

Abstract
Maintenance of genomic integrity is essential for the survival of all organisms. Homologous recombination (HR) is the major pathway for high-fidelity repair of DNA double-stranded breaks (DSBs). In addition to the classic BRCA–RAD51 pathway, another secondary HR sub-pathway dependent on RAD52 has been suggested to be functioning in mammalian cells. Importantly, RAD52 has been shown to be synthetically lethal to BRCA1/2-deficient cells, rendering RAD52 to be a desirable target in cancer therapy. In the current study, we performed a structure-based virtual screening of 47 737 drug-like compounds to identify RAD52-specific inhibitors. The top ranked virtual screening hits were further characterized using molecular dynamics simulation and biochemical and cell-based assays. We found that one compound, namely, F779-0434 specifically suppressed the growth of BRCA2-deficient cells and disrupted RAD52–ssDNA interaction in vitro. This RAD52-specific inhibitor identified in the current study is a promising compound for targeted cancer therapy, and it can also be used as a probe to study the mechanisms of DNA repair in human cells.
A novel compound named F779-0434 caused synthetic lethality in BRCA2-deficient cancer cells by disrupting RAD52–ssDNA associations.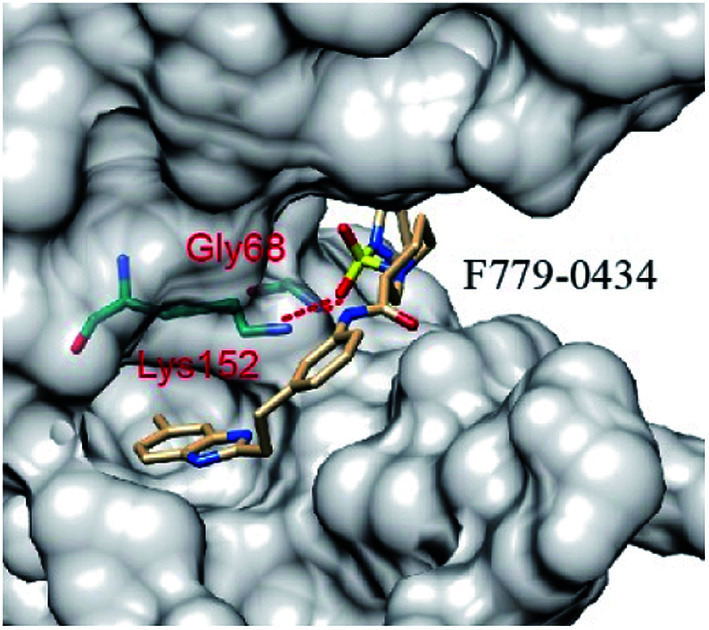
1. Introduction
Efficient and high-fidelity DNA repair mechanisms are essential for the maintenance of genome integrity in all organisms.1,2 In contrast to the DNA repair pathways in normal cells, certain DNA repair pathways in cancer cells are often impaired due to intrinsic genome instability, which causes the cancer cells to rely on the remaining alternative DNA repair mechanisms for survival.3–5 Homologous recombination (HR) is an essential DNA repair pathway for accurate repair of DNA double-stranded breaks (DSBs).6,7 HR repair of DSBs usually depends on the BRCA–RAD51 pathway and includes BRCA1, BRCA2, RAD51, PALB2, and RAD51 paralogs as the major genes.8 Heritable mutations in these key HR factors, most notably BRCA1 and BRCA2, cause genome instability and predisposition to malignant diseases such as breast and ovarian cancer.9,10
Synthetic lethality refers to situations in which the defects in two different genes or pathways together result in cell death, whereas a defect in one of the two does not affect cell viability.11,12 Thus, pharmacological targeting of tumor-specific DNA repair pathways may cause synthetic lethality in BRCA1/2-deficient cells with a significantly lower risk of side effects for personalized medicine.13,14 A notable example is the development of poly (ADP-ribose) polymerase 1 (PARP-1) inhibitors. PARP1 is involved in DNA damage signaling and repair of DNA single-stranded breaks (SSB), which is essential for viability of cancer cells that are deficient in the HR pathway.13,15 PARP1 inhibitors have been used to induce synthetic lethality in tumors with a defective BRCA–RAD51 pathway, presumably by elevation of the number of DSBs in the HR-impaired cancer cells.16
In addition to the BRCA–RAD51 pathway, another secondary HR sub-pathway dependent on RAD52 has been suggested to be functioning in mammalian cells. In yeast, RAD52 is the main recombination mediator and the central participant in single-strand annealing.17 In contrast to the severe recombination and repair phenotypes observed in yeast, deletion of RAD52 in mammalian cells and mice only causes a mild effect on DNA recombination.18–20 However, the RAD52-dependent sub-pathway becomes essential for viability of cells that are defective for the BRCA–RAD51 sub-pathway. Previous studies have demonstrated that cells deficient in BRCA1/2 or associated proteins in this pathway, including PALB2 and RAD51 paralogs, are not viable when the RAD52 protein is inactivated.21–23 Thus, these findings identify RAD52 as a potential therapeutic target against familial breast and ovarian cancer and possibly for other types of cancers with defects in BRCA1/2 and other related genes.
Although it is clear that RAD52 is critical for the survival of BRCA-deficient cancer cells, the molecular mechanism is still largely unknown. Biochemically, human RAD52 has been shown to promote the annealing of two complementary ssDNA strands.24,25 RAD52 also pairs ssDNA to complementary homologous regions in supercoiled DNA.26,27 These observations suggest that the cellular functions of the RAD52 protein largely depend on its ssDNA binding properties and therefore, the inhibition of the RAD52–ssDNA interaction should have similar consequences as those of RAD52 depletion.
Several studies have explored the efficacy of targeting RAD52 in BRCA-deficient cancer cell lines.28–31 In the current study, we used structure-based virtual computer screening and molecular dynamics simulation, followed by functional assays to identify compounds capable of exerting synthetic lethality in BRCA-deficient cells by targeting the RAD52–ssDNA interaction. We identified 30 putative RAD52 inhibitors from the 47 737 compounds that were screened. Representative compounds with higher drug-like properties were further characterized using biochemical and cell-based assays. We found one compound, namely, F779-0434, which specifically suppressed the growth of BRCA2-deficient cells and disrupted RAD52–ssDNA interaction in vitro. This RAD52-specific inhibitor identified in the current study is a promising compound for targeted cancer therapy, and it can also be used as a probe to study the mechanisms of DNA repair in human cells.
2. Results
2.1. Identification of putative RAD52 inhibitors by virtual screening
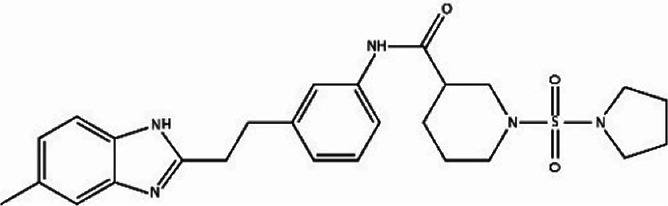
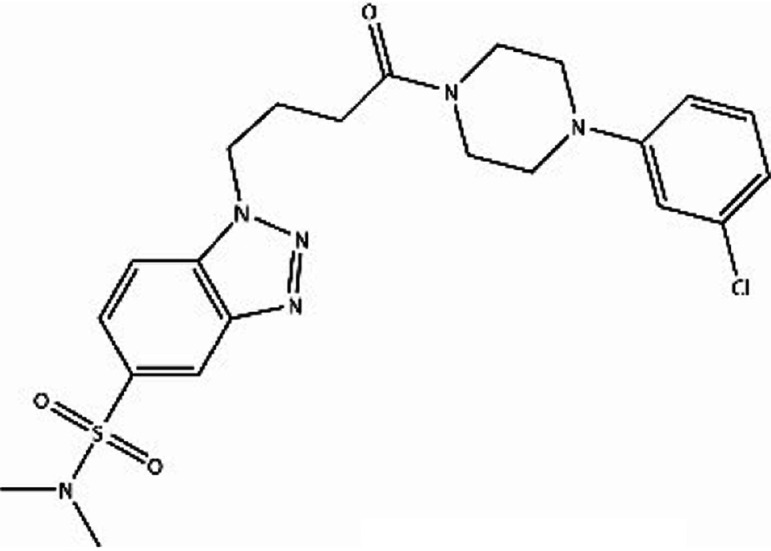
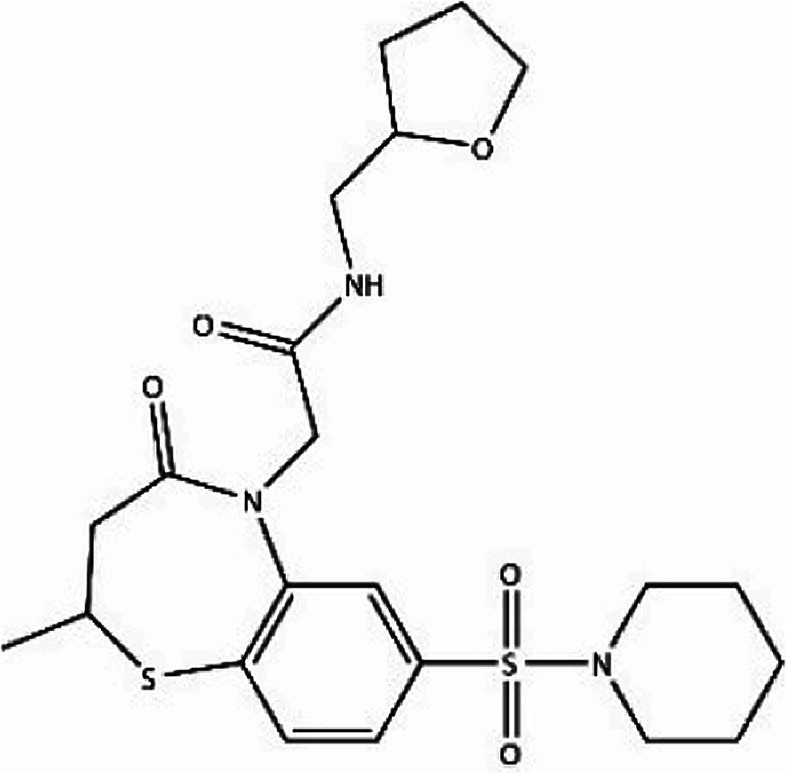
We first identified the motifs and amino acid residues that are critical for ssDNA binding in the RAD52 monomer based on previously reported biochemical and structural studies32–34 (Fig. 1A). The docking site and the size of the grid for virtual screening were then determined according to the location of these key DNA-binding amino acid residues (Fig. 1B). We screened a total of 47 737 compounds by a two-round scoring strategy. Compounds that scored in the top 30 were grouped into five classes based on similarities in their chemical structure, and they were further analyzed for AMDET properties (Table S1 and S2†). F779-0434, F848-0436, G640-1014, and D207-0130 were chosen for further analysis (Table 1) based on their drug-like properties.
Fig. 1. Human RAD52 undecameric ring structure and its DNA binding site. (A). Human Rad52 protein monomer forms an undecameric ring structure (PDB: 5JRB); the key amino acid residues reported to be associated with the ssDNA binding are shown in orange. (B) The blue spheres indicate the docking sites for virtual screening. White cubes indicate the location and size of the grid to be calculated.

Structures and docking scores of representative putative RAD52 inhibitors.
| Compound | Structure | Grid score | Amber score |
|---|---|---|---|
| F779-0434 |
|
−52.15 | −65.42 |
| F848-0436 |
|
−52.40 | −59.21 |
| G640-1014 |
|
−51.82 | −58.61 |
| D207-0130 |
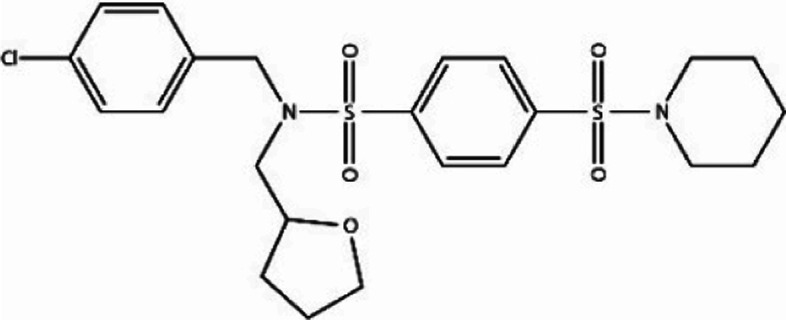
|
−51.74 | −62.97 |
Although the grid score of F779-0434 was second among those of the four representative RAD52 candidate inhibitors, the fractional difference from the lowest score exhibited by F848-0436 was only −0.25 kcal mol−1. After the second round of evaluation by the amber scoring tool, the score of F779-0434 was 65.42 kcal mol−1, which was −2.63 kcal mol−1 lower than that of the compound D207-0130, which was ranked second. The amber score enables all or a part of the receptor to be flexible for reproducing the so-called “induced-fit” and thus, the amber score is considered to be a better reflection of the receptor–ligand affinity. Therefore, based on the results of the amber score, we concluded that F779-0434 was the molecule with the highest affinity for the RAD52 protein.
2.2. Putative inhibitors associated with RAD52 through hydrogen bonding and hydrophobic interaction
All of the four putative RAD52 inhibitors could successfully dock into the pockets defined for DNA binding (Fig. 2). Except F848-0434, the other three compounds were predicted to form two hydrogen bonds with the amino acid residues of RAD52. It is noteworthy that LYS152 acted as a key amino acid residue and played an important role in RAD52-ssDNA binding, which was formed by hydrogen bonding with the oxygen atoms on F779-0434, G640-1014 and D207-0130.
Fig. 2. RAD52–ligands ((A) F779-0434, (B) F848-0436, (C) G640-1014 and (D) D207-0130) complex structures. The ligands and amino acid residues of RAD52 involved in hydrogen bonding are presented as stick models. The dashed red lines represent the hydrogen bonds predicted by the LigPlot+ software.

The key amino acid residues of RAD52 involved in DNA binding were plotted using LigPlot+ to study the mechanism of the RAD52-ligand interaction (Fig. 3). The hydrogen bonds and hydrophobic interactions between the amino acid residues and the compounds are presented by dashed lines and arcs, respectively. Compounds F779-0434, ARG55, TYR65 and ARG156 exhibited hydrophobic interactions, and LYS152 exhibited hydrogen bonding (Fig. 3A). For F848-0436, all of the key DNA-binding amino acid residues were found to be involved in hydrophobic interactions, and no hydrogen bonds were predicted to form between these residues and the compound (Fig. 3B). G640-1014, ARG55, TYR65 and ARG156 were involved in hydrophobic interactions, whereas LYS152 formed two hydrogen bonds with the compound (Fig. 3C). TYR65 and ARG153 were involved in the hydrophobic interactions for D207-0130, and LYS152 and ARG55 were involved in hydrogen bonding (Fig. 3D).
Fig. 3. 2D diagrams of the interactions between RAD52 and the four candidate inhibitors. Hydrophobic interactions are presented by red arcs. Hydrogen bonds are indicated by dashed green lines. Ligands are presented in purple. C, N, O, and Br atoms are shown in black, blue, red, and green, respectively. The green circles and ellipses highlight the key residues reported in literatures. The graph was generated using the LigPlot+ software.

2.3. Structural stability and residue flexibility of RAD52 in molecular dynamics simulation
The backbone root-mean-square deviation (RMSD) values with respect to the X-ray structure were calculated to assess the stability of the four RAD52-ligand complexes during the molecular dynamics (MD) simulation. As shown in Fig. 4, the RMSD values of RAD52-F779-0434 and RAD52-G640-1014 complexes fluctuated in the initial 5 ns of MD simulation, averaging at 0.71 nm and 0.58 nm, respectively. The RMSD values of the two complexes were found to be stable in the next 25 ns of the simulation, suggesting a stable association between RAD52 protein and the compounds. Although the average RMSD value for the RAD52-F848-0436 complex during the whole 30 ns simulation was only 0.57 nm, the fluctuation in the first 15 ns was relative high, indicating impaired binding of F848-0436 to the RAD52 protein. The RMSD value of the RAD52-D207-0130 complex failed to reach a stable status throughout the whole process of 30 ns simulation, suggesting unstable binding to the RAD52 protein.
Fig. 4. Backbone RMSD values (with respect to the X-ray structure) for RAD52-ligand complexes during 30 ns MD simulations.

To probe the flexibilities of the four RAD52–ligand complexes, the root-mean-squared fluctuation (RMSF) of the protein backbone was calculated (Fig. 5). The lower the RMSF value, the more stable the amino acid. As shown in Fig. 5A, the fluctuation trend of the backbone amino acid residues of the four complexes between amino acid residues 25 to 175 was relatively consistent, and it peaked at around resides GLY60, ASN100, GLU130. We further focused on the DNA binding-related residues ARG55, TYR65 (Fig. 5B), LYS152, ARG153, and ARG156 (Fig. 5C). ARG55 and TYR65 were located at the shoulder of the peak at GLY60, giving RMSF values of around 0.25 nm. Notably, LYS 152, ARG 153, and ARG 156 were all located at the lower end of the RMSF curve, indicating that the DNA binding pocket was more stable during MD simulation, which favored the binding of inhibitors.
Fig. 5. RMSF of the RAD52 protein backbone. (A). RMSFs observed during three equal portions of a 30 ns MD simulation of complexes of RAD52 with the putative inhibitors. (B) and (C). Enlarged presentation of the segments containing the key DNA-binding amino acid residues (ARG55, TYR65, LYS152, ARG153, and ARG156), (B) the fluctuation of amino acids from GLN45-ARG70; (C) the fluctuation of amino acids from GLY150-ASP170.

2.4. Binding free energies of the RAD52–ligand association
Based on the MD trajectory file, we calculated the total energies of the four RAD52–ligand complexes (Fig. 6). RAD52-F779-0434 complex had the lowest value of total energy, indicating that even in the dynamic state, F779-0434 was the most stable small molecule bound to RAD52 among the four candidate compounds.
Fig. 6. Comparison of the binding free energy values of the RAD52–ligand associations.

2.5. Compound F779-0434 specifically inhibited the growth of BRCA2-deficient cell line
BRCA2-deficient cell lines have been shown to be sensitive to the inhibition of the RAD52 function. To determine whether these putative RAD52 inhibitors, which were selected based on their ADMET properties and molecular dynamics simulation results, were truly effective, we tested their effect on the growth of BRCA2-proficient (BxPC3) and BRCA2-deficient (Capan-1) cells. After treating the cells for 6 days at the indicated concentrations, it was found that F779-0434 was the only compound that displayed a differential inhibition on BxPC3 and Capan-1 cell growth (Fig. 7A). F779-0434 inhibited 50% of the Capan-1 cell growth at a concentration of about 10 μM, whereas more than 90% of the BxPC3 cells survived at the same concentration. At the concentration of 20 μM, the growth of Capan-1 cells was almost completely inhibited by F779-0434 when compared with the survival of more than 60% of BxPC3 cells. Even though molecular dynamics analyses predicted relatively stable associations of the other three compounds (F848-0436, G640-1014, and D207-0130) with the RAD52 protein, these three compounds did not demonstrate strong inhibitory effects on the growth of either BxPC3 or Capan-1 cells. The concentrations needed to inhibit 50% of the cell growth were all higher than 100 μM. More importantly, there were no differential inhibitory effects of these compounds on BxPC3 and Capan-1, suggesting that these three compounds might target proteins other than RAD52.
Fig. 7. Effect of the selected compounds on the survival of Capan-1(BRCA2-) and BxPC3 (BRCA2+) cells. The experiments were repeated at least three times; error bars indicate standard error of the mean (SEM).

2.6. Compound F779-0434 inhibited RAD52-ssDNA association
Based on the results of the cell viability assay, we further analyzed the effect of compound F779-0434 on the RAD52–ssDNA interaction in vitro to confirm that the synthetic lethality effect observed resulted from the direct inhibition of the RAD52 function. Histidine-tagged human RAD52 protein was expressed in E. coli and purified to homogeneity, as seen on SDS-PAGE (Fig. 8A and B). We then performed an affinity pull-down assay to analyze the RAD52–ssDNA interaction. In this assay, RAD52 associated with ssDNA could be pulled down as a result of streptavidin–biotin affinity interaction, and the associated RAD52 could be visualized by SDS-PAGE (Fig. 8C). As shown in Fig. 8D, the addition of F779-0434 efficiently inhibited the binding of the RAD52 protein to ssDNA. When an F779-0434 solution having a concentration of 5 μM was added, around 50% of the RAD52–ssDNA association was disrupted, presumably due to the occupation of the ssDNA binding motif of RAD52 (Fig. 8E). The RAD52–ssDNA interaction was invisible on the SDS-PAGE at 20 μM F779-0434 concentration.
Fig. 8. Compound F779-0434 inhibited Rad52–ssDNA association. (A). Purified human RAD52 protein was analyzed by SDS-PAGE. (B). Western blot analysis of RAD52 expressing plasmid-transformed E. coli lysate and purified RAD52 protein with anti-Histag antibody, lane 1, uninduced total E. coli lyaste; Lane 2, 0.25 mM IPTG induced E. coli lysate; lane 3, purified Rad52 protein; (C). Schematic illustration of the pull-down assay performed to analyze RAD52–ssDNA interaction. (D). A representative SDS-PAGE of the pull-down assay result; (E). Quantification of RAD52 pulled down by ssDNA. The experiments were repeated at least three times. Error bars indicate standard error of the mean (SEM).

3. Discussion
Targeting synthetically lethal gene pairs involved in the survival of tumor cells with small molecule inhibitor has been demonstrated to be an effective way in cancer therapy, especially considering the success of PARP inhibitors.35,36 Depletion of the human DNA repair protein RAD52 leads to synthetic lethality with defects in tumor suppressors including BRCA1, BRCA2, PALB2, and RAD51 paralogs.21–23,37 In contrast to PARP1 inactivation, RAD52 causes lethality of BRCA1/2-deficient cells through different mechanisms, which are still not well understood. It is likely that the synthetic lethality of BRCA/PALB2-RAD52 is a result of the disruption of the putative RAD52 recombination mediator function and its role in single-strand annealing (SSA).4 RAD52 forms an oligomeric ring,32,33,38,39 where the ssDNA-binding motif of RAD52 is continuous around the circumference of the ring and has shallow sub-pockets that are repeated in each monomer.40,41 Since the DNA binding activity of RAD52 is essential for its proposed recombination mediator function and SSA, we designate the ssDNA-binding groove as the feature to be targeted by small molecule inhibitors, which are expected to cause synthetic lethality in BRCA-deficient cells.
Drug discovery and development are time- and resource-consuming processes. Structure-based design and screening strategies apply computational power to expedite and facilitate hit identification and hit-to-lead selection.42–44 In the current study, we performed a virtual screening of 47 737 compounds from the Targeted Diversity Library (TDL) of Chemdiv database, which signified the superposition of highly diverse chemical space on the assortment of divergent families or sub-families of targets and unique biomolecules. Our docking strategy focused on the experimentally identified key amino acid residues for RAD52–ssDNA interaction (Fig. 1). Based on the scores, we chose the top 30 compounds and grouped them into five categories based on their chemical structures (Table S1†). Even though all of the 47 737 compounds included in the TDL are drug-like, our ADMET analysis revealed the differences in their pharmacological properties (Table S2†). Four compounds, namely, F779-0434, F848-0436, G640-1014, and D207-0130, were selected based on the AMDET analysis results and their availability from the vendor for further molecular dynamics and biochemical studies. Computational docking placed all of the four predicted RAD52 inhibitors into the ssDNA-binding groove (Fig. 2). Molecular dynamics simulation indicated that three out of the four selected compounds formed hydrogen bonds with key amino acid residues involved in DNA binding, suggesting stable RAD52–ligand interactions (Fig. 3).
The root-mean-square deviation (RMSD) measures the distance of certain atoms in a molecule with respect to a reference structure, which enables the identification of time points for conformational changes. For the four analyzed compounds, RMSD clearly indicated that the association of G640-1014 and D207-0130 to RAD52 caused vigorous fluctuation in the protein backbone, suggesting unstable binding of these two compounds to RAD52 (Fig. 4). The RMSD fluctuations of F779-0434 and F848-0436 were lower, especially the RMSD fluctuation of F779-0434 was measured to be the lowest among those of the four candidates. This observation was consistent with the cell viability assay results, in which we showed that F779-0434 selectively killed cells depleted of BRCA2 with the highest cytotoxicity (Fig. 7). The root-mean-square fluctuation (RMSF) measures the deviation between the positions of certain particles to the reference position, defining the high fluctuating areas in the molecule. Our analysis revealed that all of the previously identified key amino acid residues (ARG55, TYR65, LYS152, ARG153, and ARG156) involved in the RAD52–ssDNA binding were located in the areas with lower RMSF fluctuation (Fig. 5). Our pull-down experiment results further confirmed that F779-0434 efficiently inhibited the association of ssDNA to RAD52 protein (Fig. 8), causing the synthetic lethality observed in the cell viability assay.
We analyzed the specific rotary powers of the four compounds explored in the current study. Specifically, the values are as follows: F779-0434 α20D = 0.323, G640-1014 α20D = −0.167, and D207-0130 α20D = −0.151. These values suggested that the compounds exploited for our biological assays were basically racemates. Even though the concentration required for efficient killing by the RAD52-specific inhibitor identified in the current study was relatively high, F779-0434 could be further studied as a promising compound for targeted cancer therapy. Its efficacy could be further tested in other genetic backgrounds, which is synthetically lethal with RAD52. BRCA1 and BRCA2 mutations are predisposed to malignant diseases such as breast and ovarian cancers.9,10 Mutations in PALB2 and RAD51C also contribute to hereditary breast and ovarian cancers.45,46 Replacing or supplementing standard radiation and chemotherapies with specific RAD52 inhibitors will help to decrease the toxicity associated with these treatments.
4. Materials and methods
4.1. RAD52 and compounds library preparation for docking
The file for the crystal structure of RAD52 (PDB entry: 5JRB) containing the first 212 amino acid residues with a resolution of 2.4 Å was downloaded from the RCSB Protein Data Bank database. The PDB file of RAD52 was modified to include only chain A of the elemental structure, and it was then prepared using UCSF Chimera (version 1.11.2),47 in which hydrogen atoms and standard charges were added. Commercially available chemical compounds in the Targeted_Diversity_47 737 library from the ChemDiv database were converted from the original SDF format into the mol2 format using the Openbabel software. Then, the Chimera software was used to minimize the energy of the 3D structure of the compounds.
4.2. Virtual screening for potential RAD52 inhibitors
The docking site in RAD52 protein was determined according to previous studies, which included the reported DNA binding-related key amino acid residues (ARG 55, TYR 65, LYS 152, ARG 153, ARG 156).28,32,33 The selected sphere was within a root-mean-square deviation (RMSD) of 0.6 nm of every atom in the crystal structure of the ligand.
A two-step docking was performed using the DOCK 6.5 program.48 The first step of docking was carried out using the grid-based scoring method, which allowed the ligand to be flexible and to be structurally rearranged in response to the receptor. The ranked structures of these compounds were then rescored using the amber scoring algorithm. The conformations of the top 30 compounds with the lowest amber scores were chosen, and the compounds were classified based on their structural characteristics. Furthermore, these 30 compounds were subjected to the iLOG predictor of the Swiss ADMET online tool for the in silico ADME property analyses.49
4.3. Molecular dynamic simulation
The MD simulations were performed using the GROMACS package (version 4.6.7).50 To convert the protein structure into the topology, we used pdb2gmx with the AMBER99SB force field and the TIP3P water model. General amber force field (GAFF) parameters were used for the selected compounds, and the atomic partial charges were determined by the AM1-BCC method via the Antechamber program. The parmchk program was then employed to check missing force field parameters and generate additional force field parameter files.51 To obtain the parameters and topologies for the compounds, Antechamber Python parser interface (ACPYPE) tools were employed.52 The simulations were then performed under periodic boundary conditions (PBC) with a dodecahedral periodic box and by setting the minimum distance between the protein and the edge of the box to 1 Å. The system was solvated by adding simple point charge (SPC) water molecules.53 To make the system neutral overall, Na+ and Cl− were added to mimic a physiological NaCl concentration of 0.15 M.
Energy minimization was performed by GROMACS.54 Then, 100 ps NVT (constant number of particles, volume, and temperature) and 100 ps NPT (constant number of particles, pressure, and temperature) equilibrations were carried out for each system with the RAD52 and compound position restraints. Finally, we carried out a 30 ns production simulation with a 0.002 ps time step at constant pressure (1 bar) and temperature (300 K). During the MD simulation process, all bond lengths were constrained by the LINCS algorithm,55 and the long range electrostatic interactions were calculated via the particle mesh Ewald (PME) method.56
4.4. Structural analysis of MD trajectories
Per-residue root-mean-square fluctuation (RMSF) and root-mean-square deviation (RMSD) analysis were carried out by the g_rmsf and g_rmsd programs, respectively, in the GROMACS package (version 4.6.7).50 The potential energy, kinetic energy, and total energy were calculated using the g_energy program available in GROMACS,50 and we extracted information from edr files generated during the MD simulations. In order to more intuitively compare the energy levels between the different MD systems, we normalized the three energy component values, and the normalization was performed as follows: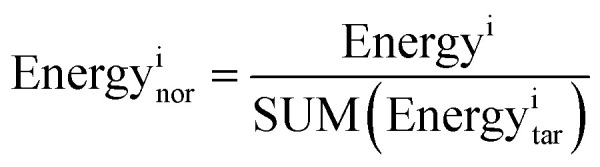
Energyi stands for pre-normalization value of a specific type of binding energy. SUM(Energyitar) stands for the sum of binding energies of the RAD52–ligand complexes.
Attractive forces (hydrogen bonds and hydrophobic interactions) between key residues identified beforehand in the RAD52 binding system were analyzed using the LigPlot+ software (version 1.4.5).57 The 3D protein–ligand complexes generated during MD simulations were flattened into 2D diagrams using the LIGPLOT algorithm.58
4.5. Chemicals, protein and DNA
The chemical compounds, F779-0434, F848-0436, G640-1014 and D207-0130, were purchased from ChemDiv (San Diego, CA, USA). The pET21-Rad52 expression plasmid was obtained from Dr Tomohiko Sugiyama from Ohio University, in which the human Rad52 ORF was cloned into pET21 plasmid for 6His-Rad52 expression. Human 6His-Rad52 were purified as described.59 The sequence of the 50-mer biotinylated oligonucleotide ssDNA was 5′-TAAATGCCAATGCTGCTGATACGTACTCGGACTGATTCGGAACTGTAACG-3′, and it was synthesized by BGI (Shenzhen China). Streptavidin MagBeads used in pull down assay was purchased from Yeasen Biotechnology (Shanghai, China)
4.6. Cell viability assay
BxPC3 cells were cultured in RPMI 1640 media supplemented with 10% FBS and 1% antibiotics (Gibco, Thermo Fisher Scientific, Waltham, MA, USA); Capan-1 cells were cultured in IMDM media containing 20% FBS and 1% antibiotics. Cells were seeded in a 96-well plate at the density of 500 cells/well. After 24 h, the cells were treated with indicated concentrations of compounds. Cell culture media containing invariant concentrations of compounds were refreshed every 3 days. After 6 days of treatment, absorbance was measured at 450 nm using CCK8 (7sea Biotechnology, Shanghai, China), according to the manufacture's protocol.
4.7. Western blotting
Human 6His-Rad52 protein was detected by western blot analysis using antibody against histidine affinity tag. Briefly, E. coli lysate or purified RAD52 was separated on 10% SDS-PAGE gel, transferred to PVDF membrane, blocked with 10% nonfat milk in TBST and probed using monoclonal mouse anti-His antibody (Proteintech, Wuhan, China). Immunoblot signal was detected using the chemiluminescence detection kit (Pierce, Thermo Fisher Scientific, Waltham, MA, USA).
4.8. RAD52 pull-down assay
The pull down assay was performed as described by Sugiyama and colleagues.59 Streptavidin MagBeads was prepared by washing with the reaction buffer (25 mM Tris–HCl, pH 7.5, 100 μM DTT, 1 mM MgCl2, 10 μg ml−1 BSA) for 3 times. RAD52 protein, biotin end-labeled ssDNA, MagBeads and various concentrations of F779-0434 were added to the reaction system, and this was incubated at 37 °C for 1 min. The beads were precipitated by setting the reaction tubes on a magnet holder for 3 min; the supernatant was removed, and the beads were washed with reaction buffer for 3 times. The washed beads were mixed with SDS-PAGE sample loading buffer, boiled for 5 minutes and analyzed by 10% SDS-PAGE gel.
Author contributions
Jian Li, Rong Sun, and Qi Zhao conceived and designed the experiments; Jian Li, Qianye Yang, Yang Zhang, and Kejia Huang performed the experiments; Jian Li, Qianye Yang, Rong Sun, Qi Zhao analyzed the data; Jian Li, Rong Sun, and Qi Zhao wrote the manuscript. All the authors went over the manuscript before submission.
Conflicts of interest
The authors declare no conflict of interest.
Supplementary Material
Acknowledgments
This study was supported by the Natural Science Foundation of China (No. 31500657), the International Collaboration Projects from the Science and Technology Bureau of Sichuan Province (No. 2017HH0097) and the Technology Foundation for Selected Overseas Chinese Scholar from Ministry of Human Resources and Social Security of the People's Republic of China to Jian Li.
Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01919c
References
- Moynahan M. E. Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010;11(3):196–207. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyama T. Wilson 3rd D. M. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair. 2013;12(8):620–636. doi: 10.1016/j.dnarep.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helleday T. DNA repair as treatment target. Eur. J. Cancer. 2011;47(suppl 3):S333–S335. doi: 10.1016/S0959-8049(11)70192-7. [DOI] [PubMed] [Google Scholar]
- Lok B. H. Powell S. N. Molecular pathways: understanding the role of Rad52 in homologous recombination for therapeutic advancement. Clin. Cancer Res. 2012;18(23):6400–6406. doi: 10.1158/1078-0432.CCR-11-3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord C. J. Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481(7381):287–294. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- Li X. Heyer W. D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18(1):99–113. doi: 10.1038/cr.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung P. Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol. 2006;7(10):739–750. doi: 10.1038/nrm2008. [DOI] [PubMed] [Google Scholar]
- Karanam K. et al., Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol. Cell. 2012;47(2):320–329. doi: 10.1016/j.molcel.2012.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King M. C. Marks J. H. Mandell J. B. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302(5645):643–646. doi: 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- Antoniou A. et al., Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am. J. Hum. Genet. 2003;72(5):1117–1130. doi: 10.1086/375033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. 2008;26(22):3785–3790. doi: 10.1200/JCO.2008.16.0812. [DOI] [PubMed] [Google Scholar]
- McLornan D. P. List A. Mufti G. J. Applying synthetic lethality for the selective targeting of cancer. N. Engl. J. Med. 2014;371(18):1725–1735. doi: 10.1056/NEJMra1407390. [DOI] [PubMed] [Google Scholar]
- Farmer H. et al., Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Kaelin Jr W. G. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer. 2005;5(9):689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- Bryant H. E. et al., Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- Yap T. A. et al., Poly(ADP-ribose) polymerase (PARP) inhibitors: Exploiting a synthetic lethal strategy in the clinic. Ca-Cancer J. Clin. 2011;61(1):31–49. doi: 10.3322/caac.20095. [DOI] [PubMed] [Google Scholar]
- Mortensen U. H. Lisby M. Rothstein R. Rad52. Curr. Biol. 2009;19(16):R676–R677. doi: 10.1016/j.cub.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Rijkers T. et al., Targeted inactivation of mouse RAD52 reduces homologous recombination but not resistance to ionizing radiation. Mol. Cell. Biol. 1998;18(11):6423–6429. doi: 10.1128/MCB.18.11.6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi-Iwai Y. et al., Homologous recombination, but not DNA repair, is reduced in vertebrate cells deficient in RAD52. Mol. Cell. Biol. 1998;18(11):6430–6435. doi: 10.1128/MCB.18.11.6430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanez R. J. Porter A. C. Differential effects of Rad52p overexpression on gene targeting and extrachromosomal homologous recombination in a human cell line. Nucleic Acids Res. 2002;30(3):740–748. doi: 10.1093/nar/30.3.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z. et al., Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. U. S. A. 2011;108(2):686–691. doi: 10.1073/pnas.1010959107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok B. H. et al., RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene. 2013;32(30):3552–3558. doi: 10.1038/onc.2012.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J. Buechelmaier E. S. Powell S. N. Rad51 paralog complexes BCDX2 and CX3 act at different stages in the BRCA1-BRCA2-dependent homologous recombination pathway. Mol. Cell. Biol. 2013;33(2):387–395. doi: 10.1128/MCB.00465-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyck E. et al., Visualization of recombination intermediates produced by RAD52-mediated single-strand annealing. EMBO Rep. 2001;2(10):905–909. doi: 10.1093/embo-reports/kve201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimme J. M. et al., Human Rad52 binds and wraps single-stranded DNA and mediates annealing via two hRad52-ssDNA complexes. Nucleic Acids Res. 2010;38(9):2917–2930. doi: 10.1093/nar/gkp1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagawa W. et al., Homologous pairing promoted by the human Rad52 protein. J. Biol. Chem. 2001;276(37):35201–35208. doi: 10.1074/jbc.M104938200. [DOI] [PubMed] [Google Scholar]
- Murfuni I. et al., Survival of the replication checkpoint deficient cells requires MUS81-RAD52 function. PLoS Genet. 2013;9(10):e1003910. doi: 10.1371/journal.pgen.1003910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan K. et al., Identification of a Small Molecule Inhibitor of RAD52 by Structure-Based Selection. eLife. 2016;11(1):e0147230. doi: 10.1371/journal.pone.0147230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F. et al., Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Res. 2016;44(9):4189–4199. doi: 10.1093/nar/gkw087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengel S. R. et al., Small-molecule inhibitors identify the RAD52–ssDNA interaction as critical for recovery from replication stress and for survival of BRCA2 deficient cells. eLife. 2016;5:e14740. doi: 10.7554/eLife.14740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandramouly G. et al., Small-Molecule Disruption of RAD52 Rings as a Mechanism for Precision Medicine in BRCA-Deficient Cancers. Chem. Biol. 2015;22(11):1491–1504. doi: 10.1016/j.chembiol.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagawa W. et al., Crystal structure of the homologous-pairing domain from the human Rad52 recombinase in the undecameric form. Mol. Cell. 2002;10(2):359–371. doi: 10.1016/S1097-2765(02)00587-7. [DOI] [PubMed] [Google Scholar]
- Singleton M. R. et al., Structure of the single-strand annealing domain of human RAD52 protein. Proc. Natl. Acad. Sci. U. S. A. 2002;99(21):13492–13497. doi: 10.1073/pnas.212449899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saotome M. et al., Structure of the human DNA-repair protein RAD52 containing surface mutations. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2016;72(Pt 8):598–603. doi: 10.1107/S2053230X1601027X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouleau M. et al., PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer. 2010;10(4):293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. et al., In Silico Screening Identifies a Novel Potential PARP1 Inhibitor Targeting Synthetic Lethality in Cancer Treatment. Int. J. Mol. Sci. 2016;17(2):258. doi: 10.3390/ijms17020258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer-Morales K. et al., Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood. 2013;122(7):1293–1304. doi: 10.1182/blood-2013-05-501072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd J. A. Forget A. L. Knight K. L. Correlation of biochemical properties with the oligomeric state of human rad52 protein. J. Biol. Chem. 2002;277(48):46172–46178. doi: 10.1074/jbc.M207262200. [DOI] [PubMed] [Google Scholar]
- Stasiak A. Z. et al., The human Rad52 protein exists as a heptameric ring. Curr. Biol. 2000;10(6):337–340. doi: 10.1016/S0960-9822(00)00385-7. [DOI] [PubMed] [Google Scholar]
- Lloyd J. A. McGrew D. A. Knight K. L. Identification of residues important for DNA binding in the full-length human Rad52 protein. J. Mol. Biol. 2005;345(2):239–249. doi: 10.1016/j.jmb.2004.10.065. [DOI] [PubMed] [Google Scholar]
- Mortensen U. H. et al., A molecular genetic dissection of the evolutionarily conserved N terminus of yeast Rad52. Genetics. 2002;161(2):549–562. doi: 10.1093/genetics/161.2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchen D. B. et al., Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discovery. 2004;3:935. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- Kapetanovic I. M. Computer-aided drug discovery and development (CADDD): in silico-chemico-biological approach. Chem.-Biol. Interact. 2008;171(2):165–176. doi: 10.1016/j.cbi.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider G. Virtual screening: an endless staircase? Nat. Rev. Drug Discovery. 2010;9:273. doi: 10.1038/nrd3139. [DOI] [PubMed] [Google Scholar]
- Antoniou A. C. et al., Breast-cancer risk in families with mutations in PALB2. N. Engl. J. Med. 2014;371(6):497–506. doi: 10.1056/NEJMoa1400382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meindl A. et al., Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat. Genet. 2010;42(5):410–414. doi: 10.1038/ng.569. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F. et al., UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Brozell S. R. et al., Evaluation of DOCK 6 as a pose generation and database enrichment tool. J. Comput.-Aided Mol. Des. 2012;26(6):749–773. doi: 10.1007/s10822-012-9565-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A. Michielin O. Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronk S. et al., GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29(7):845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. et al., Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graphics Modell. 2006;25(2):247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Sousa da Silva A. W. Vranken W. F. ACPYPE - AnteChamber PYthon Parser interfacE. BMC Res. Notes. 2012;5(1):367. doi: 10.1186/1756-0500-5-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H. J. C., et al., Interaction Models for Water in Relation to Protein Hydration, in Intermolecular Forces: Proceedings of the Fourteenth Jerusalem Symposium on Quantum Chemistry and Biochemistry Held in Jerusalem, ed. B. Pullman, Springer Netherlands, Israel, Dordrecht, 1981, pp. 331–342 [Google Scholar]
- Van Der Spoel D. et al., GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26(16):1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Hess B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008;4(1):116–122. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- Essmann U. et al., A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103(19):8577–8593. doi: 10.1063/1.470117. [DOI] [Google Scholar]
- Laskowski R. A. Swindells M. B. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011;51(10):2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- Wallace A. C. Laskowski R. A. Thornton J. M. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8(2):127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- Sugiyama T. Kantake N. Dynamic regulatory interactions of rad51, rad52, and replication protein-a in recombination intermediates. J. Mol. Biol. 2009;390(1):45–55. doi: 10.1016/j.jmb.2009.05.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.