between our two laboratories. We also thank those who followed and moved the program in the many exciting new directions: Souvagya Biswas, Alec Christian, Jaime Coelho, Jennifer Crawford, Tobias Gensch, Samantha Gross-light, Margaret Hilton, Mingyou Hu, Junqi Li, Javier Miro, Zach Niemeyer, Manuel Orlandi, Suresh Pindi, Jolene Reid, Chris Sandford, Richard Thornbury, Cheng-Che Tsai, and Eiji Yamamoto. We also thank our wonderful colleague, Prof. Scott Miller, who has made exceptional contributions to this program. Finally, M.S.S. thanks the NIH (1R35GM136271-01) for their continued support of this research. F.D.T. acknowledges the NIH (1R35GM118190) for their continued support of this research.
ABBREVIATIONS
- BA
boronic acid
- BINOL
1,1′-bi-2-naphthol
- CAPT
chiral anion phase transfer
- CPA
chiral phosphoric acid
- DAP
doubly axially chiral phosphate
- DFT
density functional theory
- EDS
enantio-determining step
- ESP
electrostatic potential map
- KIE
kinetic isotope effect
- LFER
linear free energy relationships
- MLR
multivariable linear regression
- NBO
natural bond orbital
- NCI
noncovalent interaction
- NLE
nonlinear effect
- PA
phosphate anion
- TS
transition state
Biographies
Jennifer M. Crawford received her B. A. in Chemistry and Mathematics from St. Olaf College in 2016 before beginning her Ph.D. studies with Professor Matthew Sigman at the University of Utah. Her dissertation work focused on understanding conformationally flexible organocatalysts through the application of statistical modeling tools. Currently, she is a process chemist at GlaxoSmithKline.
Cian Kingston received his B.Sc. in Medicinal Chemistry and Ph.D. with Professor Pat Guiry from University College Dublin in 2013 and 2017, respectively. Thereafter, he pursued postdoctoral studies with Professor Phil Baran at Scripps Research and Professor Matthew Sigman at the University of Utah. His current research focuses on the application of data science tools in the study of reaction mechanisms.
F. Dean Toste received his B.Sc. and M.Sc. from the University of Toronto and completed his Ph.D. studies at Stanford University under the guidance Professor Barry Trost. After a postdoctoral appointment with Professor Robert Grubbs at the California Institute of Technology, he took an Assistant Professorship at the University of California, Berkeley, in 2002. In 2006, he was promoted to Associate Professor and is currently Gerald E. K. Branch Distinguished Professor of Chemistry.
Matthew S. Sigman received a B.S. in chemistry from Sonoma State University in 1992 before obtaining his Ph.D. at Washington State University with Bruce Eaton in 1996. He then moved to Harvard University for postdoctoral work with Eric Jacobsen. In 1999, he joined the faculty of the University of Utah, where his research group has focused on the development of new synthetic methodologies with an underlying interest in reaction mechanism.
Footnotes
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.accounts.1c00285
Contributor Information
Jennifer M. Crawford, Department of Chemistry, University of Utah, Salt Lake City, Utah 84112, United States; Present Address: Chemical Development, GlaxoSmithKline, 1250 S. Collegeville Rd., Collegeville, PA, USA.
Cian Kingston, Department of Chemistry, University of Utah, Salt Lake City, Utah 84112, United States;.
F. Dean Toste, Department of Chemistry, University of California, Berkeley, California 94720, United States;.
Matthew S. Sigman, Department of Chemistry, University of Utah, Salt Lake City, Utah 84112, United States;.
REFERENCES
- (1).Milo A; Neel AJ; Toste FD; Sigman MS A data-intensive approach to mechanistic elucidation applied to chiral anion catalysis. Science 2015, 347, 737–43. [DOI] [PMC free article] [PubMed] [Google Scholar]; The application of data visualization and statistical modeling tools in conjunction with traditional physical organic experiments supports proposed noncovalent interactions (NCIs) between substrate and chiral phosphoric acid catalyst in chiral anion phase transfer (CAPT) catalysis.
- (2).Orlandi M; Coelho JAS; Hilton MJ; Toste FD; Sigman MS Parametrization of Non-covalent Interactions for Transition State Interrogation Applied to Asymmetric Catalysis. J. Am. Chem. Soc 2017, 139, 6803–6806. [DOI] [PMC free article] [PubMed] [Google Scholar]; The development and application of computed interaction energies and distances as descriptors for NCIs.
- (3).Miro J; Gensch T; Ellwart M; Han SJ; Lin HH; Sigman MS; Toste FD Enantioselective Allenoate-Claisen Rearrangement Using Chiral Phosphate Catalysts. J. Am. Chem. Soc 2020, 142, 6390–6399. [DOI] [PMC free article] [PubMed] [Google Scholar]; Statistical modeling tools, NCI analysis, transition state analysis, and physical organic experiments are used to investigate NCIs within an enantioselective allenoate-Claisen rearrangement catalyzed by doubly axially chiral phosphoric acids.
- (4).Reid JP; Sigman MS Comparing quantitative prediction methods for the discovery of small-molecule chiral catalysts. Nat. Rev. Chem 2018, 2, 290–305. [Google Scholar]
- (5).Sigman MS; Harper KC; Bess EN; Milo A The Development of Multidimensional Analysis Tools for Asymmetric Catalysis and Beyond. Acc. Chem. Res 2016, 49, 1292–301. [DOI] [PubMed] [Google Scholar]
- (6).Santiago CB; Guo J-Y; Sigman MS Predictive and mechanistic multivariate linear regression models for reaction development. Chem. Sci 2018, 9, 2398–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Satyanarayana T; Abraham S; Kagan HB Nonlinear Effects in Asymmetric Catalysis. Angew. Chem., Int. Ed 2009, 48, 456–94. [DOI] [PubMed] [Google Scholar]
- (8).Blackmond DG Kinetic Aspects of Nonlinear Effects in Asymmetric Catalysis. Acc. Chem. Res 2000, 33, 402–11. [DOI] [PubMed] [Google Scholar]
- (9).Simmons EM; Hartwig JF On the Interpretation of Deuterium Kinetic Isotope effects in C-H Bond Functionalizations by Transition-Metal Complexes. Angew. Chem., Int. Ed 2012, 51, 3066–72. [DOI] [PubMed] [Google Scholar]
- (10).Hansch C; Leo A; Taft RW A Survey of Hammett Substituent constants and Resonance and Field Parameters. Chem. Rev 1991, 91, 165–195. [Google Scholar]
- (11).Fuchs R; Carlton DM Substituent Effects in the Solvolysis and Thiosulfate Reactions of 3-, 4-and 3, 5-Substituted α-Chlorotoluenes. J. Am. Chem. Soc 1963, 85, 104–107. [Google Scholar]
- (12).Buckley N; Oppenheimer NJ Reactions of Charged Substrates. 5. The Solvolysis and Sodium Azide Substitution Reactions of Benzylpyridinium Ions in Deuterium Oxide. J. Org. Chem 1996, 61, 7360–7372. [DOI] [PubMed] [Google Scholar]
- (13).Um I-H; Han H-J; Ahn J-A; Kang S; Buncel E Reinterpretation of Curved Hammett Plots in Reaction of Nucleophiles with Aryl Benzoates: Change in Rate-Determining Step or Mechanism versus Ground-State Stabilization. J. Org. Chem 2002, 67, 8475–8480. [DOI] [PubMed] [Google Scholar]
- (14).Sandford C; Fries LR; Ball TE; Minteer SD; Sigman MS Mechanistic Studies into the Oxidative Addition of Co(I) Complexes: Combining Electroanalytical Techniques with Parameterization. J. Am. Chem. Soc 2019, 141, 18877–18889. [DOI] [PubMed] [Google Scholar]
- (15).It is important to note that the term “mechanistic break” is difficult to define because every pair of molecules reacts through a unique mechanism. In this review, a break signifies a distinct change to the “thin” mechanism of the reaction that results in a change in reaction output, see:; Nieves-Quinones Y; Singleton DA. Dynamics and the Regiochemistry of Nitration of Toluene. J. Am. Chem. Soc 2016, 138, 15167–15176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).For further discussion on how insufficient data or descriptor space can affect a poor correlation, see:; Bess EN; Bischoff AJ; Sigman MS. Designer substrate library for quantitative, predictive modeling of reaction performance. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 14698–14703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Yamamoto E; Hilton MJ; Orlandi M; Saini V; Toste FD; Sigman MS Development and Analysis of a Pd(0)-Catalyzed Enantioselective 1,1-Diarylation of Acrylates Enabled by Chiral Anion Phase Transfer. J. Am. Chem. Soc 2016, 138, 15877–15880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Maji R; Mallojjala SC; Wheeler SE Chiral phosphoric acid catalysis: from numbers to insights. Chem. Soc. Rev 2018, 47, 1142–1158. [DOI] [PubMed] [Google Scholar]
- (19).Parmar D; Sugiono E; Raja S; Rueping M Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates. Chem. Rev 2014, 114, 9047–9153. [DOI] [PubMed] [Google Scholar]
- (20).Parmar D; Sugiono E; Raja S; Rueping M Addition and Correction to Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates. Chem. Rev 2017, 117, 10608–10620. [DOI] [PubMed] [Google Scholar]
- (21).Phipps RJ; Hamilton GL; Toste FD The progression of chiral anions from concepts to applications in asymmetric catalysis. Nat. Chem 2012, 4, 603–14. [DOI] [PubMed] [Google Scholar]
- (22).Neel AJ; Milo A; Sigman MS; Toste FD Enantiodivergent Fluorination of Allylic Alcohols: Data Set Design Reveals Structural Interplay between Achiral Directing Group and Chiral Anion. J. Am. Chem. Soc 2016, 138, 3863–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Bryliakov KP Dynamic Nonlinear Effects in Asymmetric Catalysis. ACS Catal. 2019, 9, 5418–5438. [Google Scholar]
- (24).Athavale SV; Simon A; Houk KN; Denmark SE Demystifying the asymmetry-amplifying, autocatalytic behaviour of the Soai reaction through structural, mechanistic and computational studies. Nat. Chem 2020, 12, 412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Tsukamoto M; Gopalaiah K; Kagan HB Equilibrium of homochiral oligomerization of a mixture of enantiomers. Its relevance to nonlinear effects in asymmetric catalysis. J. Phys. Chem. B 2008, 112, 15361–15368. [DOI] [PubMed] [Google Scholar]
- (26).Buono F; Walsh PJ; Blackmond DG Rationalization of Anomalous Nonlinear Effects in the Alkylation of Substituted Benzaldehydes. J. Am. Chem. Soc 2002, 124, 13652–13653. [DOI] [PubMed] [Google Scholar]
- (27).Gómez-Gallego M; Sierra MA Kinetic Isotope Effects in the Study of Organometallic Reaction Mechanisms. Chem. Rev 2011, 111, 4857–4963. [DOI] [PubMed] [Google Scholar]
- (28).Westheimer FH The Magnitude of the Primary Kinetic Isotope Effect for Compounds of Hydrogen and Deuterium. Chem. Rev 1961, 61, 265–273. [Google Scholar]
- (29).Pattawong O; Mustard TJL; Johnston RC; Cheong PH-Y Mechanism and Stereocontrol: Enantioselective Addition of Pyrrole to Ketenes Using Planar-Chiral Organocatalysts. Angew. Chem., Int. Ed 2013, 52, 1420–1423. [DOI] [PubMed] [Google Scholar]
- (30).Hess RA; Hengge AC; Cleland WW Kinetic Isotope Effects for Acyl Transfer from p-Nitrophenyl Acetate to Hydroxylamine Show a pH-Dependent Change in Mechanism. J. Am. Chem. Soc 1997, 119, 6980–6983. [Google Scholar]
- (31).DelMonte AJ; Haller J; Houk KN; Sharpless KB; Singleton DA; Strassner T; Thomas AA Experimental and Theoretical Kinetic Isotope Effects for Asymmetric Dihydroxylation. Evidence Supporting a Rate-Limiting “(3 + 2)” Cycloaddition. J. Am. Chem. Soc 1997, 119, 9907–9908. [Google Scholar]
- (32).Beno BR; Houk KN; Singleton DA Synchronous or Asynchronous? An “Experimental” Transition State from a Direct Comparison of Experimental and Theoretical Kinetic Isotope Effects for a Diels–Alder Reaction. J. Am. Chem. Soc 1996, 118, 9984–9985. [Google Scholar]
- (33).Neel AJ; Hehn JP; Tripet PF; Toste FD Asymmetric Cross-Dehydrogenative Coupling Enabled by the Design and Application of Chiral Triazole-Containing Phosphoric Acids. J. Am. Chem. Soc 2013, 135, 14044–14047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wheeler SE; Bloom JWG Toward a More Complete Understanding of Noncovalent Interactions Involving Aromatic Rings. J. Phys. Chem. A 2014, 118, 6133–6147. [DOI] [PubMed] [Google Scholar]
- (35).Rücker C; Rücker G; Meringer M y-Randomization and Its Variants in QSPR/QSAR. J. Chem. Inf. Model 2007, 47, 2345–2357. [DOI] [PubMed] [Google Scholar]
- (36).Neel AJ; Hilton MJ; Sigman MS; Toste FD Exploiting non-covalent pi interactions for catalyst design. Nature 2017, 543, 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Dougherty DA The cation-pi interaction. Acc. Chem. Res 2013, 46, 885–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Zhou B; Haj MK; Jacobsen EN; Houk KN; Xue XS Mechanism and Origins of Chemo- and Stereoselectivities of Aryl Iodide-Catalyzed Asymmetric Difluorinations of beta-Substituted Styrenes. J. Am. Chem. Soc 2018, 140, 15206–15218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Wheeler SE Understanding substituent effects in noncovalent interactions involving aromatic rings. Acc. Chem. Res 2013, 46, 1029–38. [DOI] [PubMed] [Google Scholar]
- (40).Orlandi M; Toste FD; Sigman MS Multidimensional Correlations in Asymmetric Catalysis through Parameterization of Uncatalyzed Transition States. Angew. Chem., Int. Ed 2017, 56, 14080–14084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Suresh CH; Koga N Quantifying the Electronic Effect of Substituted Phosphine Ligands via Molecular Electrostatic Potential. Inorg. Chem 2002, 41, 1573–1578. [DOI] [PubMed] [Google Scholar]
- (42).Laconsay CJ; Seguin TJ; Wheeler SE Modulating Stereoselectivity through Electrostatic Interactions in a SPINOL-Phosphoric Acid-Catalyzed Synthesis of 2,3-Dihydroquinazolinones. ACS Catal. 2020, 10, 12292–12299. [Google Scholar]
- (43).Poater A; Ragone F; Mariz R; Dorta R; Cavallo L Comparing the enantioselective power of steric and electrostatic effects in transition-metal-catalyzed asymmetric synthesis. Chem. - Eur. J 2010, 16, 14348–53. [DOI] [PubMed] [Google Scholar]
- (44).It is important to note the criticism of leave-one-out Q2 at this point, see:; Golbraikh A; Tropsha A. Beware of q2! J. Mol. Graphics Modell 2002, 20, 269–276. [DOI] [PubMed] [Google Scholar]; It should also be noted that although 10 is considered to be a good value for k in k-fold cross validation, the exact requirement will depend on the data set. Hence, arbitrary values for k are often tested during model formation in our laboratory (Sigman), for further discussion see ref 45.
- (45).Kohavi R A study of cross-validation and bootstrap for accuracy estimation and model selection. IJCAI’95: Proceedings of the 14th Internation Joint Conference on Artificial Intelligence; ACM: Montreal, Canada, 1995; pp 1137–1145. [Google Scholar]
- (46).Cheong PH-Y; Legault CY; Um JM;Çelebi-Ölçüm N; Houk KN Quantum Mechanical Investigations of Organocatalysis: Mechanisms, Reactivities, and Selectivities. Chem. Rev 2011, 111, 5042–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Goerigk L; Mehta N A Trip to the Density Functional Theory Zoo: Warnings and Recommendations for the User. Aust. J. Chem 2019, 72, 563–573. [Google Scholar]
- (48).Grimme S; Schreiner PR Computational Chemistry: The Fate of Current Methods and Future Challenges. Angew. Chem., Int. Ed 2018, 57, 4170–4176. [DOI] [PubMed] [Google Scholar]
- (49).Peng Q; Duarte F; Paton RS Computing organic stereoselectivity – from concepts to quantitative calculations and predictions. Chem. Soc. Rev 2016, 45, 6093–6107. [DOI] [PubMed] [Google Scholar]
- (50).Crawford JM; Sigman MS Conformational Dynamics in Asymmetric Catalysis: Is Catalyst Flexibility a Design Element? Synthesis 2019, 51, 1021–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Davie EAC; Mennen SM; Xu Y; Miller SJ Asymmetric Catalysis Mediated by Synthetic Peptides. Chem. Rev 2007, 107, 5759–5812. [DOI] [PubMed] [Google Scholar]
- (52).Featherston AL; Shugrue CR; Mercado BQ; Miller SJ Phosphothreonine (pThr)-Based Multifunctional Peptide Catalysis for Asymmetric Baeyer–Villiger Oxidations of Cyclobutanones. ACS Catal. 2019, 9, 242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Metrano AJ; Chinn AJ; Shugrue CR; Stone EA; Kim B; Miller SJ Asymmetric Catalysis Mediated by Synthetic Peptides, Version 2.0: Expansion of Scope and Mechanisms. Chem. Rev 2020, 120, 11479–11615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Shugrue CR; Featherston AL; Lackner RM; Lin A; Miller SJ Divergent Stereoselectivity in Phosphothreonine (pThr)-Catalyzed Reductive Aminations of 3-Amidocyclohexanones. J. Org. Chem 2018, 83, 4491–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Shugrue CR; Miller SJ Phosphothreonine as a Catalytic Residue in Peptide-Mediated Asymmetric Transfer Hydrogenations of 8-Aminoquinolines. Angew. Chem., Int. Ed 2015, 54, 11173–11176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Kwon Y; Li J; Reid JP; Crawford JM; Jacob R; Sigman MS; Toste FD; Miller SJ Disparate Catalytic Scaffolds for Atroposelective Cyclodehydration. J. Am. Chem. Soc 2019, 141, 6698–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Yoon TP; Jacobsen EN Privileged Chiral Catalysts. Science 2003, 299, 1691. [DOI] [PubMed] [Google Scholar]
- (58).Reid JP; Sigman MS Holistic prediction of enantioselectivity in asymmetric catalysis. Nature 2019, 571, 343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Werth J; Sigman MS Connecting and Analyzing Enantioselective Bifunctional Hydrogen Bond Donor Catalysis Using Data Science Tools. J. Am. Chem. Soc 2020, 142, 16382–16391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Werth J; Sigman MS Linear Regression Model Development for Analysis of Asymmetric Copper-Bisoxazoline Catalysis. ACS Catal. 2021, 11, 3916–3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Durand DJ; Fey N Computational Ligand Descriptors for Catalyst Design. Chem. Rev 2019, 119, 6561–6594. [DOI] [PubMed] [Google Scholar]
- (62).Gensch T; Friederich P; Peters E; Gaudin T; Pollice R; Jorner K; Nigam A; Lindner D’Addario M; Sigman MS; Aspuru-Guzik AA; dos Passos Gomes G A Comprehensive Discovery Platform for Organophosphorus Ligands for Catalysis. ChemRxiv 2021, DOI: 10.26434/chemrxiv.12996665.v1. [DOI] [PubMed] [Google Scholar]
- (63).Shi Y; Prieto PL; Zepel T; Grunert S; Hein JE Automated Experimentation Powers Data Science in Chemistry. Acc. Chem. Res 2021, 54, 546–555. [DOI] [PubMed] [Google Scholar]
- (64).Christensen M; Yunker L; Adedeji F; Häse F; Roch L; Gensch T; dos Passos Gomes G; Zepel T; Sigman M; Aspuru-Guzik AH Data-science driven autonomous process optimization. ChemRxiv 2020, DOI: 10.26434/chemrxiv.13146404.v2. [DOI] [PMC free article] [PubMed] [Google Scholar]