

Abstract
Thermosensitive microgels consisting of a solid core of polystyrene and a shell of cross-linked poly(N-isopropylacrylamide) (PNIPA) were synthesized as nano-reactors, in which Ag–Pd bimetallic nanoparticles were prepared through simultaneous in situ reduction reaction. The spatial distribution of metallic nanoparticles in the microgels was analyzed by small angle X-ray scattering (SAXS) and the results indicated that metal nanoparticles were mainly located in the inner layer of microgels. The catalytic activity of Ag–Pd bimetallic nanoparticles was investigated using the reduction of p-nitrophenol to p-aminophenol by NaBH4 as model reaction. The data demonstrated that Ag–Pd bimetallic nanoparticles showed enhanced catalytic activity compared to each monometallic nanoparticle alone and their catalytic activity was controllable by temperature due to the volume transition of PNIPA microgels.
Thermosensitive microgels with PS core and cross-linked PNIPA shell were synthesized as nano-reactor to prepare Ag–Pd bimetallic nanoparticles.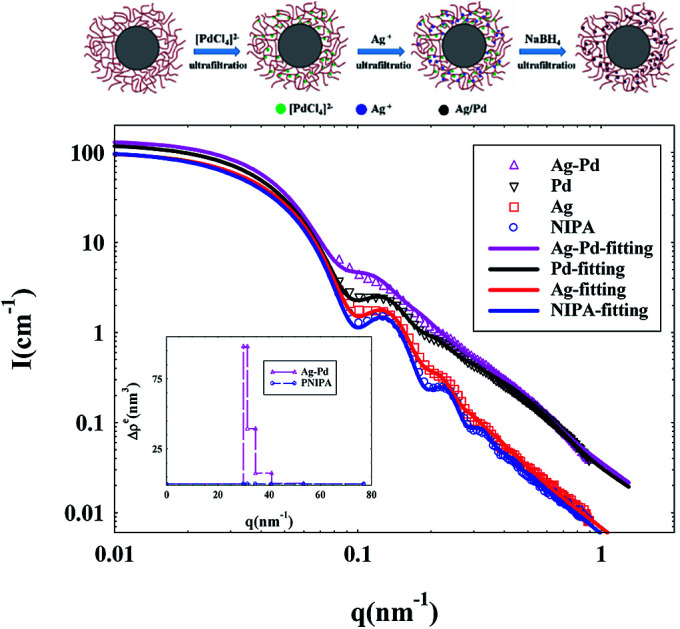
Introduction
In recent years, a variety of metallic nanoparticles1–5 have been synthesized and regarded as ideal nano-catalysts6–13 due to their high surface-to-volume ratio. Among them, bimetallic nanoparticles have attracted great interest because of their improved catalytic activity compared to monometallic nanoparticle, which is caused by the synergic effect.14–16 However, metal nanoparticles require a suitable support to prevent aggregation during the catalytic process. The metal nanoparticles is commonly protected by stabilizers like dendrimers,17–19 microgels,20,21 surfactants,22 polymers,23 and colloids,24 which also serve as carriers.
The thermosensitive microgels, which consist of solid core of polystyrene and shell of poly (N-isopropylacrylamide) (PNIPA) cross-linked by N,N′-methylenebisacrylamide (BIS),25,26 were reported as nano-reactors to prepare some metal nanoparticles.2,7,25,26 PNIPA microgels undergo reversible volume transition at 32 °C. Most of the water are expelled during the volume transition, so that PNIPA microgel networks shrink with increasing temperature to 32 °C.27 Those metallic nanoparticles embedded in PNIPA microgels are fully accessible to reactants, but above the volume transition, the marked shrinkage of networks forms a barrier for the diffusion of the reactants, which provides the theoretical basis of regulating catalytic activity of metal nanoparticles within PNIPA microgels.
The bimetallic nanoparticles were always prepared through metal ions or metal complex ions with the same kind of charge. Both Ag+ and [PdCl4]2− can be absorbed into PNIPA microgel networks and the obtained nanosized Ag and Pd particles in situ were strongly immobilized within the networks of thermosensitive microgels, respectively.2,25 The catalytic activity of metallic nanoparticles could be tested via the reduction of p-nitrophenol, which is easy to be monitored by UV/vis spectrometer. Especially when the concentration of sodium borohydride is significantly excessive, this model reaction is pseudofirst-order reaction,28 which can simplify the data processing. However, to the best of our knowledge, no report can be found on the preparation of Ag–Pd bimetallic nanoparticles by simultaneous reduction from Ag+ and [PdCl4]2− with opposite charge in PNIPA microgels.
The morphology of metal nanoparticles could be easily observed by transmission electron microscopy (TEM), but the information on spatial distribution of metallic nanoparticles in the microgels was inevitably missing due to the dry measuring state. Small angle X-ray scattering (SAXS) has been reported as a powerful method to investigate core–shell particles with narrow size distribution and high electron density contrast. SAXS is especially capable of analyzing the sample in a mild condition where is almost no destruction for samples.29–32 Moreover, SAXS could give a detailed illustration of spatial distribution of the metal nanoparticles.
In this work, the Ag–Pd bimetallic nano-catalysts were prepared through simultaneous in situ reduction reaction in the nano-reactors of PNIPA microgels. The spatial distribution of bimetallic nanoparticles in the microgels was obtained by SAXS. The catalytic activity of Ag–Pd bimetallic nano-particles was investigated by the model reduction of p-nitrophenol to p-aminophenol.
Experimental
Materials
Styrene, sodium dodecyl sulfate (SDS), potassium peroxodisulfate (KPS) and hydrochloric acid were purchased from Titans. NIPA and BIS were purchased from J & K chemical. Sodium borohydride (NaBH4), p-nitrophenol, palladium chloride and silver nitrate (AgNO3) were purchased from Aldrich. Water was purified using reverse osmosis and ion exchange in a Milli-Q system. Photoinitiator (2-[p-(2-hydroxy-2-methylpropiophenone)]-ethyleneglycol-methacrylate) (HMEM) was synthesized according to our previous publication.33 All other chemicals were used as received without further purification.
Synthesis of PS core
The PS core nanoparticles were synthesized by controlled emulsion polymerization.26,34 Typically, 0.48 g of SDS as surfactant were dissolved in 150 ml water, 6.25 g of styrene and 0.3 g of NIPA were dispersed as monomers. After the system was heated to 80 °C with stirring under nitrogen protection, the polymerization was started by adding 0.1 g of KPS as initiator and polymerized for 2 h. Then 0.3 g of photoinitiator (HMEM) dissolved in 2.4 g acetone was added in a “starved” condition (0.5 ml min−1) when the temperature cooled down to 70 °C. After another 1 h, a thin shell of photoinitiator covering the PS core was formed. The PS core latex was purified by ultrafiltration with 10 times the volume of water.
Synthesis of PNIPA microgels
The core–shell PNIPA microgels were prepared by photo-emulsion polymerization. PS core latex (50.1 g) was diluted with 150 g of water. After addition of 4.1 g NIPA and 0.42 g BIS, the mixture was mixed with vigorous stirring under a nitrogen atmosphere and reacted for 2 h in the UV reactor.2,26,34 Then the latex was purified by ultrafiltration.
Preparation of Ag and Pd mono- and bimetallic nanoparticles
The preparation of the silver nanoparticles in an aqueous solution was carried out by addition of silver nitrate to PNIPA microgel latex and subsequent reduction with sodium borohydride. In a typical run, an aqueous solution of AgNO3 (3 mM) was added to PNIPA microgel latex (100 ml). The mixture was homogenized with a magnetic stirrer for 2 h under the nitrogen atmosphere. Then a solution of sodium borohydride (0.023 g) in water (5 g) were added slowly to the system with vigorous stirring and reacted for 1 h. The obtained PNIPA microgels with silver nanoparticles were purified by ultrafiltration.
The preparation of the palladium nanoparticles was similar to that of silver nanoparticles. Firstly, chloropalladic acid was synthesized by mixing palladium chloride and hydrochloric acid with the mole ratio of 1 : 4. Then its aqueous solution (3 mM) was added to PNIPA microgel latex (100 ml) and stirred for 2 h. Finally, a solution of sodium borohydride (0.023 g) in water (5 g) were added slowly to the system with vigorous stirring under the nitrogen atmosphere and reacted for 1 h. The obtained PNIPA microgels with palladium nanoparticles were also purified by ultrafiltration.
The Ag–Pd bimetallic nanoparticles were prepared by simultaneous in situ reduction as shown in Fig. 1. It is worth noting that the chloropalladic acid was added to PNIPA latex before the addition of silver nitrate in order to prevent the precipitation of silver ions. The obtained PNIPA microgel latex with Ag–Pd bimetallic nanoparticles was also purified by ultrafiltration.
Fig. 1. Schematic representation of preparation of Ag–Pd bimetallic nanoparticles.

Characterization
The size and size distribution of PNIPA microgels were determined by dynamic light scattering (DLS) operated by PSS Nicomp 380. The concentration of metal nanoparticles immobilized in PNIPA microgels was determined by ICP-AES (IRIS 1000) under 27.12 MHZ. The morphology of metal nanoparticles was observed by TEM (JEOL 2100F). TEM was operated at an acceleration voltage of 200 kV.
The spectra change and kinetic results were obtained by UV/vis spectrometer (UV-2550). Mixed solution (3 ml) was put into a cuvette during the measurements. In a typical run, 0.01 M sodium borohydride aqueous solution was added to a solution of p-nitrophenol (0.1 mM), then different amount of PNIPA microgel latex with metal nanoparticles (Ag, Pd, or Ag–Pd) was added to start the reaction. The kinetics of the reduction reaction was monitored by measuring the extinction of solution at 400 nm as the function of time.
All small angle X-ray scattering (SAXS) experiments were performed at BL16B1 beamline in Shanghai synchrotron radiation facility (SSRF), and the detector used is MAR165 CCD. The distance from sample to detector was set as 2 m, and the scattering signals appeared among the scattering vector q-range of 0.085–1.1 nm−1.
During each SAXS measurement, 0.1–0.2 ml solution (1 wt%) was added into a sample cell by syringe, and the sample cell was a 1 mm-thick polystyrene plastic template, which was wrapped by polyimide film on both sides. After getting original data, the five-layer model was used as fitting model,32,35–40 which is widely used in this field. Through fitting, the spatial distribution of metallic nanoparticles in the PNIPA microgel layer can be obtained. Table 1 listed electron densities of related substances. Fig. 2 showed the decomposition of the scattering intensity of PNIPA microgels with metal nanoparticles.
Electron density of related substances.
| Substance | ρ (g cm−3) | ρ i (e nm−3) | Δρe (ρi − ρH2O) |
|---|---|---|---|
| H2O | 0.997 | 333.3 | 0 |
| Poly(styrene) | 1.05 | 339.7 | 6.4 |
| Poly (N-isopropyl acrylamide) | 0.879 | 289.9 | −44.5 |
| Ag | 10.49 | 2751.5 | 2418.2 |
| Pd | 12.02 | 3127.8 | 2794.5 |
Fig. 2. Decomposition of the scattering intensity of PNIPA microgel with metal nanoparticles.

The decomposition of the scattering intensity of PNIPA microgels with metal nanoparticles can be described by
| I0 = Ics + Iin + Ifluct + Imetal | 1 |
 |
2 |
where I0 represents the overall scattering intensity of a single composite particle, ICS the contribution of core–shell structure (eqn (2)), while B(q) is the scattering amplitude and Δρe(r) the excess electron density. Iin comes from the contribution of static spatial inhomogeneity which can be determined by Guinier function, namely
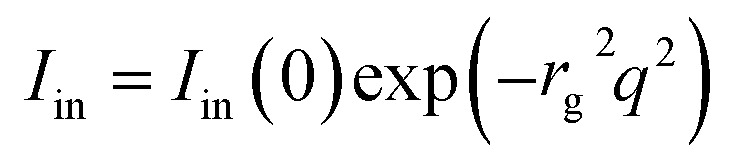 |
3 |
I
metal is the contribution of monometallic nanoparticles, which can be calculated by the same method as ICS with the shell thickness of zero, and the oscillation of the curve for this part at large q is the characteristic peaks for spherical particles. Moreover, Ifluct stems from the contribution of thermal fluctuations of network structure, and can be calculated by 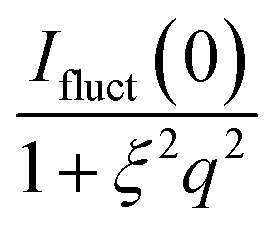 . Here Ifluct(0) is treated as adjustable parameters and ξ is the correlation length of the spatial fluctuations of the network, which is of the order of a few nanometers. It is worth noting that the experimental data used here is derived from the PNIPA microgels latex (1 wt%) loaded with metal nanoparticles. More detailed fitting process can be found in our previous publications.41–43
. Here Ifluct(0) is treated as adjustable parameters and ξ is the correlation length of the spatial fluctuations of the network, which is of the order of a few nanometers. It is worth noting that the experimental data used here is derived from the PNIPA microgels latex (1 wt%) loaded with metal nanoparticles. More detailed fitting process can be found in our previous publications.41–43
Results and discussion
Observation by DLS
The size and size distribution of PNIPA microgels before and after preparation of metal nanoparticles were observed by DLS (Fig. 3 and Table 2).
Fig. 3. Size distribution of PS core and PNIPA microgels before and after preparation of metal nanoparticles as determined by DLS.

Size and polydispersity index (PDI) of PS core and PNIPA microgels.
| PS | PNIPA | Ag@PNIPA | Pd@PNIPA | Ag–Pd@PNIPA | |
|---|---|---|---|---|---|
| Diameter (nm) | 72 | 157 | 156 | 159 | 155 |
| PDI | 0.015 | 0.037 | 0.052 | 0.056 | 0.064 |
Compared to PS core, the size of PNIPA microgels increased significantly and the size distribution became wider (Fig. 3). The thickness of the PNIPA microgel shell was about 42.5 nm (Table 2). No significant impact on the overall size can be observed after further generation of metal nanoparticles within the microgels. It seems that the interaction between the PNIPA nanogel networks and the metal nanoparticles has no remarkable influence on the spatial extensional structure of PNIPA nanogel.27 However, no information on the prepared metal nanoparticles can be obtained from DLS.
Observation by TEM
The morphology of obtained metal nanoparticles in PNIPA microgels was observed by TEM (Fig. 4). In Fig. 4, metal nanoparticles were clear and distinguishable which located around the spherical PNIPA microgels. The size of different metal nanoparticles could be estimated by TEM images. The average diameter of Ag–Pd bimetallic, Ag and Pd nanoparticles were 5.4 ± 0.8, 8.1 ± 0.5, and 4.2 ± 0.4 nm, respectively.
Fig. 4. Morphology and size distribution of metal nanoparticles prepared in PNIPA microgels. (a) Ag–Pd bimetallic nanoparticles; (b) Ag nanoparticles; (c) Pd nanoparticles. The size distribution histograms origin from the TEM images.

The concentration of obtained metal nanoparticles in PNIPA microgel latex was determined by ICP-AES and listed in Table 3. Since the initial concentrations of [PdCl4]2− and Ag+ were the same, the concentrations of Ag and Pd in the Ag–Pd bimetallic nanoparticles were only slightly less than those of monometallic nanoparticles, which means our approach to prepare bimetallic nanoparticles in PNIPA microgels simultaneously does not reduce the yield of Ag and Pd nanoparticles significantly. The reduction of yield for Pd nanoparticles (0.07 mmol L−1) is larger than that for Ag (0.04 mmol L−1), which is probably due to the stronger binding interaction between PNIPA and silver nanoparticles compared to that between PNIPA and Pd nanoparticles.44 As a result, during the purification process after preparation metal nanoparticles, more Pd nanoparticles were lost.
Concentration of metal nanoparticles in PNIPA microgel latex determined by ICP-AES.
| Samples | Ag (mmol L−1) | Pd (mmol L−1) |
|---|---|---|
| Ag@PNIPA | 0.45 | 0 |
| Pd@PNIPA | 0 | 0.51 |
| Ag–Pd@PNIPA | 0.41 | 0.44 |
Observation by SAXS
The size of PNIPA microgels measured by DLS was the hydrodynamic radius, which was mainly determined by the longest chain from core. However the data from SAXS were based on the electronic density difference of the carrier system and distribution media. The data from SAXS reflected the length of most of the chains, so that the results from SAXS were more reasonable than DLS. At the same time, the distribution of metal nanoparticles in PNIPA microgels can be analyzed by the change of electronic density.
In the SAXS measurements, the concentration of PNIPA microgels was 1 wt%, and the concentrations of metal nanoparticles were listed in Table 3. Fig. 5 demonstrated that the scattering intensity of PNIPA microgels increased significantly after generation of metal nanoparticles, which resulted from the addition of metal nanoparticles with high electron density. The significant increase of the scattering intensity indicates the success preparation of metal nanoparticles.
Fig. 5. SAXS curves of PNIPA microgels loaded with different metal nanoparticles. (solid lines represent fitting curves). The inset shows the excess electron density Δρe(r) of PNIPA microgels versus radius.

As shown in Fig. 5, the reduced oscillations of SAXS curves after generation of metal nanoparticles indicate that the size distribution of PNIPA microgels becomes wider which is consistent to the DLS results in Fig. 3. The increase in scattering intensity at high q range after preparation of Pd nanoparticles gives us a clue that some of the generated Pd nanoparticle may locate outside the PNIPA microgels. It confirms that the interactions between PNIPA and Pd are weaker than those between PNIPA and Ag,44 and some Pd nanoparticles can move out of the PNIPA microgels during SAXS measurements.
From the excess electron density profile in the inset of Fig. 5, parameters on the size of metal nanoparticles and their distribution in PNIPA microgels were obtained. It is worth noting that in order to more clearly show the difference in electron density after loading metal nanoparticles, the PNIPA electron density of each layer was set as a benchmark. As shown by the inset of Fig. 5, the electron density near the core surface is the highest and reduces outward along the radial, which manifested that metal nanoparticles mainly locate in the inner layer of PNIPA microgels. The size of PNIPA microgels is ca. 153 nm as determined by SAXS, which was less than that by DLS as expected.
Catalytic activity of mono- and bimetallic nanoparticles
The catalytic activity of the obtained Ag, Pd and Ag–Pd bimetallic nanoparticles was investigated by using the reduction of p-nitrophenol to p-aminophenol by NaBH4 as a model reaction, which was monitored by testing the change of characteristic absorption peak of p-nitrophenol.43,45,46 In Fig. 6, the absorbance at 400 nm constantly declined, which indicated the continuous decrease of p-nitrophenol concentration.
Fig. 6. Spectra of p-nitrophenol in the presence of PNIPA microgels with Ag–Pd bimetallic nanoparticles as catalysts at different time during reaction at 20 °C. The concentrations of the reactants are [p-nitrophenol] = 0.1 mmol L−1 and [NaBH4] = 10 mmol L−1.

Since that the concentration of NaBH4 was in great excess compared to 4-nitrophenol in our experiments, its concentration can be considered as constant during the reaction. Thus, the model reaction can be regarded as pseudo-first-order reaction, which can be described by eqn (4),47–49
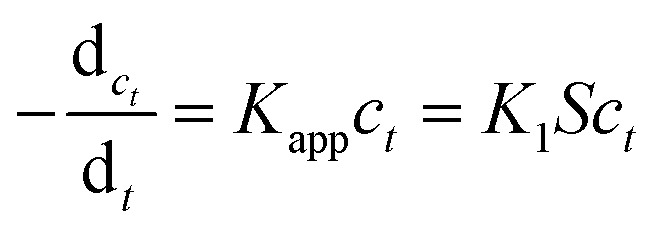 |
4 |
where Kapp is the apparent rate constant proportional to the surface area S (normalized to the unit volume of the system) of the metal nanoparticles embedded in PNIPA microgels, ct is the concentration of p-nitrophenol at time t, K1 is the rate constant normalized to surface area. According to eqn (4), Kapp could be obtained by exploring the concentration change of p-nitrophenol with reaction time (Fig. 7).
Fig. 7. Influence of bimetallic nanoparticles concentration on the reduction of p-nitrophenol. [p-nitrophenol] = 0.1 mmol L−1, [NaBH4] = 10 mmol L−1, T = 20 °C. (a) [Ag] = 1.76 mg L−1, [Pd] = 1.88 mg L−1; (b) [Ag] = 1.32 mg L−1, [Pd] = 1.41 mg L−1; (c) [Ag] = 0.88 mg L−1, [Pd] = 0.94 mg L−1; (d) [Ag] = 0.44 mg L−1, [Pd] = 0.47 mg L−1.

As shown in Fig. 7, the model reaction indeed followed the first-order kinetics and the apparent rate constants normalized to the unit surface area of bimetallic nanoparticles were obtained. The model reaction was carried out on the surface of the metal nanoparticles, so the apparent rate constant highly depended on the surface area of the metal nanoparticles. Fig. 8 showed the plot of Kappversus the surface S of the metal nanoparticles. The reaction rate constant K1 normalized to the unit surface area was 0.12 L m−2 s.
Fig. 8. The rate constant Kapp as a function of the surface area of bimetallic nanoparticles normalized to the unit volume of the system.

As comparison, the kinetics of model reaction catalyzed by Ag and Pd monometallic nanoparticles immobilized in PNIPA microgels were investigated (Fig. 9), and the apparent rate constants normalized to the unit surface area of monometallic nanoparticles were obtained and listed in Table 4.
Fig. 9. The rate constant Kapp as a function of the surface area of monometallic nanoparticles normalized to the unit volume of the system. (a) Ag nanoparticles; (b) Pd nanoparticles.

Comparison of catalytic activity of the metal nanoparticles.
As shown in Table 4, the catalytic activities of Ag and Pd were very close to those reported in literatures,2,25 and the catalytic activity of Ag was lower than Pd.17,20 Most important is that the catalytic activity of bimetallic nanoparticles was higher than that of monometallic nanoparticles.
Effect of temperature on catalytic activity of bimetallic nanoparticles
Fig. 10 showed the dependence of the reaction rate under the catalysis of Ag–Pd bimetallic nanoparticles immobilized in PNIPA microgels on temperature. When temperature was below 20 °C, the apparent rate constant increased upon increasing temperature, which stems from the fact that the selected model reaction was an endothermic reaction50 and the increase of temperature can promote the reaction rate significantly. When temperature was between 20 °C and 32 °C, apparent rate constant decreased upon increasing temperature. In fact, when the temperature was increased and approached to the volume transition temperature of PNIPA, the PNIPA microgels started to shrink. The shrinkage of PNIPA microgels prevented the reactants from entering the surface of the metal nanoparticles, and overwhelmed the positive effect on reaction rate by increasing temperature. As the temperature was above 32 °C, the reaction rate increased again upon increasing temperature since the volume transition of PNIPA microgels was fully happened at 32 °C and the positive effect of heating on reaction rate was dominated again.
Fig. 10. Effect of temperature on the catalytic activity of bimetallic nanoparticles. [Ag] = 1.76 mg L−1, [Pd] = 1.88 mg L−1.

Therefore, the catalytic activity of metal nanoparticles immobilized in PNIPA microgels can be modulated by temperature, which should be very useful to control the reactions.
Conclusions
Core–shell nanoparticles with PS core and thermosensitive PNIPA microgel shell prepared by photo-emulsion polymerization were employed as nano-reactors to generate Ag–Pd bimetallic nanoparticles in situ. The morphology of obtained Ag–Pd bimetallic nanoparticles were observed by TEM and their average size was estimated as ca. 5.4 ± 0.8 nm. As revealed by SAXS, Ag–Pd nanoparticles were mainly located near the surface of PS core, and their amount reduced outward along the radial. The catalytic activity of metal nanoparticles was tested by the reduction of p-nitrophenol to p-aminophenol by NaBH4 and the results demonstrated that the Ag–Pd bimetallic nanoparticles showed higher the catalytic activity than that of Ag and Pd monometallic nanoparticles. Due to the volume transition of PNIPA microgels upon increasing temperature to 32 °C, the catalytic activity of Ag–Pd bimetallic nanoparticles immobilized in PNIPA microgels is controllable by temperature.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We thank the financial support by National Natural Science and Foundation of China (51773061, 21476143, and 51761135128), 111 Project Grant (B08021), and Shanghai Synchrotron Radiation Facility.
References
- Lu Y. Mei Y. Schrinner M. Ballauff M. Moeller M. W. J. Phys. Chem. C. 2007;111:7676–7681. [Google Scholar]
- Mei Y. Lu Y. Polzer F. Ballauff M. Drechsler M. Chem. Mater. 2007;19:1062–1069. doi: 10.1021/cm062554s. [DOI] [Google Scholar]
- Sharma G. Mei Y. Lu Y. Ballauff M. Irrgang T. Proch S. Kempe R. J. Catal. 2007;246:10–14. doi: 10.1016/j.jcat.2006.11.016. [DOI] [Google Scholar]
- Sharma G. Ballauff M. Macromol. Rapid Commun. 2004;25:547–552. doi: 10.1002/marc.200300107. [DOI] [Google Scholar]
- Zhu Z. Guo X. Wu S. Zhang R. Wang J. Li L. Ind. Eng. Chem. Res. 2011;50:13848–13853. doi: 10.1021/ie2017306. [DOI] [Google Scholar]
- Astruc D. Lu F. Aranzaes J. R. Angew. Chem., Int. Ed. 2006;44:7852–7872. doi: 10.1002/anie.200500766. [DOI] [PubMed] [Google Scholar]
- Lu Y. Yuan J. Polzer F. Drechsler M. Preussner J. ACS Nano. 2010;4:7078–7086. doi: 10.1021/nn102622d. [DOI] [PubMed] [Google Scholar]
- Wong M. S. Alvarez P. J. J. Fang Y. L. AkçİN N. Nutt M. O. Miller J. T. Heck K. N. J. Chem. Technol. Biotechnol., Biotechnol. 2010;84:158–166. doi: 10.1002/jctb.2002. [DOI] [Google Scholar]
- Notar Francesco I. Fontaine-Vive F. Antoniotti S. J. Cheminf. 2014;6:2784–2791. [Google Scholar]
- He W. Wu X. Liu J. Hu X. Zhang K. Hou S. Zhou W. Xie S. Chem. Mater. 2010;22:2988–2994. doi: 10.1021/cm100393v. [DOI] [Google Scholar]
- Chen K. J. Lee C. F. Rick J. Wang S. H. Liu C. C. Hwang B. J. Biosens. Bioelectron. 2012;33:75–81. doi: 10.1016/j.bios.2011.12.020. [DOI] [PubMed] [Google Scholar]
- Wang R. Wu Z. Chen C. Qin Z. Zhu H. Wang G. Wang H. Wu C. Dong W. Fan W. Chem. Commun. 2013;49:8250–8252. doi: 10.1039/C3CC43948H. [DOI] [PubMed] [Google Scholar]
- Ding K. Liu L. Cao Y. Yan X. Wei H. Guo Z. Int. J. Hydrogen Energy. 2014;39:7326–7337. doi: 10.1016/j.ijhydene.2014.03.026. [DOI] [Google Scholar]
- Schrinner M. Proch S. Mei Y. Kempe R. Miyajima N. Ballauff M. Adv. Mater. 2008;20:1928–1933. doi: 10.1002/adma.200702421. [DOI] [Google Scholar]
- Kaiser J. Szczerba W. Riesemeier H. Reinholz U. Radtke M. Albrecht M. Lu Y. Ballauff M. Faraday Discuss. 2013;162:45–55. doi: 10.1039/C3FD20132E. [DOI] [PubMed] [Google Scholar]
- Shang L. Jin L. Guo S. Zhai J. Dong S. Langmuir. 2010;26:6713–6719. doi: 10.1021/la9040612. [DOI] [PubMed] [Google Scholar]
- Wu H. Liu Z. Wang X. Zhao B. Zhang J. Li C. J. Colloid Interface Sci. 2006;302:142–148. doi: 10.1016/j.jcis.2006.06.019. [DOI] [PubMed] [Google Scholar]
- Oh S. K. Kim Y. G. Heechang Ye A. Crooks R. M. Langmuir. 2003;19:10420–10425. doi: 10.1021/la0353778. [DOI] [Google Scholar]
- Lu X. Imae T. J. Phys. Chem. C. 2013;111:2416–2420. [Google Scholar]
- Nie Z. Xu S. Seo M. Lewis P. C. Kumacheva E. J. Am. Chem. Soc. 2005;127:8058–8063. doi: 10.1021/ja042494w. [DOI] [PubMed] [Google Scholar]
- Sahiner N. Yasar A. O. Fuel Process. Technol. 2013;111:14–21. doi: 10.1016/j.fuproc.2012.09.064. [DOI] [Google Scholar]
- Bol’shagin E. Y. Roldughin V. I. Colloid J. 2012;74:649–654. doi: 10.1134/S1061933X12060038. [DOI] [Google Scholar]
- Mayer A. B. R. Polym. Adv. Technol. 2015;12:96–106. doi: 10.1002/1099-1581(200101/02)12:1/2<96::AID-PAT943>3.0.CO;2-G. [DOI] [Google Scholar]
- And S. K. Bruening M. L. Chem. Mater. 2005;17:301–307. doi: 10.1021/cm048421t. [DOI] [Google Scholar]
- Lu Y. Mei Y. Drechsler M. Ballauff M. Angew. Chem. 2006;45:813–816. doi: 10.1002/anie.200502731. [DOI] [PubMed] [Google Scholar]
- Lu Y. Mei Y. Ballauff M. Drechsler M. J. Phys. Chem. B. 2006;110:3930–3937. doi: 10.1021/jp057149n. [DOI] [PubMed] [Google Scholar]
- Ballauff M. Macromol. Chem. Phys. 2010;204:220–234. doi: 10.1002/macp.200290072. [DOI] [Google Scholar]
- Gu S. Wunder S. Lu Y. Ballauff M. Fenger R. Rademann K. Jaquet B. Zaccone A. J. Phys. Chem. C. 2014;118:18618–18625. [Google Scholar]
- Ballauff M. Bolze J. Dingenouts N. Hickl P. Pötschke D. Macromol. Chem. Phys. 2010;197:3043–3066. doi: 10.1002/macp.1996.021971001. [DOI] [Google Scholar]
- Varga N. Benkő M. Sebők D. Dékány I. Colloids Surf., B. 2014;123:616–622. doi: 10.1016/j.colsurfb.2014.10.005. [DOI] [PubMed] [Google Scholar]
- Grünewald T. A. Lassenberger A. Oostrum P. D. J. V. Rennhofer H. Zirbs R. Capone B. Vonderhaid I. Amenitsch H. Lichtenegger H. C. Reimhult E. Chem. Mater. 2015;27:4763–4771. doi: 10.1021/acs.chemmater.5b01488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. Chu F. Li L. Han H. Tian Y. Wang Y. Yuan Z. Zhou Z. Guo X. J. Polym. Sci., Part B: Polym. Phys. 2016;54:405–413. doi: 10.1002/polb.23901. [DOI] [Google Scholar]
- Guo X. Weiss A. A. Ballauff M. Macromolecules. 1999;32:6043–6046. doi: 10.1021/ma990609o. [DOI] [Google Scholar]
- Pradhan N. Pal A. Pal T. Colloids Surf., A. 2002;196:247–257. doi: 10.1016/S0927-7757(01)01040-8. [DOI] [Google Scholar]
- Han H. Li L. Yang Q. Tian Y. Wang Y. Ye Z. Klitzing R. V. Guo X. J. Mater. Sci. 2018;53:3210–3224. doi: 10.1007/s10853-017-1747-5. [DOI] [Google Scholar]
- Wang W. Li L. Yu X. Han H. Guo X. J. Polym. Sci., Part B: Polym. Phys. 2015;52:1681–1688. doi: 10.1002/polb.23580. [DOI] [Google Scholar]
- Dingenouts N. Seelenmeyer S. Deike I. Rosenfeldt S. Ballauff M. Lindner P. Narayanan T. Phys. Chem. Chem. Phys. 2001;3:1169–1174. doi: 10.1039/B009104I. [DOI] [Google Scholar]
- Ballauff M. Prog. Polym. Sci. 2007;32:1135–1151. doi: 10.1016/j.progpolymsci.2007.05.002. [DOI] [Google Scholar]
- Dingenouts N. Ballauff M. Acta Polym. 2010;49:178–183. doi: 10.1002/(SICI)1521-4044(199804)49:4<178::AID-APOL178>3.0.CO;2-Q. [DOI] [Google Scholar]
- Wang W. Li L. Han H. Tian Y. Zhou Z. Guo X. Colloid Polym. Sci. 2015;293:2789–2798. [Google Scholar]
- Tian Y. Li L. Han H. Wang W. Wang Y. Ye Z. Guo X. Polymers. 2016;8(145):01–15. doi: 10.3390/polym8040145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. Li L. Henzler K. Lu Y. Wang J. Han H. Tian Y. Wang Y. Zhou Z. Lotze G. Biomacromolecules. 2017;18:1574–1581. doi: 10.1021/acs.biomac.7b00164. [DOI] [PubMed] [Google Scholar]
- Han H. Li L. Wang W. Tian Y. Wang Y. Wang J. Klitzing R. V. Guo X. Langmuir. 2017;12:9857–9865. doi: 10.1021/acs.langmuir.7b02239. [DOI] [PubMed] [Google Scholar]
- Xu H. Xu J. Zhu Z. Liu H. Liu S. Macromolecules. 2006;39:8451–8455. doi: 10.1021/ma061584d. [DOI] [Google Scholar]
- Lu Y. Wittemann A. Ballauff M. Drechsler M. Macromol. Rapid Commun. 2010;27:1137–1141. doi: 10.1002/marc.200600190. [DOI] [Google Scholar]
- Wu S. Dzubiella J. Kaiser J. Drechsler M. Guo X. Ballauff M. Lu Y. Angew. Chem., Int. Ed. 2012;51:2229–2233. doi: 10.1002/anie.201106515. [DOI] [PubMed] [Google Scholar]
- Wunder S. Polzer F. Lu Y. Mei Y. Ballauff M. J. Phys. Chem. C. 2010;114:8814–8820. [Google Scholar]
- Wunder S. Lu Y. Albrecht M. Ballauff M. ACS Catal. 2011;1:908–916. doi: 10.1021/cs200208a. [DOI] [Google Scholar]
- Lu Y. Yu M. Drechsler M. Ballauff M. Macromol. Symp. 2007;254:97–102. doi: 10.1002/masy.200750815. [DOI] [Google Scholar]
- Mandlimath T. R. Gopal B. J. Mol. Catal. A: Chem. 2011;350:9–15. doi: 10.1016/j.molcata.2011.08.009. [DOI] [Google Scholar]