

Abstract
Fibroblast growth factor 21 (FGF21) is a promising therapeutic agent for treatment of type 2 diabetes (T2D) and non-alcoholic steatohepatitis (NASH). We show that therapeutic levels of FGF21 were achieved following subcutaneous (s.c.) administration of mRNA encoding human FGF21 proteins. The efficacy of mRNA was assessed following 2-weeks repeated s.c. dosing in diet-induced obese (DIO), mice which resulted in marked decreases in body weight, plasma insulin levels, and hepatic steatosis. Pharmacokinetic/pharmacodynamic (PK/PD) modelling of several studies in both lean and DIO mice showed that mRNA encoding human proteins provided improved therapeutic coverage over recombinant dosed proteins in vivo. This study is the first example of s.c. mRNA therapy showing pre-clinical efficacy in a disease-relevant model, thus, showing the potential for this modality in the treatment of chronic diseases, including T2D and NASH.
Keywords: mRNA therapy, LNP, FGF21, insulin resistance, steatosis, DIO mice
Graphical abstract
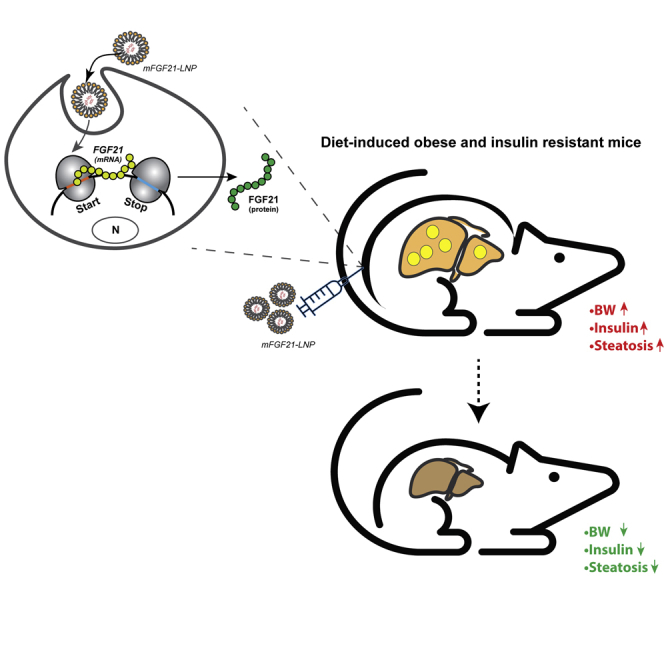
Synthetic mRNA encoding FGF21 encapsulated in LNP effectively transcribes to a functional protein in human adipocytes. In addition, repeated s.c. administration of FGF21 mRNA to DIO, insulin-resistant/steatotic mice leads to disease reversal.
Introduction
In recent years, developments in basic RNA biology and technology have brought forth a corresponding revolution in RNA-based strategies to generate new types of therapeutics, including antisense RNA, small interfering RNA, RNA aptamers, and mRNA (mRNA).1,2 In particular, mRNA has emerged as an exciting alternative to deliver therapeutic proteins.2,3 The advantages with mRNA include that (1) mRNA is translated into therapeutic protein by target cells leading to post-translational modifications that are likely to resemble the endogenous proteins; (2) compared with DNA or viral based gene therapies, mRNA does not require nuclear localization, which reduces decrease risks of genomic insertions and mutations, and (3) mRNA therapy avoids constitutive gene expression but maintains a transient expression with a defined half-life. With these advantages, mRNA could offer the delivery of secreted proteins with challenging biophysical properties, as well as intracellular and membrane proteins that, so far, rely on viral-based gene therapy.
Despite so many exciting perspectives, many barriers have prevented clinical utilization of mRNA for patients with chronic diseases. The major challenges include instability of mRNA in vivo; it is susceptible to degradation by endo- and exo-nucleases. In addition, cellular membrane lipid bilayers block entry of highly charged, high molecular weight mRNAs. Further, innate immune pattern recognition receptors, like Toll-like receptors have evolved to mount immune responses to RNA invasion.4 A new generation of modified RNA from Moderna therapeutics (Moderna, Inc) has showed improved stability and decreased immunogenicity.5 A recent example of mRNA as a therapeutic in early clinical pipeline is AZD8601.6
Lipid nanoparticles (LNPs), have emerged as an effective formulation to encapsulate mRNAs, both protecting them from degrading enzymes and enabling the entry of the RNA to cells,7 and are currently the non-viral RNA delivery platform of choice.8,9 Several LNP formulations have been evaluated in the preclinical setting.10, 11, 12, 13, 14 Up until recently, the amino lipid MC3 (DLin-MC3-DMA) was the most clinically advanced oligonucleotide delivery system, with ONPATTRO (Patisiran) awarded break through therapy in 2018 by the U.S. Food and Drug Administration for the treatment of the polyneuropathy of hereditary transthyretin-mediated amyloidosis in adults.15 With the emergency use authorization of two mRNA based-coronavirus disease 2019 (COVID-19) vaccines, Moderna’s mRNA-1273 sever acute respiratory distress syndrome coronavirus disease 2 vaccine16,17 and Pfizer-BioNTech COVID-19 vaccine,18,19 intramuscular administration of LNPs is now widely accepted and has been successfully administered to several hundred million individuals worldwide. Despite the success, an LNP formulation suitable for subcutaneous (s.c.) repeated and chronic dosing is still a gap. The s.c. administration could open the possibility of patient self-administration and hence long-term chronic treatment that might enable mRNA to be used as a novel modality of protein replacement or regenerative therapies.
In our recent publication,20 we have reported in vivo systematic characterization of functionalized LNPs that enable tolerable s.c. administration of mRNA as a viable approach to achieve systemic levels of therapeutic proteins. Moderna therapeutics have recently reported a new amino lipid (lipid 5), which has enhanced transfection efficiency and decreased immunogenicity (when dosed intravenously [i.v.]) compared with MC3-containing LNP in rodents and non-human primates (NHPs).21 In this study, we extended the application of this novel LNP to repeated s.c. delivery of mRNA constructs for treatment of metabolic disorders.
More than 400 million individuals are diagnosed with type 2 diabetes (T2D) worldwide.22 Therapeutic agents that correct the underlying pathologies of diabetes such as insulin resistance and obesity remain attractive to bring disease reversal and decrease comorbidities. Multiple studies have demonstrated that pharmacological administration of recombinant FGF21- or FGF21-mimetics in obese and diabetic animal models, including rodents and NHPs, leads to profound metabolic improvements.23,24 These effects include a decrease in body weight and liver steatosis, as well as improved insulin sensitivity and beta cell function.24
FGF21 is a hepatokine that binds and activates a cell surface receptor complex composed of FGF receptors (FGFR1c/2c/3c) and co-factor β-Klotho leading to ERK1/2 phosphorylation.25 Although FGF receptors are ubiquitously expressed, β-Klotho expression is restricted to a few tissues, including brown (BAT) and white adipose tissue, as well as the liver and central nerve system.26 The development of FGF21 analogs suitable for chronic treatment has been challenging, due to poor pharmacokinetic properties of native FGF21 including short half-life (an estimated half-life of approximately 1 h in rodents and 0.5–2 h in NHPs27) and its susceptibility to aggregation and degradation by proteases. Clinical FGF21 candidates require amino acid replacement for half-life extension and a decrease in protein aggregation, which increases the risk of immunogenicity.28
In this work, we used mRNA to express FGF21, a therapeutic protein with well-established pharmacodynamic effects, and its half-life extended Fc-analog to explore the potential of mRNA as a new modality for treatment of metabolic disorders. We demonstrate that mRNA can be delivered repeatedly via the s.c. route to express and secrete a pharmacologically active protein, thus providing a proof of concept for the treatment of a chronic disease such as T2D and non-alcoholic steatohepatitis (NASH).
Results
mFGF21 translates to functional protein in human adipocytes
To test if mRNA encoding human FGF21 (mFGF21) expresses functional FGF21 protein in vitro, equal numbers of human adipose-derived stem cells (hASCs) from healthy subjects were transfected with equal amount of mFGF21 or control mRNA encoding enhanced-GFP (meGFP) using lipofectamine. All mFGF21-transfected cells showed a significant increase in FGF21 expression as shown both by visual examination of representative picture of stained cells by immunocytochemistry and by the detection of protein levels in the media by ELISA compared with relative control cells transfected with meGFP (n = 4–8) (Figure 1A).
Figure 1.

mFGF21 translates into functional protein in human adipocytes
(A) Representative immunostaining and ELISA analysis of human FGF21 in cells transfected with control LNP and mFGF21. (B–D) Phosphorylation of ERK in adipocytes treated with recFGF21 and mmFGF21. (E) Glucose uptake in adipocytes stimulated by recFGF21 or mFGF21. (F) mFGF21 protein in conditioned media of transfected adipocytes. (G) FGF21 down-stream genes expression in human adipocytes treated with recFGF21 or mFGF21. (H) Injection site luciferase quantification performed at 6, 24, and 48 h after injection. (I) Representative image of RNA distribution at the injection site (brown signal; RNAscope) was taken at 6 h after single s.c. administration of mmLUC 0.3 mg/kg. Figures show mean ± standard error of the mean. n = 4–8, ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 versus PBS, #p < 0.05, ##p < 0.01, and ###p < 0.001 versus LNP. Ordinary one-way ANOVA was performed followed by Sidak’s multiple comparison test.
To determine the activity of mRNA-encoded FGF21 protein, terminally differentiated white human adipocytes were stimulated, with recombinant human FGF21 (recFGF21) or conditional medium containing equal amounts of mRNA encoded FGF21 protein, for 20 min. Cell lysates were subsequentially analyzed by western blot for MAP kinase pathway (phosphorylated Erk1/2 [p-Erk1/2] and total Erk1/2 [Erk1/2] expression) compared with non-transfected cells or conditioned media derived from an meGFP group (Figure 1B). To provide more compelling evidence for the activity of mRNA-encoded FGF21 protein, we also studied the dose-response (Figure 1C) and time-course (Figure 1D) of mRNA-encoded FGF21 protein-induced phosphorylation of Erk1/2 as well insulin-independent glucose uptake and compared it with recFGF21 (Figures 1C–1E). Although we could not identify any difference in potency on Erk1/2 signaling between mFGF21 and recFGF21 proteins (Figure 1C), a clear and statistically significant difference on efficacy was evident for both Erk1/2 signaling and insulin-independent glucose uptake, favoring mRNA-encoded FGF21 (Figures 1D and 1E). These in vitro data clearly demonstrate that mFGF21 effectively directs the biosynthesis and secretion of active FGF21 protein that induces MAP Kinase signaling similar to recFGF21 in mature adipocytes. Furthermore, the downstream pharmacological effects, here measured as glucose uptake, are consistent with physiologically relevant signaling.
We further evaluated the activity of FGF21 protein when transfecting cells using mFGF21 encapsulated in a LNP, as previously reported.20,21 LNP-encapsulating mFGF21 (LNP-mFGF21) was incubated with ex vivo differentiated human adipocytes, and the FGF21 downstream effector genes, e.g., DUSP4 and SPRY4, were measured as indicative of FGF21 action. The treatment of human adipocytes with LNP-mFGF21 for 24 h resulted in the secretion of approximately 500 ng/mL of FGF21 into the conditioned media, while control LNP-treated adipocytes had undetectable levels of human FGF21 (Figure 1F). The increase in FGF21 levels coincided with a robust increase in both DUSP4 and SPRY4 mRNAs, similar to that of recFGF21 protein (Figure 1G). These data demonstrate that the LNP-containing mFGF21 transfects mature human adipocytes effectively and the mRNA-derived FGF21 protein exerts autocrine effects leading to activation of downstream pathways.
Finally, to better understand the primary location and cellular expression after the s.c. administration of mRNA-formulated LNP, an imaging study (IVIS, Figure 1H) with a subsequent histological evaluation (RNA-Scope, Figure 1I) was performed using Luciferase mRNA (LNP-mLuc). Luciferase protein expression was followed over 48 h and confirmed to be confined to the site of administration (Figure 1H) as previously also reported by Davies et al.20 Furthermore, histological assessment showed that the mLuc (Figure 1I) was localized in the adipose layer.
Pharmacokinetics
First we investigated the half-life and bioavailability of recFGF21 and recFc-FGF21 proteins in lean mice. RecFGF21 showed a short terminal half-life of 1.5 h and a complete systemic bioavailability after s.c. administration (Figure 2A), while recFc-FGF21 had a terminal half-life of approximately 9 h and a systemic bioavailability of 40% after s.c. administration (Figure 2B). This is in agreement with previous literature reporting that recFGF21 is rapidly cleared after administration in vivo while conjugation with a Fc-domain extends the half-life of FGF21 (recFc-FGF21).27
Figure 2.

Plasma concentration-time profiles of FGF21 and Fc-FGF21 administered as recombinant protein or expressed by mRNA
(A) The i.v. (black, n = 4) and s.c. (blue, n = 4) administration of 0.3 mg/kg recFGF21 protein (B), i.v. (magenta, n = 8) and s.c. (turquoise, n = 20) administration of 0.74 mg/kg recFc-FGF21 protein (C), s.c. administration of 0.5 mg/kg mFGF21 (red, n = 4), (D) repeated s.c. twice daily administration of recFGF21 (blue squares, n = 10) and once daily administration of mFGF21 expressed protein (red dots, n = 8) measured after 14 days matched with the predicted concentrations (lines show simulation; blue for recFGF21 and red mFGF21) (E), single (n = 16) and once daily repeated (n = 18) s.c. administration of 0.5 mg/kg mFc-FGF21 (brown and amber lines). (A–C) lean mice, (D and E) DIO mice.
Next, we explored whether the administration of FGF21 mRNAs could increase the level and duration of FGF21 and Fc-FGF21 exposure. As shown in Figure 2C, the s.c. administration of mFGF21 resulted in a delayed peak concentration (tmax of approximately 8 h) and a longer terminal half-life of the recFGF21 (approximately 3 h) compared with recFGF21. The bioavailability of FGF21 protein when dosed as mFGF21 was calculated by: AUCmRNA expressed protein·CLprotein/DosemRNA, where AUC is the area under the curve. The administration of mFGF21 resulted in a net production of FGF21 protein that was 26 times higher than would be obtained with a corresponding molar dose of the recFGF21. The combination of an extended plasma concentration-time profile and a high net protein production from mFGF21 delivery allowed us to decrease the dosing frequency of mFGF21 to once daily instead of twice daily as needed for recFGF21 to obtain a similar average FGF21 exposure during the dosing interval. Using a pharmacokinetic (PK) model of the single dose exposure in lean mice, we also simulated the exposure profiles of recFGF21 and mFGF21 expressed protein after 14 days of repeated dosing in DIO mice (Figure 2D). The levels of plasma recFGF21 or mFGF21 expressed protein measured after 14 days matched with the predicted concentrations suggesting that PK properties of FGF21 protein and the mRNA delivery in lean and DIO mice are similar.
Encouraged by the improved exposure of FGF21 protein when dosed as mFGF21, we explored whether mFc-FGF21 could further improve the exposure of Fc-FGF21 protein (Figure 2E). As for mFGF21, a delayed peak Fc-FGF21 concentration was observed after mFc-FGF21 (tmax = 30 h) as compared with recFc-FGF21 (tmax = 6 h). However, after a single dose of mFc-FGF21, no improvement in the terminal half-life between mRNA translated Fc-FGF21 protein and recFc-FGF21 was observed. In addition, the estimated net production (c.f. bioavailability as fraction) of Fc-FGF21 protein after a single dose of mFc-FGF21 was only 0.86, indicating that the net gain of protein expression using the mFc-FGF21 construct is much less as compared with mFGF21. After repeated dosing of mFc-FGF21, a small accumulation of Fc-FGF21 protein was initially observed as expected from the half-life of the expressed protein (Figure 1E), but with continued repeated dosing, there was a small drop-off in the protein levels with time.
The s.c. injection of LNP-mFc-FGF21 results in dose-dependent increases in target engagement biomarkers
To investigate whether s.c. administered LNP-encapsulated mFc-FGF21 could exert expected FGF21 protein effects, DIO mice were dosed once daily over 2 weeks with mFc-FGF21 (0.15, 0.5, or 1.5 mg/kg/day) or with recFc-FGF21 (0.5 mg/kg/day) as a positive control (Table S1; study 2). The dose of the recFc-FGF21 used as control was based on an efficacy dose-response study performed in DIO mice (Table S1; study 1 and Figure S1). The expression of FGF21 immediate downstream target genes, Dusp4, Dusp6, and Spry4, were monitored in key target tissues including BAT (Figures 3A and 3B) and liver (Figures 3C and 3D). In both BAT and the liver, recFc-FGF21 significantly induced the expression of Dusp4, Dusp6, and Spry4. Similarly, the expression of these genes increased with mFc-FGF21 dose, while the genes for Fgfr1c and Klb (β-Klotho), as expected, remained unchanged (Figures S2A–S2D). Efficacy data on body weight lowering and plasma insulin and triglycerides levels in the final sample from this study are reported as Table S1 (study 2).
Figure 3.

mRNA encoding FGF21 translates in robust target engagement in vivo
Two weeks repeated dosing with protein (recFc-FGF21) or mRNA encoding Fc-FGF21 (mFc-FGF21) to DIO mice translates to FGF21 signal/target engagement in BAT (A and B) and liver (C and D). Figures show mean ± standard error of the mean. n = 4–8, ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 versus PBS, #p < 0.05, ##p < 0.01, and ###p < 0.001 versus LNP. Ordinary one-way ANOVA was performed followed by Sidak’s multiple comparison test.
Repeated dosing of mFGF21 or mFc-FGF21 decreased obesity and improved biomarkers of the metabolic syndrome in DIO mice
To investigate whether mFGF21 and mFc-FGF21 would result in similar therapeutic benefits as recFc-FGF21 protein, DIO mice were treated with recFc-FGF21 (0.5 mg/kg/d), mFGF21 (0.3 mg/kg/d) or mFc-FGF21 (0.5 mg/kg/d) for 2 weeks (Table S1; study 3). recFc-FGF21 was selected as positive control instead of recFGF21 to match the dosing frequency of mRNA.
Two vehicle control groups were included in this study PBS (for recombinant protein) and control LNP for the mRNA groups. There was no difference in body weight gain or plasma glucose or lipids levels between the control groups (Figures 4A, 4C, 4E, and 4F). However, plasma insulin levels were slightly higher in the LNP group compared with the PBS group (Figure 4D).
Figure 4.

mRNA encoding FGF21 improves metabolic endpoints in DIO mice
Two weeks repeated dosing with protein (recFc-FGF21) or mRNA encoding FGF21 (mFGF21) or Fc-FGF21(mFc-FGF21) to DIO mice reduces body weight (A) without affecting food intake (B) and reduces plasma glucose (C), insulin (D), TG (E) and cholesterol (F). Figures show mean ± standard error of the mean. n = 4–8, ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 versus PBS, ##p < 0.01 and ###p < 0.001 versus LNP. One-way ANOVA was performed followed by Sidak’s multiple comparison test.
All FGF21 entities, regardless of modality, decreased body weight by 5%–7.5% over the 2-week dosing period compared with respective vehicles, but did not show any significant effects on food intake (Figure 4B). In addition, recFc-FGF21 protein, as well as mFGF21 and mFc-FGF21 expressed proteins, all dramatically decreased plasma fasting glucose and insulin (Figures 4C and 4D) at the end of treatment period, suggesting improved insulin sensitivity. Plasma triglycerides (TGs) and total cholesterol were also significantly decreased by recFc-FGF21 and mFGF21 (Figures 4E and 4F).
At the end of 2 weeks treatment, both mFGF21 and mFc-FGF21 had a decreased liver weight (corrected by body weight) to a similar degree as recFc-FGF21 (Figure 5B). All FGF21 treatments markedly reduced liver steatosis as can be seen by the representative hematoxylin-eosin (HE) sections, representative lipid droplets indicated with black arrows (Figure 5A). The HE sections were scored according to the grade score scale defined by Kleiner et al.29 High fat diet feeding induced extensive liver steatosis (vehicle-treated groups, PBS, or control LNP), while all FGF21 treatments resulted in a clear and significant reversal of steatosis severity from a severity of 3–4 to a severity of 1–3 (Figure 5C). The decrease in steatosis was confirmed by measurements of total liver TG (Figure 5D). Overall, mRNA encoding for both FGF21 and Fc-FGF21 demonstrated similar therapeutic benefits in DIO mice as recFc-FGF21 following 2 weeks of treatment, thus confirming strong support for mFGF21 therapy in treating insulin resistance and non-alcoholic liver disease.24
Figure 5.

mRNA encoding FGF21 reduces liver steatosis in DIO mice
Two weeks repeated dosing with protein (recFc-FGF21) or mRNA encoding FGF21 (mFGF21) or Fc-FGF21(mFc-FGF21) to DIO mice reduces liver steatosis as shown by representative images from each treatment group (A), liver weight (B), scoring of liver fat (C) and liver TG content (D) compared with their respective controls (PBS or LNP). (B–D) Mean ± standard error of the mean. n = 4–8, ∗p < 0.05 and ∗∗p < 0.01 versus PBS, ##p < 0.01 versus LNP. One-way ANOVA was performed followed by Sidak’s multiple comparison test (B). Black arrows (A) indicate lipid droplets.
Determination of in vivo potency of FGF21 and Fc-FGF21 when dosed as recombinant protein or when expressed by mRNA
We next determined the in vivo potency of FGF21 and Fc-FGF21 on body weight reduction when expressed from mRNA or when dosed as recombinant proteins. To do this, we first performed a PK analysis where we integrate all available exposure data from all relevant PK and pharmacodynamic studies (Table S2). We used a mixed effects modelling approach, where the PK model was simultaneously fit to either the FGF21 or the Fc-FGF21 protein concentrations observed after the dosing of both recombinant protein and when expressed from mRNA (detailed in the supplementary methods). The individual fits of the model to the observed protein concentrations are shown in supplementary methods Figure S6 (FGF21) and S7 (Fc-FGF21) and the final parameter estimates are reported in Table S3.
The derived individual PK parameters were then fixed as constants in the subsequent PK/pharmacodynamic (PD) model of drug-induced body weight reduction. The FGF21 proteins were assumed to increase the energy expenditure as described by an Emax function, where the two proteins (FGF21 and Fc-FGF21) and the two modalities (recombinant protein and mRNA) were added as categorical covariates on potency (EC50). The individual fit of the model to the observed body weight change are shown in supplementary methods (Figure S8) and the estimates of the derived PD parameter are summarized in Table S4.
The results from this analysis showed that the in vivo potency for body weight reduction was identical between recFGF21 and mFGF21 (Table 1, EC50 = 0.136 nM). The conjugation of FGF21 to a Fc-domain (Fc-FGF21) had a clear negative impact on in vivo potency of FGF21, regardless of the modality used. However, mFc-FGF21 was five-fold more potent in vivo compared with recFc-FGF21 (Table 1) (EC50 = 5.46 nM vs. 26.6 nM). The maximal drug-induced increase in energy expenditure (Emax) was estimated to be 1.76-fold higher in drug-treated mice compared with vehicle-treated mice. As a result, the model predicts that, if the mice were to receive a maximal drug response, they would continue to lose weight until they would reach a minimal body weight of approximately 20 g, which is in line with the expected weight of a lean age-matched mouse (20–25 g).30 However, if left untreated, the mice would continue to gain weight and eventually reach a maximal body weight of 50–56 g.30
Table 1.
In vivo potency of drug-induced body weight reduction in DIO mice
| Parameters | FGF21 | Fc-FGF21 |
|---|---|---|
| EC50 protein (nM/L−1) | 0.136 (0.095–0.177) | 26.6 (17.3–35.8) |
| EC50 mRNA (nM/L−1) | 0.136 (0.095–0.177) | 5.46 (3.57–7.34) |
Estimates presented as mean and 95% confidence interval.
Discussion
We have designed and characterized two FGF21 mRNAs in vitro and in vivo for protein production and evaluated the PK and PD parameters. The novelty of this study is that we can repeatedly administer mRNA via the s.c. route, resulting in expected PD effects of FGF21 treatment. These results illustrate the potential of mRNA therapeutics for treatment of chronic diseases.
Currently, protein-based biopharmaceuticals account for approximately 40% of all drugs in clinical development, which has resulted in an increase in both EU and U.S. approvals.31 This trend is expected to continue over the next 5–10 years. However, manufacturing of protein-based pharmaceuticals is a complex and costly process, owing to the large molecular size, custom production and purification, and in certain instances non-native post-translational modification of the protein. Biologic production requires specially engineered cells for protein production. Subsequent purification of the therapeutic proteins requires specific conditions for each protein, resulting in additional quality control steps to ensure efficacy and safety. Given these limitations, we believe that mRNA could serve as an alternative way to treat patients while conserving validated biological processes. However, mRNA-based therapy also faces challenges, including RNA in vivo stability, innate immune recognition, and efficacious delivery.
The use of mRNA as a drug has been extensively evaluated for both therapeutic proteins and prophylactic vaccines. The most advanced mRNA drugs are currently being used for vaccination purposes with several programs in late stage clinical testing.32 The approval of two mRNA vaccines for COVID-19 has illustrated the potential of the platform.33, 34, 35, 36
There are several preclinical reports that show the potential of mRNA for treatment of genetic and inflammatory diseases; however, these have been limited to the i.v. administration of a single or a few doses.11,37 For the treatment of chronic diseases such as T2D and NASH, s.c. dosing would be the ideal route of administration for drugs that cannot be formulated for oral use.
In this report, we explored the potential of mRNA with reduced immunogenicity and improved in vivo stability to provide the therapeutic benefits of the metabolic regulator FGF21. We demonstrated that mRNA encoding native FGF21 or Fc-FGF21 translated a functional protein in ex vivo differentiated human adipocytes. The functionality of mRNA expressed protein was proven in human adipocytes by studying MAPK activation, leading to increased pERK levels and glucose uptake as good as or better than recFGF21 protein. The FGF21 produced by mFGF21 exerted autocrine effects by the induction of FGF21 downstream gene expression. Based on our LNP-mLuc in vivo experiment (Figures 1H and 1I), we speculate that adipocytes in s.c. adipose tissue are a major cell type producing the therapeutic proteins. Supporting this notion, luciferase mRNA could be detected in adipocytes at the injection site 6 h after s.c. dosing of LNP-mLuc.
Cells that are transfected by a LNP-containing mRNA will express the corresponding human protein, and will start to secrete it into the systemic circulation. The prolonged half-life of FGF21 observed following administration of mFGF21 (t½,ka mRNA = 3 h) compared with recFGF21 (t½,FGF21 = 1.5 h), indicates that the elimination of FGF21 protein after mFGF21 administration is production rate limited (c.f., an absorption rate limited elimination of a small molecule administered in an oral slow release formulation). For the Fc-FGF21 protein, no such prolongation is observed after the administration of mFc-FGF21, since the elimination rate of the expressed protein is slower (t½,Fc-FGF21 = 8.9 h) than the production rate (t½,ka mRNA = 7.1 h) (Table S2). Therefore, the observed terminal half-life for Fc-FGF21 will be the same when expressed by mFc-FGF21 or when dosed as recFc-FGF21, since in both cases it is the intrinsic elimination rate of the protein that is the rate limiting step of the observed half-life.
Interestingly, mFGF21 showed a net production of FGF21 protein that results in a 26-fold higher exposure, in terms of the area under the protein concentration-time curve, than would be obtained when dosing a corresponding molar dose of recFGF21. To our disappointment, the corresponding net production of Fc-FGF21 protein after mFc-FGF21 delivery was only 0.86. This indicates that, for each mole of mFc-FGF21 dosed, only 0.86 moles of protein are produced. However, since the bioavailability of s.c. administered recFc-FGF21 is approximately 40%, and not complete as for recFGF21, the net gain of using mFc-FGF21 compared with recFc-FGF21 is approximately two-fold (0.86/0.40). The calculation of the mRNA net protein production should not be mixed up with the absolute number of protein copies translated from each mRNA by the ribosomes. The net protein production is rather a composite parameter that includes, but is not limited to, the productive uptake (including cell transfection and endosomal escape), the repetitive translation of each mRNA sequence by the ribosomes, and the fraction of expressed protein secreted into the circulation. The calculation of net protein production is an application of the standard equation of bioavailability (F = CL·AUC/dose) on mRNA delivery. A key assumption in the calculation is that the clearance (CL) of recombinant dosed protein and the mRNA expressed protein is the same. If it is known that the proteins show major differences in protein folding or in post translational modifications when expressed in vitro (recombinant protein production) and when expressed in vivo (after mRNA transfection), one should be careful making this assumption. Since the net production of protein from mRNA ideally should be a value much higher than 1, the use of the term bioavailability may not be appropriate, since it is generally related to a fraction ranging between 0 and 1. We, therefore, suggest to use the term net production efficacy (nPE) when referring to the bioavailability of mRNA expressed proteins.
With an improved PK profile and the high nPE, once daily s.c. injections of mFGF21 at a relatively low dose are sufficient to maintain the plasma concentration of FGF21 above EC50 in vivo, while twice daily dosing at a relatively high dose is needed for recFGF21 to achieve similar PD effects (Figure S3). The mechanism behind this can be better understood by realizing that the relationship between drug concentration and drug effect (i.e., the stimulation of energy expenditure) is not linear, but is described by an Emax function (Figure S3). Consequently, when the drug concentration is increased, e.g., by two-fold, the effect of the drug may not increase to the same extent. The best leverage of the drug response is obtained at concentrations close to the EC50. At concentrations distant from the EC50, e.g., at concentrations generating more than 80% of the Emax (EC80, FGF21 = 0.54 nM/L) only small changes in the effect is obtained when increasing the drug concentration. After mFGF21 administration, the FGF21 concentration can be maintained close to the EC50 with small changes during the dosing interval. In contrast, owing to the short half-life of recFGF21, this treatment generates drug concentrations which resides at either very high drug concentrations (>EC80) or at a very low drug concentration (<EC20) during most of the daily dosing interval, where a poor leverage of the effect is obtained (Figure S4). Simulations show that the daily dose of recFGF21 in DIO mice can be decreased from 2 mg/kg (1 mg/kg b.i.d) to 0.3 mg/kg (0.05 mg/kg q6d) with maintained body weight reduction, by using a dosing regimen which keeps the drug concentrations within the efficacious EC20–EC80 concentration range (Figure S4).
Our data indicate that use of the mRNA expressing the FGF21 protein (mFGF21) is the preferred choice when comparing the four different FGF21 entities explored in this work. To draw this conclusion, it is important to simultaneously assess the PK properties (nPE, the protein half-life) and the PD effect (EC50) of the different entities. The detailed reasoning goes as follows. For FGF21, the high nPE and the improved PK profile favors the use of mFGF21 compared with recFGF21. Since the potency and clearance are the same, a 26-fold lower molar dose is required for mFGF21 compared with recFGF21. Adjusting for the approximately 16-fold difference in molecular weight the equivalent milligram per kilogram dose of mFGF21 is approximately 1.5-fold lower as compared with recFGF21. For Fc-FGF21, the mFc-FGF21 is also favored compared with recFc-FGF21, but for different reasons. Despite the disappointingly low nPE for mFc-FGF21, a 10-fold lower molar dose of mFc-FGF21 compared with recFc-FGF21 can still be used to generate the same pharmacological effect (Figure S5). This is due to the small but still two-fold higher nPE of mFc-FGF21 compared with the bioavailability of recFc-FGF21, and the five-fold higher potency determined for mFc-FGF21 expressed protein compared with recFc-FGF21. However, since the difference in molecular weight is about 10-fold between mFc-FGF21 and recFc-FGF21, it gives that the same milligram per kilogram dose will generate the same pharmacological response. When comparing the once daily dose needed for mFGF21 to generate the same body weight reduction as mFc-FGF21, the two mRNAs can be ranked by calculating the dose needed to target the corresponding EC50 as the steady state concentration during the dosing interval by using the equation: Dose = CL·EC50·τ/nPE, where τ is the duration of the dosing interval. Feeding the equation with the data derived in Tables S3 and S4, the daily molar dose of mFGF21 is 1.7-fold lower as compared with mFc-FGF21 (Table 2; 0.41 nM/kg vs. 0.72 nM/kg). It is clear that despite the 700-fold lower CL of Fc-FGF21 compared with FGF21, the advantage of using mFc-FGF21 over mFGF21 is lost owing to a 40-fold relative loss in in vivo potency and the 30-fold lower nPE. Taking the difference in molecular weight into account provides a three-fold lower milligram per kilogram dose in favor of mFGF21, which is of importance since our mRNAs are encapsulated into LNP and local tolerability depends on the lipid load of the LNP formulation. So, overall, the use of the mRNA expressing the FGF21 protein (mFGF21) is the preferred choice when comparing the four different FGF21 entities explored in this work.
Table 2.
Calculated dose targeting in vivo EC50 as average drug concentration at steady state
| Treatment | Daily dose (mg/kg) | Dose (mg/kg) | Mw (kDa) | Daily dose (nM/kg) | Dose (nM/kg) | F or nPE | CL (L/h/kg) | EC50 (nM/L) | τ (h) |
|---|---|---|---|---|---|---|---|---|---|
| recFGF21 q6d | 0.191 | 0.032 | 19.3 | 9.9 | 1.6 | 1.09 | 3.3 | 0.136 | 4 |
| mFGF21 q.d. | 0.125 | 0.125 | 302 | 0.41 | 0.41 | 26 | 3.3 | 0.136 | 24 |
| recFc-FGF21 q.d. | 0.357 | 0.357 | 47.8 | 7.5 | 7.5 | 0.402 | 0.0047 | 26.6 | 24 |
| mFc-FGF21 q.d. | 0.368 | 0.368 | 512 | 0.72 | 0.72 | 0.858 | 0.0047 | 5.46 | 24 |
Mw, molecular weight; F, bioavailability; nPE, net production efficiency; CL, clearance; EC50, potency; τ, dosing interval.
The therapeutic effects of FGF21 have been described in several pre-clinical models including obese and insulin-resistant DIO mice.38,39 The most commonly observed metabolic changes elicited by FGF21 analogs or mimetics includes a significant decrease in body weight and adiposity, which is not accompanied by a decrease in calorie intake.39, 40, 41 This effect is associated with improvements in whole body insulin sensitivity, an alleviation of liver steatosis, and improvements in various circulating metabolic markers.39,42,43 Interestingly, the recent literature proposes that, rather than functioning as a master regulator of metabolism, FGF21 instead functions as a master sensitizer of specific hormonal signals regulating metabolism.24
This study shows that mFGF21 treatment in DIO mice has a clear therapeutic benefit leading to an improved metabolic profile consistent with previous reports for FGF21 analogs or mimetics 24. The presented experiments demonstrate, for the first time, that s.c. administration of mRNA in mice, translates into a functional therapeutic protein in vivo, and shows a proof of concept for further development of s.c. delivery of mRNA therapeutics for the treatment of chronic diseases such as T2D and NASH.
Materials and methods
Isolation and culture of hASCs
Samples of adipose tissue were collected from patients undergoing elective surgery at Sahlgrenska University Hospital in Gothenburg, Sweden. All study subjects received written and oral information before giving written informed consent for the use of the tissue. The studies were approved by The Regional Ethical Review Board in Gothenburg, Sweden. hASCs were isolated from the stromal vascular fraction as described in Bartesaghi et al.44 and cultured in a growth medium DMEM/Ham’s F-12 with 10% FBS, 10 mM HEPES, 33 μM biotin, 17 μM pantothenate, 1 nM Fibroblast growth factor, all from Sigma Aldrich, 50 U/mL penicillin and 50 μg/mL streptomycin at 37 °C, 5% CO2 in air with 80% humidity. For adipocyte differentiation, 90% confluent cells were treated with DMEM/F12 with 3% FCS (PAA; Gold) supplemented with 1 μM dexamethasone (Sigma), 500 μM 3-isobutyl-1-methyxanthine (IBMX; Sigma), and 100 nM insulin. To promote white adipogenesis, 1 μM pioglitazone was included in the differentiation medium. Media were changed every other day during proliferation and differentiation, until fully differentiated (day 14). hASCs were tested for and were free of mycoplasma.
Generation of mRNA
mRNA encoding human FGF 21 (mFGF21) or Luciferase were synthesized as previously described 11. Briefly, hFGF21 mRNA was codon optimized and synthesized by in vitro transcription using T7 RNA polymerase-mediated transcription. The uridine-5′ triphosphate (UTP) was substituted with 1-methylpseudo-uridine UTP and was added to the transcription reaction in place of uridine. The mRNA was transcribed, using a linearized DNA template, which also incorporate the sequence encoded for 5′ and 3’ UTRs, the open reading frame and a poly A tail. mRNA was capped using a vaccinia enzyme in the presence of S-adenosylmethionine. A donor methyl group from S-adenosylmethionine was added to methylated capped RNA (cap-0), resulting in a cap-1 modification to increase mRNA translation efficiency These chemical modifications to the mRNA are designed to both improve protein translation and reduce immunogenicity and have been described in detail as used by Carlsson et al.45 and An et al.11 For wild-type FGF21, the amino acid sequence of the rs739320 variant (leucine→proline at position 174) was selected for design of mRNA, as it is the most common allele in human population. Several different mRNA designs all expressing the same protein were evaluated to compare the efficiency of FGF21 production by different mRNA constructs using hASCs (15,000 cells/96 well seeded the day before transfection in basal medium (BM1 [Zenbio], 3% FBS, 17 ng/mL FGF) transfected with the mRNA constructs using Lipofectamine (Life Technologies, #11668019 0.4μ L/well, diluted in 17 μL OPTI-MEM [Life Technology #31985070]; 250 ng mRNA/well, diluted in 3 μL of RNAse-free water). After 24 h of transfection, the FGF21 protein secreted into the conditioned medium was measured using an ELISA (R&D Systems #DF2100). The highest expressing mRNA constructs tested were selected for further evaluation in in vivo PK and efficacy studies.
Production of recombinant FGF21 and Fc-FGF21
The codon-optimized gene sequence coding for mature FGF21 residues His29-Ser209 (common allele rs739320 L174P variant) was subcloned in a bacterial T7-driven expression vector pET24a in fusion with an N-terminal His6 tag followed by TEV protease cleavage sequence. Protein was expressed in BL21star (DE3) using overnight autoinduction at 20°C followed by a two-step purification with IMAC. The bacterial pellet was resuspended in 50 mM Tris-HCl, pH 8.0, 1 M urea, 0.5 M NaCl, and 1 mM EDTA and lysed by passing through a cell disruptor (Constant System). The cleared supernatant was then loaded on to a column (HisTrap FF) equilibrated in buffer A (buffer A: 50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 10% glycerol, 5 mM imidazole). The protein was purified endotoxin free by adding 0.5% Triton X-114 in washing buffer A and in all subsequent steps endotoxin free buffers were used. The protein was eluted with buffer B (50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 10% glycerol, 400 mM imidazole) followed by Superdex 75 26/60 gel filtration in a PBS buffer, pH 7.4. The typical yield was 60 mg from 1 L of bacterial culture. FGF21 has a short half-life in vivo, and a Fc-fusion was designed to increase circulation levels for longer duration of action. The Fc region of human immunoglobulin G127 was codon optimized for mammalian expression and subcloned in pEBNAZ mammalian expression vector under control of a CMV promotor. The Fc fusion protein was preceded by a mouse Ig kappa chain V-III P01661 signal sequence for secretion into the medium. The Fc sequence was followed by a 15 amino acid Gly-Ser-rich linker followed by mature FGF21 sequence residues His29-Ser209, including two stabilizing mutations L98R/P171G (FGF21 numbering). Mutation L98R prevents aggregation of FGF21, whereas mutation P171G stabilizes FGF21 against proteolytic inactivation by FAPα.27 Suspension grown Expi293F cells were PEI transfected with expression plasmid and cell supernatant was harvested after 5 days. Fc-fusion protein was purified in PBS using protein A (MabSelect SuRe, GE Healthcare) affinity purification under endotoxin-free conditions. The supernatant was incubated with the resin over night at 4°C and washed with 1× PBS, pH 7.4. The protein was eluted with 0.1 M glycine, pH 3.5. The eluate was mixed with 1 M Tris-HCl, pH 9.0, to neutralize the low pH. Buffer exchange to 1× PBS, pH 7.4, was done by using PD10 columns. Typical yields were over 10 mg purified fusion protein from 100 mL culture supernatant. The amino acid sequences of recombinant proteins used in this study are reported in Table S5.
LNP formulation
LNPs were prepared by microfluidic mixing as previously described.20,21 Briefly, an ethanolic solution of the lipid components (including steroid prodrug when used) and a solution of the mRNA in 6.25 mM sodium acetate buffer (pH 5) were mixed at a ratio of 1:3, respectively, and a total flow rate of 12 mL/min using a NanoAssemblr (Precision Nanosystems). After microfluidic mixing, the LNPs were dialyzed overnight against 500× sample volume of PBS (pH 7.4) using Slide-A-Lyzer G2 dialysis cassettes with a molecular weight cut-off of 10 k (ThermoFisher Scientific). Formulations were concentrated using Amicon ultra centrifugal filters (EMD Millipore) and passed through a 0.22-μm filter, and stored at 4°C until use. All formulations were tested for particle size and mRNA encapsulation and were found to be between 60 nm and 100 nm in size, with more than 90% encapsulation. LNPs were formulated using a biodegradable, ionizable lipid (lipid 5, as previously described in Sabnis et al.21), cholesterol, distearoylphosphatidylcholine, and dimyryristoylphosphatidylcholine polyethyleneglycol 2000 at a percent molar composition of 50:38.5:10:1.5, respectively, and at a total lipid:mRNA weight ratio of either 10:1 or 20:1 (nitrogen/phosphate ratio of 3 or 6, respectively). Where specified, rofleponide or budesonide myristate (a C14 fatty acid prodrug ester of rofleponide or budesonide) was incorporated in the LNP at a steroid:mRNA weight ratio of between 1:6 and 1:150 to enable dosing of the steroid at 0.01 or 0.05 mg/kg.20 Further details of the LNP formulations used in the various studies including lipid:mRNA weight ratio and dose of steroid can be found in Table S2.
Tissue distribution studies
LNP encapsulated Luciferase mRNA (mmLUC) mRNA were administered to C57Bl/6 mice s.c. (intrascapular) at a dose of 0.3 mg/kg and a dosing volume of 5 mL/kg while under light anesthesia (isoflurane). At 8, 12, 24, and 48 h after administration, whole body scans of the mice were collected using an IVIS Spectrum in vivo Imaging System (PerkinElmer). Twenty minutes before imaging each mouse received a 150 mg/kg dose of luciferin (Rediject D-Luciferin, PerkinElmer) administered s.c. at a dosing volume of 5 mL/kg.
Immunoblotting
For western blots, cells were lysed in RIPA buffer (50 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, and 150 mM NaCl), supplemented with protease and phosphatase inhibitor cocktails according to the manufacturer’s recommendations. Protein concentrations were measured using the BCA protein assay kit. Proteins were separated by SDS-PAGE on NuPAGE Novex 4–12% Bis-Tris protein gels and transferred to nitrocellulose membranes. Specific proteins were detected with the indicated primary antibodies. Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) rabbit mAb #4370 (Cell Signaling) and p44/42 MAPK (Erk1/2) (3A7) mouse mAb #9107 (Cell Signaling), human klotho beta antibody (β-Klotho) MAB5889 (R&D System) and monoclonal anti-beta-actin (β-actin, AC-15) #A5441(Sigma). Protein-antibody interactions were detected with a horseradish peroxidase-conjugated secondary antibody, subjected to ECL chemiluminescent reagent. Secondary antibodies were from Dako.
Glucose uptake
hASC were seeded in Cytostar T 96-well plates at 15,000 cells/well density and fully differentiated in adipocyte medium (AM; Zen-Bio Inc.) with 0.05 mM IBMX, 0.1 μM dexamethasone, 10 nM insulin, and 1 μM pioglitazone (7 days); and thereafter kept in AM (changed every other day). For glucose uptake, adipocytes were starved for 3 h in DMEM/0.1% BSA, stimulated with FGF-21 for 24 h, and washed twice with KRP buffer (15 mM HEPES, pH 7.4, 118 mM NaCl, 4.8 mM KCl, 1.2 mM MgSO4, 1.3 mM CaCl2, 1.2 mM KH2PO4, 0.1% BSA), and 100 μL of KRP buffer containing 2-deoxy-d-14Cglucose (2-DOG) (0.1 μCi, 100 μM) was added to each well. Control wells contained 100 μL KRP buffer with 2-DOG (0.1 μCi, 10 mM) to monitor for nonspecificity. The uptake reaction was carried out for 1 h at 37°C, and measured using a Wallac 1450 MicroBeta counter (Perkin Elmer).
ERK phosphorylation assay
The fully differentiated human adipocytes were starved for 3 h in a low glucose 1:1 DMEM/F12 media (Invitrogen) supplemented with 0.3% BSA; cells were treated with increasing concentrations of FGF21 variants for 10 min. Cells were washed and lysates were harvested. Phosphorylated and total ERK1/2 was measured using MSD Phospho/Total ERK1/2 whole cell lysate kit (Cat# K15107D, Meso-Scale Discovery) following the manufacturer’s protocol. The amount of phosphor-ERK was normalized by dividing phosphor ERK signal with the total plus phosphor ERK signals.
RNA extraction and quantitative real-time PCR (qPCR)
Total RNA was extracted using RNAeasy (Qiagen) and reverse transcription of 1 μg of total RNA was carried out using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, ThermoFisher Scientific). Quantitative PCR was performed using TaqMan Gene Expression Assays (Table S6) on a QuantStudio 7 Flex Real-Time PCR System (Applied Biosystems, ThermoFisher Scientific). Runs were analyzed using Applied Biosystems platform software, with auto threshold and baseline, then exported and analyzed using SAS JMP v13 statistical software.
DIO mice studies
All in vivo procedures were approved by the Gothenburg region ethics committee and were performed at an AAALAC-accredited site (001560). DIO C57bl/6 male mice were purchased from Taconic Denmark at 20–23 weeks of age. Starting at 6 weeks of age, the mice were fed a high-fat diet (HFD; D12492) for approximately 13 weeks. The mice were acclimatized for at least 2 weeks before the study start and they were randomized into the different treatment groups based on body weight. At dose start, the mice weighed approximately 40 g. Body weight and food intake was measured two times a week starting the week before treatment start. The HFD was changed once weekly. The mice had free access to food and water throughout the study. All treatments where given as daily s.c. injections and the sites were rotated between five sites (two on each side of the spine and one in the neck region). The mice were treated starting at 10 am each day. Light cycle dawn 6:30–7 am, dusk 5:30–6 pm, room temperature 22°C and humidity of 40%–60%. After 2 weeks of treatment, a terminal blood sample was collected from 4-h fasted, isoflurane-anaesthetized mice. For the target engagement study, BAT and liver tissues were collected and placed in liquid nitrogen for later mRNA analysis. For the efficacy study, the livers were weighed and then placed in formalin as described below for later HE staining.
Liver histology
Liver samples were fixed overnight at 4°C in PBS-buffered 4% (wt/vol) paraformaldehyde and embedded in paraffin. Cross-sections (5 μm) were prepared for HE staining. The semiquantitative analysis for hepatocellular lipidosis was performed according to an adapted scoring protocol from Kleiner et al.29 Briefly, the livers were scored for the fraction of hepatocytes showing intracytoplasmic vacuolation compared with unaffected cells as followed: 0 = no vacuolation; 1 = <5% vacuolated hepatocytes; 2 = 5%–30% vacuolated hepatocytes; 3 = > 30%–60% vacuolated hepatocytes; 4 = >60% vacuolated hepatocytes.
Plasma analysis
Insulin was measured using a Mouse/Rat Insulin kit from Meso Scale Discovery (K152BZC-1). Enzymatic colorimetric methods, assayed on the ABX Pentra 400 (Horiba ABX), were used for analysis of: glucose (Glucose HK CP, A11A01667 Horiba ABX), TGs (Trig/GB, 11877771216, Roche Diagnostics GmbH), cholesterol (Cholesterol CP ABX Pentra, A11A01634, HORIBA ABX), β-hydroxybutyrate (RANBUT: D-3-hydroxybutyrate, RB1007, Randox) and free fatty acids (NEFA-HR[2] WaKO Chemicals GmbH). Liver tissue TG levels were assessed after homogenization of approximately 50 mg tissue in isopropanol followed by analysis of TG levels in the supernatant (TGs CP, ABX Pentra, A11A01640, Horiba ABX).
Plasma and medium concentration of FGF21 and Fc-FGF21 (ELISA)
The total human FGF21 and Fc-FGF21 in plasma was analyzed using a bead based Milliplex immunoassay kit (HLPPMAG-57K, Merck Millipore). The quantification of FGF21 was made against the kit standards. The quantification of Fc-FGF21 was made against the in-house Fc-FGF21 recombinant reference protein used in the in vivo study. To fall within the calibration range samples were diluted 50–2,000 fold using the kit assay buffer before the two-fold dilution with serum matrix specified in the kit protocol. Intact FGF21 and Fc-FGF21 in plasma were analyzed using an ELISA kit (KT 879, Epitope Diagnostics, Inc) sensitive only to N- and C-terminal intact FGF21. Quantification of FGF21 was made against the kit standards. Quantification of Fc-FGF21 was made against in-house Fc-FGF21. To fall within the calibration range, samples were diluted 5- to 400-fold using MSD diluent 9 (RB54C, Meso Scale Diagnostics). Medium concentration of human FGF21 was analyzed using an ELISA kit (R&D System, DF2100). Quantification of FGF21 was made against the kit standards. To fall within the calibration range, samples were diluted 200- to 1,000-fold with dilution buffer following the kit protocol.
PK/PD modelling to determine in vivo potencies of the different FGF21 constructs and modalities
A detailed description and related information of the modelling used to study the of effects of the different FGF21 therapeutics on body weight and exposure to determine their in vivo potencies can be found in the Supplementary methods and Tables S1–S4.
Acknowledgments
In memory of our beloved colleague Frank Seeliger, who passed away before completing the submission of the manuscript. We thank Moderna, Inc., for the provision of mRNA. We thank Ann Kjellstedt, Anette Stringer, Ida Stellert, Malin Antonsson, and Karolina Ploj for help with the in vivo studies; Liselotte Björsson, Leif Aasehaug, Ingela Ahlstedt, and Ingela Maxwell for plasma analysis; Annika Pålsson, Charlotte Pollard, and Marianna Yanez Arteta for LNP formulations; Ann-Christine Andréasson for FGF21 in vitro histology; Camilla Johansson for liver histology; Peter Konings for histology grade statistical analysis; and Magnus Söderberg for pathology support.
Author contributions
K.W., S.B., D.H., X-R.P., S.A., N. D., N.B., F.S., M.L., and M.S.W. were members of the cross-functional team within AstraZeneca that were involved in conceptualizing and designing all studies, in data analysis/interpretation and overall execution of the project. K.W., S.B., D.H., and X-R.P. were largely responsible for authoring the manuscript, with contribution from all co-authors according to area of expertise. K.W., M.S.W., M.L., and X-R.P. were responsible for the design and conduct of all in vivo studies used to support the investigations and summarizing/interpreting the results. S.B., S.W., and M.L. were responsible for the design and conduct of all in vitro studies used to support the investigations and summarizing/interpreting the results. D.H. was responsible for PK/PD modeling. F.S. was responsible for liver histology study. Nigel D. was responsible for the co-ordination, manufacture, characterization, and supply of LNP formulations. Niek D. was responsible for the co-ordination of protein engineering and constructs design. Louise B. was responsible for recombinant protein production, characterization and supply. Luis B. was responsible for design and interpretation of the biodistribution study. Sima P. and Krissy B. were responsible for biodistribution study.
Declaration of interests
L. Brito, S.P., and K.B. were at the time of study conduct employees of Moderna, Inc. All other authors were at the time of study conduct and publication all employees of AstraZeneca.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.04.010.
Supplemental information
References
- 1.Lieberman J. Tapping the RNA world for therapeutics. Nat. Struct. Mol. Biol. 2018;25:357–364. doi: 10.1038/s41594-018-0054-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chandler R.J. Messenger RNA therapy as an option for treating metabolic disorders. Proc. Natl. Acad. Sci. U S A. 2019;116:20804–20806. doi: 10.1073/pnas.1914673116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao W., Hou X., Vick O.G., Dong Y. RNA delivery biomaterials for the treatment of genetic and rare diseases. Biomaterials. 2019;217:119291. doi: 10.1016/j.biomaterials.2019.119291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Granot Y., Peer D. Delivering the right message: challenges and opportunities in lipid nanoparticles-mediated modified mRNA therapeutics-An innate immune system standpoint. Semin. Immunol. 2017;34:68–77. doi: 10.1016/j.smim.2017.08.015. [DOI] [PubMed] [Google Scholar]
- 5.Nelson J., Sorensen E.W., Mintri S., Rabideau A.E., Zheng W., Besin G., Khatwani N., Su S.V., Miracco E.J., Issa W.J., et al. Impact of mRNA chemistry and manufacturing process on innate immune activation. Sci. Adv. 2020;6:eaaz6893. doi: 10.1126/sciadv.aaz6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gan L.M., Lagerstrom-Fermer M., Carlsson L.G., Arfvidsson C., Egnell A.C., Rudvik A., Kjaer M., Collen A., Thompson J.D., Joyal J., et al. Intradermal delivery of modified mRNA encoding VEGF-A in patients with type 2 diabetes. Nat. Commun. 2019;10:871. doi: 10.1038/s41467-019-08852-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moss K.H., Popova P., Hadrup S.R., Astakhova K., Taskova M. Lipid nanoparticles for delivery of therapeutic RNA oligonucleotides. Mol. Pharm. 2019;16:2265–2277. doi: 10.1021/acs.molpharmaceut.8b01290. [DOI] [PubMed] [Google Scholar]
- 8.Kowalski P.S., Rudra A., Miao L., Anderson D.G. Delivering the messenger: advances in Technologies for therapeutic mRNA delivery. Mol. Ther. 2019;27:710–728. doi: 10.1016/j.ymthe.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reichmuth A.M., Oberli M.A., Jaklenec A., Langer R., Blankschtein D. mRNA vaccine delivery using lipid nanoparticles. Ther. Deliv. 2016;7:319–334. doi: 10.4155/tde-2016-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeRosa F., Guild B., Karve S., Smith L., Love K., Dorkin J.R., Kauffman K.J., Zhang J., Yahalom B., Anderson D.G., Heartlein M.W. Therapeutic efficacy in a hemophilia B model using a biosynthetic mRNA liver depot system. Gene Ther. 2016;23:699–707. doi: 10.1038/gt.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.An D., Schneller J.L., Frassetto A., Liang S., Zhu X., Park J.S., Theisen M., Hong S.J., Zhou J., Rajendran R., et al. Systemic messenger RNA therapy as a treatment for methylmalonic acidemia. Cell Rep. 2017;21:3548–3558. doi: 10.1016/j.celrep.2017.11.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang L., Berraondo P., Jerico D., Guey L.T., Sampedro A., Frassetto A., Benenato K.E., Burke K., Santamaria E., Alegre M., et al. Systemic messenger RNA as an etiological treatment for acute intermittent porphyria. Nat. Med. 2018;24:1899–1909. doi: 10.1038/s41591-018-0199-z. [DOI] [PubMed] [Google Scholar]
- 13.Schrom E., Huber M., Aneja M., Dohmen C., Emrich D., Geiger J., Hasenpusch G., Herrmann-Janson A., Kretzschmann V., Mykhailyk O., et al. Translation of angiotensin-converting enzyme 2 upon liver- and lung-targeted delivery of optimized chemically modified mRNA. Mol. Ther. Nucleic Acids. 2017;7:350–365. doi: 10.1016/j.omtn.2017.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y., Su H.H., Yang Y., Hu Y., Zhang L., Blancafort P., Huang L. Systemic delivery of modified mRNA encoding herpes simplex virus 1 thymidine kinase for targeted cancer gene therapy. Mol. Ther. 2013;21:358–367. doi: 10.1038/mt.2012.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adams D., Gonzalez-Duarte A., O'Riordan W.D., Yang C.C., Ueda M., Kristen A.V., Tournev I., Schmidt H.H., Coelho T., Berk J.L., et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018;379:11–21. doi: 10.1056/nejmoa1716153. [DOI] [PubMed] [Google Scholar]
- 16.Hassett K.J., Benenato K.E., Jacquinet E., Lee A., Woods A., Yuzhakov O., Himansu S., Deterling J., Geilich B.M., Ketova T., et al. Optimization of lipid nanoparticles for intramuscular administration of mRNA vaccines. Mol. Ther. Nucleic Acids. 2019;15:1–11. doi: 10.1016/j.omtn.2019.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baden L.R., El Sahly H.M., Essink B., Kotloff K., Frey S., Novak R., Diemert D., Spector S.A., Rouphael N., Creech C.B., et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021;384:403–416. doi: 10.1056/NEJMoa2035389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.PubChem [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; 2004-. PubChem Patent Summary for US-9738593-B2; Available from: https://pubchem.ncbi.nlm.nih.gov/patent/US-9738593-B2.
- 19.Walsh E.E., Frenck R.W., Jr., Falsey A.R., Kitchin N., Absalon J., Gurtman A., Lockhart S., Neuzil K., Mulligan M.J., Bailey R., et al. Safety and immunogenicity of two RNA-based covid-19 vaccine candidates. N. Engl. J. Med. 2020;383:2439–2450. doi: 10.1056/nejmoa2027906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davies N., Hovdal D., Edmunds N., Nordberg P., Dahlen A., Dabkowska A., Arteta M.Y., Radulescu A., Kjellman T., Hoijer A., et al. Functionalized lipid nanoparticles for subcutaneous administration of mRNA to achieve systemic exposures of a therapeutic protein. Mol. Ther. Nucleic Acids. 2021;24:369–384. doi: 10.1016/j.omtn.2021.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sabnis S., Kumarasinghe E.S., Salerno T., Mihai C., Ketova T., Senn J.J., Lynn A., Bulychev A., McFadyen I., Chan J., et al. A novel amino lipid series for mRNA delivery: improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol. Ther. 2018;26:1509–1519. doi: 10.1016/j.ymthe.2018.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khan M.A.B., Hashim M.J., King J.K., Govender R.D., Mustafa H., Al Kaabi J. Epidemiology of type 2 diabetes - global burden of disease and forecasted trends. J. Epidemiol. Glob. Health. 2019;10:107–111. doi: 10.2991/jegh.k.191028.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kliewer S.A., Mangelsdorf D.J. A dozen years of Discovery: insights into the physiology and pharmacology of FGF21. Cell Metab. 2019;29:246–253. doi: 10.1016/j.cmet.2019.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geng L., Lam K.S.L., Xu A. The therapeutic potential of FGF21 in metabolic diseases: from bench to clinic. Nat. Rev. Endocrinol. 2020;16:654–667. doi: 10.1038/s41574-020-0386-0. [DOI] [PubMed] [Google Scholar]
- 25.Minard A.Y., Tan S.X., Yang P., Fazakerley D.J., Domanova W., Parker B.L., Humphrey S.J., Jothi R., Stockli J., James D.E. mTORC1 is a major regulatory node in the FGF21 signaling network in adipocytes. Cell Rep. 2016;17:29–36. doi: 10.1016/j.celrep.2016.08.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fon Tacer K., Bookout A.L., Ding X., Kurosu H., John G.B., Wang L., Goetz R., Mohammadi M., Kuro-o M., Mangelsdorf D.J., Kliewer S.A. Research resource: comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol. Endocrinol. 2010;24:2050–2064. doi: 10.1210/me.2010-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hecht R., Li Y.S., Sun J., Belouski E., Hall M., Hager T., Yie J., Wang W., Winters D., Smith S., et al. Rationale-based engineering of a potent long-acting FGF21 analog for the treatment of type 2 diabetes. PLoS One. 2012;7:e49345. doi: 10.1371/journal.pone.0049345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaich G., Chien J.Y., Fu H., Glass L.C., Deeg M.A., Holland W.L., Kharitonenkov A., Bumol T., Schilske H.K., Moller D.E. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab. 2013;18:333–340. doi: 10.1016/j.cmet.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 29.Kleiner D.E., Brunt E.M., Van Natta M., Behling C., Contos M.J., Cummings O.W., Ferrell L.D., Liu Y.C., Torbenson M.S., Unalp-Arida A., et al. Nonalcoholic Steatohepatitis Clinical Research Network Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 30.Hansen H.H., Hansen G., Secher T., Feigh M., Veidal S.S., Fosgerau K., Jelsing J., Vrang N. In: Translational Research Methods in Diabetes, Obesity, and Nonalcoholic Fatty Liver Disease. Krentz A., Weyer C., Hompesch M., editors. Springer; 2019. Animal models of type 2 diabetes, obesity and nonalcoholic steatohepatitis – clinical translatability and applicability in preclinical drug development. [Google Scholar]
- 31.Lagassé H.D., Alexaki A., Simhadri V.L., Katagiri N.H., Jankowski W., Sauna Z.E., Kimchi-Sarfaty C. Recent advances in (therapeutic protein) drug development. F1000Res. 2017;6:113. doi: 10.12688/f1000research.9970.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pardi N., Hogan M.J., Porter F.W., Weissman D. mRNA vaccines - a new era in vaccinology. Nat. Rev. Drug Discov. 2018;17:261–279. doi: 10.1038/nrd.2017.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Administration, USFaD . Pfizer-BioNTech COVID-19 Vaccine. FDA Office of Media Affairs; 2020. FDA takes key action in fight against COVID-19 By issuing emergency use authorization for first COVID-19 vaccine. [Google Scholar]
- 34.Administration, USFaD . Moderna COVID-19 Vaccine. FDA Office of Media Affairs; 2020. FDA takes additional action in fight against COVID-19 By issuing emergency use authorization for second COVID-19 vaccine. [Google Scholar]
- 35.Agency E.M. BioNTech and Pfizer; 2020. EMA Recommends First COVID-19 Vaccine for Authorisation in the EU.https://www.ema.europa.eu/en/news/ema-recommends-first-covid-19-vaccine-authorisation-eu [Google Scholar]
- 36.Agency E.M. 2020. EMA Recommends COVID-19 Vaccine Moderna for Authorisation in the EU.https://www.ema.europa.eu/en/news/ema-recommends-covid-19-vaccine-moderna-authorisation-eu [Google Scholar]
- 37.Veiga N., Goldsmith M., Granot Y., Rosenblum D., Dammes N., Kedmi R., Ramishetti S., Peer D. Cell specific delivery of modified mRNA expressing therapeutic proteins to leukocytes. Nat. Commun. 2018;9:4493. doi: 10.1038/s41467-018-06936-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu C., Schonke M., Zhou E., Li Z., Kooijman S., Boon M.R., Larsson M., Wallenius K., Dekker N., Barlind L., et al. Pharmacological treatment with FGF21 strongly improves plasma cholesterol metabolism to reduce atherosclerosis. Cardiovasc. Res. 2021;118:489–502. doi: 10.1093/cvr/cvab076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu J., Lloyd D.J., Hale C., Stanislaus S., Chen M., Sivits G., Vonderfecht S., Hecht R., Li Y.S., Lindberg R.A., et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes. 2009;58:250–259. doi: 10.2337/db08-0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coskun T., Bina H.A., Schneider M.A., Dunbar J.D., Hu C.C., Chen Y., Moller D.E., Kharitonenkov A. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology. 2008;149:6018–6027. doi: 10.1210/en.2008-0816. [DOI] [PubMed] [Google Scholar]
- 41.Kolumam G., Chen M.Z., Tong R., Zavala-Solorio J., Kates L., van Bruggen N., Ross J., Wyatt S.K., Gandham V.D., Carano R.A., et al. Sustained Brown fat stimulation and insulin sensitization by a humanized bispecific antibody agonist for fibroblast growth factor receptor 1/βKlotho complex. EBioMedicine. 2015;2:730–743. doi: 10.1016/j.ebiom.2015.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kharitonenkov A., Shiyanova T.L., Koester A., Ford A.M., Micanovic R., Galbreath E.J., Sandusky G.E., Hammond L.J., Moyers J.S., Owens R.A., et al. FGF-21 as a novel metabolic regulator. J. Clin. Invest. 2005;115:1627–1635. doi: 10.1172/jci23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keinicke H., Sun G., Mentzel C.M.J., Fredholm M., John L.M., Andersen B., Raun K., Kjaergaard M. FGF21 regulates hepatic metabolic pathways to improve steatosis and inflammation. Endocr. Connect. 2020;9:755–768. doi: 10.1530/ec-20-0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bartesaghi S., Hallen S., Huang L., Svensson P.A., Momo R.A., Wallin S., Carlsson E.K., Forslow A., Seale P., Peng X.R. Thermogenic activity of UCP1 in human white fat-derived beige adipocytes. Mol. Endocrinol. 2015;29:130–139. doi: 10.1210/me.2014-1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carlsson L., Clarke J.C., Yen C., Gregoire F., Albery T., Billger M., Egnell A.C., Gan L.M., Jennbacken K., Johansson E., et al. Biocompatible, purified VEGF-A mRNA improves cardiac function after intracardiac injection 1 Week post-myocardial infarction in swine. Mol. Ther. Methods Clin. Dev. 2018;9:330–346. doi: 10.1016/j.omtm.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.