

Abstract
CRISPR-associated transposition systems allow guide RNA-directed integration of a single DNA cargo in one orientation at a fixed distance from a programmable target sequence. We define the mechanism explaining this process by characterizing the transposition regulator, TnsC, from a type V-K CRISPR-transposase system using cryo-electron microscopy (cryo-EM). Polymerization of ATP-bound TnsC helical filaments could explain how polarity information is passed to the transposase. TniQ caps the TnsC filament, establishing a universal mechanism for target information transfer in Tn7/Tn7-like elements. Transposase-driven disassembly establishes delivery of the element only to unused protospacers. Finally, structures with the transition state mimic, ADPᐧAlF3, reveals how TnsC transitions to define the fixed point of insertion. These mechanistic findings provide the underpinnings for engineering CRISPR-associated transposition systems for research and therapeutic applications.
One Sentence Summary:
Cryo-EM studies reveals the role of the AAA+ regulator TnsC for target-site selection in CRISPR-associated transposition systems.
CRISPR-associated proteins (Cas) are macromolecular machines that provide bacteria and archaea with adaptive immunity against bacteriophages and other invasive genetic elements. The RNA-guided DNA nuclease activity of CRISPR-Cas systems has been repurposed (most notably in the case of CRISPR-Cas9) for programmable genomic editing by making precise double-strand breaks (DSBs) in DNA complementary to the RNA guide(1). Although conventional CRISPR-Cas systems can generate DSBs with high fidelity at chosen DNA sites, the actual insertion of new DNA is dependent upon inefficient processes like homology-directed repair or nonhomologous end-joining. Moreover, introduction of a DSB into the host genome is dangerous as it can lead to genome instability. Excitingly there exist examples of transposons that are naturally programable for targeting: Tn7-like transposons that have co-opted type I (Cascade)(2) and type V (Cas12)(3) CRISPR-Cas systems on multiple independent occasions for guide RNA-directed transposition. These CRISPR-associated Tn7-like transposons have been shown to exhibit a single programmable DNA integration event at a precise distance and in a specific orientation with respect to the protospacer adjacent motif (PAM) site(4–6).
In both prototypic Tn7 and in the Tn7-like transposon relatives which encode CRISPR-Cas systems, the overall mechanism of transposase recruitment and insertion into a specific target site remains elusive, with little structural information to guide mechanistic studies. Despite remarkable diversity(7), every RNA-directed transposition system characterized to-date contains a CRISPR effector protein (Cas12k in this study), proteins dedicated to target capture (TniQ+TnsC), and transposase (TnsB) (Figure 1A). Previous structural studies have focused on the I-F3 Cascade-TniQ target-DNA binding complex, expressed from the element found in V. cholerae(8). While the Cascade-TniQ structure reveals the physical association between the CRISPR effector domain and TniQ, how target-DNA binding ultimately results in transposition remains mysterious.
Figure 1. ATP is crucial for both RNA-guided transposition of ShCAST, and filament assembly of AAA+ regulator TnsC.
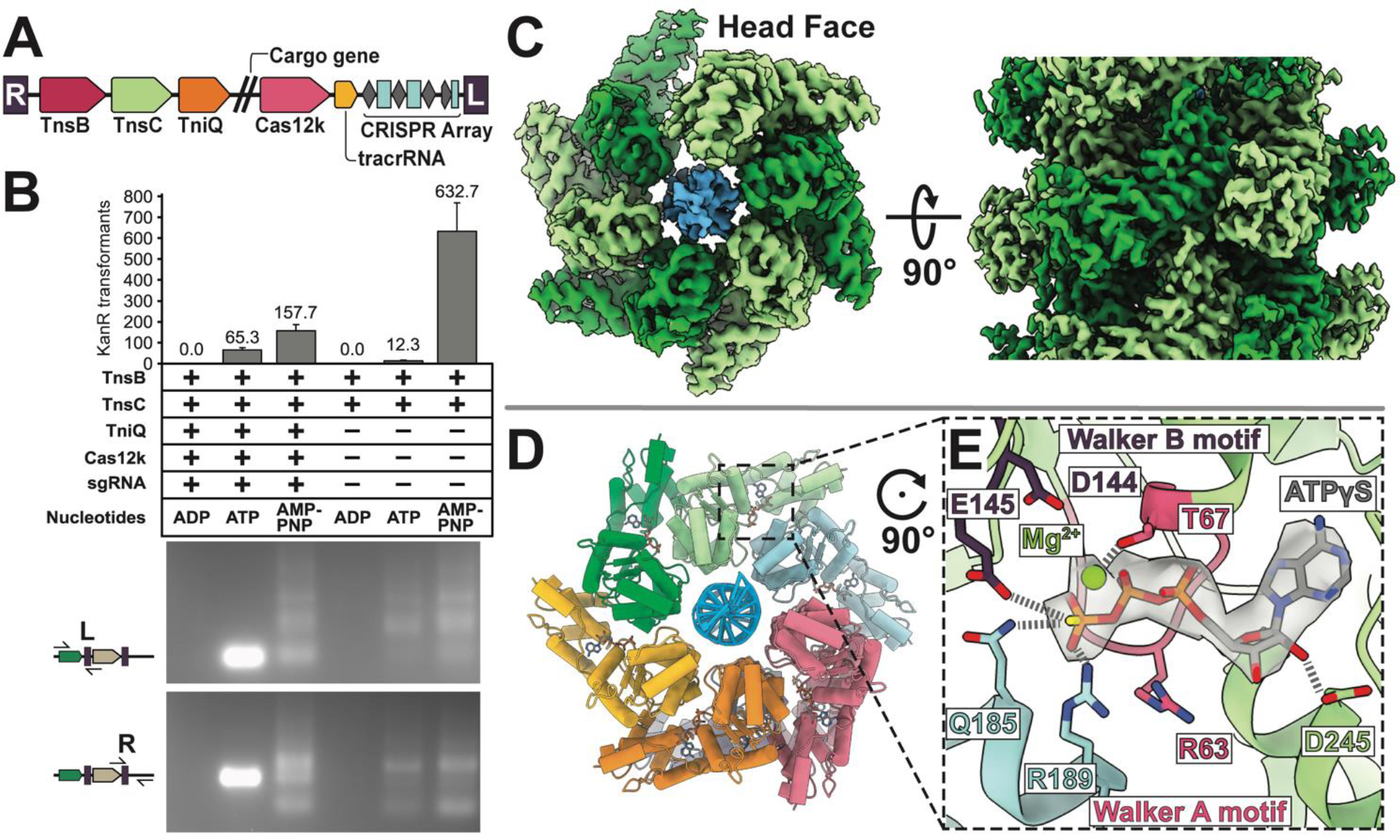
(A). The ShCAST transposon is defined by the right- (R) and left- (L) ends of the element, encoding: Cas12k RNA-binding effector, TniQ, TnsC, and TnsB, trans-activating crRNA (tracrRNA), and CRISPR array. Double slash represents the region where cargo genes are found in the transposon. (B) ATP-hydrolysis is required for targeted RNA-guided transposition. Colony counts from transformation of each deproteinated transposition reaction as a proxy for overall transposition activity are shown (one third of total reaction volume was used in transformation). Data indicate mean +/− standard deviation (n=3). Reaction mixes were additionally analyzed by PCR using a transposon end primer (L or R) along with primers flanking the target site as indicated in the schematic on the left. A high rate of on-target insertion results in a single intense band, while insertions distributed around the target plasmid results in a number of PCR products. A single representative image of n=3 replicates is shown. (C) High-resolution (3.2 Å) cryo-EM reconstruction of ATPγS-TnsC (light and dark green) forms a continuous helical filament encircling DNA (blue). The arrangement of layers (6 TnsC subunits) within the filament is referred to here as a ‘head-to-tail’ configuration. (D) Atomic model of TnsC helical filament consists of 6 subunits of TnsC arranged in a helical spiral. (E) The ATP-binding pocket follows canonical features of AAA+ proteins, with conserved Walker A (pink) and Walker B motif (purple) coordinating ATPγS-binding. The adjacent subunit (light blue) forms inter-subunit contacts that partially contribute to filament formation by forming interactions with the terminal phosphate.
CRISPR-associated transposons share crucial features with the prototypic Tn7 element. However, instead of a guide RNA complex, prototypic Tn7 uses TnsD (consisting of a TniQ domain integrated with a DNA-binding domain) to recognize a specific attachment site (attTn7) in the bacterial genome for integration. An incompletely characterized interaction between TnsD and the regulator protein TnsC (a homolog of TnsC from RNA-directed transposition systems) will recruit the core TnsA + TnsB transposase bound to the ends of the element to integrate into the target DNA(9). TnsC is a AAA+ protein that has functional parallels with MuB, from bacteriophage Mu(10). More broadly, in both Tn7/Tn7-like systems and Mu, these AAA+ proteins are important regulators of transposition: mediating both (i) transposase recruitment to the target-site and (ii) preventing multiple insertions from occurring (also referred to as target-site immunity(11)), a phenomenon also reported in CRISPR-associated transposons(4, 5).
Here, we use cryo-EM to characterize TnsC of type V-K CRISPR-associated transposase system from S. hofmanni (referred to throughout as ShCAST). We were particularly drawn to the ShCAST system because of its relative simplicity (4 protein components), and established in vitro activity(5). We discovered that in the presence of ATP, TnsC forms a continuous spiraling helical filament interacting with one DNA strand within the duplex, providing a target site search mechanism that is also capable of conducting polarity information to the transposase. We demonstrate that propagation of the TnsC filament terminates when it forms a complex with TniQ, its binding partner, establishing a target site capture mechanism for Tn7 and the extended family of Tn7-like elements. We identify a TnsB transposase-directed process that disassembles the TnsC filament, driven by ATP hydrolysis, explaining how a targeted protospacer is only used once, while future insertions are diverted to new protospacers. This same TnsB interaction with TnsC would provide a mechanism to direct the TnsB-bound transposon DNA to a TnsC-TniQ complex for transposition. Notably, we find that ADPᐧAlF3 bound TnsC collapses to a single hexamer that would be capable of conveying precise distance information from the protospacer to the point of insertion, and points to a nucleotide-based feature for stabilizing the TnsC-TniQ complex.
Results
ATP hydrolysis drives ShCAST target site selection
The TnsC regulator protein conveys essential information from the guide RNA complex to the transposase. Previous work with prototypic Tn7 and the Mu transposition system indicate that the nucleotide-bound states of the TnsC and MuB ATPases are important for target site selection. Mu is a well-studied model system for transposition: MuB ATPase forms helical filaments in the presence of ATP, and MuB disassembly is stimulated by MuA transposase (12, 13). ATP hydrolysis is required for proper target selection in both Mu(12) and prototypic Tn7(14). To test nucleotide co-factor requirements for ShCAST targeting, we performed an in vitro transposition assay (See materials and methods)(5). Clearly targeted in vitro transposition was only detected in the presence of ATP (Figure 1B). Sequencing of seven independent events indicated transposition occurred at the expected distance from the PAM with two events being simple inserts and five co-integrates consistent with previous observations(5, 15, 16) (Figure S1A). The ShCAST proteins are incapable of simple inserts on their own, these products must form by an alternate mechanism in the plasmid targets (e.g. RecA-independent recombination mechanism like template switching). Bulk analysis of the reaction products found with AMPPNP using PCR revealed a collection of products, which we could confirm resulted entirely from untargeted transposition by direct DNA sequencing (Figures 1B and S1B). Robust targeted transposition required the presence of all reaction components; however, we found considerable random transposition with TnsB, TnsC and ATP only, something not found with ADP, but stimulated with AMPPNP. This indicates that, similar to Mu and prototypic Tn7, ATP-hydrolysis (via TnsC) is required for ShCAST to select the correct target(14, 17). We also established a genetic assay that monitors full transposition events for the ShCAST system and, similar to the work of others, found a combination of RNA-targeted and off-target events (25% vs 75%, respectively, in this assay) occur with the ShCAST system (see below)(5, 18, 19). We also discovered that, similar to MuB ATPase(20), ShCAST TnsC forms helical filaments in the presence of ATP, AMPPNP, and ATPγS. To investigate the structural basis of these observations, we pursued the cryo-EM structure of TnsC.
The atomic structure of TnsC possesses a canonical AAA+ fold and forms helical filaments
We discovered that TnsC is able to adopt a helical filament (Figure S2) with a 61 screw axis encircling DNA in the ATP (or ATPγS) bound states (Figure 1C,S3), on average ~220 Å in length or ~5 full-turns (Figure S2). Therefore, each TnsC layer has two potential polymerization interfaces, which we refer to as the ‘head’ and the ‘tail’ face (Figure 1C). Helical search of the cryo-EM images (using IHRSR) defines in a rise of 6.82 Å and a twist of 60° (see materials and methods), consistent with helical layer-line analysis (Figure S2). The ATPγS-bound state is of higher resolution overall (3.2 Å vs 3.6 Å for ATPγS vs ATP, respectively, Figure S4) and with a more uniform distribution as assessed by local resolution estimates (3.0 – 4.0 Å, ATPγS vs 3.5 – 6 Å, ATP Figure S5). A full-length model (257 of 276 residues) was built into the ATPγS cryo-EM map, starting from a homology model based on distantly related AAA+ YME1 (15% sequence identity, see materials and methods)(21) (Figure 1D).
ShCAST TnsC and prototypic Tn7 TnsC each have a conserved AAA+ domain, but ShCAST TnsC lacks the N- and C-terminal extensions of prototypic Tn7 which mediate its interactions with other Tns proteins(22) (Figure S6). As sequence analysis suggests, the structure follows most of the features of the initiator clade of AAA+ proteins. A DALI(23) search reveals strong structural resemblance to the N-terminal portion of Cdc6 (global rmsd is 2.7 Å)(24) (Figure S7), highlighting the high degree of conservation within the AAA+ domain, even among ATPases of highly divergent function. Correspondingly, the highly conserved Walker A motif (G60ESRTGKT67), and Walker B motif (M140LIIDE145) (Figure 1E, S6 & S8) form a pocket for ATP binding, and mutation of the catalytic glutamate (E145) almost completely abrogates in vitro and in vivo transposition activity (Figure S9). In addition to these intra-subunit contacts, the ATP-binding pocket is completed by highly conserved R189 (the arginine finger) and Q185 (Figure S8) from an adjacent subunit. These residues form hydrogen-bonding interactions with the terminal phosphate of ATP, both recognizing ATP and stabilizing inter-subunit contacts (Figure 1E). Mutation of these residues (R189A and Q185A) causes reduced transposition activity which may be related to an impaired ability to form helical filaments (as visualized by EM) (Figure S9A, C). Interestingly, the R189A mutant allowed transposition at wild type levels in vitro, but targeting to the protospacer was lost (Figure S9A,B). Notably, D144 appears too distant (4.6 Å) from the magnesium ion to facilitate a nucleophilic attack on the gamma-phosphate of ATP (Figure 1E), suggesting that a conformational change (possibly brought about by the transposase, TnsB) is required to carry out ATP hydrolysis (see below and discussion). Taken together, these observations explain why TnsC, which is predominantly a monomer in solution, requires ATP to oligomerize into the observed helical filament, but can be readily disassembled upon ATP-hydrolysis.
Unidirectional TnsC filamentation provides a mechanism for establishing insertion polarity
ATP-bound TnsC forms a right-handed helical filament wrapping around the DNA duplex, forming a spiral ‘ladder’ of interactions with the sugar-phosphate backbone (Figure 2A). Each TnsC subunit contributes two amino-acid contacts: K103 and T121, to interact with two adjacent backbone phosphates (Figure 2A), which suggests ShCAST TnsC most likely exhibits little to no DNA-sequence specificity, similar to MuB(20) and TnsC from prototypic Tn7(25). In the ShCAST system, these protein-DNA contacts distort duplex DNA, similar to the AAA+ transposition protein IstB(26). In this case, ShCAST TnsC distorts DNA to match the helical symmetry of the filament (Figure 2B). Strikingly, these interactions are formed preferentially with one strand of the DNA duplex (Figure 2A), and the local resolution of the complementary strand of DNA is substantially worse than the bound strand (Figure S5–B,D,F). This suggests that TnsC can form filaments on single-stranded DNA that resemble filaments assembled on double-stranded DNA, a hypothesis we confirmed using EM (Figure S10). We found in vivo transposition was nearly abolished with the single K103A and T121A mutants, but unexpectedly the double mutant was reduced only 65% (Figure S9D–E). In vitro, K103A+T121A mutant transposition activity was ~10-times wild-type levels, but only 20% of these were on-target insertions (Figure S9A–B). The lack of specific DNA interactions may in part be compensated by the highly basic surface formed within the pore of the TnsC filament (Figure S11). The findings suggest that protein-DNA interactions at K103 and T121 are important for restraining transposition and directing targeting information from the effector complex to the transposase.
Figure 2. Structural analysis of TnsC-DNA interactions reveals how TnsC distorts DNA; coupled with cryo-EM based time-course experiments these data suggest that TnsC polymerizes in the 5’ to 3’ direction.

(A) Each TnsC subunit makes two major contacts with the DNA sugar-phosphate backbone (blue, sticks), forming a ladder of interactions selectively with one strand of DNA. (B) TnsC-DNA interactions result in DNA distortions. One full turn of B-form DNA spans 34 Å (gray), however the spacing between layers (41 Å) stretches the DNA, distorting one full turn of the duplex DNA to match TnsC’s helical spacing. (C) The fraction of filament collisions, otherwise referred to as head-to-head filaments, observed is influenced by both nucleotide (ATP vs ATPγS) and increase over time (more are present after 12 hours vs 10 min). Relevant 2D class averages for each sample are shown with 100 Å scale bar.
Protein filament polymerization is generally a unidirectional process. Consistent with this, TnsC filaments reconstituted with non-hydrolyzable analog ATPγS or frozen immediately upon reconstitution (with ATP) exhibit uniform polarity to cover the entire DNA substrate used in our analysis (i.e. each ‘head’ of TnsC interacts with the ‘tail’ of the adjacent TnsC layer, termed ‘head-to-tail’) (Figure 2C). While ATP-dependent filaments were stable over short timeframes, they appeared to be more dynamic with prolonged incubation. For example, when samples are incubated overnight, we observe a substantial number of two converging filaments, forming head-to-head filament structures where they meet (20% vs none, for 12 hours and 10 min reconstitution respectively, Figure 2C&S12). This is consistent with a dynamic process where the single filament we initially observed coating the entire DNA substrate could presumably partially disassociate allowing new converging filaments to form (see materials and methods for detailed explanation, Figure S12). Therefore, we hypothesize that polar growth of TnsC filaments in the 5’ to 3’ direction of the bound DNA strand is the searching mechanism that enables TnsC to search for its target site, which is defined by TniQ and Cas12k.
TniQ interacts with TnsC to define the target-site
In prototypic Tn7 and Tn7-like systems, target information is conveyed from a TniQ domain family protein called TnsD to TnsC(27). By contrast, in the RNA-guided transposition systems, the target site is chosen by a TniQ-associated guide-RNA complex. In the case of I-F3 systems TniQ is positioned at the programed insertion site via its association with cascade(8). We propose here that TnsC filaments are perpetually searching for a target-site via directional growth. This directional searching of TnsC filaments, until collision with an appropriately positioned TniQ, could explain how insertions occur only on one side of an effector complex. Therefore, diverse targeting mechanisms may have evolved by fusing TniQ to different DNA-binding domains, and in the case of guide RNA-directed systems, by associating with CRISPR-effector proteins. We believe this would serve as a unifying model accounting for diverse targeting mechanisms spanning both prototypic Tn7 and Tn7-like elements with and without CRISPR-Cas systems. In order to explore this further, we reconstituted a simplified, minimal system to probe the possible role of TniQ to act as a target-site selection factor.
To directly visualize the interaction between TnsC and TniQ and their possible roles in target selection, we incubated TnsC and TniQ together in the presence of ATP and DNA, examining the resulting complexes using high-resolution cryo-EM reconstructions (3.9 Å resolution, Figure S4). We find that TniQ selectively engages with the polymerizing face, capping the TnsC filaments (Figure 3B), consistent with the idea that the 5’ to 3’ directional propagation of the filament leads to productive interactions with the Cas12k-TniQ complex. We observe a total of two TniQ monomers; each copy interacts with two TnsCs (the TniQ-TnsC interface buried surface area is 1367.5 Å2 vs 1051.75 Å2 for adjacent TnsC subunits along the body of the TnsC filament) (Figure 3B), even though sterically three TniQ can be bound to the advancing TnsC filament. Each TniQ monomer also appears to be interacting with DNA, contacting the DNA strand that is not bound by TnsC (Figure S13A). Despite the high overall quality of the cryo-EM reconstruction, the local resolution of TniQ is too low for de novo model building (6–8 Å, see materials and methods, Figure S13B). Nevertheless, homology models of TniQ’s functional domains (helix-turn-helix and zinc-finger or HTH and ZnF, respectively, Figure S13C) built from the I-F3 TniQ crystal structure (PDB ID: 6V9P)(28) explain the cryo-EM density well (see materials and methods, Figure 3C). Both the HTH and ZnF motifs appear to interact with the same region of TnsC. In the type I-F3 system TniQ associates as a homo-dimer, however in the ShCAST system TniQ is naturally found as essentially a ‘minimal’ TniQ domain, lacking a dimerization interface (Figure 3A). Correspondingly, we do not see substantial protein-protein interactions between the two copies of TniQ. However, reminiscent of the I-F3 system, the two copies of TniQ are oriented such that the N-terminus of one TniQ monomer is close to the C-terminus of the other (Figure 3B). The ATPγS TnsC atomic model explains the remaining cryo-EM density well, indicating that TniQ binding itself does not change TnsC helical parameters.
Figure 3. Cryo-EM structure of TniQ-TnsC reveals how target-site selector protein, TniQ, binding at the target-site can interact with polymerizing TnsC.

(A) TniQ from ShCAST is truncated with respect to I-F3 TniQ. Numbers indicate residue positions. Functional domains corresponding to the helix-turn-helix (HTH, orange) motif and zinc-finger ribbon (ZnF, pink) motif are indicated. The light blue domain (only in I-F3) corresponds to the C-terminal winged helix-turn-helix motif, and is missing in ShCAST TniQ. (B) Two copies of TniQ (orange/pink) interact with the head interface of the ATP-bound TnsC filament. Each monomer of TniQ interacts with two subunits (light/dark green) of TnsC. The Cryo-EM map shown is filtered according to local-resolution estimates (Bsoft). The N-terminus of TniQ is labeled ‘N’. (C) Homology models of the helix-turn-helix (HTH) and Zinc-finger (ZnF) motifs fit well with the observed cryo-EM density map. Cryo-EM density for ShCAST TniQ is shown, TnsC (green) and DNA (blue) are displayed in ribbon.
The selective interaction of TniQ with only the advancing end of TnsC filaments explains how TniQ, likely also associated with Cas12k during the guide RNA-directed process, selects target-site insertion polarity. With this model of ShCAST TnsC-TniQ interaction in hand, we speculate on the possible higher order assembly of a guide RNA-directed target-site selection complex. Superimposing our docked ShCAST TniQ model onto the type I-F3 Cascade-TniQ structure (PDB 6PIJ) reveals that the spatial organization of TniQ’s functional domains is conserved (global RMSD is 2.5 Å, Figure S13D). Our model additionally reveals a possible path for the double-stranded DNA downstream of the R-loop (Figure S13E), which was not visualized in previous structures(8).
The TnsC ADPᐧAlF3 structure represents a target-capture state and contains spacing information
A notable feature of Tn7 and Tn7-like elements, including guide RNA-directed systems, is that the point of insertion is displaced a fixed distance from the actual machinery of target recognition, and no particular sequence is required for end joining on the target DNA. The TnsC filaments we identify here would provide a mechanism to offset the point of transposase association and point of insertion from the recognized target sequence. However, how can the precise spacing that is a hallmark of Tn7 and Tn7-like elements be dictated by an extended, continuous filament?
In prototypic Tn7 and Mu, TnsC (MuB) oligomers are disassembled by ATP-hydrolysis, stimulated by the transposase TnsB (MuA)(13, 29). We discovered that this feature is conserved in the ShCAST system: ATP bound TnsC filaments are disassembled upon addition of TnsB whereas AMPPNP bound filaments are not (Figure S14). We predict the interactions between TnsC and TnsB could be reminiscent of MuB-MuA interactions(30–32), and would therefore be located near the tail face of TnsC. This immediately suggested to us a link between ATP-hydrolysis and the precise insertional spacing from the PAM site observed for all guide RNA-directed transposition systems to-date(4, 5). While a continuous TnsC filament would be incompatible with the insertional preferences observed (i.e. fixed spacing from the PAM site), TnsC filament ‘trimming’ by TnsB may result in a specific oligomeric configuration that neatly encodes spacing information (see discussion). The TniQ association across protomers of TnsC and with DNA could additionally physically resist dissociation, or act in an allosteric fashion to allow TnsC to resist hydrolysis.
To investigate the hydrolytic state, we determined the cryo-EM structure of TnsC using a nucleotide analog that represents a hydrolysis transition state mimic, ADPᐧAlF3. Our 3.9 Å cryo-EM reconstruction (Figure 4A, S4) revealed that ADPᐧAlF3 bound TnsC assembles only in an asymmetric structure that can be described as two hexamers oriented in a head-to-head configuration, similar to the configuration found when converging filaments meet (Figure 2C, S15A). Although the same length of DNA substrate was used for reconstitution of ADPᐧAlF3 and ATP-bound TnsC (60 bp in both cases), the ADPᐧAlF3 particles were significantly shorter (the DNA-binding footprint is 22 nucleotides total, Figure S15B&C). This indicates that the ADPᐧAlF3 complex represents a conformational state of TnsC that is different from the continuous helical filament.
Figure 4. Cryo-EM structure of ADP•AlF3 bound TnsC adopts a closed-off hexameric structure that is unable to support propagation of the filament.
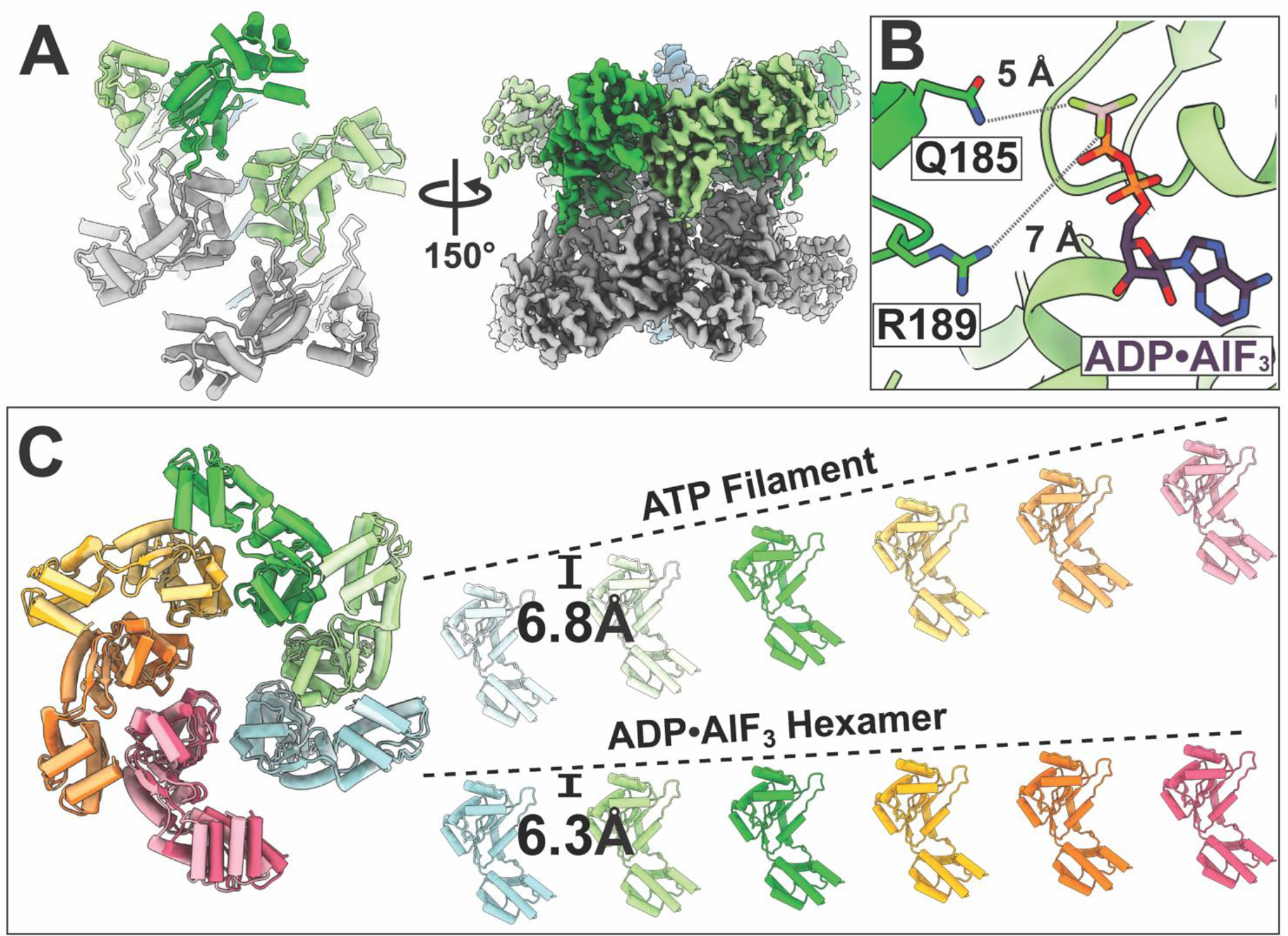
(A) The ADP•AlF3 3.9 Å cryo-EM consensus density map reveals a head-to-head configuration of hexamers bound to duplex DNA. The cartoon (left) shows the relative orientation of each hexamer. This structure cannot support the formation of more than one helical turn, indicated by the triangle symbol (representing a conformational change within the hexamer compared to the filament model in Figure 1C) and the bars to indicate that polymerization is inhibited. (B) TnsC subunits are repositioned such that the terminal phosphate is too far (5 – 7 Å) to be coordinated by inter-subunit contacts: Q185 and R189. (C) The difference in subunit position results in a smaller rise (indicated by dotted lines) in the ADP•AlF3 hexamer compared to the ATP-bound TnsC filament (6.3 vs 6.8 Å per subunit) resulting in a ‘closed’ configuration that cannot accommodate another subunit to propagate the helical filament.
The structural configuration had obvious implications for relating the distance from the protospacer to the point of integration. In the ATP-binding pocket of ADPᐧAlF3 structure, the lack of a gamma phosphate results in a loss of inter-subunit contacts (Q185 and R189 are 5–7 Å from ADPᐧAlF3), which results in an altered TnsC subunit organization (Figure 4B) and higher conformational flexibility (Movie S1). This altered binding-site configuration propagates to result in an overall smaller helical rise in the ADPᐧAlF3 state (6.3 Å vs 6.8 Å for ADPᐧAlF3 vs ATP, respectively Figure 4C). We believe this represents the conformational changes that occur upon filament disassembly. The lack of TnsC filaments in the ADPᐧAlF3 sample also suggests that, upon ATP hydrolysis, the head-to-head configuration we observe is more stable against disassembly compared to the filament. This is intriguing because the interface between the two TnsC subunits (representing a total surface area of 1332.5 Å2) corresponds to the previously identified TniQ binding-site (Figure 3B). We speculate that the observed head-to-head interface is substituting for the interface between TnsC and TniQ above and the Cas12k-TniQ complex found during bona-fide transposition. Thus, it is possible that one TnsC hexamer may remain stably bound to the Cas12k-TniQ complex after TnsB-stimulated ATP-hydrolysis.
Discussion
Previous biochemical characterization of the ShCAST system(5) and work presented here indicates that programmed insertion of the DNA element occurs at a fixed distance from the protospacer in a single orientation. From these structural studies, we form a comprehensive picture that reconciles ShCAST TnsC’s seemingly disparate proposed roles in target-site selection (Figure 5 and Movie S2) which draws strong mechanistic parallels with MuB(30, 33). Our cryo-EM structures of ATP-bound TnsC reveals filaments that polymerize unidirectionally in the 5’ to 3’ direction. We hypothesize that such filaments represent a ‘searching’ state that would encounter the Cas12k-TniQ defined target-site, with a specific polarity, on the PAM distal side of the effector. Our cryo-EM structure of TniQ-TnsC reveals the potential nature of this association at the target-site: only one face of TnsC forms productive interactions with TniQ. Enticingly, an ability of TnsB (possibly bound to the transposon ends needed for integration) to ‘follow’ TnsC to the chosen target-site could draw the element to the integration site marked by TniQ, as a similar process drives plasmid partitioning systems using ATPases(34). Interestingly, our model also accounts for the ‘immunity’ process previously reported for the ShCAST system that prevents multiple insertions from occurring at the same protospacer (5). In the post-hydrolysis state, we observe that TnsC is incapable of forming a filament. While the exact form of TnsC in the active integration complex remains to be resolved, our results suggest how TnsC filaments interact with a Cas12k-TniQ complex with the right polarity and how TnsB-mediated ATP hydrolysis defines a shortened, integration-competent state.
Figure 5. Mechanistic model describing TnsC’s role in target-site selection.
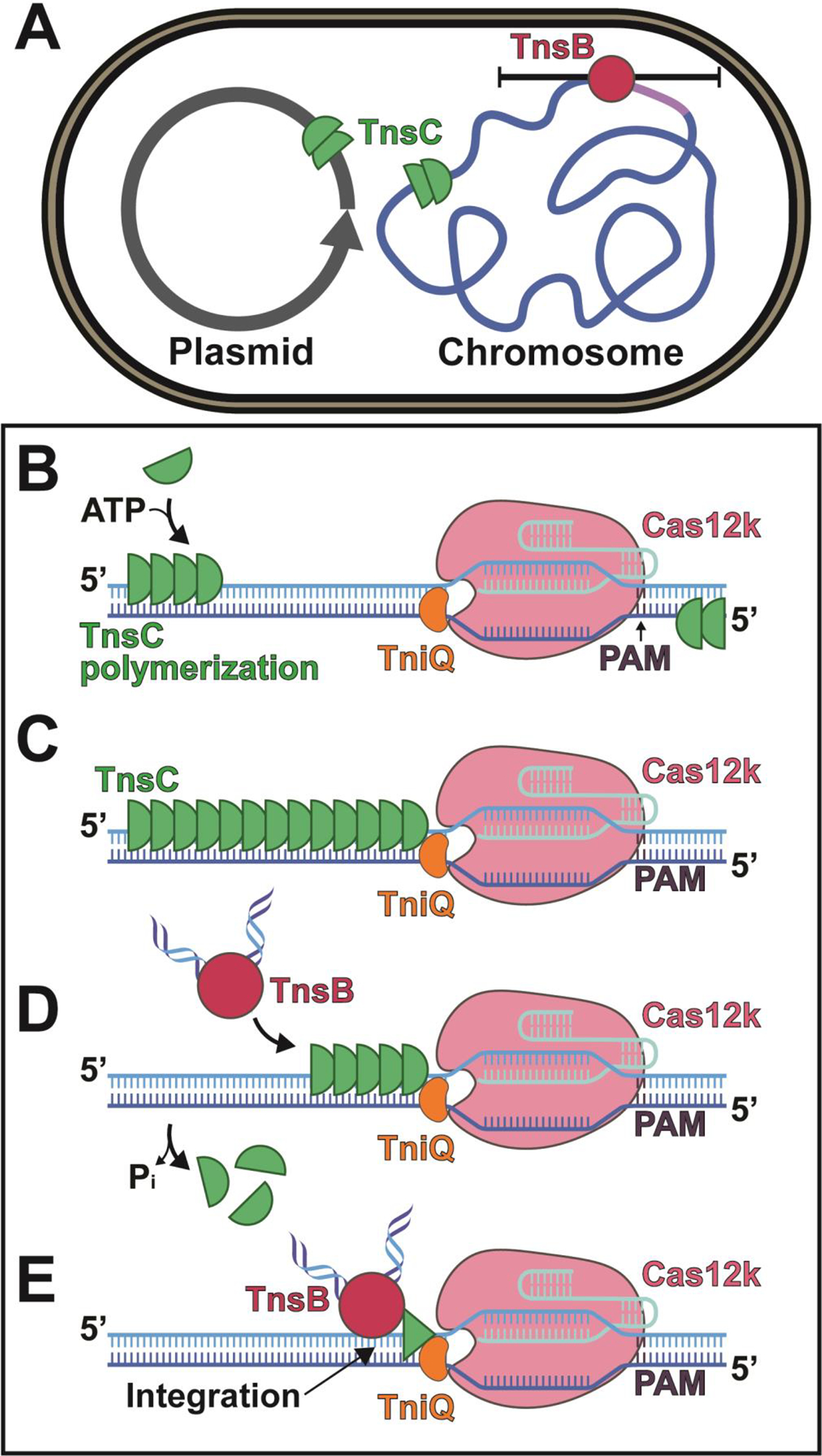
TnsC promotes exploration of alternative target-sites by polymerizing along DNA. (A) ShCAST elements may exist in mobile plasmids or in attachment sites (indicated by purple segment) within bacterial chromosomes. TnsB around the previous insertion sites (red circle) triggers TnsC depolymerization (indicated with black bars) thereby rendering sites ‘immune’ to insertion. (B-E) Results are summarized using a conceptual cartoon describing TnsC function. Movie S2 summarizes the same information using the reported structures. (B) TnsC polymerizes unidirectionally along DNA (green semi-circles) on either strand in the presence of ATP in the 5’ to 3’ direction. (C) Once TnsC encounters Cas12k-TniQ it is prevented from polymerizing further and forms a complex with TniQ. (D) TnsB (bound to the terminal ends of the transposon) is able to stimulate TnsC depolymerization and simultaneously be recruited to the target-site. (E) TnsC is disassembled to a finite oligomeric assembly (indicated by green triangle that represents a conformational change that is unable to support a continuous helical filament), which allows integration a fixed distance from the protospacer.
We have also shown that TnsC induces a distortion in its DNA substrate by enforcing the helical parameters of the TnsC filament onto the DNA. Although the implications of this slight unwinding of the DNA requires further exploration, it is tempting to speculate that this distortion could be crucial for its function. DNA distortions are a generally utilized driving force for integrases(35). As such, the high potential energy stored in the distorted DNA can be harnessed by the TnsB transposase machinery in order to facilitate forward transposition, or alternatively, may play a role in ensuring stable binding of TnsC at the target-site. In prototypic Tn7, it has been established that TnsABC transposes at a specific position in a single orientation using a DNA distortion induced by TnsD(TniQ)(36). Interestingly, a similar spacing paradigm has been proposed with the prototypic Tn7 system, but where a different mechanism is used to accomplish the same spacing feature and distribution of binding interfaces on opposite sides of TnsC(37). In the absence of TnsD, a DNA distortion associated with triplex DNA is sufficient to initiate TnsC recruitment(38), suggesting that TnsC presents a pre-distorted DNA to the TnsB transposase to accommodate target DNA. Our results indicate that in the Cas12k system TnsC introduces distortions in DNA upon binding, suggesting that the roadblock of the TniQ-Cas12k complex may constrain this distortion information and passing it to the TnsB transposase to allow access to target DNA to license transposition.
. The target immunity process found with some transposons protects the element and the surrounding region from subsequent insertion events due to a local high concentration of the transposase. In the case of CRISPR-associated transposons, the process would also serve to divert insertions to unused protospacers. The results presented here suggest that filamentation and disassembly of the AAA+ regulators in these systems is the structural basis of target immunity as described in other transposition systems with both Mu and prototypic Tn7. The physical association between TnsC and TniQ revealed by our cryo-EM reconstruction immediately explains how target-site selection information can be conveyed between the TnsC regulator and the RNA-binding CRISPR effector domain to result in programmable insertion. It is likely that analogous physical interactions occur in prototypic Tn7 and type I-F3 guide RNA-directed systems.
Tn7-like transposons, including the prototypic Tn7 and guide RNA-directed transposition systems, display a remarkable diversity of targeting modalities which predominantly rely on proteins with TniQ domains. This diversity in targeting pathways suggests a remarkable adaptability for TniQ, which defines the target site for the core transposition system(2, 3, 39, 40). Notably, both TnsC and TniQ in ShCAST are significantly smaller than their equivalents from other systems (in prototypic Tn7 and other Tn7-like systems), containing only the highly conserved AAA+ core of TnsC and the HTH and ZnF motifs of the TniQ domain. Thus, we suspect that the structure visualized here likely represents the conserved, minimal functional interactions that are required between TniQ and TnsC. The interactions between ShCAST TnsB transposase and its regulator TnsC, however, remain to be determined. Therefore, the TnsC-TniQ structure provides an excellent starting point for engineering new links and interactions between new target DNA recognition modules and the core transposase for more sophisticated genome-editing applications.
Supplementary Material
Acknowledgements:
We gratefully acknowledge the Cornell Center for Materials Research facility (CCMR), as well as Katherine Spoth and Mariena Silvestry-Ramos, for maintenance of electron microscopes used for this research (NSF-DMR1715879). We additionally acknowledge XSEDE for computational resources used for image processing (MCB200090 to E.H.K). We additionally thank Amanda Byer, Nozomi Ando, Gira Bhabha, and Seychelle Vos for advice on the use of nucleotide analogs, as well as members of the Ke and Peters group for helpful and stimulating discussions. We thank Lisa Eshun-Wilson, Eric Alani, and Brooks Crickard for valuable advice throughout this project. Last but not least, we thank Nancy Craig and Alba Guarné for valuable feedback on the manuscript.
Funding:
This research is supported by the NIH: R00-GM124463 to E.H.K., R01GM129118 to J.E.P, R21AI148941 to J.E.P, and GM118174 to A.K.
Footnotes
Competing Interests: The Peters lab has corporate funding for research that is not directly related to the work in this publication. Cornell University has filed patent applications with J.E.P. as inventor involving CRISPR-Cas systems associated with transposons that are not directly related to this work.
Data and materials availability: Atomic models are available through the Protein Data Bank (PDB) with accession codes 7M99 (ATPγS TnsC), 7M9A (ADPᐧAlF3 TnsC consensus), 7M9C (ADPᐧAlF3 TnsC open), 7M9B (ADPᐧAlF3 TnsC closed), and 7N6I (TniQ-bound TnsC); all cryo-EM reconstructions are available through the EMDB with accession codes EMD-23724 (ATP TnsC head-to-tail), EMD-23725 (ATP TnsC head-to-head), EMD-23720 (ATPγS TnsC), EMD-23721 (ADPᐧAlF3 TnsC consensus), EMD-23722 (ADPᐧAlF3 TnsC open), EMD-23723 (ADPᐧAlF3 TnsC closed), and EMD-23726 (TniQ-TnsC). NGS data is available from the NCBI Sequence Read Archive (Bioproject : PRJNA737449).
References
- 1.Knott GJ, Doudna JA, CRISPR-Cas guides the future of genetic engineering. Science 361, 866–869 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peters JE, Makarova KS, Shmakov S, Koonin EV, Recruitment of CRISPR-Cas systems by Tn7-like transposons. Proc Natl Acad Sci U S A 114, E7358–E7366 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Faure G et al. , CRISPR-Cas in mobile genetic elements: counter-defence and beyond. Nat Rev Microbiol 17, 513–525 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klompe SE, Vo PLH, Halpin-Healy TS, Sternberg SH, Transposon-encoded CRISPR-Cas systems direct RNA-guided DNA integration. Nature 571, 219–225 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Strecker J et al. , RNA-guided DNA insertion with CRISPR-associated transposases. Science 365, 48–53 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saito M et al. , Dual modes of CRISPR-associated transposon homing. Cell, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peters JE, Targeted transposition with Tn7 elements: safe sites, mobile plasmids, CRISPR/Cas and beyond. Mol. Microbiol. 112, 1635–1644 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halpin-Healy TS, Klompe SE, Sternberg SH, Fernández IS, Structural basis of DNA targeting by a transposon-encoded CRISPR-Cas system. Nature 577, 271–274 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Peters JE, Tn7. Microbiol Spectr 2, (2014). [DOI] [PubMed] [Google Scholar]
- 10.Mizuuchi K, Transpositional recombination: mechanistic insights from studies of mu and other elements. Annu Rev Biochem 61, 1011–1051 (1992). [DOI] [PubMed] [Google Scholar]
- 11.Skelding Z, Queen-Baker J, Craig NL, Alternative interactions between the Tn7 transposase and the Tn7 target DNA binding protein regulate target immunity and transposition. EMBO J 22, 5904–5917 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baker TA, Mizuuchi M, Mizuuchi K, MuB protein allosterically activates strand transfer by the transposase of phage Mu. Cell 65, 1003–1013 (1991). [DOI] [PubMed] [Google Scholar]
- 13.Greene EC, Mizuuchi K, Direct observation of single MuB polymers: evidence for a DNA-dependent conformational change for generating an active target complex. Mol. Cell 9, 1079–1089 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Bainton RJ, Kubo KM, Feng JN, Craig NL, Tn7 transposition: target DNA recognition is mediated by multiple Tn7-encoded proteins in a purified in vitro system. Cell 72, 931–943 (1993). [DOI] [PubMed] [Google Scholar]
- 15.Rice PA, Craig NL, Dyda F, Comment on “RNA-guided DNA insertion with CRISPR-associated transposases”. Science 368, (2020). [DOI] [PubMed] [Google Scholar]
- 16.Vo PLH, Acree C, Smith ML, Sternberg SH, Unbiased profiling of CRISPR RNA-guided transposition products by long-read sequencing. bioRxiv, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamauchi M, Baker TA, An ATP-ADP switch in MuB controls progression of the Mu transposition pathway. EMBO J 17, 5509–5518 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vo PLH et al. , CRISPR RNA-guided integrases for high-efficiency, multiplexed bacterial genome engineering. Nat Biotechnol, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benjamin E Rubin SD, Cress Brady F., Crits-Christoph Alexander, He Christine, Xu Michael, Zhou Zeyi, Smock Dylan C., Tang Kimberly, Owens Trenton K., Krishnappa Netravathi, Sachdeva Rohan, Deutschbauer Adam M., Banfield Jillian F., Doudna Jennifer A., Targeted Genome Editing of Bacteria Within Microbial Communities. bioRxiv, (2020). [Google Scholar]
- 20.Mizuno N et al. , MuB is an AAA+ ATPase that forms helical filaments to control target selection for DNA transposition. Proc Natl Acad Sci U S A 110, E2441–2450 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Puchades C et al. , Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science 358, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi KY, Spencer JM, Craig NL, The Tn7 transposition regulator TnsC interacts with the transposase subunit TnsB and target selector TnsD. Proc. Natl. Acad. Sci. U. S. A. 111, E2858–2865 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holm L, DALI and the persistence of protein shape. Protein Sci 29, 128–140 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J et al. , Structure and function of Cdc6/Cdc18: implications for origin recognition and checkpoint control. Mol Cell 6, 637–648 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Gamas P, Craig NL, Purification and characterization of TnsC, a Tn7 transposition protein that binds ATP and DNA. Nucleic Acids Res. 20, 2525–2532 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arias-Palomo E, Berger JM, An Atypical AAA+ ATPase Assembly Controls Efficient Transposition through DNA Remodeling and Transposase Recruitment. Cell 162, 860–871 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitra R, McKenzie GJ, Yi L, Lee CA, Craig NL, Characterization of the TnsD-attTn7 complex that promotes site-specific insertion of Tn7. Mob DNA 1, 18 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jia N, Xie W, de la Cruz MJ, Eng ET, Patel DJ, Structure-function insights into the initial step of DNA integration by a CRISPR-Cas-Transposon complex. Cell Res 30, 182–184 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skelding Z, Sarnovsky R, Craig NL, Formation of a nucleoprotein complex containing Tn7 and its target DNA regulates transposition initiation. EMBO J 21, 3494–3504 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baker TA, Mizuuchi M, Mizuuchi K, MuB protein allosterically activates strand transfer by the transposase of phage Mu. Cell 65, 1003–1013 (1991). [DOI] [PubMed] [Google Scholar]
- 31.Wu Z, Chaconas G, Characterization of a region in phage Mu transposase that is involved in interaction with the Mu B protein. J Biol Chem 269, 28829–28833 (1994). [PubMed] [Google Scholar]
- 32.Mizuno N et al. , MuB is an AAA+ ATPase that forms helical filaments to control target selection for DNA transposition. Proc. Natl. Acad. Sci. U. S. A. 110, E2441–2450 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greene EC, Mizuuchi K, Target immunity during Mu DNA transposition. Transpososome assembly and DNA looping enhance MuA-mediated disassembly of the MuB target complex. Mol Cell 10, 1367–1378 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Hwang LC et al. , ParA-mediated plasmid partition driven by protein pattern self-organization. EMBO J 32, 1238–1249 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arinkin V, Smyshlyaev G, Barabas O, Jump ahead with a twist: DNA acrobatics drive transposition forward. Curr Opin Struct Biol 59, 168–177 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuduvalli PN, Rao JE, Craig NL, Target DNA structure plays a critical role in Tn7 transposition. EMBO J 20, 924–932 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yao Shen JG-B, Petassi Michael T., Peters Joseph E., Ortega Joaquin, Guarné Alba, Structural basis for DNA targeting by the Tn7 transposon. bioRxiv, (2021). [DOI] [PubMed] [Google Scholar]
- 38.Rao JE, Miller PS, Craig NL, Recognition of triple-helical DNA structures by transposon Tn7. Proc Natl Acad Sci U S A 97, 3936–3941 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shan-Chi Hsieh JEP, Tn7-CRISPR-Cas12K elements manage pathway choice using truncated repeat-spacer units to target tRNA attachment sites. bioRxiv, (2021). [Google Scholar]
- 40.Petassi MT, Hsieh SC, Peters JE, Guide RNA Categorization Enables Target Site Choice in Tn7-CRISPR-Cas Transposons. Cell 183, 1757–1771 e1718 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bainton R, Gamas P, Craig NL, Tn7 transposition in vitro proceeds through an excised transposon intermediate generated by staggered breaks in DNA. Cell 65, 805–816 (1991). [DOI] [PubMed] [Google Scholar]
- 42.Peters JE, in Methods for General and Molecular Microbiology, 3rd edition. (2007), chap. 31. [Google Scholar]
- 43.Lander G, Herzik MA, Wu M Jr, Lander GC, Setting up the Talos Arctica electron microscope and Gatan K2 direct detector for high-resolution cryogenic single-particle data acquisition. Protocol Exchange, (2017). [Google Scholar]
- 44.Herzik MA Jr., Setting Up Parallel Illumination on the Talos Arctica for High-Resolution Data Collection. Methods Mol. Biol. 2215, 125–144 (2021). [DOI] [PubMed] [Google Scholar]
- 45.Mastronarde DN, Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Tegunov D, Cramer P, Real-time cryo-electron microscopy data preprocessing with Warp. Nat. Methods 16, 1146–1152 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA, cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Zivanov J et al. , New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He S, Scheres SHW, Helical reconstruction in RELION. J Struct Biol 198, 163–176 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tan YZ et al. , Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat Methods 14, 793–796 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Punjani A, Fleet DJ, 3D Variability Analysis: Directly resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM images. Cold Spring Harbor Laboratory, (2020). [DOI] [PubMed] [Google Scholar]
- 52.Nakane T, Kimanius D, Lindahl E, Scheres SH, Characterisation of molecular motions in cryo-EM single-particle data by multi-body refinement in RELION. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE, The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Emsley P, Lohkamp B, Scott WG, Cowtan K, Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frenz B, Walls AC, Egelman EH, Veesler D, DiMaio F, RosettaES: a sampling strategy enabling automated interpretation of difficult cryo-EM maps. Nat. Methods 14, 797–800 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pettersen EF et al. , UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 57.André I, Modeling the Structure of Helical Assemblies with Experimental Constraints in Rosetta. Methods Mol. Biol. 1764, 475–489 (2018). [DOI] [PubMed] [Google Scholar]
- 58.Afonine PV et al. , Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr D Struct Biol 74, 531–544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Echols N et al. , Graphical tools for macromolecular crystallography in PHENIX. J Appl Crystallogr 45, 581–586 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pettersen EF et al. , UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 61.Ko J, Park H, Heo L, Seok C, GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res 40, W294–297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Williams CJ et al. , MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci 27, 293–315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Edgar RC, MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA, Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A 98, 10037–10041 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Khlebnikov A, Datsenko KA, Skaug T, Wanner BL, Keasling JD, Homogeneous expression of the P(BAD) promoter in Escherichia coli by constitutive expression of the low-affinity high-capacity AraE transporter. Microbiology (Reading) 147, 3241–3247 (2001). [DOI] [PubMed] [Google Scholar]
- 66.Waddell CS, Craig NL, Tn7 transposition: two transposition pathways directed by five Tn7-encoded genes. Genes Dev 2, 137–149 (1988). [DOI] [PubMed] [Google Scholar]
- 67.Peters JE, Craig NL, Tn7 transposes proximal to DNA double-strand breaks and into regions where chromosomal DNA replication terminates. Mol Cell 6, 573–582 (2000). [DOI] [PubMed] [Google Scholar]
- 68.Kovach ME et al. , Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166, 175–176 (1995). [DOI] [PubMed] [Google Scholar]
- 69.Cronan JE, A family of arabinose-inducible Escherichia coli expression vectors having pBR322 copy control. Plasmid 55, 152–157 (2006). [DOI] [PubMed] [Google Scholar]
- 70.Pettersen EF et al. , UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci 30, 70–82 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.