

Abstract
Ovarian cancer (OvCa) is a leading cause of mortality from gynecologic malignancy due to its disseminated peritoneal metastasis. The tumor microenvironment dominates epithelial–mesenchymal transition (EMT) development and impacts cancer metastasis as well as mediates drug resistance. Tumour cell interaction with the collagen I matrix is critical in OvCa development. To better understand the role of the collagen matrix and the underlying mechanisms in the early stage of OvCa invasion, we developed a three-dimensional (3D) culture model in vitro by embedding OvCa cells within collagen I to recreate the architecture of a solid tumour. Our results showed that tumour spheroids formed in the 3D collagen model displayed good viability and decreased growth rates, which partly recapitulated the growth behavior of in vivo tumour cells. Collagen I enhanced the OvCa cell motility/invasion capability by up-regulating the expression of MMPs and α5β1 integrin. Moreover, highly invasive OvCa cells in collagen showed the overexpression of mesenchymal markers (N-cadherin, vimentin and fibronectin) and transcriptional factors (Snail and Slug). EMT-associated TGF-β1/Smad4 and Wnt5b/β-catenin signaling pathways were significantly up-regulated accordingly. Additionally, a remarkably enhanced drug resistance to chemotherapeutics was also detected in the 3D cultures. Collectively, the bioengineered 3D collagen models could recapitulate the in vivo tumour-like microenvironment and reflect some biological characteristics of human OvCa more accurately. The collagen I matrix promoted local invasion via EMT and enhanced the multidrug resistance in OvCa. This system might serve as a comprehensive in vitro model to better understand the manifold mechanisms of OvCa metastasis and also provide a robust tool for screening new anti-ovarian cancer therapeutics.
OV-NC and OV-206 cells cultured in collagen I hydrogel scaffolds, could gradually generate multicellular spheroids.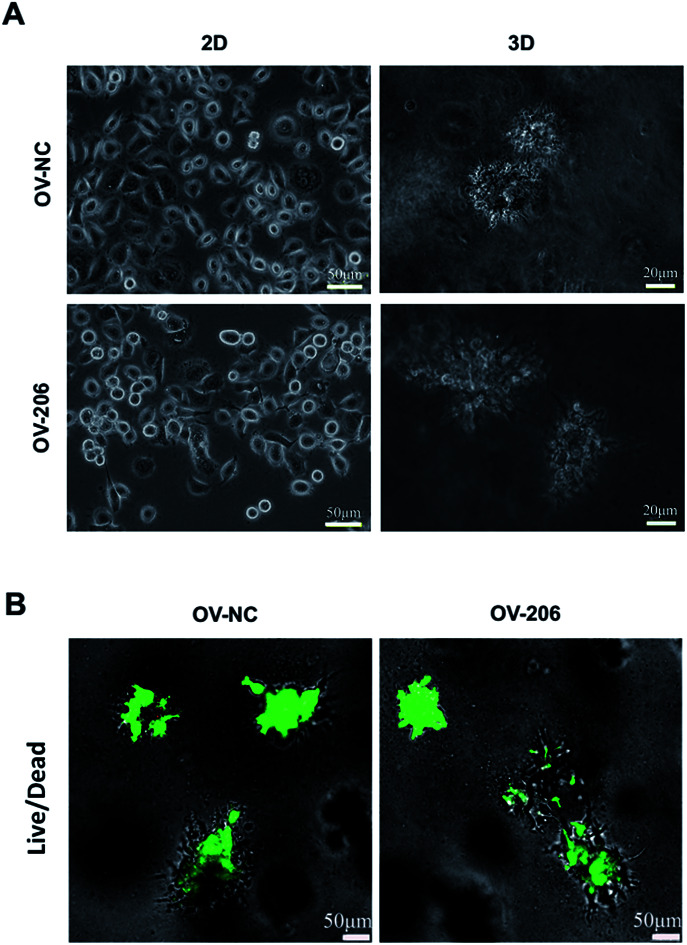
Introduction
Ovarian cancer (OvCa) is the fourth leading cause of gynecological cancer-related death among women worldwide.1 The high lethality of OvCa is primarily due to the presence of disseminated peritoneal metastasis. As most patients with OvCa are diagnosed at an advanced stage,2 the identification of the critical factors regulating the metastatic process of OvCa would have tremendous clinical significance. Recent studies have shown that the tumour microenvironment (that is composed of stromal cells, extracellular matrix (ECM) and soluble growth factors) plays an important role in affecting cancer metastasis as well as mediating drug resistance.3 The primary microenvironment for the peritoneal metastasis of OvCa is predominantly comprised of interstitial collagen I, which is the most abundant component of the ovarian ECM.4 It has been demonstrated that collagen I enhances the migration of multiple OvCa cell lines and primary OvCa cells preferentially adhere to collagen I.5,6 In addition, collagen I plays a novel role in inducing chemoresistance by up-regulating tau, which has been associated with paclitaxel resistance in OvCa.7 Therefore, a better understanding of the mechanisms of the interactions of ovarian tumour cells with the collagen I matrix will provide us with new targets for the retardation of the peritoneal metastatic spread.
In the past few decades, conventional two-dimensional (2D) culture has been widely accepted for exploring the mechanisms of tumorigenesis and metastasis. However, it cannot provide the microenvironmental clues that regulate the in vivo behaviour of tumour cells. In this regard, a bioengineering three-dimensional (3D) culture model in vitro is needed for the study of the metastasis of the disease. Such 3D scaffolds can provide 3D architectural space for cell growth, emulate the native tumour microenvironment ECM in vivo (recapitulation of cell–matrix interactions) and provide more accurate drug efficacy analyses.8 In addition, cancer cells in 3D culture tend to form multicellular aggregates, termed spheroids, which are similar to those observed in micrometastasis or poorly vascularized regions in solid tumours.9,10 The biocompatibility and 3D architecture of collagen I hydrogels are suitable properties for reproducing the solid tumour microenvironmental conditions in vivo. As a result, collagen I, as a superior biomaterial was chosen in this study to construct a 3D OvCa culture model and simulate the peritoneal metastasis microenvironment for the in vitro OvCa studies.
Epithelial–mesenchymal transition (EMT) is a critical step in the early stage of tumour metastasis, which is closely linked to invasion, recurrence, and therapy resistance.3 EMT is generally characterized by a change in the cell morphological specificity due to the loss of epithelial marker (E-cadherin) and up-regulation of mesenchymal markers such as vimentin, N-cadherin and fibronectin, leading to the loss of cell–cell interaction and increased cell motility.11 In addition, EMT is also associated with chemoresistance, emphasizing its relevance with respect to the recurrence of the disease.12 The EMT process of transformed cells can be induced typically by the activation of numerous signaling pathways, such as the TGF-β or Wnt pathway.13 For example, TGF-β1, a homodimeric cytokine, binds to cell surface receptors to activate Smad 2/3 proteins, which form the complex with Smad 4 and then promotes the expression of various transcription factors including Snail and Slug.14 Wnt5b protein, a secreted growth factor, binds to the cell surface receptor Frizzled, leading to stabilization of β-catenin,15 and then up-regulates the target genes such as Snail and Twist to induce EMT.14,16 In addition to soluble proteins that initiate EMT, ECM molecules have been shown to induce similar changes in epithelial cells. Recent studies have shown that interactions with collagen I can induce EMT in pancreatic cancer and lung cancer.17,18 However, to date, little is known about the role of the collagen I matrix in regulating the process of OvCa EMT and its underlying mechanisms. We therefore developed a 3D culture model by embedding the invasive OvCa cells (OV-NC and OV-206) in collagen gel and detected the growth profile, histological features, invasion capability, EMT-related gene expression and drug susceptibility of 3D cultured cancer cells to identify the effects of collagen I on EMT and drug resistance in the invasion procedure. This 3D collagen culture model will complement the current cell culture models used for the study of OvCa to enhance our overall understanding of the very early steps of ovarian cancer metastasis as well as to evaluate the efficacy of anticancer drugs.
Materials and methods
Cell culture and transfection
In our previous studies, 50 cases of ovarian cancer patient specimens were obtained, of which 25 were platinum-resistant and 25 were platinum-sensitive. Interestingly, we found that miR-206 was highly expressed in platinum-resistant OvCa patients by high-throughput microRNA analysis. Therefore, miR-206 was transfected into the human ovarian cancer cell line OV-2008 (OV-NC, platinum-sensitive) to generate the platinum-resistant cell line OV-2008-206 (OV-206, platinum-resistant), which stably expresses miR-206.
OV-NC cells were cultured in RPMI-1640 medium (Hyclone, USA) supplemented with 10% fetal bovine serum (Sciencell, USA), 100 units per mL penicillin and 100 units per mL streptomycin (Hyclone, USA) at 37 °C with 5% CO2 as reported previously.19 The miR-206 mimics were designed and chemically synthesized by Ambion (Life Technologies Corporation, Denmark).20 Lipofectamine 2000 (Invitrogen, USA) was incubated with pre-miR-206 (miRNA mimic) or their scrambled negative controls at a concentration of 90 nmol L−1 and incubated in serum-free RPMI1640 for 20 min before being added to OV-2008 cells. Cells were incubated at 37 °C for 4 h before 10% FBS was replaced. Protein and RNA were harvested 48 h after transfection.21
Casting of collagen hydrogel for 3D culture of ovarian cancer cells
Rat tail collagen solution (BD Biosciences, San Jose, CA) at 3.4 mg mL−1 was diluted with 0.1% HAc (v/v) to maintain the final concentration at 2 mg mL−1 for all experiments. Before casting the 3D cultures in cell culture plates, the bottom of each well was pre-coated with the diluted collagen solution at 37 °C for 2 h. To generate the 3D scaffold, OV-NC and OV-206 cells were trypsinized and resuspended as individual cells at 2 × 106 cells per mL. The cell suspension was mixed with 2 mg mL−1 collagen hydrogel and 10 × DMEM, using 0.1% (v/v) NaOH to adjust to neutral pH. According to the previous preparation techniques of our laboratory, we constructed a 3D culture model of OvCa cells. The final cell concentration in the model was 2.5 × 105 cells per mL. Once in plate, the collagen gel solution was kept in a humidity box at 37 °C for 2 h to allow gel polymerization via thermal cross-linking; then, 1.5 mL fresh cell culture medium was gently added to each well. The medium was refreshed every other day.
Cell viability and proliferation
Cell viability was assessed by calcein-AM/EthD-1 staining (Invitrogen, USA) as described previously.22 Only live cells with intracellular esterase activity can digest non-fluorescent calcein-AM into fluorescent calcein. Dead or dying cells containing damaged membranes allow the entrance of EthD-1 to stain the nuclei. Images were captured by a confocal laser scanning microscope (Leica DM4000B, Germany). To determine cell proliferation on the 3D collagen hydrogel, these two types of cells were cultured in 96-well plates under 2D and 3D culture conditions. At each time point, 10% CCK-8 (Dojindo, Japan) in cell culture media was added to each well and the plate was incubated for 4 h at 37 °C before being measured with micro-plate fluorometer at 450 nm (Thermo 354, USA).
Histology and immunohistochemistry staining
Hematoxylin and eosin (H&E) staining
Collagen-embedded OV-NC and OV-206 cells were harvested and fixed with 4% paraformaldehyde and dehydrated in graded concentrations of ethanol. Then, the paraffin-embedded sections were stained. Images were captured for microscopic evaluation (Leica TCS SP8, Germany).
Whole mount staining
Pretreatment of the collagen-cancer samples was similar to that of H&E before paraffin-embedding. The samples were transparentized with dimethylbenzene and then stained with carmine dyes.
Scanning electron microscopy (SEM) observation
OV-NC cells and collagen scaffolds were collected, washed, and fixed in 2.5% glutaraldehyde. After immobilization, the samples were washed with PBS for 10 min and then immobilized with 1% osmium acid, which was pre-cooled at 4 °C for 2 h. Samples were dehydrated in gradient ethanol for 15 min. Finally, the samples were dried and electrochemically sprayed. The ultrastructure of the samples was observed under SEM.
Immunohistochemistry (IHC) staining
Culture samples were collected at day 7 of culture and fixed in 4% paraformaldehyde for 12 h. The PFA-fixed and paraffin embedded 3D tumour tissue blocks were sectioned to 5 μm-thickness and attached to adhesive-coated slides. The sections were deparaffinized and rehydrated and then, antigen retrieval was performed by boiling the slides in 10 mM sodium citrate buffer. The samples were treated with 3% hydrogen peroxide and then with a non-immune goat serum, followed by incubation with the primary antibody of MMP-9 (dilution, 1 : 200; Proteintech, China) or MMP-10 (dilution, 1 : 500; Sigma-Aldrich) overnight at 4 °C. The sections lacking incubation with specific primary antibodies were used as negative controls. Following primary antibody incubation, the samples were incubated with a cocktail of biotinylated goat anti-rabbit or anti-mouse secondary antibody for 15 min and then incubated with avidin/biotin/peroxidase complex for 15 min. The samples were rinsed, developed with diaminobenzidine, counterstained with hematoxylin, and finally dehydrated and mounted.
Migration and invasion assays
For migration and invasion assays, the cells were serum-starved for 12 h before performing the assays and then digested from the plate dish and collagen hydrogel. Boyden chamber invasion assays were performed using the 24-well (8 μm pore size) Transwell chamber (Corning, USA) coated with BD Matrigel basement membrane matrix. The cells that did not migrate through the membrane were removed with a cotton swab. The cells that penetrated the matrigel to invade the lower surface of the membrane were fixed in methanol and stained with crystal violet as described for the migration assay. For the wound-healing assay, an artificial wound was created. To visualize migrated cells and wound healing, images were taken at 0, 24 and 48 h. Migrating or invading cells on the surface of the membranes were counted manually using Image-Pro Plus version 6.0 (Media Cybernetics, Inc).
Quantitative polymerase chain reaction (qRT-PCR)
Cells were harvested, total RNA was isolated using Trizol reagent (ambion Life Technologies, Denmark), and 1 mg RNA was reverse transcribed using a cDNA synthesis kit (TaKaRa Bio, Dalian). For qRT-PCR, the relative gene expression was determined using SYBR® Premix Ex Taq™II Kit (Takara Bio, Dalian) and the Agilent Technologies System. Primers are shown in Table 1 and the conditions used were described previously.23 Amplification was performed for 40 cycles of 3 min each at 95 °C, 30 s each at 58 °C, and 3 min each at 72 °C. The obtained results were analyzed using the 2−ΔΔCT method and normalized to the β-actin data. Each sample was analyzed in triplicate.
The primer sequences for qRT-PCR analysis.
| Genes | Primer sequences | T m (°C) |
|---|---|---|
| mmp-1 | F: AATTTGCCGACAGAGATGAAGTC | 48.4 |
| R: TCAGAAAGAGCAGCATCGATATG | ||
| mmp-2 | F: TGATGGTGTCTGCTGGAAAG | 46.6 |
| R: AGCAAACCTCGAACAGATGC | ||
| mmp-3 | F: TGATGGACCTGGAAATGTTTTG | 46 |
| R: AGGGAGTGGCCAATTTCATG | ||
| mmp-9 | F: TCTTCCCCTTCACTTTCCTG | 43.8 |
| R: TGTCGCTGTCAAAGTTCGAG | ||
| mmp-10 | F: GAAGATGCATCAGGCACCAA | 46.6 |
| R: ATCTTGCGAAAGGCGGAACT | ||
| mmp-13 | F: GGACCCTGGAGCACTCATGT | 45.3 |
| R: CTTGCCCTCTACAACCAACA | ||
| TGF-β1 | F: CATTTGTCTGGCGTTTTTGGA | 48.7 |
| R: CTTGCGACCCACGTAGTAGA | ||
| Smad4 | F: GCATTCCAGCCTCCCATTT | 51.2 |
| R: TCCAGAAGGGTCCACGTATCC | ||
| Wnt5b | F: CGCCAATGATGAACATCTC | 47.8 |
| R: ATTGGGATGGGTTGAGGC | ||
| β-catenin | F: AAGGTGTGGCGACATATGCA | 50.2 |
| R: CAAGTCCAAGATCAGCAGTCTCA | ||
| Snail | F: CTGCGTGGGTTTTTGTATCC | 46.6 |
| R: TCTGTCAGCCTTTGTCCTGTAG | ||
| Slug | F: AGCCTTTTTCTTGCCCTCACT | 49.7 |
| R: GGACTCACTCGCCCCAAAG | ||
| Fibronectin | F: CCTGGAGCAAGAAGGATAATCG | 49.7 |
| R: GGTAAACAGCTGCACGAACATC | ||
| Vimentin | F: GAAATTGCAGGAGGAGATGC | 46.6 |
| R: CGCATTGTCAACATCCTGTC | ||
| E-cadherin | F: CTGACACACCCCCTGTTGGT | 47.3 |
| R: CCATTGGATCCTCAACTGCAT | ||
| N-cadherin | F:TAGCTAATCTAACTGTGACCGATAAGG | 53.4 |
| R: TTGGTTTGACCACGGTGACTAA | ||
| Integrin α5 | F: ATGCTCCATGTAGATCACAAGAT | 55.7 |
| R:TTCCCAAAGTCCTTGCTGCT | ||
| Integrin β1 | F: CTTGCCCTCTACAACCAACA | 53.4 |
| R: TGCCCCGGTACGTGATATTG | ||
| β-actin | F: GGACTATGACTTAGTTGCGTTAC | 48.4 |
| R: TTTGCATTACATAATTTACACGA |
Western blotting
The cells under different conditions were harvested and protein was extracted. After quantification by BCA assay kit (KeyGEN, China), each protein sample (45 μg) was separated on a 10% SDS polyacrylamide gel, transferred to polyvinylidene difluoride polyscreen membrane (millipore, Germany) and then incubated with anti-MMP-9 (1 : 1000; Proteintech, China), anti-MMP-10 (1 : 500; Sigma-Aldrich, USA), anti-E-cadherin (1 : 500; Proteintech, China) anti-vimentin (1 : 300; Proteintech, China) and anti-β-actin (1 : 1000, Proteintech, China) antibodies overnight at 4 °C. Secondary anti-rabbit antibody (Proteintech, China) was added relative to the primary antibody and incubated for 2 h at room temperature. The blots were finally developed using an ECL western blot protocol (Lumilight enhanced chemiluminescence, Roche). Protein signals were visualized. Quantification of immune reactivity was performed by densitometric scanning using the Quantity One-1-Danalysis software (Bio-Rad, Hercules, CA).
Drug sensitivity changes within 2D and 3D cell cultures
Dose responses for chemotherapeutic agents were evaluated in 2D and 3D cultures. OvCa cells were seeded in a 96-well plate and cultured for 2 days on a monolayer and 7 days on a scaffold before treatment. Then, the culture medium was replaced with fresh medium containing different drugs (carboplatin, 5-fluorouracil, and paclitaxel) at various concentrations and the cells were incubated with the drugs for 48 h. Cell viability was measured as the percentage of viable cells relative to an untreated control. IC50 was assayed as the drug concentration that caused a 50% decrease in absorbance at 450 nm as compared to the untreated control.
Statistical analysis
SPSS 17.0 (Chicago, IL, USA) was used for all quantitative analyses. Results are expressed as the mean ± S.D. Student's t-test was used to analyze the difference between the groups. P < 0.05 was considered statistically significant.
Results
Growth profile of two ovarian cancer cell lines in the 3D collagen scaffold
The two OvCa cell lines cultured in 3D collagen scaffold exhibited morphologies that were different from the cells in the 2D monolayer culture (Fig. 1). In the monolayer culture, both OV-NC and OV-206 cells grew adherently in short spindle shapes. However, after being seeded in the scaffolds for three days, OV-NC cells formed stereoscopic ovoid cell masses. Similar to OV-NC cells, OV-206 cells also formed cell clusters in the 3D culture. After 7 days in culture, numerous cellular spheroids were generated in the collagen scaffolds but not in the 2D culture plates (Fig. 1A). The spheroids had the typical morphologies of the tumour-like tissue grown in vivo. Furthermore, these spheroids were evenly distributed in the scaffold and the size of the spheroids increased progressively with time until the 7th day. The majority of OV-NC and OV-206 cells in 3D culture exhibited good viability and very few cancer cells underwent apoptosis and necrosis after 7 days of cultivation (Fig. 1B). The data suggest that compared to the 2D culture, the 3D collagen culture model, a simple cell encapsulating methodology, more effectively mimics the in vivo microenvironment of OvCa cells.
Fig. 1. Growth profile and viability of OV-NC and OV-206 cells in 2D and 3D cultures. Morphology images showing morphological characteristics of the structures formed by OV-NC and OV-206 cells grown in different culture conditions for 7 days (A). Viability of OV-NC and OV-206 cells in 3D scaffolds detected by live/dead staining (green, live cells) during 15 day cultivation (B).

Proliferation of ovarian cancer cells in the 3D collagen scaffold
The proliferation of OV-NC cells in collagen increased gradually during 13 days of cultivation and the proliferation rate was relatively higher within the initial 7 days, indicating that the cells keep proliferating during spheroid formation (Fig. 2 left). In contrast, the growth of OV-NC cells in monocultures quickly reached the peak after 5 days of culture. A similar growth trend was observed in OV-206 cells in 3D and monolayer cultures (Fig. 2 right). It is noteworthy that OV-NC and OV-206 cells grew more slowly in 3D collagen culture than in monolayer culture, which is similar to the proliferation pattern of solid tumours in vivo. These results suggest that the collagen matrix as a biological scaffold material has good biocompatibility and can better support the growth of OvCa cells in comparison with the 2D culture method.
Fig. 2. Proliferation kinetics of OV-NC and OV-206 cells in 2D and 3D cultures. OV-NC and OV-206 cell proliferation in 2D and 3D culture was determined by CCK-8 assay at different time points. Results are shown as the mean ± SD.

Morphological characteristics of ovarian cancer cells in 3D collagen scaffolds
Next, we carried out whole mount staining to have a global view of the tumour cells in collagen. The results showed that the cells aggregated into multicellular spheroids, which is consistent with the phase-contrast microscopy results (Fig. 3A). Notably, the growth patterns of OV-NC and OV-206 cells were different in the 3D scaffolds. OV-NC cells formed round and regular cell spheroids, while the spheroids of OV-206 were more irregular and had more apparent cell processes.
Fig. 3. Morphological characteristics of OvCa cells in 3D culture by whole mount, H&E staining and SEM. OV-NC and OV-206 cells in 3D culture with whole mount staining (A). OV-NC and OV-206 cells in 3D collagen culture by H&E staining (B). Collagen I meshwork and OV-NC cells in collagen scaffolds imaged by SEM (C). Yellow arrows indicate OV-NC cell clusters.

H&E staining demonstrated that the cancer cells aggregated and formed cell clusters in the collagen scaffolds (Fig. 3B). OV-NC cells were densely-compacted and had small protrusions, while OV-206 cells were more loosely aggregated. This confirmed a cluster growth profile of OvCa cells in collagen gel. The observed different morphological characteristics indicated that OV-206 cells may have more malignant biological behaviour than OV-NC cells.
We also performed scanning electron microscopy (SEM) to further reveal the ultra-structure of OvCa cells in collagen I and the collagen I scaffold. The collagen scaffolds were fibrous networks. OV-NC cells were embedded within the collagen fibers and successfully generated cell clusters. The surface of the cells was covered with a large amount of extracellular matrix (Fig. 3C). All of these observations demonstrated that our collagen-based 3D culture system provides a similar microenvironment to that observed in vivo for the growth of OvCa cells.
Collagen I enhanced the motility capability of OvCa cells in 3D culture
In order to confirm the impact of the collagen matrix on the invasion and metastasis potential of OvCa cells, we performed wound healing and Transwell invasion assays to determine the motility of OV-NC and OV-206 cells under two different culture conditions. After cell cultivation for 48 h, the scratches in the 3D culture healed more quickly than in the corresponding 2D culture of both OV-NC and OV-206 cells and the area of cell migration was significantly increased in the 3D cultures (P < 0.05) (Fig. 4A and C). However, when the cells were cultured for only 24 h, a significant difference in migration areas between the 2D and 3D culture was only found in OV-206 cells (P < 0.05), indicating that 3D collagen culturing can promote the migration activity of OvCa cells, particularly of OV-206 cells.
Fig. 4. Comparison of cell migration and metastasis activity in OvCa cells in different cultures. The migration rates of OV-NC and OV-206 cells in different groups were compared by the wound healing assay (A). Microscopic observation was recorded at 0, 24 and 48 h after scratching the cell layer. Graph showing the distance of migration for these two types of cells in different culture groups (C). Invasive ability of OvCa cells was assessed by Matrigel invasion assay (B). The graph of invasion cells at 36 and 48 h in different groups (D). Each bar represents the cell numbers adherent to the lower membrane. Values are expressed as the mean ± SD of three replicates. *P < 0.05. n = 3 independent experiments.

Transwell invasion assay results were similar to those of the wound healing assay (Fig. 4B and D). After cell cultivation for 36 and 48 h, the mean number of the two types of OvCa cells penetrating the Transwell membrane in the 3D group significantly increased (P < 0.05) when compared to the 2D model, indicating that the invasion properties of OvCa cells were enhanced in response to the collagen matrix.
MMP-mediated proteolysis and integrin-mediated adhesion are required for the penetration of the collagen matrix
The matrix metalloproteinase (MMP) family is the main hydrolase in the remodeling of ECM.24 To investigate whether the proteolytic dependence for collagen matrix barrier removal is a type of MMP-mediated invasion, the gene expression of MMPs was detected by real-time PCR. We found that the expression of mmp-1, -2, -3, -9, -10, -13 had different degrees of up-regulation in the 3D culture system (P < 0.05). Nevertheless, the two cancer cell lines exhibited different MMP gene expression patterns in 3D culture. For example, mmp-2 and mmp-9 gene expression increased at a higher rate in OV-NC cells than in OV-206 cells (P < 0.05), while mmp-10 (P = 0.025) and mmp-13 (P = 0.047) gene expression increased at a higher rate in OV-206 than in OV-NC cells; no statistical difference for mmp-1 and mmp-3 gene expression was found between them (P > 0.05) (Fig. 5C). In accordance with the results of gene expression, the protein levels of MMP-9 and MMP-10 clearly increased in the 3D culture (P < 0.05) (Fig. 5A and B). These results suggest that the highly expressed MMPs facilitate the degradation of collagen I to enhance tumour cell invasion.
Fig. 5. Comparison of invasion-related MMPs expression between different culture conditions. Protein expression of MMP-9 and MMP-10 for OV-NC and OV-206 cells in the 2D and 3D cultures was detected by IHC staining (A) and western blotting (B). Gene levels of MMPs in the two OvCa cell lines in different cultures were examined by qRT-PCR (C). Expression data was normalized against β-actin. Each bar represents the mean ± SD. *P < 0.05. n = 3 independent experiments.

The adhesion of OvCa cells to the ECM via the integrin transmembrane receptors is of great importance for cancer progression; αv and β1 are the most expressed integrin subunits.25 Our data indicate that adhesion molecules α5 and β1 integrin subunits were significantly increased in 3D cultured OvCa cells (P < 0.05) and the up-regulation of the β1 subunit was more prominent in 3D cultured OV-206 cells (Fig. 6B). The data suggest that the adhesion molecule integrin α5β1 was involved in cancer cell adhesion to the collagen matrix and contributed to the penetration of the collagen, particularly for more malignant cells.
Fig. 6. Protein and gene expression levels of EMT-related genes in OvCa in different cultures. Protein expression of E-cadherin and vimentin in OV-NC and OV-206 cells in 2D and 3D culture systems were detected by western blotting (A). Expression level of EMT-related genes for OV-NC and OV-206 cells in different culture systems were examined via qRT-PCR (B). Expression data was normalized against β-actin. Each bar represents the mean ± SD. *P < 0.05. n = 3 independent experiments.

Collagen I-induced EMT was mediated by TGF-β and Wnt pathways in OvCa cells
To explore the role of collagen I on EMT in OvCa development, we first analyzed the gene expression of epithelial and mesenchymal markers in EMT by real-time RT-PCR. The relative mRNA expression levels of mesenchymal markers vimentin, N-cadherin and fibronectin were significantly increased in 3D cultured OV-NC and OV-206 cells (P < 0.05), (Fig. 6B). In particular, significant up-regulation (>6-fold) of these mesenchymal markers was identified in OV-206 cells. Unlike the mesenchymal markers, the expression of epithelial marker E-cadherin was markedly decreased in 3D cultured cancer cells as compared to that in the 2D monolayer culture (P < 0.05). Consistent with real-time RT-PCR results, the protein level of vimentin was up-regulated (P < 0.05) and the protein level of E-cadherin was down-regulated (P < 0.05) in 3D cultured cells (Fig. 6A). Both OV-NC and OV-206 cells acquired more typical mesenchymal traits at the molecular level in 3D culture, indicating that collagen I was capable of inducing the EMT process in OvCa cells.
To explore the underlying mechanism on collagen-induced EMT, the mRNA levels of critical molecules in EMT-related TGF-β and Wnt signaling pathways were further tested. As shown in Fig. 6B, TGF-β1/Smad4 and Wnt5b/β-catenin were significantly up-regulated in 3D cultured OV-NC and OV-206 cells when compared to those in the 2D cultured cells (P < 0.05). In addition, the mRNA levels of EMT-related transcriptional factors, namely, Snail and Slug were also increased in 3D cultured cells (P < 0.05) (Fig. 6B). These data demonstrated that collagen-induced EMT in OvCa cells might be ascribed to the co-activation of TGF-β and Wnt signaling pathways, leading to the up-regulation of Snail and Slug.
Chemosensitivity of ovarian cancer cells in different culture models
Carboplatin (CBP), 5-fluorouracil (5-FU) and paclitaxel (PTX) are the commonly used chemotherapeutic drugs for OvCa patients. To determine if the collagen matrix has an effect on chemoresistance, we evaluated the drug susceptibility of the two OvCa cell lines under different culture conditions using the AlamarBlue assay. The half-maximal inhibitory concentration (IC50) values of the three drugs were markedly higher in 3D cultured cells, as compared to those in 2D cultured cells (Table 2). For example, in OV-NC cells, IC50 of CBP was 27 ± 0.4 μmol L−1 in 2D cultured cells, but was 92 ± 3.1 μmol L−1 in 3D cultured cells, thus indicating a 3.4-fold increase in 3D cultured cells. Similarly, the IC50 values of 5-FU and PTX in OV-NC cells increased by 12- and 10-fold, respectively, in 3D cultured cells as compared to 2D cultured cells (5-FU: 71 ± 0.4 vs. 6 ± 0.1 μmol L−1, PTX: 2 ± 5.8 vs. 0.2 ± 1.0 μmol L−1 (3D vs. 2D)). Compared with 2D cultured cells, 3D cultured OV-206 cells were even more resistant to these three drugs. The IC50 values of CBP, 5-FU, and PTX in 3D cultured OV-206 cells increased by 3.8, 4.1, and 5.3-fold in 3D cultured cells when compared with the corresponding IC50 values in 2D cultured cells (CBP: 154 ± 5.9 vs. 41 ± 1.8 μmol L−1, 5-FU: 336 ± 1.3 vs. 82 ± 0.4 μmol L−1, PTX: 132 ± 9.4 vs. 25 ± 5.0 μmol L−1 (3D vs. 2D)). In brief, the IC50 values of these three drugs significantly increased in the two OvCa cell lines in the 3D collagen culture, suggesting that the collagen matrix promoted chemotherapy resistance.
Cytotoxicity assay of the anticancer drugs against ovarian cancer cellsa,b.
| IC50 | CBP (μmol L−1) | 3D/2D | 5-FU (μmol L−1) | 3D/2D | PTX (μmol L−1) | 3D/2D |
|---|---|---|---|---|---|---|
| 2D NC | 27 ± 0.4 | 1 | 6 ± 0.1 | 1 | 0.2 ± 1.0 | 1 |
| 3D NC | 92 ± 3.1 | 3.4 | 71 ± 0.4 | 12 | 2 ± 5.8 | 10 |
| 2D 206 | 41 ± 1.8 | 1 | 82 ± 0.4 | 1 | 25 ± 5.0 | 1 |
| 3D 206 | 154 ± 5.9 | 3.8 | 336 ± 1.3 | 4.1 | 132 ± 9.4 | 5.3 |
Abbreviation: carboplatin (CBP), 5-fluorouracil (5-FU), and paclitaxel (PTX).
IC50: mean of the concentration of anticancer drug achieving a 50% inhibition of cell viability.
Discussion
The development of 3D in vitro cell culture models for studying the complex mechanisms of tumourigenesis plays an essential role in advancing fundamental cancer research. This culture system recapitulates aspects of the in vivo tumour microenvironment and has many advances in studying the dynamics of tumour progression, and drug responsiveness. Tumour cells cultured within a 3D in vitro environment have the ability to acquire morphologies/phenotypes and respond to stimuli similar to in vivo solid tumours. In the present study, OV-NC or OV-206 cells cultured in collagen I hydrogel scaffolds could gradually generate multicellular spheroids, demonstrating cell–cell/–matrix interactions that encourage cell signalling and a phenotype representative of in vivo tumour biological systems. The cell proliferation rates in 3D culture increased progressively during two weeks of cultivation, similar to the proliferation of solid tumours in vivo. Morphological analysis confirmed that the two types of OvCa cells cultured in collagen displayed good viability and a more tissue-like morphology. These characteristics of the 3D cultured OvCa cells suggested that collagen I, as a biological scaffold material, has good biocompatibility, supports unconfined cellular proliferation and can better imitate the developmental process of OvCa in vivo from single cell to multicellular spheroid.
The successful invasion process requires motility, adhesion and degradation of the matrix. Several reports have shown that cell motility is enhanced in metastatic OvCa cells through the interaction with the 3D collagen matrix.26,27 Consistent with these previous reports, our findings indicate that the migration and invasion capacity of OvCa cells are enhanced through their interaction with 3D collagen matrix, suggesting a microenvironment relevant to the OvCa metastatic process. It is noteworthy that the migration activity of OV-206 cells is stronger than OV-NC cells, indicating that up-regulation of miR-206 expression enhances the invasive behaviour of OvCa cells; the potential targets and mechanisms need to be further investigated. The adhesion of tumour cells to ECM components is a pivotal step in developing peritoneal dissemination of intra-abdominal malignancy. As a major membrane receptor of fibrillar collagen, integrins play an important role in tumour progression. It has been reported that ECM affects cancer invasion by regulating αvβ1 integrin downstream signaling.28 Increased expression of α5β1 integrin was demonstrated to enhance tumour cell invasion in the 3D collagen matrix through the generation of a higher contractile force.29 Our results show that α5β1 integrin was significantly up-regulated in 3D cultured OvCa cells, suggesting that α5β1 integrin-mediated adhesion to collagen I is required for the invasive process. It is worth pointing out that in addition to the α5β1 integrin, another important subtype of integrin, i.e., α2β1 integrin has also been reported to be able to promote adhesion, proliferation, and migration of pancreatic cancer cells cultured on collagen.30 Thus, we analyzed that the β1-subunit of integrin might be one of the indispensable molecules in mediating cell-ECM adhesion, while the involvement of α-subunits might be varied from different cell types and their culture microenvironment. In addition to metastasis-related adhesion molecules, MMPs, which are the major enzymes, are involved in the remodeling of ECM and contribute to the malignant potential of tumour cells.24 The study reported by Sodek et al. reported that the capability of OvCa cells to penetrate through the collagen I barrier could be prevented by a broad range MMP inhibitor, GM6001.31 This suggests that the proteolytic activity of MMPs plays a pivotal role in promoting tumour cell invasion into the surrounding collagen matrices. Consistently, we found that the gene expression of collagen-cleaving MMPs (MMP-1 and -13), gelatin (denatured collagen)-cleaving MMPs (MMP-2 and -9) as well as the MMPs that degraded a broad spectrum of ECM proteins (MMP-3 and -10) were all up-regulated in the 3D cultures of OvCa cells compared to their 2D cultures. The data further supported that collagen I could facilitate MMP mediated hydrolysis to enhance the invasion capability of malignant OvCa cells penetrating the matrix. Interestingly, mmp-2 and mmp-9 gene expression, unlike the mmp-1, -3, -10 and -13 expression patterns, increased much higher in OV-NC cells than in OV-206 cells. This might be because up-regulation of miR-206 in OV-206 reduced the expression of mmp-2 and mmp-9 to some extent, which is consistent with the previous study.32 Collectively, collagen I, as the substrate for the attachment and migration of OvCa cells, stimulates invasive behaviour by increased MMP activity, which promotes peritoneal tumour formation.
Recently, EMT has been found to play a critical role in the cancer metastasis, drug resistance and poor prognosis of OvCa.33 The process of EMT, controlled by various pro-invasion signals from the tumour microenvironment, promotes cancer cell invasion and dissemination.34 In particular, collagen I has been described to induce EMT of lung cancer cells and pancreatic cancer cells through different pathways.17,18,35 Herein, our study demonstrated that OvCa cell lines are able to respond to the 3D microenvironment provided by collagen I and display the enhanced EMT phenotype evidenced by their up-regulation of vimentin, N-cadherin and fibronectin and decreased expression of E-cadherin. This further supports that collagen I might contribute to the reduced cell–cell connection of OvCa cells and increased tumour cell motility. Diverse signaling pathways are involved in the collagen I-induced EMT process. In human lung cancer cells, collagen I promotes EMT-like changes through increasing the production of active TGF-β and the activation of the Smad pathway.18 Similarly, collagen I binds to its receptor DDR2 and activates downstream SRC/ERK2 to stabilize Snail in breast tumour cells, thus maintaining the EMT phenotype and facilitating continued tumour cell invasion in collagen I-rich ECM.36 In this study, we found that TGF-β1 expression of OvCa cells in 3D culture was significantly up-regulated in accordance with the increase in Smad4 expression. Moreover, Wnt5b expression was significantly elevated in 3D cultured cells accompanied by up-regulation of β-catenin. The up-regulated TGF-β and Wnt pathways could mediate EMT individually or cooperate with each other. Activation of Smad or β-catenin leads to the up-regulation of EMT-related transcription factors Snail and Slug, which in turn, repress E-cadherin transcription, subsequently promoting early dissemination of cells from the tumour mass.37 We conclude that collagen I could be an independent extracellular stimulating factor to induce EMT by up-regulating TGF-β/Smad and Wnt/β-catenin signaling pathways, thus enhancing the invasion capacity of OvCa. Therefore, inhibition of collagen I-induced EMT might be beneficial for the treatment of OvCa metastasis.
The microenvironment at metastatic sites could affect tumour cells, leading to the development of drug resistance. For example, significant differences in the efficacy of chemotherapeutic agents has been found for 2D cultured cancer cells and the clinical application of these drugs.38 It was also demonstrated that tumour cells grown on 3D scaffolds developed multidrug resistance in comparison with those grown as monolayers.9 In our study, both OvCa cell lines displayed 3-12-fold increase in drug resistance to CBP, 5-FU and PTX when grown as spheroids in 3D culture, suggesting that collagen I has a critical role in OvCa chemoresistance. Drug resistance of 3D cultured cancer cells may be ascribed to the limited delivery of drugs into the core of tumour spheroids as well as increased cell survival in spheroids.39 Moreover, the collagen matrix can limit the effect of chemotherapy by activating specific signaling pathways, which may contribute to chemoresistance via EMT. For example, activation of EMT by the TGF-β pathway is a signature indicative of resistance to platinum-based chemotherapy.40 Haslehurst et al. also showed that EMT transcription factors Snail and Slug contribute to cisplatin or paclitaxel resistance in OvCa.41 The chemoresistance mediated by Snail and Slug is associated with p53-mediated pro-survival signaling.42 In addition to signaling pathways, aberrant expression of miRNAs also plays a significant role in OvCa chemoresistance.43 In this study, OV-206 cells displayed a greater drug resistance than OV-NC, indicating that miR-206 was involved in the development of multidrug resistance of OvCa. In contrast to our findings, Chen et al. found that miR-206 overexpression reduced cisplatin resistance and EMT in lung cancer.44 The mechanisms of miR-206 in governing the drug resistance of OvCa cells require additional and more detailed studies in the future.
Conclusions
The bioengineered 3D culture model of OvCa recapitulates the tissue-like morphology of in vivo solid tumours and provides a robust tool for dissecting complex functions of collagen I in tumour progression. The enhanced invasion and capacity for metastasis of malignant cells respond to collagen I and require MMP-mediated hydrolysis and cell-matrix adhesion. Moreover, the collagen matrix induced EMT procedure in OvCa cells, which is mediated by TGF-β and Wnt signaling pathways, leads to increased expression of Snail and Slug. In addition, 3D collagen models enhance chemotherapy resistance and provide a valuable in vitro research platform for screening new chemotherapeutic agents for invasive OvCa. These results extend our understanding of the critical role of collagen I in OvCa peritoneal metastasis, which may aid in the development of useful therapeutic strategies to target cancer progression.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This study was supported by the Natural Science Foundation of China (31470952 and 31771068) and Natural Science Foundation of Liaoning Province (2014023003).
References
- Siegel R. L. Miller K. D. Jemal A. Ca-Cancer J. Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- Ukaji T. Lin Y. Banno K. Okada S. Umezawa K. PLoS One. 2015;10:e0137663. doi: 10.1371/journal.pone.0137663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drak Alsibai K. Meseure D. Dev. Dyn. 2018;247:405–431. doi: 10.1002/dvdy.24548. [DOI] [PubMed] [Google Scholar]
- Witz C. A. Montoya-Rodriguez I. A. Cho S. Centonze V. E. Bonewald L. F. Schenken R. S. J. Soc. Gynecol. Invest. 2001;8:299–304. doi: 10.1177/107155760100800508. [DOI] [PubMed] [Google Scholar]
- Shen Y. Shen R. Ge L. Zhu Q. Li F. Int. J. Gynecol. Cancer. 2012;22:1316–1324. doi: 10.1097/IGC.0b013e318263ef34. [DOI] [PubMed] [Google Scholar]
- Moser T. L. Pizzo S. V. Bafetti L. M. Fishman D. A. Stack M. S. Int. J. Cancer. 1996;67:695–701. doi: 10.1002/(SICI)1097-0215(19960904)67:5<695::AID-IJC18>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Gurler H. Yu Y. Choi J. Kajdacsy-Balla A. A. Barbolina M. V. Int. J. Mol. Sci. 2015;16:3419–3433. doi: 10.3390/ijms16023419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong E. L. Lamhamedi-Cherradi S. E. Burdett E. Ramamoorthy V. Lazar A. J. Kasper F. K. Farach-Carson M. C. Vishwamitra D. Demicco E. G. Menegaz B. A. Amin H. M. Mikos A. G. Ludwig J. A. Proc. Natl. Acad. Sci. U. S. A. 2013;110:6500–6505. doi: 10.1073/pnas.1221403110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard Y. K. Wang C. Ravi S. Howell M. C. Mallela J. Alibrahim M. Green R. Hellermann G. Mohapatra S. S. Mohapatra S. PLoS One. 2013;8:e75345. doi: 10.1371/journal.pone.0075345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman A. Sanchez Alemany V. Nguyen Y. Zhang C. R. Kaufman L. J. Biomaterials. 2017;115:19–29. doi: 10.1016/j.biomaterials.2016.11.014. [DOI] [PubMed] [Google Scholar]
- Tellez C. S. Juri D. E. Do K. Bernauer A. M. Thomas C. L. Damiani L. A. Tessema M. Leng S. Belinsky S. A. Cancer Res. 2011;71:3087–3097. doi: 10.1158/0008-5472.CAN-10-3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery J. P. Nat. Rev. Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- De Wever O. Pauwels P. De Craene B. Sabbah M. Emami S. Redeuilh G. Gespach C. Bracke M. Berx G. Histochem. Cell Biol. 2008;130:481–494. doi: 10.1007/s00418-008-0464-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber M. A. Kraut N. Beug H. Curr. Opin. Cell Biol. 2005;17:548–558. doi: 10.1016/j.ceb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Xu K. Buchsbaum R. J. J. Visualized Exp. 2012;62:e3760. doi: 10.3791/3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. Weinberg R. A. Dev. Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Shintani Y. Hollingsworth M. A. Wheelock M. J. Johnson K. R. Cancer Res. 2006;66:11745–11753. doi: 10.1158/0008-5472.CAN-06-2322. [DOI] [PubMed] [Google Scholar]
- Shintani Y. Maeda M. Chaika N. Johnson K. R. Wheelock M. J. Am. J. Respir. Cell Mol. Biol. 2008;38:95–104. doi: 10.1165/rcmb.2007-0071OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H. Bi T. Qu Z. Jiang J. Cui S. Wang Y. Oncol. Rep. 2014;32:1003–1012. doi: 10.3892/or.2014.3311. [DOI] [PubMed] [Google Scholar]
- Zhao H. Yu X. Ding Y. Zhao J. Wang G. Wu X. Jiang J. Peng C. Guo G. Z. Cui S. Oncotarget. 2016;7:53254–53268. doi: 10.18632/oncotarget.10736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H. Liu S. Wang G. Wu X. Ding Y. Guo G. Jiang J. Cui S. Oncol. Rep. 2015;33:591–598. doi: 10.3892/or.2014.3640. [DOI] [PubMed] [Google Scholar]
- Wang X. Wang W. Ma J. Guo X. Yu X. Ma X. Biotechnol. Prog. 2006;22:791–800. doi: 10.1021/bp050386n. [DOI] [PubMed] [Google Scholar]
- Han H. W. Hsu S. H. Acta Biomater. 2016;42:157–167. doi: 10.1016/j.actbio.2016.06.014. [DOI] [PubMed] [Google Scholar]
- Brinckerhoff C. E. Rutter J. L. Benbow U. Clin. Cancer Res. 2000;6:4823–4830. [PubMed] [Google Scholar]
- Davidson B. Goldberg I. Gotlieb W. H. Kopolovic J. Risberg B. Ben-Baruch G. Reich R. Cancer Metastasis Rev. 2003;22:103–115. doi: 10.1023/A:1022272204045. [DOI] [PubMed] [Google Scholar]
- Barbolina M. V. Adley B. P. Kelly D. L. Fought A. J. Scholtens D. M. Shea L. D. Stack M. S. Lab. Invest. 2008;88:602–614. doi: 10.1038/labinvest.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G. G. Risteli J. Puistola U. Kauppila A. Risteli L. Cancer Res. 1993;53:5028–5032. [PubMed] [Google Scholar]
- Maschler S. Wirl G. Spring H. Bredow D. V. Sordat I. Beug H. Reichmann E. Oncogene. 2005;24:2032–2041. doi: 10.1038/sj.onc.1208423. [DOI] [PubMed] [Google Scholar]
- Mierke C. T. Frey B. Fellner M. Herrmann M. Fabry B. J. Cell Sci. 2011;124:369–383. doi: 10.1242/jcs.071985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzesiak J. J. Bouvet M. Br. J. Cancer. 2006;94:1311–1319. doi: 10.1038/sj.bjc.6603088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodek K. L. Brown T. J. Ringuette M. J. BMC Cancer. 2008;8:223. doi: 10.1186/1471-2407-8-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H. Cao Y. D. Ye W. X. Sun Y. Y. Tumori. 2010;96:751–755. doi: 10.1177/030089161009600518. [DOI] [PubMed] [Google Scholar]
- Deng J. Wang L. Chen H. Hao J. Ni J. Chang L. Duan W. Graham P. Li Y. Oncotarget. 2016;7:55771–55788. doi: 10.18632/oncotarget.9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R. Weinberg R. A. J. Clin. Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menke A. Philippi C. Vogelmann R. Seidel B. Lutz M. P. Adler G. Wedlich D. Cancer Res. 2001;61:3508–3517. [PubMed] [Google Scholar]
- Zhang K. Corsa C. A. Ponik S. M. Prior J. L. Piwnica-Worms D. Eliceiri K. W. Keely P. J. Longmore G. D. Nat. Cell Biol. 2013;15:677–687. doi: 10.1038/ncb2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabe H. J. Biochem. 2011;149:633–639. doi: 10.1093/jb/mvr044. [DOI] [PubMed] [Google Scholar]
- Quiros R. M. Valianou M. Kwon Y. Brown K. M. Godwin A. K. Cukierman E. Gynecol. Oncol. 2008;110:99–109. doi: 10.1016/j.ygyno.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y. J. Hsu S. H. Biomaterials. 2014;35:10070–10079. doi: 10.1016/j.biomaterials.2014.09.010. [DOI] [PubMed] [Google Scholar]
- Marchini S. Fruscio R. Clivio L. Beltrame L. Porcu L. Fuso Nerini I. Cavalieri D. Chiorino G. Cattoretti G. Mangioni C. Milani R. Torri V. Romualdi C. Zambelli A. Romano M. Signorelli M. di Giandomenico S. D'Incalci M. Eur. J. Cancer. 2013;49:520–530. doi: 10.1016/j.ejca.2012.06.026. [DOI] [PubMed] [Google Scholar]
- Haslehurst A. M. Koti M. Dharsee M. Nuin P. Evans K. Geraci J. Childs T. Chen J. Li J. Weberpals J. Davey S. Squire J. Park P. C. Feilotter H. BMC Cancer. 2012;12:91. doi: 10.1186/1471-2407-12-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurrey N. K. Jalgaonkar S. P. Joglekar A. V. Ghanate A. D. Chaskar P. D. Doiphode R. Y. Bapat S. A. Stem Cells. 2009;27:2059–2068. doi: 10.1002/stem.154. [DOI] [PubMed] [Google Scholar]
- Park S. M. Gaur A. B. Lengyel E. Peter M. E. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q. Y. Jiao D. M. Wang J. Hu H. Tang X. Chen J. Mou H. Lu W. Oncotarget. 2016;7:24510–24526. doi: 10.18632/oncotarget.8229. [DOI] [PMC free article] [PubMed] [Google Scholar]