

Abstract
A novel series of asymmetrically substituted bis-thioureas has been synthesized in an effective pattern via the reaction of diamines with isothiocyanates utilizing the ultrasonic irradiation as a sustainable energy source. This reaction performs well at mild conditions to give the products in quantitative yields for a broad scope of substrates. The bis-thiourea derivatives are used for the design of unprecedented series of bis-2-iminothiazolidin-4-ones promoted by the ultrasonic irradiation. The reaction affords the formation of regioselective products, which depends on the pKas of the diamines. The diamine linked to the thiourea possessing lower pKa is involved in the imino part, and the diamine having higher pKa is a part of the other heterocyclic nitrogen. Moreover, this new strategy has excellent environmental parameters, demonstrating that this protocol is a green and sustainable process.
A series of bis-thioureas was synthesized as viable precursors for the design of bis-2-iminothiazolidin-4-ones utilizing the ultrasonic irradiation as a sustainable energy source.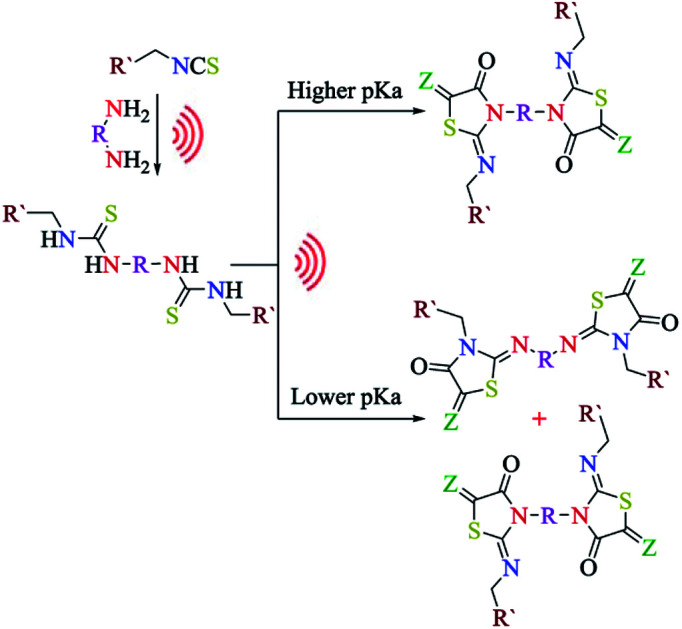
Introduction
Both symmetrical and asymmetrical thiourea derivatives represent a class of significant compounds in drug design.1 These derivatives have been utilized in many therapeutics such as anti-bacterial,2 anti-HIV,3 anti-inflammatory,4 anti-cancer,5 antioxidant,6 anti-tubercular,7 anti-parasitic,8 antifungal9 and rodenticide agents.10 Furthermore, the thiourea derivatives have been utilized as agrochemicals, such as herbicides,11 and as a regulator for the growth of insects.12 They have additionally wide applications in the preparation of heterocyclic compounds and other organic molecules.13 Recently, thioureas have been employed as ligands and organocatalysts in a diversity of asymmetric organic syntheses.14 Moreover, they have been used in the manufacture of polymer15 and as sensors for heavy metal detection.16 In addition, the applications of thiourea derivatives have been extended to analytical chemistry; they are used for the metal ions preconcentration.17 These fundamental characteristics of thiourea derivatives make them versatile building blocks in synthetic organic chemistry.18 Thus, great endeavors have been directed to the evolution of novel synthetic protocols for these important building blocks.19 These reported strategies have some disadvantages comprising the requirements of harsh reaction conditions, poisonous reagents, and the fact that these strategies are mainly restricted to prepare symmetrically substituted thioureas.
As a privileged moiety, thiazole is an important structural motif that has acquired an important standing in newfangled heterocyclic chemistry essentially because of its presence in a broad variety of biologically active compounds.20 Due to the high degree of biological activity of thiazole derivatives, numerous procedures of their preparation have been reported.21 In spite of these methods, newer protocols for the generation of thiazole derivatives remain in demand.
Recently, focusing on green chemistry utilizing eco-friendly chemicals and reaction conditions is a standout among the most intriguing advancements in the preparation of broadly utilized organic compounds.22 The ultrasound-assisted organic reactions have attracted much attention due to the greater selectivity, simplicity of procedure, repression of the generation of by-product, and fast preparation of different organic compounds when compared with traditional thermal heating.23 Solvent, the main contributor of the ecological performance of the reaction, is another factor that ought to be taken into consideration in a green methodology.24 Despite water being a predominantly attractive solvent, most organic compounds do not adequately dissolve in it.25 Alternatively, ethanol performs well as the reaction medium since it is comparatively non-hazardous to the environment.24 Motivated by the previously mentioned actualities and as a part of our interest in developing economic and simple reactions promoted by ultrasound to synthesize bis-heterocyclic compounds,22 herein, we wish to report a facile strategy for the fast preparation of new and asymmetrically substituted bis-thioureas as building blocks for the synthesis of bis-2-imino-4-thiazolinone derivatives in superb yields mediated by the ultrasonic irradiation.
Results and discussion
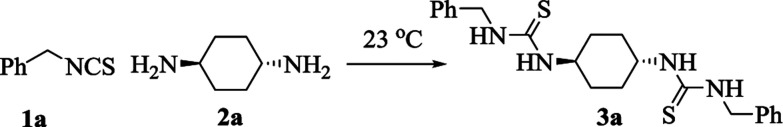
In our initial studies based on the preparation of asymmetrically substituted bis-thioureas, benzyl isothiocyanate 1a and trans-1,4-diaminocyclohexane 2a were chosen as the model reagents for the optimization of the reaction conditions. To test the possibility of our envisioned protocol, in a model reaction, to a solution of benzyl isothiocyanate 1a (1 mmol) in acetonitrile (MeCN, 10 mL), trans-1,4-diaminocyclohexane 2a (0.5 mmol) was added pinch wise over a period of 5 min under stirring at ambient temperature (23 °C). The stirring process was continued for 120 min until a sticky solid was formed. MeOH (2 mL) was added to this mixture with continuous stirring for another 15 min. The formation of a novel product along with other minor side products was observed together with the retention of some starting materials. Isolation by a usual workup procedure was followed by purification (45% isolated yield, Table 1, entry 1), and the characterization revealed the product to be 1,1′-[(1R,4R)-cyclohexane-1,4-diyl]bis(3-benzylthiourea) 3a. Further optimization such as increasing the amount of benzyl isothiocyanate to 2.5 equiv. ameliorated the isolated yield (3a) up to 58%. Next, the above-mentioned model reaction was carried out under ultrasonic irradiation (40 MHz) for 5 min at ambient temperature (23 °C). Thin layer chromatography displayed the complete transformation of the starting materials to the product 3a. Interestingly, the obtained bis-thiourea derivative 3a was isolated instantly after the substrates were mixed, and it recrystallized from EtOH in an excellent yield (92%, Table 1, entry 3). Screening of some other solvents all afforded inferior outcomes except dichloromethane (DCM), which afforded 3a in 88% yield (Table 1, entry 4). For instance, EtOH and water delivered 3a in only 21% and 25% yields, respectively (Table 1, entries 5 and 6). A polar aprotic solvent such as acetone also afforded a poor reaction yield (Table 1, entry 7). Interestingly, performing the aforesaid model reaction under a solvent-free condition furnished the required product 3a in 97% yield (Table 1, entry 8). Reducing the reaction time to 3 min led to the incomplete conversion to product 3a (Table 1, entry 9). Consequently, the ultrasonic irradiation of 1a (1 mmol) and 2a (0.50 mmol) at room temperature for 5 min were selected as the optimum conditions to produce the derivative 3a in 97% yield (Table 1, entry 8).
Optimization of the reaction conditions for the preparation of 1,1′-[(1R,4R)-cyclohexane-1,4-diyl]bis(3-benzylthiourea) 3aa.
| ||||
|---|---|---|---|---|
| Entry | Solvent | Method | Time (min) | Yield (%)b |
| 1 | MeCN | Stirring | 120 | 45 |
| 2 | MeCN | Stirring | 120 | 58c |
| 3 | MeCN | US | 5 | 92 |
| 4 | DCM | US | 5 | 88 |
| 5 | EtOH | US | 5 | 21 |
| 6 | H2O | US | 10 | 25 |
| 7 | Acetone | US | 5 | 15 |
| 8 | — | US | 5 | 97 |
| 9 | — | US | 3 | 82 |
The reaction was conducted with 1a (1.0 mmol) and 2a (0.5 mmol).
Isolated yields.
The reaction was conducted with 1a (1.5 mmol) and 2a (0.5 mmol).
After verifying the practicality of the protocol, the generality and scope of this reaction were assessed by using different types of isothiocyanates (1a, b) and diamines (2a, b) (Scheme 1). Better yields were observed in the case of trans-1,4-diaminocyclohexane (2a), for example, 3a (97%) and 3b (96%). The suggested structures of the derivatives 3a–d were deduced from their satisfactory spectral data. The IR spectra of the compounds 3a–d displayed the characteristic stretching bands of NH and C S groups at 3271–3207 and 1262–1250 cm−1, respectively. The 1H NMR spectra of the derivatives 3a–d showed the NH protons of the thiourea functionality (S CNHR) at the region δ = 7.89–7.25 ppm. The other protons showed up at the expected chemical shifts (ESI†).
Scheme 1. An ultrasound-assisted protocol for the synthesis of asymmetrically substituted bis-thioureas 3a–d.26.

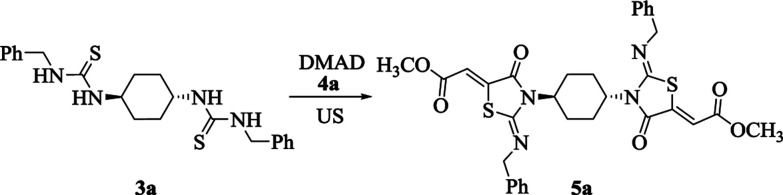
The asymmetrically substituted thiourea frameworks have been demonstrated to be advantageous precursors for diversified functional compounds including thiazoles, imidazoles and pyrimidines. Thus, bis-thioureas 3a–d represent versatile building blocks for the preparation of various bis-heterocycles such as bis-thiazolidines. For our initial investigation that aimed to synthesize bis-thiazolidines, asymmetrically substituted bis-thiourea 3a and dimethyl acetylenedicarboxylate (DMAD, 4a) were taken as the model substrates. We commenced this study by conducting the reaction in EtOH at room temperature under ultrasonic irradiation, which exclusively produced the compound 5a within 7 min (Table 2, entry 1). The obtained results encouraged us to further investigate the influence of the solvent and temperature on the model reaction. As displayed in Table 2, different outcomes of the target product 5a (46–88%) were obtained when the mixture of 3a with DMAD was stirred under given conditions in diverse solvents. The reaction using MeOH and water gave the corresponding product 5a in satisfactory yields (Table 2, entries 2 and 3). In contrast, the results with other solvents such as MeCN and DCM were unsatisfactory (Table 2, entries 4 and 5). Likewise, due to the issue of isolating the solid products, poor yields occurred when dioxane and DMF were employed as solvents in the model reaction (Table 2, entries 6 and 7). From the environmental and economical standpoint and considering the toxicity of MeOH, water was selected as the reaction medium for this series of reactions. Moreover, on performing the corresponding solvent-free reaction, an unfruitful result was obtained (Table 2, entry 8). Similarly, increasing the operation temperature of the reaction did not enhance the reaction yields (Table 2, entries 9 and 10). Nevertheless, slight improvement in the reaction yield was observed when the time was increased to 25 minutes (Table 2, entries 11 and 12). Thus, the best reaction conditions were optimized in water at room temperature under ultrasonic irradiation for 10 min (Table 2, entry 11).
Optimization of the reaction conditions for the synthesis of dimethyl 2,2′-[(2Z,2′Z)-((1R,4R)-cyclohexane-1,4-diyl]bis(2-(benzylimino)-4-oxothiazolidin-3-yl-5-ylidene))(2Z,2′Z)-diacetate 5a.
| ||||
|---|---|---|---|---|
| Entry | Solvent | Temp. (°C) | Time (min) | Yield (%) |
| 1 | EtOH | 23 | 7 | 84 |
| 2 | MeOH | 23 | 7 | 88 |
| 3 | H2O | 23 | 7 | 86 |
| 4 | MeCN | 23 | 7 | 65 |
| 5 | DCM | 23 | 7 | 60 |
| 6 | Dioxane | 23 | 7 | 46 |
| 7 | DMF | 23 | 7 | 55 |
| 8 | — | 23 | 10 | 67 |
| 9 | H2O | 50 | 7 | 75 |
| 10 | H2O | 70 | 7 | 63 |
| 11 | H 2 O | 23 | 10 | 88 |
| 12 | H2O | 23 | 25 | 89 |
Having successfully achieved an optimized procedure for this new protocol, we moved to establish the generality of the abovementioned reaction. The developed protocol operated effectively with a broad range of substrates, tolerating a variety of bis-thioureas (3a–d, Scheme 2) and dialkyl acetylenedicarboxylates (4a, b). It was noticed that the derivatives 5a–d comprising cyclohexyl moiety participated in the reaction smoothly with higher yields when compared with the derivatives 6 and 7 comprising p-xylenyl moiety. Interestingly, the derivatives 5–7 could also be acquired by one-pot synthesis without the isolation of the intermediates 3a–d. Hence, upon the completion of the reaction between alkyl isothiocyanates (1a, b) and diamines (2a, b) under ultrasonic irradiation (monitored by TLC), the bis-thioureas 3a–d produced were suspended in water followed by the addition of dialkyl acetylenedicarboxylates (4a, b), and the resulting mixture was sonicated at room temperature for additional 10 min to give the desired products (5–7, Scheme 2, method B).
Scheme 2. An ultrasound-assisted protocol for the synthesis of 5–7.27.

The formation of 2-imino-4-oxothiazolidin-5-ylidene acetates (5–7) proceeded via an addition reaction followed by cyclization and hence, the amine substituent on both sides of thiourea moiety was anticipated to play a significant role on the regioselectivity of the products. Singh and his co-workers28 found an acceptable correlation between the regioselectivity of the N-acylation pathway and the pKa values of the precursor amines in thiourea. The greater the difference between the pKa values of the precursor amines linked to thiourea, the higher is the regioselectivity of N-acylation with the preferential acylation occurring to the amine with a lower pKa. An analogous result was also obtained during the preparation of both 2-iminothiazolines and thiazole-2-imines.29
When asymmetrical thioureas 3c and 3d (Scheme 2) containing benzyl and allyl moieties, respectively, linked to the thiourea reacted with dialkyl acetylenedicarboxylates (4a, b), a mixture of bis-2-imino-4-oxothiazolidines 6a–d and 7a–d, respectively, were obtained (6a, b : 7a, b = 48 : 52 and 6c, d : 7c, d = 50 : 50). The measured pKa values of p-xylylenediamine, benzyl amine and allyl amine were 9.46, 9.34 and 9.49, respectively. The 1H NMR spectra of these products displayed two sets of peaks corresponding to the methylene and methine groups and multiple peaks for aromatic protons (see ESI†). While a larger difference in the pKas of the precursor amines linked to thiourea was observed, one product was exclusively formed as demonstrated in the derivatives 5a and 5b having trans-1,4-diaminocyclohexane (pKa 10.80) and benzyl amine (pKa 9.34). This was further demonstrated with other derivatives 5c and 5d having trans-1,4-diaminocyclohexane (pKa 10.80) attached to one side and allyl amine (pKa 9.49) attached to the other side in thioureas. The 1H NMR spectra of these products (5a–d) displayed only one peak corresponding to the methylene and methine protons (see ESI†). Consequently, we could deduce that in asymmetrical bis-thioureas, the amine having a higher value of pKa was a part of the heterocyclic nitrogen of the five-membered ring, and the amine linked to the thiourea possessing a lower value of pKa was a part of the imino component.
Cyclization of the asymmetrically substituted bis-thiourea derivatives 3a–d with ethyl chloroacetate was also studied under ultrasonic irradiation. Initially, the reaction was carried out at a mild temperature of 30 °C for 30 minutes in EtOH containing catalytic amount of NaOAc. Then, the reaction was proceeded with a considerable amount of starting materials remaining unreacted. However, by increasing the operating temperature to 60 °C, the required products 8–10 were obtained in moderate yields (≈60%) with some unconsumed starting materials still present. Next, the reaction was studied at different temperatures, viz., 70 °C, 80 °C and 90 °C for 30 min, affording the products in ≈75%, 88% and 89% yields, respectively. Following this study, the best result was obtained at 90 °C, and this temperature condition yielded the products in ≈90% under sonication for 30 min. Likewise, in the case of the cyclization of asymmetrical bis-thioureas 3c, d, a mixture of regioisomeric products may be possible. However, with all asymmetrical bis-thioureas 3a, b used, only one regioisomeric product was obtained in high yields under the present reaction conditions. The formation of the compounds 8–10 was established by the disappearance of the thiourea NHs and C S absorption bands and the appearance of the characteristic lactam C O bands at 1720–1716 cm−1 in the IR spectra. The 1H NMR spectra of the products displayed the presence of a singlet at δ = 4.04–4.02 (8a and 8b) and a doublet at δ = 4.12–4.07 (9 and 10), which was assigned to the methylene protons of the thiazolidinone ring and confirmed the ring closure. The presence of the doublet might have been due to the mixture of regioisomeric products (9a : 10a = 49 : 51 and 9b : 10b = 50 : 50). Condensation of bis-thiazolidin-4-ones 8a, b with 4-chlorobenzaldehyde in EtOH buffered with NaOAc under ultrasonic irradiation afforded bis-5-arylidene-2-imino-4-thiazolidinones 11a, b (Scheme 3). The suggested structures of 11a, b were confirmed by elemental analyses and spectroscopic data. The derivatives 11a, b existed as E and Z geometrical isomers, respectively; the Z conformation of the exocyclic C C double bond was assigned on the basis of 1H NMR spectra, which revealed the characterized down-field signals corresponding to their methine-group protons when compared to that of the E-isomer.30 For instance, the 1H NMR spectrum of the derivative 11a displayed only one type of methine proton that was deshielded by the adjacent C O and was detected at δ = 7.82 ppm. Then, we investigated a one-pot three-component reaction of bis-thioureas 3a, b with ethyl chloroacetate and 4-chlorobenzaldehyde, respectively, under ultrasonic irradiation; the required products 11a, b, respectively, were obtained in good yields under irradiation for 30 min at 90 °C. Additionally, the derivatives 8–10 could also be synthesized through the reactions of bis-thioureas 3a–d (1 mmol) with chloroacetyl chloride (2.1 mmol) under ultrasonic irradiation (Scheme 3, solvent-free and catalyst-free conditions). These products were identical in all respects (IR and NMR) to the products obtained using ethyl chloroacetate. However, this procedure could not be applied as the one-pot protocol for the syntheses of 11a, b since chloroacetyl chloride is quite reactive towards the base utilized.
Scheme 3. An ultrasound-assisted protocol for the synthesis of the derivatives 8–11.31.

From the green chemistry point of view, it would be valuable to estimate the “greenness” of our novel aforementioned protocols. Diverse metrics32 that have been applied to estimate and compare the ‘greenness’ of methodologies and products were calculated for some selected compounds (3a, 5a and 8a). These metrics included Carbon Efficiency (CE), Atom Economy (AE), E-factor, Reaction Mass Efficiency (RME) and Process Mass Intensity (PMI). To assess the possibility of the scale-up of the present strategies, the selected reactions were carried out on gram-scale to afford the required compounds 3a, 5a and 8a in 97.21, 90.19 and 94% yields, respectively (Scheme 4). An excellent combination of green metrics such as higher values of AE (100–75%), CE (100–86.66%) and RME (97.21–70.5%) and lower values of both PMI (1.41–1.03) and E-factor (0.098–0.028) confirmed the eco-friendly nature of the present protocols.
Scheme 4. Scaled-up synthesis of derivatives 3a, 5a and 8a.

Conclusions
In summary, we have successfully developed an operational and convenient protocol for the effective sonochemical syntheses of asymmetrical bis-thioureas. These synthesized compounds are subjected to a variety of reagents for the preparation of a novel series of regioselective bis-iminothiazolines. We have found a correlation between the regioselectivity and pKa of the amine. In comparison with the existing arduous protocols for the preparation of iminothiazolines,21 our methodology is superior in terms of simplicity, environmental friendliness, convenience, and applicability on a gram-scale.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Electronic supplementary information (ESI) available: Experimental procedures for the synthesis of bis-thioureas and bis-2-iminothiazolidin-4-ones and NMR spectra. See DOI: 10.1039/c8ra01253a
Notes and references
- Shakeel A. Altaf A. A. Qureshi A. M. Badshah A. Journal of Drug Design and Medicinal Chemistry. 2016;2:10–20. doi: 10.11648/j.jddmc.20160201.12. [DOI] [Google Scholar]
- Venkatesh P. Pandeya S. Int. J. ChemTech Res. 2009;1:733–741. [Google Scholar]
- Buran K. Durmaz I. Cetin R. Onen-Bayram F. E. Med. Chem. Commun. 2015;1:90–93. [Google Scholar]
- Sondhi S. Sharma V. K. Singhal N. Verma R. Shukla R. Raghubir R. Dubey M. Phosphorus, Sulfur Silicon Relat. Elem. 2000;156:21–33. doi: 10.1080/10426500008044991. [DOI] [Google Scholar]
- Huang X. C. Wang M. Pan Y. M. Yao G. Y. Wang H. S. Tian X. Y. Qin J. K. Zhang Y. Eur. J. Med. Chem. 2013;69:508–520. doi: 10.1016/j.ejmech.2013.08.055. [DOI] [PubMed] [Google Scholar]
- Sudzhaev A. R. Rzaeva I. A. Nadzhafova R. A. Safarov Y. S. Allakhverdiev M. A. Russ. J. Appl. Chem. 2011;84:1394–1397. doi: 10.1134/S1070427211080167. [DOI] [Google Scholar]
- Sriram D. Yogeeswari P. Dinakaran M. Irumurugan R. J. J. Antimicrob. Chemother. 2007;59:1194–1196. doi: 10.1093/jac/dkm085. [DOI] [PubMed] [Google Scholar]
- Mahajan A. Yeh S. Nell M. van Rensburg C. E. J. Chibale K. Bioorg. Med. Chem. Lett. 2007;17:5683–5685. doi: 10.1016/j.bmcl.2007.07.049. [DOI] [PubMed] [Google Scholar]
- Wu J. Shi Q. Chen Z. He M. Jin L. Hu D. Molecules. 2012;17:5139–5150. doi: 10.3390/molecules17055139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter C. P. J. Am. Med. Assoc. 1945;129:927–931. doi: 10.1001/jama.1945.02860480007002. [DOI] [PubMed] [Google Scholar]
- Ke S.-Y. Xue S.-J. ARKIVOC. 2006;10:63–68. [Google Scholar]
- Tunaz H. Uygun N. Turk. J. Agric. For. 2004;28:377–456. [Google Scholar]
- Kodomari M. Suzuki M. Tanigawa K. Aoyama T. Tetrahedron Lett. 2005;46:5841–5843. doi: 10.1016/j.tetlet.2005.06.135. [DOI] [Google Scholar]; Rambo R. S. Jacoby C. G. da Silva T. L. Schneider P. H. Tetrahedron: Asymmetry. 2015;26:632–637. doi: 10.1016/j.tetlet.2005.06.135. [DOI] [Google Scholar]
- Barman M. K. Sinha A. K. Nembenn S. Green Chem. 2016;18:2534–2541. doi: 10.1039/C5GC02545A. [DOI] [Google Scholar]; Zhang Z. Schreiner P. R. Chem. Soc. Rev. 2009;38:1187–1198. doi: 10.1039/C5GC02545A. [DOI] [PubMed] [Google Scholar]; Jiang X. Shi X. Wang S. Sun T. Cao Y. Wang R. Angew. Chem., Int. Ed. Engl. 2012;51:2084–2087. doi: 10.1039/C5GC02545A. [DOI] [PubMed] [Google Scholar]
- Goffin E. Lamoral-Theys D. Tajeddine N. De Tullio P. Mondin L. Lefranc F. Gailly P. Rogister B. Kiss R. Pirotte B. Eur. J. Med. Chem. 2012;54:834–844. doi: 10.1016/j.ejmech.2012.06.050. [DOI] [PubMed] [Google Scholar]
- Abdolmaleki A. J. Appl. Polym. Sci. 2012;124:2508–2512. doi: 10.1002/app.33644. [DOI] [Google Scholar]
- Zhang S. Pu Q. Liu P. Sun Q. Su Z. Anal. Chim. Acta. 2002;452:223–230. doi: 10.1016/S0003-2670(01)01476-3. [DOI] [Google Scholar]
- Paul S. Gupta M. Gupta R. Loupy A. Synthesis. 2002:75–78. doi: 10.1016/j.tet.2004.08.031. [DOI] [Google Scholar]; Heinelt U. Schultheis D. Jager S. Lindenmaire M. Pollex A. Beckmann H. S. G. Tetrahedron. 2004;60:9883–9888. doi: 10.1016/j.tet.2004.08.031. [DOI] [Google Scholar]
- Štrukil V. Igrc M. D. Fábián L. Eckert-Maksić M. Childs S. L. Reid D. G. Duer M. J. Halasz I. Mottillo C. Friščić T. Green Chem. 2012;14:2462–2473. doi: 10.1039/C2GC35799B. [DOI] [Google Scholar]; Đud M. Magdysyuk O. V. Margetić D. Štrukil V. Green Chem. 2016;18:2666–2674. doi: 10.1039/C2GC35799B. [DOI] [Google Scholar]; Chau C.-M. Chuan T.-M. Liu K.-M. RSC Adv. 2014;4:1276–1282. doi: 10.1039/C2GC35799B. [DOI] [Google Scholar]; Mane M. Balaskar R. Gavade S. Pabrekar P. Mane D. Arabian J. Chem. 2013;6:423–427. doi: 10.1039/C2GC35799B. [DOI] [Google Scholar]; Maddani M. R. Prabhu K. R. J. Org. Chem. 2010;75:2327–2332. doi: 10.1039/C2GC35799B. [DOI] [PubMed] [Google Scholar]
- Banothu J. Vaarla K. Bavantula R. Crooks P. A. Chin. Chem. Lett. 2014;25:172–175. doi: 10.1016/j.cclet.2013.10.001. [DOI] [Google Scholar]; Moulard T. Lagorce J. F. Thomes J. C. Raby C. J. Pharm. Pharmacol. 1993;45:731–735. doi: 10.1016/j.cclet.2013.10.001. [DOI] [PubMed] [Google Scholar]; Faermark I. F. Shvarts G. Grizik S. I. Gravik V. G. Khim.-Farm. Zh. 1990;24:35. doi: 10.1016/j.cclet.2013.10.001. [DOI] [Google Scholar]
- Li Z. Jin L. Cai C. Org. Chem. Front. 2017;4:2039–2043. doi: 10.1039/C7QO00396J. [DOI] [Google Scholar]; Behbehani H. Ibrahim H. M. Molecules. 2012;17:6362–6385. doi: 10.1039/C7QO00396J. [DOI] [PMC free article] [PubMed] [Google Scholar]; Mohareb R. M. Al-Omran F. Steroids. 2012;77:1551–1559. doi: 10.1039/C7QO00396J. [DOI] [PubMed] [Google Scholar]; Reichelt A. Bailis J. M. Bartberger M. D. Yao G. Shu H. Kaller M. R. Allen J. G. Weidner M. F. Keegan K. S. Dao J. H. Eur. J. Med. Chem. 2014;80:364–382. doi: 10.1039/C7QO00396J. [DOI] [PubMed] [Google Scholar]; Hantzsch A. Weber J. H. Ber. Dtsch. Chem. Ges. 1887;20:3118–3132. doi: 10.1039/C7QO00396J. [DOI] [Google Scholar]; Aguilar E. Meyers A. I. Tetrahedron Lett. 1994;35:2473–2476. doi: 10.1039/C7QO00396J. [DOI] [Google Scholar]; Ochiai M. Nishi Y. Hashimoto S. Tsuchimoto Y. Chen D. W. J. Org. Chem. 2003;68:7887–7888. doi: 10.1039/C7QO00396J. [DOI] [PubMed] [Google Scholar]; Cook A. H. Heilbron I. MacDonald S. F. Mahadevan A. P. J. Chem. Soc. 1949:1064–1068. doi: 10.1039/C7QO00396J. [DOI] [Google Scholar]; Kikelj D. Urleb U. Synthesis. 2002:627–833. doi: 10.1039/C7QO00396J. [DOI] [Google Scholar]; Botta M. Castagnolo D. Pagano M. Synlett. 2009:2093–2096. doi: 10.1039/C7QO00396J. [DOI] [Google Scholar]; Lingaraju G. S. Swaroop T. R. Vinayaka A. C. Synthesis. 2012;44:1373–1379. doi: 10.1039/C7QO00396J. [DOI] [Google Scholar]
- Wael A. A. A. Raafat M. S. ARKIVOC. 2016;iii:187–201. doi: 10.3987/COM-16-13479. [DOI] [Google Scholar]; Wael A. A. A. Ahmed F. A. ARKIVOC. 2017;v:327–340. doi: 10.3987/COM-16-13479. [DOI] [Google Scholar]; Wael A. A. A. Raafat M. S. Saleh A. R. Heterocycles. 2016;92:1224–1243. doi: 10.3987/COM-16-13479. [DOI] [Google Scholar]
- Divya V. Vikash S. Singh O. G. Shubha J. Green Chem. 2017;19:5885–5899. doi: 10.1039/C7GC03279J. [DOI] [Google Scholar]
- Capello C. Fischer U. Hungerbühler K. Green Chem. 2007;9:927–934. doi: 10.1039/B617536H. [DOI] [Google Scholar]
- Chanda A. Fokin V. V. Chem. Rev. 2009;109:725–748. doi: 10.1021/cr800448q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- General procedure for the synthesis of bis-thioureas (3a–d). Isothiocyanates (1a, b, 0.5 mmol) and diamines (2a, b, 1.0 mmol) were mixed in a 25 mL round-bottomed flask. The reaction mixture was sonicated in water bath (23 °C) of an ultrasonic cleaner for 5 min as indicated in Table 1. After completion of the reaction (the reaction was monitored by TLC, silica gel; DCM : petroleum ether = 5 : 2 v/v), the reaction mixture was triturated with 2 mL chilled EtOH, and the precipitate was filtered, affording the crude product, which was purified by recrystallization from EtOH (95–97%).
- General methods for the synthesis of derivatives 5–7. Method A: to a solution of 1 mmol of bis-thioureas 3a–d and 10 mL of deionized water in a 50 mL round-bottom flask, 2 mmol of dialkyl acetylenedicarboxylate (DAADs, 4a, b) was added dropwise at room temperature with continuous stirring. After the complete addition of DAADs, the reaction mixture was sonicated for 10 min (TLC, DCM : MeOH = 9 : 1 v/v); during the reaction, a solid appeared in the solution. After completion of the reaction, excess solvent was removed by filtration, and the residue was recrystallized from ethanol, affording the desired product as colorless to light yellow crystals. Method B: isothiocyanates (1a, b, 0.5 mmol) were added dropwise to diamines (2a, b, 1.0 mmol) under ultrasonic irradiation for 5 min. To the resulting residue, 10 mL of water was added followed by dropwise addition of 2 mmol of dialkyl acetylenedicarboxylate (DAADs, 4a, b) at room temperature. After the complete addition of DAADs, the reaction mixture was sonicated for 10 min (TLC, DCM : MeOH = 9 : 1 v/v); during the reaction, a solid appeared in the solution. After completion of the reaction, the excess solvent was removed by filtration, and the residue was recrystallized from ethanol, affording the desired product as colorless to light yellow crystals (88–92%).
- Singh C. B. Ghosh H. Murru S. Patel B. K. J. Org. Chem. 2008;73:2924–2927. doi: 10.1021/jo702628g. [DOI] [PubMed] [Google Scholar]
- Singh C. B. Muru S. Kavala V. Patel B. K. Org. Lett. 2006;8:5397–5399. doi: 10.1039/B807775D. [DOI] [PubMed] [Google Scholar]; Yella R. Ghosh H. Patel B. K. Green Chem. 2008;10:1307–1312. doi: 10.1039/B807775D. [DOI] [Google Scholar]; Wagh Y. B. Kuwar A. S. Patil D. R. Tayade Y. A. Jangale A. D. Terdale S. S. Trivedi D. R. Gallucci J. Dalal D. S. Ind. Eng. Chem. Res. 2015;54:9675–9682. doi: 10.1039/B807775D. [DOI] [Google Scholar]
- Hatem A. A. Heba S. A. Kamal M. D. Eur. J. Med. Chem. 2010;45:2427–2432. doi: 10.1016/j.ejmech.2010.02.026. [DOI] [PubMed] [Google Scholar]
- General procedures for the preparation of compounds 8–10. Method A; from ethyl chloroacetate: the appropriate bis-thioureas 3a–d (1 mmol), ethyl chloroacetate (2 mmol) and sodium acetate (0.16 g, 2 mmol) were irradiated under ultrasonic conditions in ethanol (25 mL) at 90 °C for 30 min (TLC, DCM : MeOH = 9 : 1 v/v). The resulting solid was filtered, dried and recrystallized from ethanol containing few drops of dioxane to afford the corresponding bis-thiazolidine derivatives 8–10. Method B; from chloroacetyl chloride: To bis-thioureas 3a–d (1 mmol) placed in a 10 mL round bottom flask, chloroacetyl chloride (1.2 mmol) was added drop wise under ultrasonic irradiation. After complete addition of chloroacetylchloride, acetonitrile (5 mL) was added to the reaction mixture, and the mixture was stirred continuously for 5 min (TLC, DCM : MeOH = 9 : 1 v/v). The resulting solid was filtered, dried and recrystallized from ethanol containing few drops of dioxane to afford the corresponding bis-thiazolidine derivatives 8–10 (93–88%). Syntheses of derivatives 11a, b. A one-pot three component mixture of the appropriate bis-thioureas 3a, b (1 mmol), ethyl chloroacetate (2 mmol), 4-chlorobenzaldehyde (2 mmol) and sodium acetate (5 mmol) were mixed in ethanol (15 mL). The reaction mixture was sonicated at 90 °C for 30 min (TLC, DCM : MeOH = 8 : 2 v/v). The resulting residue was filtered and recrystallized from dioxane to afford the corresponding bis-thiazolidine derivatives 11a, b in 86–90% yield.
- Curzons D. Constable D. J. C. Mortimer D. N. Cunningham V. L. Green Chem. 2001;3:1–6. doi: 10.1039/B007871I. [DOI] [Google Scholar]; Jimen Pez-Gonzalez C. Constable D. J. C. Ponder C. S. Chem. Soc. Rev. 2012;41:1485–1498. doi: 10.1039/B007871I. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.