

Abstract
Nitrobenzothiazinone (BTZ) is a promising scaffold with potent activity against M. tuberculosis by inhibiting decaprenylphosphoryl-beta-d-ribose 2′-oxidase (DprE1). But unfavorable durability poses a challenge to further development of this class of agents. Herein, a series of BTZs bearing a variety of different substituents at the C-2 position were designed and synthesized. Compounds were screened for their antimycobacterial activity against Mycobacterium tuberculosis H37Ra and were profiled for metabolic stability, plasma protein-binding capacity and pharmacokinetics in vivo. In general, these new BTZs containing N-piperazine, N-piperidine or N-piperidone moiety have excellent antitubercular activity and low cytotoxicity. Several of the compounds showed improved microsomal stability and lower plasma protein-binding, opening a new direction for further lead optimization. And we obtained compound 3o, which maintained good anti-tuberculosis activity (MIC = 8 nM) and presented better in vitro ADME/T and in vivo pharmacokinetic profiles than reported BTZ compound PBTZ169, which may serve as a candidate for the treatment of tuberculosis.
Nitrobenzothiazinone (BTZ) is a promising scaffold with potent activity against M. tuberculosis by inhibiting decaprenylphosphoryl-beta-d-ribose 2′-oxidase (DprE1).
1. Introduction
Tuberculosis (TB) is one of the top ten causes of death worldwide, a major disease causing 1.3 million deaths every year and represents the leading cause of mortality resulting from a bacterial infection.1 Furthermore, with the sprawl of multi-drug resistant–TB (MDR–TB) and rifampicin resistant–TB (RR–TB), it causes nearly 50% mortality in treated individuals, and this disease threatens people's health seriously (http://www.who.int/tb/publications/global_report/archive/en/). The currently recommended treatment for cases of drug-susceptible TB is a 6-month regimen of four first-line drugs: isoniazid (INH), rifampicin (RIF), ethambutol (EMB) and pyrazinamide (PZA), which is followed by a longer phase of treatment with RIF and INH to eradicate the remaining bacilli which entered into a dormant, slowly replicating latent phase.2 While, the latest data reported by WHO showed the treatment success rate for MDR–TB is 54%, reflecting high rates of treatment failure.3
Nitrobenzothiazinone (BTZ), represented by BTZ043 (Fig. 1a) and PBTZ169 (Fig. 1b), is one of the most promising scaffolds in the antitubercular drug pipelines with nanomolar concentrations against both sensitive-TB and MDR–TB.4–6 The nitro group at position 8 of BTZ is critical for antituberculosis activity.7 BTZs undergo nitroreduction to form a nitroso derivative, which then covalently reacts with cysteine 387 (C387) residue of decaprenylphosphoryl-β-d-ribose (DPR) 2′-oxidase (DprE1). DprE1 enzyme catalyzes biosynthesis of a prenylphosphoryl-β-d-arabinose (DPA), which is an essential process for the mycobacteria cell wall assembly.8–10BTZ043 and PBTZ169 are undergoing extensive preclinical studies and phase II clinical trials, respectively.2,11
Fig. 1. (a) BTZ043 (b) PBTZ169 (c) hit compound M-1.

Pharmacokinetic profile showed positive correlation with in vivo efficacy.12 Effort to improve pharmacokinetic properties in the early drug discovery process is essential to minimize the attrition rate during the pre-clinical and clinical development phases. It has been discovered that BTZ043 and PBTZ169 displayed similar pharmacokinetic properties,5 and exhibited unfavorable metabolic stability in our tests (Table 4). It poses a challenge to drug formulation as well as other aspects of clinical studies during the continuing development of this promising class of antitubercular agents. Some work to improve the pharmacokinetic properties of BTZ043 and PBTZ169 has been reported.13–15 And in our previous study, we synthesized a series of 4-carbonyl piperazine substituted 1,3-benzothiazin-4-one derivatives and most of these compounds exhibited good antitubercular activities and favorable aqueous solubility. However, the bioavailability of hit compound M-1 (Fig. 1c) (F 8.6%) was not improved compared with BTZ043 (F 29.5%), which may result from hydrolysis of the amide group. Therefore, we synthesized new series of BTZs with alternative groups at position 2. Our focus was to improve the metabolic stability of this kind of scaffold and maintain good in vitro potency by medicinal chemistry strategy simultaneously. Here, we present results of the SAR analyses along with our efforts toward elucidation of the improvement of pharmacokinetic profiles.
Pharmacokinetic parameters for compound 3o and PBTZ169 in ICR mice mollowing intravenous and oral administrationa.
| Parameters | 3o-IV | 3o-PO | PBTZ169-IV | PBTZ169-PO | ||||
|---|---|---|---|---|---|---|---|---|
| Location | Plasma | Tissue | Plasma | Tissue | Plasma | Tissue | Plasma | Tissue |
| Dose (mg kg−1) | 10 | 10 | 100 | 100 | 10 | 10 | 100 | 100 |
| Terminal T1/2 (h) | 2.62 | 1.75 | 2.40 | 2.84 | 2.67 | 3.16 | 2.09 | 4.23 |
| T max (h) | 1.75 | 0.08 | 2.00 | 2.00 | 0.08 | 0.08 | 2.00 | 0.50 |
| C max (ng mL−1) | 5674.10 | 8872.56 | 2648.48 | 4578.33 | 15 600.40 | 4208.74 | 2996.34 | 811.35 |
| AUC0–∞ (ng mL−1 h) | 3035.87 | 5335.01 | 13 611.26 | 24 194.36 | 9074.85 | 15 146.56 | 18 882.08 | 7841.88 |
| V ss (mL kg−1) | 4524.65 | — | — | — | 1154.42 | — | — | — |
| Cl (L h−1 kg) | 3.29 | 1.87 | — | — | 1.10 | 0.66 | — | — |
| MRT0–∞ (h) | 1.37 | 1.08 | 3.73 | 4.19 | 1.05 | 4.50 | 3.70 | 6.50 |
| F (%) | — | — | 44.83 | 45.35 | — | — | 20.81 | 5.18 |
Values are the mean from three animals. Dash indicates that the value was not measured or was not relevant.
2. Results and discussion
2.1. Synthesis
2.1.1. An overview of nitrobenzothiazinone synthesis
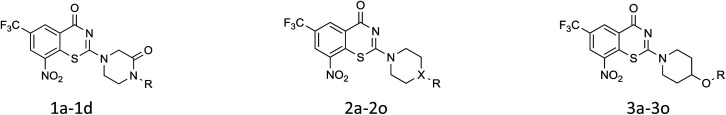
The structural variation on hit compound M-1 was divided into coupling with four series linkers. The synthesis of core nitrobenzothiazinone was achieved by utilizing efficient one-pot three-step reaction as outline in Scheme 1. A reaction of 2-chloro-3-nitro-5-(trifluoromethyl) benzoic acid in dichloromethane with oxalyl chloride and catalyst DMF gave 2-chloro-3-nitro-5-(trifluoromethyl)benzoyl chloride. The intermediate was prepared from 2-chloro-3-nitro-5-(trifluoromethyl)benzoyl chloride and ammonium thiocyanate in dichloromethane using 18-Crown-6 as a phase-transfer catalyst (PCT). Next, the appropriate heterocyclic amines nucleophilic attacked on the isothiocyanato group. Due to the presence of two electron-withdrawing groups in the para and ortho position of the chlorine atom, respectively, it was then cyclization to the desired benzothiazine-4-ones from the sulfur heterocyclization (Scheme 1).
Scheme 1. Synthesis of nitrobenzothiazinone derivative: (a) cat. DMF, oxalyl chloride, DCM, rt; (b) cat. 18-crown-6, NH4SCN, DCM, rt; (c) tert-butyl piperazine-1-carboxylate, DCM, rt; (d) cat. TFA, DCM, rt; (h) cat. K2CO3, ester, dry DMF, 60 °C, 3 h; (e) 1a5-1d5, DCM, rt; (f) DCM, rt. (g) cat. K2CO3, ester, dry DMF, 60 °C 3 h.

2.1.2. Synthesis of amine derivatives
Detailed synthetic pathways to amine derivatives 21–22, 41–48 and 49–58 which are commercially unavailable are depicted in Scheme 2. For rapid SAR exploration, a parallel synthesis route was designed that would enable preparation of various analogues with minimum purification. Coupling of 1–4 with Boc-glycine yielded 9–12 in the presence of N-(3-dimethylaminopropyl)-N′-ethylcarbodimide hydrochloride diethyl azodicarboxylate (EDCI) and 1-hydroxybenzotriazole (HOBT) followed by treatment with trifluoroacetic acid in dichloromethane. 4-Nitrobenzenesulfonyl chloride was dropped into a suspension of the trifluoroacetate and trimethylamine at 0 °C bath in dichloromethane to give the desired acetamide 13–16, then the piperazinones (17–20) were formed by further reaction with 1,2-dibromoethane. Treatment of 17–20 with methylmercaptopropionate in acetonitrile/DMSO (v/v 49/1) at 50 °C gave the desired compounds (21–24) in 69.06% overall yield starting from 1–4. Analogue 41–48 was synthesized from the corresponding benzoyl chloride (25–32) in 73.02% yield over two steps as shown in Scheme 2via nucleophilic substitution of compounds 25–32 with 1-Boc-4-hydroxypiperidine followed by deprotection the Boc-group. Reaction mixture evaporated, dissolved in methyl alcohol. Triethylamine was added to the solution followed by removal of the solvent in vacuo to give yellow solid, which was used to synthesis of 2h–2o.
Scheme 2. Synthesis of amine derivatives: (h) cat. EDCI, HOBT, Boc-glycine, DCM, rt.12 h; (i) cat. TFA, DCM, rt, 1 h; (j) cat. TEA, 4-nitrobenzenesulfonyl chloride, 0 °C, 0.5 h, rt, 6 h; (k) cat. K2CO3, 1,2-dibromoethane, DMF, 60 °C; (l) cat. LiOH, methyl-mercaptopropionat, CAN/DMSO (v/v = 49/1), 50 °C, 6 h; (m) cat. TEA, DCM, rt; (n) cat. TFA, DCM, 0 °C, 0.5 h, rt, 1.5–4 h; reagents and conditions: (o) cat. PPh3, DEAD, THF, rt; (p) cat. TFA, DCM, rt; (q) cat. NaH, dry DMSO, rt, 0 °C, 0.5 h, rt; (r) cat. NaH, dry DMF, 0 °C, 0.5 h, rt.

Coupling of 1-Boc-4-hydroxypiperidine with phenol (49–58) in the presence of diethyl azodicarboxylate (DEAD) and PPh3 followed by treatment with trifluoroacetic acid (TFA) in dichloromethane gave 69–78 in 5–70% yield.
Intermediates 83, 84 were obtained from 2-bromopyridine in two steps. First, condensation of 1-Boc-4-hydroxypiperidine with sodium hydride in dry DMF, and then treatment of 2-bromopyridine (79, 80) furnished 81 and 82. The Boc-group was deprotected by TFA (trifluoroacetic acid) to afford 83 and 84. Similarly, this condition was also used to produce 91–93 with acyl chloride (85–87). All target compounds and intermediates were characterized by 1HNMR and LC-MS.
2.2. Biological evaluation
2.2.1. In vitro antimycobacterial activity
Compounds prepared in this manner were tested the against M.tb H37Ra medium using the standard broth micro dilution method and minimum inhibitory concentration (MIC) values determined at 7 days.16 Kinetic solubility of the test compounds was determined in different pH conditions/mediums, and apparent permeability (Papp) of test compounds was determined by using MDKI-MDCKII cells.17 Metabolic stabilities of selected compounds in human, rat, and mouse liver microsomes were assessed by incubating for 30 min and measuring the percentage of compound remaining. CYP inhibition potential was determined using the following recombinant microsomes:18 CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4. SD rats were used to determine Pharmacokinetic parameters and the experimental data was analyzed by WinNonlin V6.2.1.
Initial medicinal chemistry strategy was to replace amido bond with C–N bond for improving the metabolic stability. First the piperazinone derivatives were synthesized as lactam is the main metabolite of cyclic amine. The introduction of oxygen atom also served as a hydrogen-bond acceptor19 (Table 1). Cyclopentyl piperazinone 1a showed best anti-M.tb activity (MIC = 13 nM). To further explore the effect of the ring size, we synthesized a 6-membered aliphatic cyclic which led to a slightly loss of activity. No anti-M.tb activity difference was observed in homologs cyclohexyl (2b) and cyclohexylmethyl (2c) substitution. A replacement of cycloalkyl-substitution with linear butyl group 2d caused a drastic increase on aqueous solubility (49.56 μg mL−1) resulted from molecular flexibility enhancement.20 However, further investigation of butyl substitute 2d was suspended because of the potential metabolic instability of alkyl group in vivo.21
SAR of analoguesa.
| |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compd | X | R | MIC (nM) | clog P | Aqueous solubility (μg mL−1) | CC50 (Vero) μMb | Compd | X | R | MIC (nM) | clog P | Aqueous solubility (μg mL−1) | CC50 (Vero) μMb |
| 1a | — |
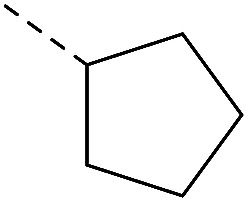
|
13 | 3.52 | 7.37 | >10 | 2n | C |
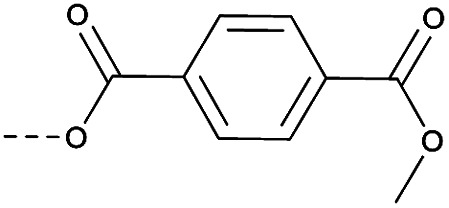
|
63 | 4.15 | ND | >10 |
| 1b | — |
|
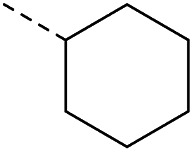
17 | 4.08 | 1.63 | >10 | 2o | C |
|
31 | 4.66 | ND | >10 |
| 1c | — |
|
17 | 4.79 | 3.57 | >10 | 3a | — |
|
16 | 4.21 | 0.5 | >10 |
| 1d | — |
|
34 | 49.56 | 3.57 | >10 | 3b | — |
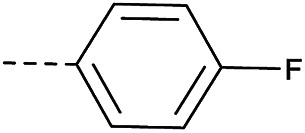
|
≤2 | 4.42 | <1 | >10 |
| 2a | C |
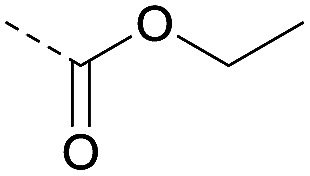
|
8 | 2.78 | ND | >10 | 3c | — |
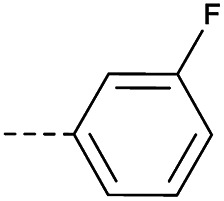
|
31 | 4.42 | ND | >10 |
| 2b | N |
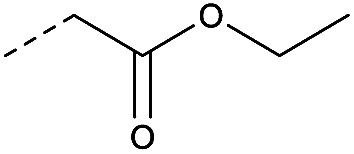
|
20 | 3.23 | ND | >10 | 3d | — |
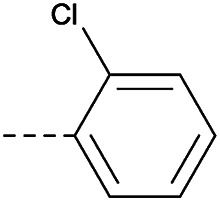
|
31 | 4.76 | ND | >10 |
| 2c | C |
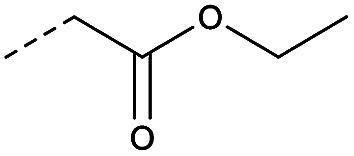
|
16 | 3.40 | ND | >10 | 3e | — |
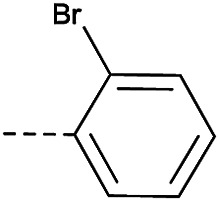
|
31 | 4.91 | ND | >10 |
| 2d | N |
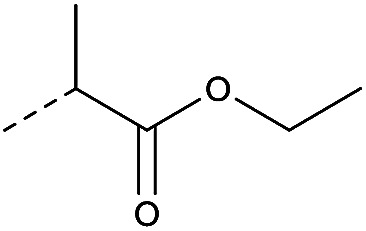
|
8 | 3.54 | ND | >10 | 3f | — |
|
4 | 5.26 | ND | >10 |
| 2e | N |
|
31 | 4.07 | ND | >10 | 3g | — |
|
16 | 4.63 | ND | >10 |
| 2f | N |
|
64 | 1.76 | ND | >10 | 3h | — |
|
8 | 5.33 | ND | >10 |
| 2g | N |
|
16 | 3.40 | ND | >10 | 3i | — |
|
16 | 4.71 | 1 | >10 |
| 2h | C |
|
≤2 | 4.33 | ND | >10 | 3j | — |
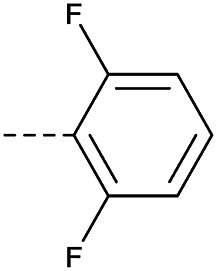
|
125 | 4.21 | ND | >10 |
| 2i | C |
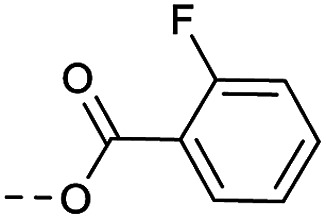
|
4 | 3.89 | ND | >10 | 3k | — |
|
31 | 3.54 | ND | >10 |
| 2j | C |
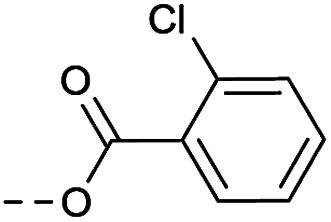
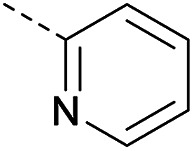
|
8 | 4.46 | ND | >10 | 3l | — |
|
31 | 3.72 | ND | >10 |
| 2k | C |
|
8 | 5.07 | ND | >10 | 3m | — |
|
31 | 3.21 | 2.39 | >10 |
| 2l | C |
|
8 | 4.36 | ND | >10 | 3n | — |
|
4 | 3.63 | 1.73 | >10 |
| 2m | C |
|
63 | 4.12 | ND | >10 | 3o | — |
|
8 | 2.87 | 5.14 | >10 |
| BTZ043 | 4 | 2.45 | <1 | >10 | |||||||||
ND: not determined. aclog P was calculate by Chembiodraw Ultra version 11.0. bCytotoxicityagainst kidney epithelial cells extracted from an African green monkey (VERO). Dash indicates that the value was not relevant.
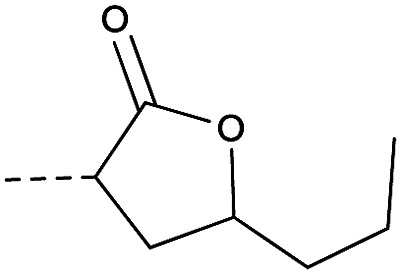
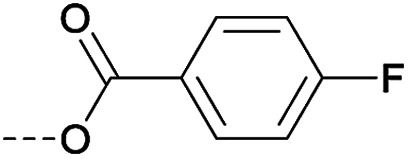
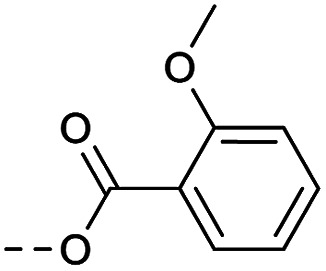
Then piperazine and piperidine derivatives were also designed and synthesized (Table 2). The alkyl esters exhibited good anti-M.tb activity and acceptable solubility. The compound 2a ethyl formate showed good anti-M.tb activity (8 nM). The analogues of 2a with four-carbon atoms ester linker (2b) showed a reduced activity (MIC = 20 nM). Interestingly, this effect could be balanced with an introduction of methyl on α-C of the carbonyl group (2d, MIC = 8 nM). Compound 2d maintained anti-M.tb activity with dramatic improvement on solubility (27.07 μg mL−1 in pH 6.5, 84.16 μg mL−1 in pH 2.0) compared with BTZ043, probably due to the disruption of molecule planarity.22 A significant reduced activity could be observed when the 2-methyl was changed to 2-ethyl (2e, MIC = 31 nM) or lactone (2f, MIC = 64 nM). And introduction of alkyl on the lactone ring led to obvious enhanced potency (2gvs.2f). Compound 2c showed the same range of activity (MIC = 16 nM) as compound 2b (MIC = 20 nM), suggesting similar effect of piperidine and piperazine on activity. Next, we turned our attention to the aryl esters. Aryl analogues with small halogens (2h, 2i, and 2j) were all maintained good anti-M.tb activity, in particular compound 2i with a low value of MIC ≤2 nM. para-Methoxy-substitution (2l) was favored over ortho (2m) substituted analogues. While, bulk groups such as ester group (2n, MIC = 63 nM) and dimethylamino (2o, MIC = 31 nM) resulted in a significant drop in activity.
Properties of leading compoundsa.
| Compd | HLM/RLM/MLM T1/2 (min)b | Human PPB (% bound)c | Mean Papp (10–6 cm s−1) | Efflux ratio | |
|---|---|---|---|---|---|
| A to B | B to A | ||||
| 1a | 72/15.6/9.4 | 90.7 | 10.59 | 7.23 | 0.68 |
| 2d | 29.7/9.4/6.9 | 92.5 | 9.46 | 5.04 | 0.53 |
| 2h | 0.6/17.4/0.6 | 99.8 | 0.10 | 0.15 | 1.59 |
| 3b | 10.1/27.5/28.9 | >99 | 1.01 | 0.93 | 0.92 |
| 3c | 7.7/15.4/19.0 | >99 | 0.42 | 0.37 | 0.88 |
| 3n | 7.5/8.9/16.7 | 99.4 | 5.14 | 4.55 | 0.89 |
| 3o | 61.4/100.9/104.7 | 98.6 | 9.67 | 3.58 | 0.37 |
| BTZ043 | 46.1/22.0/16.7 | 99.4 | 9.02 | 7.17 | 0.79 |
| PBTZ169 | 11.1/14.7/14.4 | 99.6 | 0.74 | 0.74 | 1 |
Permeability was correlated with the apparent permeability coefficient Papp (cm s−1), which was interpreted as follows: low permeability (Papp ≤ 1.0 × 10−6 cm s−1), moderate permeability (1.0 × 10−6 < Papp < 5.5 × 10−6 cm s−1) or high permeability (Papp ≥ 5.5 × 10−6 cm s−1).
HLM = human liver microsomes, RLM = rat liver microsomes, MLM = mouse liver microsomes.
PPB = plasma protein binding.
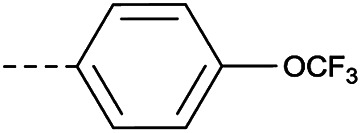
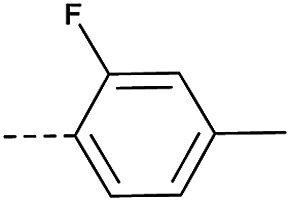
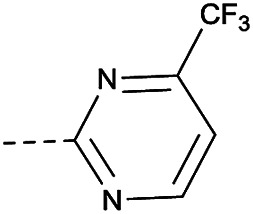
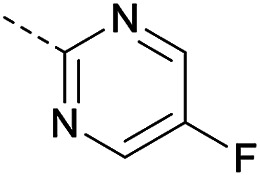
In consideration of metabolic instability of ester group,23 compounds with 4-oxypiperidyl moiety 3a–3o were designed and synthesized (Table 3). Similarly, we initially explored the effect of adding small halogens to the benzene ring. Benzyl analogue with fluoro-substitution (3a) was favored over chlorine-substitution (3d) and bromine-substitution (3e) analogues. Furthermore, meta substitution (3c) showed the best anti-M.tb activity (MIC ≤ 2 nM) relative to ortho (3a, MIC = 16 nM) and para (3b, MIC = 4 nM) substitution. The methyl, trifluoromethyl and trifluoromethoxy derivatives (3g, 3f and 3h) showed good activities and disubstituted groups (3i, 3g) were mainly detrimental to activity. Potentially decreased aqueous solubility may be caused by high hydrophobic functionality of benzene ring, so the strategy of bioisostere replacement was used to replacement of benzene ring with hydrophilic group. Pyridine and pyrimidine were designated as high metabolic stability and good hydrophilicity.24 Pyrimidine-substitution demonstrated stronger activity than pyridine-substitution. meta Trifluoromethyl-substitution (3n) showed best anti-M.tb activity in this group of inhibitors (MIC = 4 nM) but unacceptable aqueous solubility (1.73 μg mL−1). Replacement of the ortho-trifluoromethyl group with a para-fluorine (3o) was investigated to maintain good activity (MIC = 8 nM) with an increased solubility (5.14 μg mL−1). All compounds tested in this series were not toxic to VERO cells up to 10 μ M (CC 50 > 10 μM). Based on the above studies 1a, 2d, 2h, 3b, 3c, 3n and 3o were selected as representative examples for subsequent validation through resynthesis, retesting, and ADME profiling.
Pharmacokinetic parameters for compound 2d, 3n and 3o in SD rat following intravenous and oral administrationa.
| Parameters | 2d | 3n | 3o | |||
|---|---|---|---|---|---|---|
| IV | Oral | IV | Oral | IV | Oral | |
| Dose (mg kg−1) | 1 | 10 | 1 | 10 | 1 | 10 |
| Terminal T1/2 (h) | 0.78 | 0.93 | 10.40 | 5.01 | 10.69 | 9.86 |
| T max (h) | 0.08 | 0.50 | 0.50 | 2.00 | 0.58 | 2.00 |
| C max (ng mL−1) | 1057.10 | 494.83 | 2310.20 | 285.40 | 4179.64 | 8423.01 |
| AUC0–∞ (ng mL−1 h) | 421.60 | 498.60 | 12 670.46 | 1657.85 | 22 666.86 | 65 818.33 |
| V z (L kg−1) | 3.48 | — | 1.19 | — | 0.69 | — |
| Cl (L h−1 kg) | 3.39 | — | 0.08 | — | 0.05 | — |
| MRT0–∞ (h) | 0.44 | 1.86 | 12.71 | 6.55 | 12.36 | 12.58 |
| F (%) | — | 11.78 | — | 1.5 | — | 29.35 |
Values are the mean from three animals. Dash indicates that the value was not measured or was not relevant.
2.2.2. In vitro pharmacokinetic studies
Changes in metabolic stability, aqueous solubility and cell permeability/efflux of compound may modulate activity in addition to any changes in target engagement, leading to enormous fluctuations on the bioavailability of drug in vivo.25In vitro pharmacokinetic properties of these selected analogs are summarized in Table 4. As we expected, solubility profiling data of 1a, 3n, 3o indicated that the introduction of ionization center and hydrogen-bond acceptor resulted in a 2–7 fold increase in aqueous solubility relative to BTZ043. Compound 2d showed the prominent aqueous solubility (27.07 μg mL−1 in pH = 6.5 and 84.16 μg mL−1 in pH = 2.0) of all compounds. However, no solubility increase of 2h, 3b and 3c compared with BTZ043 were observed, which may be caused by the high hydrophobicity of benzene ring. Further in vitro permeability profiling showed that 1a, 3n and 3o had high permeability with Papp > 5.5 × 10−6 in MDRI-MDCKII assay, while the permeability of 2h and 3c can't be acceptable. With poor aqueous solubility and low permeability, 2h and 3c were classified into the fourth group according to Biopharmaceuticals Classification System (BCS). Hence, no further study on these two compounds was carried. Meanwhile, the stabilities of these compounds in microsomes were also be profiled. Compound 2h showed poor metabolic stability in human liver microsomes (elimination T1/2 = 0.6 min), rat liver microsomes (elimination T1/2 = 17.4 min) and mice liver microsomes stability (100.9 min) of 3b (elimination T1/2 = 10.1 min) suggests that ester hydrolysis is the main route of metabolism.
Also, the elimination T1/2 (29.7 min) of compound 2d in HLM was 50-fold higher than that of compound 2h (0.6 min), suggesting the introduction of methyl group in the vicinity of ester bond may improve metabolic stability. Exhilaratingly, compound 3o showed excellent anti-M.tb activity, and good stability across all three species could be noticed with the elimination T1/2 in the presence of human liver microsomes (61.4 min), rat microsomes (100.9 min) and mice microsomes (104.7 min). Furthermore, we selected five human CYP450 isoforms CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 for the inhibitory test. Inhibition on CYPs can be detected of compound 1a (IC50 = 8.6 μM for CYP2C19; IC50 = 5.33 μM for CYP2A4), 3b (IC50 = 6.6 μM for CYP2C19) and 3c (IC50 = 8.6 μM for CYP2C19) (Table S1†). Generally, compounds 1a, 3c, 3n and 3o exhibited lower protein binding (PPB) compared to PBTZ169 (Table S2†). The three most promising compounds in in vitro pharmacokinetic studies, 2d, 3n and 3o, were selected for the progression into mouse PK studies.
2.2.3. In vivo pharmacokinetic studies
With the good anti-M.tb activity and favorable druggability in vitro, intravenous and oral PK studies – of 2d, 3n and 3o in SD rats were characterized (Table 3). Compound 2d displayed markedly inferior PK properties (Table 3) with high clearance (Cl = 3.39 L h−1 kg) and short terminal T1/2 (0.78 h IV, 0.93 h PO). The poor PK is possibly due to its instability in liver microsomes. A long terminal T1/2 (5.01 h) when dosed orally indicated an improvement in metabolic stability of 3n. However, this modification resulted in poor absorption (F 1.54%). Compound 3o displayed encouraging PK profile in SD rats (Table 3). It exhibited the longest terminal T1/2 (9.86 h) among all three compounds, indicating good metabolic stability in SD rats. 3o displayed good exposure with Cmax of 2000-fold above the MIC90 when dose orally (10 mg kg−1). These results suggested that this modification improved metabolic stability, and resulted in good PK profile.
Then Compound 3o and PBTZ169 were subjected to intravenous (10 mg mL−1) and oral (100 mg mL−1) mouse PK (Table 4, Fig. 2a–d). The absorption of 3o was very good with a bioavailability (F) of 44.83% compared with PBTZ169 (20.81%) (Table 4, S8†). The excellent stability in liver microsomes and lower PPB of 3o were speculated as important reasons which led the good bioavailability. Furthermore, compound 3o exhibited more favorable lung enrichment than PBTZ169. Generally, compound 3o displayed near parallel concentration–time profile in plasma (Fig. 2a) and lung tissue (Fig. 2b) whatever intravenous (Tables S3 and S5†) or oral administration. (Tables S4 and S6†), suggesting a rapid equilibrium between these two tissues. 3o showed almost 4-folds higher absorption in lung than PBTZ169 (45.35% vs. 5.18%) (Tables 4 and S6 and S10†). As the Vss and lung distribution could give an initial indication about a compound's potential for in vivo efficacy,12 the two parameters of compound 3o were analyzed. Compound 3o displayed a higher volume of distribution (Vss = 4524.65 mL kg−1) than that of PBTZ169 (Vss = 1154.42 mL kg−1) resulting in pronounced penetration into tissues (LAUC/PAUC 1.8 and LCmax/PCmax 1.7) (Tables 4 and S5, S7 and S9†). The duration during which the drug concentration of lungs are above MIC (T > MIC) was selected as an important parameter to estimate in vivo efficiency as BTZs are time dependent antimicrobial agents.4 Compound 3o displayed T > MIC 100%, which supported to in vivo efficiency. The lung concentration–time data following PO administration showed a double peak-shaped curve (Fig. 2b), indicating the possibility of enterohepatic recirculation of 3o in ICR mice.
Fig. 2. (a) Mean plasma concentration–time profiles of 3o after IV and PO dose in ICR mice (n = 3) (mean ± sd). (b) Mean lung concentration–time profiles of 3o after IV and PO dose in ICR mice (n = 3) (mean ± sd). (c) Mean plasma concentration–time profiles of PBTZ169 after IV and PO dose in ICR mice (n = 3) (mean ± sd). (d) Mean lung concentration–time profiles of PBTZ169 after IV and PO dose in ICR mice (n = 3) (mean ± sd).

3. Conclusion
Optimization of original hit compound M-1 at position 2 gave series compounds with improved metabolic stability. It provides us a new direction for lead optimization as this very promising scaffold. Screening of these compounds based on PK profile led to the identification of a potent hit compound 3o, 2-(4-(5-fluoropyrimidin-2-yl)piperidin-1-yl)-6-nitro-8-(trifluoromethyl)-4H-benzo[e][1,3]thia-zin-4-one. This compound maintained the good anti-M.tb activity in vitro (MIC = 8 nM) and good metabolic stability in SD rats. The results of PK studies in mice model suggested that 3o is a promising compound with good exposure and favourable absorptions both in plasma and lung tissue. These foundations may translate to excellent in vivo efficacy in the further study of compound 3o in the TB-mice model. Therefore, compound 3o was identified as a pre-clinical candidate worthy of further pharmacology and toxicology study in vivo in the future.
4. Experimental
4.1. Chemistry
4.1.1. General chemistry
Chemical and reagents were analytical or chemically pure reagents. Organic solvents were dried using the dry solvent system or dried over activated molecular sieves 3 Å. 1H NMR spectra were recorded on Bruker AVANCEIII 400 MHz spectrometer. (chemical shifts are given in ppm (δ) relative to TMS (Me4Si) as the internal standard, coupling constants (J) are in hertz (Hz), and signals are designated as follows: s, singlet; d, doublet; t, triplet; m, multiplet; br s, broad singlet, etc.). The residual peak of the solvent DMSO-d6 (δ H 2.50 ppm) or CDCl3 (δ H 7.26 ppm) was internal standard for 1H. Mass spectral data were obtained on a Waters Quattro Premier XE spectrometer. TLC was performed on Silica gel 60 F254 with detection by UV light unless staining solution is mentioned.
4.1.1.1. General procedure for the synthesis of compound 21–24
A solution of appropriate amine (1–4) (40 mol, eq.), Boc-glycine (7.001 g, 40 mol), HOBt (5400 g, 40 mol) and EDCI (11.502 g, 60 mmol) in dichloromethane (30 mL) was stirred for 12 h. Aqueous hydrochloric acid solution (4 N, 5 mL) was added to the reaction followed by 10 min stir and filtered. The filtrate was washed with a saturated potassium carbonate solution (50 mL × 2). The organic layer was separated, washed by brine, and dried over MgSO4. After filter and removal of the solvent in vacuo, the solid was redissolved in trifluoroacetic acid and stirred for 1 h at room temperature. Then, the solvent removed in vacuo to yield corresponding trifluoroacetate (93.37%). To a suspension of the trifluoroacetate in dichloromethane (30 mL) and trimethylamine (3 eq.) bath was added 4-nitrobenzenesulfonyl chloride (1.0 eq.) dropwise at 0 °C over a period of 30 min, then stirred for 6 h without cooling. The solvent was evaporated under reduced pressure and added 30 mL water, the mixture was stirred for 30 min and filtered to yield a white solid. Then the solid was dissolved in 10% aqueous sodium hydroxide solution and filtered, the pH was adjusted to 2–3 using a hydrochloric acid solution (6 N). After cooling to room temperature, the solution was filtered and the filter residue (corresponding acetamide) was dried under reduced pressure (85.33%).
Then a solution of the filter residue (1.0 eq.) and potassium carbonate (10.0 eq.) in DMF was stirred at 60 °C for 0.5 h, followed by addition of 1,2-dibromoethane (10 eq.) and the reaction was stirred for another 24 h. The solvent was evaporated under reduced pressure and added 30 mL water, the mixture was stirred for 30 min and filtered to yield a yellow solid (86.68%). (Piperazinone)
Lithium hydroxide (3.0 eq.) was added to a solution of methyl-mercaptopropionate (2.5 eq.) in acetonitrile/DMSO (v/v 49/1), and the mixture was stirred at 50 °C for 30 min. Then, a solution of piperazinone (1.0 eq.) in acetonitrile/DMSO (v/v 49/1) was added dropwise and the reaction was stirred for another 6 h. The solvent removed in vacuo to yield crude oil, purification by column chromatography to afford 21–24 (43–74%).
21 (70.51% yield) 1H NMR (400 MHz, CDCl3, Me4Si) δ 5.03–4.94 (m, 1H), 3.55 (s, 2H), 3.23 (s, 2H), 3.07 (s, 2H), 1.85–1.45 (m, 8H).
24 (71.05% yield) 1H NMR (400 MHz, CDCl3, Me4Si) δ 4.46 (dd, J = 11.4, 3.6 Hz, 1H), 3.54 (s, 2H), 3.27–3.18 (m, 2H), 3.10–3.00 (m, 2H), 1.79 (s, 2H), 1.76–1.29 (m, 8H).
4.1.1.2. General procedure for the synthesis of compound tert-butyl 4-(8-nitro-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazin-2-yl)piperazine-1-carboxylate
To 3 g of 2-chloro-3-nitro-5-(trifluoromethyl)benzoic acid in dichloromethane 2.64 mL of oxalyl chloride and 0.05 mL DMF were added. The reaction mixture was stirred for 2 h at room temperature. The solvent is removed under reduced pressure, and the residue was dissolved in 25 mL dry dichloromethane. The mixture and 0.2 g of PEG-400 was added to 1.57 g of ammoniumthiocyanate dropwise. It was then stirred at room temperature for 1.5 h. The reaction mixture was filtered. And the filtrate was added to 1.49 g of Boc-piperazine, and it was stirred at room temperature for 40 min. The solvent was evaporated under reduced pressure, and crude product was purified by column chromatography to give yellow solid tert-butyl 4-(8-nitro-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazin-2-yl)piperazine-1-carboxylate (45%).
mp 229.5–232.2 °C. 1H-NMR (400 MHz, DMSO, Me4Si): δ 9.15 (s, 1H), 9.13 (s, 1H), 3.94 (br s, 4H), 3.53 (br s, 4H), 1.44 (s, 9H); 13C-NMR (DMSO, 400 MHz, Me4Si): δ 164.72, 161.66, 153.61, 145.33, 144.18, 136.24, 128.83, 125.93, 123.80, 79.47, 45.55, 41.91, 27.98. LC-MS(ESI): m/z 438.1 (M + H)+.
8-Nitro-2-(piperazin-1-yl)-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazin-4-one was prepared from tert-butyl 4-(8-nitro-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazin-2-yl)piperazine-1-carboxylate by utilizing TFA in DCM. And then evaporated, extracted by DCM (15 mL × 3). The combined organic layers were washed with a saturated sodium bicarbonate solution and brine, dried over sodium sulfate, and evaporated to afford product 8-nitro-2-(piperazin-1-yl)-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazin-4-one (85%). LC-MS(ESI): m/z = 347. 0 (M + H)+.
4.1.1.3. General procedure for the synthesis of compound 41–48
To a solution of 1-Boc-4-hydroxypiperidine (1.0 eq.) in DCM was dropped slowly appropriate benzoyl chloride (1.1 eq.), followed by addition of trimethylamine (0.1 eq.). The reaction mixture was stirred at room temperature for 1.5–4 h, extracted by a saturated sodium bicarbonate solution. The organic layer was dried over sodium sulfate and evaporated to oil 33–40. Compounds 41–48 was prepared from 33–40 by utilizing TFA in DCM, and evaporated, dissolved in methyl alcohol. Triethylamine was added to the solution followed by removal of the solvent in vacuo to give yellow solid, which used to synthesis of 41–48.
4.1.1.4. General procedure for the synthesis of compound 69–78
A solution of 1-Boc-4-hydroxypiperidine, appropriate phenol, triphenylphosphine and diethyl azodicarboxylate was stirred in tetrahydrofuran at room temperature for 5 h, evaporated, purified by column chromatography to compounds 59–68. Compound 69–78 was prepared from 59–68 by utilizing TFA in DCM. Then the solvent was evaporated under reduced pressure, and extracted with DCM. The combined organic layers were washed with a saturated sodium bicarbonate solution and brine, dried over sodium sulfate, filtered and evaporated to afford 69–78.
4.1.1.5. General procedure for the synthesis of compound 83, 84, 91–93
To a mixture of 1-Boc-4-hydroxypiperidine and sodium hydride was added dry DMSO (10 mL) at 0 °C bath, then stirred for 30 min. A solution of appropriate 2-bromopyridine in dry DMSO was added dropwise at 0 °C, then stirred for 6 h without cooling. The reaction was quenched by adding water (10 mL) and the product extracted with DCM (2 × 10 mL). The combined organic layers were washed with a saturated sodium bicarbonate solution and brine, dried over sodium sulfate, filtered evaporated, and purified by column chromatography to give intermediate 81 and 82.
83 and 84 were prepared from 81 and 82 by utilizing TFA in DCM. Then the solvent was evaporated under reduced pressure, and extracted with DCM. The combined organic layers were washed with a saturated sodium bicarbonate solution and brine, dried over sodium sulfate, filtered and evaporated to afford 83 and 84.
Similarly, this condition and appropriate 2-chloropyrimidines, dry N,N-dimethylformamide were also used for the synthesis of 91–93.
4.1.1.6. 2-(4-Cyclopentyl-3-oxopiperazin-1-yl)-8-nitro-6(trifluoromethyl)-4H-benzo[e]-[1,3]thiazin-4-one (1a)
Method 4.1.2 starting from 1 (54% yield, yellow powder). 1H NMR (400 MHz, DMSO, Me4Si) δ 9.13 (s, 1H), 8.81 (s, 1H), 4.97 (m, 1H), 4.50 (s, 2H), 4.29 (s, 2H), 3.47 (s, 2H), 1.93 (m, 2H), 1.76 (m, 2H), 1.65 (m, 2H), 1.51 (m, 2H). LC-MS(ESI) LC-MS(ESI): m/z 465.1 (M + Na)+. HPLC purity 99%.
4.1.1.7. 2-(4-Cyclohexyl-3-oxopiperazin-1-yl)-8-nitro-6-(trifluoromethyl)-4H-benzo[e]-[1,3]thiazin-4-one (1b)
Method 4.1.2 starting from 2 (51% yield, yellow powder). 1H NMR (400 MHz, CDCl3CDCl3, Me4Si) δ 9.13 (d, J = 2.1 Hz, 1H), 8.81 (d, J = 2.0 Hz, 1H), 4.59–4.18 (m, 4H), 3.50 (d, J = 25.1 Hz, 2H), 3.33 (d, J = 7.2 Hz, 1H), 1.75–1.16 (m, 10H). LC-MS(ESI) LC-MS(ESI): m/z 457.1 (M + H)+. HPLC purity 98%.
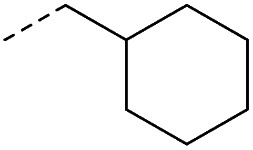
4.1.1.8. 2-(4-(Cyclohexylmethyl)-3-oxopiperazin-1-yl)-8-nitro-6-(trifluoromethyl)-4H-benzo-[e][1,3]thiazin-4-one (1c)
Method 4.1.2 starting from 3 (48% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.13 (d, J = 2.1 Hz, 1H), 8.81 (d, J = 2.1 Hz, 1H), 5.30 (s, 1H), 4.60–4.21 (m, 4H), 3.47 (s, 2H), 1.93–1.34 (m, 12H). LC-MS(ESI): m/z 493.1 (M + Na)+. HPLC purity 99%.
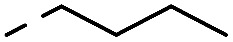
4.1.1.9. 2-(4-Butyl-3-oxopiperazin-1-yl)-8-nitro-6-(trifluoromethyl)-4H-benzo[e][1,3]-thiazin-4-one (1d)
Method 4.1.2 starting from 4 (62% yield, yellow powder). 1H NMR (400 MHz, DMSO-d6) δ 9.12 (d, J = 6.6 Hz, 1H), 8.81 (d, J = 7.3 Hz, 1H), 4.57 (d, J = 48.4 Hz, 2H), 4.31 (s, 2H), 3.48 (dd, J = 19.1, 11.7 Hz, 4H), 1.66–1.53 (m, 2H), 1.35 (p, J = 7.4, 6.5 Hz, 2H), 1.08–0.88 (m, 3H). LC-MS(ESI): m/z 431.1 (M + H)+. HPLC purity 99%.
4.1.1.10. Ethyl 1-(8-nitro-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazin-2-yl)piperidine-4-carboxylate (2a)
Method 4.1.2 starting from ethyl 4-piperidinecarboxylate (60% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.10 (d, J = 2.1 Hz, 1H), 8.77 (d, J = 2.1 Hz, 1H), 4.11 (q, J = 7.1 Hz, 4H), 3.45 (p, J = 5.0 Hz, 1H), 2.75 (m, 4H), 1.24 (t, J = 6.6, 3H). LC-MS(ESI): m/z 454.1 (M + Na)+. HPLC purity 99%.
4.1.1.11. 2Ethyl 2-(4-(8-nitro-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazin-2-yl)-piperazin-1-yl)acetate (2b)
Method 4.1.2 starting from 1-(ethoxycarbonylmethyl)piperazine (54% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.10 (d, J = 2.1 Hz, 1H), 8.77 (d, J = 2.1 Hz, 1H), 4.21 (q, J = 7.1 Hz, 4H), 3.32 (s, 2H), 2.80 (s, 4H), 1.27 (s, 3H). LC-MS(ESI): m/z 447.1 (M + H)+. HPLC purity 98%.
4.1.1.12. 3Ethyl 2-(1-(8-nitro-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazin-2-yl)-piperidin-4-yl)acetate (2c)
Method 4.1.2 starting from ethyl 1-(ethoxycarbonylmethyl)piperidine (47% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.20 (d, J = 2.3 Hz, 1H), 8.80 (d, J = 1.9 Hz, 1H), 4.45 (m, 6H), 3.02 (m, 1H), 2.50 (s, 4H), 1.40 (t, J = 6.6 Hz, 3H). LC-MS(ESI): m/z 446.1 (M + H)+. HPLC purity 96%.
4.1.1.13. Ethyl 2-(4-(8-nitro-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazin-2-yl)-piperazin-1-yl)propanoate (2d)
Ethyl 2-bromopropionate (1.5 eq.) was added to a mixture of compound 11(1.0 eq.) and potassium carbonate in dry DMF, the reaction was heated at 60 °C for 3 h followed by pouring in 20 mL water. The reaction mixture was then extracted with DCM (10 mL × 3). The combined organic layers were washed with brine. Dried over sodium sulfate, and the solvent removed in vacuo to yield a yellow solid, purification by chromatography to afford 2d (68%, yellow solid). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.03 (d, J = 2.0 Hz, 1H), 8.72 (d, J = 2.0 Hz, 1H), 4.31 (t, J = 7.8 Hz, 2H), 4.22–3.79 (m, 4H), 3.62 (t, J = 7.8 Hz, 2H), 3.18 (s, 4H). ESI HRMS exact mass calcd. For (C18H19F3N4O5S + Na)+ requires m/z 483.09, found m/z 483.0888. HPLC purity 96%.
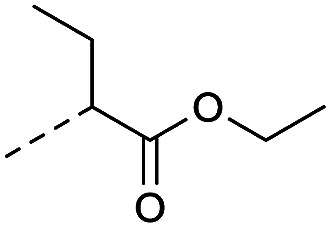
4.1.1.14. Ethyl 2-(4-(8-nitro-4-oxo-6-(trifluoromethyl)-4H-benzo[e]-[1,3]thiazin-2-yl)-piperazin-1-yl) butanoate (2e)
Method 4.1.24 starting from 2-bromobutanoic acid ethyl ester (58% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.04 (s, 1H), 8.72 (s, 1H), 4.90 (dd, J = 7.0, 5.0 Hz, 1H), 4.16 (qd, J = 7.1, 2.5 Hz, 3H), 4.07–3.37 (m, 8H), 1.85 (dt, J = 14.4, 7.3 Hz, 2H), 1.22 (t, J = 7.1 Hz, 3H), 0.95 (t, J = 7.4 Hz, 3H). LC-MS(ESI): m/z 475.1 (M + H)+. HPLC purity 95%.
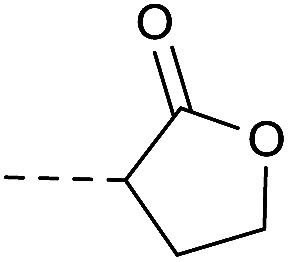
4.1.1.15. 8-Nitro-2-(4-(2-oxotetrahydrofuran-3-yl)piperazin-1-yl)-6-(tri-fluoromethyl)-4H-benzo[e][1,3]thiazin-4-one (2f)
Method 4.1.24 starting from 2-bromo-4-butanolide (63% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.20 (d, J = 2.1 Hz, 1H), 8.80 (d, J = 2.1 Hz, 1H), 4.50–3.95 (m, 6H), 3.77–3.60 (m, 1H), 2.66–2.32 (m, 4H), 1.85–1.68 (m, 2H). LC-MS(ESI): m/z 445.1 (M + H)+. HPLC purity 99%.
4.1.1.16. 8-Nitro-2-(4-(2-oxo-5-propyltetrahydrofuran-3-yl)piperazin-1-yl)-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazin-4-one (2g)
Method 4.1.24 starting from 3-bromo-5-propyldihydrofuran-2(3H)-one (43% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.03 (d, J = 2.1 Hz, 1H), 8.70 (d, J = 2.1 Hz, 1H), 4.32–3.84 (m, 5H), 3.69–3.57 (m, 1H), 2.78–2.25 (m, 4H), 1.88–1.66 (m, 2H), 1.57 (ddd, J = 13.9, 9.4, 5.3 Hz, 2H), 1.30–1.00 (m, 2H), 0.90 (dd, J = 8.4, 6.4 Hz, 3H). LC-MS(ESI) m/z 487.1 (M + H)+. HPLC purity 98%.
4.1.1.17. 1-(8-Nitro-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazin-2-yl)piperidin-4-yl 4-fluorobenzoate (2h)
Method 4.1.2 starting from 25 (44% yield, yellow powder).1H NMR (400 MHz, CDCl3, Me4Si) δ 9.04 (dd, J = 2.1, 0.7 Hz, 1H), 8.70 (dd, J = 2.1, 0.7 Hz, 1H), 7.90 (td, J = 7.6, 1.9 Hz, 1H), 7.49 (dddd, J = 8.3, 7.4, 4.9, 1.9 Hz, 1H), 7.17 (dd, J = 7.6, 1.1 Hz, 1H), 7.14–7.03 (m, 1H), 5.39 (p, J = 4.4 Hz, 1H), 4.30–3.79 (m, 4H), 2.00 (d, J = 21.8 Hz, 4H). ESI HRMS exact mass calcd. For (C21H15F4N3NaO5S + Na)+ requires m/z 520.06, found m/z 520.0565. HPLC purity 99%.
4.1.1.18. 1-(8-Nitro-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazin-2-yl)piperidin-4-yl 2-fluorobenzoate (2i)
Method 4.1.2 starting from 26 (41% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.04 (d, J = 2.2 Hz, 1H), 8.70 (d, J = 2.1 Hz, 1H), 7.89 (tt, J = 7.3, 1.8 Hz, 1H), 7.58–7.45 (m, 1H), 7.14–7.04 (m, 1H), 5.39 (q, J = 4.6 Hz, 1H), 4.41–3.88 (m, 4H), 2.00 (d, J = 24.3 Hz, 4H). LC-MS(ESI): m/z = 498.1(M + H)+. HPLC purity 99%.
4.1.1.19. 1-(8-Nitro-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazin-2-yl)piperidin-4-yl 2-chlorobenzoate (2j)
Method 4.1.24 starting from 27 (46% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.04 (d, J = 2.1 Hz, 1H), 8.70 (d, J = 2.1 Hz, 1H), 7.86–7.74 (m, 1H), 7.50–7.37 (m, 2H), 7.29 (ddd, J = 8.5, 6.4, 2.2 Hz, 1H), 5.38 (q, J = 4.9 Hz, 1H), 4.02 (d, J = 11.6 Hz, 4H), 2.05 (s, 4H). LC-MS(ESI) m/z 436.0 (M + Na)+. HPLC purity 99%.
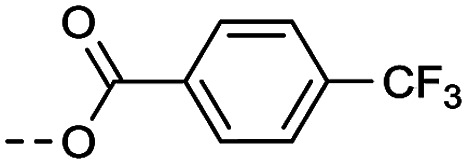
4.1.1.20. 1-(8-Nitro-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazin-2-yl)piperidin-4-yl 4-(trifluoromethyl)benzoate (2k)
Method 4.1.2 starting from 28 (41% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.10 (s, 1H), 8.78 (s, 1H), 7.40 (m, 2H), 7.07 (dd, J = 8.6, 1.1 Hz, 2H), 5.35 (m, 1H), 3.58 (m, 2H), 3.01 (m, 2H), 2.30 (m, 2H), 2.06 (m, 3H). LC-MS(ESI) m/z 548.1 (M + H)+. HPLC purity 98%.
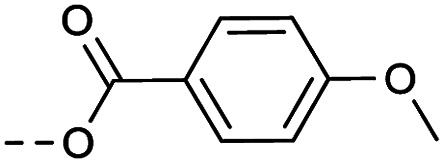
4.1.1.21. 1-(8-Nitro-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazin-2-yl)piperidin-4-yl 4-methoxybenzoat (2l)
Method 4.1.2 starting from 29 (43% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.10 (dd, J = 2.1, 0.8 Hz, 1H), 8.80–8.73 (m, 1H), 7.02–6.94 (m, 2H), 6.93–6.84 (m, 2H), 5.10–4.27 (m, 2H), 3.80 (s, 3H), 3.56 (t, J = 12.2 Hz, 2H), 2.99 (tt, J = 9.9, 4.2 Hz, 1H), 2.26 (m, 2H), 2.09–1.99 (m, 2H). LC-MS(ESI) m/z 510.1 (M + H)+. HPLC purity 97%.
4.1.1.22. 1-(8-Nitro-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazin-2-yl)piperidin-4-yl 2-methoxybenzoate (2m)
Method 4.1.2 starting from 30 (42% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.12 (dd, J = 2.1, 0.7 Hz, 1H), 8.80–8.75 (m, 1H), 7.82 (dd, J = 8.0, 1.8 Hz, 1H), 7.54–7.49 (m, 1H), 7.05–6.98 (m, 2H), 5.41 (q, J = 4.4 Hz, 1H), 4.03 (m, 2H), 3.92 (s, 3H), 2.07 (m, 4H), 1.64 (m, 2H). LC-MS(ESI) m/z 510.1 (M + H)+. HPLC purity 99%.
4.1.1.23. Methyl (1-(8-nitro-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazin-2-yl)-piperidin-4-yl) terephthalate (2n)
Method 4.1.2 starting from 31 (46% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.05 (dd, J = 2.1, 0.7 Hz, 1H), 8.74–8.69 (m, 1H), 8.09–8.00 (m, 4H), 5.34 (dq, J = 7.0, 3.5 Hz, 1H), 4.22–3.94 (m, 4H), 3.89 (s, 3H), 2.04 (d, J = 55.5 Hz, 4H). LC-MS(ESI) m/z 538.1(M + H)+. HPLC purity 98%.
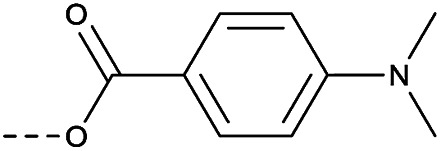
4.1.1.24. 1-(8-Nitro-4-oxo-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazin-2-yl)piperidin-4-yl 4-(dimethylamino)benzoate (2o)
Method 4.1.2 starting from 32 (40% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.11 (s, 1H), 8.77 (s, 1H), 7.49–7.41 (m, 1H), 7.34 (m, 1H), 6.99 (dt, J = 8.2, 4.2 Hz, 2H), 5.26 (m, 1H), 4.13–3.92 (m, 4H), 3.34–3.21 (m, 2H), 3.03 (m, 2H), 2.12 (s, 6H). LC-MS(ESI) m/z 523.1(M + H)+. HPLC purity 99%.
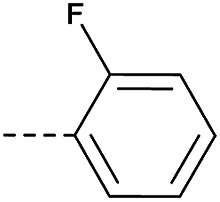
4.1.1.25. 2-(4-(2-Fluorophenoxy)piperidin-1-yl)-8-nitro-6-(trifluorom-ethyl)-4H-benzo[e][1,3]-thiazin-4-one (3a)
Method 4.1.2 starting from 49 (52% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.04 (dd, J = 2.1, 0.7 Hz, 1H), 8.70 (dd, J = 2.1, 0.7 Hz, 1H), 7.90 (td, J = 7.6, 1.9 Hz, 1H), 7.49 (dddd, J = 8.3, 7.4, 4.9, 1.9 Hz, 1H), 7.17 (dd, J = 7.6, 1.1 Hz, 1H), 7.14–7.03 (m, 1H), 5.39 (p, J = 4.4 Hz, 1H), 4.30–3.79 (m, 4H), 2.00 (d, J = 21.8 Hz, 4H). LC-MS(ESI) m/z 493.1(M + Na)+. HPLC purity 99%.
4.1.1.26. 2-(4-(4-Fluorophenoxy)piperidin-1-yl)-8-nitro-6-(trifluoro-methyl)-4H-benzo[e][1,3]-thiazin-4-one (3b)
Method 4.1.2 starting from 50 (47% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.12 (dd, J = 2.1, 0.8 Hz, 1H), 8.78 (dd, J = 2.1, 0.8 Hz, 1H), 8.12–8.03 (m, 2H), 7.19–7.10 (m, 2H), 5.38 (tt, J = 6.9, 3.6 Hz, 1H), 4.34–3.68 (m, 4H), 2.11 (d, J = 49.2 Hz, 4H). ESI HRMS exact mass calcd. For (C20H19F3N4O5S + Na)+ requires m/z 492.06, found m/z 492.0620. HPLC purity 99%.
4.1.1.27. 2-(4-(3-Fluorophenoxy)piperidin-1-yl)-8-nitro-6-(trifluorome-thyl)-4H-benzo[e][1,3]-thiazin-4-one (3c)
Method 4.1.2 starting from 51 (55% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.04 (d, J = 2.0 Hz, 1H), 8.70 (d, J = 2.0 Hz, 1H), 7.25–7.19 (m, 1H), 6.70–6.54 (m, 3H), 4.63 (t, J = 4.2 Hz, 1H), 4.17–3.48 (m, 4H), 1.98 (d, J = 10.3 Hz, 4H). LC-MS(ESI): m/z 493.1 (M + Na)+. HPLC purity 99%.
4.1.1.28. 2-(4-(2-Chlorophenoxy)piperidin-1-yl)-8-nitro-6-(trifluo-romethyl)-4H-benzo[e][1,3]thiazin-4-one (3d)
Method 4.1.2 starting from 52 (51% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.04 (d, J = 2.1 Hz, 1H), 8.70 (d, J = 2.1 Hz, 1H), 7.86–7.74 (m, 1H), 7.50–7.37 (m, 2H), 7.29 (ddd, J = 8.5, 6.4, 2.2 Hz, 1H), 5.38 (q, J = 4.9 Hz, 1H), 4.02 (d, J = 11.6 Hz, 4H), 2.05 (s, 4H). LC-MS(ESI): m/z 486.0 (M + H)+. HPLC purity 96%.
4.1.1.29. 2-(4-(2-Bromophenoxy)piperidin-1-yl)-8-nitro-6-(trifluo-romethyl)-4H-benzo[e][1,3]thiazin-4-one (3e)
Method 4.1.2 starting from 53 (46% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.03 (d, J = 2.1 Hz, 1H), 8.69 (d, J = 2.1 Hz, 1H), 7.51 (dd, J = 7.9, 1.6 Hz, 1H), 7.26–7.19 (m, 1H), 6.94–6.79 (m, 2H), 4.74 (s, 1H), 2.17–1.84 (m, 4H), 1.55 (s, 4H). LC-MS(ESI) m/z 552.0 (M + Na)+. HPLC purity 97%.
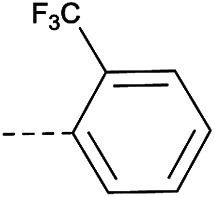
4.1.1.30. 8-Nitro-6-(trifluoromethyl)-2-(4-(2-(trifluoromethyl)pheno-xy)piperidin-1-yl)-4H-benzo[e][1,3]thiazin-4-one (3f)
Method 4.1.2 starting from 54 (54% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.04 (d, J = 2.1 Hz, 1H), 8.69 (d, J = 2.1 Hz, 1H), 7.55 (dd, J = 7.8, 1.6 Hz, 1H), 7.44 (td, J = 7.9, 1.7 Hz, 1H), 7.03–6.90 (m, 2H), 4.83 (s, 1H), 2.24–1.84 (m, 4H), 1.54 (s, 4H). LC-MS(ESI) m/z 520.1 (M + H)+. HPLC purity 97%.
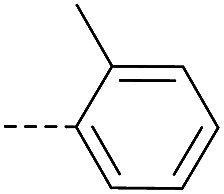
4.1.1.31. 8-Nitro-2-(4-(o-tolyloxy)piperidin-1-yl)-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazin-4-one (3g)
Method 4.1.2 starting from 55 (57% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.04 (d, J = 2.1 Hz, 1H), 8.69 (d, J = 2.1 Hz, 1H), 7.17–7.03 (m, 2H), 6.88–6.72 (m, 2H), 4.67 (s, 1H), 3.88 (s, 4H), 2.19 (s, 3H), 1.99 (s, 4H). LC-MS(ESI) m/z 466.1 (M + H)+. HPLC purity 97%.
4.1.1.32. 8-Nitro-2-(4-(4-(trifluoromethoxy)phenoxy)piperidin-1-yl)-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazin-4-one (3h)
Method 4.1.2 starting from 56 (48% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.03 (d, J = 2.1 Hz, 1H), 8.70 (d, J = 2.1 Hz, 1H), 7.16–7.06 (m, 2H), 6.92–6.82 (m, 2H), 4.62 (t, J = 4.1 Hz, 1H), 4.37–3.63 (m, 4H), 2.00 (s, 4H). LC-MS(ESI) m/z 536.1 (M + H)+. HPLC purity 96%.
4.1.1.33. 2-(4-(2-Fluoro-4-methylphenoxy)piperidin-1-yl)-8-nitro-6-(tri-fluoromethyl)-4H-benzo[e][1,3]thiazin-4-one (3i)
Method 4.1.2 starting from 57 (54% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.04 (d, J = 2.1 Hz, 1H), 8.69 (d, J = 2.1 Hz, 1H), 6.92–6.76 (m, 3H), 3.91 (d, J = 25.6 Hz, 4H), 3.42 (s, 1H), 2.23 (s, 3H), 1.94 (s, 4H). LC-MS(ESI) m/z 506.1 (M + Na)+. HPLC purity 98%.
4.1.1.34. 2-(4-(2,6-Difluorophenoxy)piperidin-1-yl)-8-nitro-6-(trifle-oromethyl)-4H-benzo-[e][1,3]thiazin-4-one (3j)
Method 4.1.2 starting from 58 (49% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.04 (d, J = 2.1 Hz, 1H), 8.69 (d, J = 2.1 Hz, 1H), 7.00–6.80 (m, 3H), 4.50 (s, 1H), 3.98 (s, 4H), 1.97 (d, J = 42.4 Hz, 4H). LC-MS(ESI) m/z 510.1 (M + Na)+. HPLC purity 99%.
4.1.1.35. 8-Nitro-2-(4-(pyridin-2-yloxy)piperidin-1-yl)-6-(trifluoro-methyl)-4H-benzo[e][1,3]thiazin-4-one (3k)
Method 4.1.2 starting from 79 (52% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.11 (d, J = 2.3 Hz, 1H), 8.78 (d, J = 2.3 Hz, 1H), 8.00 (d, J = 3.4 Hz, 1H), 7.30 (d, J = 8.7 Hz, 1H), 7.15 (d, J = 8.3 Hz, 1H), 7.03(dd, J = 8.6, 3.1 Hz, 1H), 4.72 (p, J = 4.6 Hz, 1H), 4.00 (dt, J = 13.6, 6.3 Hz, 2H), 2.06 (d, J = 16.0 Hz, 4H), 1.35–1.24 (m, 2H). LC-MS(ESI): m/z 475.1 (M + Na)+. HPLC purity 95%.
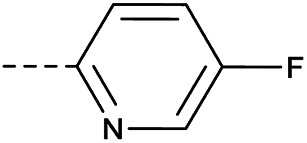
4.1.1.36. 2-(4-((5-Fluoropyridin-2-yl)oxy)piperidin-1-yl)-8-nitro-6-(tri-fluoromethyl)-4H-benzo[e][1,3]thiazin-4-one (3l)
Method 4.1.2 starting from 80 (45% yield, yellow powder).1H NMR (400 MHz, CDCl3, Me4Si) δ 9.11 (d, J = 2.1 Hz, 1H), 8.78 (d, J = 2.1 Hz, 1H), 8.11 (d, J = 3.1 Hz, 1H), 7.43 (d, J = 8.7 Hz, 1H), 7.15 (dd, J = 8.7, 3.1 Hz, 1H), 4.72 (p, J = 4.3 Hz, 1H), 4.00 (dt, J = 13.6, 6.7 Hz, 2H), 2.06 (d, J = 15.5 Hz, 4H), 1.35–1.24 (m, 2H). LC-MS(ESI): m/z 471.1 (M + Na)+. HPLC purity 98%.
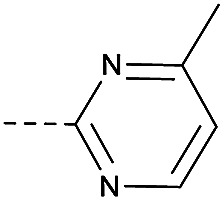
4.1.1.37. 2-(4-((4-Methylpyrimidin-2-yl)oxy)piperidin-1-yl)-8-nitro-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazin-4-one (3m)
Method 4.1.2 starting from 85 (58% yield, yellow powder). 1H NMR (400 MHz, CDCl3, Me4Si) δ 9.11 (d, J = 2.1 Hz, 1H), 8.77 (d, J = 2.1 Hz, 1H), 8.35 (dd, J = 9.2, 5.0 Hz, 1H), 6.85 (d, J = 5.0 Hz, 1H), 4.34 (dd, J = 17.2, 10.2 Hz, 1H), 4.11 (p, J = 6.8 Hz, 4H), 2.48 (d, J = 7.4 Hz, 4H), 1.60 (s, 3H). LC-MS(ESI) m/z 490.1 (M + Na)+. HPLC purity 97%.
4.1.1.38. 8-Nitro-6-(trifluoromethyl)-2-(4-((4-(trifluoromethyl)pyrimidin-2-yl)oxy)piperidin-1-yl)-4H-benzo[e][1,3]thiazin-4-one (3n)
Method 4.1.2 starting from 88 (52% yield, yellow powder).1H NMR (400 MHz, CDCl3, Me4Si) δ 9.11 (d, J = 2.1 Hz, 1H), 8.79 (dd, J = 13.5, 3.5 Hz, 2H), 7.33 (d, J = 4.9 Hz, 1H), 5.50 (p, J = 4.7 Hz, 1H), 4.67–3.90 (m, 4H), 2.17 (s, 4H). ESI HRMS exact mass calcd. For (C19H13F6N5O3S + Na)+ requires m/z 544.05, found m/z 544.0482. HPLC purity 97%.
4.1.1.39. (4-((4-Chloropyrimidin-2-yl)oxy)piperidin-1-yl)-8-nitro-6-(trifluoromethyl)-4H-benzo[e][1,3]thiazin-4-one (3o)
Method 4.1.2 starting from 87 (47% yield, yellow powder).1H NMR (400 MHz, CDCl3, Me4Si) δ 9.12 (d, J = 2.1 Hz, 1H), 8.78 (d, J = 2.1 Hz, 1H), 8.36 (d, J = 5.7 Hz, 1H), 6.70 (d, J = 5.7 Hz, 1H), 5.57 (tt, J = 6.8, 3.6 Hz, 1H), 4.12 (q, J = 7.2 Hz, 4H), 2.22–2.01 (m, 4H). ESI HRMS exact mass calcd. For (C18H13F4O3S + Na)+ requires m/z 494.05, found m/z 494.0526. HPLC purity 99%.
5. Biology
5.1. Mycobacterium tuberculosis H37Ra MIC assay
The MICs against Mycobacterium tuberculosis H37Ra were detected by the microplate Alamar blue assay. Compounds were dissolved in dimethyl sulfoxide (DMSO) with concentrations of 1 to 0.002 μmol L−1. M. tuberculosis was grown to late log phase in Middlebrook 7H9 medium supplemented with 0.05% Tween 80, 0.2% (vol/vol) glycerol, and 10% (vol/vol) oleic acid-albumin-dextrose-catalase. Cultures were then centrifuged, washed, and resuspended in phosphate-buffered saline. Suspensions were filtrated through an 8 μm-pore-size filter. Then the aliquots were frozen at −80 °C. The number of CFU was determined by plating on 7H11 agar plates. Two-fold dilutions of compounds were prepared in Middlebrook 7H12 medium in a volume of 100 μL in 96-well microplates. M.tb (100 μL inoculums of 106 CFU mL−1) was added, the plates were incubated at 37 °C. On the 10th day, add 20 μL 0.01% Alamar Blue and 12.5 μL of 20% Tween 80 to each well. After 24 hours the fluorescence of each well is measured at excitation 530 nm; emission 590 nm.
The MICs were defined as the lowest concentration effecting a reduction in fluorescence of ≥90% relative to the controls. Reported MICs are an average of two or three individual measurements.
5.2. The inhibition to cytochrome P450
CYP inhibition potential of compounds were determined using the following recombinant microsomes: CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4. Each incubation (T0, T5, T20, T30, T60, NCF60) which contained recombinant microsomes (80 μL), compounds (10 μL), and extra phosphate buffer (100 mM, pH = 7.4) was added in NCF60. Incubation mixtures were preincubated at 37 °C for 5 minutes, after incubation, 25 mL of NADPH regenerating system solution was added to each well and reactions was started immediately at 37 °C. The reactions were terminated with 300 μL after 60 minutes terminal solution (tolbutamide 100 ng mL−1 and labetalol 100 ng mL−1). After centrifugation, the supernatant was analyzed by LC-MS/MS.
5.3. Plasma protein binding rate (PPB) determining
The extent of plasma protein binding for each test compounds was determined by equilibrium dialysis. A 96-well Rapid Equilibrium Dialysis (RED) kit (ThermoFisher Scientific) with cassettes of compounds (pooling of compounds predialysis) was used to perform measurement (each run in triplicate). The plasma was centrifuged (4 minutes, 3500 rpm) and adjusted to pH = 7.4 + 0.1 with 1% phosphoric acid and 1 N sodium hydroxide. Test compound was added to the plasma which was mixed and heated (1 μM, 1% [vol/vol] DMSO, 37 °C). Regenerated cellulose membranes (5000 Daltons, Harvard Apparatus) were soaked in phosphate buffer for 5 minutes and placed within Fast Micro-Equilibrium Dialyzers (Harvard Apparatus). Subsequently, the plasma containing compounds was added to the donor side of the single-use RED plate. Buffer was added to the other side. Equilibrium dialysis was undertaken by incubation (18 hours, 37 °C) and samples were removed from each compartment for LC-MS/MS analysis.
5.4. Pharmacokinetic study
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of “Experimental Animal management regulations in China” and approved by the Animal Ethics Committee of “Institutional Animal Care and Treatment Committee of Sichuan University, China”. SD rats and mice were used in the pharmacokinetic study. Every treatment group contains 3 rats. Animals were dosed with compounds suspension (i.v. and p.o.). Compounds were dissolved in 20% hydroxypropyl-b-cyclodextrin (pH = 3.0) for intravenous injection and suspended in 0.1% Tween 80 with 0.5% CMC–Na (pH = 3.0) for oral administration. Blood was collected from the jugular vein of each animal at the following times after administration of drugs: 0.083, 0.25, 0.5, 1, 2, 4, 6, 8 and 24 h after a single IV dosing for SD rats; 0.25, 0.5, 1, 2, 4, 6, 8, 12 and 24 h after a single oral dosing for SD rats; 0.083, 0.25, 0.5, 1, 2, 4, 6, 8 and 24 h after a single IV and oral dosing for ICR mice; all blood samples were centrifuged at 3000 rpm for 10 min to obtain serum which was then stored at −20 °C. 150 μL of the serum was added to 500 μL of acetonitrile and the mixture was centrifuged at 13 000 rpm for 10 min to remove protein. The supernatant was dried and dissolve in 100 μL of acetonitrile, the solution was centrifuged at 13 000 rpm for 10 min.
The supernatant was moved to a sample bottle for HPLC analysis. Pharmacokinetics profile samples were determined directly from the experimental data using WinNonlin V6.2.1.
5.5. Permeability assay
MECK cells were cultured and screened as previous report.26 Transport assays were conducted using 0.3 mL of apical (AP) donor solution and 1 mL of basolateral (BL) acceptor solution (transport medium). All compounds were tested in three replicate monolayers. Monolayers were incubated with donor and acceptor solutions for 60 min at 37 °C, 95% humidity, with 30 rpm reciprocal shaking. BL compartments were sampled at 15, 30, and 60 min. AP compartments were sampled at 60 min. The quality control compound set (a pool of fenoterol, propranolol and digxin) was assayed as a pool under the same conditions as the test compounds, using a donor solution containing 100 μM of each compound in transport medium (pH 7.4). Most compounds, including the quality control compound set, were quantified by HPLC.
Calculations-Papp (apparent permeability) values were calculated according to the following equation:Papp = (dQ/dt) × (1/C0) × (1/A)dQ/dt = the permeability rate (calculated by plotting the percent of initial AP drug mass (peak area) found in the BL compartment versus time and determining the slope of the line.)
C 0 = the initial concentration in the donor compartment.
A = surface area of the filter.
5.6. Stability in microsomes assay
Previously published incubation procedures using HLM, RLM and MLM were used with modifications.27 Appropriate substrates and PBTZ169 (positive controls) were added to incubation tubes. The compounds were individually added in concentration of 10 μM to separate incubation tubes. The incubation mixtures contained 100 mM phosphate buffer (pH 7.5). The compounds were preincubated with HLMs, RLMs and MLMs (without the index substrates) for 10 minutes at 37 °C, and then followed by 60 min incubation at 37 °C with the substrates (10 μL NADPH). All incubations were performed in duplicate. 300 μL of acetonitrile with (100 ng mL−1 tolbutamide and 100 ng mL−1 labetalol solution) was used to stop the reactions. After centrifugation, the supernatant was transferred to HPLC vials for HPLC-UV or HPLC-fluorescence analysis.
5.7. Solubility experiment
Previously published method was used with modifications.28 For solubility experiments with water, an excess amount of solid compounds substance was added to microcentrifuge tubes containing 500 μL of the medium (water with different pH). Reliability of the protocol was confirmed as similar solubilities (RSD < 5%) were obtained in 500 μL medium (microcentrifuge tubes) and 5 mL medium (test tubes) (data not shown). The microcentrifuge tubes were placed at 37 °C in a prewarmed shaking incubator to reach equilibrium. After incubation, samples were centrifuged for 10 min at 14 000 rpm min−1 and 37 °C. Subsequently, 100 μL of the supernatant was mixed with 100 μL of methanol (protein precipitation) and centrifuged for 5 min at 14 000 rpm min−1 and 37 °C. The supernatant was diluted into the mobile phase used during the subsequent high-performance liquid chromatography (HPLC) analysis.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by the Natural Science Foundation of China (81703570), China Postdoctoral Science Foundation (2017M612962) and National S&T Major Special Project on Major New Drug Innovations (2018ZX09721001).
Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra00720a
References
- Giller D. B. Giller B. D. Giller G. V. Shcherbakova G. V. Bizhanov A. B. Enilenis I. I. Glotov A. A. Eur. J. Cardio Thorac. Surg. 2017 doi: 10.1093/ejcts/ezx447. [DOI] [Google Scholar]
- Villemagne B. Crauste C. Flipo M. Baulard A. R. Deprez B. Willand N. Eur. J. Med. Chem. 2012;51:1–16. doi: 10.1016/j.ejmech.2012.02.033. [DOI] [PubMed] [Google Scholar]
- Burki T. K. Respir. Med. 2018;6:13. doi: 10.1016/S2213-2600(17)30468-X. [DOI] [PubMed] [Google Scholar]
- Makarov V. Manina G. Mikusova K. Mollmann U. Ryabova O. Saint-Joanis B. Dhar N. Pasca M. R. Buroni S. Lucarelli A. P. Milano A. De Rossi E. Belanova M. Bobovska A. Dianiskova P. Kordulakova J. Sala C. Fullam E. Schneider P. McKinney J. D. Brodin P. Christophe T. Waddell S. Butcher P. Albrethsen J. Rosenkrands I. Brosch R. Nandi V. Bharath S. Gaonkar S. Shandil R. K. Balasubramanian V. Balganesh T. Tyagi S. Grosset J. Riccardi G. Cole S. T. Science. 2009;324:801–804. doi: 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov V. Lechartier B. Zhang M. Neres J. van der Sar A. M. Raadsen S. A. Hartkoorn R. C. Ryabova O. B. Vocat A. Decosterd L. A. Widmer N. Buclin T. Bitter W. Andries K. Pojer F. Dyson P. J. Cole S. T. EMBO Mol. Med. 2014;6:372–383. doi: 10.1002/emmm.201303575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon R. Nath M. Mini Rev. Med. Chem. 2017;17:549–570. doi: 10.2174/1389557516666160606204639. [DOI] [PubMed] [Google Scholar]
- Piton J. Foo C. S. Cole S. T. Drug discovery today. 2017;22:526–533. doi: 10.1016/j.drudis.2016.09.014. [DOI] [PubMed] [Google Scholar]
- Trefzer C. Rengifo-Gonzalez M. Hinner M. J. Schneider P. Makarov V. Cole S. T. Johnsson K. J. Am. Chem. Soc. 2010;132:13663–13665. doi: 10.1021/ja106357w. [DOI] [PubMed] [Google Scholar]
- Batt S. M. Jabeen T. Bhowruth V. Quill L. Lund P. A. Eggeling L. Alderwick L. J. Futterer K. Besra G. S. Proc. Natl. Acad. Sci. U. S. A. 2012;109:11354–11359. doi: 10.1073/pnas.1205735109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brecik M. Centarova I. Mukherjee R. Kolly G. S. Huszar S. Bobovska A. Kilacskova E. Mokosova V. Svetlikova Z. Sarkan M. Neres J. Kordulakova J. Cole S. T. Mikusova K. ACS Chem. Biol. 2015;10:1631–1636. doi: 10.1021/acschembio.5b00237. [DOI] [PubMed] [Google Scholar]
- Kwon Y. S. Jeong B. H. Koh W. J. Curr. Opin. Pulm. Med. 2014;20:280–286. doi: 10.1097/MCP.0000000000000045. [DOI] [PubMed] [Google Scholar]
- Lakshminarayana S. B. Boshoff H. I. Cherian J. Ravindran S. Goh A. Jiricek J. Nanjundappa M. Nayyar A. Gurumurthy M. Singh R. Dick T. Blasco F. Barry 3rd C. E. Ho P. C. Manjunatha U. H. PLoS One. 2014;9:e105222. doi: 10.1371/journal.pone.0105222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv K. You X. Wang B. Wei Z. Chai Y. Wang B. Wang A. Huang G. Liu M. Lu Y. ACS Med. Chem. Lett. 2017;8:636–641. doi: 10.1021/acsmedchemlett.7b00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov V. Neres J. Hartkoorn R. C. Ryabova O. B. Kazakova E. Sarkan M. Huszar S. Piton J. Kolly G. S. Vocat A. Conroy T. M. Mikusova K. Cole S. T. Antimicrob. Agents Chemother. 2015;59:4446–4452. doi: 10.1128/AAC.00778-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karoli T. Becker B. Zuegg J. Möllmann U. Ramu S. Huang J. X. Cooper M. A. J. Med. Chem. 2012;55:7940–7944. doi: 10.1021/jm3008882. [DOI] [PubMed] [Google Scholar]
- Maitre T. Aubry A. Veziris N. N. Engl. J. Med. 2017;377:2403–2404. doi: 10.1056/NEJMc1714218. [DOI] [PubMed] [Google Scholar]
- Chong S. Dando S. A. Morrison R. A. Pharmaceut. Res. 1997;14:1835–1837. doi: 10.1023/A:1012112820371. [DOI] [PubMed] [Google Scholar]
- Aggarwal A. Parai M. K. Shetty N. Wallis D. Woolhiser L. Hastings C. Dutta N. K. Galaviz S. Dhakal R. C. Shrestha R. Wakabayashi S. Walpole C. Matthews D. Floyd D. Scullion P. Riley J. Epemolu O. Norval S. Snavely T. Robertson G. T. Rubin E. J. Ioerger T. R. Sirgel F. A. van der Merwe R. van Helden P. D. Keller P. Bottger E. C. Karakousis P. C. Lenaerts A. J. Sacchettini J. C. Cell. 2017;170:249–259. doi: 10.1016/j.cell.2017.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guengerich F. P. Johnson W. W. Biochemistry. 1997;36:14741–14750. doi: 10.1021/bi9719399. [DOI] [PubMed] [Google Scholar]
- Walker M. A. Bioorg. Med. Chem. Lett. 2017;27:5100–5108. doi: 10.1016/j.bmcl.2017.09.041. [DOI] [PubMed] [Google Scholar]
- Nakajima M. zasshi Y. J. Pharm. Soc. Jpn. 2017;137:697–705. doi: 10.1248/yakushi.16-00250-5. [DOI] [PubMed] [Google Scholar]
- Fray M. J. Bull D. J. Carr C. L. Gautier E. C. L. Mowbray C. E. Stobie A. J. Med. Chem. 2001;44:1951–1962. doi: 10.1021/jm001124p. [DOI] [PubMed] [Google Scholar]
- Breitenlechner C. B. Wegge T. Berillon L. Graul K. Marzenell K. Friebe W.-G. Thomas U. Schumacher R. Huber R. Engh R. A. Masjost B. J. Med. Chem. 2004;47:1375–1390. doi: 10.1021/jm0310479. [DOI] [PubMed] [Google Scholar]
- Peglion J.-L. Goument B. Despaux N. Charlot V. Giraud H. Nisole C. Newman-Tancredi A. Dekeyne A. Bertrand M. Genissel P. Millan M. J. J. Med. Chem. 2002;45:165–176. doi: 10.1021/jm010975+. [DOI] [PubMed] [Google Scholar]
- Manjunatha U. H. Smith P. W. Bioorg. Med. Chem. 2015;23:5087–5097. doi: 10.1016/j.bmc.2014.12.031. [DOI] [PubMed] [Google Scholar]
- Irvine J. D. Takahashi L. Lockhart K. Cheong J. Tolan J. W. Selick H. E. Grove J. R. J. Pharmaceut. Sci. 1999;88:28–33. doi: 10.1021/js9803205. [DOI] [PubMed] [Google Scholar]
- Greenblatt D. J. Harmatz J. S. Walsh J. K. Luthringer R. Staner L. Otmani S. Nedelec J. F. Francart C. Parent S. J. Staner C. Biopharm Drug Dispos. 2011;32:489–497. doi: 10.1002/bdd.773. [DOI] [PubMed] [Google Scholar]
- Annaert P. Brouwers J. Bijnens A. Lammert F. Tack J. Augustijns P. Eur. J. Pharmaceut. Sci. 2010;39:15–22. doi: 10.1016/j.ejps.2009.10.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.