

Summary
Large offspring syndrome (LOS) and Beckwith-Wiedemann syndrome are similar epigenetic congenital overgrowth conditions in ruminants and humans, respectively. We have reported global loss-of-imprinting, methylome epimutations, and gene misregulation in LOS. However, less than 4% of gene misregulation can be explained with short range (<20kb) alterations in DNA methylation. Therefore, we hypothesized that methylome epimutations in LOS affect chromosome architecture which results in misregulation of genes located at distances >20kb in cis and in trans (other chromosomes). Our analyses focused on two imprinted domains that frequently reveal misregulation in these syndromes, namely KvDMR1 and IGF2R. Using bovine fetal fibroblasts, we identified CTCF binding at IGF2R imprinting control region but not KvDMR1, and allele-specific chromosome architecture of these domains in controls. In LOS, analyses identified erroneous long-range contacts and clustering tendency in the direction of expression of misregulated genes. In conclusion, altered chromosome architecture is associated with LOS.
Subject areas: Biological sciences, Genetics, molecular biology
Graphical abstract
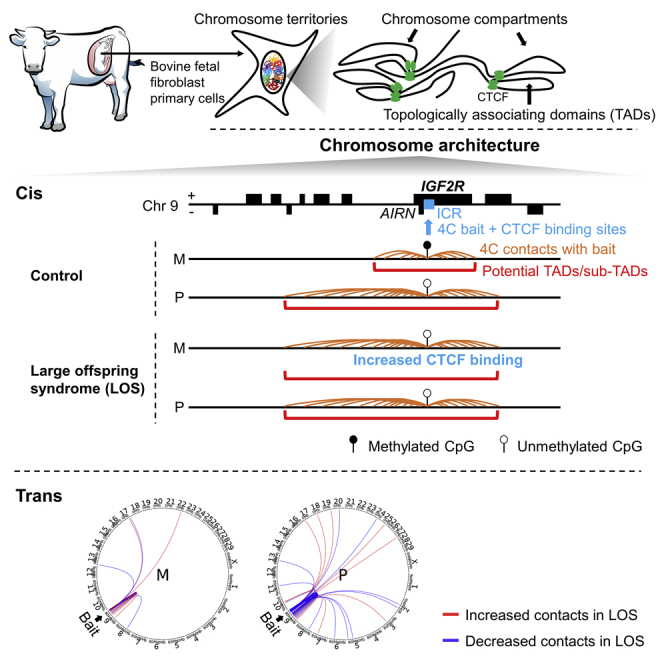
Highlights
-
•
IGF2R imprinted domain has allele-specific chromosome architecture in bovines
-
•
In bovines, CTCF binds at IGF2R imprinting control region but not at KvDMR1
-
•
Bovine large offspring syndrome (LOS) shows altered chromosome architecture at IGF2R
-
•
Misregulated genes in LOS exhibit genomic location-based clustering tendency
Biological sciences; Genetics; Molecular biology
Introduction
Large/abnormal offspring syndrome (LOS/AOS) is a naturally occurring congenital overgrowth syndrome in ruminants and its incidence increases with the use of assisted reproductive technologies (ART) (Rivera et al., 2021). Frequently observed abnormal phenotypes in LOS include macrosomia, omphalocele, and abnormal organ development (Rivera et al., 2021). Gestational problems associated with the dam include dystocia and in some cases fetal and/or maternal death (Rivera et al., 2021). In humans, a similar congenital overgrowth syndrome, Beckwith-Wiedemann syndrome (BWS, OMIM #130650), shares phenotypes and molecular aberrations with LOS (Brioude et al., 2018; Li et al., 2019a).
Previous studies from our laboratory have reported global alterations of DNA methylation and global loss-of-imprinting, including at the KCNQ1 (KvDMR1) and IGF2R imprinted domains in LOS fetuses (Chen et al., 2013, 2015, 2017), signatures also observed in BWS (Rossignol et al., 2006). Genomic imprinting is an epigenetic phenomenon which controls parental-allele-specific expression of a subset of genes involved in fetal development in mammals by regulating allele-specific DNA methylation status at their regulatory regions (i.e., imprinting control region; ICR) (Verona et al., 2003). We also reported global misregulation of protein-coding genes and non-coding RNAs in these LOS fetuses (Chen et al., 2017). In addition, we found global misregulation of microRNA genes in several tissues of LOS fetuses and human tongue of BWS patients (Li et al., 2019a). However, a limited number of misregulated genes (<4%) located within 20 kb (short range) of regions with aberrant DNA methylation showed associations in LOS (Chen et al., 2017; Li et al., 2019a).
It is well established that DNA methylation can affect gene expression at remote regulatory regions (i.e., kilobases/megabases away and also interchromosomal) by regulating chromosomal architecture (Ong and Corces, 2014). Chromosomal architecture defines spatial organization of the genome during interphase and includes topologically associating domains (TADs), chromosome compartments, and chromosome territories (Dixon et al., 2012). TADs are self-interacting genomic regions within a chromosome that range from kilobases to megabases in length. The formation and maintenance of TADs primarily relies on architectural proteins such as CCCTC-binding factor (CTCF) and cohesin protein complex (Nora et al., 2017; Rao et al., 2017), although CTCF-free TAD boundaries exist in mice and humans (Kagey et al., 2010). Binding of CTCF can be inhibited by DNA methylation (Rodriguez et al., 2010). TADs can either facilitate spatial interactions between chromosomal loci by reducing distance through looping or block spatial interactions by physical insulation of the TADs from the rest of the genome (Doyle et al., 2014; Lupiáñez et al., 2015). TADs also serve as regulators of gene expression, including imprinted genes, by orchestrating physical interactions of regulatory regions with gene promoters (Llères et al., 2019), and their disruption can lead to aberrant expression of genes and cause severe diseases or malformations (Lupiáñez et al., 2015).
In this study, we hypothesized that IGF2R ICR and KvDMR1 have allele-specific chromosome architecture in control bovine fetuses and disrupted chromosome architecture in LOS fetuses. We used fibroblast cells derived from skin of day 105 Bos taurus indicus x Bos taurus taurus F1 hybrid control and LOS fetuses to perform circular chromosome conformation capture (4C) sequencing and detected allele-specific and condition-specific chromosome architectures which are associated with allele-specific DNA methylation status and global gene misregulation.
Results
We investigated the potential contribution of chromosome architecture in LOS by querying two of its commonly misregulated imprinted domains, namely IGF2R ICR and KvDMR1 using control (n = 4) and LOS (n = 4) fibroblasts.
Identification of paternal genomic variants
Genomic DNA of the bull’s spermatozoa that sired all fetuses was sequenced to determine parental origin of fetal DNA. Bovine genome assembly ARS-UCD1.2 (Rosen et al., 2020) was used for all sequencing alignments. In total, ∼ 17.6 million raw short variants were identified from which ∼17.5 million were retained after filtering and used as reference for whole genome bisulfite sequencing (WGBS) data analyses.
Global determination of DNA methylation status
WGBS was conducted for control and LOS fibroblasts to determine their global DNA methylation status. Read alignment information may be found in Table S1A. Group comparison between control and LOS identified 9,634 significantly differentially methylated regions (DMRs) on chromosomes 1–29 (Table S1B). Loss-of-methylation in LOS was observed for IGF2R ICR (Chr9:96220970-96221849 and 96222010-96223969, 26.8% and 50.6% decrease, respectively) and KvDMR1 (Chr29:48907678-48909917, 24.7% decrease) (Figure 1).
Figure 1.

Differentially methylated regions identified within IGF2R ICR and KvDMR1 in LOS when compared with controls
(A–C) Data are represented as box plots with dots indicating individual samples. Y-axis shows average CpG DNA methylation level (not allelic).
Allele-specific DNA methylation of a 385 bp region (Chr29:48908122-48908506) within the identified DMR of KvDMR1 was also determined through bisulfite PCR, cloning, and Sanger sequencing with primers previously used by us (Chen et al., 2013). A SNP at Chr29:48908415 (maternal = C, paternal = G, missing in LOS #4) was used to assign parental-allele identity. Fibroblast and muscle samples from Control #2 and #4 showed >94% maternal allele methylation and 0% paternal allele methylation. For the LOS group, only LOS #3 showed <50% methylation on the maternal alleles for this region (Figure S1).
CTCF binding site prediction
In silico prediction identified 32,732 potential CTCF binding sites in the bovine genome with two localizing within the IGF2R ICR (Chr9:96223387-96223405 and 96223413-96223431) and two within KvDMR1 (Chr29:48908185-48908198 and 48908388-48908396). Comparisons between predicted CTCF binding sites and global DMRs showed that 125 CTCF sites overlap with 122 DMRs (Table S1C).
Confirmation of CTCF binding to IGF2R ICR and KvDMR1
Binding of CTCF protein to the in silico predicted CTCF binding sites was investigated by chromatin immunoprecipitation (ChIP). For this, PCR primers were designed to include the putative CTCF binding sites and a SNP for IGF2R ICR and KvDMR1. The SNP for IGF2R ICR is located at Chr9:96223677 (maternal = C, paternal = T) and for KvDMR1 is located at Chr29:48908415 (maternal = C, paternal = G). In addition, PCR primers were designed for a region of IGF2R’s intron 3 which contained no predicted CTCF binding site to set background levels, which could be caused by cohesin bound CTCF during chromatin extrusion activity (Davidson et al., 2019b; Sanborn et al., 2015). For IGF2R ICR, a PCR amplicon was visible for the CTCF ChIP, positive controls 2% input DNA and Histone H3 ChIP, but not for the negative control (rabbit IgG) in all control and LOS fibroblasts (Figure 2A). The ratio of CTCF ChIP to input was significantly higher (p < 0.00001) for IGF2R ICR (∼143.39% on average) than for IGF2R intron 3 (∼38.85% on average), indicating specific binding of CTCF at IGF2R ICR (Figure 2B). There was more CTCF bound at the IGF2R ICR in LOS when compared to controls (p < 0.01). For KvDMR1, the ratio of CTCF to input was ∼18.65% on average and was not higher than the background level identified using IGF2R intron 3, suggesting lack of CTCF binding at KvDMR1 (Figure 2B). We also determined the ratio of the parental alleles in the CTCF ChIP PCR amplicons based on the Sanger sequencing florescence intensity of the SNP, and found a higher ratio of maternal alleles in LOS than controls (Figures 2C and 2D).
Figure 2.

Validation of CTCF binding by chromatin immunoprecipitation (ChIP)
(A) PCR amplifications of ChIP products and input genomic DNA of predicted CTCF binding sites within the IGF2R ICR and KvDMR1 and within a region of IGF2R with no predicted CTCF binding site, namely intron 3. PCR amplicons were visualized on a 7% acrylamide gels. 2% input = input genomic DNA after micrococcal nuclease digestion without ChIP; Histone H3 ChIP = positive control; Rabbit IgG ChIP = negative control (for unspecific binding).
(B) Band intensity ratio between CTCF ChIP and 2% input DNA from (A) indicating increased presence of CTCF at IGF2R ICR in LOS and no binding at KvDMR1. Data are represented as mean ± SD. P-values were from t-test.
(C) Allele-specific binding of CTCF at IGF2R ICR shown by Sanger sequencing. Peaks show the intensity of florescence signal for each nucleotide. The nucleotide enclosed in a box denotes a SNP between the maternal (C, blue) and paternal (T, red) alleles.
(D) Increased maternal allele binding of CTCF in LOS samples. Maternal allele ratio of CTCF ChIP and 2% input DNA, and corresponding CTCF/2% input ratio calculated from (C). The high ratio of maternal allele in the 2% input could be caused by micrococcal nuclease digestion during the ChIP procedure, as its efficiency is known to be affected by the status of chromatin compression (Grewal and Elgin, 2002). Data are represented as mean ± SD. P-value was from t-test.
4C assay design and sequencing
To determine whether alteration of chromosome architecture occurs in LOS or not, we designed and performed 4C sequencing, a technique used to identify contacts between a target region (referred to as bait) and other regions of the genome (Krijger et al., 2020). 4C assays were designed for IGF2R ICR which included the CTCF binding sites within the bait region. It is known that in 4C sequencing the use of different restriction enzymes has impact on the results (Krijger et al., 2020; van deWerken et al., 2012). This is because the recognition sequence of each enzyme has a different prevalence in the genome, thus resulting in length differences of digested and ligated DNA fragments during sequencing library preparation. Very short DNA fragments are difficult to uniquely align back to the genome, and the sequencing quality and successfulness are decreased for long fragments (Tan et al., 2019). In this study we used two restriction enzymes for the second digestion to maximize the identification of contacts, and the assays were named IGF2R_MseI (4 base cutter) and IGF2R_BsrI (5 base cutter). We present the data generated from the MseI digestion in the main manuscript and for BsrI in the supplement.
For KvDMR1, even though no CTCF binding was detected, we were still interested in identifying potential loss-of-methylation related changes in genome architecture at this domain, this assay was named KvDMR1. For this assay, we only included one LOS sample (i.e., #3) because our bisulfite sequencing results only showed this individual as having maternal loss-of-methylation. For these assays, the previously mentioned SNPs were included in the sequencing reads to differentiate the parental alleles and only one enzyme for the second digestion was used (i.e., Tsp45I) because no other specificity was available.
In addition to the control and LOS groups, fibroblasts from the control group were treated with 0.5 uM Decitabine (5-aza-2'-deoxycytidine) for 96 h to serve as a positive control for loss-of-methylation and named as DC group. Alignment information for all assays and samples are found in Table S2A.
General TAD structure identified by 4C sequencing
We first compared the overall (not allelic) pattern of chromosomal interactions with each bait regions using a running window size of 50 NlaIII restriction fragments, because we were interested in identifying large-scale structural changes in LOS. For IGF2R, the control group of IGF2R_MseI and IGF2R_BsrI assays showed reads enriched regions of ∼ 273 kb (chr9:96033799-96306553) and ∼ 174kb (chr9:96121421-96295401) identified by software fourSig (Williams et al., 2014), respectively (Figures S2A and S2B). These interaction enriched regions around the bait are considered to be sub-TADs (Lupiáñez et al., 2015). We identified that both the LOS and DC groups have a large number of differentially interacting regions when compared with controls identified by software DESeq2 (Love et al., 2014) (Table S3A). Of note, for the DC-induced global demethylation group, we only focused on the cis results to characterize local topology as far-cis and trans results could be affected by additional demethylation events. In addition, statistical analyses for read enrichment and differences between groups were also conducted with a second software 4Cker as corroboration (Raviram et al., 2016) (Figure S3). However, because of 4Cker’s lack of consistency among different assays in identifying far-cis contacts (>± 500 kb on the same chromosome) and inability to perform trans (interchromosomal) analysis, fourSig and DESeq2 results were reported as main results and used for downstream analyses.
For cis contacts (± 500 kb flanking the bait), the IGF2R_MseI assay identified gain of contact in 14 merged windows and loss of contact in 3 merged windows in LOS when compared to controls (Figure S2A, Tables S2 and S3A) and the IGF2R_BsrI assay identified gain of contact in 33 merged windows and loss of contact in 4 merged windows in LOS when compared to controls (Figure S2B, Tables S2 and S3A). Both assays showed that the DC group mainly identified gain of cis contacts (Tables S2 and S3A). For visual comparison of sub-TADs between assays and treatments please refer to Figure S4. The read enrichment in each individual sample can be found in Figure S5. For far-cis contacts (>± 500 kb on the same chromosome), the IGF2R_MseI assay identified gain of contact in 91 merged windows and loss of contact in 64 merged windows in LOS when compared to controls (Figure S2D, Tables S2 and S3A) whereas the IGF2R_BsrI assay identified gain of contact in 81 merged windows and loss of contact in 29 merged windows in LOS when compared with controls (Figure S2E, Tables S2 and S3A). For trans contacts (interchromosomal), the IGF2R_MseI assay identified gain of contact in 19 merged windows and loss of contact in 19 merged windows in LOS when compared to controls (Figure S2F, Tables S2 and S3A) whereas the IGF2R_BsrI assay identified gain of contact in 107 merged windows and loss of contact in 9 merged windows in LOS when compared to controls (Figure S2G, Tables S2 and S3A). Of relevance, several change of contact regions identified by IGF2R_MseI and IGF2R_BsrI assays colocalized with predicted CTCF binding sites or DMRs (Tables S2 and S3A).
For the KvDMR1 assay, the control group showed a cis reads enriched region of ∼ 635 kb (chr29:48610455-49245209) (Figure S2C, Tables S2 and S3A). Statistical comparisons with LOS were not done because only one fetus was analyzed. DC group showed gain of contact in 14 merged windows in cis when compared to controls (Tables S2 and S3A).
Allele specific sub-TAD in control group
Parental allele specific analyses of 4C sequencing data were conducted based on the SNP retained in the sequencing reads, as previously mentioned. It should be noted that allele specificity only applies to the bait sequence but not the interacting regions. We first compared the paternal allele to the maternal allele in the control group for the three 4C assays. A higher percentage of 4C reads were captured by the paternal allele bait in all three datasets (Figure S6), indicating that the unmethylated paternal alleles of KvDMR1 and IGF2R ICR have higher frequency of physical interactions with other chromosomal loci. For IGF2R domain, the paternal allele had a larger sub-TAD than the maternal allele, for example the IGF2R_MseI assay identified sub-TADs of ∼149 kb (chr9:96134602-96283672) and ∼ 265 kb (chr9:96041522-96306553), for the maternal and paternal alleles, respectively (Figure 3A, Tables S3B and S4A). The paternal sub-TAD incorporates an additional four protein-coding genes, namely TCP1, PNLDC1, MRPL18, and MAS1, and two small nucleolar RNA genes, namely LOC112448166 and LOC112448168. For far-cis and trans contacts, two contacts are different between the alleles (Figures 3D and 3E). In addition, as expected, the enrichment of contacts is different between the IGF2R_BsrI and IGF2R_MseI assays, nonetheless, they identified similar pattern of contacts around the bait (Figures S7A and S4, Tables S3B and S4B).
Figure 3.

4C identified allele-specific cis and trans contacts with IGF2R ICR
Shown are data for the IGF2R_MseI assay.
(A) Comparison of cis contacts between the paternal and maternal alleles in controls. Track ‘4C CPM’ shows the mean normalized count of reads aligned to the genome indicating physical contacts with the bait. Track ‘Peaks’show regions with statistically significant contacts with the bait identified by fourSig software within a group. Track ‘Gain’ (red line) and ‘Loss’ (blue line) indicate regions with statistically significant difference in contacts with the bait regions identified by DESeq2 between alleles. Track ‘CTCF’ shows predicted CTCF binding sites on the sense (gold line) or antisense (black line) strand. The gene annotation is at the bottom of the figure. Mb = megabases. CPM = counts per million reads. M = maternal allele. P = paternal allele.
(B and C) Comparison of allele-specific cis contacts between control, LOS, and DC. Shown are the comparison of LOS and DC groups vs controls. Track ‘Gain’ (red line) and ‘Loss’ (blue line) indicate regions with statistically significant difference in contacts with the bait regions identified by DESeq2 between groups. Track ‘DMR’ shows non-allelic differentially methylated regions identified between the LOS and the control group with the red line indicating increased and blue line indicating decreased methylation levels. All other track information as in (A).
(D and E) Comparison of contacts in far-cis and trans between parental alleles in controls. (D) far-cis contacts (chromosome 9) and (E) trans contacts (interchromosomal) in controls. Circos plots showing DESeq2-identified statistically different contacts with the bait in the paternal vs the maternal allele. Red line indicates increased contacts and blue line indicates decreased contacts.
For KvDMR1, the maternal and paternal alleles in the control group showed reads enriched regions of ∼575 kb (chr29:48634040-49209049) and ∼ 640 kb (chr29:48605573-49245209), respectively (Figure 4A, Tables S3B and S4C). About equal number of increased (6 merged windows) and decreased (7 merged windows) far-cis contacts were identified on chromosome 29 for paternal alleles when compared with maternal alleles (Figure 4D). Trans contacts identified only two differences with chromosome 11 (increase in paternal) and chromosome 14 (decrease in paternal; Figure 4E). The read enrichment in each individual sample can be found in Figures S8 and S9. 4Cker results show general gain of cis contacts for all three assays (Figures S10–S12A).
Figure 4.

4C identified allele-specific cis and trans contacts with KvDMR1
(A) Comparison of cis contacts between the paternal and maternal alleles in controls. Track ‘4C CPM’ shows the mean normalized count of reads aligned to the genome indicating physical contacts with the bait. Track ‘Peaks’show regions with statistically significant contacts with the bait identified by fourSig software within a group. Track ‘Gain’ (red line) and ‘Loss’ (blue line) indicate regions with statistically significant difference in contacts with the bait regions identified by DESeq2 between alleles. Track ‘CTCF’ shows predicted CTCF binding sites on the sense (gold line) or antisense (black line) strand. The gene annotation is at the bottom of the figure. Mb = megabases. CPM = counts per million reads. M = maternal allele. P = paternal allele.
(B and C) Comparison of allele-specific cis contacts between control, LOS, and DC. Shown are the comparison of LOS and DC groups vs controls. Track ‘Gain’ (red line) and ‘Loss’ (blue line) indicate regions with statistically significant difference in contacts with the bait regions identified by DESeq2 between groups. Track ‘DMR’ shows non-allelic differentially methylated regions identified between the LOS and the control group with the red line indicating increased and blue line indicating decreased methylation levels. All other track information as in (A).
(D and E) Comparison of contacts in far-cis and trans between parental alleles in controls. (D) far-cis contacts (chromosome 29) and (E) trans contacts (interchromosomal) in controls. Circos plots showing DESeq2-identified statistically different contacts with the bait in the paternal vs the maternal allele. Red line indicates increased contacts and blue line indicates decreased contacts.
Altered allele specificity of sub-TAD in LOS
Next, we compared the unmethylated paternal allele between LOS and controls. For the IGF2R domain, both IGF2R_BsrI and IGF2R_MseI assays showed similar sub-TAD structure between treatments (Figures 3B, S7B, and S4, Tables S3C and S4). The two assays identified similar number of gain of far-cis contacts but IGF2R_MseI showed more loss of far-cis contacts in LOS compared to controls (Figures 5A and S7E) and IGF2R_BsrI assay captured more gain of trans contacts and less loss of trans contacts than IGF2R_MseI assay for LOS (Figures 5D and S7H). The DC group showed extended sub-TAD and more gain of contacts at surrounding regions, which could be because of the impacts of global loss-of-methylation on chromosome architecture (Figures 3B and S7B). For KvDMR1, the paternal allele behaved similarly between groups (Figure 4B, Tables S3C and S4). 4Cker results for the three assays can be found in Figures S10–S12.
Figure 5.

Distribution of altered IGF2R ICR far-cis and trans contact across various genomic contexts
Shown are data for the IGF2R_MseI assay. (A, D, G, and I) Circos plots showing DESeq2-identified statistically different contacts with the bait in LOS vs. controls. Red line indicates increased contacts and blue line indicates decreased contacts. (A and D) Paternal and (G and I) maternal allele-specific comparisons. (A and G) far-cis contacts (chromosome 9) and (D and I) trans contacts (interchromosomal). P = paternal allele. M = maternal allele. (B-C, E-F, and H) Figures show the total number of altered far-cis (B-C and H) or trans (E-F) contacts identified and the number and percent of increased (B, E, and H) and decreased (C and F) contacts over each genomic context. For example, ∼55% of increased far-cis contacts in the paternal allele overlap with repetitive sequences (n = 17) and genes (n = 17). In addition, the figures include the number and percent of altered contacts that overlap differentially methylated regions (DMR) and within 100kb of differentially expressed genes (DEG) reported in this work. Analyses were only conducted for conditions with greater than five altered contacts.
Lastly, we determined the effects of loss-of-methylation on the maternal allele in LOS. For the IGF2R domain, we observed that the sub-TAD structure of the maternal allele in LOS resembles the paternal allele of controls for both IGF2R_MseI (chr9:96087833-96303166) and IGF2R_BsrI (chr9:96121486-96295401) assays which is likely the result of loss-of-methylation, as the sub-TAD structure is the same in the DC and LOS maternal alleles (Figures 3C, S7C, and S4, Tables S3D and S4). Furthermore, this allele gained more far-cis and trans contacts in LOS for both assays (Figures 5G, 5I, S7F, and S7I). For KvDMR1, the maternal allele behaved similarly between groups (Figure 4C, Tables S3D and S4). 4Cker results for the three assays can be found in Figures S10–S12.
Next, we analyzed the genomic contexts (i.e., predicted CTCF binding sites, repetitive sequences, promoters, gene bodies, exons, introns and CpG islands, shores, and shelves) of the IGF2R ICR’s far-cis and trans contacts which are significantly different between LOS and control (Figures 5 and S14). Further, in order to determine the density of genes in the regions overlapping the contacts, we calculated the number of genes/million bases (Figures S13 and S15). The specificity of the enrichment of different genomic contexts over changed contacts was determined by a permutation test by shuffling each region ten thousand times. For this, the shuffle in cis included ± 500 kb of the bait, the far-cis shuffle included the chromosome without the baits’ cis region, and the trans shuffle was performed with all chromosomes except the chromosome containing the bait region (Figures S13 and S15). For both IGF2R_MseI and IGF2R_BsrI assays, the increased far-cis contacts in LOS show enrichment for CpG islands, shores, and shelfs, especially for shores, and depletion of repetitive sequences (Figures S13A, S13E, S15A, and S15D). This enrichment of CpG shelfs could be partially due to the uneven distribution of CpG islands on chromosome 9 because there is an enrichment around chr9:101000000-105000000. This is not the case for repetitive sequences as they do not show uneven distribution on chromosome 9.
Gene expression and its association with 4C identified interactions
Transcriptome analyses identified differences between LOS and controls (Table S5A). As expected, genes associated with extracellular matrix, including collagen, vimentin, thrombospondin, tenascin, fibronectin, and filamin were highly expressed in fibroblasts of both groups. In total, there were 548 differentially expressed genes (DEG) between controls and LOS, including IGF2R with ∼3.5 folds downregulation in LOS (Figure 6 and Table S5A), similar to the results observed in muscle by quantitative RT-PCR (∼3.3 folds downregulation in LOS). Enriched signaling pathways include lysosome, glycan degradation, and glycosaminoglycan degradation (Table S5B).
Figure 6.

Location based clustering tendency of differentially expressed genes indicates global alteration of chromosome architecture in LOS
Shown are the genomic locations of differentially methylated regions (DMRs), differentially expressed genes (DEG) and predicted CTCF binding sites that overlap LOS DMRs. In addition, gene density and log10 transformed gene expression level per million bases are shown. The vertical location indicates level of misregulation for DMR and DEG, and sense (external) or antisense (internal) strands of CTCF binding sites. Mb = megabases. Note: track three (expression level) shows that for the most part the level of expression of genes globally is similar between LOS and controls.
Genome coordinate based analyses identified clustering tendency of DEG. In total, 149 (27.2%) DEGs were found within 200 kb of another DEG, which is the average size of sub-TADs (Montefiori et al., 2018) (Figure 6 and Table S5C). Most of the clustered DEGs reveal the same direction of misregulation. Given that the distribution of genes in the genome is not even, we conducted a permutation test to confirm this clustering tendency. The test was repeated ten thousand times, and for each time 548 genes (same number as DEGs) were randomly picked from the 15,042 expressed genes in our sample and the number of clustered genes in 200kb was calculated. The identified 149 clustered DEGs is significantly higher (p = 0) than the mean of permutation tests (91.46 with standard deviation 8.49) (Figure S16).
Furthermore, DMRs were identified within the promoter region of 0.9% DEGs (n = 5). These showed an inverse correlation between DNA methylation and transcript abundance (Table S5D). When associating DEGs within a 100 kb flanking region from the gain/loss of contacts identified by 4C sequencing, eight (1.5%) DEGs were found for the IGF2R_MseI assay (RAB8B, MMP2, LOC100848985, FAM126A, EPHA5, RGS17, QKI, and RFNG) and eight (1.5%) DEGs were found for the IGF2R_BsrI assay (DCLK1, FJX1, PYCR1, LOC112442278, MIR2887-1, ENTPD1, ASH1L, and QKI; Tables S2 and S4).
Discussion
In this study, we used primary fibroblast cell lines derived from bovine control and LOS fetuses to determine imprinted domain chromosome architecture. Our results demonstrate that chromosome architecture at IGF2R imprinted domain is different between the parental alleles, a conformation disrupted in LOS. The alteration of the maternal sub-TAD in LOS was very likely associated with loss-of-methylation at the IGF2R ICR because similar changes were observed in the DC group, which served as the positive control for loss-of-methylation. We observed biallelic binding of CTCF protein to the predicted binding sites within IGF2R ICR in control and LOS fibroblasts, with increased maternal allele preference in LOS. Biallelic binding of CTCF has been reported in mouse embryos (Marcho et al., 2015).
From the 4C sequencing data, we were not able to conclude whether the CTCF binding sites in the 4C bait were involved in the formation of the observed sub-TADs at the IGF2R domain. For instance, each of these two binding sites may serve as the boundary for one of two neighboring sub-TADs. Alternatively, the boundaries of the observed sub-TAD could be defined by one of the predicted CTCF binding sites around 95.96 and 96.3 Mb, thus the binding site within the bait could be involved in formation of far-cis or trans contacts, a known function of CTCF (Ling et al., 2006). Although most of the far-cis and trans interacting regions did not colocalize with predicted CTCF binding sites, they still served as indicators for spatial closeness as the maintenance of chromosome architecture is a highly dynamic process (Nora et al., 2017).
Loss-of-imprinting is defined as biallelic silencing/expression of imprinted genes, a phenomenon correlated with loss or gain of methylation at their ICR (Verona et al., 2003). Methylation on the maternal allele of the IGF2R ICR prevents the expression of the long non-coding RNA (lncRNA) AIRN (Sleutels et al., 2002), which when expressed, silences IGF2R by attracting Polycomb repressive complexes to the locus (Schertzer et al., 2019). Consistent with this, we observed a ∼3.5 fold decrease of IGF2R transcript in the LOS group which is associated with hypomethylation on the maternal allele’s ICR. The extended sub-TAD on the maternal allele in LOS harbors six non-imprinted genes. It is possible that their regulatory regions (i.e. enhancer/silencers), alter the expression of IGF2R, as we previously showed biallelic AIRN in the muscle of day 105 bovine fetuses (Chen et al., 2017). Future studies will determine the validity of this hypothesis.
Initially we only expected to see alteration of chromosome architecture on the maternal allele of IGF2R ICR in LOS because this is the allele that suffers loss-of-methylation. As expected, some gain and loss of interactions were detected in LOS. Unexpectedly, the normally unmethylated paternal allele showed a larger number of altered far-cis and trans contacts in LOS than in controls, indicating changes of the three-dimensional shape of chromosome 9 and spatial changes within the nucleus. We could not find a reference on our search of the literature on this phenomenon, but this is an observation we intend to follow up on as this can potentially extend the definition of loss-of-imprinting.
For the KvDMR1 domain, none of the imprinted genes (CDKN1C, KCNQ1, PHLDA2) were differentially expressed in LOS. Even though allelic comparison in controls identified hundreds of differentially interacting regions in cis, few allelic differences were detected between the DC and control groups, suggesting limited regulatory effects of DNA methylation at this locus. In mice, CTCF-driven chromosome architecture at KvDMR1 regulates Cdkn1c expression during myoblast differentiation, a regulation affected by DNA methylation (Battistelli et al., 2014). However, we did not detect CTCF binding at this locus perhaps indicating species-specific regulation of KvDMR1.
For the five misregulated genes reversely correlated with DMRs, four were protein-coding genes and one was a lncRNA. The downregulated LOC535280, also known as neuroblastoma suppressor of tumorigenicity 1-like (NBL1/DAN), is a tumor suppressor (Cui et al., 2016). The downregulated gene charged multivesicular body protein 6 (CHMP6) plays roles in plasma membrane receptor downregulation and recycling (Yorikawa et al., 2005). Downregulated LOC101907348 is a lncRNA of unknown function. The downregulated gene ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1) has immunosuppression functions via degradation of adenosine triphosphate (Feng et al., 2011). Lastly, paternally expressed 10 (PEG10) is an imprinted gene that was upregulated in the LOS group. PEG10 has roles in promoting cell proliferation and its upregulation has been reported in BWS, Wilms tumor, and hepatoblastoma, which are two frequent tumors in BWS (Berland et al., 2021; Cairo et al., 2008; Jiménez Martín et al., 2021).
When analyzing clustering tendency of misregulated genes, a 200 kb distance was chosen based on the average sub-TAD size found in human (∼185-208 kb (Montefiori et al., 2018)), analyses identified 149 clustered misregulated genes, many of which had the same direction of misregulation. Of these, two clusters involve detected DMRs at CTCF binding sites in LOS. This could however be an underestimation of the effect of DNA methylation on CTCF regulation because the statistical methods utilized identified a DMR only when 10 contiguous CpG sites in a running window had altered methylation.
When we queried the flanking 100 kb of the IGF2R-associated altered contacts, we found that the paternal allele has association with eight dysregulated genes whereas the maternal allele has only one association with QKI. Several of these genes have been reported to be involved in development and tumorigenesis. For example, the downregulated matrix metallopeptidase 2 (MMP2) functions in cleaving extracellular matrix components and signal molecules (Bauvois, 2012). Mutation of MMP2 in human has been reported in several syndromes showing muscle and bone malformation (Rouzier et al., 2006). The downregulated EPH receptor A5 (EPHA5) is the membrane-bound receptor for Ephrin and its downregulation has also been reported in tumorigenesis (Li et al., 2015). The upregulated gene regulator of G protein signaling 17 (RGS17) is an oncogene and shows increased expression in several tumor types (James et al., 2009; Sokolov et al., 2011).
In summary, our study characterized allele specific chromosome architecture at IGF2R and KVDMR1 imprinted domains. In addition, we determined clustering tendency of LOS misregulated genes indicating genome-wide location-based cause of misregulation. Importantly, architectural changes at IGF2R occur in both the maternal and paternal alleles in LOS. We conclude that altered chromosome architecture is associated with LOS.
Limitations of the study
First, in this study, we used skin fibroblast primary cells to characterize chromosome architecture of control and LOS fetuses. Given that chromosome architecture could be tissue/cell-type specific (Fraser et al., 2015), the patterns identified in fibroblasts may not in its entirety recapitulate what happened in other cell types. Second, we identified changes of allele-specific chromosome architecture at IGF2R imprinted domain in LOS, which was coupled with alterations in DNA methylation level, CTCF binding, and IGF2R expression. However, we could not determine whether the changed architecture in LOS was involved in genomic imprinting regulation and/or whether the altered chromosome contacts were the cause or result of DNA methylation defects. Future studies would need to be done to clarify this point using genome editing tools. Third, with 4C sequencing, we could not determine whether there was only one or multiple sub-TADs included in the read enriched regions around the bait. Ongoing Hi-C studies will address this question.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CTCF | Cell Signaling Technology | Cat#3418; RRID: AB_2086791 |
| Histone H3 | Cell Signaling Technology | Cat#4620; RRID: AB_1904005 |
| Normal Rabbit IgG | Cell Signaling Technology | Cat#2729; RRID: AB_1031062 |
| Bacterial and virus strains | ||
| DH10B Competent Cells | Thermo Scientific | EC0113 |
| Biological samples | ||
| Bovine fetal tissues | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM | Gibco | 11885084 |
| Fetal bovine serum | Atlanta Biologicals | S11150H |
| Antibiotic-antimycotic | Gibco | 15240062 |
| HEPES | Sigma-Aldrich | H4034 |
| 0.05% trypsin-EDTA | Gibco | 25300054 |
| DMSO | Sigma-Aldrich | D2650 |
| Decitabine | Sigma-Aldrich | A3656 |
| Phenol:Chloroform:Isoamyl Alcohol | Sigma-Aldrich | P3803 |
| TRIzol™ Reagent | Invitrogen | 15596026 |
| RQ1 RNase-Free DNase | Promega | M6101 |
| random hexamers | Promega | C1181 |
| formaldehyde | Electron Microscopy Sciences | 157-4 |
| T4 DNA Ligase | New England Biolabs | M0202L |
| Proteinase K | Fisher | BP1700 |
| RNase A | Roche | 10109142001 |
| NlaIII | New England Biolabs | R0125S |
| Tsp45I | New England Biolabs | R0583S |
| MseI | New England Biolabs | R0525S |
| BsrI | New England Biolabs | R0527S |
| Critical commercial assays | ||
| EZ DNA Methylation-Direct™ Kit | ZYMO RESEARCH | D5021 |
| SuperScript® IV Reverse Transcriptase | Invitrogen | 18090010 |
| GoTaq® Flexi DNA Polymerase | Promega | M8295 |
| Wizard® SV Gel and PCR Clean-Up System | Promega | A9282 |
| CloneJET PCR Cloning Kit | Thermo Scientific | K1231 |
| Qubit dsDNA HS Assay Kit | Invitrogen | Q32851 |
| QIAquick PCR Purification Kit | QIAGEN | 28104 |
| Platinum Taq DNA Polymerase High Fidelity | Invitrogen | 11304-011 |
| AxyPrep MAG PCR Clean-Up Kit | Axygen | MAG-PCR-CL-5 |
| NEBNext Library Quant Kit for Illumina | New England Biolabs | E7630S |
| SimpleChIP Enzymatic Chromatin IP Kit | Cell Signaling Technology | 9003S |
| Deposited data | ||
| RNA-seq, WGBS, 4C-seq, and DNA-seq | This study | GEO: GSE197130 |
| Experimental models: Cell lines | ||
| Bovine fetal fibroblast primary cells | This study | N/A |
| Oligonucleotides | ||
| GE_KvDMR1_F1 | Integrated DNA Technologies | 5’-AATCCGATCGCAAGGGT |
| GE_KvDMR1_R1 | Integrated DNA Technologies | 5’-GCTTCTCGGTGAGGAGAG |
| GE_IGF2R_ICR_F | Integrated DNA Technologies | 5’-GGGGGAGGGTCTTTAAGGTTG |
| GE_IGF2R_ICR_R | Integrated DNA Technologies | 5’-TGGCTTTCAGGCTCCATAGAA |
| BI_KvDMR1_F | Integrated DNA Technologies | 5’-GTGAGGAGTATGGTATTGAGG |
| BI_KvDMR1_R | Integrated DNA Technologies | 5’-CCCCTACAAACTATCCAATCAACT |
| 4C_KvDMR1_F | Integrated DNA Technologies | 5’- TACACGACGCTCTTCCGATCT/CT CAGCGCCCAGCTTAC |
| 4C_KvDMR1_R | Integrated DNA Technologies | 5’- CAGACGTGTGCTCTTCCGATCT/ TCACGACTTGGCTCTTCTC |
| 4C_IGF2R_ICR_F | Integrated DNA Technologies | 5’- TACACGACGCTCTTCCGATCT/ TTTAGGCGCGGAAGAACGAT |
| 4C_IGF2R_ICR_R | Integrated DNA Technologies | 5’-CAGACGTGTGCTCTTCCGATCT/ GTGCGCACAGCCGCCAGAA |
| GE_KvDMR1_F2 | Integrated DNA Technologies | 5’-GCACACCGCTTTCCACACC |
| GE_KvDMR1_R2 | Integrated DNA Technologies | 5’-GCACTGAGGTGACTGCGG |
| GE_IGF2R_INT3_F | Integrated DNA Technologies | 5’-CTCTGGAGGGTTTCAGCGTC |
| GE_IGF2R_INT3_R | Integrated DNA Technologies | 5’-AGGGAATACGCTTTCCCACG |
| Software and algorithms | ||
| 4Cker | Open source | https://github.com/rr1859/R.4Cker |
| BBMap | Open source | https://jgi.doe.gov/data-and-tools/bbtools/bb-tools-user-guide/bbmap-guide/ |
| bedtools | Open source | https://bedtools.readthedocs.io/en/latest/ |
| bismark | Open source | https://www.bioinformatics.babraham.ac.uk/projects/bismark/ |
| BisSNP | Open source | https://github.com/dnaase/Bis-tools |
| bowtie2 | Open source | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| bwa-mem2 | Open source | https://github.com/bwa-mem2/bwa-mem2 |
| circular | Open source | https://cran.r-project.org/web/packages/circular/index.html |
| CTCFBSDB2.0 | Open source | https://insulatordb.uthsc.edu/ |
| DAVID Bioinformatics Resources | LHRI | https://david.ncifcrf.gov/ |
| DESeq2 | Open source | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| edgeR | Open source | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| fourSig | Open source | https://sourceforge.net/projects/foursig/ |
| GATK | Open source | https://gatk.broadinstitute.org/hc/en-us |
| ggplot2 | Open source | https://cran.r-project.org/web/packages/ggplot2/index.html |
| HISAT2 | Open source | http://daehwankimlab.github.io/hisat2/ |
| HTSeq | Open source | https://htseq.readthedocs.io/en/master/ |
| hummingbird | Open source | https://www.bioconductor.org/packages/release/bioc/html/hummingbird.html |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Integrative Genomics Viewer | Open source | https://software.broadinstitute.org/software/igv/ |
| JASPAR2020 | Open source | https://bioconductor.org/packages/release/data/annotation/html/JASPAR2020.html |
| picard | Open source | https://broadinstitute.github.io/picard/ |
| Samtools | Open source | http://www.htslib.org/ |
| StringTie | Open source | https://ccb.jhu.edu/software/stringtie/ |
| Sushi | Open source | https://bioconductor.org/packages/release/bioc/html/Sushi.html |
| TFBSTools | Open source | https://bioconductor.org/packages/release/bioc/html/TFBSTools.html |
| Trimmomatic | Open source | http://www.usadellab.org/cms/?page=trimmomatic |
| Other | ||
| Custom code | This Study | Zenodo: https://doi.org/10.5281/zenodo.6449167 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Rocío Melissa Rivera (riverarm@missouri.edu).
Materials availability
This study did not generate new unique reagents. Commercially available reagents are indicated in the key resources table.
Experimental model and subject details
Animal tissues
Day 105 Bos taurus indicus (B. t. indicus; Brahman breed) x Bos taurus taurus (B. t. taurus; Angus breed) F1 hybrid fetuses were generated by our laboratory in 2019 as reported before (Chen et al., 2013; Rivera and Hansen, 2001) and used as tissue donors. This breeding strategy aimed to introduce genetic variants, including single-nucleotide polymorphisms (SNPs), to differentiate parental alleles. The control group was generated using artificial insemination (AI) and the ART group was generated by in vitro production procedures. The LOS group was defined as individuals from the ART group with body weight greater than 97th centile of controls. On day 105 of pregnancy, conceptuses were collected by caesarean section to maintain nucleic acid integrity. The identifier, original ID, sex, and body weight of fetuses used in this study were as follows; 1) control fetuses: Control#1 (original ID 533, female, 388g), Control#2 (647, female, 396g), Control#3 (640, male, 448g), and Control#4 (648, male, 466g); 2) LOS fetuses: LOS#1 (656, female, 704g), LOS#2 (602, male, 752g), LOS#3 (604B, female, 986g), and LOS#4 (664, male, 1080g).
All the animal procedures were approved by University of Missouri Animal Care and Use Committee under protocol 9455. Trained personnel and Veterinarians performed all animal handling and surgeries.
Establishment of skin fibroblast primary cell line, cell culture, and Decitabine treatment
Fibroblast cells were chosen since they originate from mesoderm and share lineage with skeletal muscle and kidney (Chan et al., 2016; Davidson et al., 2019a; Vodyanik et al., 2010). Muscle mass is a main contributor to the increased birth weight in LOS, and Wilms tumor of the kidney and rhabdomyosarcoma are tumors observed in BWS (Brioude et al., 2018; Li et al., 2019b). We reasoned that the molecular aberrations in skeletal muscle and kidney would be conserved in fibroblast if they occurred during early embryo development. We showed this to be the case for expression of IGF2R and DNA methylation at KvDMR1 in this study. Further, using fibroblast cells will allow comparison of findings with the human counterpart syndrome BWS, since skin fibroblasts is one of the frequently obtained tissue samples from these patients and is being used to characterize the syndrome (Brioude et al., 2018; Naveh et al., 2021).
Fetal skins were collected to establish fibroblast primary cell line using protocol adapted from https://animal.ifas.ufl.edu/hansen/lab_protocol_docs/bovine_fetal_fibrobasts.pdf (Dobbs et al., 2013). Briefly, approximately one square centimeter piece of skin was collected from each fetus during fetal collection and incubated in 1 mL fresh bovine embryonic fibroblast medium (BEF; 89% (v/v) DMEM (Gibco, 11885084), 10% (v/v) fetal bovine serum (FBS; Atlanta Biologicals, S11150H), and 1% (v/v) Antibiotic-antimycotic (10000 units/mL penicillin, 10000 ug/mL streptomycin, and 25 ug/mL Gibco Amphotericin B; Gibco, 15240062)) containing 25mM HEPES (Sigma-Aldrich, H4034) at 38.5°C until further processing within 12 hours. The skin pieces were washed in homemade DPBS containing 1% (v/v) antibiotic-antimycotic three times, diced into smaller pieces, and transferred into a well of a 12 well plate containing 1 mL BEF medium. The skin pieces were cultured at 38.5°C (body temperature of cattle) with 5% CO2 and 1 mL fresh BEF medium was added every two days. When outgrowing fibroblast cells reached confluency, the skin pieces were removed, and cells were trypsinized with 0.05% trypsin-EDTA (Gibco, 25300054) and transferred to T75 flasks (MIDSCI, TP90076) with 12 mL BEF medium. The medium was changed every two days until cells reached 80–90% confluency and were cryopreserved to keep them at low passage number (i.e., n < 10). For cryopreservation, cells were trypsinized, counted with a hemacytometer, centrifuged at 250 × g, resuspended in BEF medium containing 10% (v/v) DMSO (Sigma-Aldrich, D2650) at a concentration of ∼1-2 million cells/mL, and transferred to a cryotube (MIDSCI, CM-4). The cryotubes were kept at −80°C overnight and then transferred into liquid nitrogen for long term storage. When recovering from cryopreservation, the cells were thawed at 38.5°C, centrifuged and washed with BEF medium to remove remaining DMSO, and cultured as described above.
To induce loss of DNA methylation, a subgroup of fibroblasts derived from control fetuses were treated with 0.5 uM Decitabine (5-aza-2’-deoxycytidine) for 96 hours. BEF medium with 0.5 uM Decitabine were changed every 24 hours to maintain the concentration of Decitabine. The identifier of Decitabine treated control samples are DC#1 to DC#4 corresponding to Control#1 to Control#4.
Method details
All the chromosomal coordinates in this manuscript refer to bovine genome assembly ARS-UCD1.2 unless otherwise specified (Rosen et al., 2020).
Genomic DNA extraction
Fibroblasts, semen of the sire of the fetuses (JDH MR. MANSO 7 860958 154BR599 11200 EBS/INC CSS 2), or fetal tissue samples were lysed in lysis buffer (0.05 M Tris-HCl (pH 8.0), 0.1 M EDTA, and 0.5% (w/v) SDS) with proteinase K (Fisher BioReagents, BP1700) at 55°C for four hours (cells and semen) or overnight (tissue). Genomic DNA was extracted with Phenol:Chloroform:Isoamyl Alcohol (SIGMA, P3803) following the manufacturer’s instructions. The concentration of DNA was measured by using a NanoDrop® ND-1000 Spectrophotometer (Thermo Fisher Scientific) and DNA integrity was confirmed by electrophoresis on a 0.7% agarose gel. Genomic DNA samples were stored at −20°C.
Bisulfite conversion of genomic DNA
Genomic DNA was bisulfite converted with EZ DNA Methylation-Direct™ Kit (ZYMO RESEARCH, D5021) following the manufacturer’s instructions. Bisulfite converted DNA samples were stored at −80°C.
RNA isolation
Total RNA from cultured fibroblast cells and tissue samples was isolated using TRIzol™ Reagent (Invitrogen, 15596026) following the manufacturer’s instructions. The concentration of RNA samples was measured by using the NanoDrop® ND-1000 Spectrophotometer. RNA samples were stored at −80°C.
Reverse transcription of mRNAs
Total RNA samples were treated with RQ1 Rnase-Free Dnase (Promega, M6101) following the manufacturer’s instructions to remove genomic DNA contamination. cDNA was synthesized using SuperScript® IV Reverse Transcriptase (Invitrogen, 18090010) with random hexamers (Promega, C1181) following the manufacturer’s instructions. cDNA samples were stored at −20°C.
Polymerase chain reaction (PCR), molecular cloning, and sanger sequencing
GoTaq® Flexi DNA Polymerase (Promega, M8295) was used for end-point PCR following the manufacturer’s instructions. Genomic primer GE_KvDMR1_F1 (5’-AATCCGATCGCAAGGGT, Chr29: 48907972-48907988) and GE_KvDMR1_R1 (5’-GCTTCTCGGTGAGGAGAG, Chr29: 48908541-48908558) were used to amplify KvDMR1 region to identify SNP, and the thermocycler conditions were: 95°C 2 min; 95°C 30s, touchdown from 71.8°C to 61.8°C by 1°C per cycle 30s, 72°C 38s, 40 cycles; 72°C 5min. Genomic primer GE_IGF2R_ICR_F (5’-GGGGGAGGGTCTTTAAGGTTG, Chr9: 96223334-96223354) and GE_IGF2R_ICR_R (5’-TGGCTTTCAGGCTCCATAGAA, Chr9: 96223732-96223752) were used to amplify IGF2R ICR to identify SNP, and the thermocycler conditions were: 95°C 2min; 95°C 30s, 64°C 30s, 72°C 30s, 35 cycles; 72°C 5min.
Bisulfite primer BI_KvDMR1_F (5’-GTGAGGAGTATGGTATTGAGG, Chr29: 48908486-48908506) and BI_KvDMR1_R (5’-CCCCTACAAACTATCCAATCAACT, Chr29: 48908205-48908229) were used to amplify KvDMR1 region to determine DNA methylation status, and the thermocycler conditions were: 95°C 2 min; 95°C 30s, 58.7°C 30s, 72°C 30s, 35 cycles; 72°C 5min.
The PCR products were resolved on a 2% (w/v) agarose gel and visualized using ethidium bromide. Bands of expected sizes were cut and DNA was retrieved from the gel using Wizard® SV Gel and PCR Clean-Up System (Promega, A9282). Sanger sequencing for the retrieved DNA was performed at the University of Missouri Genomics Technology Core.
Molecular cloning was performed using CloneJET PCR Cloning Kit (Thermo Scientific, K1231) and DH10B Competent Cells (Thermo Scientific, EC0113) to determine allelic DNA methylation level or allelic CTCF ChIP enrichment following the manufacturer’s instructions.
Quantitative Reverse-transcriptase PCR for IGF2R
Quantitative RT-PCR of IGF2R was done using TaqMan® probes (ThermoFisher Scientific, Hanover Park, IL) and a QuantStudio 3 Real-Time PCR System (Applied BioSystems, Waltham, Massachusetts). The mRNA level of each target transcript was normalized to the geometric mean of three endogenous normalizers, namely NUCKS1, RBM39, SF3B1. Amplifications were performed in at least duplicates. Each group’s cycle threshold difference and 2-Delta Delta Ct was calculated to determine the fold difference in transcript levels.
CTCF binding site prediction in bovine genome
Potential CTCF binding sites were predicted globally for bovine genome assembly ARS-UCD1.2 (Rosen et al., 2020) using TFBSTools 1.26.0 (Tan and Lenhard, 2016) with database JASPAR2020 (Fornes et al., 2020). CTCF motifs of ‘vertebrates’ were used for prediction and min.score was set to 90%. CTCFBSDB2.0 (Ziebarth et al., 2012) was also used for CTCF binding sites prediction in local regions.
Circular chromosome conformation capture (4C) library preparation and sequencing
The 4C library preparation procedure mainly followed the Krijger protocol (Krijger et al., 2020) with some adaptations from other published protocols (Gheldof et al., 2012; Splinter et al., 2012; van deWerken et al., 2012). Briefly, for each sample, six million fibroblast cells were fixed with 2% (v/v) formaldehyde (Electron Microscopy Sciences, 157-4) at a concentration of two million cells per mL. Fixed cells were lysed, washed, and underwent first restriction enzyme (RE) digestion overnight as described in Krijger protocol (Krijger et al., 2020). Note: the specifics of the REs are explained below. On day two, after confirming good digestion efficiency by 0.7% agarose gel electrophoresis (downwards shift of the DNA smear), the first RE was inactivated according to manufacturer’s instructions and samples were diluted and ligated with T4 DNA Ligase (New England Biolabs, M0202L) overnight. On day three, after confirming good ligation efficiency by 0.7% agarose gel electrophoresis (upwards shift of the DNA smear), the samples were treated with Proteinase K (Fisher, BP1700) overnight to reverse the formaldehyde cross-links between protein and DNA. On day four, samples were treated with Rnase A (Roche, 10109142001) and ligated DNA was extracted using Phenol-Chloroform. Next, ligated DNA samples were digested with the second RE overnight. On day five, after confirming good digestion efficiency by electrophoresis, the second RE was inactivated by heating or removed by Phenol-Chloroform extraction if heat insensitive. DNA concentration was measured by Qubit dsDNA HS Assay Kit (Invitrogen, Q32851) and these DNA samples were ligated with T4 DNA Ligase overnight at a concentration of 5 ng/ul. On day six, after confirming good ligation efficiency by electrophoresis, DNA was ethanol precipitated and purified with QIAquick PCR Purification Kit (QIAGEN, 28104). Concentration of the purified DNA, in other words 4C template, was measured by Qubit assay. The 4C templates were stored at −20°C.
Primers for the two PCR steps were designed as described in Krijger protocol (Krijger et al., 2020). Platinum Taq DNA Polymerase High Fidelity (Invitrogen, 11304-011) was used for 4C PCR following the manufacturer’s instructions. Following each PCR step, the products were purified with AxyPrep MAG PCR Clean-Up Kit (Axygen, MAG-PCR-CL-5) to remove remaining primers, primer dimers, and self-ligation bands. 4C libraries were stored at −20°C. Concentration of the 4C libraries were measured by Qubit assay and the integrity of 4C libraries were confirmed by NEBNext Library Quant Kit for Illumina (New England Biolabs, E7630S). Average DNA fragment size of 4C libraries were measured by fragment analyses using Fragment Analyzer Systems (Agilent) at University of Missouri Genomics Technology Core. Molar concentration of 4C libraries were calculated based on Qubit concentration and average DNA fragment size, and equal molar amount of 4C libraries were pooled. A 15% spike-in of PhiX Control v3 (Illumina, FC-110-3001) was included in the final library to increase base diversity and improve color balance (proportions and distribution of dyes used to report different nucleotides) during sequencing for the bait (4C target region in the genome) sequence. The final library was sequenced on the NovaSeq platform for 250bp paired-end reads at University of Missouri Genomics Technology Core.
KvDMR1 and IGF2R ICR were selected for 4C sequencing. Fibroblast cells of Control#1 to Control#4 and LOS#3, and Decitabine treated fibroblasts DC#1 to DC#4 were used for KvDMR1. For IGF2R ICR, all the samples Control#1 to #4, LOS#1 to #4, and DC#1 to #4 were used.
In total, three 4C assays were designed, one for KvDMR1 and two for IGF2R ICR. For each region, the 4C bait contains predicted CTCF binding sites and a SNP. In order to include the SNP in the sequencing reads, one of the two restriction enzyme (RE) digestion sites has to be adjacent to the SNP which limited the choice of RE. For KvDMR1, NlaIII (New England Biolabs, R0125S) and Tsp45I (New England Biolabs, R0583S) were selected for the first and second RE digestion, respectively. For IGF2R ICR, two different RE were used as the second RE, which resulted in two 4C assays, namely IGF2R_MseI and IGF2R_BsrI. NlaIII (first RE) and MseI (New England Biolabs, R0525S) were used for IGF2R_MseI, and NlaIII (first RE) and BsrI (New England Biolabs, R0527S) were used for IGF2R_BsrI. For the first round of PCR, primer 4C_KvDMR1_F (5’- TACACGACGCTCTTCCGATCT/CTCAGCGCCCAGCTTAC, Chr29: 48907934-48907950, ‘/’ indicates the split site where sequences on the left side are complementary to Illumina_i5 or i7 primers, and sequences on the right side are complementary to the genome) and 4C_KvDMR1_R (5’- CAGACGTGTGCTCTTCCGATCT/TCACGACTTGGCTCTTCTC, Chr29: 48908379-48908397) were used for KvDMR1, and the thermal conditions were: 94°C 2min; 94°C 15s, 65.3°C 1min, 68°C 3min, 16 cycles; 68°C 5min. Primer 4C_IGF2R_ICR_F (5’- TACACGACGCTCTTCCGATCT/TTTAGGCGCGGAAGAACGAT, Chr9: 96223648-96223667) and 4C_IGF2R_ICR_R (5’-CAGACGTGTGCTCTTCCGATCT/GTGCGCACAGCCGCCAGAA, Chr9: 96223397-96223415) were used for IGF2R_MseI and IGF2R_BsrI, and the thermal conditions were: 94°C 2min; 94°C 15s, 62.1°C 1min, 68°C 3min, 16 cycles; 68°C 5min. For the second round of PCR, 17 pairs of Illumina_i5 (5’-AATGATACGGCGACCACCGAGATCTACAC-index-ACACTCTTTCCCTACACGACGCTCTTCCGATCT) and Illumina_i7 (5’- CAAGCAGAAGACGGCATACGAGAT-index-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT) primers were designed with different index sequences (UDI0001-UDI0017 in the official manual ‘Illumina Adapter Sequences v16’) and assigned to different samples. For IGF2R_MseI and IGF2R_BsrI assays, UDI0001-12 were used for control #2, #4, #1, #3, LOS #2, #1, #4, #3, DC #2, #4, #1, and #3, respectively. For KvDMR1 assay, UDI0001, 4, 7, 10, 13-17 were used for control #2, #4, LOS #3, DC #2, control #1, #3, DC #1, #3, and #4, respectively. For some indexes, the three samples from different 4C assays that shared the index can be separated based on bait sequence in the reads, which served as a secondary barcode. The thermal conditions for Illumina_i5 and Illumina_i7 were: 94°C 2min; 94°C 15s, 65.5°C 1min, 68°C 3min, 20 cycles; 68°C 5min. These designs resulted in three 4C datasets after sequencing.
4C data analyses
Raw sequencing reads were first sorted into the three 4C datasets (KvDMR1, IGF2R_MseI, and IGF2R_BsrI) based on the bait sequences. This was accomplished by aligning the bait part of the reads to the bait sequences using bowtie2 (Langmead and Salzberg, 2012) and filtering for unique alignments. Next, the parental alleles were assigned to each read pairs based on the SNP in the bait sequences. Then the bait sequences were removed from the reads and reads were trimmed for adapter sequences and low quality bases using trimmomatic 0.39 (Bolger et al., 2014) with parameters ‘ILLUMINACLIP:adapter_seq:2:30:10:1:true LEADING:20 TRAILING:20 AVGQUAL:20 MAXINFO:0:0.5’. The trimmed reads were split into multiple fragments by the recognition sites of corresponding first and second RE used. The first fragment on the 5’ side of each read was kept and read pairs were combined and aligned to genome as single-end reads using bowtie2 with parameters ‘-N 1-L 15 –no-unal’. Aligned reads with mapping quality less than 20 were filtered out using Samtools 1.13 (Li et al., 2009).
The genome was fragmented by NlaIII recognition site from hereunto referred as NlaIII restriction fragments. Read coverage was calculated for NlaIII restriction fragments using bedtools 2.30.0 with parameters ‘coverage-sorted-counts’ (Quinlan and Hall, 2010). Self-ligation reads aligned to the NlaIII restriction fragments covering the bait region and one up/downstream fragment were excluded for statistical analyses. 4C peaks were called using fourSig with parameters ‘cis.only = FALSE, window.size = 50, iterations = 1000, fdr = 0.01, fdr.prob = 0.05, only.mappable = FALSE’ (Williams et al., 2014). fourSig reported 4C peaks for individual samples were converted to NlaIII restriction fragments, and the restriction fragments present in at least two samples in a group were reported as the 4C peaks for the group.
Statistical comparison between groups to identify change of 4C interactions were conducted using a running window approach which has been widely used for 4C analyses (Simonis et al., 2006; van deWerken et al., 2012). The sum read coverage of a running window of 50 NlaIII restriction fragments were calculated and used as input for statistical comparison by DESeq2 (Love et al., 2014). A prefilter for low coverage window using ‘rowSums(counts>2) ≥ 2’ were conducted to make the input list manageable. Default settings of DESeq2 were used for other steps. False discovery rate (FDR) was controlled at 0.05 for DESeq2 results (labelled as Padj) by the Benjamini-Hochberg method. The coordinates of the 13th restriction fragment in each running window were used to indicate that window. Overlapped and continuous significant window with the same direction of changes were merged. In addition, statistical analyses for read enrichment and differences between groups were also conducted with a second software 4Cker as corroboration (Raviram et al., 2016). Parameters used for 4Cker functions are k = 5 for ‘nearBaitAnalysis’, k = 10 for ‘cisAnalysis’, and pval = 0.05 for ‘differentialAnalysis’ as recommended by the manual. Trans analyses were not performed with 4Cker according to the software manual recommendation and also due to very low efficiency in processing multiple samples as a group.
Genomic content related analyses and permutation test
Information of gene annotation was obtained from NCBI (GCF_002263795.1_ARS-UCD1.2_genomic.gff) (O’Leary et al., 2016). Per million base gene density was calculated based on the annotation. Repeated and overlapped exons were merged for each gene, and introns were calculated based on merged exons. Promoters (1kb) were calculated based on transcription start sites annotation and only included protein coding genes and long non-coding RNAs. Annotation of CpG islands and repeated sequences were obtained from UCSC Genome Browser (Kent et al., 2002). Locations of CpG shores (flanking 2kb from CpG islands) and shelves (flanking 2-4kb from the CpG island) were calculated based on CpG island annotation. Bedtools and custom Perl scripts were used for permutation test and identify overlapped genomic location and make tables (Quinlan and Hall, 2010). R package Sushi, circular, and ggplot2 were used for making figures (Lund et al., 2017; Phanstiel et al., 2014; Wickham, 2011). Integrative Genomics Viewer was also used for visualization (Robinson et al., 2011).
Bull semen genomic sequencing and data analyses
Genomic sequencing for semen DNA of the bull used to sire all the fetuses in this study was conducted by University of Missouri Genomics Technology Core. Information on library preparation and sequencing obtained from the Core is as follows: The library was constructed following the manufacturer’s protocol with reagents supplied in Illumina’s TruSeq DNA PCR-Free sample preparation kit (#FC-121-3001). Briefly, DNA was sheared using standard Covaris methods to generate average fragmented sizes of 350 bp. The resulting 3’ and 5’ overhangs were converted to blunt ends by an end repair reaction which uses a 3’ to 5’ exonuclease activity and polymerase activity. A single adenosine nucleotide was added to the 3’ ends of the blunt fragment followed by the ligation of Illumina indexed paired-end adapters. The adaptor ligated library was purified twice with AxyPrep Mag purification beads. The purified library was quantified using KAPA library quantification kit (KK4824) and library fragment size confirmed by Fragment Analyzer (Agilent Technologies, Inc.). Libraries were diluted and sequenced according to Illumina’s standard sequencing protocol for the NovaSeq 6000.
For genomic sequencing data analyses, we followed the pipeline for 1000 bull genome project (Hayes and Daetwyler, 2019). Briefly, raw sequencing reads were trimmed for adapter sequences and low quality bases using trimmomatic 0.39 (Bolger et al., 2014) with parameters ‘ILLUMINACLIP:adapter_seq:2:30:10:1:true LEADING:20 TRAILING:20 AVGQUAL:20 SLIDINGWINDOW:3:15’. Trimmed reads were aligned to the bovine genome using bwa-mem2 (Vasimuddin et al., 2019) with default parameters. Samtools 1.13 (Li et al., 2009) was used to convert, sort, filter, and index bam files. Aligned reads with mapping quality (MAPQ) less than 20 were excluded from downstream analyses. Read groups were added using AddOrReplaceReadGroups function of picard 2.25.5 (Broad Institute, 2018). The dataset of known variants in bovine was acquired from the 1,000 bull genome project, namely ARS1.2PlusY_BQSR_v3.vcf.gz. GATK 4.2.1.0 (Van der Auwera and O’Connor, 2020) was used to recalibrate base quality and identify variants in the genomic sequencing data with the known variant dataset as reference. Parameters used for BaseRecalibrator and HaplotypeCaller were ‘–bqsr-baq-gap-open-penalty 45’ and ‘–pcr-indel-model NONE’, respectively. Raw variants were scored using 2D model of CNNScoreVariants function of GATK with parameter ‘-tensor-type read_tensor’. Scored variants were filtered using FilterVariantTranches function of GATK with parameter ‘–info-key CNN_2D –invalidate-previous-filters –snp-tranche 99.95 –indel-tranche 99.4’.
Whole genome bisulfite sequencing (WGBS) and data analyses
WGBS for cultured fibroblast cells was conducted by CD Genomics. Information on library preparation and sequencing obtained from the company is as follows: For WGBS library preparation, 1 ug of genomic DNA was fragmented by sonication to a mean size of approximately 200–400 bp. Fragmented DNA was end-repaired, 5′-phosphorylated, 3′-dA-tailed and then ligated to methylated adapters. The methylated adapter-ligated DNAs were purified using 0.8× Agencourt AMPure XP magnetic beads and subjected to bisulfite conversion by ZYMO EZ DNA Methylation-Gold Kit (zymo). The converted DNAs were then amplified using 25 μL KAPA HiFi HotStart Uracil + ReadyMix (2X) and 8-bp index primers with a final concentration of 1 μM each. The constructed WGBS libraries were then analyzed by Agilent 2100 Bioanalyzer and quantified by a Qubit fluorometer with Quant-iT dsDNA HS Assay Kit (Invitrogen), and finally sequenced on Illumina Hiseq X ten sequencer. 0.1–1% lambda DNA were added during the library preparation to monitor bisulfite conversion rate.
For WGBS data analyses, duplicated reads generated during PCR and sequencing were removed from raw sequencing reads using the clumpify function of BBMap 38.90 (Bushnell, 2021). The remaining raw reads were trimmed for adapter sequences and low quality bases using trimmomatic 0.39 (Bolger et al., 2014) with parameters ‘ILLUMINACLIP:adapter_seq:2:30:10:1:true LEADING:20 TRAILING:20 AVGQUAL:20 MAXINFO:0:0.5’. Trimmed reads were aligned to the bovine genome using bismark 0.23.0 (Krueger and Andrews, 2011) with parameters ‘-X 900 –unmapped –ambiguous –non_bs_mm’. Trimmed reads were also aligned to lambda phage genome to determine bisulfite conversion rates. Samtools 1.13 (Li et al., 2009) was used to convert, sort, filter, and index bam files. MarkDuplicates function of picard 2.25.5 (Broad Institute, 2018) was used to further remove duplicated reads after alignment. Read groups were added for each samples using AddOrReplaceReadGroups function of picard. Variants identified in bull semen genomic sequencing data and the previously mentioned variants acquired from the 1,000 bull genome project served as known variants to identify genomic variants in WGBS data. Indel realignment was performed using RealignerTargetCreator and IndelRealigner functions of BisSNP 1.0.1 (Liu et al., 2012). Base quality recalibration was carried out using BisulfiteCountCovariates and BisulfiteTableRecalibration functions of BisSNP 0.82.2 since these functions are missing in version 1.0.0 and 1.0.1. Parameters used for BisulfiteCountCovariates were ‘-cov ReadGroupCovariate-cov QualityScoreCovariate-cov CycleCovariate-baqGOP 30’. Genomic variants were identified using BisSNP 1.0.1 with default setting expect that ‘-bsRate’ was changed to bisulfite conversion rate observed from lambda phage genome alignment for each sample. BisSNP identified variants were filtered by its VCFpostprocess function with parameter ‘-windSizeForSNPfilter 0’. Additionally, genomic variants were identified using BS-SNPer 1.0 (Gao et al., 2015) with parameters ‘-minhetfreq 0.1 –minhomfreq 0.85 –minquali 15 –mincover 5 –maxcover 1,000 –minread2 2 –errorate 0.02 –mapvalue 20’. M-bias plots were generated using bismark and the first 3 bases of R1 reads and the first 4 bases of R2 reads showed biased CpG methylation level, thus these bases were excluded from downstream analyses. CpG methylation information were extracted from the bam files using bismark_methylation_extractor function of bismark with parameters ‘-p –ignore 3 –ignore_r2 4 –comprehensive –no_header –gzip –bedGraph –buffer_size 50%--cytosine_report’. Statistical analyses were conducted using R package hummingbird (Ji, 2019) with parameter ‘minCpGs = 10, minLength = 100, maxGap = 300’ to identify differentially methylated regions (DMRs) between LOS and Control groups. DMRs with at least 15% difference in methylation level (both gain and loss of methylation) and at least 2 mean read coverage at CpG sites were reported. The sex chromosomes were not analyzed to circumvent confounding created by X chromosome inactivation associated DNA methylation.
Chromatin immunoprecipitation (ChIP) for CTCF protein
ChIP for CTCF protein was conducted in fibroblasts derived from control and LOS fetuses to verify in silico predicted CTCF binding site within the region used as bait for the 4C assay. SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology, 9003S) and CTCF (D31H2) XP Rabbit mAb (Cell Signaling Technology, 3418S) were used for this experiment following the manufacturer’s instructions. Briefly, three ChIP assays with different antibodies (CTCF, Histone H3 [positive control (Cell Signaling Technology, 4620S)], and Normal Rabbit IgG [negative control (Cell Signaling Technology, 2729S)] were conducted for each sample. For each sample, 12 million fibroblast cells, equivalent to 4 million per ChIP assay, were fixed with 1% (v/v) formaldehyde (Electron Microscopy Sciences, 157-4) at a concentration of 0.5 million cells per mL. Fixed cells were washed, lysed, digested with 0.5 ul of micrococcal nuclease, and sonicated to break nuclear membrane as described in the manual. Once good digestion efficiency (about 150–900 bp DNA fragments, equivalent to 1-5 nucleosomes) was confirmed by 0.7% agarose gel electrophoresis, ChIP buffer and antibodies of recommended amount (1:50 dilution for CTCF and Histone H3 and 1.5 ug for Rabbit IgG) were added into each sample and incubated overnight at 4°C. On day two, ChIP-Grade Protein G Magnetic Beads were used to pull down antibodies and bound protein and DNA. After washing, elution, and reversing crosslink, the DNA was eventually purified with spin columns.
To confirm in silico predicted CTCF binding sites, genomic primers were designed to amplify a short region covering the binding sites. The size of amplicon used in this study was longer than recommended by the manual since we included the SNPs in the amplicon to differentiate parental alleles. For IGF2R ICR, primer GE_IGF2R_ICR_F and GE_IGF2R_ICR_R were used and the thermal conditions were: 95°C 2min; 95°C 30s, 64°C 30s (0.3°C/s ramp temperature), 72°C 30s, 35 cycles; 72°C 5min. As allelic bias during PCR is possible, we also report results using 1M betaine (to relax secondary structures; Sigma B2629). For KvDMR1, primer GE_KvDMR1_F2 (5’-GCACACCGCTTTCCACACC, Chr29: 48908151-48908169) and GE_KvDMR1_R2 (5’-GCACTGAGGTGACTGCGG, Chr29: 48908477-48908494) were used and the thermal conditions were: 95°C 2min; 95°C 30s, 67.3°C 30s, 72°C 30s, 35 cycles; 72°C 5min. In addition, primer GE_IGF2R_INT3_F (5’-CTCTGGAGGGTTTCAGCGTC, Chr9: 96229536-96229555) and GE_IGF2R_INT3_R (5’-AGGGAATACGCTTTCCCACG, Chr9: 96229935-96229954) were used to amplify a region of IGF2R’s intron 3 which contained no predicted CTCF binding site to set background levels for traveling CTCF, and the thermal conditions were: 95°C 2min; 95°C 30s, 64°C 30s, 72°C 30s, 35 cycles; 72°C 5min. PCR amplicons were visualized on 7% acrylamide gel and the intensity of bands were measured using ImageJ (Schneider et al., 2012). T-test was conducted using online T-Test Calculator (https://www.socscistatistics.com/tests/studentttest/default2.aspx).
RNA sequencing (RNA-seq) and data analyses
RNA-seq for cultured fibroblast cells was conducted by BGI. Information on library preparation and sequencing obtained from the company is as follows: mRNA molecules were purified from total RNA using oligo (dT)-attached magnetic beads. mRNA molecules were fragmented into small pieces using fragmentation reagent after reaction a certain period in proper temperature. First strand cDNA was generated using random hexamer primed reverse transcription, followed by a second strand cDNA synthesis. The synthesized cDNA was subjected to end repair and then was 3’ adenylated. Adapters were ligated to the ends of these 3’ adenylated cDNA fragments. PCR was used to amplify the cDNA fragments with adapters from previous step. PCR products were purified with Ampure XP Beads (AGENCOURT) and dissolved in EB solution. Library was validated on the Agilent Technologies 2100 bioanalyzer. The double stranded PCR products were heat denatured and circularized by the splint oligo sequence. The single strand circle DNA (ssCir DNA) were formatted as the final library. The library was amplified with phi29 to make DNA nanoball (DNB) which had more than 300 copies of one molecular. The DNBs were load into the patterned nanoarray and pair end 100 bases reads were generated in the way of combinatorial Probe Anchor Synthesis (cPAS).
Reads were aligned to the Bos taurus reference genome, ARS-UCD1.2, using HISAT2 with the –dta flag to allow for downstream transcript assembly (Pertea et al., 2016). Reads aligned to the genome were assembled into transcripts using StringTie and all transcripts merged (Pertea et al., 2016). Transcript abundance was estimated using HTSeq with the following flags –order = pos,--idattr = gene, and –stranded = no (Anders et al., 2015). To note that the gene symbols were in accordance with “gene_id” column instead of “gene” column in GCF_002263795.1_ARS-UCD1.2_genomic.gtf file (from NCBI) since the former differentiates repeated genes from different genomic location by adding a “_X” tag. Statistical comparison between Control and LOS groups were conducted using DESeq2. A prefilter for low abundance genes were conducted using ‘rowSums(cpm(counts) > 0.2) ≥ 3’which resulted in 15,042 identified as expressed in this study (Robinson et al., 2010). Default settings of DESeq2 were used for other steps. FDR was controlled at 0.05. DE genes enriched signaling pathways were identified using DAVID Bioinformatics Resources 6.8 (Huang et al., 2009). Per million base gene expression level for control and LOS groups were calculated as the sum of the group mean CPM of all genes detected.
Quantification and statistical analysis
A two-tailed t-test was used for ChIP related comparisons and p less than 0.05 was considered as significant. fourSig was used to identify significant 4C contacts with parameters ‘cis.only = FALSE, window.size = 50, iterations = 1,000, fdr = 0.01, fdr.prob = 0.05, only.mappable = FALSE’ (Williams et al., 2014). DESeq2, which performs a Wald test, was used to detect significant differences in 4C contacts and gene expression with false discovery rate controlled at 0.05 using the Benjamini-Hochberg method (Love et al., 2014). 4Cker was also used to identify significant 4C contacts and significant differences in 4C contacts with parameters k = 5 for ‘nearBaitAnalysis’, k = 10 for ‘cisAnalysis’, and pval = 0.05 for ‘differentialAnalysis’ (Raviram et al., 2016). Hummingbird, which is based on a Bayesian Hidden Markov Model, was used to identify significantly differentially methylated regions with parameter ‘minCpGs = 10, minLength = 100, maxGap = 300’ (Ji, 2019). Signaling pathway analyses were conducted using DAVID Bioinformatics Resources 6.8 (Huang et al., 2009). For permutation tests (10,000 shuffles), the p values were calculated as p = n(|Exp - mean(Exp)| ≥ |Obs - mean(Exp)|)/10,000.
Acknowledgments
This work was supported by Agriculture and Food Research Initiative (grant AFRI - 2018-67015-27598); National Science Foundation (grant IOS1545780); Genomics Technology Core, University of Missouri (MUGTC, Tier1 Block Grant); College of Agriculture, Food & Natural Resources, University of Missouri (Dr. Roger L. Morrison Scholarship to Y.L.); and the William and Nancy Thompson professorship (J.C.). We thank Dr. Robert Schnabel for assistance with the DNA sequencing analyses, Astrid Roshealy Brau for IGF2R qRT-PCR, and MUGTC staff for 4C sequencing assistance.
Author contributions
Y.L. and R.M.R. designed research; Y.L. performed research; Y.L., F.B., M.R.H., J.C., and D.H. analyzed data; Y.L. and R.M.R. wrote the paper; all authors edited the paper.
Declaration of interests
The authors declare no competing interests.
Published: May 20, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104269.
Supplemental information
Data and code availability
-
•
All raw data of RNA-seq, WGBS, 4C-seq, and DNA-seq reported in this paper are publicly available in the GEO database with accession numbers GSE197130 as of the date of publication. Original gel images reported in this paper are available from the lead contact upon request.
-
•
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
All additional information required to reanalyze the data reported in this paper are available from the lead contact upon request.
References
- Anders S., Pyl P.T., Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battistelli C., Busanello A., Maione R. Functional interplay between MyoD and CTCF in regulating long-range chromatin interactions during differentiation. J. Cell Sci. 2014;127:3757–3767. doi: 10.1242/jcs.149427. [DOI] [PubMed] [Google Scholar]
- Bauvois B. New facets of matrix metalloproteinases MMP-2 and MMP-9 as cell surface transducers: outside-in signaling and relationship to tumor progression. Biochim. Biophys. Acta (BBA)-Rev. Cancer. 2012;1825:29–36. doi: 10.1016/j.bbcan.2011.10.001. [DOI] [PubMed] [Google Scholar]
- Berland S., Rustad C.F., Bentsen M.H.L., Wollen E.J., Turowski G., Johansson S., Houge G., Haukanes B.I. Double paternal uniparental isodisomy 7 and 15 presenting with Beckwith-Wiedemann spectrum features. Mol. Case Stud. 2021:a006113. doi: 10.1101/mcs.a006113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics (Oxford, England) 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brioude F., Kalish J.M., Mussa A., Foster A.C., Bliek J., Ferrero G.B., Boonen S.E., Cole T., Baker R., Bertoletti M., et al. Expert consensus document: Clinical and molecular diagnosis, screening and management of Beckwith–Wiedemann syndrome: an international consensus statement. Nat. Rev. Endocrinol. 2018;14:229–249. doi: 10.1038/nrendo.2017.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad Institute . Broad Institute; 2018. Picard Toolkit. [Google Scholar]
- Bushnell B. 2021. BBMap. [Google Scholar]
- Cairo S., Armengol C., De Reyniès A., Wei Y., Thomas E., Renard C.-A., Goga A., Balakrishnan A., Semeraro M., Gresh L., et al. Hepatic stem-like phenotype and interplay of Wnt/β-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell. 2008;14:471–484. doi: 10.1016/j.ccr.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Chan S.K., Chan S.S.-K., Hagen H.R., Swanson S.A., Stewart R., Boll K.A., Aho J., Thomson J.A., Kyba M. Development of bipotent cardiac/skeletal myogenic progenitors from MESP1+ mesoderm. Stem Cell Rep. 2016;6:26–34. doi: 10.1016/j.stemcr.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Hagen D.E., Elsik C.G., Ji T., Morris C.J., Moon L.E., Rivera R.M. Characterization of global loss of imprinting in fetal overgrowth syndrome induced by assisted reproduction. Proc. Natl. Acad. Sci. U S A. 2015;112:4618–4623. doi: 10.1073/pnas.1422088112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Hagen D.E., Ji T., Elsik C.G., Rivera R.M. Global misregulation of genes largely uncoupled to DNA methylome epimutations characterizes a congenital overgrowth syndrome. Sci. Rep. 2017;7:12667. doi: 10.1038/s41598-017-13012-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Robbins K.M., Wells K.D., Rivera R.M. Large offspring syndrome: a bovine model for the human loss-of-imprinting overgrowth syndrome Beckwith-Wiedemann. Epigenetics. 2013;8:591–601. doi: 10.4161/epi.24655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C., Zhang H., Guo L.-N., Zhang X., Meng L., Pan X., Wei Y. Inhibitory effect of NBL1 on PDGF-BB-induced human PASMC proliferation through blockade of PDGFβ-p38MAPK pathway. Biosci. Rep. 2016;36 doi: 10.1042/bsr20160199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson A.J., Lewis P., Przepiorski A., Sander V. Seminars in Cell & Developmental Biology. Elsevier; 2019. Turning mesoderm into kidney; pp. 86–93. [DOI] [PubMed] [Google Scholar]
- Davidson I.F., Bauer B., Goetz D., Tang W., Wutz G., Peters J.-M. DNA loop extrusion by human cohesin. Science. 2019;366:1338–1345. doi: 10.1126/science.aaz3418. [DOI] [PubMed] [Google Scholar]
- Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbs K.B., Khan F.A., Sakatani M., Moss J.I., Ozawa M., Ealy A.D., Hansen P.J. Regulation of pluripotency of inner cell mass and growth and differentiation of trophectoderm of the bovine embryo by colony stimulating factor 2. Biol. Reprod. 2013;89 doi: 10.1095/biolreprod.113.113183. [DOI] [PubMed] [Google Scholar]
- Doyle B., Fudenberg G., Imakaev M., Mirny L.A. Chromatin loops as allosteric modulators of enhancer-promoter interactions. PLoS Comput. Biol. 2014;10:e1003867. doi: 10.1371/journal.pcbi.1003867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L., Sun X., Csizmadia E., Han L., Bian S., Murakami T., Wang X., Robson S.C., Wu Y. Vascular CD39/ENTPD1 directly promotes tumor cell growth by scavenging extracellular adenosine triphosphate. Neoplasia. 2011;13:206-IN2. doi: 10.1593/neo.101332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornes O., Castro-Mondragon J.A., Khan A., Van der Lee R., Zhang X., Richmond P.A., Modi B.P., Correard S., Gheorghe M., Baranašić D., et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020;48:D87–D92. doi: 10.1093/nar/gkz1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser J., Ferrai C., Chiariello A.M., Schueler M., Rito T., Laudanno G., Barbieri M., Moore B.L., Kraemer D.C., Aitken S., et al. The FANTOM Consortium Hierarchical folding and reorganization of chromosomes are linked to transcriptional changes in cellular differentiation. Mol. Syst. Biol. 2015;11:852. doi: 10.15252/msb.20156492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S., Zou D., Mao L., Liu H., Song P., Chen Y., Zhao S., Gao C., Li X., Gao Z., et al. BS-SNPer: SNP calling in bisulfite-seq data. Bioinformatics. 2015;31:4006–4008. doi: 10.1093/bioinformatics/btv507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheldof N., Leleu M., Noordermeer D., Rougemont J., Reymond A. Gene Regulatory Networks. Springer; 2012. Detecting long-range chromatin interactions using the chromosome conformation capture sequencing (4C-seq) method; pp. 211–225. [DOI] [PubMed] [Google Scholar]
- Grewal S.I., Elgin S.C. Heterochromatin: new possibilities for the inheritance of structure. Curr. Opin. Genet. Dev. 2002;12:178–187. doi: 10.1016/s0959-437x(02)00284-8. [DOI] [PubMed] [Google Scholar]
- Hayes B.J., Daetwyler H.D. 1000 bull genomes project to map simple and complex genetic traits in cattle: applications and outcomes. Annu. Rev. Anim. Biosci. 2019;7:89–102. doi: 10.1146/annurev-animal-020518-115024. [DOI] [PubMed] [Google Scholar]
- Huang D.W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- James M.A., Lu Y., Liu Y., Vikis H.G., You M. RGS17, an overexpressed gene in human lung and prostate cancer, induces tumor cell proliferation through the cyclic AMP-PKA-CREB pathway. Cancer Res. 2009;69:2108–2116. doi: 10.1158/0008-5472.can-08-3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji T. A Bayesian hidden Markov model for detecting differentially methylated regions. Biometrics. 2019;75:663–673. doi: 10.1111/biom.13000. [DOI] [PubMed] [Google Scholar]
- Jiménez Martín O., Schlosser A., Furtwängler R., Wegert J., Gessler M. MYCN and MAX alterations in Wilms tumor and identification of novel N-MYC interaction partners as biomarker candidates. Cancer Cell Int. 2021;21:1–15. doi: 10.1186/s12935-021-02259-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagey M.H., Newman J.J., Bilodeau S., Zhan Y., Orlando D.A., van Berkum N.L., Ebmeier C.C., Goossens J., Rahl P.B., Levine S.S., et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijger P.H., Geeven G., Bianchi V., Hilvering C.R., de Laat W. 4C-seq from beginning to end: a detailed protocol for sample preparation and data analysis. Methods. 2020;170:17–32. doi: 10.1016/j.ymeth.2019.07.014. [DOI] [PubMed] [Google Scholar]
- Krueger F., Andrews S.R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Zhu Y., Ma C., Qiu Z., Zhang X., Kang Z., Wu Z., Wang H., Xu X., Zhang H., et al. Downregulation of EphA5 by promoter methylation in human prostate cancer. BMC Cancer. 2015;15:1–10. doi: 10.1186/s12885-015-1025-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Donnelly C.G., Rivera R.M. Overgrowth syndrome. Vet. Clin. Food Anim. Pract. 2019;35:265–276. doi: 10.1016/j.cvfa.2019.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Hagen D.E., Ji T., Bakhtiarizadeh M.R., Frederic W.M., Traxler E.M., Kalish J.M., Rivera R.M. Altered microRNA expression profiles in large offspring syndrome and Beckwith-Wiedemann syndrome. Epigenetics. 2019;14:850–876. doi: 10.1080/15592294.2019.1615357. 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling J.Q., Li T., Hu J.F., Vu T.H., Chen H.L., Qiu X.W., Cherry A.M., Hoffman A.R. CTCF mediates interchromosomal colocalization between Igf2/H19 and Wsb1/Nf1. Science. 2006;312:269–272. doi: 10.1126/science.1123191. [DOI] [PubMed] [Google Scholar]
- Liu Y., Siegmund K.D., Laird P.W., Berman B.P. Bis-SNP: combined DNA methylation and SNP calling for Bisulfite-seq data. Genome Biol. 2012;13:R61. doi: 10.1186/gb-2012-13-7-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llères D., Moindrot B., Pathak R., Piras V., Matelot M., Pignard B., Marchand A., Poncelet M., Perrin A., Tellier V., et al. CTCF modulates allele-specific sub-TAD organization and imprinted gene activity at the mouse Dlk1-Dio3 and Igf2-H19 domains. Genome Biol. 2019;20 doi: 10.1186/s13059-019-1896-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund U., Agostinelli C., Agostinelli M.C. Repository CRAN; 2017. Package ‘circular’. [Google Scholar]
- Lupiáñez D.G., Kraft K., Heinrich V., Krawitz P., Brancati F., Klopocki E., Horn D., Kayserili H., Opitz J.M., Laxova R., et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161:1012–1025. doi: 10.1016/j.cell.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcho C., Bevilacqua A., Tremblay K.D., Mager J. Tissue-specific regulation of Igf2r/Airn imprinting during gastrulation. Epigenet. Chromatin. 2015;8:10. doi: 10.1186/s13072-015-0003-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montefiori L.E., Sobreira D.R., Sakabe N.J., Aneas I., Joslin A.C., Hansen G.T., Bozek G., Moskowitz I.P., McNally E.M., Nóbrega M.A. A promoter interaction map for cardiovascular disease genetics. Elife. 2018;7:e35788. doi: 10.7554/elife.35788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naveh N.S.S., Deegan D.F., Huhn J., Traxler E., Lan Y., Weksberg R., Ganguly A., Engel N., Kalish J.M. The role of CTCF in the organization of the centromeric 11p15 imprinted domain interactome. Nucleic Acids Res. 2021;49:6315–6330. doi: 10.1093/nar/gkab475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora E.P., Goloborodko A., Valton A.-L., Gibcus J.H., Uebersohn A., Abdennur N., Dekker J., Mirny L.A., Bruneau B.G. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell. 2017;169:930–944.e22. doi: 10.1016/j.cell.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D., et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44:D733–D745. doi: 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong C.-T., Corces V.G. CTCF: an architectural protein bridging genome topology and function. Nat. Rev. Genet. 2014;15:234–246. doi: 10.1038/nrg3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M., Kim D., Pertea G.M., Leek J.T., Salzberg S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016;11:1650–1667. doi: 10.1038/nprot.2016.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanstiel D.H., Boyle A.P., Araya C.L., Snyder M.P. Sushi. R: flexible, quantitative and integrative genomic visualizations for publication-quality multi-panel figures. Bioinformatics. 2014;30:2808–2810. doi: 10.1093/bioinformatics/btu379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S.S., Huang S.-C., Glenn St Hilaire B., Engreitz J.M., Perez E.M., Kieffer-Kwon K.-R., Sanborn A.L., Johnstone S.E., Bascom G.D., Bochkov I.D., et al. Cohesin loss eliminates all loop domains. Cell. 2017;171:305–320.e24. doi: 10.1016/j.cell.2017.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raviram R., Rocha P.P., Müller C.L., Miraldi E.R., Badri S., Fu Y., Swanzey E., Proudhon C., Snetkova V., Bonneau R., Skok J.A. 4C-ker: a method to reproducibly identify genome-wide interactions captured by 4C-Seq experiments. PLoS Comput. Biol. 2016;12:e1004780. doi: 10.1371/journal.pcbi.1004780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera R.M., Hansen P.J. Development of cultured bovine embryos after exposure to high temperatures in the physiological range. Reproduction (Cambridge, England) 2001;121:107–115. doi: 10.1530/rep.0.1210107. [DOI] [PubMed] [Google Scholar]
- Rivera R.M., Donnelly C.G., Patel B.N., Li Y., Soto-Moreno E.J. Abnormal offspring syndrome. Bov. Reprod. 2021:876–895. doi: 10.1002/9781119602484.ch71. [DOI] [Google Scholar]
- Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics (Oxford, England) 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez C., Borgel J., Court F., Cathala G., Forné T., Piette J. CTCF is a DNA methylation-sensitive positive regulator of the INK/ARF locus. Biochem. Biophys. Res. Commun. 2010;392:129–134. doi: 10.1016/j.bbrc.2009.12.159. [DOI] [PubMed] [Google Scholar]
- Rosen B.D., Bickhart D.M., Schnabel R.D., Koren S., Elsik C.G., Tseng E., Rowan T.N., Low W.Y., Zimin A., Couldrey C., et al. De novo assembly of the cattle reference genome with single-molecule sequencing. Gigascience. 2020;9:giaa021. doi: 10.1093/gigascience/giaa021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol S., Steunou V., Chalas C., Kerjean A., Rigolet M., Viegas-Pequignot E., Jouannet P., Le Bouc Y., Gicquel C. The epigenetic imprinting defect of patients with Beckwith—Wiedemann syndrome born after assisted reproductive technology is not restricted to the 11p15 region. J. Med. Genet. 2006;43:902–907. doi: 10.1136/jmg.2006.042135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzier C., Vanatka R., Bannwarth S., Philip N., Coussement A., Paquis-Flucklinger V., Lambert J.-C. A novel homozygous MMP2 mutation in a family with Winchester syndrome. Clin. Genet. 2006;69:271–276. doi: 10.1111/j.1399-0004.2006.00584.x. [DOI] [PubMed] [Google Scholar]
- Sanborn A.L., Rao S.S.P., Huang S.-C., Durand N.C., Huntley M.H., Jewett A.I., Bochkov I.D., Chinnappan D., Cutkosky A., Li J., et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. 2015;112:E6456–E6465. doi: 10.1073/pnas.1518552112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schertzer M.D., Braceros K.C., Starmer J., Cherney R.E., Lee D.M., Salazar G., Justice M., Bischoff S.R., Cowley D.O., Ariel P., et al. lncRNA-induced spread of polycomb controlled by genome architecture, RNA abundance, and CpG island DNA. Mol. Cell. 2019;75:523–537.e10. doi: 10.1016/j.molcel.2019.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonis M., Klous P., Splinter E., Moshkin Y., Willemsen R., De Wit E., Van Steensel B., De Laat W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture–on-chip (4C) Nat. Genet. 2006;38:1348–1354. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- Sleutels F., Zwart R., Barlow D.P. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature. 2002;415:810–813. doi: 10.1038/415810a. [DOI] [PubMed] [Google Scholar]
- Sokolov E., Iannitti D.A., Schrum L.W., McKillop I.H. Altered expression and function of regulator of G-protein signaling-17 (RGS17) in hepatocellular carcinoma. Cell Signal. 2011;23:1603–1610. doi: 10.1016/j.cellsig.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splinter E., de Wit E., van de Werken H.J., Klous P., de Laat W. Determining long-range chromatin interactions for selected genomic sites using 4C-seq technology: from fixation to computation. Methods. 2012;58:221–230. doi: 10.1016/j.ymeth.2012.04.009. [DOI] [PubMed] [Google Scholar]
- Tan G., Lenhard B. TFBSTools: an R/bioconductor package for transcription factor binding site analysis. Bioinformatics (Oxford, England) 2016;32:1555–1556. doi: 10.1093/bioinformatics/btw024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan G., Opitz L., Schlapbach R., Rehrauer H. Long fragments achieve lower base quality in Illumina paired-end sequencing. Sci. Rep. 2019;9:2856–2857. doi: 10.1038/s41598-019-39076-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Werken H.J., de Vree P.J., Splinter E., Holwerda S.J., Klous P., de Wit E., de Laat W. Methods in Enzymology. Elsevier; 2012. 4C technology: protocols and data analysis; pp. 89–112. [DOI] [PubMed] [Google Scholar]
- Van der Auwera G.A., O’Connor B.D. 2020. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra (O’Reilly Media) [Google Scholar]
- Vasimuddin M., Misra S., Li H., Aluru S. 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS) IEEE; 2019. Efficient architecture-aware acceleration of BWA-MEM for multicore systems; pp. 314–324. [Google Scholar]
- Verona R.I., Mann M.R., Bartolomei M.S. Genomic imprinting: intricacies of epigenetic regulation in clusters. Annu. Rev. Cell Dev. Biol. 2003;19:237–259. doi: 10.1146/annurev.cellbio.19.111401.092717. [DOI] [PubMed] [Google Scholar]
- Vodyanik M.A., Yu J., Zhang X., Tian S., Stewart R., Thomson J.A., Slukvin I.I. A mesoderm-derived precursor for mesenchymal stem and endothelial cells. Cell Stem Cell. 2010;7:718–729. doi: 10.1016/j.stem.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011;3:180–185. doi: 10.1002/wics.147. [DOI] [Google Scholar]
- Williams R.L., Jr., Starmer J., Mugford J.W., Calabrese J.M., Mieczkowski P., Yee D., Magnuson T. fourSig: a method for determining chromosomal interactions in 4C-Seq data. Nucleic Acids Res. 2014;42:e68. doi: 10.1093/nar/gku156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorikawa C., Shibata H., Waguri S., Hatta K., Horii M., Katoh K., Kobayashi T., Uchiyama Y., Maki M. Human CHMP6, a myristoylated ESCRT-III protein, interacts directly with an ESCRT-II component EAP20 and regulates endosomal cargo sorting. Biochem. J. 2005;387:17–26. doi: 10.1042/bj20041227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebarth J.D., Bhattacharya A., Cui Y. CTCFBSDB 2.0: a database for CTCF-binding sites and genome organization. Nucleic Acids Res. 2012;41:D188–D194. doi: 10.1093/nar/gks1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All raw data of RNA-seq, WGBS, 4C-seq, and DNA-seq reported in this paper are publicly available in the GEO database with accession numbers GSE197130 as of the date of publication. Original gel images reported in this paper are available from the lead contact upon request.
-
•
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
All additional information required to reanalyze the data reported in this paper are available from the lead contact upon request.