

Abstract
Herein, we present the synthesis of Ag–Au bimetallic nanocomposites stabilized with organic–inorganic hybrid microgels. The aim is to get both the surface plasmon resonance (SPR) and catalytic performance of the composite material can be changed in response to external stimuli. Ag@poly(N-isopropylacrylamide-co-3-methacryloxypro-pyltrimethoxysilane) (Ag@P(NIPAM-co-MAPTMS)) hybrid microgels were synthesized by seed-emulsion polymerization using Ag nanoparticles (NPs) as the core and NIPAM/MAPTMS as monomers. Ag–Au@P(NIPAM-co-MAPTMS) bimetallic hybrid microgels were prepared by a galvanic replacement (GR) reaction between Ag NPs and HAuCl4, with the composition and structure of these bimetallic nanocomposites being determined by the amount of added HAuCl4. The highly porous organic–inorganic microgel layer provided confined space for the GR reaction, effectively preventing the aggregation of Ag–Au NPs. The shell layer of P(NIPAM-co-MAPTMS) three-dimensional network chains not only enhanced nanocomposite dispersity and stability, but also provided highly porous gel microdomains that could increase the diffusion of the substrate and hence enhanced catalytic activity. Additionally, the SPR and catalytic properties of Ag–Au@P(NIPAM-co-MAPTMS) are reversibly sensitive to external temperature. With increase of temperature, the maximum absorption peak of bimetallic nanocomposites shifted to longer wavelengths, and the catalytic activity of these composites for the reduction of 4-nitrophenol by NaBH4 remarkably increased. The features above mentioned are related to presence of the thermosensitive PNIPAM chains and the highly porous structure constructed by rigid MAPTMS segments intersected between NIPAM chains.
Ag–Au bimetallic nanocomposites stabilized with organic–inorganic hybrid microgels allowed the mass transfer of reactants to be controlled by temperature modulation.
Introduction
Bimetallic nanoparticles (NPs) have been widely used in catalysis, sensing, and biomedical applications1–3 owing to their multi-functionality originating from the synergistic effects of the two components, exhibiting an increased number of active sites due to the enhanced effect of the electromagnetic field on bimetallic interfaces.4–7 In addition, the above NPs exhibit enhanced catalytic activity and selectivity owing to their ability to improve electron transfer rates and change original electronic structures.8–10 Meanwhile, the regulation of bimetallic NPs morphology/crystal structure can greatly enhance surface plasmon resonance (SPR) properties and hence improve catalytic performance, optical absorption, and surface-enhanced Raman scattering.11–13 Among the multitude of bimetallic NPs, Ag–Au ones have attracted increased attention due to the high extinction coefficient and electronegativity of Ag NPs and the high chemical stability and biological activity of Au NPs.14,15 The similarity between the lattice spacings of Ag and Au allows the fabrication of Ag–Au bimetallic nanocrystals with tunable morphology,16 and the optical performance of Ag–Au NPs can be regulated by adjusting their structure and the relative contents of Ag and Au.17,18
Compared to the co-reduction approach, the galvanic replacement (GR) reaction strategy had received more attention due to offering facile reaction conditions and allowing the fabrication of bimetallic NPs with tunable structures that can be controlled by adjusting the concentration ratio of metal precursor salts.19–21 The unique porous structure of Ag–Au bimetallic NPs is characterized by an increased number of surface active sites and a large void volume, which not only promotes electron transfer on the nanocatalyst surface, but also accelerates the reaction of the substrate in contact with the catalyst and thus improves catalytic activity.22–24
The surface of bimetallic NPs is commonly coated with chemically inert protective layers (SiO2, dendrimers, polymers, etc.) to improve dispersity and stability.25–27 In particular, environment-sensitive polymer microgels with three-dimensional network structures can not only effectively inhibit the aggregation of metal NPs, but also make their optical and catalytic properties sensitive to changes of environmental conditions such as temperature, pH, and ionic strength.28–31 Among the variety of environment-sensitive polymer microgels, poly(N-isopropylacrylamide) (PNIPAM) has received considerable attention due to its low critical solution temperature (LCST) being close to human body temperature.32 For example, Liz-Marzán et al. have developed Au-PNIPAM core–shell nanocomposites, showing that the catalytic activity of encapsulated Au NPs is affected by both temperature and shell composition. Importantly, the diffusion of reactants toward the catalytic core can be tuned by the closure/opening of the PNIPAM network in response to temperature changes.33 Lu et al. synthetized yolk–shell Au-PNIPAM hybrid microgels wherein Au NPs were encapsulated in a hollow PNIPAM shell, with the porosity and hydrophobicity being effectively controlled by temperature changes. Moreover, the above Au NPs exhibited tunable catalytic activity and selectivity in the reduction of 4-nitrophenol (4-NP) and nitrobenzene (NB) in aqueous solution.34 Wu et al. reported a one-pot synthesis of core–shell Au@polyvinylpyrrolidone (PVP) hybrid nanogels, revealing that embedded Au NPs demonstrated both pH-modulated catalytic activity and anti-aggregation properties upon recycling.35 Zha et al. reported a novel type of three-component composite microgels (AuNR@(AgNPs/PNIPAM)), showing that they exhibit light-controllable catalytic activity for the reduction of 4-NP by NaBH4 that can be modulated by varying the power density of visible and near-infrared (NIR) light.36 Ahmed and Farooqi have reported Ag NPs were introduced within the copolymerization of NIPAM with acrylamide (AAm), or 2-hydroxyethylmethacrylate (HEMA) and acrylic acid (AAc) hybrid microgels. It was showed that these polymer microgels do not only stabilize Ag NPs in polymer network but also enhance the mass transport of hydrophilic reactant 4-NP from outside to inside the polymer network.37–39
Compared to pure PNIPAM network chains, the swelling and shrinkage of poly(N-isopropylacrylamide-co-3-methacryloxypropyltrimethoxysilane) (P(NIPAM-co-MAPTMS)) organic–inorganic hybrid microgels with temperature was partly limited because the thermosensitive PNIPAM chains were separated by rigid MAPTMS segments.40,41 Furthermore, the rigid MAPTMS segments could enhance the three-dimensional porous inflexibility of P(NIPAM-co-MAPTMS), which is favor for the diffusion and mass transfer. We herein propose a novel route for the fabrication of porous Ag–Au bimetallic nanocomposites stabilized by organic–inorganic hybrid microgels with temperature sensitivity, and these structural features are expected to enhance the catalytic reactivity of bimetallic nanoparticles by intensification of the mass transformation. Based on above considerations, Ag@P(NIPAM-co-MAPTMS) hybrid microgels were synthesized by seed-emulsion polymerization using Ag NPs as the core and NIPAM/MAPTMS as monomers. Furthermore, Ag–Au bimetallic nanocomposites stabilized with P(NIPAM-co-MAPTMS) organic–inorganic hybrid microgels were prepared by a GR reaction between Ag NPs and HAuCl4, exhibiting SPR and catalytic properties being sensitive to external temperature. The shell layer of P(NIPAM-co-MAPTMS) three-dimensional network chains not only enhanced nanocomposite dispersity and stability, but also provided highly porous gel microdomains that could increase the diffusion of the substrate and hence enhanced catalytic activity. These expectations were proved through the reduction of 4-NP by NaBH4 catalyzed by Ag–Au@P(NIPAM-co-MAPTMS) bimetallic hybrid microgels.
Experimental
Materials
NIPAM (TCI) was purified by recrystallization from n-hexane/benzene (40/60, v/v) before use. MAPTMS was supplied by Alfa Aesar. PVP (Mw ≈ 30 000), sodium citrate, glucose, silver nitrate (AgNO3), NH3·H2O (25 wt%), sodium dodecyl sulfate (SDS), N,N′-methylenebisacrylamide (MBA), ammonium persulfate (APS), chloroauric acid (HAuCl4, 99.3%), absolute ethanol, sodium borohydride (NaBH4), 4-NP, and sodium hydroxide (NaOH) were purchased from Sinopharm Chemical Reagent Co. (China) and used as received. Doubly distilled water was used in all experiments.
Synthesis of Ag@P(NIPAM-co-MAPTMS) hybrid microgels
Ag NPs stabilized by P(NIPAM-co-MAPTMS) organic–inorganic hybrid microgels were prepared in two steps.
In the first step, Ag NPs were prepared using glucose as a reductant and sodium citrate/PVP as stabilizers.42 Typically, a solution of 0.151 g of PVP, 0.048 g of sodium citrate, and 0.082 g of glucose in 30 mL of water was transferred into a 100 mL three-necked round-bottom flask and stirred at 400 rpm while being purged with N2 to remove oxygen. The deoxygenated solution was heated to 100 °C, and fresh [Ag(NH3)2]+ was dropwise added within 30 min to obtain a green-yellow Ag NP dispersion that was repeatedly centrifuged with doubly distilled water at 9500 rpm for 30 min. Finally, the obtained NP dispersion was diluted with water to 30 mL.
Ag NPs encapsulated in P(NIPAM-co-MAPTMS) organic–inorganic hybrid microgels were synthesized by seeded emulsion polymerization using Ag NPs modified with NIPAM as core seeds. Typically, 10 mL of Ag NP dispersion was added to a solution of 0.08 g of NIPAM and 0.018 g of SDS in 50 mL of water, and the mixture was stirred for 24 h, being subsequently heated to 70 °C under a flow of N2 while stirring at 400 rpm in a 150 mL three-necked round-bottom flask equipped with a condenser. In the next step, an aqueous solution of an appropriate amount of NIPAM and 0.003 g of MBA cross-linker was added to the reaction flask. After 1 h, 28 μL of aqueous APS (0.95 M) and 0.005 g of MAPTMS were injected into the reaction system, and polymerization was performed for 4 h. The produced yellow Ag@P(NIPAM-co-MAPTMS) hybrid microgels were repeatedly centrifuged at 9500 rpm, sequentially washed with alternately water and absolute ethanol, and dispersed in 50 mL of water before use. To determine the effect of NIPAM loading on the dispersity of the Ag@P(NIPAM-co-MAPTMS) hybrid microgel shell layer, P(NIPAM-co-MAPTMS) organic–inorganic hybrid microgels were fabricated using different masses of NIPAM (0.02, 0.04, 0.06, and 0.08 g) while keeping the amounts of MAPTMS, MBA, APS, and SDS constant.
Synthesis of Ag–Au@P(NIPAM-co-MAPTMS) bimetallic hybrid microgels
Core–shell Ag–Au@P(NIPAM-co-MAPTMS) bimetallic hybrid microgels were controllably synthesized by a GR reaction between Ag NPs and HAuCl4 using the former as a sacrificial template (Fig. 1). Typically, 50 mL of Ag@P(NIPAM-co-MAPTMS) aqueous dispersion was added into a 150 mL three-necked round-bottom flask upon 30 min stirring (400 rpm) at 25 °C. Subsequently, a certain volume of aqueous HAuCl4 (0.002 M) was added into the flask using a constant-pressure funnel. After 6 h, 1.6 mL of aqueous ammonia was added into the flask, and the reaction was continued for 30 min. The obtained Ag–Au@P(NIPAM-co-MAPTMS) bimetallic hybrid microgels were centrifuged (9000 rpm), sequentially washed alternately with water and absolute ethanol for several times, and freeze-dried (FD-1A-80, Beijing Boyikang Experimental Equipment Company).
Fig. 1. Schematic illustration for synthesis of core–shell Ag–Au@P(NIPAM-co-MAPTMS) bimetallic hybrid microgels.

Ag–Au@P(NIPAM-co-MAPTMS) bimetallic hybrid microgels with different contents of Ag and Au were controllably prepared by changing the volume of HAuCl4 solution (1.0, 3.0, 6.0, and 9.0 mL), with the utilized nomenclature presented in Table 1.
Naming of Ag–Au@P(NIPAM-co-MAPTMS) bimetallic hybrid microgels prepared using different amounts of HAuCl4.
| Samples | Volume of HAuCl4 |
|---|---|
| Ag@P(NIPAM-co-MAPTMS) | 0 mL |
| Ag–Au@P(NIPAM-co-MAPTMS)-1 | 1 mL |
| Ag–Au@P(NIPAM-co-MAPTMS)-2 | 3 mL |
| Ag–Au@P(NIPAM-co-MAPTMS)-3 | 6 mL |
| Ag–Au@P(NIPAM-co-MAPTMS)-4 | 9 mL |
Characterization
The morphology and size dispersion of hybrid microgels were probed by transmission electron microscopy (TEM; JEM-2100, JEOL, Japan) at an accelerating voltage of 200 kV. Elemental mapping and high-angle annular dark field scanning TEM (HAADF-STEM) imaging were conducted using field emission transmission electron microscopy (FE-TEM) coupled with energy-dispersive X-ray spectroscopy (EDX) (Tecnai G2 F20, FEI, USA). UV-Vis spectra were recorded on a U-3900 UV-Vis spectrophotometer equipped with a temperature controller (Hitachi, Japan). Infrared spectra were recorded on an Avatar 360 Fourier transform infrared (FT-IR) spectrometer (Nicolet, USA) using the KBr pellet technique. Thermal stabilities were determined using a Q1000DSC+LNCS+FACS Q600SDT thermogravimetric analyzer (TA, USA) at a heating rate of 10 °C min−1 in an atmosphere of N2. Swelling behavior was characterized by dynamic light scattering measurements (Nano-ZS90, Malvern, UK) in the temperature range of 25–45 °C. Crystal structures were determined by X-ray diffraction (XRD) analysis (D/MAX-3C, Rigaku, Japan) performed using Cu Kα radiation at 35 kV and 40 mA. Surface compositions were determined by X-ray photoelectron spectroscopy (XPS; AXIS ULTRA, Kratos Analytical Ltd., Japan) using monochromatic Al Kα radiation.
Catalytic reduction of 4-NP
The catalytic activity of hybrid microgels was systematically evaluated for the reduction of 4-NP by excess NaBH4 as a model reaction, and the reaction progress was monitored in situ by measuring changes of 4-NP absorbance at 400 nm. Typically, a 0.003 g catalyst sample was dissolved in 20 mL of water. Separately, 100 μL of aqueous 4-NP (2.0 × 10−3 M) and 3.0 mL of aqueous NaBH4 (0.10 M) were added into a standard quartz cuvette (solution pH was adjusted to 10 by NaOH), and 80 μL of the above catalyst dispersion was injected. The absorption spectra of the obtained mixture were acquired in situ in the range of 250–500 nm at 1 min intervals for different reaction temperatures, and apparent reaction rate constants (kapp) were obtained from temporal changes of the absorption peak intensity at 400 nm by plotting ln(At/A0) vs. reduction time (At and A0 are absorbances at time t and 0, respectively).43
Results and discussion
Morphology and crystal structure of Ag NPs
TEM imaging showed that the prepared bright yellow Ag NPs exhibited a quasi-spherical shape (Fig. 2a and b) with an average diameter of ∼42 nm (Fig. 2c) and a maximum absorption wavelength of 423 nm (Fig. 2d), which was attributed to their SPR.44 Furthermore, the narrow half peak width of the UV-Vis absorption peak confirmed that Ag NPs exhibited strong SPR. High-resolution TEM imaging allowed the lattice spacing d to be determined as ∼0.235 nm (Fig. 2e), which was assigned to the (111) crystal planes of face-centered cubic Ag.45Fig. 2f shows a typical XRD pattern of Ag NPs, with diffraction peaks at 38.2°, 44.4°, 64.5°, and 77.6° corresponding to the (111), (200), (220), and (311) planes of face-centered cubic Ag,46 respectively. The dominance of the (111) peak suggested that the obtained Ag NPs mainly featured (111) facets.45
Fig. 2. TEM images (a and b), size distribution (c), UV-Vis spectrum (d), HR-TEM image (e), and XRD pattern (f) of Ag NPs.

Morphology of Ag@P(NIPAM-co-MAPTMS) hybrid microgels
Ag NPs stabilized with P(NIPAM-co-MAPTMS) organic–inorganic hybrid microgels were synthesized by seed-emulsion polymerization using NIPAM-modified Ag NPs as core seeds, with the amount of the NIPAM monomer directly affecting the dispersity and uniformity of the P(NIPAM-co-MAPTMS) coating layer. At a NIPAM loading of 0.02 g, the obtained surface layer was uneven, and the Ag NPs were prone to aggregation (ESI, Fig. S1a and b†). Conversely, a much better coating layer was observed at a NIPAM loading of 0.04 g (Fig. 3a and b), and the thus obtained Ag@P(NIPAM-co-MAPTMS) hybrid microgels exhibited good dispersity and a well-pronounced core–shell structure. The average hydrodynamic diameter of the above microgels was determined as ∼243 nm by dynamic light scattering measurements (Fig. 3c). However, at increased NIPAM loadings (0.06 and 0.008 g), large numbers of individual P(NIPAM-co-MAPTMS) particles were formed due to the monomers being present in excess to Ag NPs (ESI, Fig. S1c–f†).
Fig. 3. TEM images (a and b) and hydrodynamic diameter distribution (c) of Ag@P(NIPAM-co-MAPTMS) hybrid microgels prepared at a NIPAM loading of 0.04 g.

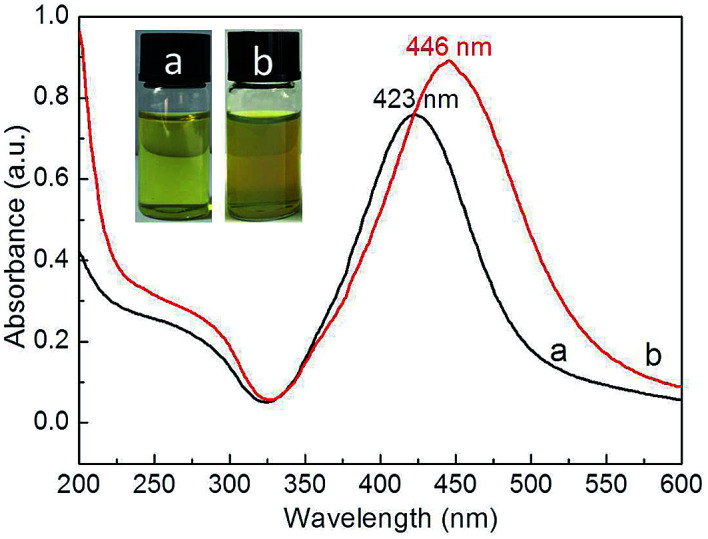
Compared to that of Ag NPs (423 nm), the absorption peak maximum of Ag@P(NIPAM-co-MAPTMS) hybrid microgels was red-shifted to 446 nm (Fig. 4) due to the cross-linked structure of P(NIPAM-co-MAPTMS) shell layers altering the refractive index of coated Ag NPs and hence enhancing their Rayleigh scattering and inducing a red shift of the SPR absorption peak.47
Fig. 4. UV-Vis spectra and photographs of Ag NPs (a) and Ag@P(NIPAM-co-MAPTMS) hybrid microgels (b).
The presence of PNIPAM and MAPTMS in organic–inorganic hybrid microgels was confirmed by FT-IR spectroscopy (ESI, Fig. S2†). Peaks at 2877, 2935, and 2935 cm−1 were assigned to C–H stretching vibrations and –CH2 and –CH3 groups of PNIPAM,48 while those at 1649 and 1547 cm−1 were assigned to the C O stretch of PNIPAM amide (I) and (II) groups. The peak at 1387 cm−1 was attributed to the deformation of PNIPAM –C(CH3)2 methyls, and that at 1126 cm−1 was assigned to the –Si–O–Si– stretching vibration of MAPTMS.49
Morphology and structure of Ag–Au@P(NIPAM-co-MAPTMS) bimetallic hybrid microgels
Ag NPs covered with P(NIPAM-co-MAPTMS) organic–inorganic hybrid microgels were used as sacrificial templates to synthesize Ag–Au bimetallic nanocomposites via a GR reaction between Ag NPs and HAuCl4. Herein, we chose ammonia for removing AgCl produced as a by-product during the formation of Au.50 To explore the effect of HAuCl4 dosage on the structure of Ag–Au bimetallic nanocomposites, Ag–Au@P(NIPAM-co-MAPTMS) hybrid microgels were prepared using different volumes of 0.002 M aqueous HAuCl4 (1.0, 3.0, 6.0, and 9.0 mL), with the corresponding samples denoted as Ag–Au@P(NIPAM-co-MAPTMS)-1, Ag–Au@P(NIPAM-co-MAPTMS)-2, Ag–Au@P(NIPAM-co-MAPTMS)-3, and Ag–Au@P(NIPAM-co-MAPTMS)-4, respectively (Table 1).
The contents of Ag and Au and the structure of Ag–Au bimetallic nanocomposites were significantly affected by the dosage of HAuCl4, e.g., the morphology of the above nanocomposites changed from porous to hollow with increasing HAuCl4 solution volume. Thus, when 1.0 mL of HAuCl4 was used, porous structures were observed (Ag–Au@P(NIPAM-co-MAPTMS)-1; Fig. 5a and b), whereas sharp edges appeared when the volume equaled 3.0 mL (Ag–Au@P(NIPAM-co-MAPTMS)-2; Fig. 5c and d). As the volume of HAuCl4 increased to 6.0 mL, the holes in the porous structure of Ag–Au@P(NIPAM-co-MAPTMS)-3 bimetallic hybrid microgels became larger (Fig. 5e and f), and a well-pronounced hollow structure was finally obtained for Ag–Au@P(NIPAM-co-MAPTMS)-4 (Fig. 5g and h).
Fig. 5. TEM images of Ag–Au@P(NIPAM-co-MAPTMS) bimetallic hybrid microgels obtained at different volumes of HAuCl4 solution. (a and b) 1.0 mL, (c and d) 3.0 mL, (e and f) 6.0 mL, (g and h) 9.0 mL.

To determine the structures of Ag and Au in Ag–Au@P(NIPAM-co-MAPTMS)-2, the above nanocomposite was probed by elemental mapping (HAADF-STEM), which showed that Ag and Au elements were uniformly distributed in the core region of Ag–Au@P(NIPAM-co-MAPTMS)-2 bimetallic hybrid microgels (ESI, Fig. S3†).
As mentioned above, the structure of Ag–Au bimetallic nanocomposites embedded in P(NIPAM-co-MAPTMS) hybrid microgels was regulated by changing the amount of added HAuCl4, which was rationalized as follows. During the GR reaction between Ag NPs and HAuCl4, AuCl4− ions acted as electron donors and were adsorbed on the surface of Ag NPs. The addition of a small amount of Au initially led to the formation of pits on the Ag surface that subsequently evolved into voids of Ag–Au bimetallic nanocomposites due to the dissolution of Ag in the particle interior. Thus, at increased amounts of AuCl4− ions, Au–Ag bimetallic nanocomposites with hollow interiors and porous walls formed as a result of Ag dealloying from Au–Ag alloyed walls. The formation of these hollow structures was attributed to the difference of Ag and Au diffusion rates, which is known as the Kirkendall effect,21 with the overall mechanism schematically illustrated in Fig. 6.
Fig. 6. The schematic illustration of the possible mechanism for Ag–Au bimetallic nanocomposites with pore structure by GR reaction.

The crystal structures of Ag–Au@P(NIPAM-co-MAPTMS) bimetallic hybrid microgels with different Ag and Au contents were determined by HR-TEM. Based on the crystal lattice spacings in different regions of Ag–Au@P(NIPAM-co-MAPTMS) (ESI, Fig. S4†), their two lattice spacing were determined as 0.202 and 0.233 nm, being similar to those between Ag and Au (200) and (111).51 In addition, the Fast Fourier Transform (FFT) maps of different Ag–Au@P(NIPAM-co-MAPTMS) bimetallic hybrid microgels showed polycrystalline rings and thus indicated that the formed nanocomposites exhibited a polycrystalline structure.9
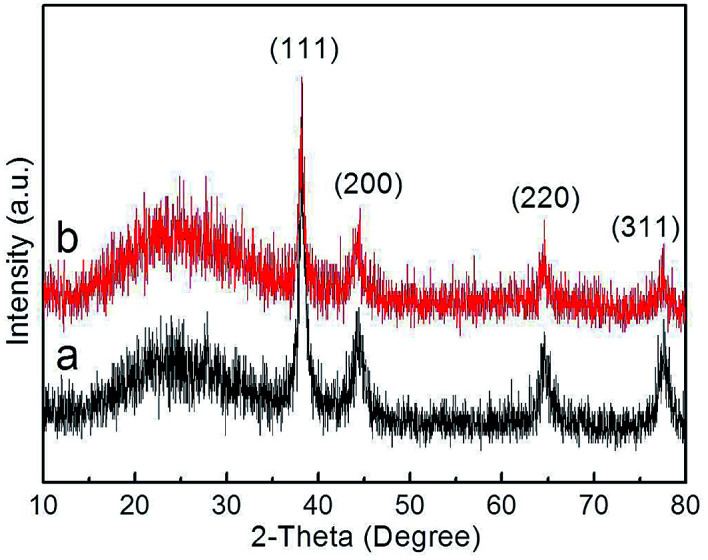
The crystal structures of composite materials were investigated by XRD (Fig. 7). For Ag@P(NIPAM-co-MAPTMS), the broad peak at ∼20° was assigned to amorphous P(NIPAM-co-MAPTMS) shell layers. Compared to those of Ag@P(NIPAM-co-MAPTMS) (Fig. 7a), the peaks of Ag–Au@P(NIPAM-co-MAPTMS) at 38.1°, 44.3°, 64.4°, and 77.4° corresponding to Au or Ag NPs (face-centered cubic, JCPDF 04-783 and 04-784) and the diffraction peaks of Au and Ag were obviously weakened (Fig. 7b), which indicated the occurrence of Ag–Au alloying.52
Fig. 7. XRD patterns of the hybrid microgels. (a) Ag@P(NIPAM-co-MAPTMS), (b) Ag–Au@P(NIPAM-co-MAPTMS)-3.
The chemical compositions of the prepared microgels were further probed by XPS. Both Ag@P(NIPAM-co-MAPTMS) (Fig. 8A-a) and Ag–Au@P(NIPAM-co-MAPTMS)-3 showed typical signals of C, N, O, Ag, and Si, with an additional signal of Au observed for the latter microgel (Fig. 8A-b). The two peaks at 373.5 and 367.5 eV in the high-resolution XPS spectrum of Ag–Au@P(NIPAM-co-MAPTMS)-3 were ascribed to Ag 3d3/2 and Ag 3d5/2 transitions, respectively (Fig. 8B). The splitting of the Ag 3d doublet was estimated as 6.0 eV, confirming the metallic nature of Ag.53 The two peaks at 87.4 and 83.7 eV were assigned to Au 4f5/2 and Au 4f7/2 transitions, respectively, with the splitting of the Au 4f doublet (3.7 eV; Fig. 8C) also confirming the metallic nature of Au.54
Fig. 8. Survey XPS spectra of hybrid microgels (A) and Ag 3d (B) and Au 4f (C) spectra of Ag–Au@P(NIPAM-co-MAPTMS)-3. (a) Ag@P(NIPAM-co-MAPTMS), (b) Ag–Au@P(NIPAM-co-MAPTMS)-3.

Comparison of Ag 3d and Au 4f spectra of Ag–Au@P(NIPAM-co-MAPTMS) bimetallic hybrid microgels showed that the binding energies of the corresponding peaks decreased with increasing Au content (Fig. 9). Thus, when the volume of HAuCl4 increased from 3 to 9 mL, the binding energies of Ag 3d5/2 and Ag 3d3/2 peaks decreased from 368 to 367.5 eV and from 374 to 373.5 eV, respectively (Fig. 9A), with the binding energies of Au 4f7/2 and Au 4f5/2 peaks decreasing from 84 to 83.5 eV and from 87.7 to 87.2 eV, respectively (Fig. 9B). XPS analyses of Ag–Au@P(NIPAM-co-MAPTMS) bimetallic hybrid microgels with different Ag–Au contents showed that with increasing HAuCl4 volume, the Au content of the Ag–Au alloy increased, while that of Ag0 decreased and that of Ag+ increased.55 The observed binding energy changes of Ag and Au peaks in the spectra of Ag–Au@P(NIPAM-co-MAPTMS) bimetallic hybrid microgels were in accordance with the mechanism of the redox reaction between Ag NPs and HAuCl4 (ESI, Table S1†).
Fig. 9. Ag 3d (A) and Au 4f (B) XPS spectra of Ag–Au@P(NIPAM-co-MAPTMS) bimetallic hybrid microgels prepared using different volumes of HAuCl4. (a) 3.0 mL, (b) 6.0 mL, (c) 9.0 mL.

Thermosensitivity of hybrid microgels
Hydrodynamic diameter variations of Ag@PNIPAM and Ag@P(NIPAM-co-MAPTMS) were measured by dynamic light scattering at 20–50 °C. Data were acquired after 5 min sample dispersion stabilization at each temperature, with all measurements repeated five-fold and the final results reported as averages. The swelling degree of temperature-induced of Ag@P(NIPAM-co-MAPTMS) hybrid microgels was less than that of Ag@PNIPAM microgels (Fig. 10). Thus, Ag@PNIPAM microgels featured a hydrodynamic diameter of 270.3 nm at 20 °C, with the significant decrease to 231.1 nm observed at 32 °C (Fig. 10a) rationalized by the fact that the latter temperature corresponded to the LCST of PNIPAM, above which the PNIPAM chains exist in a shrinked state.56
Fig. 10. Variations of microgel hydrodynamic diameter with temperature. (a) Ag@PNIPAM, (b) Ag@P(NIPAM-co-MAPTMS).
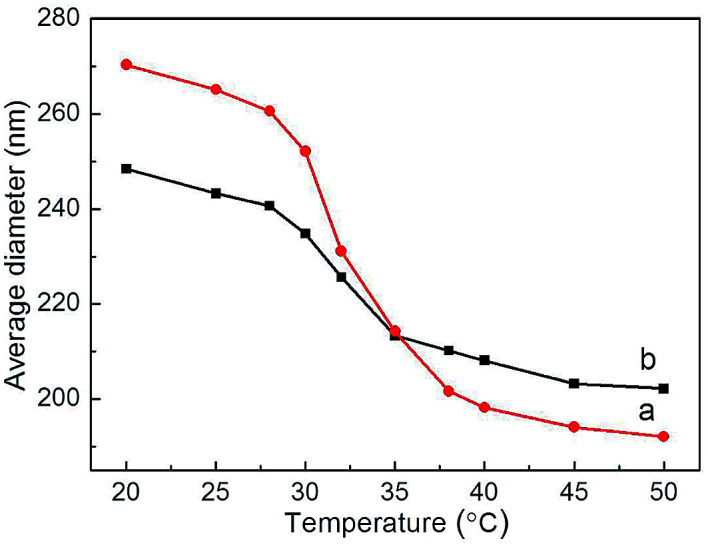
The average hydrodynamic diameter of Ag@P(NIPAM-co-MAPTMS) equaled ∼248.5 nm at 20 °C, decreasing to 225.7 nm at 32 °C (Fig. 10b). Thus, the swelling and shrinkage degrees of the above microgel were obviously reduced by the introduction of rigid MAPTMS segments into polymer chains, which inhibited the swelling and shrinkage of the shell layer.40 The thermosensitivity of Ag@P(NIPAM-co-MAPTMS) hybrid microgels also decreased after MAPTMS introduction, confirming that the shell layer comprised organic–inorganic network chains.
SPR properties of hybrid microgels
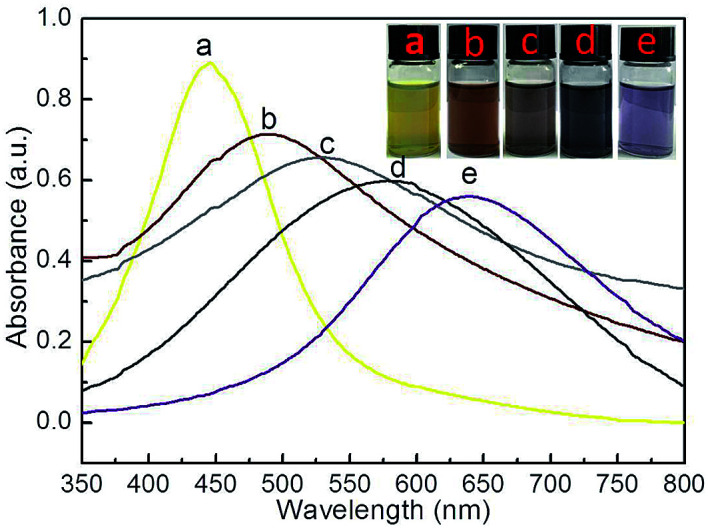
The UV-Vis spectra of Ag–Au@P(NIPAM-co-MAPTMS) bimetallic hybrid microgels obtained at different dosages of HAuCl4 revealed that the absorption peak maximum at 446 nm (Fig. 11a) was broadened and shifted to longer wavelengths with increasing HAuCl4 loading (488, 531, 578, and 638 nm at 1, 3, 6, and 9 mL, respectively; Fig. 11b–e), with the concomitant dispersion color change from bright yellow to light purple reflecting the formation of Ag–Au alloy nanocomposites at the atomic level.57
Fig. 11. UV-Vis spectra and photographs of Ag–Au@P(NIPAM-co-MAPTMS) hybrid microgels obtained using different volumes of HAuCl4. (a) 0 mL, (b) 1 mL, (c) 3 mL, (d) 6 mL, (e) 9 mL.
To study the effect of solvent on the SPR of Ag@P(NIPAM-co-MAPTMS) and Ag–Au@P(NIPAM-co-MAPTMS)-3, the UV-Vis absorption spectra of these composites were recorded in water, acetone, tetrahydrofuran, and carbon disulfide (Fig. 12), with the former composite showing absorption maxima at 446, 454, 460, and 467 nm, respectively (Fig. 12a). Similarly, the absorption maxima of Ag–Au@P(NIPAM-co-MAPTMS)-3 progressively shifted to longer wavelengths (Fig. 12b) with increasing solvent refractive index (H2O 1.333, acetone 1.358, tetrahydrofuran 1.405, carbon disulfide 1.627).58 In addition, the shrinkage of P(NIPAM-co-MAPTMS) in different solvents also changed the refractive index of the micro-environment surrounding Ag or Ag–Au NPs and hence shifted the position of the absorption peak.
Fig. 12. UV-Vis spectra of hybrid microgels in different solvents. (a) Ag@P(NIPAM-co-MAPTMS), (b) Ag–Au@P(NIPAM-co-MAPTMS)-3.

The Ag–Au nanocomposites exhibited the environmental regulated LSPR that endowed from the thermosensitivity of P(NIPAM-co-MAPTMS)-based organic–inorganic hybrid microgels, as the temperature was raised from 20 to 50 °C (with complete microgel collapse observed at the latter temperature), the LSPR peak positions of Ag@P(NIPAM-co-MAPTMS) and Ag–Au@P(NIPAM-co-MAPTMS)-3 shifted from 446 to 452 nm and from 578 to 585 nm, respectively (Fig. 13a and b), which was ascribed to the collapse of P(NIPAM-co-MAPTMS) polymer shells resulting in enhanced Rayleigh scattering due to increasing the refractive index contrast with the solvent. The above red shifts of Ag and Ag–Au NPs peaks with increasing temperature were attributed only to the local refractive index increase resulting from water expulsion during heating-induced volume phase transitions.28,47 Therefore, it was concluded that the optical properties of Ag–Au bimetallic nanocomposites stabilized by P(NIPAM-co-MAPTMS) organic–inorganic hybrid network structures can be regulated by solvent and temperature changes. Moreover, the variation of the SPR band position further confirmed the successful encapsulation of Ag and Ag–Au NPs by the polymer microgel shell.
Fig. 13. UV-Vis spectra of hybrid microgels recorded at different temperatures (a and b) and extinction peak wavelength change as the hybrid microgels solution was heated up to 50 °C followed by cooling to 20 °C for multiple cycles (c and d). (a and c) Ag@P(NIPAM-co-MAPTMS), (b and d) Ag–Au@P(NIPAM-co-MAPTMS)-3.

Fig. 13c shows the values of surface plasmon resonance wavelength of the Ag@P(NIPAM-co-MAPTMS) hybrid microgels as the temperature increases from 20 °C to 50 °C, followed by cooling down to 20 °C, and then increases again. Initially, at 20 °C, a single peak at 445 nm of the extinction spectrum shifts to 451 nm upon heating up to 50 °C. After cooling back to 20 °C, the peak at 451 nm returns to the previous 445 nm. This change is related to the fact that the hybrid microgel at 20 °C is in a swelling state, while that at 50 °C is in a shrinking state. Generally, the shrinkage makes refractive index increase, resulting in absorption spectrum red shift. It is this reason that the SPR peak of Ag NPs shows red shift as the temperature increase from 20 °C to 50 °C. Similarly, Ag–Au@P(NIPAM-co-MAPTMS)-3 hybrid microgels also showed SPR variation with temperature change (Fig. 13d). In fact, the shrinkage of the microgel could lead to a decrease of distance between Ag NPs or Ag–Au NPs, but this shrinkage is not enough to make Ag NPs or Ag–Au NPs aggregate due to Ag NPs and Ag–Au NPs immobilized in the networks of the hydrogel. Generally, the aggregation of metal nanoparticles is irreversible. Here, the reversible variation of SPR with temperature change is possibly related to the reversible change of refractive index rather than the aggregation. The similar explanation for this phenomenon has been proposed in literature.59
Catalytic activity of hybrid microgels
The catalytic reduction of 4-NP by NaBH4 was monitored in situ by measuring the intensity of the absorbance peak of 4-NP at 400 nm. In the absence of the catalyst, no absorption intensity change was observed after 30 min (ESI, Fig. S5†), indicating that the non-catalytic reduction of 4-NP hardly occurred.
When the reduction of 4-NP was performed in the presence of Ag–Au@P(NIPAM-co-MAPTMS) nanocatalysts with different Au contents (catalyst concentration = 0.15 g L−1, temperature = 25 °C), the above absorption intensity gradually decreased, and a new absorption band of 4-aminophenol appeared at 315 nm (Fig. 14).60 Simultaneously, the color of the reaction mixture changed from yellow to colorless. The time required for the complete reduction of 4-NP catalyzed by Ag@P(NIPAM-co-MAPTMS) equaled ∼42 min (Fig. 14a), decreasing to 24 and 17 min in cases of Ag–Au@P(NIPAM-co-MAPTMS)-2 and Ag–Au@P(NIPAM-co-MAPTMS)-3, respectively (Fig. 14b and c). Thus, the reaction rate was greatly improved by increasing the Au content of Ag–Au bimetallic nanocomposites.61,62
Fig. 14. UV-Vis absorption spectra recorded during the reduction of 4-NP catalyzed by different nanocomposite catalysts (a–c) and plots of ln(At/A0) vs. time (d). (a) Ag@P(NIPAM-co-MAPTMS), (b) Ag–Au@P(NIPAM-co-MAPTMS)-2, (c) Ag–Au@P(NIPAM-co-MAPTMS)-3.

The kinetics of the above catalytic reduction was investigated by plotting ln(At/A0) vs. reaction time (Fig. 14d) (At and A0 denote absorbances at 400 nm at times t and 0, respectively). Taking into account the fact that NaBH4 was present in large excess with respect to 4-NP, a pseudo-first-order model with respect to the latter was applied. The apparent rate constants (kapp) determined from the linear plot of ln(At/A0) vs. reduction time at 25 °C equaled 0.062, 0.123, and 0.174 min−1 for Ag@P(NIPAM-co-MAPTMS), Ag–Au@P(NIPAM-co-MAPTMS)-2 and Ag–Au@P(NIPAM-co-MAPTMS)-3, respectively, confirming that the catalytic activity of Ag–Au@P(NIPAM-co-MAPTMS) bimetallic hybrid microgels significantly increased with increasing relative content of Au. Moreover, the porous Ag–Au bimetallic nanocomposites provided a large surface area for catalytic reaction, which significantly enhanced the reaction rate.63 Notably, the P(NIPAM-co-MAPTMS) shell not only improved the stability and dispersity of Ag–Au bimetallic nanocomposites, but also provided a restricted reaction space for increasing the local concentration of the reaction substrate. Therefore, the temperature-dependent swelling and shrinkage of the above shell can be used to regulate catalytic activity.
To verify the temperature sensitivity of the reduction of 4-NP with NaBH4 in the presence of Ag–Au@P(NIPAM-co-MAPTMS)-3 bimetallic hybrid microgels, their catalytic performances were evaluated at different temperatures. The corresponding UV-Vis absorption spectra for Ag–Au@P(NIPAM-co-MAPTMS)-3 (ESI, Fig. S6†) indicated that the reaction completion times equaled 17, 16, 19, 12, and 10 min at 25, 30, 32, 35, and 40 °C, respectively, with the absence of a monotonous reaction rate increase implying that catalysis by Ag–Au@P(NIPAM-co-MAPTMS)-3 was inhibited at a certain temperature due to the thermosensitivity of P(NIPAM-co-MAPTMS) microgels.
After a certain incubation time, kapp values were determined at different reaction temperatures (Fig. 15a), equaling 0.174, 0.190, 0.162, 0.282, and 0.320 min−1 for reduction catalyzed by Ag–Au@P(NIPAM-co-MAPTMS)-3 at 25, 30, 32, 35, and 40 °C, respectively (Fig. 15b). Interestingly, the change of reaction rate with increasing temperature could not be well described by the Arrhenius equation, rather comprising three distinct stages. In the first stage, when the temperature was increased from 25 to 30 °C, PNIPAM existed in an extended state, favoring the diffusion of reactants into the domains of Ag–Au bimetallic nanocomposites. As a result, kapp increased, and the catalytic reaction was accelerated. In the second stage, at temperatures (30 to 32 °C) close to the LCST of PNIPAM, PNIPAM segments existed in a compact state, therefore inhibiting the contact of reactants with Ag–Au and decreasing kapp. In the third stage (32 to 40 °C), the increase of temperature overrode the above inhibitory effect,34 and the value of kapp continued to increase. Thus, the above kapp–temperature correlation indicated that the chemical environment of Ag–Au nanocomposites in Ag–Au@P(NIPAM-co-MAPTMS)-3 was significantly changed upon crossing the LCST threshold. Based on these results, we concluded that catalysis by Ag–Au@P(NIPAM-co-MAPTMS)-3 could be effectively controlled by changing the reaction temperature.
Fig. 15. Plots of ln(At/A0) vs. time for the reduction of 4-NP catalyzed by Ag–Au@P(NIPAM-co-MAPTMS)-3 at different temperatures (a) and plot of kapp as a function of temperature (b).

In order to further explore the effect of temperature on the kapp, the activation energy (Ea), Arrhenius factor (A), enthalpy (ΔH) and entropy (ΔS) of activation for catalytic reduction of 4-NP before and after LCST was calculated according to Arrhenius and Eyring equation.64–66 As can be seen, the Ea of before LCST and after LCST are 1.25 × 104 J mol−1 and 1.80 × 104 J mol−1, respectively (ESI Table S2†). The value of A and Ea is greater at below LCST as compared to that at above LCST, which shows that catalytic reduction is more favourable at high temperatures as compared to low temperatures. The value of activation parameters at below LCST are ΔH = 0.97 × 104 J mol−1 and ΔS = −260.89 J mol−1 K−1, and that above LCST are ΔH = 1.39 × 104 J mol−1, ΔS = −244.60 J mol−1 K−1. It should be noted that the high value of ΔH and ΔS at above LCST suggests that reaction is more favourable at high temperatures as compared to that at low temperatures (below LCST). Above 35 °C, the collapsed P(NIPAM-co-MAPTMS) organic–inorganic gel shell could be envisaged to require more energy for the reactant to close to nanocatalysts in the core part. It suggests that reaction is more favourable thermodynamically at high temperatures.
Mechanism of catalytic activity regulation
To explore the catalytic activity regulation mechanism of Ag–Au bimetallic nanocomposites loaded with P(NIPAM-co-MAPTMS) organic–inorganic hybrid microgels, we compared the activities of Ag–Au@PNIPAM and Ag–Au@P(NIPAM-co-MAPTMS)-3 with identical contents of Ag–Au nanocomposites at temperatures of 25, 32, and 40 °C. As a result, the times required for the Ag–Au@PNIPAM catalyzed reduction of 4-NP to proceed to completion were determined as 19, 21, and 13 min at 25, 32, and 40 °C, respectively, with kapp values determined as described above (ESI, Fig. S7†).
Fig. 16 shows the temperature dependence of kapp for Ag–Au@PNIPAM and Ag–Au@P(NIPAM-co-MAPTMS)-3, demonstrating that the latter catalyst exhibited higher activity than the former at identical temperatures, which showed that the introduction of the rigid MAPTMS into PNIPAM chains enhanced substrate mass transfer.
Fig. 16. Variation of kapp with temperature for the reduction of 4-NP catalyzed by Ag–Au@P(NIPAM-co-MAPTMS)-3 and Ag–Au@PNIPAM.
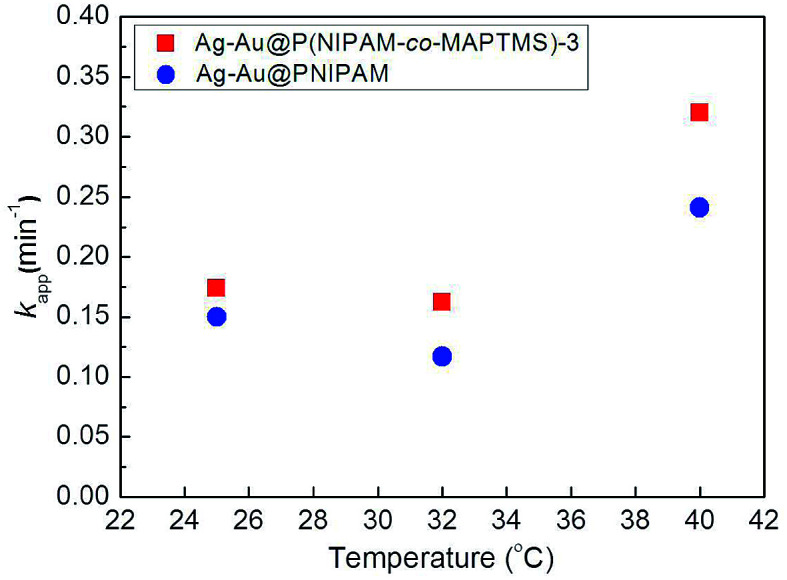
Upon a temperature increase from 25 to 32 °C, the kapp of Ag–Au@PNIPAM decreased from 0.150 to 0.117 min−1, while that of Ag–Au@P(NIPAM-co-MAPTMS)-3 decreased from 0.174 to 0.162 min−1. When the temperature was raised to 40 °C, the reduction rate significantly increased in the case of Ag–Au@P(NIPAM-co-MAPTMS). Compared to pure PNIPAM network chains, the swelling and shrinkage of P(NIPAM-co-MAPTMS) organic–inorganic hybrid microgels was limited by the introduced rigid MPTMS segments. Close to its LCST, the PNIPAM shell layer encapsulating Ag–Au bimetallic nanocomposites existed in a completely collapsed state, hindering contact between reactants and the metal catalyst. In Ag–Au@P(NIPAM-co-MAPTMS), the thermosensitive PNIPAM chains were separated by rigid MAPTMS fragments, which inhibited the shrinkage of the former and allowed the facile diffusion of 4-NP and its contact with the Ag–Au nanocatalyst. When the temperature was further increased to 40 °C, P(NIPAM-co-MAPTMS) hybrid microgels could easily maintain their highly porous structure, which favored substrate diffusion and increased its local concentration. Correspondingly, the enhanced permeability of the P(NIPAM-co-MAPTMS) organic–inorganic layer shell could provide microdomains for the diffusion and mass transfer of the substrate to Ag–Au bimetallic nanocomposites. Therefore, Ag–Au@P(NIPAM-co-MAPTMS) exhibited higher catalytic activity than Ag–Au@PNIPAM, with the above mechanism schematically illustrated in Fig. 17.
Fig. 17. The diagram of a possible catalytic activity regulation mechanism with Ag–Au@P(NIPAM-co-MAPTMS)-3 and Ag–Au@PNIPAM.

The comparison of apparent rate constant and intrinsic rate constant of our nanocatalytic systems to the literature recently reported on silver nanoparticles loaded hybrid microgels was presented in ESI (Table S3†). Comparing to other reports, our prepared Ag–Au bimetallic nanocomposites stabilized with P(NIPAM-co-MAPTMS) organic–inorganic hybrid microgels exhibited higher catalytic activity for reduction of 4-NP below and above LCST. The reason is that three-dimensional structure of the P(NIPAM-co-MAPTMS) could provide microdomains for substrate diffusion and mass transfer due to the introduction of the rigid MAPTMS into PNIPAM network chains.
Reusability of composite catalysts
The stability of Ag–Au bimetallic nanocatalysts was explored by determining the catalytic conversion of Ag–Au@P(NIPAM-co-MAPTMS)-3 under typical conditions at 25 °C after re-cycling. The obtained results showed that although the above conversion decreased after four re-use cycles, it was still above 89% (ESI, Fig. S8†), with this slight reduction ascribed to the partial blockage of active sites.67 Thus, Ag–Au@P(NIPAM-co-MAPTMS) bimetallic hybrid microgel composites could serve as stable, efficient, and reusable catalysts.
Conclusions
In this study, we herein propose a novel route for the fabrication of porous Ag–Au bimetallic nanocomposites stabilized by organic–inorganic hybrid microgels with temperature sensitivity, and these structural features are expected to enhance the catalytic reactivity of nanometal particles by intensification of the mass transfer. To verify the feasibility mentioned above, Ag–Au bimetallic nanocomposites stabilized with P(NIPAM-co-MAPTMS) organic–inorganic hybrid microgels were prepared, and the reduction of 4-NP by NaBH4 catalyzed by the bimetallic hybrid microgels was performed. Based on the experiment result, the following conclusions can be drawn. (1) Introduction of rigid PMAPTMS segments into PNIPAM microgel could enhance the three-dimensional porous inflexibility of P(NIPAM-co-MAPTMS), which is favor for the diffusion and mass transfer; (2) P(NIPAM-co-MAPTMS) could immobilize Ag–Au bimetallic nanoparticles into network to inhibit the aggregation of metal nanoparticles; (3) both SPR and catalytic properties of Ag–Au@P(NIPAM-co-MAPTMS) are reversibly sensitive to external temperature due to presence of temperature sensitive PNIPAM chains. All conclusions aforementioned are significant to construction of hybrid microgels used in catalysis.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China (21173141), Key Industry Project of Shaanxi Province of China (2011K08-14), National 111 Project of China (B14041), and Changjiang Scholars and Innovative Research Team in University of China (IRT1070).
Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01343h
Notes and references
- Tan R. L. S. Song X. H. Chen B. Chong W. H. Fang Y. Zhang H. Wei J. Chen H. Y. Nanoscale. 2016;8:3447–3453. doi: 10.1039/C5NR07763J. [DOI] [PubMed] [Google Scholar]
- Wu Y. R. Sun X. J. Yang Y. Li J. M. Zhang Y. Qin D. Acc. Chem. Res. 2017;50:1774–1784. doi: 10.1021/acs.accounts.7b00216. [DOI] [PubMed] [Google Scholar]
- Yin Z. Wang Y. Song C. Q. Zheng L. H. Ma N. Liu X. Li S. W. Lin L. L. Li M. Z. Xu Y. Li W. Z. Hu G. Fang Z. Y. Ma D. J. Am. Chem. Soc. 2018;140:864–867. doi: 10.1021/jacs.7b11293. [DOI] [PubMed] [Google Scholar]
- Zhang H. Jin M. Xia N. Y. Chem. Soc. Rev. 2012;41:8035–8049. doi: 10.1039/C2CS35173K. [DOI] [PubMed] [Google Scholar]
- Wang A. Q. Liu X. Y. Mou C. Y. Zhang T. J. Catal. 2013;308:258–271. doi: 10.1016/j.jcat.2013.08.023. [DOI] [Google Scholar]
- Gilroy K. D. Ruditskiy A. Peng H. C. Qin D. Xia Y. N. Chem. Rev. 2016;116:10414–10472. doi: 10.1021/acs.chemrev.6b00211. [DOI] [PubMed] [Google Scholar]
- Ahn J. W. Wang D. Ding Y. Zhang J. W. Qin D. ACS Nano. 2018;12:298–307. doi: 10.1021/acsnano.7b06353. [DOI] [PubMed] [Google Scholar]
- Wang D. S. Li Y. D. Adv. Mater. 2011;23:1044–1060. doi: 10.1002/adma.201003695. [DOI] [PubMed] [Google Scholar]
- Menezes W. G. Neumann B. Zielasek V. Thiel K. Bäumer M. Phys. Chem. Chem. Phys. 2013;14:1577–1581. doi: 10.1002/cphc.201201100. [DOI] [PubMed] [Google Scholar]
- Monga A. Pal B. New J. Chem. 2015;39:304–313. doi: 10.1039/C4NJ01419G. [DOI] [Google Scholar]
- Cortie M. B. McDonagh A. M. Chem. Rev. 2011;111:3713–3735. doi: 10.1021/cr1002529. [DOI] [PubMed] [Google Scholar]
- Fan M. K. Lai F. J. Chou H. L. Lu W. T. Hwang B. J. Brolo A. G. Chem. Sci. 2013;4:509–515. doi: 10.1039/C2SC21191B. [DOI] [Google Scholar]
- Li C. X. Wang C. Han Q. Y. Wu Y. N. Zheng H. R. Plasmonics. 2017;12:1373–1379. doi: 10.1007/s11468-016-0396-x. [DOI] [Google Scholar]
- Wu D. J. Liu X. J. Appl. Phys. Lett. 2010;97:61904–61911. doi: 10.1063/1.3479053. [DOI] [Google Scholar]
- Ganguly M. Jana J. Palc A. Pal T. RSC Adv. 2016;6:17683–17703. doi: 10.1039/C5RA26430H. [DOI] [Google Scholar]
- Njoki P. N. Wu W. J. Zhao H. Hutter L. Schiff E. A. Maye M. M. J. Am. Chem. Soc. 2011;133:5224–5227. doi: 10.1021/ja200650w. [DOI] [PubMed] [Google Scholar]
- Bansal A. Sekhon J. S. Verma S. S. Plasmonics. 2014;9:143–150. doi: 10.1007/s11468-013-9607-x. [DOI] [Google Scholar]
- Liu H. P. Liu T. Z. Zhang L. Han L. Gao C. B. Yin Y. D. Adv. Funct. Mater. 2015;25:5435–5443. doi: 10.1002/adfm.201502366. [DOI] [Google Scholar]
- Sun Y. G. Xia N. Y. Science. 2002;298:2176–2179. doi: 10.1126/science.1077229. [DOI] [PubMed] [Google Scholar]
- Sun Y. G. Xia Y. N. J. Am. Chem. Soc. 2004;126:3892–3901. doi: 10.1021/ja039734c. [DOI] [PubMed] [Google Scholar]
- Moreau L. M. Schurman C. A. Kewalramani S. Shahjamali M. M. Mirkin C. A. Bedzyk M. J. J. Am. Chem. Soc. 2017;139:12291–12298. doi: 10.1021/jacs.7b06724. [DOI] [PubMed] [Google Scholar]
- Zhu C. Z. Du D. Eychmüller A. Lin Y. H. Chem. Rev. 2015;115:8896–8943. doi: 10.1021/acs.chemrev.5b00255. [DOI] [PubMed] [Google Scholar]
- Tsao Y. C. Rej S. Chiu C. Y. Huang M. H. J. Am. Chem. Soc. 2014;136:396–404. doi: 10.1021/ja410663g. [DOI] [PubMed] [Google Scholar]
- Yang L. H. Lv J. Sui Y. M. Fu W. Y. Zhou X. M. Ma J. W. Su S. Zhang W. J. Lv P. Wu D. Mu Y. N. Yang H. B. CrystEngComm. 2014;16:2298–2304. doi: 10.1039/C3CE42052C. [DOI] [Google Scholar]
- Sánchez-Muñoz O. L. Salgado J. Martínez-Pastor J. Jiménez-Villar E. Nanosci. Nanotechnol. 2012;2:1–7. doi: 10.5923/j.nn.20120201.01. [DOI] [Google Scholar]
- Lin M. Wang Y. Q. Sun X. Y. Wang W. H. Chen L. X. ACS Appl. Mater. Interfaces. 2015;7:7516–7525. doi: 10.1021/acsami.5b01077. [DOI] [PubMed] [Google Scholar]
- Contreras-Caceres R. Alonso-Cristobal P. Mendez-Gonzalez D. Laurenti M. Maldonado-Valdivia A. Garcia-Blanco F. Cabarcos E. L. Fernandez-Barbero A. Lopez-Romero J. M. Rubio-Retama J. Langmuir. 2014;30:15560–15567. doi: 10.1021/la503864f. [DOI] [PubMed] [Google Scholar]
- Dong X. Zou X. B. Liu X. Y. Lu P. Yang J. M. Lin D. L. Zhang L. Zha L. S. Colloids Surf., A. 2014;452:46–50. doi: 10.1016/j.colsurfa.2014.03.090. [DOI] [Google Scholar]
- Li G. L. Tai C. A. Neoh K. G. Kang E. T. Yang X. L. Polym. Chem. 2011;2:1368–1374. doi: 10.1039/C1PY00054C. [DOI] [Google Scholar]
- Chen S. Y. Wang L. Y. Dong X. Liu X. Y. Zhou J. F. Yang J. M. Zha L. S. RSC Adv. 2016;6:48479–48488. doi: 10.1039/C6RA04247C. [DOI] [Google Scholar]
- Xiao C. F. Wu Q. S. Chang A. P. Peng Y. H. Xu W. T. Wu W. T. J. Mater. Chem. A. 2014;2:9514–9523. [Google Scholar]
- Schild H. G. Prog. Polym. Sci. 1992;17:163–249. doi: 10.1016/0079-6700(92)90023-R. [DOI] [Google Scholar]
- Carregal-Romero S. Buurma N. J. Pérez-Juste J. Liz-Marzán L. M. Hervés P. Chem. Mater. 2010;22:3051–3059. doi: 10.1021/cm903261b. [DOI] [Google Scholar]
- Wu S. Dzubiella J. Kaiser J. Drechsler M. Guo X. H. Ballauff M. Lu Y. Angew. Chem., Int. Ed. 2012;51:2229–2233. doi: 10.1002/anie.201106515. [DOI] [PubMed] [Google Scholar]
- Xiao C. F. Chen S. M. Zhang L. Y. Zhou S. Q. Wu W. T. Chem. Commun. 2012;48:11751–11753. doi: 10.1039/C2CC36002K. [DOI] [PubMed] [Google Scholar]
- Li S. Y. Lin D. L. Zhou J. F. Zha L. S. J. Phys. Chem. C. 2016;120:4902–4908. [Google Scholar]
- Begum R. Farooqi Z. H. Ahmed E. Naseem K. Ashraf S. Sharif A. Rehan R. Appl. Organomet. Chem. 2017;31:1–8. doi: 10.1002/aoc.3563. [DOI] [Google Scholar]
- Begum R. Farooqi Z. H. Butt Z. Wu Q. S. Wu W. T. Irfan A. J. Environ. Sci. doi: 10.1016/j.jes.2017.12.003. [DOI] [PubMed] [Google Scholar]
- Farooqi Z. H. Khan S. R. Begum R. Mater. Sci. Technol. 2017;33:129–137. doi: 10.1080/02670836.2016.1170396. [DOI] [Google Scholar]
- Zhao J. Liu L. F. Zhang Y. Acta Phys.-Chim. Sin. 2015;31:1549–1558. [Google Scholar]
- Wang M. Y. Niu R. Huang M. Zhang Y. Sci. China: Chem. 2015;45:76–89. [Google Scholar]
- Li A. H. Liu J. Q. Liu G. Z. Zhang J. Z. Feng S. Y. J. Polym. Sci., Part A: Polym. Chem. 2014;52:87–95. doi: 10.1002/pola.26974. [DOI] [Google Scholar]
- Hervés P. Pérez-Lorenzo M. Liz-Marzán L. M. Dzubiella J. Lu Y. Ballauff M. Chem. Soc. Rev. 2012;41:5577–5587. doi: 10.1039/C2CS35029G. [DOI] [PubMed] [Google Scholar]
- Bastus N. G. Merkoci F. Piella J. Puntes V. Chem. Mater. 2014;26:2836–2846. doi: 10.1021/cm500316k. [DOI] [Google Scholar]
- Hong X. Wang D. S. Yu R. Yan H. Sun Y. He L. Niu Z. Q. Peng Q. Li Y. D. Chem. Commun. 2011;47:5160–5162. doi: 10.1039/C1CC10903K. [DOI] [PubMed] [Google Scholar]
- Dong Y. Ma Y. Zhai T. Y. Shen F. G. Zeng Y. Fu H. B. Yao J. N. Macromol. Rapid Commun. 2007;28:2339–2345. doi: 10.1002/marc.200700483. [DOI] [Google Scholar]
- Contreras-Cáceres R. Sánchez-Iglesias A. Karg M. Pastoriza-Santos I. Pérez-Juste J. Pacifico J. Hellweg T. Fernández-Barbero A. Liz-Marzán L. M. Adv. Mater. 2008;20:1666–1670. doi: 10.1002/adma.200800064. [DOI] [Google Scholar]
- Zhang K. Ma J. Zhang B. Zhao S. Li Y. P. Xu Y. X. Yu W. X. Wang J. Y. Mater. Lett. 2007;61:949–952. doi: 10.1016/j.matlet.2006.06.021. [DOI] [Google Scholar]
- Zhang F. Wang C. C. Colloid Polym. Sci. 2008;286:889–895. [Google Scholar]
- Choi Y. Hong S. Liu L. C. Kim S. K. Park S. Langmuir. 2012;28:6670–6676. doi: 10.1021/la202569q. [DOI] [PubMed] [Google Scholar]
- Sau T. K. Murphy C. J. J. Am. Chem. Soc. 2004;126:8648–8649. doi: 10.1021/ja047846d. [DOI] [PubMed] [Google Scholar]
- Garcia A. G. Lopes P. P. Gomes J. F. Pires C. Ferreira E. B. Lucena R. G. M. Gasparotto L. H. S. Tremiliosi-Filho G. New J. Chem. 2014;38:2865–2873. doi: 10.1039/C4NJ00041B. [DOI] [Google Scholar]
- Park J. Kim Y. Oh C. Shin S. Kim Y. Oh S. Kong S. Mater. Res. Bull. 2005;40:271–280. doi: 10.1016/j.materresbull.2004.10.012. [DOI] [Google Scholar]
- Li J. Liu C. Y. Liu Y. J. Mater. Chem. 2012;22:8426–8430. doi: 10.1039/C2JM16386A. [DOI] [Google Scholar]
- Sun J. Wu H. X. Jin Y. D. Nanoscale. 2014;6:5449–5457. doi: 10.1039/C4NR00445K. [DOI] [PubMed] [Google Scholar]
- Silva C. S. O. Baptista R. P. Santos A. M. Martinho J. M. G. Cabral J. M. S. Taipa M. A. Biotechnol. Lett. 2006;28:2019–2025. doi: 10.1007/s10529-006-9188-2. [DOI] [PubMed] [Google Scholar]
- Alissawi N. Zaporojtchenko V. Strunskus T. Kocabas I. Chakravadhanula V. S. K. Kienle L. Garbe-Schönberg D. Faupe F. Gold Bull. 2013;46:3–11. doi: 10.1007/s13404-012-0073-6. [DOI] [Google Scholar]
- Guo W. S. Pleixats R. Shafir A. Chem.–Asian J. 2015;10:2437–2443. doi: 10.1002/asia.201500290. [DOI] [PubMed] [Google Scholar]
- Qian Z. X. Guye K. N. Masiello D. J. Ginger D. S. J. Phys. Chem. B. 2017;121:1092–1099. doi: 10.1021/acs.jpcb.7b00013. [DOI] [PubMed] [Google Scholar]
- Menumerov E. Hughes R. A. Neretina S. Nano Lett. 2016;16:7791–7797. doi: 10.1021/acs.nanolett.6b03991. [DOI] [PubMed] [Google Scholar]
- Liu X. W. Wang D. S. Li Y. D. Nano Today. 2012;7:448–466. doi: 10.1016/j.nantod.2012.08.003. [DOI] [Google Scholar]
- Jiang H. L. Akita T. Ishida T. Haruta M. Xu Q. J. Am. Chem. Soc. 2011;133:1304–1306. doi: 10.1021/ja1099006. [DOI] [PubMed] [Google Scholar]
- Liu K. Bai Y. C. Zhang L. Yang Z. B. Fan Q. K. Zheng H. Q. Yin Y. D. Gao C. B. Nano Lett. 2016;16:3675–3681. doi: 10.1021/acs.nanolett.6b00868. [DOI] [PubMed] [Google Scholar]
- Khan S. R. Farooqi Z. H. Zaman W. Ali A. Begum R. Kanwal F. Siddiq M. Mater. Chem. Phys. 2016;171:318–327. doi: 10.1016/j.matchemphys.2016.01.023. [DOI] [Google Scholar]
- Begum R. Rehan R. Farooqi Z. H. Butt Z. Ashraf S. J. Nanopart. Res. 2016;18:231. doi: 10.1007/s11051-016-3536-5. [DOI] [Google Scholar]
- Demirci S. Sahiner N. Water, Air, Soil Pollut. 2015;226:64. doi: 10.1007/s11270-015-2332-7. [DOI] [Google Scholar]
- Davoodnia A. Khojastehnezhad A. J. Chin. Chem. Soc. 2012;4:57. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.