

Abstract
Aging-induced alterations to the blood-brain barrier (BBB) are increasingly being seen as a primary event in chronic progressive neurological disorders that lead to cognitive decline. With the goal of increasing delivery into the brain in hopes of effectively treating these diseases, a large focus has been placed on developing BBB permeable materials. However, these strategies have suffered from lack of specificity towards regions of disease progression. Here we report on the development of a nanoparticle (C1C2-NP) that targets regions of increased claudin-1 expression that reduces BBB integrity. Using dynamic contrast enhanced magnetic resonance imaging (DCE-MRI) we find that C1C2-NP accumulation and retention is significantly increased in brains from 12-month-old mice as compared to non-targeted NPs and brains from 2-month-old mice. Furthermore, we find C1C2-NP accumulation in brain endothelial cells with high claudin-1 expression, suggesting target-specific binding of the NPs, which was validated through fluorescence imaging, in vitro testing, and biophysical analyses. Our results further suggest a role of claudin-1 in reducing BBB integrity during aging and show altered expression of claudin-1 can be actively targeted with NPs. These findings could help develop strategies for longitudinal monitoring of tight junction protein expression changes during aging as well as be used as a delivery strategy for site-specific delivery of therapeutics at these early stages of disease development.
Keywords: BBB, NPs, TJs, MRI, fluorescence imaging, C1C2
The blood-brain barrier (BBB) plays a crucial role in the central nervous system (CNS) by creating a barrier between the neural tissue and the blood to prevent contamination from foreign substances. Aging is linked with decreased BBB integrity as well as functional impairment of transporters and can aggravate BBB responses to any CNS injury and systemic inflammatory stimuli1–2 across multiple different brain regions including the cortex,3 hippocampus,3–4 and corpus callosum.5 This is a result of age-induced alterations in gene expression, mitochondrial dysfunction, or abnormal protein accumulation in the CNS.6–7 Moreover, these alterations have been implicated in promoting neurodegeneration, declined cognitive function, reduced cerebral blood flow, and vasculopathies;8–10 thus, BBB disruption can initiate during normal aging and lead to mild cognitive impairment and progression to Alzheimer’s Disease and other dementias.4, 11 In fact, BBB breakdown has recently been proposed as an early biomarker of neurocognitive decline.
The function of the BBB is controlled through a complex and not completely understood interaction with the neurovascular unit (NVU), but BBB function relies on adequate assembly of tight junction (TJ) proteins on the surface of brain endothelial cells.12 These transmembrane proteins play a vital role in the formation and maintenance of TJ function.12–15 Among TJ transmembrane protein members, claudin-5 and occludin constitute TJs at the BBB.13 Claudins and occludin have four membrane-spanning helices and two extracellular segments that facilitate interactions between other TJ proteins to ultimately form TJ barriers.14–15 TJ protein expression is altered during aging and leads to the observed age-related leakiness of the BBB.16–18 Indeed, claudin-5 expression decreases during aging and disease.19 In addition to claudin-5, brain endothelial cell surface expression of claudin-1 has been found to increase in response to injury, resulting in increased leakiness of the BBB.20 Furthermore, claudin-1 is transcriptionally regulated by Sirt1,21 where age-associated reductions in expression are observed,22 which would be expected to increase expression of claudin-1, and may play a causative role in BBB hyperpermeability in aging and disease.23–25 In addition, inflammatory stimuli such as those observed during normal aging,26–27 have been shown to increase expression of claudin-1 in astrocytes, which play a major protective role in the BBB.28 This increased presentation of claudin-1 may impair claudin-5’s BBB function through disruption to claudin-5 interactions with other TJ proteins. Therefore, claudin-1 represents a strong cell surface receptor candidate for specific targeting of the early stages of BBB breakdown prior to neurocognitive decline.
Nanoparticles (NPs) offer the advantage of multifunctionality whereby they can be engineered to display a targeting agent on their surface to promote binding to specific cells, provide contrast in various imaging modalities, and carry a therapeutic payload to increase target engagement. There has been significant effort placed on developing NPs that can cross the BBB to improve brain delivery for various neurological disorders.29–31 Strategies typically involve passive strategies that rely on increased leakiness of the BBB such as with a brain tumor32 or following trauma,33–42 or through active strategies with the so-called “trojan horse” method that exploits transporters expressed on brain endothelial cells to hijack the native transport mechanism into the brain.43–47 These include targets such as the transferrin transporters, amino acid transporters, glucose transporters, etc. However, this strategy suffers from the lack of specificity to target diseased brain regions as these transporters are typically ubiquitously expressed. Furthermore, the transporters can become downregulated during the disease process thus reducing uptake in regions where delivery is desired. Therefore, our goal has been to identify brain endothelial cell targets that become overexpressed on the luminal side of the BBB to provide active targeting at the earliest stages of the disease process specifically where these alterations in the brain are occurring.
Here we report our development of a claudin-1 targeted NP that directly binds purified claudin-1 and has the ability to accumulate and be retained in regions of the brain vasculature with aging-induced increases in claudin-1 expression. We utilize a Gd NP core that provides contrast in magnetic resonance imaging (MRI) to observe NP accumulation and retention in the brain as well as fluorophore modification for fluorescence imaging of NP binding to regions of high claudin-1 expression. We find that 12-month-old mice (corresponding to middle age in humans) have higher claudin-1 expression in brain endothelial cells than their 2-month-old (corresponding to adolescence in humans) counterparts, and that this higher claudin-1 expression correlates with higher NP accumulation and retention as measured by MRI.
RESULTS AND DISCUSSION
Nanoparticle synthesis and characterization
BBB dysfunction has been shown with aging and age-related neurodegenerative conditions such as Alzheimer’s disease.4 This BBB dysfunction represents a possible target for delivery to the aging brain using multifunctional NPs. Here we utilized ultrasmall 3.5 nm Gd NPs labeled with AF647 to provide multimodal imaging through MRI and fluorescence. PEG diacid was used to coat the synthesized NPs. The addition of PEG to NP surfaces can reduce clearance by the reticuloendothelial system (RES) and increase circulation time.48–50 Not only can the PEG coating prevent aggregation, but it can also increase solubility in serum because of repeating the hydrophilic ethylene glycol units.51–52 The high-resolution TEM images of PEG diacid coated Gd NP cores revealed the average NP diameter was around 3.5 nm verifying their ultrasmall size (Figure 1A). From DLS measurements, the average hydrodynamic size of the nanoparticle was 10.5 nm (Figure 1B), and the zeta potential was −16.1 mV (Figure 1C). The PEG diacid coating was confirmed using FTIR. We compared PEG diacid coated Gd NPs with free PEG diacid and observed that the C = O stretch was red-shifted by ~ 165 cm−1 from that (= 1740 cm−1) of a free PEG diacid (Figure 1D), confirming the attachment of -COOH group to the NPs as commonly observed in metal oxide NPs coated with -COOH group containing ligands.53–54
Figure 1.
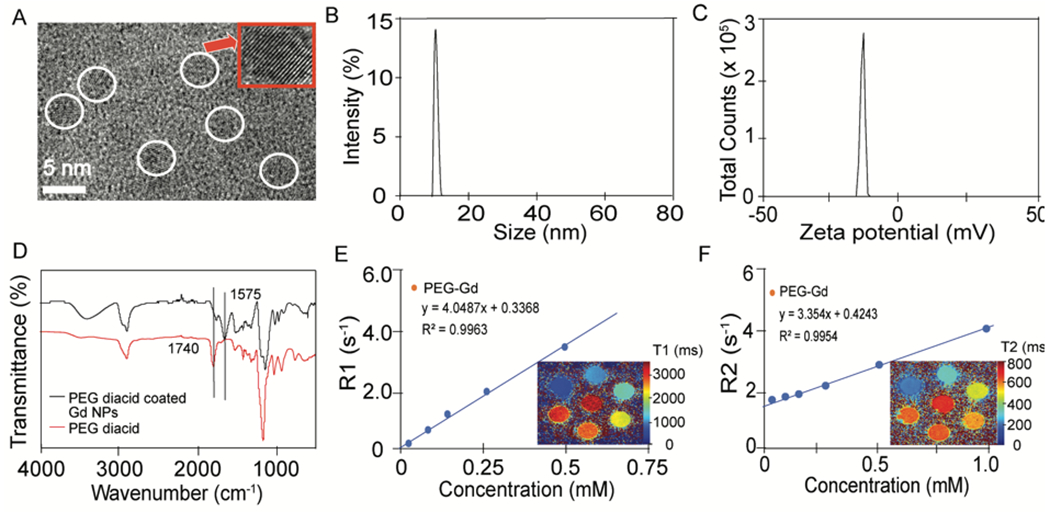
NP characterizations. a) HRTEM images of the PEG diacid coated ultrasmall Gd nanoparticles. The corresponding circle shows the diameter of the nanoparticle, which is 3.5 nm. b) From DLS, the average size of the nanoparticle is 10.5 nm. c) The zeta potential value is −16.1 mV for Gd nanoparticles. d) FT-IR absorption spectra of (I) free PEG diacid (red line), (II) PEG diacid coated ultrasmall Gd nanoparticles (black line). e) R1 relaxivity and T1 map images, f) R2, and T2 map images of Gd nanoparticles.
To determine the MRI enhancing properties of the NPs, both R1 and R2 were measured at 9.4 T and plotted as a function of Gd concentration. Longitudinal (r1) water proton relaxivities were estimated from the corresponding slopes, giving a value of 4.05 s−1mM−1 and transverse (r2) water proton relaxivities were estimated from the corresponding slopes, giving a value of 3.35 s−1mM−1 for PEG-Gd NPs. T1 and T2 map images (Figure 1E and 1F) show apparent dose-dependent contrast enhancement.
Peptides were conjugated to PEG the surface of the NPs through EDC-NHS chemistry. Peptide density on the surface of the NP was between 15–19 peptides per NP (Table 1) depending on the peptides, indicating similar peptide densities of around 2.02, 2.14, 2.56 and 2.02 nm2/ peptide, respectively. The hydrodynamic sizes and zeta potentials of all peptide conjugated NPs were similar (Table 1) reducing the chances of confounding cell binding and uptake effects caused by different physicochemical properties of the NPs.55–57
Table 1.
Properties of NPs and peptides
| Name | Sequence of peptides | Hydrodynamic size (nm) | No. of peptides Per NP | Zeta Potential, mV (with NPs) |
|---|---|---|---|---|
| Cldn146-NP | QEFYDPLTPINARYE | 13.94±0.08 | 19 | 1.62±0.13 |
| Cldn53-NP | SCVSQSTGQIQCKVFDSLLNLNSTLQAT | 13.8±0.11 | 18 | 2.24±0.18 |
| Ocldn207-NP | GSQIYMICNQFYTPGGTG | 14.23±0.11 | 15 | 3.65±0.19 |
| C1C2-NP | SSVSQSTGQIQSKVDSLLNLNSTQATR | 14.5±0.13 | 19 | 7.81±0.38 |
MRI assessment of brain accumulation and retention
BBB permeability is typically investigated utilizing small molecule Gd-based contrast agents for DCE-MRI. We have recently extended the use of this DCE-MRI method for use with contrast-enhancing NPs to compare the permeabilities of different NPs.33 DCE-MRI represents a robust method for assessing NP accumulation and retention in the brain as compared to more static, non-invasive methods such as concentration mapping or fluorescence imaging at single time points, which cannot distinguish between NP in the blood or that which has been transferred to the tissue compartment. While DCE-MRI has typically been used to measure permeability of tissues, we have recently reported a method for comparing the permeabilities of NPs within a tissue using MRI to identify the permeability coefficient Ktrans.33–34 The spatial resolution of this method does not enable direct observation of NP escaping from the vasculature into the parenchyma, but the observation of accumulation over time in a region may suggest this extravasation is occurring. NPs may, alternatively, be retained on the luminal side of the BBB, but in that case, their accumulation may suggest engagement with cellular receptors such that local concentrations at the BBB are higher than those in the general circulation. The effect of peptide targeting on NP accumulation and retention in the brains of 12-month-old mice was assessed in brain regions known to be affected by BBB breakdown during aging (i.e., corpus callosum,5 hippocampus,4 hypothalamus,58 cortex59–60) and in regions thought to be minimally affected by aging (i.e., muscle). In the corpus callosum (Figure 2), NPs targeted with the C1C2 peptide against claudin-1 (C1C2-NPs) showed a significant increase in Ktrans in 12-month-old mice as compared to other peptide modified and control NPs as well as compared to 2-month-old mice suggesting preferential binding to upregulated surface claudin-1 induced by aging. Similarly in the hippocampus (Figure 3), C1C2-NPs had a trending increase in Ktrans that was not significant because of high variability of Ktrans values in these 12-month-old mice that could be a result of differential claudin-1 expression between mice. A summary of Ktrans results for each brain region is shown in Table 2. In the corpus callosum of 12-month-old mice, the mean Ktrans value for control NPs was 0.0017 min−1, similar to that observed in 12–16 month old mice using Magnevist as the contrast agent.5 The mean Ktrans value for C1C2-NPs was significantly greater at 0.0058 min−1, suggesting active binding to increased claudin-1 expression. In the hippocampus of 12-month-old mice, the mean Ktrans value for control NPs was 0.0018 min−1, similar to a Ktrans of ~0.001 min−1 observed for middle aged humans of around 40 years of age using MultiHance as the contrast agent.4 For C1C2-NPs, a Ktrans value of 0.0040 min−1 was observed in the hippocampus. The highest Ktrans value in the cortex and hypothalamus of 2-month-old or/and 12-month-old mice was 0.0148 min−1 and 0.0102 min−1 for C1C2-NPs (Supplementary Figures 1–2). On the other hand, the highest Ktrans value in the muscle was observed for control mice as 0.0079 min−1 (Supplementary Figure 3).
Figure 2.
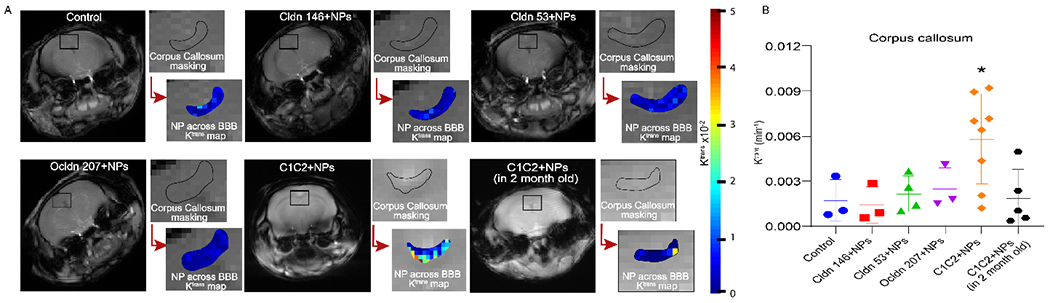
NP accumulation in the corpus callosum. a) Ktrans maps showing NPs uptake in corpus callosum, b) Mean Ktrans values of different peptide conjugated NPs in corpus callosum. Data are presented as an average of mice (3-8 mice) experiments with standard deviations, *p < 0.05. Significance as compared to all other treatments was determined using two-way ANOVA with Dunnett’s post-hoc test.
Figure 3.
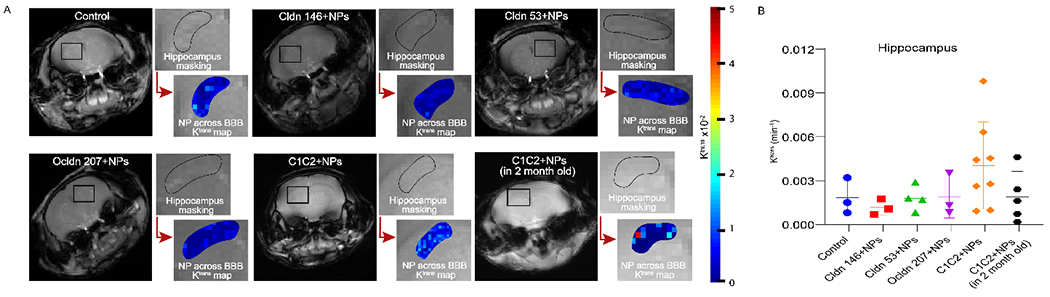
NP accumulation in the hippocampus. a) Ktrans maps showing NPs uptake in hippocampus, b) Mean Ktrans values of different peptide conjugated NPs in hippocampus. Data are presented as an average of all mice (3-8 mice). Significance as compared to all other treatments was determined using two-way ANOVA with Dunnett’s post-hoc test.
Table 2.
Brain region specific accumulation and retention of NPs targeted with various peptides as measured using the permeability coefficient, Ktrans (min−1), with MRI. CC, corpus callosum; HC, hippocampus; CTX, cortex; HT, hypothalamus.
| NP Type | |||||||
|---|---|---|---|---|---|---|---|
| Control (x 10−4) | Cldn146-NP (x 10−4) | Cldn53-NP (x 10−4) | Ocldn207-NP (x 10−4) | C1C2-NP (x 10−4) | C1C2-NP (in 2-month-old mice) (x 10−4) | ||
| Brain Region | CC | 17±14 | 14±12 | 22±12 | 25±14 | 58±30 | 19±19 |
| HC | 18±12 | 12±5 | 18±85 | 19±14 | 40±30 | 19±17 | |
| CTX | 16±10 | 3±3 | 26±9 | 20±21 | 148±67 | 22±15 | |
| HT | 12±15 | 8±2 | 26±13 | 37±13 | 102±56 | 26±17 | |
| Muscle | 79±22 | 16±13 | 9±13 | 33±7 | 57±22 | 28±29 | |
Specific binding and affinity of C1C2-NP to claudin-1
To determine whether C1C2-NPs bind claudin-1 specifically, mouse brain microvascular endothelial (bEnd.3) cells were exposed to the AF-647 modified NPs in vitro. Control cells showed no binding to C1C2-NPs as expected as claudin-1 expression is low under normal culture (Figure 4A–C). We found claudin-1 expression was induced by exposure to ethanol (the mechanism of which needs further study), and C1C2-NPs showed much higher binding to cells, suggesting specific binding to mouse claudin-1. To determine if C1C2-NPs could also bind to human claudin-1, bEnd.3 cells were transfected to express human claudin-1 and exposed to C1C2-NPs. High NP binding was observed in this condition suggesting C1C2-NPs can bind to both human and mouse claudin-1.
Figure 4.
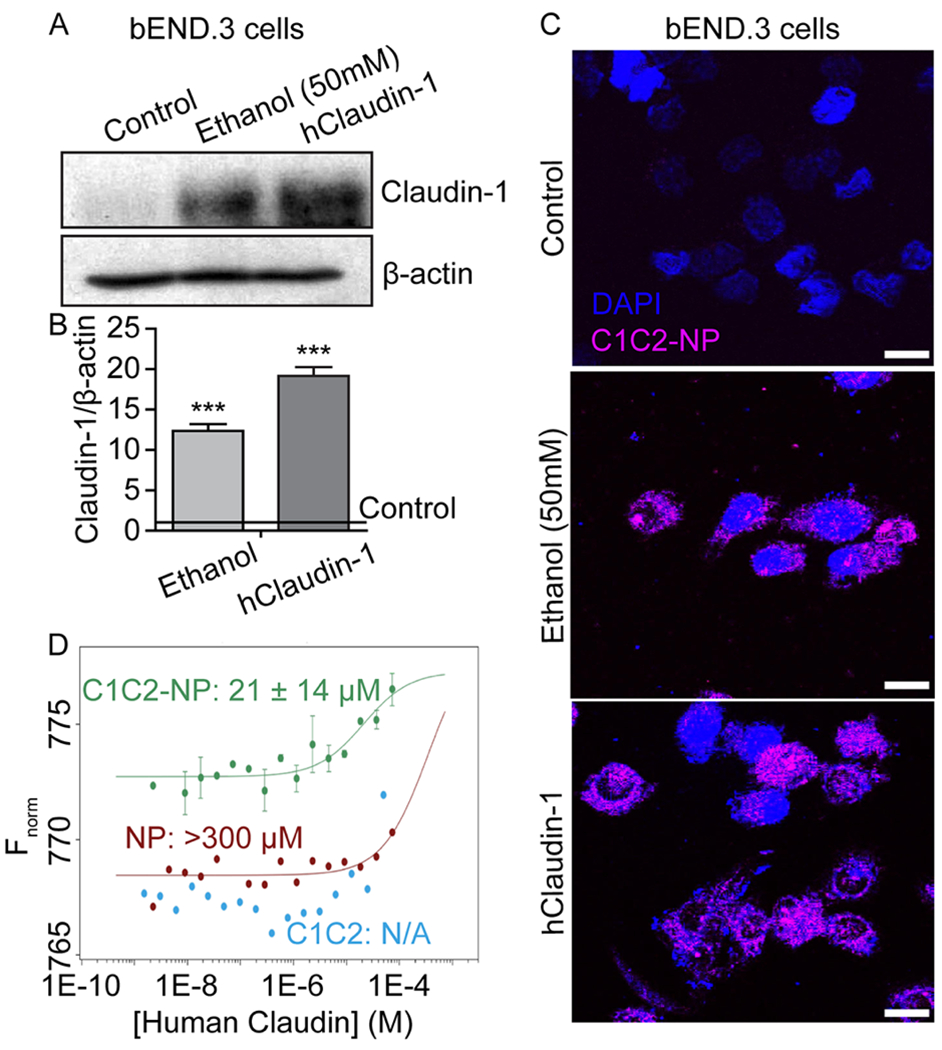
C1C2-NP specifically binds to both mouse and human Claudin-1. A-C) In vitro immunofluorescence of NPs binding claudin-1 in mouse brain endothelial (bEnd.3) cells. A) Western blot analysis of claudin-1 in bEnd.3 cells and those induced to express claudin-1 through ethanol exposure or exogenous expression through plasmid transfection. B) Quantification of claudin-1 expression relative to β-actin. *** indicates a statistical difference (p < 0.0001) in claudin-1 expression as compared to control cells. C) C1C2-NP (magenta) binding to ethanol induced mouse claudin-1 and exogenously expressed human claudin-1. Scale bar represents 10 μm. D) Dissociation constants (KD) for C1C2, C1C2-NP, and NP complex formation with human claudin-1. Binding curves for the complex formation between human claudin-1 and C1C2-NP (green), NP (red), and C1C2 peptide (blue) using microscale thermophoresis. The concentration of human claudin 1 was varied from 74 μM – 0.2 nM in all three experiments. The concentrations of C1C2-NP, NP, and peptide were fixed at 70 nM, 2 μM and 1 μM, respectively. The buffer used for the measurements contained 10 mM Tris HCl, pH 8.0, 100 mM NaCl, 4% glycerol and 0.04% b-DDM. The experimental data points with human claudin-1 and C1C2-NP are reported as standard deviations from two independent measurements. The estimated value of the dissociation constant is 21 ± 14 μM. Human claudin-1 does not show significant binding to the non-targeted NP as the dissociation constant is very high >300 μM. The data with the peptide alone does not fit at all indicating little binding with free peptide in solution.
To assess the binding affinity of C1C2-NPs to human claudin-1, we labeled C1C2-NPs with AF647 and quantified binding to recombinant human claudin-1 using MST. The dissociation constant (KD) of C1C2-NPs to human claudin-1 was 21 ± 14 μM (Figure 4D). Measuring the binding of empty NPs to human claudin-1 we found a KD of >300 μM while for C1C2 peptides alone no binding to human claudin-1 was detected. The lack of C1C2 binding to human claudin-1 observed in this assay could be caused by the conformational freedom of the peptide free in buffer, which could disrupt specific C1C2-claudin-1 interactions. Anchoring the peptide to the surface of a NP would reduce the conformational freedom of the peptide and increase specific C1C2-claudin-1 interactions. In addition, the lower KD of C1C2-NPs could be caused by a multivalent effect where multiple peptides on the surface of a NP are involved in claudin-1 interactions. In order to rule out non-specific binding of the AF-C1C2-NP to the β-DDM micelle, the concentration of β-DDM was varied and the concentration of AF-C1C2-NP was fixed at 1 μM. No significant binding could be observed within the range of β-DDM concentration used for the measurement with human claudin 1 (data not shown). The specificity of the binding of AF647-C1C2-NP was verified by measuring its binding to the human claudins −3, −4 and −9. Human claudin-3 does not show any binding. While there is evidence for claudin-1/claudin-3 cis compatibility as determined by colocalization in vivo using fluorescence microscopy,61 our MST results indicate C1C2-NP does not bind to claudin-3 in vitro. This difference may be a result of weakened interactions in vitro when there is an absence of larger claudin assemblies, or a result of the only 69% sequential similarity between claudin-1 and claudin-3 within the C1C2 binding region. The data points for human claudins 4 and 9 could not be fit to the sigmoidal dose response curve suggesting that the binding of AF647-C1C2-NP to human claudins −4 and −9, is quite weak (Supplementary Figure 4A, B). However, both human claudins −4 and −9 bind to AF647-NP (Supplementary Figure 4C, D) and this does not represent non-specific binding of the nanoparticles to the detergent micelles. These data indicate that the presence of C1C2 peptide makes the nanoparticle-peptide conjugate specific for binding to human claudin-1 as well as mouse claudin-1 from our in vitro experiment.
NPs were observed in regions of high claudin-1 expression
As we found higher Ktrans value of NPs in the 12-month-old as compared to the 2-month-old mice, we performed immunofluorescence staining of claudin-1 and CD31 to observe the co-expression of claudin-1 in brain endothelial cells and the localization of C1C2-NPs with respect to these regions (Figure 5). We found colocalization of claudin-1, CD31, and nanoparticles in the 12-month-old mice, but this was not observed in the the 2-month-old mice. To support this observation, we quantify the colocalization of CD31 and claudin-1 using the JACoP extension in ImageJ (Supplementary Figure 5). Mander’s colocalization coefficient showed a significant increase of CD31 and claudin-1 colocalization in the brains of 12-month-old mice as compared to 2-month-old mice. This strongly supports the idea that the C1C2-NPs are able to bind to regions of increased claudin-1 on the aging BBB. Furthermore, ICP-MS analysis of peripheral organs 1 hr post-injection showed increased C1C2-NP accumulation in the kidney (Supplementary Figure 6), further supporting the claim of C1C2-NP binding to claudin-1. In addition, claudin-1, which is expressed on Bowman’s capsule in the kidney,62 has higher expression levels during aging,18 and we observed higher C1C2-NP accumulation in kidneys from 12-month-old mice as compared to 2-month-old mice.
Figure 5.
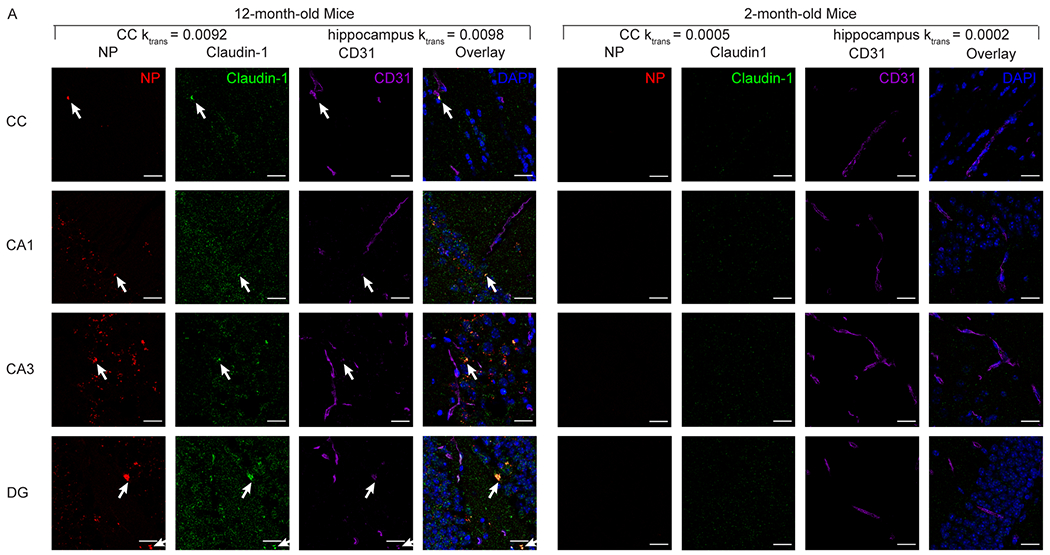
Representative images of in vivo C1C2-NP (red) accumulation in the corpus callosum and hippocampus colocalize with the immunofluorescence staining for claudin-1 (green) and CD31 (purple). Blue DAPI staining indicates nuclei. Scale bar is 20 μm. NP accumulation and high claudin-1 co-localization with CD31 can be observed in the 12-month-old mice, but not in the 2-month-old mice.
The significantly higher Ktrans observed in the corpus callosum using C1C2 NPs compared with the control non-targeted NPs (Figure 2) indicates significantly higher accumulation and retention when claudin-1 is targeted with the C1C2 peptide as compared to other peptides and peptides against occludin. This result is in accordance with previous work that found following BBB disruption occludin and claudin-5 in the brain endothelium are no longer present and instead moves away and colocalizes with astrocytes.63 There are trending differences in the hippocampus but without significance (Figure 3), but NP accumulation in the hippocampus is observed with higher claudin-1 expression on brain endothelial cells in 12-month-old mice (Figure 5, Supplementary Figure 5C and D), which might suggest higher biological variability in claudin-1 expression in the hippocampus as compared to the corpus callosum. Our findings of limited accumulation in 2-month-old mice support the previous investigation from Montagne et al.4 where BBB leakiness is lower in younger adults. The differences between corpus callosum and hippocampus may be reflective of characteristic differences between white matter and grey matter regions. Previous work from Montagne et al. suggests higher permeability in white matter regions of both humans and mice based on Ktrans quantification with the small molecule Magnevist and deposits of extravascular fibrinogen and hemosiderin.4–5 Grey matter however has shown an age-dependent increase in permeability in humans that was absent in subcortical white matter tracts.4 These previous results, however, were based on the non-targeted, small-molecule Magnevist. The results presented here suggest the importance of claudin-1 in determining uptake of C1C2 NPs, which may overcome the effects that governed permeation in the small-molecule case. The high Ktrans values in the cortex and hypothalamus with C1C2 NPs, which were unremarkable with the control NPs, may be a result of the claudin-1 interactions facilitated by the targeting peptide, though further work should be carried out to investigate regional permeability increases based on peptide targeting.
The presence of increased claudin-1 on the BBB is correlated with reduced BBB function although the specific role of claudin-1 in the normal function of the BBB is still under investigation. The question of whether increased claudin-1 expression in the brain endothelium is utilized in BBB repair following injury or disease or if increased claudin-1 expression itself leads to increased BBB leakiness requires further investigation. Claudin-1 may be required for embryonic development of the BBB and replaced by claudin-5 upon complete maturation of the BBB as claudin-1 is involved in neural tube closure in chick embryos.64 However, Tran et al., found that β-catenin activates the claudin-1 promoter and that inducible knockout of β-catenin in mice resulted in reduced claudin-1 expression and increased BBB leakiness even though claudin-5 mRNA levels were not affected,65 which suggests claudin-1 may have a role in maintaining BBB integrity. Nevertheless, these results may be affected by other mechanisms by which β-catenin helps maintain BBB integrity as Leibner et al., showed that claudin-1 expression is not under control of Wnt/β-catenin signaling.66 This idea is bolstered by Sladojevic et al. that found following injury to the BBB caused by stroke, increased claudin-1 expression correlated with reduced repair and increased leakiness of the BBB, and that decreasing cell surface expression of claudin-1 resulted in improved BBB repair.20 The detrimental effects of claudin-1 were thought to be caused by claudin-1-zona occludin and claudin-1-claudin-5 interactions that reduced claudin-5-mediated TJ integrity. Conversely, Pfeiffer et al. found that increased claudin-1 expression correlated with improved BBB integrity following autoimmune encephalomyelitis injury.67 It is thought that claudin-1 is stored in intracellular microvessels that quickly traffic to the cell membrane following injury to promote sealing of the BBB. This claudin-1 would then eventually be replaced by de novo synthesis of claudin-5 to complete regeneration of the BBB. Combined, this suggests claudin-1 may act as an initial scaffold for subsequent complete sealing of the TJ by claudin-5 in the formation of the BBB. In aging or disease, however, chronic cell surface expression of claudin-1 may be an early event in BBB disruption and chronic progression of BBB dysfunction by chronically inhibiting normal claudin-5 interactions that would completely seal the BBB. Indeed, our Western blot and Mander’s colocalization coefficient results (Supplementary Figure 5) support these previous findings. We did not observe a difference in the protein levels of claudin-5 and claudin-1 between 2-month-old and 12-month-old mice. We did, however, observe increased claudin-1 colocalization with CD31 in the brains from 12-month-old mice as compared to 2-month-old mice. Claudin-11 also plays a major role in maintaining BBB integrity and is downregulated in multiple sclerosis,68 yet it is unclear if claudin-1 and claudin-11 directly interact. Our results in 2-month-old and 12-month-old mice (Figure 5, Supplementary Figure 5C and D) suggest abnormal claudin-1 cell surface expression at the BBB is an age-related event that can be actively targeted by NPs both as a tool to study age related BBB dysfunction and as a delivery vehicle for site specific delivery of therapeutics.
We showed attachment of C1C2 to NPs could increase accumulation specifically in brains with increased C1C2 expression in endothelial cells, but also observed accumulation in other regions of the body where claudin-1 plays a major role as a tight junction protein (Supplementary Figure 6), which further supports the utility in C1C2-NPs in targeting claudin-1. However, future studies utilizing C1C2-NPs for therapeutic delivery must take into account possible effects of delivered therapeutics into these organs, especially those where claudin-1 expression is increased during aging,18 such as Bowman’s capsule in the kidney62 as we observed higher C1C2-NP accumulation in the kidney in 12-month-old as compared to 2-month-old mice (Supplementary Figure 6).
While our current study does not look at the downstream effects of C1C2-NP binding to the BBB in 12-month-old mice, C1C2 peptide has been shown to promote resealing of the BBB.20 C1C2 binds to the first extracellular loop of claudin-1, which weakens trans- and cis-claudin-1 interactions and reduces claudin-1/ZO-1 interactions. Similarly, we showed C1C2-NP actively bind to both mouse and human claudin-1 in brain endothelial cells induced to express claudin-1 through ethanol exposure or exogenously express human claudin-1 through plasmid transfection (Figure 4). Furthermore, the multivalent effect of multiple C1C2 peptides on the surface of the C1C2-NP resulted in a high binding affinity to claudin-1 with a dissociation constant of 21 μM. Previous reports have indicated that the permeability of the tight junctions in Caco II and MDCK II cell lines can be changed using C1C2 peptide at ~200 μM.20, 69 This corresponds to saturating concentrations of the peptide with respect to the Kd. Therefore, both the multivalency effect as well as the anchoring of the C1C2 peptide to the surface of a solid NP may help increase the binding affinity towards claudin-1.
Therefore, since claudin-1/claudin-5 are considered incompatible,70 reducing these claudin-1 interactions at the cell surface may allow the more robust claudin-5 interactions to predominate for tighter BBB sealing. NP-bound C1C2 may further promote claudin-1 internalization from the increased wrapping energy induced by the NP core.71–73 This could then lead to increased cytosolic degradation of claudin-1.69 Thus, specific delivery into the endothelial cells with abnormal TJ protein expression would make for an ideal treatment strategy if the NPs were modified to also deliver a therapeutic that was shown to normalize TJ protein expression. These conjectures are highly deserving of future study in the role claudin-1 plays in the early breakdown of the BBB as well the use of C1C2-NP as a tool to study and interact with the changes. The fate of NP after binding to claudin-1 (e.g., internalized, transcytosed, surface bound, etc.) will be an important consideration in future utility of our reported delivery strategy. Our results strongly support claudin-1-specific binding of the C1C2-NPs with higher NP binding in mice with higher claudin-1 colocalizion with brain endothelial cells (Figure 5).
CONCLUSIONS
BBB integrity is central to maintaining brain health and aging-induced alterations in TJ protein expression can lead to chronic leakiness of the BBB, which is directly correlated with cognitive impairment. With the eventual goal of establishing a targeting method for promoting NP delivery specifically to regions of the brain with altered function, we developed a claudin-1 targeted NP (C1C2-NP) to target this TJ protein that appears to be involved in the chronic impairment of BBB integrity. We find that C1C2-NP has high accumulation and retention in brain vasculature in aged mice. Furthermore, C1C2-NP accumulation and retention are observed in regions of high claudin-1 expression on the brain endothelium. Overall, our findings support the idea of increased claudin-1 expression in brain endothelial cells during normal aging and that these regions of altered expression can be targeted with a NP. Therefore, these C1C2-NPs offer utility as a tool to monitor alterations in TJ protein expression that may cause BBB leakiness through non-invasive MR imaging as well as a targeted delivery vehicle to improve site-specific target engagement of delivered therapeutics.
METHODS
Materials.
Gadolinium chloride hydrate (GdCl3·xH2O, 99.9%), europium (III) nitrate hydrate (Eu(NO3)3·5H2O, 99.9%), triethylene glycol (TEG, 99%), sodium hydroxide (NaOH), Poly(ethylene glycol) diacid (Mn 600), ethyl-3-(3-dimethylaminopropyl)- carbodiimide hydrochloride (EDC, ≥ 98%), and N-hydroxysuccinimide (NHS, 98%), molecular weight cutoff dialysis membranes (Flot-A-Lyzer, 20 kDa) were purchased from Sigma-Aldrich. Alexa Fluor 647 cadaverine (AF647, Mn~ 1000) was purchased from Thermo Fisher Scientific. Cldn1-146,74 cldn1-53,74 occludin-207,75 and C1C220 were purchased from Genscript Corporation, and they are all amidated at the C-terminal and acetylated at the N-terminal. Deionized water in the experiments was obtained by using a Millipore water purification system. All other chemicals and solvents used in this work were high-performance liquid chromatography (HPLC)-grade.
Synthesis of Gd NPs.
1 mmol of GdCl3.xH2O was added into 30 mL of triethylene glycol containing 100 mL three-necked flask. The mixture was heated to 80°C and magnetically stirred until the precursors were completely dissolved in the solvent. Then, 3 mmol NaOH was added and continued stirring for 4 h at 180°C. To coat the hydrophobic NPs, 2 mmol PEG-diacid was added and continued the reaction with stirring for 12 h at 150°C. After completely cooling, the synthesized nanoparticles were washed 3 times using deionized water.
Conjugation of NPs.
To conjugate NPs to the peptide, 1 mL of 0.1 mg/mL NPs were taken in 0.9 mL PBS. To the NPs 4 mg of EDC and 2 mg of NHS were added in a stepwise fashion with continuous stirring. Peptide (0.1 mg/mL) was then added and continuously stirred for 2 h. A float-a-lyzer dialysis kit was used to remove unconjugated peptides. The whole experiment was conducted in the dark.
To make the NPs fluorescent for microscale thermophoresis measurements, AF647 was attached to the Gd NPs using the EDC-NHS coupling reaction. Briefly, 0.1 mL of 1 mg/mL Gd NPs were taken into 0.9 mL PBS. Then 2 mg of EDC and 1 mg of NHS were added in a stepwise fashion with continuous stirring. Alexa Fluor 647 (0.1 mg/mL) was then added, and the mixture was continuously stirred for 2 h. Unconjugated fluorophore was removed with a Float-a-lyzer dialysis kit. The whole experiment was conducted in the dark.
Characterization.
A high voltage transmission electron microscope (TEM) (Tecnai Osiris TM, 200 kV) was used to measure particle diameters of PEG diacid coated Gd NPs. A copper grid (PELCO mesh size 400, TED PELLA, INC.) covered with an amorphous carbon membrane was placed onto a filter paper. Then, a sample solution diluted in triply distilled water was dropped over the copper grid by using a micropipette (Eppendorf, 2 - 20 μL). Dynamic light scattering (DLS) studies of the NPs were conducted using a Malvern Instruments Zetasizer Nano series instrument. Solutions of the NPs were prepared in DPBS (pH 7.4) at a concentration of 0.05 mM. The resulting solutions were filtered with 0.22 μm filters before the measurement. The NP concentration was determined by using an inductively coupled plasma mass spectrometer (ICPMS) (Agilent 7500 cx). To determine this, ~ 0.5 mL of the NP solution was taken out and treated with HNO3 to dissolve nanoparticles in the solution completely. A Fourier transform-infrared (FT-IR) (Nicolet AVATAR 380 FT-IR) was used to verify the surface coating. To record the FT-IR absorption spectrum (400–4000 cm−1), the powder sample was prepared. Peptide attachment to NPs was confirmed using BCA assay. Attaching efficiency was confirmed using the equation, Conjugation Efficiency of peptide (%) = Amount of peptide in NPs/ Initial amount of peptide x 100.
To calculate the number of peptides per NP we determined the ratio of molarities of peptides to NP cores. Molarity of the peptides were determined from the conjugation efficiency and molecular weight of the peptides (cldn146, cldn53, ocldn207 and C1C2 is 1897.06, 3027.4, 1978.22 and 2891.12 g/mol, respectively). To calculate the molarity of the NP, we calculated the mass of a NP where m = ρ x V; where ρ is the density of the NP (7.4 g/cm3 for Gd oxide) and V is the calculated volume of the NP (V= 4/3πr3, where r is the radius of the NP). The number of NP in colloidal solution is N = C/m, where C is the concentration of metal in NP solution (0.08 mg/mL) measured by ICP-MS and m is the mass of a NP calculated above. The molarity of NP is MNP = N/V x 1/NA, where N is the number of particles calculated before, V is the volume of NP solution and NA is Avogadro’s number.
R1 and R2 Relaxivity and R1 and R2 Map Image Measurements.
Both R1 and R2 map images, as well as both T1 and T2 relaxation times, were measured by using a 9.4 T MRI instrument (Varian 9.4 T) equipped with a 4 cm Millipede RF imaging probe with triple-axis gradients (100 G/cm max). A series of five aqueous solutions of different concentrations (1.0, 0.5, 0.25, 0.125, 0.0625, and 0.03125 mM Gd) were prepared by diluting each MRI solution with PBS. Then, both map images and relaxation times were measured by using these solutions. The R1 and R2 relaxivities were then estimated from the slopes in the plots of 1/T1 and 1/T2 versus NPs concentration, respectively. The measurement parameters for the fast spin-echo T1 mapping sequence were as follows: the external MR field (H) = 9.4 T, the temperature = 22ºC, the number of acquisition (NEX) = 1, the field of view (FOV) = 25×25 mm2, the matrix size = 128×128 voxels, echo train length = 16, echo spacing = 8.1 ms, slice thickness = 2 mm, seven different repetition times (TRs) were used in linear increments from 200-2000 ms, and the echo time (TE) = 32.42 ms. Signal was fit to the following equation using MATLAB to find T1:
Where, S is the signal for a given voxel, and S0 is the signal of that voxel at saturation. T2 mapping was carried out using a multi-echo scan with the same parameters as the T1 scan with the following exceptions: the number of echoes = 10, 10 TEs linearly spaced from 10-100 ms, and TR = 3000 ms. T2 mapping was performed using Osirix and linear fitting followed in MATLAB.
Animals.
Both male and female 2-month-old C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME) and 12-month-old C57BL/6J and CX3CR1-GFP (Jackson Laboratory, Bar Harbor, ME) from a C57BL/6J background mice were used. Mice were housed in a 12 h/12 h day/night cycle with ad libitum access to standard mouse chow and water. All animal work was approved by and performed under the guidance of the University of Nebraska-Lincoln IACUC. The animal experiments were run in two separate experiments using two independent preparations of C1C2-NPs. An initial experiment with n = 3–4 mice in each group (control, Cldn146, Cldn53, Ocldn, and C1C2 all in 12 month old mice) was followed by a second experiment with n = 4–5 mice in each group (C1C2 in 12 month old mice and 2 month old mice) to verify our results.
Ktrans MRI.
In vivo NP assessment consisted of dynamic contrast-enhanced (DCE)-MRI using a 9.4T MRI system (Varian) to compare targeting agent effect on the uptake as previously described.34 Briefly, mice were induced with about 2% isoflurane gas and maintained at a concentration sufficient to achieve 50 to 80 breaths per minute. Baseline T1 maps were generated using a gradient-echo sequence and the variable flip angle method with two angles, 10° and 30°.76 Mice were injected with 100 μL of a 0.5mM NP solution via tail vein catheter followed by 100 μL PBS to flush all remaining NP solution. A flip angle of 30° was used for all post-contrast scans, which occurred for 1 h following injection. TR was between 54 and 84 ms, TE between 2.73 and 4.24 ms, matrix size between 128×128 and 256×256, FOV between 20×20×10 mm3 and 25×25×10 mm3 depending on the experiment. NEX varied with matrix size to maintain temporal resolution, with 128×128, NEX = 5 for all scans, with 256×256, NEX = 4 for baseline and 1 for post-contrast scans.
Following serial image acquisition, resulting in about 100 post-contrast scans per animal, R1, concentration, and Ktrans mapping were performed using custom MATLAB script. R1 mapping was performed using the variable flip angle method based on the following equation:77
where SSPGR is signal intensity, α is FA, E1 is exp(TR/T1), and M0 is a proportionality factor related to longitudinal magnetization. E1 describes the linear relationship between the two signal intensity ratios, taking the slope, m, of that line enables calculation of T1 as:
Concentration maps were then generated by comparison of baseline R1 maps with post-contrast R1 maps using the following equation:
where C(t) is the concentration at time t, R1(t) is the post contrast R1-value at t, R1(t0) is baseline R1, and r1 is relaxivity of the contrast agent. Ktrans, the contrast extravasation rate constant, mapping was then performed using the Patlak model:
where C is the tissue concentration, Ca is the arterial plasma concentration, vp is the plasma volume fraction, and ⨂ is convolution. The Patlak model was chosen based on its previous use to assess subtle changes in BBB permeability associated with aging.78–79 MATLAB was used to execute a least squares curve fitting routine to calculate Ktrans for each voxel in the brain.
Microscale thermophoresis (MST).
Human claudin-1 was expressed and purified as reported previously.80 MST measurements were performed using a Monolith instrument (NanoTemper Technologies). For experiments with human claudin-1, the assay buffer contained 10 mM Tris HCl, pH 8.0, 100 mM NaCl, 4% glycerol, and 0.04% n-Dodecyl-β-D-Maltopyranoside (β-DDM). NPs and peptides were labeled with AF647 prior to MST analysis as described above. For the binding experiments the concentration of human claudin-1 was varied from 0.2-74000 nM and the AF647 labeled C1C2-NP was fixed at 70 nM, while the AF647 labeled NPs and AF647 labeled C1C2 peptide were fixed at 1 μM and 0.3 μM, respectively. Binding of the AF647 labeled C1C2-NP to human claudin-3, −4, and −9 were tested using 0.4-14000 nM, 0.8-28000 nM, and 0.6-20000 nM, respectively; with the AF647 labeled C1C2-NP fixed at 70 nM. This assay buffer was similar, except β-DDM was replaced with 0.1% n-Undecyl-β-D-Maltopyranoside. In order to check non-specific binding of β-DDM micelle to the AF647 labeled C1C2-NP, the concentration of ß-DDM was varied from 0.97 mM – 0.2 μM and the concentration of the AF647-labeled C1C2-NP was fixed at 1 μM. A similar experiment was performed wherein the concentration of UDM was varied from 10 mM – 0.7 μM and the concentration of AF647-labeled control NP was fixed at 1 μM. The non-specific binding of human claudins 4 and 9 to the NP was examined by varying the concentrations of human claudins 4 and 9 from 0.1 nM - 20.1 μM and 0.09 nM −31.1 μM, respectively. The human claudin sub-dilutions were mixed with the NPs or peptides and loaded in standard monolith NT.115 capillary tubes for measurement. All experiments were conducted at 23°C using the nano-red channel of the Monolith instrument and the data were analyzed by using either Monolith analysis software or Graph Pad Prism, version 9 (Graph Pad Software, San Diego, California).
In-vitro analysis over C1C2-NP binding towards Claudin-1.
bEnd.3 cells were cultured according to the ATCC culture conditions in DMEM. These cells were subjected to ethanol (50mM) treatment to induce claudin-1 expression and incubated with C1C2-NP for 4 hrs. For determining attachment of the C1C2-NP to human claudin-1, human claudin-1 encoded PCMV script plasmid was overexpressed in bEnd.3 cells and then incubated with C1C2-NP for 4 hrs.
Fluorescence imaging.
A fluorescence microscope (LSM800, Zeiss) was used to take the fluorescence image of mice brain. Mice were transcardially perfused after 2h of MRI with 4% paraformaldehyde (PFA) in Dulbecco’s phosphate-buffered saline (DPBS, Thermo Fisher Scientific, Waltham, MA). We did not perform a PBS perfusion prior to PFA because of the likelihood of washing out materials weakly bound to the vasculature or within the leaky BBB. While this experimental design does increase the chance of a false positive caused by NP uptake by peripheral macrophages that become fixed in circulation in the brain, the timeline for our imaging session and subsequent brain collection (~2hrs post-injection) is much shorter than the timeline for this peripheral macrophage uptake and trafficking to occur (e.g., it takes 24–48 hrs following a traumatic brain injury for the first circulating immune cells to arrive).81–83 Brain tissue was collected, trimmed, and fixed in 4% buffered PFA for 24 h. The brains were moved into 30% sucrose in DPBS for 3 d at 4°C for cryoprotection. The brains were then embedded in 2.6% carboxymethylcellulose (CMC, C4888, Sigma-Aldrich, St. Louis, MO), frozen on dry ice, and sliced coronally at a 15 μm thickness with cryotome (Leica Biosystems, Wetzlar, Germany). The brain slices were laid on poly-L-lysine coated microscope slides (6776215, Epredia, Kalamazoo, MI) and dried overnight at RT. Sections were washed with DPBS thrice for 5 min each to remove the CMC. The brain slices were blocked with 3% normal donkey serum, 0.3% triton X-100, and 0.1% sodium azide in DPBS for 1 h at RT. The primary and secondary antibody (Ab) were diluted in the blocking buffer. The brain sections were incubated with 1:250 dilution of rat anti-CD31 (ab56299, Abcam, Cambridge, UK) and 1:100 dilution of rabbit anti-claudin-1 (51-9000, Invitrogen, Carlsbad, CA) overnight at 4 °C then washed thrice for 5 min each with blocking buffer before the sections were incubated with a 1:250 dilution of an AF488 labeled goat anti-rat secondary Ab (ab150157, Abcam) and AF555 labeled donkey anti-rabbit secondary Ab (ab150074, Abcam) for 2 h at RT. The brain sections were again washed thrice for 5 min each with the blocking buffer before being stained with DAPI for 5 min, washed with DPBS followed by water, and mounted with ProLong™ Gold Antifade Mountant (Thermo Fisher Scientific).
Images were acquired with confocal microscopy at 40x objective lens magnification. Quantitative image analysis of claudin-1 and CD31 colocalization was performed with JACoP and ImageJ software on at least three randomly selected viewing fields for each region for each mouse.
Western blot.
Mice were transcardially perfused with ice-cold DPBS. The brains were extracted; cortex and hippocampus were separated. Cortex and hippocampus were then lyzed in RIPA lysis buffer and 1 mM phenylmethylsulfonyl fluoride. Protein concentration was measured using Pierce™ BCA protein assay (Thermo Fisher Scientific). 30 μg of extract protein was resolved by 12% SDS-PAGE and transferred onto PVDF membranes (Bio-Rad). Membranes were washed with TBS and were incubated with 3% QuickBlocker (G-Biosciences, St. Louis, MO) in TBS for 1 h RT. The membranes were incubated overnight at 4 °C with 1:500 dilution of rabbit anti-claudin-1 (51-9000, Invitrogen), 1:500 dilution of mouse anti-claudin-5 (35-2500, Invitrogen), or 1:1000 dilution of mouse anti-β-actin (A5441, Sigma) antibody in TTBS containing 3% QuickBlocker. Membranes were washed with TTBS before being incubated for 1 h at room temperature with 1:3000 dilution of HRP-conjugated goat anti-rabbit or anti-mouse secondary antibody (Bio-Rad) in TTBS containing 3% QuickBlocker. Membranes were then washed thrice with TTBS and antibody binding visualized by chemiluminescence (Clarity Western ECL Substrate, Bio-Rad) and quantified using the ChemiDoc system running the Image Lab software package (Bio-Rad).
Biodistribution.
Mice were transcardially perfused with ice-cold DPBS 1 hr following injection of control or C1C2 conjugated NPs (100 μL of 0.5 mM NPs) and heart, lung, liver, kidneys, spleen, colon, and intestine were collected and weighed. Three 2 month old and three 12 month old mice were used for this experiment. Gd concentrations were determined by digesting tissues with 600 uL HNO3 at 100 °C for 120 minutes prior to filtering and dilution for measurement using inductively coupled plasma mass spectrometry (ICP-MS).
Statistical analysis.
All the data in this study were expressed as mean ± standard error of the mean (SEM). A p < 0.05 was considered statistically significant. Data fitting for MST measurements were performed with GraphPad Prism 9.
Supplementary Material
ACKNOWLEDGMENTS
We thank P. Sollars and G. Pickard for supplying the CX3CR1-GFP mice. We acknowledge grant funding from the National Institute of Neurological Disorders and Stroke of the National Institutes of Health (R01NS109488) to FK and BX002086 (VA merit) to PD. This investigation is solely the responsibility of the authors and does not necessarily represent the official views of NINDS or NIH. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under the grant no. DGE-1610400 to HM. We thank B. McDonald in assistance for brain extraction. We acknowledge the Nano-Engineering Research Core facility supported by Nebraska Research Initiative fund for use of the confocal microscope.
Footnotes
Supporting Information Available: Supporting information is available online and includes Ktrans measurements in the cortex, hypothalamus, and muscle; microscale thermophoresis analysis of C1C2-NP binding to claudins 3, 4, and 9; comparison of claudin-1 expression and localization in brains from 2-month-old and 12-month old mice; biodistribution of control NPs and C1C2-NPs in 2-month-old and 12-month old mice.
REFERENCES
- 1.Nation DA; Sweeney MD; Montagne A; Sagare AP; D’Orazio LM; Pachicano M; Sepehrband F; Nelson AR; Buennagel DP; Harrington MG; Benzinger TLS; Fagan AM; Ringman JM; Schneider LS; Morris JC; Chui HC; Law M; Toga AW; Zlokovic BV, Blood-Brain Barrier Breakdown Is an Early Biomarker of Human Cognitive Dysfunction. Nat. Med 2019, 25, 270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Erickson MA; Banks WA, Age-Associated Changes in the Immune System and Blood(−)Brain Barrier Functions. Int. J. Mol. Sci 2019, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elahy M; Jackaman C; Mamo JC; Lam V; Dhaliwal SS; Giles C; Nelson D; Takechi R, Blood-Brain Barrier Dysfunction Developed During Normal Aging Is Associated with Inflammation and Loss of Tight Junctions but Not with Leukocyte Recruitment. Immun. Ageing 2015, 12, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Montagne A; Barnes SR; Sweeney MD; Halliday MR; Sagare AP; Zhao Z; Toga AW; Jacobs RE; Liu CY; Amezcua L; Harrington MG; Chui HC; Law M; Zlokovic BV, Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron 2015, 85, 296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Montagne A; Nikolakopoulou AM; Zhao Z; Sagare AP; Si G; Lazic D; Barnes SR; Daianu M; Ramanathan A; Go A; Lawson EJ; Wang Y; Mack WJ; Thompson PM; Schneider JA; Varkey J; Langen R; Mullins E; Jacobs RE; Zlokovic BV, Pericyte Degeneration Causes White Matter Dysfunction in the Mouse Central Nervous System. Nat. Med 2018, 24, 326–337. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.Brunk UT; Terman A, The Mitochondrial-Lysosomal Axis Theory of Aging: Accumulation of Damaged Mitochondria as a Result of Imperfect Autophagocytosis. Eur. J. Biochem 2002, 269, 1996–2002. [DOI] [PubMed] [Google Scholar]
- 7.Liang WS; Dunckley T; Beach TG; Grover A; Mastroeni D; Walker DG; Caselli RJ; Kukull WA; McKeel D; Morris JC; Hulette C; Schmechel D; Alexander GE; Reiman EM; Rogers J; Stephan DA, Gene Expression Profiles in Anatomically and Functionally Distinct Regions of the Normal Aged Human Brain. Physiol. Genomics 2007, 28, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cabeza R; Albert M; Belleville S; Craik FIM; Duarte A; Grady CL; Lindenberger U; Nyberg L; Park DC; Reuter-Lorenz PA; Rugg MD; Steffener J; Rajah MN, Maintenance, Reserve and Compensation: The Cognitive Neuroscience of Healthy Ageing. Nat. Rev. Neurosci 2018, 19, 701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gupta A; Nair S; Schweitzer AD; Kishore S; Johnson CE; Comunale JP; Tsiouris AJ; Sanelli PC, Neuroimaging of Cerebrovascular Disease in the Aging Brain. Aging Dis. 2012, 3 , 414–425. [PMC free article] [PubMed] [Google Scholar]
- 10.Wyss-Coray T, Ageing, Neurodegeneration and Brain Rejuvenation. Nature 2016, 539, 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farrall AJ; Wardlaw JM, Blood-Brain Barrier: Ageing and Microvascular Disease--Systematic Review and Meta-Analysis. Neurobiol. Aging 2009, 30, 337–352. [DOI] [PubMed] [Google Scholar]
- 12.Abbott NJ; Patabendige AA; Dolman DE; Yusof SR; Begley DJ, Structure and Function of the Blood-Brain Barrier. Neurobiol. Dis 2010, 37, 13–25. [DOI] [PubMed] [Google Scholar]
- 13.Paris L; Tonutti L; Vannini C; Bazzoni G, Structural Organization of the Tight Junctions. Biochim. Biophys. Acta 2008, 1778, 646–659. [DOI] [PubMed] [Google Scholar]
- 14.Hartsock A; Nelson WJ, Adherens and Tight Junctions: Structure, Function and Connections to the Actin Cytoskeleton. Biochim. Biophys. Acta 2008, 1778, 660–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolburg H; Lippoldt A, Tight Junctions of the Blood-Brain Barrier: Development, Composition and Regulation. Vascular Pharmacology 2002, 38, 323–337. [DOI] [PubMed] [Google Scholar]
- 16.Mooradian AD; Haas MJ; Chehade JM, Age-Related Changes in Rat Cerebral Occludin and Zonula Occludens-1 (ZO-1). Mech. Ageing Dev 2003, 124, 143–146. [DOI] [PubMed] [Google Scholar]
- 17.Virgintino D; Errede M; Robertson D; Capobianco C; Girolamo F; Vimercati A; Bertossi M; Roncali L, Immunolocalization of Tight Junction Proteins in the Adult and Developing Human Brain. Histochem. Cell Biol 2004, 122, 51–59. [DOI] [PubMed] [Google Scholar]
- 18.D’Souza T; Sherman-Baust CA; Poosala S; Mullin JM; Morin PJ, Age-Related Changes of Claudin Expression in Mouse Liver, Kidney, and Pancreas. J. Gerontol., Ser. A 2009, 64, 1146–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feng S; Cen J; Huang Y; Shen H; Yao L; Wang Y; Chen Z, Matrix Metalloproteinase-2 and −9 Secreted by Leukemic Cells Increase the Permeability of Blood-Brain Barrier by Disrupting Tight Junction Proteins. PLoS ONE 2011, 6, e20599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sladojevic N; Stamatovic SM; Johnson AM; Choi J; Hu A; Dithmer S; Blasig IE; Keep RF; Andjelkovic AV, Claudin-1-Dependent Destabilization of the Blood-Brain Barrier in Chronic Stroke. J. Neurosci 2019, 39, 743–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hasegawa K; Wakino S; Simic P; Sakamaki Y; Minakuchi H; Fujimura K; Hosoya K; Komatsu M; Kaneko Y; Kanda T; Kubota E; Tokuyama H; Hayashi K; Guarente L; Itoh H, Renal Tubular Sirt1 Attenuates Diabetic Albuminuria by Epigenetically Suppressing Claudin-1 Overexpression in Podocytes. Nat. Med 2013, 19, 1496–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buler M; Andersson U; Hakkola J, Who Watches the Watchmen? Regulation of the Expression and Activity of Sirtuins. FASEB J. 2016, 30, 3942–3960. [DOI] [PubMed] [Google Scholar]
- 23.Stamatovic SM; Martinez-Revollar G; Hu A; Choi J; Keep RF; Andjelkovic AV, Decline in Sirtuin-1 Expression and Activity Plays a Critical Role in Blood-Brain Barrier Permeability in Aging. Neurobiol. Dis 2019, 126, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen T; Dai SH; Li X; Luo P; Zhu J; Wang YH; Fei Z; Jiang XF, Sirt1-Sirt3 Axis Regulates Human Blood-Brain Barrier Permeability in Response to Ischemia. Redox Biol. 2018, 14, 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hattori Y; Okamoto Y; Maki T; Yamamoto Y; Oishi N; Yamahara K; Nagatsuka K; Takahashi R; Kalaria RN; Fukuyama H; Kinoshita M; Ihara M, Silent Information Regulator 2 Homolog 1 Counters Cerebral Hypoperfusion Injury by Deacetylating Endothelial Nitric Oxide Synthase. Stroke 2014, 45, 3403–3411. [DOI] [PubMed] [Google Scholar]
- 26.Zhang G; Li J; Purkayastha S; Tang Y; Zhang H; Yin Y; Li B; Liu G; Cai D, Hypothalamic Programming of Systemic Ageing Involving IKK-Beta, NF-KappaB and GnRH. Nature 2013, 497, 211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Minhas PS; Latif-Hernandez A; McReynolds MR; Durairaj AS; Wang Q; Rubin A; Joshi AU; He JQ; Gauba E; Liu L; Wang C; Linde M; Sugiura Y; Moon PK; Majeti R; Suematsu M; Mochly-Rosen D; Weissman IL; Longo FM; Rabinowitz JD; et al. , Restoring Metabolism of Myeloid Cells Reverses Cognitive Decline in Ageing. Nature 2021, 590, 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horng S; Therattil A; Moyon S; Gordon A; Kim K; Argaw AT; Hara Y; Mariani JN; Sawai S; Flodby P; Crandall ED; Borok Z; Sofroniew MV; Chapouly C; John GR, Astrocytic Tight Junctions Control Inflammatory CNS Lesion Pathogenesis. J. Clin. Invest 2017, 127, 3136–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bony BA; Kievit FM, A Role for Nanoparticles in Treating Traumatic Brain Injury. Pharmaceutics 2019, 11, 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furtado D; Bjornmalm M; Ayton S; Bush AI; Kempe K; Caruso F, Overcoming the Blood-Brain Barrier: The Role of Nanomaterials in Treating Neurological Diseases. Adv. Mater 2018, 30, e1801362. [DOI] [PubMed] [Google Scholar]
- 31.Gonzalez-Carter D; Liu X; Tockary TA; Dirisala A; Toh K; Anraku Y; Kataoka K, Targeting Nanoparticles to the Brain by Exploiting the Blood-Brain Barrier Impermeability to Selectively Label the Brain Endothelium. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 19141–19150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Golombek SK; May JN; Theek B; Appold L; Drude N; Kiessling F; Lammers T, Tumor Targeting via EPR: Strategies to Enhance Patient Responses. Adv. Drug Deliv. Rev 2018, 130, 17–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bony BA; Miller HA; Tarudji AW; Gee CC; Sarella A; Nichols MG; Kievit FM, Ultrasmall Mixed Eu-Gd Oxide Nanoparticles for Multimodal Fluorescence and Magnetic Resonance Imaging of Passive Accumulation and Retention in TBI. ACS Omega 2020, 5, 16220–16227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller HA; Magsam AW; Tarudji AW; Romanova S; Weber L; Gee CC; Madsen GL; Bronich TK; Kievit FM, Evaluating Differential Nanoparticle Accumulation and Retention Kinetics in a Mouse Model of Traumatic Brain Injury via K(Trans) Mapping with MRI. Sci. Rep 2019, 9, 16099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu JL; Ypma M; Chiarelli PA; Park J; Ellenbogen RG; Stayton PS; Mourad PD; Lee D; Convertine AJ; Kievit FM, Theranostic Oxygen Reactive Polymers for Treatment of Traumatic Brain Injury. Adv. Funct. Mater 2016, 26, 4124–4133. [Google Scholar]
- 36.Yoo D; Magsam AW; Kelly AM; Stayton PS; Kievit FM; Convertine AJ, Core-Cross-Linked Nanoparticles Reduce Neuroinflammation and Improve Outcome in a Mouse Model of Traumatic Brain Injury. ACS Nano 2017, 11, 8600–8611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bharadwaj VN; Lifshitz J; Adelson PD; Kodibagkar VD; Stabenfeldt SE, Temporal Assessment of Nanoparticle Accumulation after Experimental Brain Injury: Effect of Particle Size. Sci. Rep 2016, 6, 29988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bharadwaj VN; Rowe RK; Harrison J; Wu C; Anderson TR; Lifshitz J; Adelson PD; Kodibagkar VD; Stabenfeldt SE, Blood-Brain Barrier Disruption Dictates Nanoparticle Accumulation Following Experimental Brain Injury. Nanomedicine 2018, 14, 2155–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bharadwaj VN; Copeland C; Mathew E; Newbern J; Anderson TR; Lifshitz J; Kodibagkar VD; Stabenfeldt SE, Sex-Dependent Macromolecule and Nanoparticle Delivery in Experimental Brain Injury. Tissue. Eng. Part A 2020, 26, 688–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kudryashev JA; Waggoner LE; Leng HT; Mininni NH; Kwon EJ, An Activity-Based Nanosensor for Traumatic Brain Injury. ACS Sens. 2020, 5, 686–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kwon EJ; Skalak M; Lo Bu R; Bhatia SN, Neuron-Targeted Nanoparticle for siRNA Delivery to Traumatic Brain Injuries. ACS Nano 2016, 10, 7926–7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boyd BJ; Galle A; Daglas M; Rosenfeld JV; Medcalf R, Traumatic Brain Injury Opens Blood-Brain Barrier to Stealth Liposomes via an Enhanced Permeability and Retention (EPR)-Like Effect. J. Drug Target 2015, 23, 847–853. [DOI] [PubMed] [Google Scholar]
- 43.Mousazadeh M; Palizban A; Salehi R; Salehi M, Gene Delivery to Brain Cells with Apoprotein E Derived Peptide Conjugated to Polylysine (apoEdp-PLL). J. Drug Target 2007, 15, 226–230. [DOI] [PubMed] [Google Scholar]
- 44.Neves AR; Queiroz JF; Lima SAC; Reis S, Apo E-Functionalization of Solid Lipid Nanoparticles Enhances Brain Drug Delivery: Uptake Mechanism and Transport Pathways. Bioconjug. Chem 2017, 28, 995–1004. [DOI] [PubMed] [Google Scholar]
- 45.Zhao H; Bao XJ; Wang RZ; Li GL; Gao J; Ma SH; Wei JJ; Feng M; Zhao YJ; Ma WB; Yang Y; Li YN; Kong YG, Postacute Ischemia Vascular Endothelial Growth Factor Transfer by Transferrin-Targeted Liposomes Attenuates Ischemic Brain Injury after Experimental Stroke in Rats. Hum. Gene Ther 2011, 22, 207–215. [DOI] [PubMed] [Google Scholar]
- 46.Allen DD; Lockman PR, The Blood-Brain Barrier Choline Transporter as a Brain Drug Delivery Vector. Life Sci. 2003, 73, 1609–1615. [DOI] [PubMed] [Google Scholar]
- 47.Wu LP; Ahmadvand D; Su J; Hall A; Tan X; Farhangrazi ZS; Moghimi SM, Crossing the Blood-Brain-Barrier with Nanoligand Drug Carriers Self-Assembled from a Phage Display Peptide. Nat. Commun 2019, 10, 4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Vlerken LE; Vyas TK; Amiji MM, Poly(Ethylene Glycol)-Modified Nanocarriers for Tumor-Targeted and Intracellular Delivery. Pharm. Res 2007, 24, 1405–1414. [DOI] [PubMed] [Google Scholar]
- 49.Suk JS; Xu Q; Kim N; Hanes J; Ensign LM, PEGylation as a Strategy for Improving Nanoparticle-Based Drug and Gene Delivery. Adv. Drug. Deliv. Rev 2016, 99, 28–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jokerst JV; Lobovkina T; Zare RN; Gambhir SS, Nanoparticle PEGylation for Imaging and Therapy. Nanomedicine (London, U. K.) 2011, 6, 715–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kwon GS, Polymeric Micelles for Delivery of Poorly Water-Soluble Compounds. Crit. Rev. Ther. Drug Carrier Syst 2003, 20, 357–403. [DOI] [PubMed] [Google Scholar]
- 52.Kanaras AG; Kamounah FS; Schaumburg K; Kiely CJ; Brust M, Thioalkylated Tetraethylene Glycol: A New Ligand for Water Soluble Monolayer Protected Gold Clusters. Chem. Commun. (Cambridge, U. K.) 2002, 2294–2295. [DOI] [PubMed] [Google Scholar]
- 53.Soderlind F; Pedersen H; Petoral RM Jr.; Kall PO; Uvdal K, Synthesis and Characterisation of Gd2O3 Nanocrystals Functionalised by Organic Acids. J. Colloid Interface Sci 2005, 288, 140–148. [DOI] [PubMed] [Google Scholar]
- 54.Hug SJ; Bahnemann D, Infrared Spectra of Oxalate, Malonate and Succinate Adsorbed on the Aqueous Surface of Rutile, Anatase and Lepidocrocite Measured with in Situ ATR-FTIR. J. Electron Spectrosc. Relat. Phenom 2006, 150, 208–219. [Google Scholar]
- 55.Patel T; Zhou J; Piepmeier JM; Saltzman WM, Polymeric Nanoparticles for Drug Delivery to the Central Nervous System. Adv. Drug Deliv. Rev 2012, 64, 701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Georgieva JV; Kalicharan D; Couraud PO; Romero IA; Weksler B; Hoekstra D; Zuhorn IS, Surface Characteristics of Nanoparticles Determine Their Intracellular Fate in and Processing by Human Blood-Brain Barrier Endothelial Cells in Vitro. Mol. Ther 2011, 19, 318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kumagai AK; Eisenberg JB; Pardridge WM, Absorptive-Mediated Endocytosis of Cationized Albumin and a Beta-Endorphin-Cationized Albumin Chimeric Peptide by Isolated Brain Capillaries. Model System of Blood-Brain Barrier Transport. J. Biol. Chem 1987, 262, 15214–15219. [PubMed] [Google Scholar]
- 58.Mullier A; Bouret SG; Prevot V; Dehouck B, Differential Distribution of Tight Junction Proteins Suggests a Role for Tanycytes in Blood-Hypothalamus Barrier Regulation in the Adult Mouse Brain. J. Comp. Neurol 2010, 518, 943–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cullen KM; Kocsi Z; Stone J, Pericapillary Haem-Rich Deposits: Evidence for Microhaemorrhages in Aging Human Cerebral Cortex. J. Cere.b Blood Flow Metab 2005, 25, 1656–1667. [DOI] [PubMed] [Google Scholar]
- 60.Zipser BD; Johanson CE; Gonzalez L; Berzin TM; Tavares R; Hulette CM; Vitek MP; Hovanesian V; Stopa EG, Microvascular Injury and Blood-Brain Barrier Leakage in Alzheimer’s Disease. Neurobiol. Aging 2007, 28, 977–986. [DOI] [PubMed] [Google Scholar]
- 61.Furuse M; Sasaki H; Tsukita S, Manner of Interaction of Heterogeneous Claudin Species within and between Tight Junction Strands. J. Cell Biol 1999, 147, 891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu AS, Claudins and the Kidney. J. Am. Soc. Nephrol 2015, 26, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang Y; Estrada EY; Thompson JF; Liu W; Rosenberg GA, Matrix Metalloproteinase-Mediated Disruption of Tight Junction Proteins in Cerebral Vessels Is Reversed by Synthetic Matrix Metalloproteinase Inhibitor in Focal Ischemia in Rat. J. Cereb. Blood Flow Metab 2007, 27, 697–709. [DOI] [PubMed] [Google Scholar]
- 64.Baumholtz AI; Simard A; Nikolopoulou E; Oosenbrug M; Collins MM; Piontek A; Krause G; Piontek J; Greene NDE; Ryan AK, Claudins Are Essential for Cell Shape Changes and Convergent Extension Movements during Neural Tube Closure. Dev. Biol 2017, 428, 25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tran KA; Zhang X; Predescu D; Huang X; Machado RF; Gothert JR; Malik AB; Valyi-Nagy T; Zhao YY, Endothelial Beta-Catenin Signaling Is Required for Maintaining Adult Blood-Brain Barrier Integrity and Central Nervous System Homeostasis. Circulation 2016, 133, 177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liebner S; Corada M; Bangsow T; Babbage J; Taddei A; Czupalla CJ; Reis M; Felici A; Wolburg H; Fruttiger M; Taketo MM; von Melchner H; Plate KH; Gerhardt H; Dejana E, Wnt/Beta-Catenin Signaling Controls Development of the Blood-Brain Barrier. J. Cell Biol 2008, 183 (3), 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pfeiffer F; Schafer J; Lyck R; Makrides V; Brunner S; Schaeren-Wiemers N; Deutsch U; Engelhardt B, Claudin-1 Induced Sealing of Blood-Brain Barrier Tight Junctions Ameliorates Chronic Experimental Autoimmune Encephalomyelitis. Acta Neuropathol. 2011, 122, 601–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Uchida Y; Sumiya T; Tachikawa M; Yamakawa T; Murata S; Yagi Y; Sato K; Stephan A; Ito K; Ohtsuki S; Couraud PO; Suzuki T; Terasaki T, Involvement of Claudin-11 in Disruption of Blood-Brain, -Spinal Cord, and -Arachnoid Barriers in Multiple Sclerosis. Mol. Neurobiol 2019, 56, 2039–2056. [DOI] [PubMed] [Google Scholar]
- 69.Staat C; Coisne C; Dabrowski S; Stamatovic SM; Andjelkovic AV; Wolburg H; Engelhardt B; Blasig IE, Mode of Action of Claudin Peptidomimetics in the Transient Opening of Cellular Tight Junction Barriers. Biomaterials 2015, 54, 9–20. [DOI] [PubMed] [Google Scholar]
- 70.Koval M, Claudin Heterogeneity and Control of Lung Tight Junctions. Annu. Rev. Physiol 2013, 75, 551–567. [DOI] [PubMed] [Google Scholar]
- 71.Chithrani BD; Chan WC, Elucidating the Mechanism of Cellular Uptake and Removal of Protein-Coated Gold Nanoparticles of Different Sizes and Shapes. Nano Lett. 2007, 7, 1542–1550. [DOI] [PubMed] [Google Scholar]
- 72.Yuan HY; Zhang SL, Effects of Particle Size and Ligand Density on the Kinetics of Receptor-Mediated Endocytosis of Nanoparticles. Appl. Phys. Lett 2010, 96. [Google Scholar]
- 73.Chithrani BD; Ghazani AA; Chan WC, Determining the Size and Shape Dependence of Gold Nanoparticle Uptake into Mammalian Cells. Nano Lett. 2006, 6, 662–668. [DOI] [PubMed] [Google Scholar]
- 74.Mrsny RJ; Brown GT; Gerner-Smidt K; Buret AG; Meddings JB; Quan C; Koval M; Nusrat A, A Key Claudin Extracellular Loop Domain Is Critical for Epithelial Barrier Integrity. Am. J. Pathol 2008, 172, 905–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chung NP; Mruk D; Mo MY; Lee WM; Cheng CY, A 22-Amino Acid Synthetic Peptide Corresponding to the Second Extracellular Loop of Rat Occludin Perturbs the Blood-Testis Barrier and Disrupts Spermatogenesis Reversibly in Vivo. Biol. Reprod 2001, 65, 1340–1351. [DOI] [PubMed] [Google Scholar]
- 76.Deoni SC; Rutt BK; Peters TM, Rapid Combined T1 and T2 Mapping Using Gradient Recalled Acquisition in the Steady State. Magn. Reson. Med 2003, 49, 515–526. [DOI] [PubMed] [Google Scholar]
- 77.Deoni SC; Peters TM; Rutt BK, High-Resolution T1 and T2 Mapping of the Brain in a Clinically Acceptable Time with Despot1 and Despot2. Magn. Reson. Med 2005, 53, 237–241. [DOI] [PubMed] [Google Scholar]
- 78.Barnes SR; Ng TS; Montagne A; Law M; Zlokovic BV; Jacobs RE, Optimal Acquisition and Modeling Parameters for Accurate Assessment of Low Ktrans Blood-Brain Barrier Permeability Using Dynamic Contrast-Enhanced MRI. Magn. Reson. Med 2016, 75, 1967–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Heye AK; Thrippleton MJ; Armitage PA; Valdes Hernandez MDC; Makin SD; Glatz A; Sakka E; Wardlaw JM, Tracer Kinetic Modelling for DCE-MRI Quantification of Subtle Blood-Brain Barrier Permeability. Neuroimage 2016, 125, 446–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vecchio AJ; Stroud RM, Claudin-9 Structures Reveal Mechanism for Toxin-Induced Gut Barrier Breakdown. Pro.c Natl. Acad. Sci. U. S. A 2019, 116, 17817–17824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li Z; Sun L; Zhang Y; Dove AP; O’Reilly RK; Chen G, Shape Effect of Glyco-Nanoparticles on Macrophage Cellular Uptake and Immune Response. ACS Macro Lett. 2016, 5, 1059–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Clark RS; Schiding JK; Kaczorowski SL; Marion DW; Kochanek PM, Neutrophil Accumulation after Traumatic Brain Injury in Rats: Comparison of Weight Drop and Controlled Cortical Impact Models. J. Neurotraum 1994, 11, 499–506. [DOI] [PubMed] [Google Scholar]
- 83.Simon DW; McGeachy MJ; Bayir H; Clark RS; Loane DJ; Kochanek PM, The Far-Reaching Scope of Neuroinflammation after Traumatic Brain Injury. Nat. Rev. Neurol 2017, 13, 171–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.