

Abstract
The potential of adoptive cell therapy with regulatory T cells (Treg) to promote transplant tolerance is under active exploration. However, the impact of specific transplant settings and protocols on Treg manufacturing is not well-delineated. Here, we compared the use of peripheral blood mononuclear cells (PBMC) from patients before or after liver transplantation to the use of healthy control PBMC to determine their suitability for Treg manufacture using ex vivo costimulatory blockade with belatacept. Despite liver failure or immunosuppressive therapy, the capacity for Treg expansion during the manufacturing process was preserved. These experiments did not identify performance or quality issues that disqualified use of post-transplant PBMC, the currently favored protocol design. However, as Treg input correlated with output, significant CD4-lymphopenia in both pre- and post-transplant patients limited Treg yield. We therefore turned to leukapheresis post-transplant to improve absolute yield. To make deceased donor use feasible, we also developed protocols to substitute splenocytes for PBMC as allostimulators. In addition to demonstrating that this Treg expansion strategy works in a liver transplant context, this preclinical study illustrates how characterizing cellular input populations and their performance can both inform and respond to clinical trial design and Treg manufacturing requirements.
Introduction
Allogeneic liver transplantation outcomes have improved significantly, due in part to more efficacious immunosuppressive therapy (IST). However, pharmacologic IST utilization is accompanied by substantial rates of cardiovascular and renal damage, diabetes, infection, and therapy-related malignancies as well as socioeconomic costs of chronic medication dependence.1–6 Graft survival remains a problem.4,7 Therefore, alternatives producing similarly low rates of acute and/or chronic graft rejection while minimizing toxicities of chronic pharmacologic IST are desirable.8
Interest in using immunosuppressive immune populations to induce or support transplant tolerance8–12 has encouraged development of methods for expanding regulatory T cells (Treg), generally defined by a CD4+CD25hiCD127loFoxp3+ phenotype, for administration to organ transplant recipients.9,10,12–25 We and others have demonstrated that: 1) potent, alloantigen-specific Treg from healthy donors can be generated using antibodies to CD80 and CD86 or first (abatacept) or second (belatacept) generation CTLA4Ig fusion proteins to induce costimulatory blockade (CSB) during an ex vivo mixed lymphocyte reaction (MLR)14,20,23; and 2) CSB-MLR supports Treg expansion using peripheral blood mononuclear cell (PBMC) responders from end-stage renal failure patients and healthy control (HC) or HLA-mismatched healthy donor PBMC stimulators.13,15,18,26
Liver transplantation (LTx) presents an attractive option for Treg administration.8–10,17,19,27,28 The liver itself supports an immunosuppressive state,29–31 and LTx recipients can achieve persistent operational tolerance, at low frequency but higher than with other solid organs.8,27,31–34 Nonetheless, translational development of Treg adoptive transfer for LTx has been challenging. In comparison to HC donors of PBMC used to develop cell manufacturing strategies, end-stage liver failure (ESLD) patients are frequently leukopenic, lymphocytopenic, and demonstrate immunologic abnormalities potentially influenced by ESLD etiology and duration.35–41 Conditions accompanying ESLD, such as coagulopathy and ascites, can complicate PBMC collection, adversely affecting both patient safety and product characteristics.42,43 The unknowns of donor availability and timing pose relevant issues for study procedures, including consent, cell collection and manufacturing. Post-LTx patients, whose donor type, ESLD etiology, IST, alloantigen exposure and infections can modulate immune function,1–3,5–7,44–52 also present challenges related to collection and storage of liver donor stimulators for subsequent manufacturing, PBMC collection, and potential intercurrent rejection.
In summary, context-specific medical issues, responder and stimulator cell properties, and procedures for cell collection and storage have potential to impact the outcome of cell manipulation strategies established using healthy volunteers. We therefore investigated the suitability of PBMC collected from patients prior to (pre) or post-LTx as substrates for manufacturing Treg using ex vivo CSB and examined what methodologic modifications could address the challenges of deceased donor use and improving Treg yield.
Material and Methods
Study Participants.
Individuals with ESLD, pre- or post-LTx, were recruited at Massachusetts General Hospital (MGH) from 02/2016 to 05/2019 and 02-06/2021. To support an Investigational New Drug application for an Immune Tolerance Network study “A Phase I/II Drug Withdrawal Study of Alloantigen-Specific Tregs in Liver Transplantation” (LITTMUS, ITN073ST), eligibility largely paralleled that protocol, prioritizing individuals meeting standard LTx criteria, aged 18–70 years, seronegative for HIV-1/2, within 8 months post-LTx (the anticipated manufacturing window) and excluded those with autoimmune etiology. During the COVID-19 pandemic, this was relaxed to accommodate decreased clinic visits by post-LTx patients. All participants provided written informed consent (Partners Human Research Committee Institutional Review Board (IRB)-approved study 2016P000214).
In total, 122 patients enrolled and provided peripheral blood (PB) samples. Nineteen with inadequate cell yield and 5 post-LTx patients beyond the 8-month window were excluded. As only 4 post-LTx patients received IST including sirolimus (RAPA), their results were excluded from overall results and reported separately. Table 1 shows characteristics of the remaining 94 patients. The single patient with both pre- and post-LTx samples collected is counted in each group. Because of cytopenia and/or protocol limitations on sample volume, some samples contained insufficient PBMC for all determinations. Rejection episodes had occurred post-LTx in 20% (10/49). HC PBMC were obtained from deidentified volunteer PB or platelet-donor pheresis collars (DF/Partners Cancer Center IRB approved protocol 05–321). Spleens were obtained from deceased organ donors (MGH IRB approved protocol 2013P000392/1). Except for the clinical trial patient, the spleen donor differed from the future (for pre-LTx) or actual (for post-LTx) liver donor.
Table 1.
Demographic and clinical characteristics of study patients
| Pre-LTx patients* | Post-LTx | |
|---|---|---|
| Number of patients | 45 | 49 |
| Age [Median ± SD (range)] | 58 ± 9 (35–69) | 57 ± 11.5 (21–69) |
| Gender (Male : Female) | 32 : 13 | 35 : 14 |
| Race (% Caucasian) | 93% | 92% |
| Diagnosis | ||
| Alcoholic cirrhosis/hepatitis | 15 | 6 |
| NASH | 6 | 5 |
| Viral hepatitis | 2 | 5 |
| PSC/PBC | 3 | 2 |
| HCC# | 11 | 25 |
| Other## | 8 | 6 |
| Immunosuppressive Therapy | ||
| Tac + MMF + P | 32 | |
| Tac + MMF | 6 | |
| Tac + Evr | 1 | |
| Tac + P | 4 | |
| Tac | 2 | |
| CsA + MMF + P | 3 | |
| CsA + Az + P | 1 | |
| Time from LTx | ||
| ≤ 1month (30 days) | 15 | |
| > 1 – 3 months (31–91 days) | 17 | |
| ≥ 3 – 8 months (92–244 days) | 14 | |
| > 8 months | 3 |
One patient provided a pre and post-LTx sample.
HCC patients’ second diagnoses (if any): pre-LTX - NASH 5, alcoholic cirrhosis/hepatitis 3, viral hepatitis 1; post-LTx NASH 8, alcoholic cirrhosis/hepatitis 5, viral hepatitis 8, congenital biliary atresia 1
Other diagnosis: pre-LTx - cryptogenic 4, and 1 each lipodystrophy plus alpha1 antitrypsin deficiency, nodular regenerative hyperplasia, neuroendocrine tumor, substance abuse; post-LTx - 1 each cryptogenic, autoimmune hepatitis, Wilson’s disease, common variable immunodeficiency, cholangiocarcinoma, Waldenstrom’s macroglobulinemia
Abbr: Pre-LTx: pre liver transplant, post-LTx: post liver transplant, NASH: non-alcoholic steatohepatitis, PSC: primary sclerosing cholangitis; PBC: primary biliary cholangitis, HCC: hepatocellular carcinoma, Tac: tacrolimus, CsA: cyclosporine A, MMF: mycophenolate mofetil; P: prednisone, Az:azathioprine; Evr: everolimus
Cell collection and preparation.
PBMC were isolated by gradient centrifugation using Ficoll-Paque (GE Healthcare, Piscataway, NJ), as previously described15,23 and splenocytes (SPL) were isolated from deceased donor spleens. Details are provided in Methods S1.
Ex vivo belatacept-mediated costimulatory blockade (CSB) during MLR.
MLRs with belatacept-mediated CSB were established as described previously15,23 with minor modifications as described in Methods S2.
Flow Cytometry
Measurement of Treg frequency.
Treg frequencies were determined in unmanipulated PBMCs (baseline) and after 3 days in CSB-MLR by flow cytometry (gating strategy, Fig. S2; antibodies, Table S1) as described previously15 and detailed in Methods S3.
Intracellular cytokine staining.
Interleukin-2 (IL-2) response to T cell stimulation was assessed using flow cytometry as described in Methods S4.
TCR signaling assessment by phospho-flow cytometry.
PBMC were stimulated with 1 μg/mL anti-CD3 (clone UCTH1) and anti-CD28 (clone CD28.2) and then processed following BD Phosflow protocol for human PBMCs (BD Bioscience) as detailed in Methods S5.
FOXP3 Treg-specific demethylated region (TSDR) methylation analysis.
Treg were isolated from HC and post-LTx responder PBMC at baseline (unstimulated) or derived from primary CSB-MLRs. Untouched CD4+T cells were pre-enriched using EasySep Human CD4+ T cell negative selection kit (StemCell Technologies, St. Louis, MO) then stained with CD4, CD25, and CD127 antibodies and a 7-AAD viability dye. Viable Treg (7AAD-CD4+CD25hiCD127−) and control conventional (CD4+CD25−) T cells (Tconv) were sorted (Sony SH800z Cell Sorter, Sony Biotechnology, San Jose, CA), flash frozen in liquid nitrogen and submitted to EpigenDx Inc. (Hopkinton MA) for TSDR assay (Assay ID: ADS783FS2). For female samples, values were corrected by a factor of 2 as one TSDR allele is methylated due to X-inactivation.53
Real-time PCR.
Total RNA was isolated from PBMC and analyzed as detailed in Methods S6.
Treg-mediated Suppression.
Treg-mediated suppression was assessed by adding purified CSB-Treg to a fresh in vitro MLR followed by flow cytometry analysis. CSB-Treg were purified from new (modified) CSB-MLRs using HC or post-LTx patient responder PBMC with EasyStep Human CD4+CD127low CD25+Regulatory T cell Isolation kit (StemCell Technologies, Vancouver, USA), per manufacturer’s instructions. By flow cytometry, isolated Treg purity was always above 90%. Fresh untreated autologous responder PBMCs were labeled with CellTrace™ (CTV) Proliferation Kit (Life Technologies) prior to co-culture, per manufacturer’s instructions. Purified CSB-Treg were added to an in vitro MLR containing 105 CTV-labeled autologous responder PBMCs and equal numbers of γ-irradiated stimulators (i.e. same stimulator used to generate the CSB-Treg) in U-bottom 96-well plates. Treg:responder ratios of 1:5 and 1:10 were evaluated. Triplicate wells were set up for each condition. After 7–8 days, cells were stained and analyzed using flow cytometry (antibodies, Table S1) to assess CD8+ T cell proliferation (CTV dilution), activation (CD25 and/or HLA-DR expression) and cytotoxicity (perforin+granzymeB expression). Preliminary experiments using HLA-A2 discordant responder:stimulator pairs confirmed the absence of live stimulators at day 7–8. Percentage suppression was calculated as 100 × [1 – (untreated responders + Treg)/(untreated responders alone)].
Statistics
Statistical analysis was performed using GraphPad Prism v8 (GraphPad, La Jolla, CA). Two-tailed unpaired or paired t tests were performed throughout. Unpaired t tests did not assume equal variance. Welch’s correction was applied where appropriate. F test was used to compare variances. Pearson’s r was calculated to assess correlation. A p value of <0.05 was used to reject the null hypothesis.
Results
Quantitative and qualitative characteristics of PB lymphocytes and Treg in individuals with before and after liver transplantation
Our prior Treg expansion studies mostly used responder PBMC from healthy (stem cell transplant or HC) donors unlikely to have significant T cell defects. Studies report quantitative and qualitative immune abnormalities in individuals with ESLD.29,35–41 Post-LTx patients experience immunomodulation via IST, alloantigen exposure, potential rejection and/or infection.1,6,7,44,46,47,50,54,55 To determine whether T cells from individuals pre- and/or post-LTx were suitable for our manufacturing strategy, we first evaluated lymphocyte yield, phenotype and several relevant parameters of T cell immunity.
Pre- and post-LTx patients have decreased T cell and Treg counts.
Pre- and post-LTx populations had similar median PBMC concentrations falling within expected HC range (Fig. 1A).56 In comparison to HC (median x106 cells/mL 0.862 and 95% CI 0.751–1.103), pre- (0.403, 0.356–0.560) and post-LTx (0.465, 0.396–0.648) samples exhibited significant lymphopenia (Fig. 1B). All groups had similar CD19+B cell concentrations (Fig. 1C). However, in comparison to HC (median x106 cells/mL 0.593, 95% CI 0.519–0.792), pre- (0.231, 0.199–0.338) and post-LTx (0.230, 0.194–0.348) samples had significantly decreased CD3+T cell concentrations (Fig.1C). Relative to HC (median x106 cells/mL 0.371, 95% CI 0.302–0.441), pre- (0.160, 0.143–0.247) and post-LTx (0.130, 0.129–0.219) samples had decreased CD4+T cells (Fig. 1D). Relative to HC (median x106 cells/mL 0.191, 95% CI 0.159–0.237), pre- (0.038, 0.041–0.082) and post-LTx (0.055, 0.050–0.125) samples also had lower CD8+T cell concentrations. Pre- and post-LTx concentrations of CD3+, CD4+ and CD8+T cells were similar.
Figure 1. Pre- and post-LTx patients show T cell lymphopenia and decreased absolute numbers of Treg.
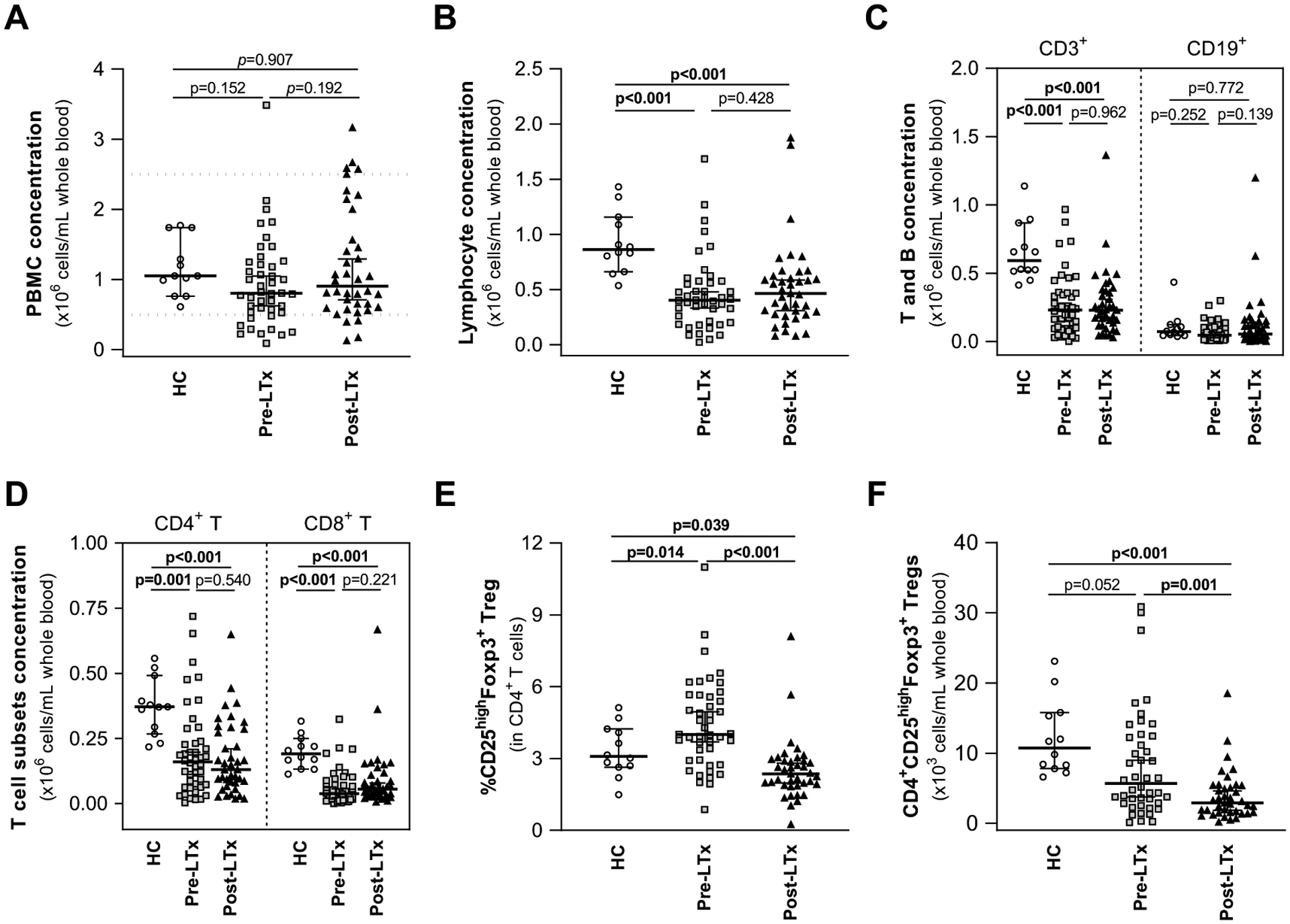
PBMC were isolated from whole blood of pre-LTx (n=44) and post-LTx patients (n=39), and HC donors (n=12).
(A) The absolute number of PBMC/mL was calculated using trypan blue staining and manual cell count. Values were similar for all groups. Dotted lines represent the normal range of healthy adult subjects.72
(B-F) Flow cytometry was used to assess the concentration (cells/ml of whole blood) of (B) lymphocytes, (C), CD3+ and CD19+ lymphocytes, (D) CD4+ and CD8+ T cells. Pre- and post-LTx patients demonstrated significant lymphopenia, decreased CD3+, CD4+ and CD8+ T cells in comparison to HC. Differences in (E) CD25highFoxp3+ Treg frequency were observed. (F) The concentration (cells/ml of whole blood) of CD25high Foxp3+ Treg within CD3+CD4+ T cells in pre- and post-LTx patients was decreased in comparison to HC.
Data show the median +/− 95% CI. P-values were calculated using unpaired t-test with Welch’s correction. Significant p-values are highlighted in bold. LTx: liver transplant; HC: healthy controls; PBMC: peripheral blood mononuclear cells; Treg: regulatory T cell.
Treg frequency (Fig. 1E) was significantly greater in pre- (median 4.47, 95%CI 4.029–5.181) and lower in post-LTx (median, 2.29, 95%CI 2.032–2.896) samples than in HC (median 3.085, 95%CI 2.642–3.998). Post-LTx samples also had lower Treg frequency than pre-LTx. However, reflecting their CD4+ lymphopenia, median Treg concentration (x103 cells/mL, Fig. 1F) was decreased in pre- (median 5.67, 95%CI 5.93–10.55) and post-LTx (median 2.920, 95%CI 2.747–5.025) compared to HC (median 10.75, 95%CI 8.774–15.59). This was significant for post-LTx samples with a trend observed for pre-LTx. Post-LTx Treg concentration was decreased in comparison to pre-LTx. Notably, the variance in the Treg frequency (Fig. 1E) was greater in pre-LTx (1.917) than post-LTx (1.351) samples (p=0.0285) as was the variance in Treg concentration (Fig. 1F) (pre-LTx 7.855 vs post-LTx 3.738, p<0.0001).
T cell receptor (TCR) signaling and IL-2 production are preserved in PBMC from pre- and post-LTx patients.
As MLR depends on TCR and costimulatory signaling,57,58 we used anti-CD3/CD28 antibodies to provide stimulation and assessed several key signaling molecules by phospho-flow cytometry. T cells in all groups responded with increased phosphorylation of proximal (Akt) and distal (Erk and S6) TCR pathway signaling molecules (Fig. 2). Results in HC and post-LTx were similar with a median 2–3-fold increase in phosphorylation in CD4+ and CD8+T cells. Median increase in phosphorylation was greater (2–6-fold) in pre-LTx samples. This was significant for all comparisons in CD4+T cells. In CD8+T cells, there was no significant difference between pre-LTx and HC, while the pre- vs post-LTx difference reached significance for pAkt and pErk but not pS6.
Figure 2. T cell receptor signaling, and IL-2 production are preserved in PBMC from pre- and post-LTx patients.
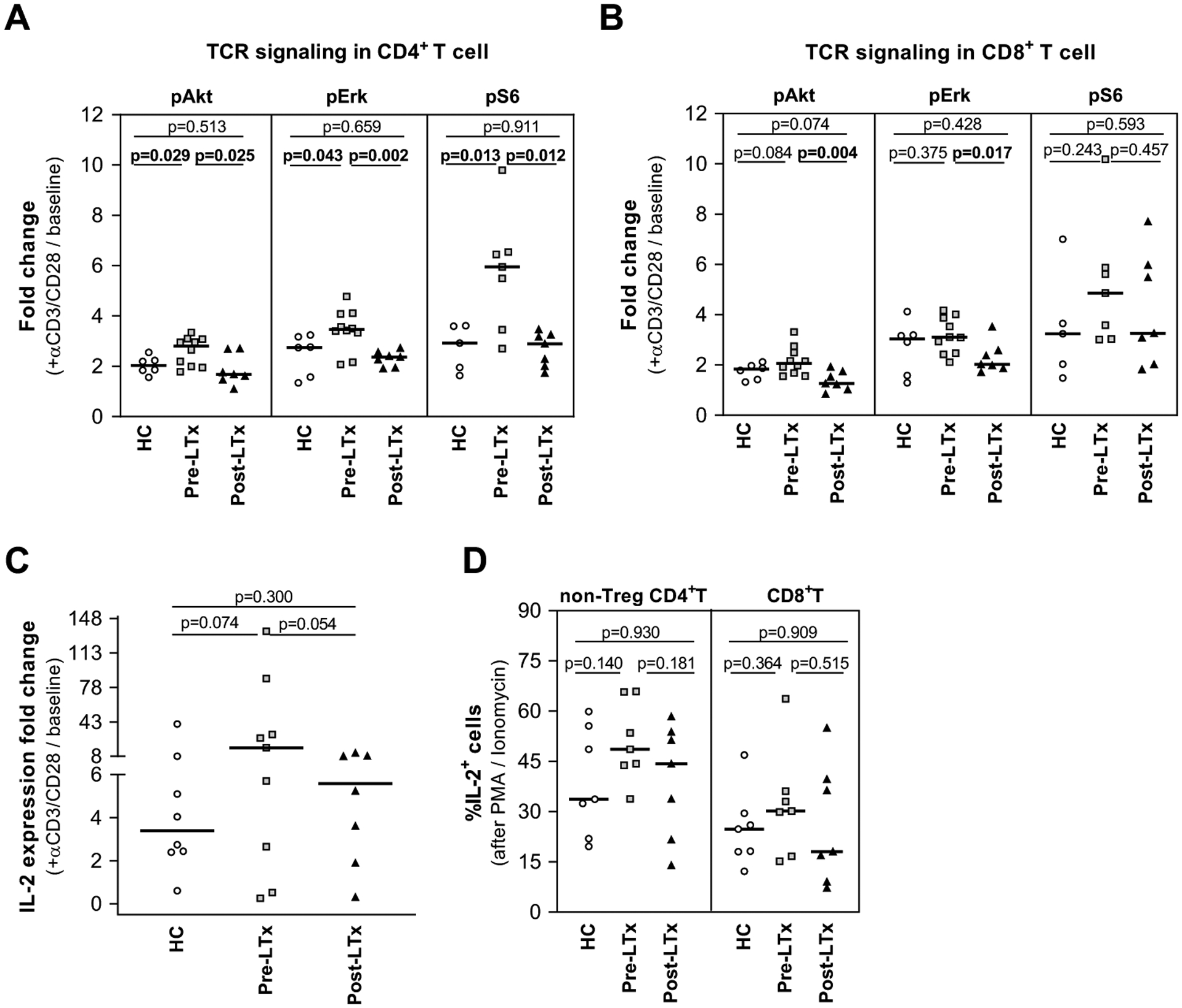
(A-C) PBMC were stimulated with anti-CD3/CD28 antibodies for 6h. (A-B) The integrity of TCR signaling was studied using phospho-flow cytometry to assess phosphorylated Akt, Erk, and S6 signaling molecules, in (A) CD4+ and (B) CD8+ T cells. In all cases, HC and post-LTx cells generated similar results whereas values in pre-LTx were consistently higher in CD4+, but not CD8+, pre-LTx cells. Values represent the fold change to baseline of each stimulated responder (HC=6, pre-LTx=10, post-LTx=7). (C) IL-2 production was evaluated using real time PCR. The IL-2 fold change vs unstimulated cells demonstrated similar results for all groups, with a trend to greater increase in pre-LTx (HC=8, pre-LTx=9, post-LTx=7). (D) PBMC stimulated for 4–6h with PMA/Ionomycin were analyzed by flow cytometry to determine the frequency of IL-2+ in non-Treg CD4+ and CD8+T cells. Results were equivalent in all groups (n=7/group).
Results represent the median. P-values were calculated using unpaired t-test with Welch’s correction. Significant p-values are highlighted in bold. HC: healthy controls; PBMC: peripheral blood mononuclear cells; TCR: T cell receptor; LTx: liver transplant.
IL-2 production is critical to Treg expansion and function.58–64 By RT-qPCR, baseline IL-2 expression by PBMC (median, range: HC 0.33, 0.11–1.01; pre-LTX 0.33, 0.08–8.23; post-LTx 0.30 0.14–1.52) showed no significant differences (HC vs pre-LTx p=0.3516, HC vs post-LTx p=0.2164, pre-LTx vs post-LTx p=0.4796). IL-2 expression rose in response to anti-CD3/anti-CD28 antibody stimulation in all groups (median, range: HC 2.06, 0.17–6.42; pre-LTX 4.31, 0.25–18.39; post-LTx 1.097, 0.26–12.25). The fold increase (Fig. 2C) was similar overall with trend for higher response in pre-LTx samples. We also evaluated the frequency of IL-2 producing cells after PMA/ionomycin stimulation. As CD4+ Treg do not produce IL-2,65,66 we assessed non-Treg CD4+ and CD8+T cells (Fig. 2D). IL-2 producing non-Treg CD4+T cell frequency (median, 95%CI) was similar in all groups: HC (33.70, 24.00–53.70), pre- (48.60, 39.8–61.80) and post-LTx (44.30, 23.91–55.29). IL-2 producing CD8+T cell frequency) was also similar: HC (24.80, 14.68–35.49), pre- (30.20, 17.27–46.96) and post-LTx (19.20, 10.77–46.28). There were no significant differences in pre- vs post-LTx non-Treg CD4+ or CD8+ IL-2 producing cell frequency.
Belatacept-mediated CSB-MLR reduced alloproliferation and supported Treg expansion by both HC and post-LTx PBMC responders
The most likely LITTMUS trial design favored Treg production from recipient PBMC collected post-LTx. As no contraindications to using post-LTx responders for Treg manufacturing had emerged, we next compared effects of belatacept-mediated CSB-MLR on HC and post-LTx PBMC responders. Notably, despite post-LTx IST, alloproliferation of post-LTx PBMC responders in MLRs without CSB was very similar to HC (p=0.7508). Addition of belatacept reduced alloproliferation in both HC and post-LTx responders (Fig. 3A) with significantly greater inhibition using post-LTx (median 82.7%, range 66.2–95.6) than HC (median 71.0%, range 39.4–97.95) PBMC.
Figure 3. Belatacept-mediated CSB during primary MLR reduces alloproliferation and supports Treg expansion by post-LTx PBMC responders.
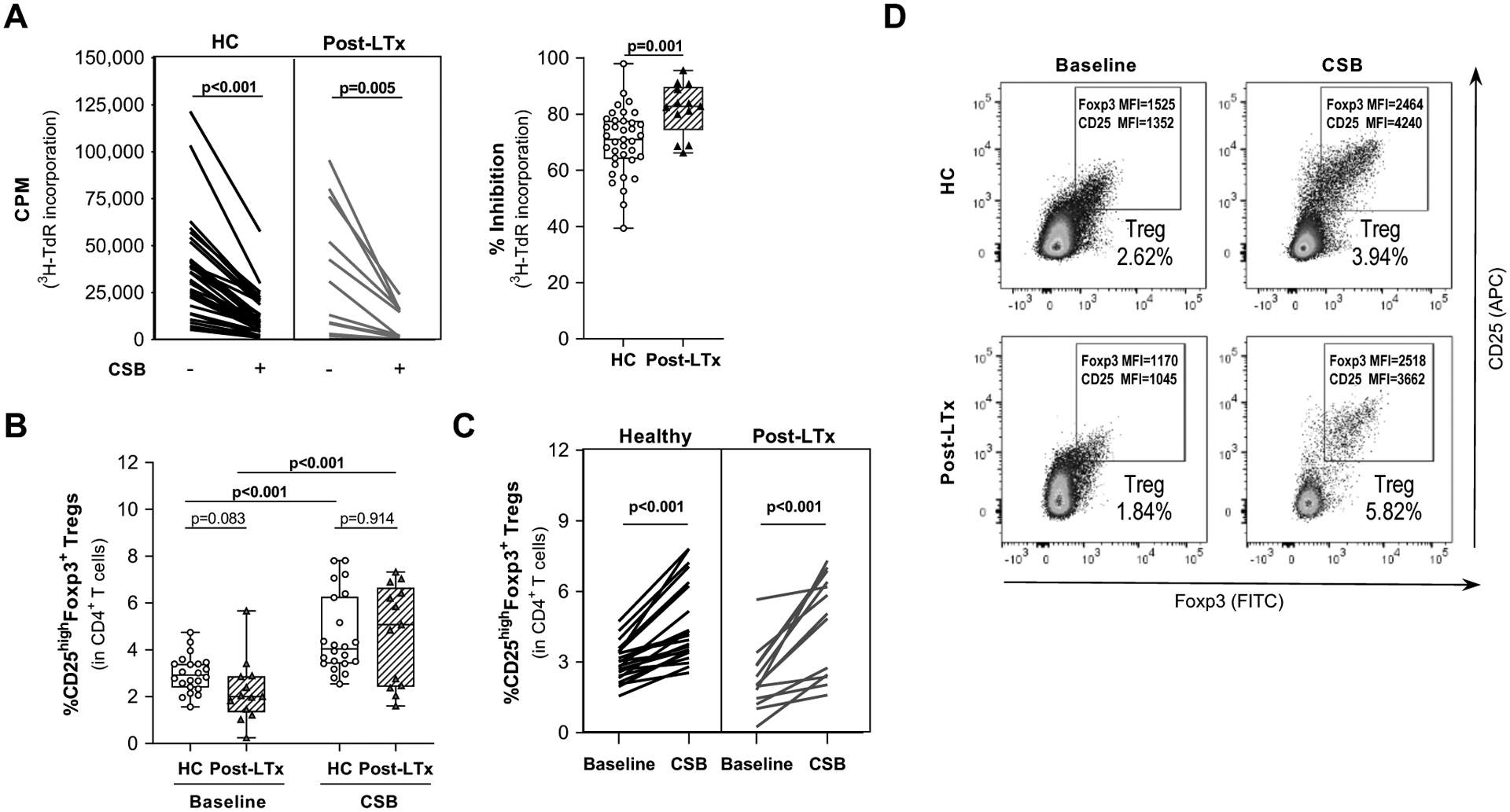
Responder PBMC from HC and post-LTx patients were co-cultured with irradiated PBMC stimulators in the presence or absence of belatacept-mediated CSB.
(A) Proliferation was measured by 3H-thymidine incorporation on day 6 of primary MLR. Left panel shows pairwise proliferation data for each sample in the presence (+) or absence (−) of belatacept-mediated CSB. Right panel shows aggregate data with median % inhibition of proliferation. CSB inhibited proliferation in both groups. The median degree of inhibition was greater in post-LTx samples (HC=34, post-LTx=22).
(B-D) Frequency of Treg (CD25highFoxp3+) within CD3+CD4+ T cells was assessed by flow cytometry prior to MLR and after primary CSB-MLR (denoted CSB) All panels: HC=22, post-LTx n=13. (B) The median Treg increase was similar for HC and post-LTx samples as was the final proportion of Treg in CD4+ cells. (C) The expansion of Treg for each pair is shown. (D) Representative flow cytometry plots demonstrated the marked increase in Foxp3 and CD25 MFI after CSB.
Data show the median +/− maximum/minimum range. P-values were calculated using paired t-test or unpaired t-test with Welch’s correction. Significant p-values are highlighted in bold. HC: healthy controls; PBMC: peripheral blood mononuclear cells; CSB: costimulatory blockade; MLR: mixed lymphocyte reaction; LTx: liver transplant; cpm: counts per minute; Treg: regulatory T cell; MFI: mean fluorescence intensity
Treg frequency at baseline (Fig. 3B,C) was lower in post-LTx (median 2.04%, range 0.24–7.86) than HC (median 2.92%, range 1.56–4.75) CD4+ PBMC. After CSB-MLR, Treg frequency increased significantly in both groups (Fig. 3B,C) with no significant difference in final frequency between HC (median 4.04%, range 2.54–7.81) and post-LTx (median 5.07%, range 1.6–7.32). Baseline Treg frequency correlated with the frequency after CSB-MLR in both HC and post-LTx responders (r=0.8308 with p<0.001, and r=0.6236 with p=0.0228, respectively). There was no correlation between baseline values and fold-change (HC: r=0.1153 with p=0.6093; post-LTx: r=−0.2861 with p=0.3674). Treg from both groups expressed increased surface CD25 and intracellular Foxp3 (Fig. 3D). Pre-LTx PBMC produced similar results (Fig. S3), although median (1.27-fold) Treg expansion by pre-LTx responders was significantly less than HC (1.53, p=0.015) or post-LTx (2.38, p=0.047).
Adapting and optimizing belatacept-mediated CSB as a manufacturing strategy for Treg generation in the setting of liver transplantation
Most LTx are performed using deceased donor organs, making collection of PBMC stimulators for Treg manufacturing problematic. We therefore evaluated thawed allogeneic splenocytes (SPL, processed and cryopreserved after organ procurement) as stimulators. We also determined whether altering CSB-MLR conditions could enhance Treg production.
SPL can be substituted for PBMC stimulators in CSB-MLR generation of Treg.
Primary MLRs and CSB-MLRs were set up using HC PBMC responders and either HC PBMC or SPL as stimulators (stimulators came from different individuals as paired acquisition was not feasible from HC or deceased donors). CSB-mediated inhibition of proliferation using SPL or HC PBMC was comparable (Fig. 4A: SPL median 79.23%, range 55.97–92.30 vs PBMC median 70.39%, range 39.40–108.0). SPL stimulators also supported similar increases in Treg frequency (Fig. 4B: SPL median1.96-fold increase (range 0.77–2.430) vs PBMC median 1.54-fold increase (range 1.11–3.50) (p=0.207).
Figure 4. The use of splenocytes as stimulators supports CSB-mediated expansion of Treg in healthy PBMCs.
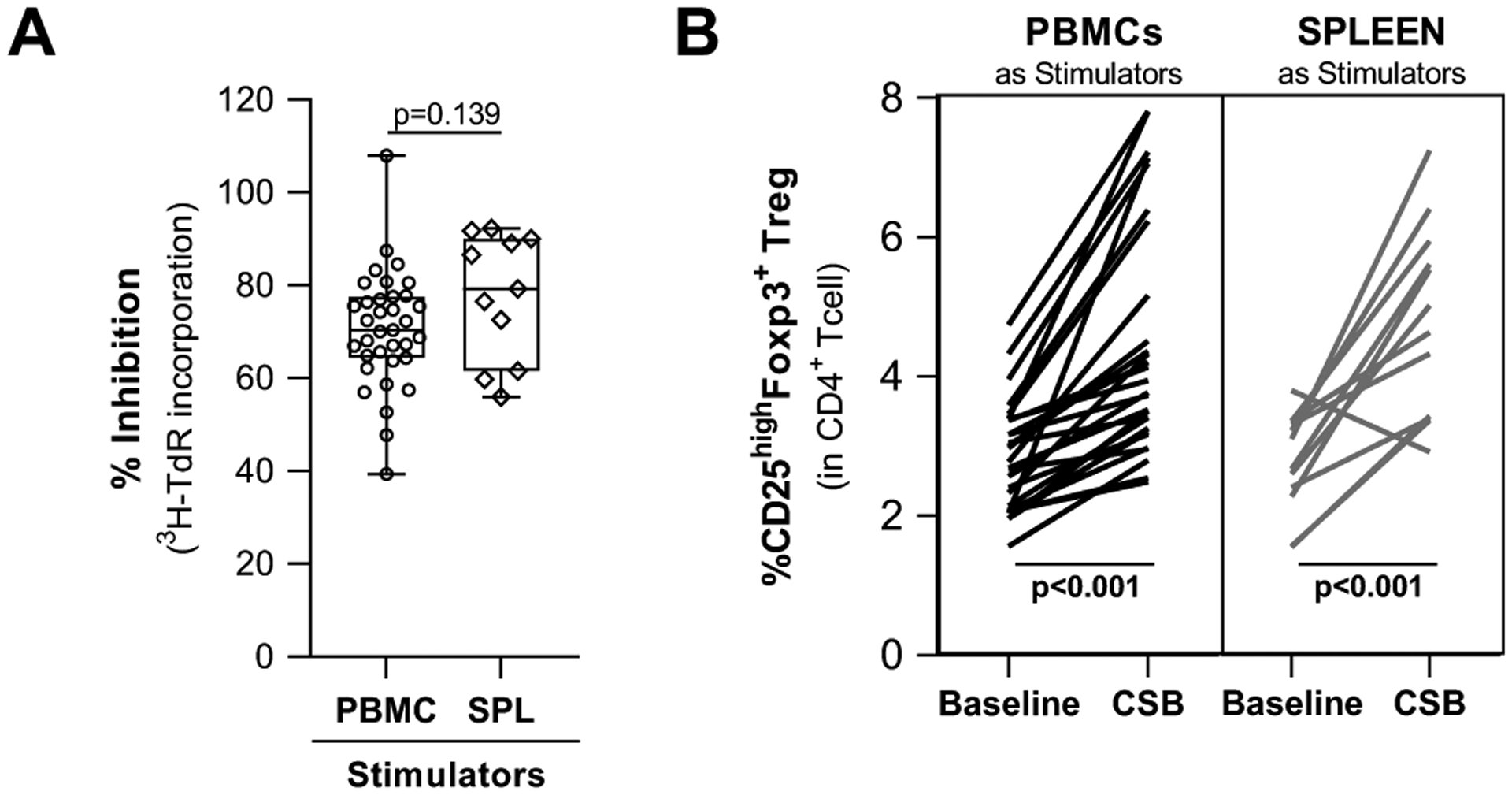
HC were co-cultured with irradiated PBMC or SPL as stimulators in the presence of belatacept-mediated CSB.
(A) Percent inhibition of proliferation (measured by 3H-thymidine incorporation) on day 6 of primary CSB-MLR was comparable using PBMCs or SPL as stimulators. (HC=35, post-LTx=11)
(B) Frequencies of CD25highFoxp3+ Treg expressed as percentage of CD3+CD4+ cells prior to (baseline) and after primary CSB-MLR (denoted CSB) indicated similar Treg expansion using PBMC or SPL as stimulators. (HC=28, post-LTx=12)
Horizontal lines are medians, boxes are 25th and 75th percentiles, and whiskers are maximum and minimum values. P-values were calculated using unpaired t-test with Welsh’s correction (A) and paired-t test (B) with significant p-values highlighted in bold. HC: healthy controls; MLR: mixed lymphocyte reaction; CSB: costimulatory blockade; SPL: splenocytes; Treg: regulatory T cell; MFI: mean fluorescence intensity
Modified CSB-MLR conditions augment Treg yield.
Production of sufficient Treg in a feasible, cost-effective manner required reduction of CSB-MLR coculture volume and increased yield from a given cell input. Increases in cell density (from 1×106 to 5×106 cells/mL) and responder:stimulator ratio (1:1 to 2:1) were screened to determine the most promising (“new”) conditions (1.5×106 total cells/mL; 2:1). No significant difference in CSB-mediated inhibition of MLR proliferation was observed using standard or new conditions with PBMC or SPL stimulators (Fig. 5A).
Figure 5. New CSB-MLR coculture conditions support scale up of Treg expansion in healthy PBMC.
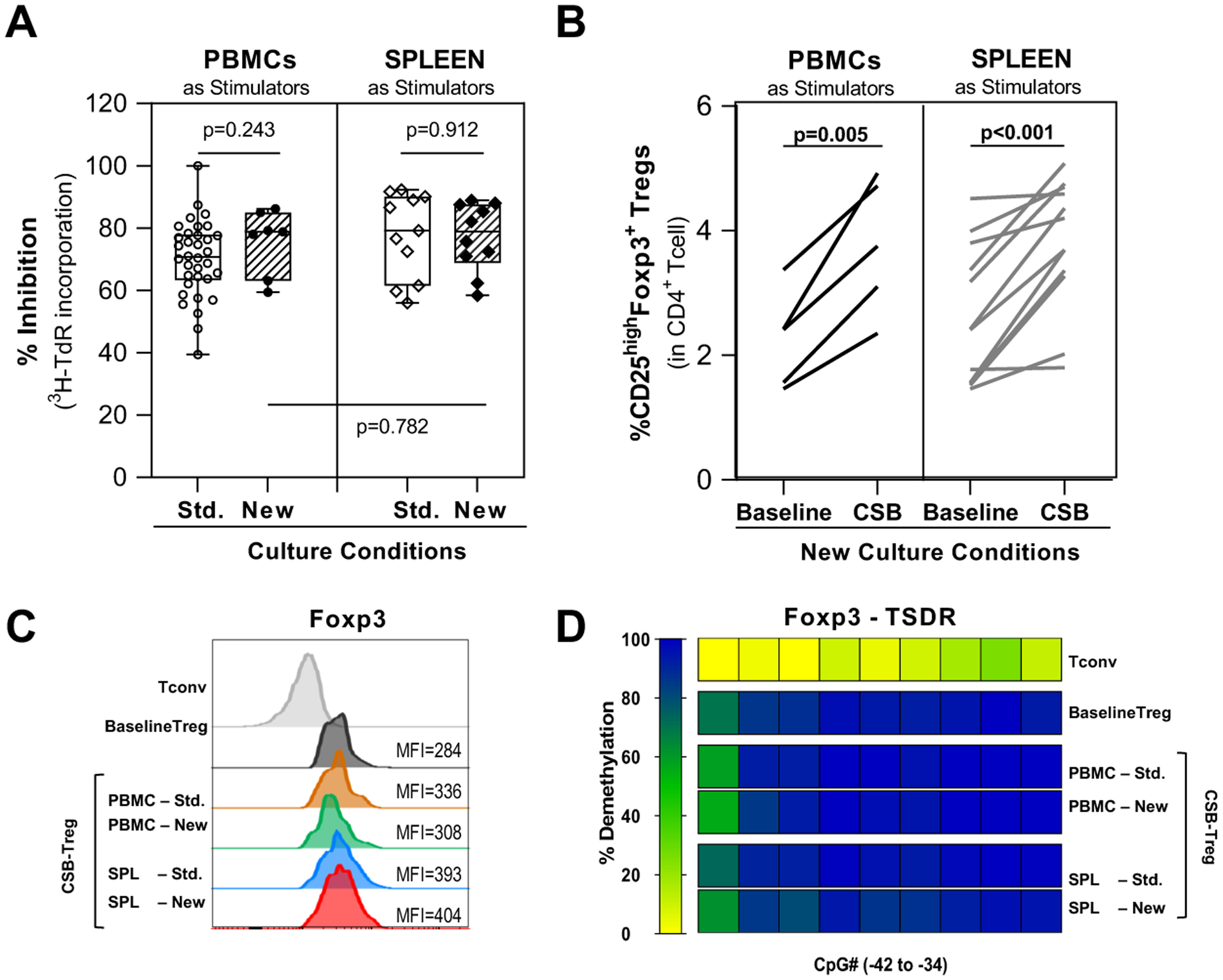
HC PBMC were co-cultured with irradiated PBMC or SPL stimulators in the presence of belatacept-mediated CSB using standard culture conditions (1×106 cells/mL and 1R:1S ratio) or new optimized conditions (1.5×106 cells/mL and 2R:1S ratio)
(A) Percent inhibition of proliferation (measured by 3H-thymidine incorporation) on day 6 of primary CSB-MLR was not significantly different using standard or new CSB-MLR co-culture conditions with either PBMC (left) or SPL (right) as stimulators. (n=10–34)
(B) Under new CSB-MLR culture conditions, measurement of CD25highFoxp3+ Treg frequency, expressed as percentage of CD3+CD4+ T cells, prior to MLR (baseline) and after primary CSB-MLR (CSB) demonstrated similar Treg expansion using PBMCs (n=5) or SPL (n=10) as stimulators.
(C-D) Characterization of CSB-MLR Treg manufactured using different culture conditions. Conventional CD4 T cells (Tconv) and natural Treg without CSB (baseline Treg) were used as controls. (C) Representative histograms and MFI values of the Treg marker Foxp3 in CSB-Treg (CD4+CD25highFoxp3+) showed no differences in Foxp3 expression. (D) TSDR demethylation in CSB-Tregs isolated by multicolor fluorescence-activated cell sorting (CD4+CD25highCD127−). The demethylation rates were translated into a color code from yellow (0%) through green (50%) up to blue (100%). Each rectangle represents the demethylation of one CpG motif. All CSB-Tregs showed comparable high demethylation pattern. (Tconv n=1; baseline Treg n=5; CSB-Treg n=2–5)
Horizontal lines are medians, boxes are 25th and 75th percentiles, and whiskers are maximum and minimum values. The p-values were calculated using unpaired T test with Welch’s correction (A) and paired t-test (B) with significant p-values highlighted in bold. HC: healthy controls; MLR: mixed lymphocyte reaction; CSB: costimulatory blockade; SPL: splenocytes; Std: standard culture conditions; New: new optimized culture conditions; MFI: mean fluorescence intensity; Tconv: conventional CD4 T cells; TSDR: Treg-specific demethylated region; Treg: regulatory T cell
Treg expansion under new conditions (Fig. 5B) was comparable using either PBMC or SPL stimulators (median 1.61-fold increase [range, 2.03–1.40] vs 1.51 [range, 2.37–0.89], respectively, p=0.36). Treg produced under both conditions using either stimulator had similar Foxp3 MFI, a correlate of Treg suppressive capacity (Fig. 5C).67 Moreover, high TSDR demethylation status was sustained in Treg isolated after CSB-MLR under both conditions and using either stimulator (Fig. 5D). Natural Treg isolated before CSB-MLR had highly demethylated FOXP3 TSDR (median 93.6%, range 78.5–93.9, n=5). The TSDR in CSB-Treg remained highly demethylated (condition:stimulator cell type: Standard:PBMC [98.4%, range 94.9–101.8 (n=2)]; Standard:SPL [94.8%, range 88.9–100.7 (n=2)]; New:PBMC [95.0%, range 88.0–100.3 (n=5)]; New:SPL [90.4.0%, range 61.1–102.3 (n=5)]).
New CSB-MLR coculture conditions support Treg expansion in post-LTx PBMC.
To evaluate these modifications in the trial context, CSB-MLRs using new conditions were conducted with three post-LTx PBMC responders. CSB produced consistent inhibition of MLR proliferation (Fig. 6A: median 74%, range 66–88%), increased CD25hiFoxp3+ Treg frequency (Fig. 6B: median increase 1.74-fold, range 1.15–2.32) and increased Treg surface CD25 (Fig. 6C). As observed in HC (Fig. 5C), intracellular Foxp3 MFI in CSB-Treg from post-LTx PBMC responders was increased (Fig. 6D). These CSB-Treg also demonstrated high TSDR demethylation (Fig. 6E) comparable to CSB-Treg from HC [post-LTx 94.0%, range 86.7–102.0 (n=3) vs HC 90.4.0%, range 61.1–102.3 (n=5); p-value = 0.3977].
Figure 6. New CSB-MLR co-culture conditions with SPL as stimulators support Treg expansion in post-LTx PBMCs.
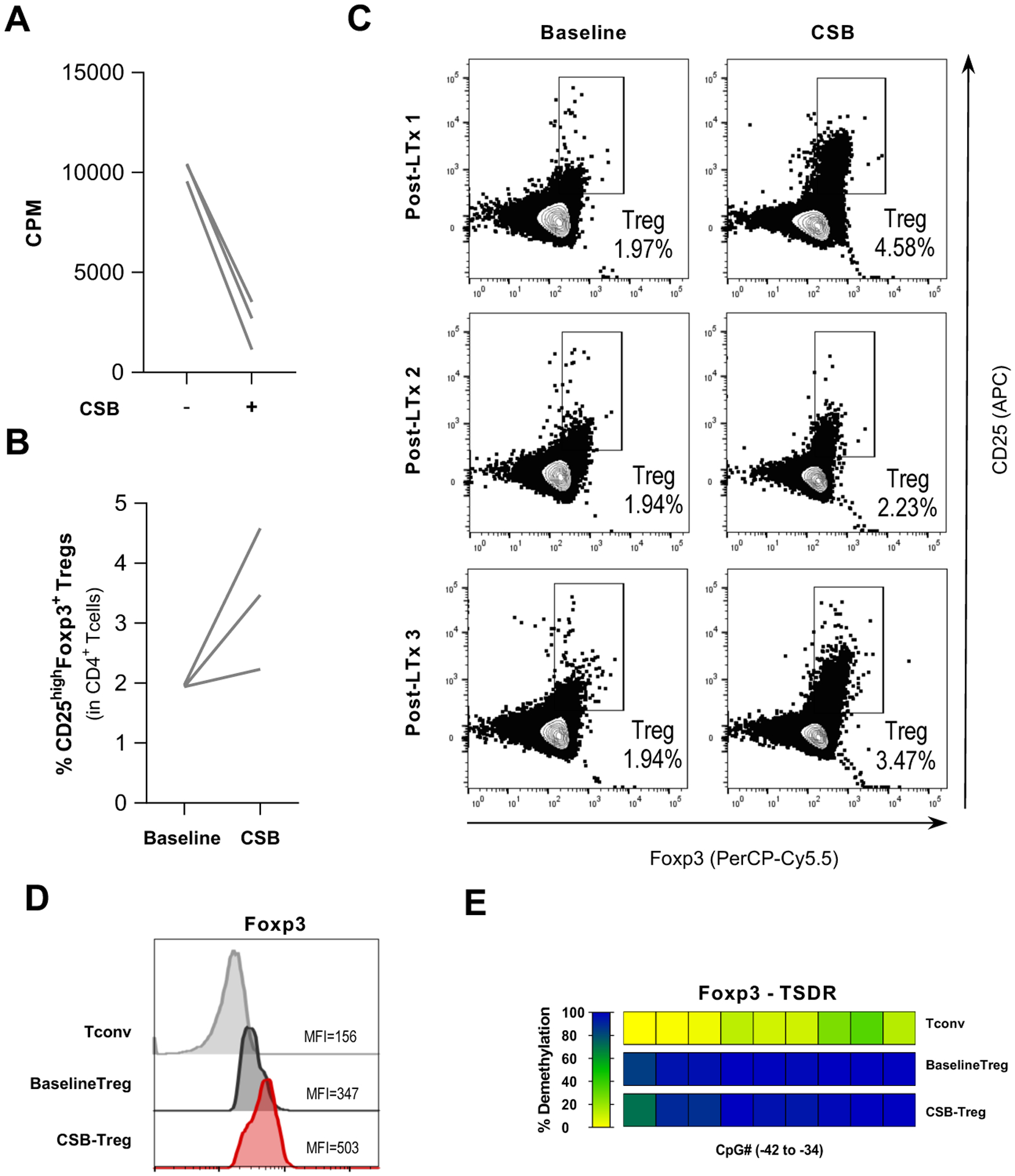
PBMCs from post-LTx patients were co-cultured using new MLR conditions (1.5×106 cells/mL and 2R:1S ratio) with irradiated SPL as stimulators in the presence of belatacept-mediated CSB.
(A) CSB-MLR demonstrated expected decreases in proliferation, measured by 3H-thymidine incorporation on day 6, showed an overall median percentage inhibition of 76% (range, 88 – 66%).
(B) Frequencies of CD25highFoxp3+ Tregs expressed as percentage of CD3+CD4+ T cells at day 0 (baseline) and day 3 CSB-MLR (CSB) showed an overall 1.9-fold median Treg expansion.
(C) Individual flow cytometry plots demonstrate the increase in CD25 expression and the frequency of CD25highFoxp3+ Tregs within CD3+CD4+ gate prior to MLR (baseline) and at day 3 after primary CSB-MLR culture (CSB).
(D-E) Characterization of CSB-MLR Treg from post-LTx PBMC samples. Conventional CD4+ T cells (Tconv) and natural Tregs without CSB (baseline Treg) from post-LTx patients were used as controls. (D) Representative histograms and MFI values of the Treg marker Foxp3 in post-LTx CSB-Tregs (CD4+CD25highFoxp3+) showed high Foxp3 expression. (E) TSDR demethylation in CSB-Treg isolated by multicolor fluoresce-activated cell sorting (CD4+CD25highCD127−). The demethylation rates were translated into a color code from yellow (0%) through green (50%) up to blue (100%). Each rectangle represents the demethylation of one CpG motif. Post-LTx CSB-Treg showed retention of high TSDR demethylation. (Tconv n=2; baseline Treg n=3; CSB-Treg n=3).
HC: healthy controls; PBMC: peripheral blood mononuclear cells; CSB: costimulatory blockade; MLR: mixed lymphocyte reaction; post-LTx: post liver transplant; SPL: splenocytes; cpm: counts per minute; Treg: regulatory T cell. MFI: mean fluorescence intensity; Tconv: conventional CD4+ T cells; TSDR: Treg-specific demethylated region; Treg: regulatory T cell
More limited characterization of CSB-Treg generation using PBMC from 4 patients on RAPA-containing IST (Fig. S4) was consistent with the larger dataset.
CSB-Treg from HC and post-LTx PBMC effectively suppress autologous CD8+ T cells.
Alloreactive T cells, particularly cytotoxic CD8+ T cells, are implicated in immune-mediated tissue damage and graft rejection and can be modulated by Treg.68–71 To assess the functionality of CSB-Treg generated from post-LTx (using the new conditions), we evaluated their capacity to suppress autologous CD8+ T using an in vitro MLR between the original (autologous) responder and γ-irradiated stimulator SPL pair. We observed negligible proliferation of CTV-labelled CD8+ T cells within post-LTx responder PBMC alone whereas brisk proliferation of CD8+ T cells was observed in MLR co-cultures with corresponding increases in the proportion expressing activation (CD25+ and/or HLA-DR+) and cytotoxicity-associated (granzymeB+perforin+) markers (Fig. 7A, columns R, RS). Similar results were observed using HC PBMC responders and stimulators (Fig. 7B, columns R, RS).
Figure 7. CSB-Treg from post-LTx and HC PBMC effectively suppress autologous CD8+ T cells.
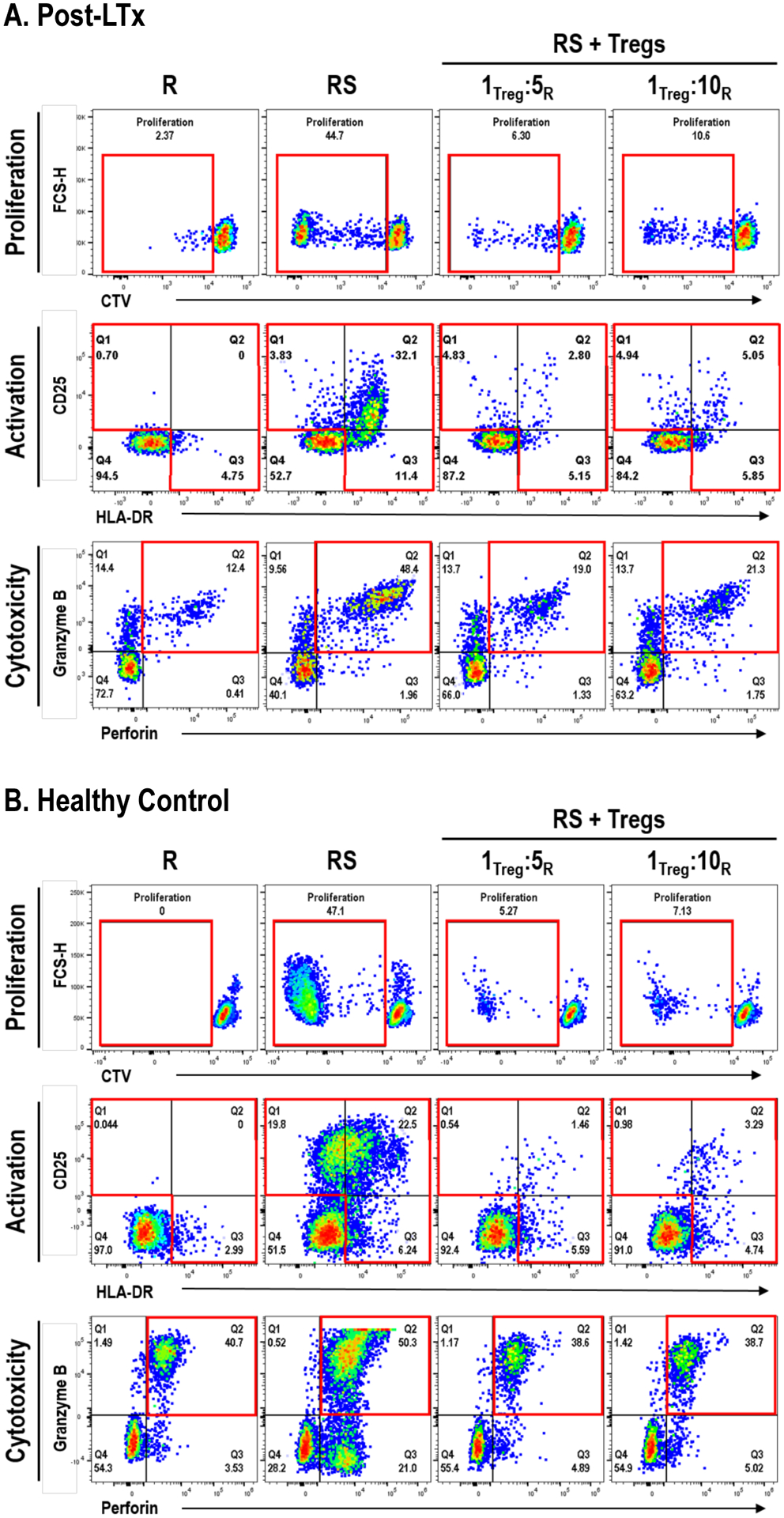
CSB-Treg from post-LTx patients (A) or HC samples (B) generated using the new conditions were evaluated for their capacity to suppress autologous CD8+ T cells using an in vitro MLR between the original (autologous) CTV-labeled responder PBMCs and the γ-irradiated stimulator pair. Different ratios of Treg:Responder were evaluated. At day 7, autologous responder PBMC cells were stained and analyzed using flow cytometry to assess CD8+ T cell proliferation (CTV dilution), as well as frequency of CD8+ T cells expressing activation (CD25+ and/or HLA-DR+) and cytotoxic markers (granzyme B+perforin+). Data is shown in representative flow cytometry plots from single experiments (independent triplicate experiments were performed for each condition, using n=3 post-LTx and n=3 HC samples). All samples were gated within CD3+CD8+ viable lymphocytes. Gates of interest are highlighted in red. Resting responder cells (R) cultured for the same period were used as a control.
(A) CD8+ T cells from post-LTx samples showed little proliferation whereas they demonstrated significant proliferation, and an increased proportion expressed activation markers and cytotoxic markers after MLR (RS). Addition of CSB-Tregs to the MLR (Treg + RS) was associated with robust suppression of autologous responder CD8+ T cell proliferation and a decrease CD8+ T cells expressing activation and cytotoxic molecules. (B) Similar results to (A) were obtained using HC PBMC responders, stimulators and HC CSB-Treg.
R: responder PBMC; S: Stimulator SPL or PBMCs; RS: responder PBMCs cocultured with stimulator cells (SPL cells for post-LTx and PBMC for HC); RS+Tregs: RS plus autologous Treg (isolated from a CSB-MLR using the same R and S) at either 1Treg:5R or 1Treg:10R ratios. HC: healthy controls; PBMC: peripheral blood mononuclear cells; CSB: costimulatory blockade; post-LTx: post liver transplant; SPL: splenocytes; Treg: regulatory T cell; CTV: CellTraceTM dye; MLR: mixed lymphocyte reaction
Addition of post-LTx CSB-Treg to MLRs at a ratio of 1Treg:5Responders results in robust suppression of autologous responder CD8+ T cells (Fig. 7A) with decreases (median, range) in proliferation (74.7%, 63.4–85.9%) and the percent expressing activation (73.0%, 73.0–73.1%) and cytotoxic (66.4%, 60.7–72.1%) markers. These results were comparable to those observed with addition of HC CSB-Treg to HC responders [decreases in proliferation (78.6%, range 38.4–88.8%) and the percent expressing activation (84.4%, range 66.4–86.4%) and cytotoxic (49.6%, range 23.3–75.2%) markers] (Fig 7B). Similar but slightly decreased suppression was observed when responder PBMC from LTx patients or HCs were cultured at 1Treg:10Responders, suggesting a dose-response effect (Fig. 7 A,B). These data show that in the in vitro setting CSB-Treg derived from post-LTx PBMC provided potent suppression of autologous alloreactive cytotoxic CD8+ T cell responses.
Use of oxygen permeable tissue culture bags permits scaled up manufacture of CSB-MLR Treg.
In all studies above, CSB-MLRs were performed in culture flasks or plates.15 As considerable volume is required to manufacture Treg in greater numbers, we evaluated use of oxygen permeable tissue culture bags with 32–510 ml/bag capacity. CSB-MLR were conducted in either flasks or bags (Fig. S5) using same HC responder:stimulator pairs and standard conditions (1×106 cells/mL; 1:1 responder:stimulator ratio; PBMC stimulators). Pair-to-pair comparison resulted in similar CD4+ responder cell viability, Treg frequency within CD4+, as well as Foxp3 MFI expression. We next evaluated the new conditions (1.5×106 cells/mL; 2:1 responder:stimulator ratio; SPL stimulators) in 32 ml tissue culture bags, using HC PBMC responders. Frequencies of Treg of expected phenotype increased during CSB-MLR.
Optimized CSB-MLR with post-LTx leukapheresis PBMC produces a Treg product suitable for clinical administration.
When using post-LTx responders, we of necessity relied on stimulator PBMC or SPL that were not identical to the liver donor. Here, we briefly describe the manufacturing outcome of the first patient enrolled in the LITTMUS trial, which used post-LTx PBMC responders from the organ recipient and banked SPL stimulators from the organ donor. Recipient responder PBMC were obtained 5 months post-LTx by leukapheresis, isolated by density gradient in CMCF, and used fresh. Using 17 510C culture bags, 9×109 recipient PBMC were cocultured with 4×109 40 Gy-irradiated donor SPL (total 1.5× 106/mL; 1.7:1 responder:stimulator ratio) in the presence of belatacept. 1.3-fold Treg expansion was measured in the pooled washed CSB-MLR product (Fig. 8). Surface CD25 and intracellular Foxp3 MFI increased, and 93.6% of expanded CD25hiCD127− Treg expressed Foxp3. Despite significant losses during column-based Treg selection, final Treg yield increased 1–2 log compared to standard conditions using venipuncture-acquired PBMC in a renal transplant setting.18
Figure 8. CSB-Treg manufacturing.
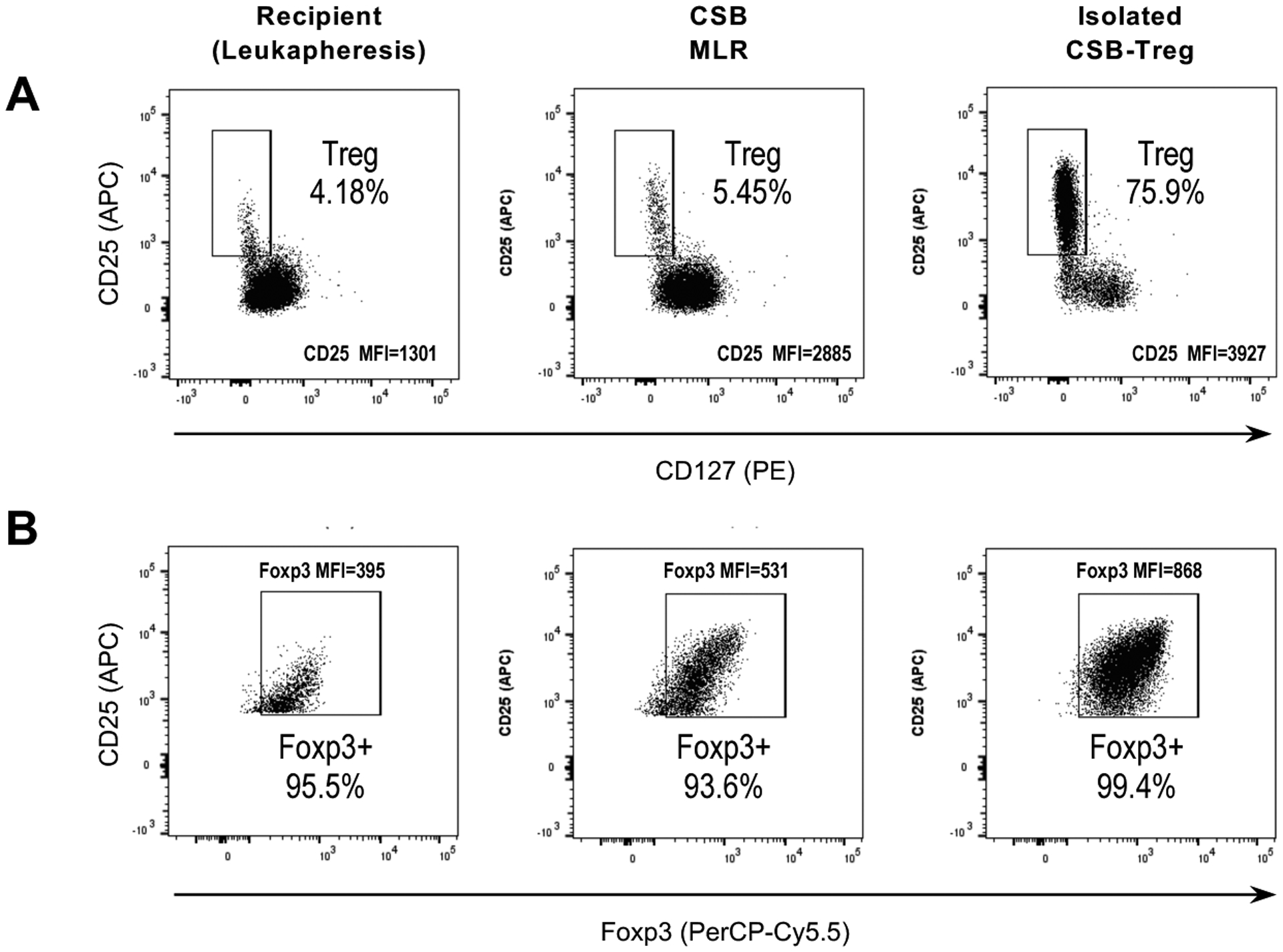
Flow cytometry plots show the frequency of CD25highCD127− Tregs within CD3+CD4+ T cells (A) and the frequency of Foxp3+ cells within CD25highCD127− Treg (B), in the organ recipient PBMCs prior to MLR (baseline), after primary CSB-MLR culture (CSB), and in the final Treg product after Treg isolation using depletion and selection columns and magnetic beads. MLR: mixed lymphocyte reaction; CSB: costimulatory blockade; MFI: mean fluorescence intensity; Treg: regulatory T cell.
Discussion
Studies detailing variable abnormalities in PBMC from ESLD patients pre- and post-LTx1–3,5–7,35–41,44–52,72 provide insufficient basis for predicting whether these characteristics would compromise CSB-MLR-mediated Treg manufacturing. Therefore, we evaluated pre- and post-LTx PBMC, from baseline characteristics through CSB-MLR manufacturing conditions, to assess feasibility and then improve outcome of CSB-MLR as a Treg production strategy for LTx.
Many animal models and in vitro studies describe IST effects, but fewer detail effects of in vivo chronic post-transplant IST exposure on human PBMC.3,32,46,50,54,55,72–75 We therefore used pre/post-LTx PBMC to examine immune functions, including response to activation and IL-2 expression, present in HC cells in which our strategy was developed and related to mechanisms by which: 1) CSB exerts specificity and engenders anergy, and 2) Treg expand and persist.57–64 We found that individuals pre-LTx or on IST post-LTx could respond to anti-CD3/anti-CD28 or PMA/ionomycin stimulation and proliferate to alloantigen. Indeed, responses closely resembled those of HC. Our IL-2 expression data is consistent with Hartel et al76 who found PBMC from HC and post-kidney transplant patients on IST demonstrated similar IL-2 mRNA expression at baseline and after anti-CD3/anti-CD28 stimulation. Notably, peak IL-2 expression was delayed in post-transplant samples, an analysis we could not perform as our samples were a volume-limited convenience sample whereas they acquired larger samples timed to IST administration. However, their work suggests additional timepoints should be adopted for future studies.
Our short duration CSB-MLR manufacturing process has many benefits – as previously described, it is fast, simple, produces Treg with significant allospecificity14,23 and is relatively economical as it requires few special reagents or tools. However, unlike longer ex vivo expansion approaches,9,10,12,13,16–19,21,22,24,25 the shorter process provides less time for Treg expansion and likely underlies the direct correlation between baseline values and final Treg yield. Lower baseline Treg concentrations were not associated with decreased relative expansion. This suggested greater input could compensate for the lower expansion, prompting a decision to obtain responder PBMC by leukapheresis rather than phlebotomy. During CSB-MLR, Treg expansion from post-LTx samples was significantly greater than observed with pre-LTx and comparable to HC. As persuasive clinical considerations favored use of post-LTx PBMC, and absent clear contraindications to their use, we prioritized examination of post-LTx samples in process modification. Interestingly, while the trial and manufacturing strategy are very different, Sanchez-Fueyo et al17 recently reported improved Treg production using PBMC obtained by leukapheresis from patients on IST post-LTx rather than by venipuncture pre-LTx. Notably, we observed multiple pre-LTx assessments were more variable than were post-LTx, seemingly reflecting the inconsistent Treg quantitation reported in ESLD.35,39–41 Pursuing this observation was lateral to our aims but may require investigation in some manufacturing settings.
The decision to use leukapheresis engendered manufacturing changes, including transitions to large oxygen-permeable culture bags and moderate increments in cell concentration and ratio. Larger deviations resulted in increased cell death and decreased Treg yield. Similarly, use of deceased donors required rethinking the stimulator population and evaluating how SPL preparation processes affected manufacturing outcome. While selection or depletion of SPL subpopulations might improve results, the goal was to design a rapid and simple process that could minimize cost and effort, avert manufacturing failures potentiated by longer, more complex approaches and facilitate dissemination. Modified CSB-MLR conditions reproduced prior results15 with both HC and post-LTx samples yielding Treg with increased Foxp3 MFI and highly demethylated TSDR signatures, characteristics associated with suppressive activity and functional stability. HC and post-LTx CSB-Treg also demonstrated significant capacity to suppress autologous responder proliferation and expression of activation and cytotoxic molecules.
In summary, together with many other considerations favoring their use, these studies provide support for use of post-LTx PBMC as a manufacturing substrate to generate Treg via CSB-MLR. Moreover, this procedure can be adapted to accommodate the practical demands of using deceased donors and improving Treg yield without compromising its attractive simplicity and rapidity. This stepwise translational process illustrates the importance of assessing the demands of each cell therapy setting and reconfiguring manufacturing processes to address the identified limitations and opportunities.
Supplementary Material
Acknowledgments:
We would like to acknowledge the technical contributions of Kenneth Janec, the professional and collaborative effort of the Connell O’Reilly Cell Manipulation Core Facility and the dedication of all MGH staff who helped to procure the samples used in this work. We would also like to thank the patients and families without whose generosity this study could not have been performed.
Funding and support:
Research reported in this publication was performed as a project of the Immune Tolerance Network and supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health under Award Number UM1AI109565. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Belatacept was received from Bristol Myers Squibb as a gift to conduct this research project.
Abbreviations:
- Az
azathioprine
- CM
complete media
- CsA
cyclosporine
- CSB
costimulatory blockade
- ESLD
end-stage liver disease
- Evr
everolimus
- HC
healthy control
- IL-2
interleukin-2
- IST
immunosuppressive therapy
- LTx
liver transplantation
- MLR
mixed lymphocyte reaction
- MMF
mycophenolate mofetil
- MTX
methotrexate
- P
prednisone
- PB
peripheral blood
- PBMC
peripheral blood mononuclear cells
- RAPA
sirolimus
- SPL
splenocyte
- TAC
tacrolimus
- Tconv
conventional T cells
- TCR
T cell receptor
- Treg
regulatory T cells
- TSDR
Treg-specific demethylated region
Footnotes
Disclosure: LT is employed by and holder of equity in Rubius Therapeutics and a founder of and holder of equity in Rheos Medicines. The remaining authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Supporting Information: Additional supporting information may be found online in the Supporting Information section at the end of the article.
Data Availability:
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
References
- 1.Bardou FN, Guillaud O, Erard-Poinsot D, et al. Tacrolimus exposure after liver transplantation for alcohol-related liver disease: Impact on complications. Transpl Immunol. 2019;56:101227. [DOI] [PubMed] [Google Scholar]
- 2.Bianchi G, Marchesini G, Marzocchi R, Pinna AD, Zoli M. Metabolic syndrome in liver transplantation: Relation to etiology and immunosuppression. Liver Transplantation. 2008;14(11):1648–1654. [DOI] [PubMed] [Google Scholar]
- 3.Cangemi M, Montico B, Faè DA, Steffan A, Dolcetti R. Dissecting the Multiplicity of Immune Effects of Immunosuppressive Drugs to Better Predict the Risk of. Front Oncol. 2019;9:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fine RN. Tolerance in Solid-Organ Transplant. Exp Clin Transplant. 2016;14(Suppl 3):1–5. [PubMed] [Google Scholar]
- 5.Laish I, Braun M, Mor E, Sulkes J, Harif Y, Ben Ari Z. Metabolic syndrome in liver transplant recipients: prevalence, risk factors, and association with cardiovascular events. Liver Transpl. 2011;17(1):15–22. [DOI] [PubMed] [Google Scholar]
- 6.Manzia TM, Angelico R, Gazia C, et al. malignancies after liver transplantation: The effect of immunosuppression-personal data and review of literature. World J Gastroenterol. 2019;25(35):5356–5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Russo MW. The Care of the Postliver Transplant Patient. J Clin Gastroenterol. 2017;51(8):683–692. [DOI] [PubMed] [Google Scholar]
- 8.Rickert CG, Markmann JF. Current state of organ transplant tolerance. Current Opinion in Organ Transplantation. 2019;24(4):441–450. [DOI] [PubMed] [Google Scholar]
- 9.Romano M, Fanelli G, Albany CJ, Giganti G, Lombardi G. Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity. Front Immunol. 2019;10:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang Q, Vincenti F. Transplant trials with Tregs: perils and promises. J Clin Invest. 2017;127(7):2505–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trzonkowski P, Bacchetta R, Battaglia M, et al. Hurdles in therapy with regulatory T cells. Sci Transl Med. 2015;7(304):304ps318. [DOI] [PubMed] [Google Scholar]
- 12.Bluestone JA, Tang Q. Treg cells-the next frontier of cell therapy. Science. 2018;362(6411):154–155. [DOI] [PubMed] [Google Scholar]
- 13.Berglund D, Korsgren O, Lorant T, Schneider K, Tufveson G, Carlsson B. Isolation, expansion and functional assessment of CD4+CD25+FoxP3+ regulatory T cells and Tr1 cells from uremic patients awaiting kidney transplantation. Transpl Immunol. 2012;26(1):27–33. [DOI] [PubMed] [Google Scholar]
- 14.Davies JK, Nadler LM, Guinan EC. Expansion of allospecific regulatory T cells after anergized, mismatched bone marrow transplantation. Sci Transl Med. 2009;1(1):1ra3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guinan EC, Cole GA, Wylie WH, et al. Ex Vivo Costimulatory Blockade to Generate Regulatory T Cells From Patients Awaiting Kidney Transplantation. Am J Transplant. 2016;16(7):2187–2195. [DOI] [PubMed] [Google Scholar]
- 16.Safinia N, Vaikunthanathan T, Fraser H, et al. Successful expansion of functional and stable regulatory T cells for immunotherapy in liver transplantation. Oncotarget. 2016;7(7):7563–7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sánchez-Fueyo A, Whitehouse G, Grageda N, et al. Applicability, safety, and biological activity of regulatory T cell therapy in liver transplantation. Am J Transplant. 2020;20(4):1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sawitzki B, Harden PN, Reinke P, et al. Regulatory cell therapy in kidney transplantation (The ONE Study): a harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet. 2020;395(10237):1627–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Todo S, Yamashita K. Anti-donor regulatory T cell therapy in liver transplantation. Hum Immunol. 2018;79(5):288–293. [DOI] [PubMed] [Google Scholar]
- 20.Watanabe M, Kumagai-Braesch M, Yao M, et al. Ex Vivo Generation of Donor Antigen-Specific Immunomodulatory Cells: A Comparison Study of Anti-CD80/86 mAbs and CTLA4-lg Costimulatory Blockade. Cell Transplant. 2018;27(11):1692–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whitehouse GP, Hope A, Sanchez-Fueyo A. Regulatory T-cell therapy in liver transplantation. Transpl Int. 2017;30(8):776–784. [DOI] [PubMed] [Google Scholar]
- 22.Zheng SG, Meng L, Wang JH, et al. Transfer of regulatory T cells generated ex vivo modifies graft rejection through induction of tolerogenic CD4+CD25+ cells in the recipient. Int Immunol. 2006;18(2):279–289. [DOI] [PubMed] [Google Scholar]
- 23.Davies JK, Barbon CM, Voskertchian A, Nadler LM, Guinan EC. Ex vivo alloanergization with belatacept: a strategy to selectively modulate alloresponses after transplantation. Cell Transplant. 2012;21(9):2047–2061. [DOI] [PubMed] [Google Scholar]
- 24.Mathew JM, HV J, LeFever A, et al. A Phase I Clinical Trial with Ex Vivo Expanded Recipient Regulatory T cells in Living Donor Kidney Transplants. Sci Rep. 2018;8(1):7428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seay HR, Putnam AL, Cserny J, et al. Expansion of Human Tregs from Cryopreserved Umbilical Cord Blood for GMP-Compliant Autologous Adoptive Cell Transfer Therapy. Mol Ther Methods Clin Dev. 2017;4:178–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berglund D, Karlsson M, Biglarnia AR, et al. Obtaining regulatory T cells from uraemic patients awaiting kidney transplantation for use in clinical trials. Clin Exp Immunol. 2013;173(2):310–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clavien PA, Muller X, de Oliveira ML, Dutkowski P, Sanchez-Fueyo A. Can immunosuppression be stopped after liver transplantation? Lancet Gastroenterol Hepatol. 2017;2(7):531–537. [DOI] [PubMed] [Google Scholar]
- 28.Todo S, Yamashita K, Goto R, et al. A pilot study of operational tolerance with a regulatory T-cell-based cell therapy in living donor liver transplantation. Hepatology. 2016;64(2):632–643. [DOI] [PubMed] [Google Scholar]
- 29.Heymann F, Tacke F. Immunology in the liver--from homeostasis to disease. Nat Rev Gastroenterol Hepatol. 2016;13(2):88–110. [DOI] [PubMed] [Google Scholar]
- 30.Crispe IN. Immune tolerance in liver disease. Hepatology. 2014;60(6):2109–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng S, Bucuvalas J. Tolerance after liver transplantation: Where are we? Liver Transpl. 2017;23(12):1601–1614. [DOI] [PubMed] [Google Scholar]
- 32.Benítez C, Londoño MC, Miquel R, et al. Prospective multicenter clinical trial of immunosuppressive drug withdrawal in stable adult liver transplant recipients. Hepatology. 2013;58(5):1824–1835. [DOI] [PubMed] [Google Scholar]
- 33.Lerut J, Sanchez-Fueyo A. An appraisal of tolerance in liver transplantation. Am J Transplant. 2006;6(8):1774–1780. [DOI] [PubMed] [Google Scholar]
- 34.Reyes J, Zeevi A, Ramos H, et al. Frequent achievement of a drug-free state after orthotopic liver transplantation. Transplant Proc. 1993;25(6):3315–3319. [PMC free article] [PubMed] [Google Scholar]
- 35.Li TY, Yang Y, Zhou G, Tu ZK. Immune suppression in chronic hepatitis B infection associated liver disease: A review. World J Gastroenterol. 2019;25(27):3527–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liaskou E, Hirschfield GM. Cirrhosis-associated immune dysfunction: Novel insights in impaired adaptive immunity. EBioMedicine. 2019;50:3–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sipeki N, Antal-Szalmas P, Lakatos PL, Papp M. Immune dysfunction in cirrhosis. World J Gastroenterol. 2014;20(10):2564–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duddempudi AT. Immunology in alcoholic liver disease. Clin Liver Dis. 2012;16(4):687–698. [DOI] [PubMed] [Google Scholar]
- 39.Choi YS, Lee J, Lee HW, et al. Liver injury in acute hepatitis A is associated with decreased frequency of regulatory T cells caused by Fas-mediated apoptosis. Gut. 2015;64(8):1303–1313. [DOI] [PubMed] [Google Scholar]
- 40.Xu D, Fu J, Jin L, et al. Circulating and liver resident CD4+CD25+ regulatory T cells actively influence the antiviral immune response and disease progression in patients with hepatitis B. J Immunol. 2006;177(1):739–747. [DOI] [PubMed] [Google Scholar]
- 41.Zhang H, Jiang Z, Zhang L. Dual effect of T helper cell 17 (Th17) and regulatory T cell (Treg) in liver pathological process: From occurrence to end stage of disease. Int Immunopharmacol. 2019;69:50–59. [DOI] [PubMed] [Google Scholar]
- 42.Intagliata NM, Argo CK, Stine JG, et al. Concepts and Controversies in Haemostasis and Thrombosis Associated with Liver Disease: Proceedings of the 7th International Coagulation in Liver Disease Conference. Thromb Haemost. 2018;118(8):1491–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liou IW. Management of end-stage liver disease. Med Clin North Am. 2014;98(1):119–152. [DOI] [PubMed] [Google Scholar]
- 44.Charlton M, Levitsky J, Aqel B, et al. International Liver Transplantation Society Consensus Statement on Immunosuppression in Liver Transplant Recipients. Transplantation. 2018;102(5):727–743. [DOI] [PubMed] [Google Scholar]
- 45.Fussner LA, Heimbach JK, Fan C, et al. Cardiovascular disease after liver transplantation: When, What, and Who Is at Risk. Liver Transpl. 2015;21(7):889–896. [DOI] [PubMed] [Google Scholar]
- 46.Kim JM, Kwon CHD, Joh JW, Choi GS, Kang ES, Lee SK. Comparative Peripheral Blood T Cells Analysis Between Adult Deceased Donor Liver Transplantation (DDLT) and Living Donor Liver Transplantation (LDLT). Ann Transplant. 2017;22:475–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miloh T, Barton A, Wheeler J, et al. Immunosuppression in pediatric liver transplant recipients: Unique aspects. Liver Transpl. 2017;23(2):244–256. [DOI] [PubMed] [Google Scholar]
- 48.Watt KD. Keys to long-term care of the liver transplant recipient. Nat Rev Gastroenterol Hepatol. 2015;12(11):639–648. [DOI] [PubMed] [Google Scholar]
- 49.Watt KD, Pedersen RA, Kremers WK, Heimbach JK, Charlton MR. Evolution of causes and risk factors for mortality post-liver transplant: results of the NIDDK long-term follow-up study. Am J Transplant. 2010;10(6):1420–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gronert Álvarez A, Fytili P, Suneetha PV, et al. Comprehensive phenotyping of regulatory T cells after liver transplantation. Liver Transpl. 2015;21(3):381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pageaux GP, Faure S, Bouyabrine H, Bismuth M, Assenat E. Long-term outcomes of liver transplantation: diabetes mellitus. Liver Transpl. 2009;15 Suppl 2:S79–82. [DOI] [PubMed] [Google Scholar]
- 52.Plotogea O, Ilie M, Sandru V, Chiotoroiu A, Bratu O, Diaconu C. Cardiovascular and Metabolic Consequences of Liver Transplantation: A Review. Medicina (Kaunas). 2019;55(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434(7031):400–404. [DOI] [PubMed] [Google Scholar]
- 54.Furukawa A, Wisel SA, Tang Q. Impact of Immune-Modulatory Drugs on Regulatory T Cell. Transplantation. 2016;100(11):2288–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Levitsky J, Miller J, Wang E, et al. Immunoregulatory profiles in liver transplant recipients on different immunosuppressive agents. Hum Immunol. 2009;70(3):146–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Riedhammer C, Halbritter D, Weissert R. Peripheral Blood Mononuclear Cells: Isolation, Freezing, Thawing, and Culture. Methods Mol Biol. 2016;1304:53–61. [DOI] [PubMed] [Google Scholar]
- 57.Gimmi CD, Freeman GJ, Gribben JG, Gray G, Nadler LM. Human T-cell clonal anergy is induced by antigen presentation in the absence of B7 costimulation. Proc Natl Acad Sci U S A. 1993;90(14):6586–6590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boussiotis VA, Freeman GJ, Berezovskaya A, Barber DL, Nadler LM. Maintenance of human T cell anergy: blocking of IL-2 gene transcription by activated Rap1. Science. 1997;278(5335):124–128. [DOI] [PubMed] [Google Scholar]
- 59.Almeida AR, Legrand N, Papiernik M, Freitas AA. Homeostasis of peripheral CD4+ T cells: IL-2R alpha and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J Immunol. 2002;169(9):4850–4860. [DOI] [PubMed] [Google Scholar]
- 60.Brandenburg S, Takahashi T, de la Rosa M, et al. IL-2 induces in vivo suppression by CD4(+)CD25(+)Foxp3(+) regulatory T cells. Eur J Immunol. 2008;38(6):1643–1653. [DOI] [PubMed] [Google Scholar]
- 61.Gasteiger G, Kastenmuller W. Foxp3+ Regulatory T-cells and IL-2: The Moirai of T-cell Fates? Front Immunol. 2012;3:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Malek TR, Castro I. Interleukin-2 receptor signaling: at the interface between tolerance and immunity. Immunity. 2010;33(2):153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ross SH, Cantrell DA. Signaling and Function of Interleukin-2 in T Lymphocytes. Annu Rev Immunol. 2018;36:411–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155(3):1151–1164. [PubMed] [Google Scholar]
- 65.Ono M, Yaguchi H, Ohkura N, et al. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature. 2007;446(7136):685–689. [DOI] [PubMed] [Google Scholar]
- 66.Wu Y, Borde M, Heissmeyer V, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126(2):375–387. [DOI] [PubMed] [Google Scholar]
- 67.Chauhan SK, Saban DR, Lee HK, Dana R. Levels of Foxp3 in regulatory T cells reflect their functional status in transplantation. J Immunol. 2009;182(1):148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ronca V, Wootton G, Milani C, Cain O. The Immunological Basis of Liver Allograft Rejection. Front Immunol. 2020;11:2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Choy JC. Granzymes and perforin in solid organ transplant rejection. Cell Death Differ. 2010;17(4):567–576. [DOI] [PubMed] [Google Scholar]
- 70.McNally A, Hill GR, Sparwasser T, Thomas R, Steptoe RJ. CD4+CD25+ regulatory T cells control CD8+ T-cell effector differentiation by modulating IL-2 homeostasis. Proc Natl Acad Sci U S A. 2011;108(18):7529–7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Taubert R, Pischke S, Schlue J, et al. Enrichment of regulatory T cells in acutely rejected human liver allografts. Am J Transplant. 2012;12(12):3425–3436. [DOI] [PubMed] [Google Scholar]
- 72.Benitez C, Lozano JJ, Fueyo AS. Gene expression profiling and transplantation tolerance in the clinic. Transplantation. 2009;88(3 Suppl):S50–53. [DOI] [PubMed] [Google Scholar]
- 73.Norero B, Serrano CA, Sanchez-Fueyo A, et al. Conversion to mycophenolate mofetil monotherapy in liver recipients: Calcineurin inhibitor levels are key. Ann Hepatol. 2017;16(1):94–106. [DOI] [PubMed] [Google Scholar]
- 74.In ‘t Veld AE, Grievink HW, Saghari M, et al. Immunomonitoring of Tacrolimus in Healthy Volunteers: The First Step from PK- to PD-Based Therapeutic Drug Monitoring? Int J Mol Sci. 2019;20(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Masri MA. The mosaic of immunosuppressive drugs. Mol Immunol. 2003;39(17–18):1073–1077. [DOI] [PubMed] [Google Scholar]
- 76.Hartel C, Fricke L, Schumacher N, Kirchner H, Muller-Steinhardt M. Delayed cytokine mRNA expression kinetics after T-lymphocyte costimulation: a quantitative measure of the efficacy of cyclosporin A-based immunosuppression. Clin Chem. 2002;48(12):2225–2231. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.