

Abstract
Background:
Allergen-induced airway hyperresponsiveness (AHR) in neonatal mice, but not adult mice, is caused by elevated innervation and consequent cholinergic hyperstimulation of airway smooth muscle (ASM). Whether this inflammation-independent mechanism contributes to ASM hypercontraction in childhood asthma warrants investigation.
Objective:
We aim to establish the functional connection between cholinergic stimulation and ASM contractility in different human age groups.
Methods:
First, we employed a neonatal mouse model of asthma to identify age-related mediators of cholinergic deregulation of ASM contractility. Next, we conducted validation and mechanistic studies in primary human ASM cells and precision-cut lung slices (PCLSs) from young (<5 years old) and adult (>20 years old) donor lungs. Finally, we evaluated the therapeutic potential of the identified cholinergic signaling mediators using culture models of human ASM hypercontraction.
Results:
We have discovered that ASM hypercontraction due to cholinergic deregulation in early postnatal life requires CD38. Mechanistically, cholinergic signaling activates the PI3K/Akt pathway in immature ASM cells to upregulate CD38 levels, thereby augmenting the Ca2+ response to contractile agonists. Strikingly, this early life, CD38-mediated ASM hypercontraction is not alleviated by the β-agonist, formoterol.
Conclusions:
Our findings identify the acetylcholine-PI3K/Akt-CD38 axis as a critical mechanism of AHR in early postnatal life. Targeting this axis may provide a tailored treatment for children at high risk for allergic asthma.
Keywords: airway smooth muscle (ASM), airway hyperresponsiveness (AHR), precision-cut lung slice (PCLS), CD38, cholinergic innervation, childhood asthma, PI3K, Akt
Graphical Abstract
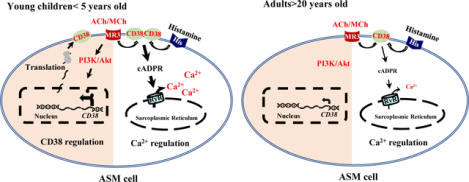
Akt (A serine/threonine kinase), ASM (Airway smooth muscle), cADPR (Cyclic ADP-ribose), PI3K (Phosphoinositide 3-kinase), RyR (Ryanodine receptor), ACh (Acetylcholine), MCh (methacholine), MR3 (muscarinic receptor 3), His R (Histamine receptor).
Capsule summary:
The acetylcholine-PI3K/Akt-CD38 pathway is a potential anti-AHR target in asthmatic children.
Introduction
Allergic asthma often initiates from early childhood exposure to environmental insults, such as allergens, respiratory virus, cigarette smoke, pollutant, and ozone. About 50% of cases of childhood asthma progress into adulthood (1, 2). Intriguingly, airway hyperresponsiveness (AHR), the key feature of asthma, can proceed even in the absence of overt inflammation (3, 4). Due to limited access to lung samples from young children, mechanisms underlying this inflammation-independent AHR remain poorly understood. Such limitation in our knowledge contributes to suboptimal control of symptoms in severe asthmatics and a lack of therapeutic intervention that modulates the progression of AHR from childhood to adulthood.
Utilizing mouse models of allergic asthma at different ages, we have recapitulated a persistent AHR phenotype following allergen exposure during the neonatal time window (5, 6). We have discovered that AHR in neonatal mice can be induced by cholinergic hyperinnervation of airway smooth muscle (ASM) and maintained long after the resolution of inflammation. Such cholinergic hyperinnervation is caused by elevated levels of nerve growth factors during allergic inflammation (5). In contrast, similar allergen exposure in adult mice has no effect on cholinergic innervation of ASM and causes transient AHR (6). Strikingly, methacholine (MCh) treatment to simulate cholinergic hyperinnervation is sufficient to induce a hypercontractile phenotype of ASM cells in pups but not in adult mice (6). Together, these findings suggest that unique contractile regulation may operate exclusively in immature ASM cells, rendering the early postnatal life a time window of susceptibility to the development of AHR by environmental stimuli.
ASM cells are the ultimate effector of airway narrowing in response to contractile stimuli and play an essential role in the pathogenesis of AHR in asthma (7, 8). In accord with ASM hyperinnervation in animal models of prolonged AHR following neonatal exposure, antibody staining of biopsy samples from adult asthmatic patients display significant increases in ASM innervation by sensory and cholinergic nerves (9, 10). In addition, clinical studies have reported that children at high risk for allergic asthma have elevated levels of nerve growth factors in the airway (11), and sustained levels of nerve growth factors positively correlate with disease severity (10, 12). Building upon the established role of nerve growth factors in the development of the nervous system in the lung (5, 13), ASM hyperinnervation may occur in young children at high risk for allergic asthma and drive persistent AHR into adulthood.
When allergic asthma develops in early childhood, ASM cells undergo a dynamic process of functional maturation. As such, mechanisms that regulate the contractile phenotype of immature ASM cells may differ from those in mature ASM cells in adults. Following a substantial transition from tonic contraction in utero to phasic contraction at birth, ASM cells continue to modify their contractile phenotype by reducing their shortening velocity and increasing their contractile force in early postnatal life (14, 15). These phenotypical changes are associated with an increase in the expression of contractile machinery components, such as smooth muscle myosin heavy chain (SMMHC), calponin, smooth muscle alpha-actin (αSMA), and caveolins (16). Using agonist-induced Ca2+ response as a measurement of contractility, we have shown that the contractile phenotype of ASM cells matures around postnatal 3 weeks (P21) in mice (6). The age when human ASM cells become functionally mature has yet to be precisely determined. Based on histological markers in the postnatal developing lung, P21 in mice is approximately 5–8 years in human (17), an age when allergic asthma is diagnosed.
ASM contraction is regulated by intracellular Ca2+. Intracellular Ca2+, in turn, is mediated through two independent pathways (18, 19). First is the phospholipase C-InsP3 receptor (PLC-IP3R) pathway that is activated through the G protein-coupled receptor that links to PLC and triggers Ca2+ release from the sarcoplasmic reticulum (SR) via IP3R. The second is the CD38-RyR pathway that involves the ADP-ribosyl cyclase activity of CD38 that biosynthesizes cADPR as a second messenger to interact with the ryanodine receptor (RyR) on the SR membrane to release Ca2+. Both the PLC-IP3R and CD38-RyR pathways contribute to ASM contraction in adults. Whether these two pathways play a distinct or redundant role in immature ASM cells has not been well studied (20).
Here, using precision-cut lung slices (PCLSs) and primary ASM cells from human donors of different ages, we have identified CD38 as a critical mediator of cholinergic deregulation of ASM contractility in early postnatal life and delineated the mechanism underlying this age-specific CD38 regulation.
Methods
Mice:
All mouse experiments were approved by the Institutional Animal Care and Use Committee at Brigham and Women’s Hospital. A double transgenic mouse line (αSMA-GFP; NG2-dsRed) was provided by Dr. Alan Fine at the Boston University School of Medicine (21). Lungs from CD38−/− mice were provided by Dr. Alonso Guedes at the University of Minnesota.
Human donor lungs:
Lungs from de-identified and deceased human donors were purchased from IIAM (International Institute for the Advancement of Medicine). These donor lungs were declined for transplantation and met the criteria of no smoking exposure or any underlying airway diseases. Because the research involved no intervention or interaction with living individuals or provided any identifiable private information about the donor, the project is deemed not human subject research by Partners Human Research Committee at Brigham and Women’s Hospital.
Preparation of the precision-cut lung slice (PCLS) and airway contraction assay:
Human PCLSs were prepared from donor lungs at 250 μm in thickness and cryopreserved using a published protocol (22). Frozen human PCLSs were thawed prior to experimental treatment. Fresh mouse PCLSs were prepared from wild-type and CD38−/− mice at P21. For airway contraction assay, PCLSs were stimulated with histamine or endothelin at increasing concentrations, and airway images were captured by an inverted phase-contrast microscope (Nikon Eclipse TS 100; Nikon, Tokyo, Japan) equipped with a Nikon DS-Ri2 camera. The area of airway lumen was measured using NIH Image J (National Institutes of Health, Bethesda, MD). Agonist-induced airway constriction was determined by normalizing the reduction in lumen area to the baseline value. For pretreatment with an agonist, PCLSs were incubated with the agonist in the culture media (DMEM/F12, Fisher Scientific, MT-10-092-CM) supplemented with anti-anti (Thermo Fisher Scientific, 15240-062) for 2.5 days. PCLSs were then washed to remove the agonist and recovered overnight, followed by the airway contraction assay. For the adult age group, human airways of 300–1000 μm in diameter and mouse airways of 100–300 μm in diameter were assayed. For the developmental age group, airways of 100–300 μm in diameter in young children and 50–150 μm in mouse pups were assayed.
Isolation of primary human ASM cells:
Segmental bronchi of donor lungs were dissected and cut open. The airway epithelium on the luminal surface was scraped off using cotton swabs. Connective tissues on the outside surface of the bronchi were removed. Airway smooth muscle strips in the bronchi were dissected out, cut into small pieces, and placed in a culture plate with the luminal side facing down. A small amount of culture media (DMEM/F12 with 10% FBS) was applied to facilitate the attachment of the smooth muscle pieces to the culture plate. After overnight, fresh culture media were added and changed every 3 days. ASM cells grew out of the muscle pieces and started to proliferate over the course of 7–14 days. Primary ASM cells within the first 3 passages were used for experiments.
Isolation of mouse ASM cells:
Neonatal and adult, αSMA-GFP; NG2-dsRed mice were subjected to ovalbumin (OVA) sensitization and challenge, followed by the isolation of GFP+ ASM cells by cell sorting using protocols as described previously(21). In this double fluorescent mouse line, ASM cells are single GFP+ and vascular smooth muscle cells were GFP+dsRed+ (21). Briefly, to induce allergic inflammation in neonatal mice, sensitization with intraperitoneal injections of 10 μg OVA (A5503; Sigma-Aldrich, St. Louis, MO, USA) in Imject alum (77161; ThermoScientific, Waltham, MA, USA) was performed on postnatal day 5 (P5) and P10. Mice were challenged with 3% aerosolized OVA solution on P18, P19, and P20 before ASM cell isolation. To induce allergic inflammation in adult mice, we sensitized 3-month-old mice by intraperitoneal injection of 25 ug OVA in Imject alum and then challenged them with 3% aerosolized OVA solution(6). Controls were challenged with PBS. Mice were sacrificed the next day after the last challenge. Lungs were dissociated with a commercially available Lung Dissociation Kit (Miltenyi Biotech, Auburn, CA), followed by the separation of ASM cells (GFP+) and vascular smooth muscle cells (GFP+dsRed+) using a Moflo cell sorter (Beckman Coulter, Fullerton, CA). ASM cells were pooled from 5–7 lungs as one sample for gene expression analysis.
siRNA transfection of primary human ASM cells:
Primary cultures of ASM cells from young children were transfected with siRNA against human CD38 mRNA (Thermo Fisher Scientific, s2657) (100nM) using lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific, 13778030) following manufacturer’s protocol. Scrambled control siRNA was similarly transfected as the negative control. The efficacy of CD38 knockdown was assessed by real-time PCR and Western blot. Cells transfected with control siRNA and CD38 siRNA were subjected to MCh treatment.
In vitro treatment of PCLS and primary human ASM cells:
Mouse and human PCLSs were treated with 10μM MCh in DMEM/F-12 with anti-anti for 2.5 days. Primary human ASM cells were maintained in DMEM/F12 with 0.5% FBS and treated with 10μM MCh for 2.5 days. To block PI3K, 10μM ly294002 (EMD Millipore, 19–142) or 100nM wortmannin (Sigma-Aldrich, W3144) was applied to cultures simultaneously with MCh. For β adrenergic agonist treatment, human PCLSs were treated with MCh and 5nM Formoterol (Sigma-Aldrich, F9552). Culture media were changed every 24 hours. To block CD38-cADPR, 100μM 8-Br-cADPR (Enzo Life Sciences, BML-CA417-0500) was added to ASM cultures 30 minutes prior to Ca2+ imaging.
Ca2+ imaging:
Primary ASM cells were incubated with 10μM Fluo-4 AM (Thermo Fisher Scientific, F14201) in HBSS for 1 hour at 37°C. Intracellular fluorescence was recorded at a rate of 1 image per second with an inverted phase-contrast fluorescence microscope (Nikon Eclipse TS 100; Nikon, Tokyo, Japan) equipped with a Nikon DS-Ri2 camera. The fluorescence intensity was analyzed by NIH Image J and expressed as Ft/F0, a ratio of the fluorescence intensity at a particular time (Ft) normalized to the average baseline fluorescence (F0) measured from 5 images prior to agonist stimulation.
Western blot:
Primary HASM cells were homogenized in RIPA buffer supplemented with a complete protease inhibitor cocktail (Roche, 11697498001) and phosphatase inhibitor cocktail (Millipore, 524627) followed by western blot assays. Primary antibodies include rabbit anti-CD38 (1: 400, Biorbyt, orb10319), rabbit anti-pan Akt (1:500, Cell Signaling Technology, 4691s), rabbit anti-phospho-Akt (1:500, Cell Signaling Technology, 4060s), mouse anti-phospho-p42/44 ERK (1:400, Cell Signaling Technology, 9106s), rabbit anti-p42/44 ERK (1:400, Cell Signaling Technology, 9102), mouse anti-β-actin (1:2000, Sigma Aldrich, A5441). HRP-conjugated secondary antibodies include goat anti-mouse (1:2000; BD Biosciences, 554002) and goat anti-rabbit (1:1000 BD Santa Cruz, sc-2004). The antigen-antibody complex was detected by SuperSignal Pico Plus Chemiluminescent Substrate (Thermo Fisher Scientific, 34577).
Antibody staining:
Paraffin sections of human lungs were stained for CD38 using a mouse anti-CD38 antibody (1:50, Thermo Fisher Scientific, MA1-19316) following an established immunohistochemistry staining protocol. Sections were counterstained with Hematoxylin QS (Vector Laboratories, H-3404). For antibody staining of the PCLS, the PCLS was fixed with 4% paraformaldehyde overnight. After washes in PBS, the PCLS was incubated with Cy3-conjugated mouse anti-α-smooth muscle actin (1:100, Sigma-Aldrich, C6198) at 4°C overnight. Images were captured by the Nikon inverted fluorescence microscope (Nikon Eclipse TS 100; Nikon, Tokyo, Japan). For quantification of α-smooth muscle actin, the pixel numbers of the fluorescent signal were measured by NIH Image J and normalized to the circumference of the airway.
Gene expression analysis:
Total RNA was extracted from mouse or human airway smooth muscle cells using a RNeasy kit (QIAGEN, 74106) followed by reverse transcription using Superscript III Reverse Transcriptase (Thermo Fisher Scientific, 18080-044). Real-time PCR was performed with CFX96 real-time system (Bio-Red). TaqMan primers (Thermo Fisher Scientific, Hs01120071 for human CD38, Hs99999901_s1 for 18S rRNA) were purchased from Thermo Fisher Scientific. The relative level of gene expression was measured by normalizing to 18S rRNA using ΔΔCt (cycle threshold difference).
Statistical Analysis:
For airway contraction assay, one airway in each PCLS was recorded as an individual observation. A minimum of 3 airways from each donor and 2–3 donors from each age group were quantified. For the Ca2+ signaling assay, each ASM cell was recorded as an observation, and a total of 15–80 ASM cells from 2–3 donors were quantified. For western blot and gene expression assays, two independent experiments were performed with primary human ASM cells (Passage 2–3) from each donor and cells from 2 donors of each age group were assayed. Results were analyzed using GraphPad Prism software (San Diego, CA). All data were reported as mean ± SEM from 2–3 independent experiments. For multiple comparisons, one or two-way ANOVA followed by Tukey’s post-hoc test was used. For the comparison between two experimental groups, an unpaired Student’s t-test was used. P value less than 0.05 was considered statistically significant.
Results
CD38 is required for MCh-induced ASM hypercontraction in neonatal mice.
We have previously shown that exposure to allergens (ovalbumin and house dust mite) in neonatal mice, but not in adult mice, causes cholinergic hyperstimulation that induces a hypercontractile phenotype of ASM cells (6). To identify the mediator of cholinergic induction of AHR in this neonatal allergic model, we purified ASM cells from saline and allergen-exposed, neonatal and adult mice and compared the expression of selected contractile genes with age. We employed an αSMA-GFP; NG2-dsRed mouse line. In this mouse line, ASM cells were single GFP+, and vascular smooth muscle cells were GFP+dsRed+, thereby enabling the isolation of ASM cells by sorting (21). Since allergic inflammation was induced in both neonatal and adult mice following allergen exposure, genes that were uniquely altered in allergic neonates were candidate mediators of cholinergic deregulation of ASM contraction. We found that CD38 and ryanodine receptor 3 (RyR3) mRNA levels were significantly elevated in ASM cells following allergen exposure in neonatal mice; however, they were unchanged in adult ASM cells (Fig. 1A and B). Other selected genes, such as Mylk, Ormdl3, Cav1, RhoA, and Rock2, were not affected by allergen exposure regardless of age. The upregulation of ASM expression of CD38 in this neonatal model is consistent with an augmented ASM Ca2+ response in a MCh exposure model in neonatal mice (Fig. E1), and airway hyperconstriction compared to neonatal mice with saline exposure (6). Based on these findings, we speculate that increased levels of CD38 in immature ASM cells triggered by cholinergic hyperstimulation may promote agonist-induced Ca2+ mobilization from SR to the cytoplasm via RyR, and ultimately, cause airway hyperconstriction.
Figure 1. CD38 is required to augment mouse airway contractility in early life by MCh treatment.
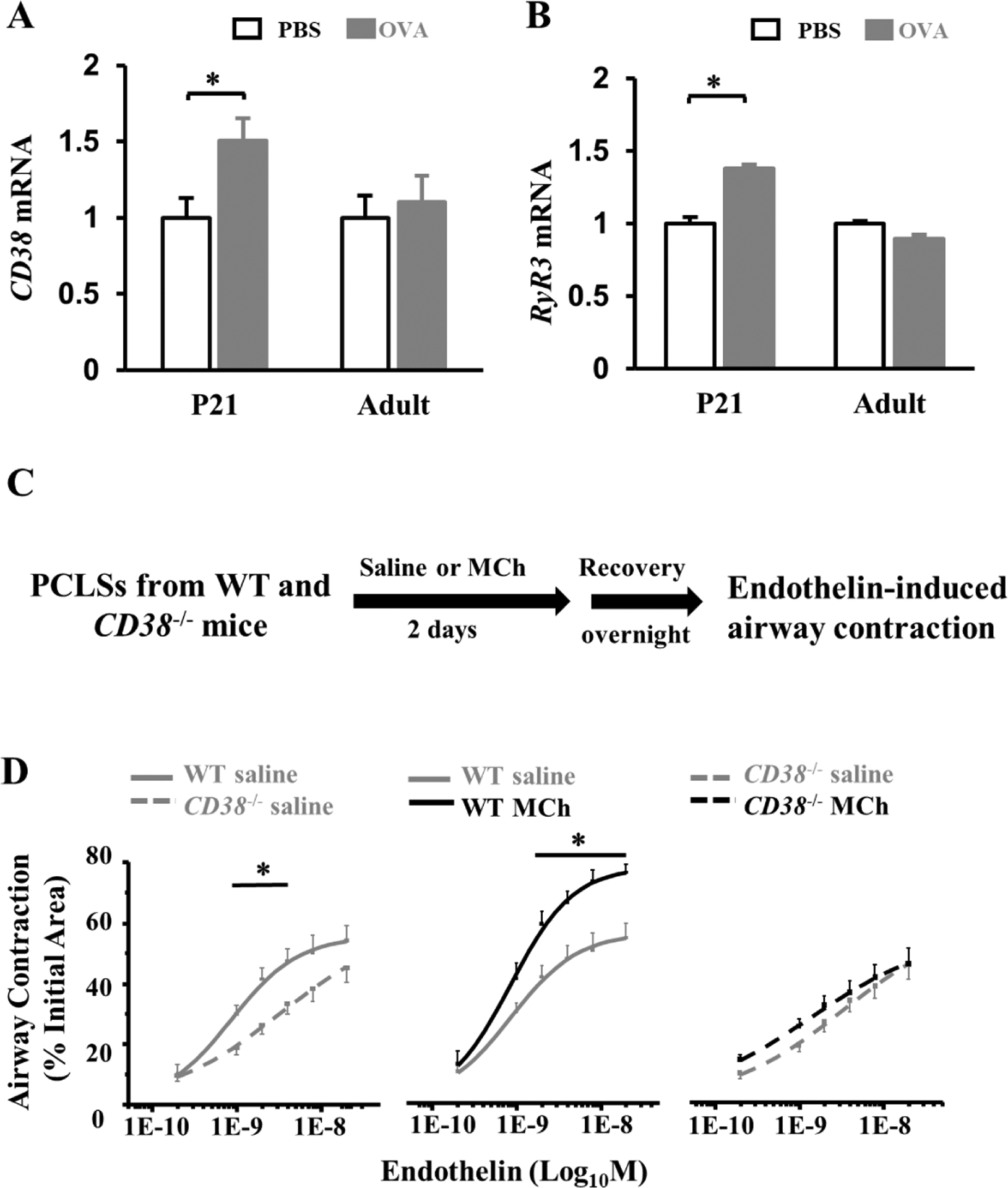
(A, B) Gene expression analyses of CD38 and ryanodine receptor 3 (RyR3) in isolated ASM cells of neonatal and adult mouse models of ovalbumin (OVA) exposure. ASM cells were pooled from 5–7 mice as one sample. Mice subjected to saline exposure were controls. For neonatal exposure, mice were analyzed at postnatal day 21 (P21). The bar graph represents the average ± SEM of 2–3 independent samples. *p<0.05 by unpaired Student’s t-test. (C) The saline and MCh treatment scheme of wild-type (WT) and CD38 −/− mouse PCLSs prior to the airway contraction assay. (D) The dose curves of saline- and MCh-treated WT and CD38−/− airways in response to endothelin stimulation. Each point represented 16–18 PCLSs from 4 CD38−/− mice or 8 slices from 2 wild-type mice at P21. *p<0.05 by two-way ANOVA followed by Tukey’s post-hoc test.
CD38 is broadly expressed in the airway by ASM cells, epithelial cells, and immune cells. To study tissue-specific function of CD38, previous studies have utilized bone marrow transplantation in adult mouse models of allergic inflammation (23). However, a bone marrow transplant is not technically feasible in neonatal mice. To circumvent this issue, we prepared neonatal murine PCLS from wild-type and CD38−/− mice at postnatal day 21 (P21), treated them with MCh, and examined whether CD38 was required for MCh-induced airway hyperconstriction in response to acute endothelin stimulation. In the absence of MCh treatment, CD38−/− PCLSs showed less contraction to endothelin (Fig. 1D), which validates the role of CD38 in the contractility of ASM cells as previously reported (24). In addition, MCh treatment promoted endothelin-induced airway contraction in wild-type PCLSs, but not in CD38−/− PCLSs (Fig. 1D). Therefore, CD38 is required for MCh-induced ASM hypercontraction in neonatal mice.
CD38 expression in human ASM cells decreases with age.
To explore whether CD38 played a similar age-related role in the contractile regulation of ASM in humans, we evaluated CD38 expression in the human airway at different ages. Non-asthmatic human lungs from deceased donors were divided into three separate age groups conforming to distinct stages of postnatal lung development in human: young children (<5 years old), teenagers (5–20 years old), and adults (>20 years old) (Table S1). CD38 was detected by antibody staining in endothelial cells, the airway epithelium, and ASM cells in human lung sections (Fig. 2A), as may be expected, given the multifaceted roles of CD38 in the vascular system, the immune system, and airway structural cells (25–27). CD38 staining in ASM and airway epithelial cells was found to be more prominent in young children than adults (Fig. 2A). To quantify the age-related difference in CD38 expression in human ASM, we derived primary ASM cells from segmental bronchi of human donor lungs. In low-passage cultures, ASM cells from young children expressed higher levels of CD38 at mRNA (~6 fold) and protein (~2 fold) levels compared to adult ASM cells (Fig. 2B–2D). These findings indicate that CD38 expression in human ASM cells decreases with age.
Figure 2. CD38 expression differs with age in human ASM cells.
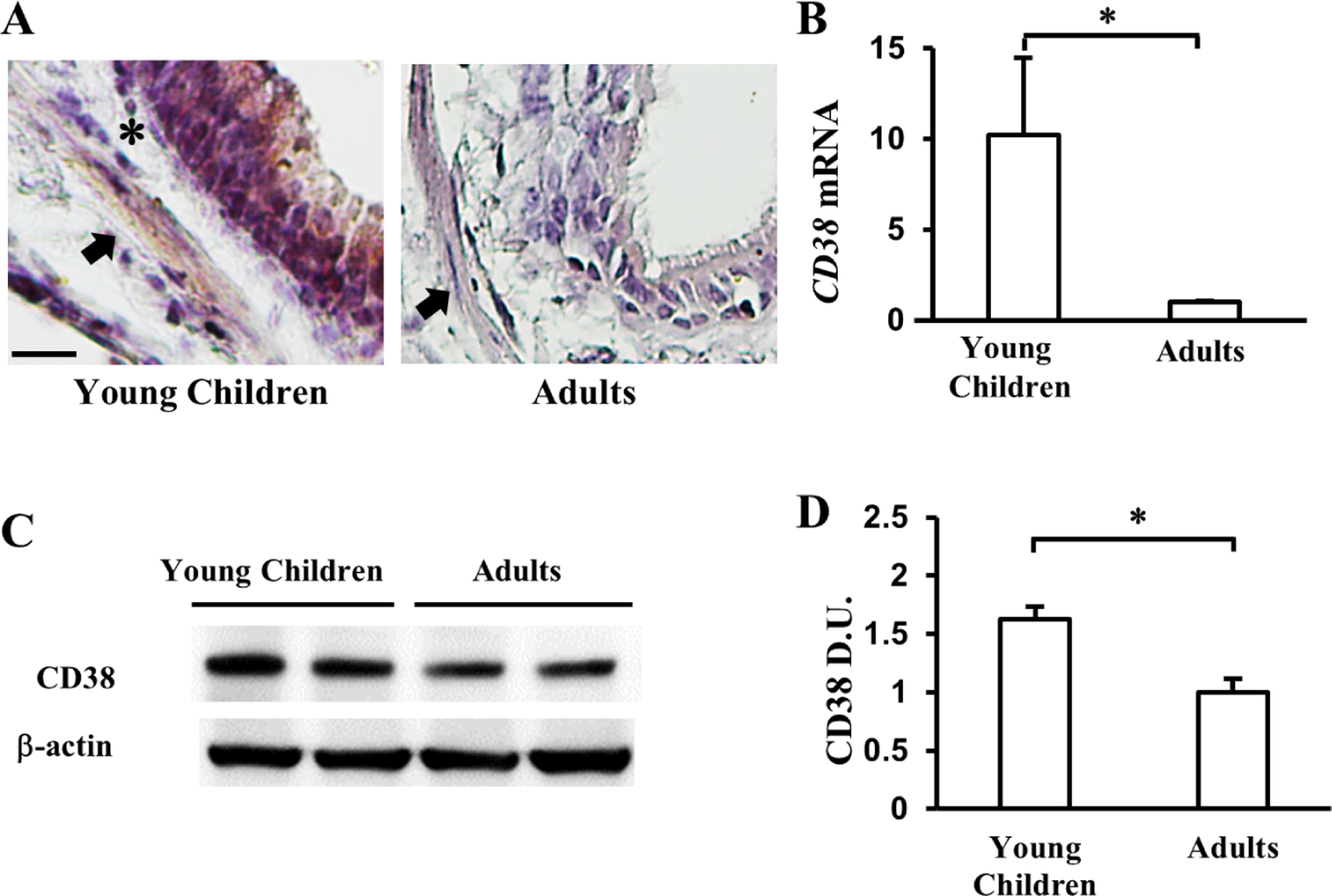
(A) Representative images of CD38 staining in the airways of young children and adults. Arrows mark CD38+ ASM cells. * marks CD38− cells in the basement membrane. Scale bar, 20μm. (B) qPCR analyses of CD38 mRNA levels in primary ASM cells isolated from young children and adult lungs. (C) The level of CD38 protein expression in primary ASM cells from young children and adults. (D) Quantification of the CD38 protein level in primary ASM cells by densitometry. CD38 mRNA and protein level of ASM in young children group were expressed in ratio to adult ones. The bar graph represents the mean ± SEM of 2 independent experiments of primary ASM cells from each donor and a total of 2 donors of each age group. *p<0.05 by unpaired Student’s t-test.
MCh treatment elevates CD38 expression and Ca2+ responses in immature ASM cells.
To test if MCh may similarly elevate CD38 expression in ASM cells in humans as in neonatal mice (Fig. 1), we treated low-passage cultures of ASM cells from young children and adults with 10μM MCh for 2.5 days following an established protocol (6) (Fig. 3A). After washing with saline and incubating with the culture medium for 4 hours, we assayed CD38 levels by Western blot assay (Fig. 3A). MCh treatment almost doubled the level of CD38 in ASM cells from young children (Fig. 3B) while having no effect on CD38 expression in adult ASM cells (Fig. 3C).
Figure 3. MCh treatment increases the expression of CD38 and the Ca2+ response in ASM cells from young children.
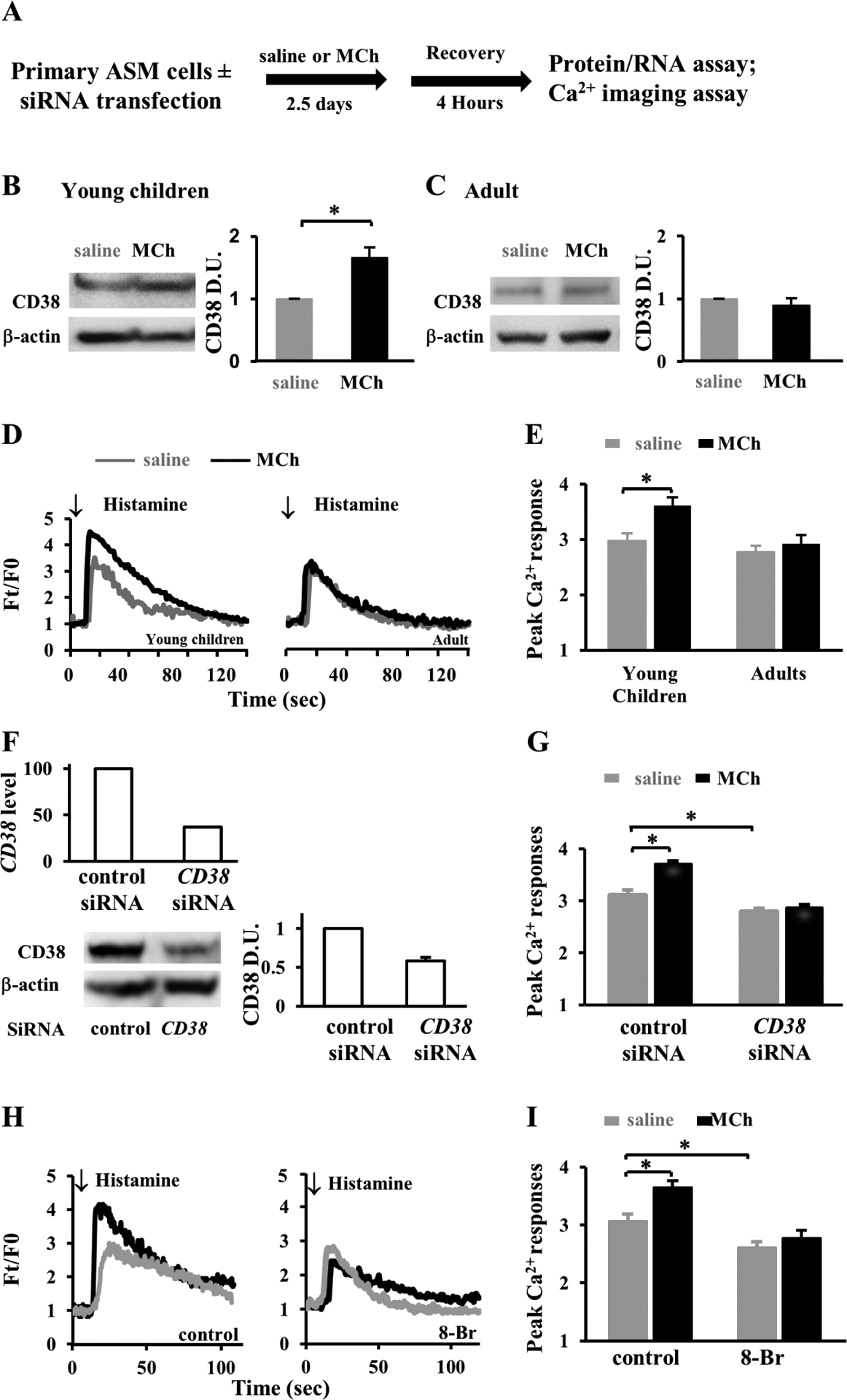
(A) Experimental scheme of treating primary ASM cells from young children and adults with 10μM MCh or saline control for 2.5 days followed by 4-hour recovery and then subjecting them to gene, protein assay, and Ca2+ imaging. For CD38 knockdown experiment, primary ASM cells were transfected with siRNA for 1.5 days before saline and MCh treatment. (B, C) Western blot assay and quantification by densitometry of CD38 protein expression in saline- and MCh-treated ASM cells from young children (B) and adults (C). Each bar graph represents the mean ± SEM of 2 independent experiments of ASM cells per donor and a total of 3 donors at each group. Data were normalized to saline treatment. (D) Representative traces and (E) the peak level of Ca2+ in response to 10μM histamine stimulation in saline- and MCh-treated young children and adult ASM cells. Each bar represents 30–32 cells from 2 children and 15–18 cells from 2 adults. (F) Assessment of CD38 knockdown in primary ASM cells from young children by qPCR of CD38 mRNA levels and by western blot assay of CD38 protein levels. Data was normalized to scrambled siRNA control. (G) The peak Ca2+ response to 10μM histamine in CD38 knockdown ASM cells. Each bar represents 65–75 cells from 2 donors. (H) Ca2+ traces and (I) peaks to 10μM histamine stimulation in saline or MCh-treated young children’s ASM cells, in the presence or absence of 100μM 8-Br-cADPR (8-Br). Each bar represents at least 15 cells from 2 donors. *p<0.05 by unpaired Student’s t-test.
We then tested whether the age-related increase in CD38 expression following MCh treatment was correlated to changes in the Ca2+ response in ASM cells. ASM cells were loaded with a fluorescent Ca2+ dye, Fluo-4 AM, before acute stimulation with 10μM histamine. Histamine quickly increased the intracellular fluorescence (Ft) from the resting level (F0) to the peak before the fluorescence intensity slowly reverted to the resting level over 2 minutes (Fig. 3D). Compared to untreated controls, MCh treatment significantly increased the peak of fluorescence in ASM cells from young children (3.61±0.15 vs. 2.98±0.12, p<0.05) but not in ASM cells from adult donors (Fig. 3D and E).
To functionally link elevated CD38 expression and augmented the Ca2+ response in immature ASM cells, we employed two different approaches to block the CD38-RyR pathway in primary cultures of ASM cells from young children. As the first approach, we used siRNA transfection to knockdown CD38 expression. Compared to scrambled siRNA, CD38 siRNA reduced CD38 expression by 50–60% assayed by qPCR and Western blot assays (Fig. 3F) and prevented MCh-augmented Ca2+ response to acute agonist stimulation (Fig. 3G). As the second approach, we suppressed the CD38 pathway by blocking the interaction of cADPR and RyR with a competitive inhibitor, 8-Bromo-cADPR (100μM), for 30 min before Ca2+ imaging. Of note, cADPR is biosynthesized through the enzymatic activity of CD38. 8-Bromo-cADPR decreased agonist-induced Ca2+ response in saline-treated groups and completely abolished the augmentation of the peak Ca2+ response by MCh (Fig. 3H and I).
To complement these loss-of-function approaches, we overexpressed wild-type CD38 (wtCD38) tagged with a green fluorescent protein (GFP) in primary ASM cells from young children (Fig. E2A). An enzymatically inactive mutant CD38 (mutCD38) was similarly transfected as control. The transfected (GFP+) ASM cells and un-transfected (GFP−) cells in the same culture were assayed for the Ca2+ response to acute histamine stimulation. Compared to un-transfected and mutCD38-transfected controls, wtCD38 overexpression significantly increased the peak Ca2+ response (Fig. E2B and C).
Taken together, we conclude that MCh treatment enhances CD38 expression and thereby increases the peak Ca2+ response to histamine via the CD38-RyR pathway.
MCh treatment enhances airway constriction in PCLSs from young donors.
We assessed the MCh-CD38 axis in the regulation of airway constriction using frozen-thawed human PCLSs(22). The PCLS retains native cell types and ASM-parenchymal interactions and thus offers an ex vivo model to study airway contraction. PCLSs from young children and adult groups (Table. S1) were treated with 10μM MCh or saline (control) for 2.5 days following an established protocol (6), washed with saline, incubated in the culture medium overnight, and examined for airway constriction in response to histamine (Fig. 4A). To avoid the confounding effect of airway size on the contractile response, we compared airways with a similar luminal area between saline- and MCh-treated PCLSs of each age group (0.068 ± 0.03 mm2 vs. 0.063 ± 0.03 mm2 in PCLSs from young children and 0.16 ± 0.04 mm2 vs. 0.16 ± 0.03 mm2 in adult PCLSs). MCh treatment by itself had no effect on the airway luminal size (ratio of post- to pretreatment luminal area, 0.99 ± 0.01 in the young children group and 1.00 ± 0.02 in the adult group). Moreover, MCh treatment did not alter airway constriction in PCLSs from adults (Fig. 4B and C) and teenagers (Fig. E3). However, it significantly enhanced airway constriction in response to histamine in PCLSs of young children (Fig. 4B and C). We found that 8-Br-cADPR completely blocked MCh-induced airway hyperconstriction in PCLSs from young children (Fig. 4D). In addition, 8-Br-cADPR elicited a more robust airway relaxation response in MCh-treated PCLSs from young children than PCLSs from adults (Fig. E4). In the relaxation assay, the airway was pre-contracted by histamine before the addition of 8-Br-cADPR. Lastly, antibody staining for αSMA revealed no change in ASM mass of young children’s airways following MCh treatment (Fig. 4E and F). Therefore, cholinergic stimulation exerts an age-specific effect on human ASM to enhance airway contraction in early childhood. Such an effect of MCh stimulation is likely mediated through the CD38 pathway and independent of ASM hyperplasia in the human airway, which is similar to our findings in primary human ASM cells (Fig. 3) and in neonatal mice (Fig. 1) (6).
Figure 4. MCh treatment induces airway hypercontraction in PCLSs from young children.
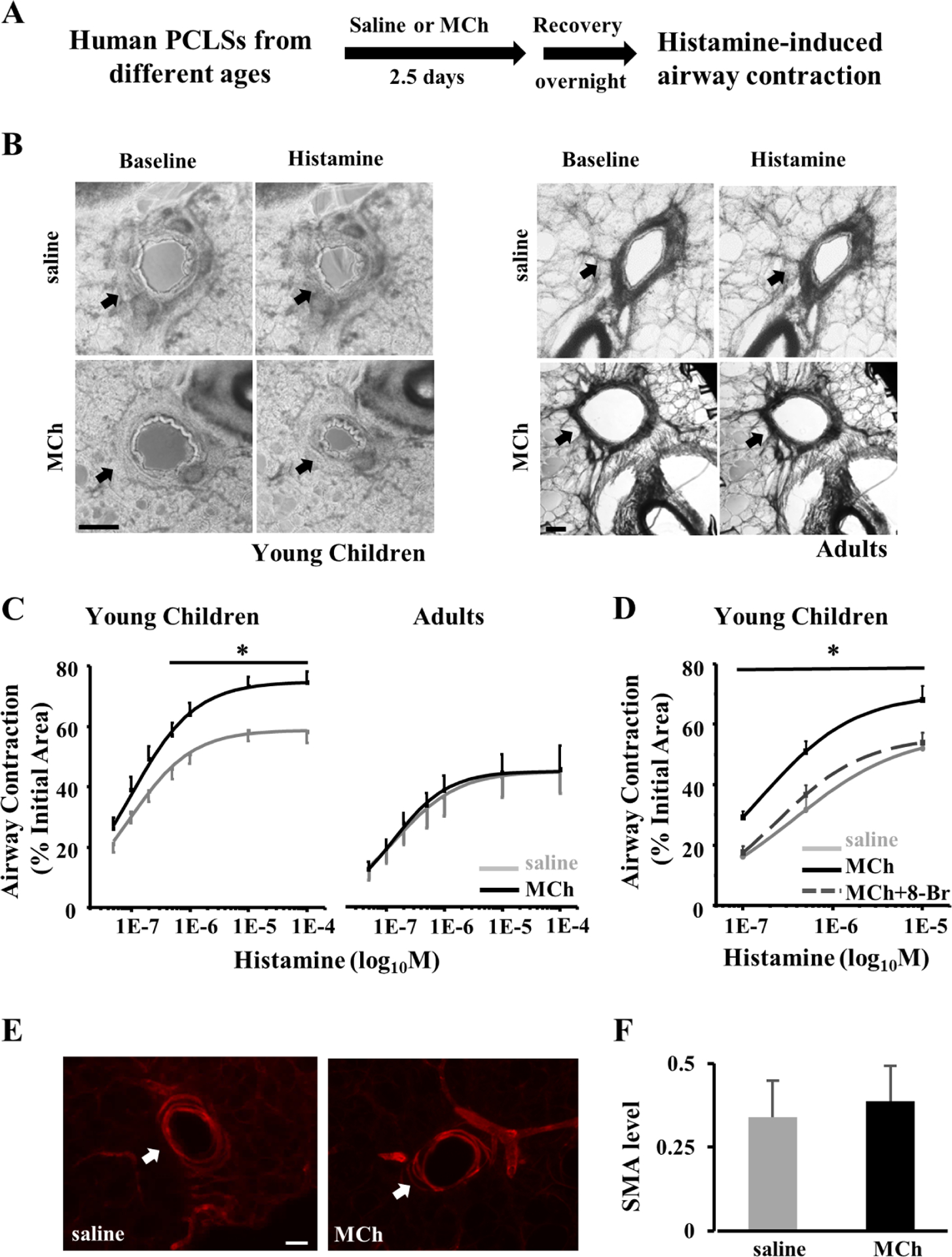
(A) Experimental scheme of MCh treatment of human PCLSs followed by recovery and contraction assays in response to histamine stimulation. PCLSs were collected from the lungs of deceased young children and adults. Saline treatment was the control. (B) Representative images of PCLSs at baseline and following 10μM histamine stimulation. Arrows mark airways in PCLSs. Scale bars, 200μm. (C) The dose-response of saline- and MCh-treated human airways to histamine stimulation in PLCSs from different age groups. Airway contraction was quantified by normalizing the reduction in the airway area to the baseline level. Each point represents the average ± SEM of 24 PCLSs from 3 young children and 11 PCLSs from 3 adults. (D) The dose-response of human airways to histamine stimulation following saline or MCh ± 8-Br-cADPR (8-Br, 100μM) treatment. 8-Br was applied 30 minutes prior to histamine stimulation. Each point represents the average ± SEM of 8–11 PCLSs from 3 young children. (E) Representative images of αSMA staining in saline- and MCh-treated PCLSs from deceased young children. Scale bars, 50 μm. The αSMA immunoreactivity around the airway was quantified in (F). The bar graph represents the average ± SEM of 8 PCLSs from 2 young children in each condition. * p<0.05 by two-way ANOVA followed by Tukey’s post-hoc test when comparing (C) MCh-treated to saline-treated airways or (D) MCh treated airways in the presence to absence of 8-Br.
MCh promotes CD38 expression by activating PI3K-Akt in immature ASM cells.
After showing that the regulation of CD38-mediated Ca2+ signaling is essential for cholinergic deregulation of ASM contractility in early postnatal life, we focused on mechanisms further upstream. CD38 expression in ASM cells is known to be subject to transcriptional and post-transcriptional regulation through multiple pathways, such as the activation of Erk1/2, NFκB and PI3K (25). In ASM cells from young children, however, we found that MCh exposure had no impact on Erk phosphorylation (Fig. E5A). In addition, we found no age-related differences in the effect of MCh on NFκB levels in ASM cells (Fig. E5B). Therefore, we tested whether age-specific CD38 regulation by cholinergic stimulation was mediated via the PI3K pathway.
It is well-established that MCh signals via the muscarinic receptor type 3 in ASM to activate the intracellular PI3K-Akt pathway (28, 29). It is also known that the PI3K-Akt pathway participates in CD38 regulation in ASM (30). We hypothesized accordingly that MCh treatment could activate PI3K-Akt, and thereby elevate CD38 expression. To test this hypothesis, we assayed the activity of the PI3K-Akt pathway in primary human ASM cells using phosphorylated Akt (pAkt) as a surrogate of activation. Compared to saline treatment, MCh treatment for 6 hours increased the relative abundance of pAkt by ~60% in immature ASM cells, while it had no effect on pAkt levels in adult ASM cells (Fig. 5A and B). We then blocked PI3K-Akt signaling with a specific PI3K inhibitor, Wortmannin. We validated the efficacy of 0.1μM Wortmannin in preventing Akt phosphorylation during a 6-hour MCh treatment (Fig. 5C and D). At this effective concentration, Wortmannin completely abolished the increase in CD38 expression in immature ASM cells following MCh treatment (Fig. 5E and F). A similar inhibition on MCh-induced CD38 upregulation was found using a second PI3K inhibitor, LY294003 (Fig. 5E and F). We conclude that MCh treatment age-dependently activates the PI3K-Akt pathway in immature ASM of young children, and this activated pathway leads to the elevation of CD38 expression.
Figure 5. MCh upregulates CD38 expression by activating the PI3K-Akt pathway in primary ASM cells from young children.
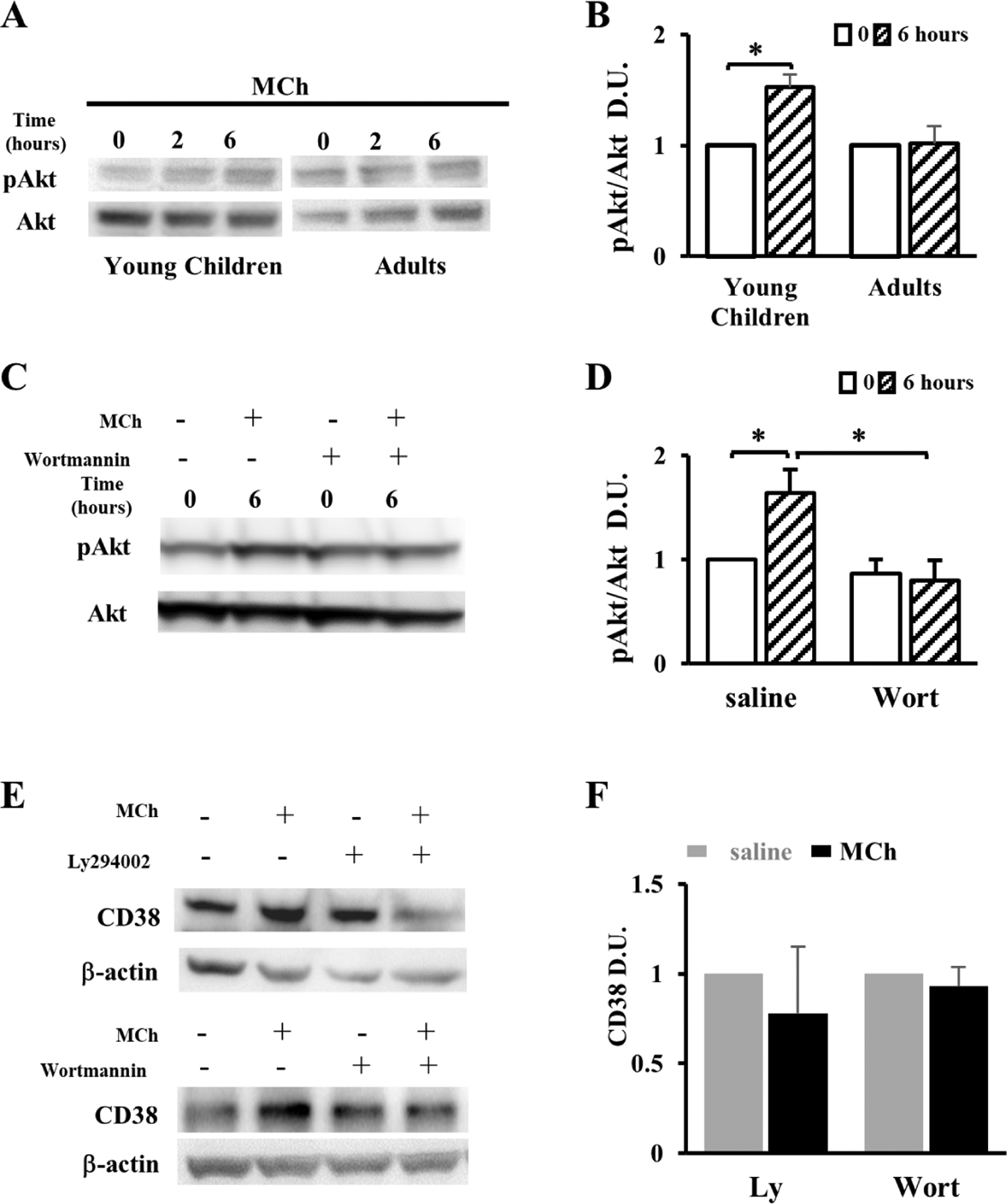
(A) Western blot assay and (B) densitometric quantification of the phosphorylated Akt (pAkt) and the total Akt in primary ASM cells from young children and adults following 10μM MCh treatment for 0, 2, and 6 hours. The extent of pAkt was expressed as the ratio to Akt. Data were normalized to 0-hour MCh treatment. (C) Western blot assay and (D) densitometric quantification of 6-hour MCh-induced Akt phosphorylation with and without the PI3K/Akt inhibitor Wortmannin (Wort, 0.1μM). Data were normalized to 0-hour MCh treatment in the absence of Wort. (E) Western blot analyses of CD38 expression in primary ASM cells from young children treated with 10μM MCh and the PI3K/Akt inhibitor, alone or in combination. β-actin is the loading control. The level of CD38 in each experimental group was quantified by densitometry in (F). Bar graphs represent the mean ± SEM of 2 independent experiments per donor and a total of 2 donors of each age group. * p<0.05 by unpaired Student’s t-test.
MCh-induced hypercontractility of ASM cells from young children requires PI3K-Akt signaling.
To test whether blockade of PI3K-Akt signaling blunted MCh-induced hypercontractility of immature ASM cells, we treated primary ASM cells in culture and PCLSs from young children with MCh and the PI3K inhibitor, separately and in combination (Fig. 6A). Wortmannin (0.1μM) and LY294002 (10μM) had no effect on the Ca2+ response and airway constriction at baseline (Fig. 6B and C). However, following MCh treatment, both PI3K inhibitors prevented the augmentation of the Ca2+ response in immature ASM cells (Fig. 6B) and abolished AHR to acute histamine stimulation (Fig. 6C). LY294002 (10μM) also diminished the increase in airway constriction in PCLSs from neonatal mice following MCh treatment (Fig. E6). These findings show that activation of the PI3K-Akt pathway by MCh treatment is required for the development of airway hypercontraction at an early age.
Figure 6. PI3K inhibitors, but not β adrenergic agonist, block ASM hypercontractility in PCLSs from young children following MCh stimulation.
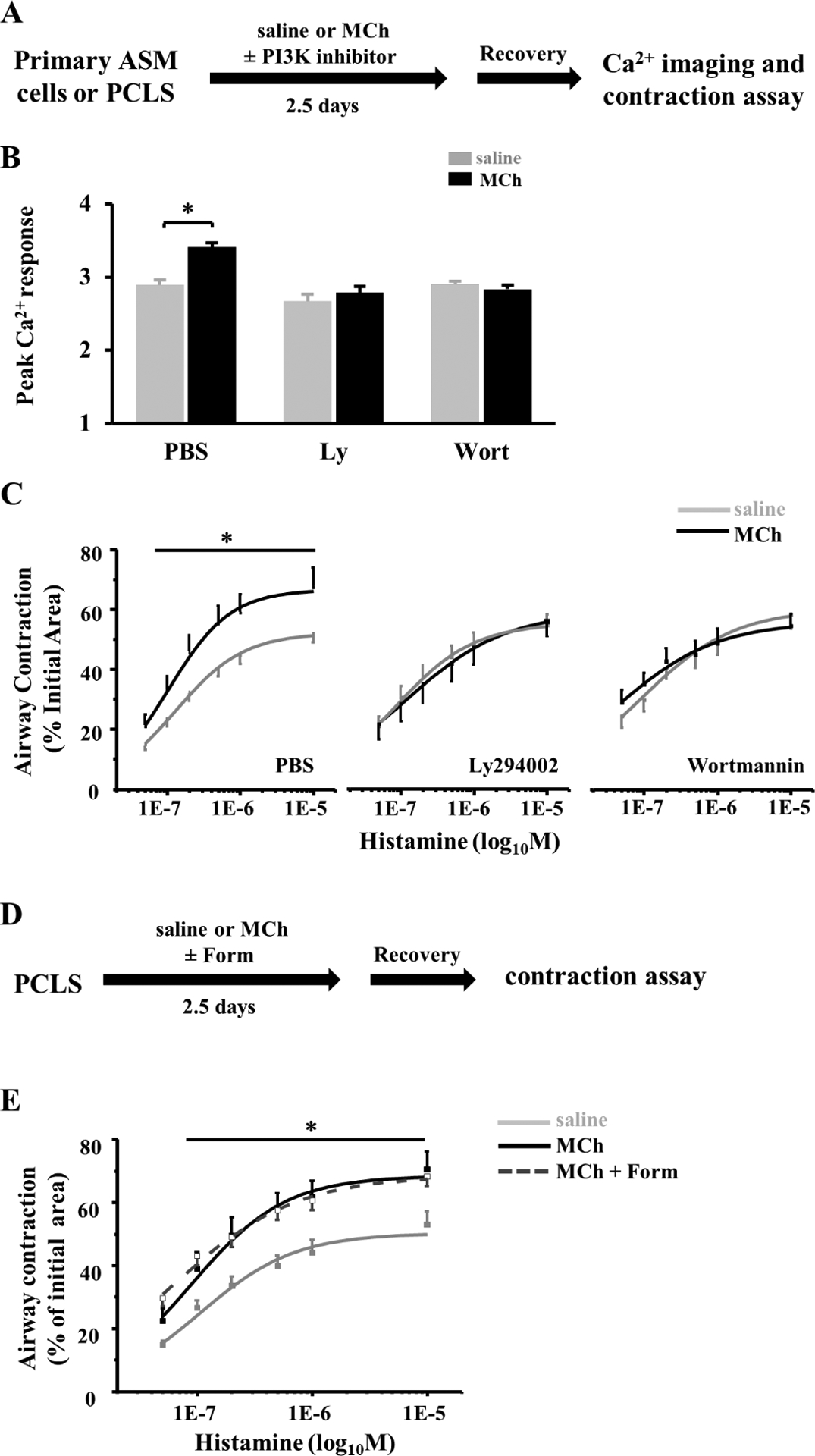
(A) Scheme of MCh treatment of ASM cells and PCLSs with and without the presence of 10μM Ly294002 (Ly) or 0.1μM Wortmannin (Wort). After recovery, ASM cells were subjected to Ca2+ imaging, and PCLSs were subjected to airway contraction assay. (B) The peak Ca2+ response to 10μM histamine in primary ASM cells from young children in each experimental group. Data represent the mean ± SEM of 70–80 cells from 2 donors in each group. *p<0.05 by unpaired Student’s t-test. (C) Airway contraction in response to histamine stimulation of PCLSs from young children with and without the treatment of MCh and the PI3K/Akt inhibitor. (D) Scheme of MCh treatment of PCLSs with and without the presence of 5nM Formoterol (Form). (E) Airway contraction in response to histamine stimulation of PCLSs from young children with and without the treatment of MCh and Formoterol. Each point represents the results of 12–13 PCLSs from 3 donors. * p<0.05 by two-way ANOVA followed by Tukey’s post-hoc test in (C) and (E).
β2 adrenergic agonist has no effect on MCh-induced AHR in PCLSs from young children.
β2 adrenergic agonists, such as formoterol, primarily reverse the ASM contraction by activating the cyclic adenosine monophosphate-protein kinase A (cAMP-PKA) pathway and thus suppressing bronchoconstrictor-induced Ca2+ responses (31). To test whether β2 agonists could prevent MCh-induced ASM hypercontractility in early life, we treated PCLSs from young children with a combination of 5nM Formoterol and MCh (Fig 6D). The dose of formoterol was chosen based on its high efficiency in airway relaxation as shown in a previous study (32). We found that formoterol treatment had no effect on airway hypercontraction induced by MCh treatment (Fig. 6E). Therefore, despite a potent airway relaxant effect, β2 adrenergic agonists have no therapeutic benefit to impede the development of AHR in PCLSs from young children following cholinergic hyperstimulation.
Discussion
This study has uncovered an age-related, cholinergic signaling mechanism that deregulates the contractile phenotype of ASM cells in young children. This mechanism, mediated by elevated expression and increased activities of CD38, is associated with upstream PI3K-Akt signaling and downstream Ca2+ signaling, leading to ASM hypercontraction and AHR. Our study has employed MCh treatment for an extended time period (2.5 days) to induce ASM hypercontraction. Although MCh elicits relatively short-term airway contraction, we have provided evidence that MCh exerts a prolonged effect on the PI3K-Akt pathway and gene expression regulation in ASM cells. Our findings, in conjunction with similar observations in neonatal mice that MCh treatment is sufficient to induce AHR (6), highlight the susceptibility of immature ASM cells to the development of a hypercontractile phenotype. Given the evidence of elevated levels of nerve growth factors in young children at high risk for asthma (11) and aberrant increases in cholinergic ASM innervation in adult patients with persistent allergic asthma (9), cholinergic hyperstimulation of immature ASM cells may serve as a pathogenic mechanism underlying AHR. We also show that the β2 agonist (formoterol), the first-line bronchodilator prescribed for children with wheezing and allergic asthma, has no effect on cholinergic deregulation of CD38 and induction of ASM hypercontractility in young children. In light of these new findings, targeting acetylcholine-CD38 axis and upstream PI3K-Akt signaling may be considered as a tailored treatment for patients at a young age. Such treatment may also blunt the progression of AHR from childhood into adulthood.
Sensory afferents and cholinergic efferents are connected through the respiratory center neurons located in the brainstem to form a neurocircuitry that innerves ASM (33, 34). Sensory afferents can respond to a variety of environmental changes and inflammatory signals and subsequently trigger the firing of cholinergic efferents and the release of acetylcholine from nerve endings. The relay from environmental sensing to cholinergic stimulation of ASM may explain why a variety of environmental insults can similarly induce wheezing and AHR in young children.
The contraction of ASM cells is dependent on the elevation of the intracellular Ca2+ concentration through the activation of the PLC-IP3R pathway and the CD38-RyR pathway. Previous studies have shown that the CD38-RyR pathway plays a marginal role in mature ASM cells following agonist stimulation (32, 35). We report here that this pathway contributes significantly to the regulation of the contractility of immature ASM cells. Intriguingly, despite a higher expression level of CD38 in ASM in young children than in adults, we observed no difference in baseline Ca2+ response to histamine in human ASM cells between age groups. This finding may reflect a compensatory effect on Ca2+ mobilization through the activated PLC-IP3R pathway in response to a high dose of histamine (10μM) tested in our assays. Similar functional compensation by the PLC-IP3R pathway was also observed in airway contraction assay comparing wild-type and CD38−/− mouse PCLSs when the agonist was applied at higher concentrations along the dose curve. However, the role of the CD38 pathway in ASM contractility becomes more prominent when the age-related difference in CD38 expression is exaggerated by MCh treatment of immature ASM cells.
The key determinant of age-related, cholinergic deregulation of ASM contractility is the activation of the intracellular PI3K-Akt pathway only in immature ASM cells, which in turn promotes CD38 expression. Previous studies have reported that the muscarinic 3 receptor itself is insufficient to alter the PI3K pathway in adult ASM cells. Instead, co-activation of tyrosine kinase receptor signaling is required to augment PI3K activities (28, 36–38). Whether and how the muscarinic 3 receptor interacts with a tyrosine kinase receptor to activate PI3K in immature ASM cells require future investigation. To block the PI3K-Akt pathway, our study has employed two different inhibitors, Ly294002 and Wortmannin that generate similar results. This approach has significantly reduced the concern of non-specific effects of Ly294002 on the Ca2+ response (39, 40). Of note, Wortmannin is not known to inhibit Ca2+ signaling.
Inhibitors of the PI3K-Akt pathway have been tested for therapeutic application in allergic asthma. For example, TG100–115(41), an inhaled PI3Kδ/γ inhibitor, was previously shown to reduce inflammation and AHR in a murine model of allergic asthma. A clinical trial of repeated inhalation of Nemiralisib (42), another highly selective PI3Kδ inhibitor in adult patients with persistent, poorly controlled asthma, has demonstrated safety and tolerability, although clinical improvement of the patients following the 28-day trial is not satisfactory. Based on our findings that the PI3K/Akt pathway may operate preferentially in young children to promote AHR, specific PI3K inhibitors may be more effective as an adjuvant treatment for children with allergic asthma than adult asthmatic patients. Along a similar line, specific blockers against the muscarinic 3 receptor and CD38 may also uniquely benefit children at high risk for and with allergic asthma.
Our study has several limitations. First, the sample size in each age group is relatively small. This caveat is offset by parallel studies in mice that identify a similar, age-related effect of MCh treatment on CD38 expression and ASM contractility. Second, there is a wide age gap between the young children and the adult groups. In addition, the available lung samples reflect only a few ages younger than 20 years of age. As such, the time window of postnatal maturation of human ASM cells needs to be further defined. Despite these limitations, clinical studies have identified the association of persistent asthma in adults with AHR apparent at an early age (3, 43, 44), which precisely is the focus of this study. Third, although our previous studies in neonatal mice have demonstrated persistent ASM hypercontractility following MCh treatment, whether cholinergic stimulation has a long-term impact on the contractile phenotype of ASM in young children warrants future investigation. Fourth, our experimental models exclude inflammatory signals that may interact with cholinergic signaling in ASM cells to regulate the contractile phenotype. Fifth, using a single concentration and continuous exposure of MCh to simulate cholinergic hyperstimulation may be oversimplified. Furthermore, precisely how long MCh needs to be active to induce AHR in vivo is difficult to assess. Our rationale for choosing 2.5 days are: (i) it has been previously established by similar assays using neonatal mouse precision-cut lung slices (6), (ii) while the acute contractile effect of MCh resolves within hours (45), based on the time course of Akt activation (6 hours), the effect of MCh on the PI3K-Akt pathway likely outlasts its acute contractile effect, and (iii) induction of a sustained, hypercontractile phenotype of ASM likely involves changes in gene expression (such as CD38) that may require a relatively long exposure period, in the order of days. Notably, the current MCh treatment protocol has reproduced key features of ASM hypercontractility in neonatal mouse models of allergic asthma, and thus, serves as a relevant approach for studying the human ASM contraction in early life. Finally, our human PCLS study of combinatorial formoterol treatment was focused on one dose and one treatment regime. While follow-up studies are planned, we wish to point out that formoterol treatment in the mouse model of early life onset asthma similarly failed to reverse AHR and improve lung function in adult mice that were exposed to allergens as neonates (6).
In summary, our study has demonstrated that as ASM continues to mature during the postnatal development of the lung, mechanisms that regulate the contractile phenotype of ASM and, by extension, AHR, differ with age. Our findings highlight the deregulated acetylcholine-CD38 axis in the development of AHR in early childhood. Targeting this axis may modify the progression of airway hyperreactivity from childhood into adulthood.
Supplementary Material
Key Messages:
In early postnatal life, human ASM hypercontraction can be elicited by CD38-mediated cholinergic stimulation, independent of inflammation.
Specifically, cholinergic signaling activates the PI3K/Akt pathway to increase CD38 expression and CD38-mediated Ca2+ responses in ASM cells.
PI3K inhibitors, but not β2 adrenergic agonists, impede the development of ASM hypercontractility following cholinergic stimulation in early postnatal life.
Acknowledgment
We would like to thank Dr. Kruti Patel for assistance with the mouse model of MCh treatment, Drs. Lee & Zhao for providing CD38 plasmids, Ai lab members for assisting the preparation of human PCLSs.
This work is supported by NIH grants, K08135443 (Y.B), 1R01HL132991 (X.A), and R21HL151695 (R.K.).
Abbreviations:
- AHR
Airway hyperresponsiveness
- Akt
A serine/threonine kinase, also known as Protein kinase B
- ASM
Airway smooth muscle
- cADPR
Cyclic ADP-ribose
- cAMP-PKA
Cyclic adenosine monophosphate-protein kinase A
- Cav1
Caveolin-1 gene
- Erk
Extracellular signal-regulated kinase
- MCh
Methacholine
- Mylk
Myosin light chain kinase gene
- NFκB
Nuclear factor kappa B
- OVA
Ovalbumin
- Ormdl3
ORMDL sphingolipid biosynthesis regulator 3 gene
- PCLS
Precision-cut lung slice
- PLC-IP3R
Phospholipase C-inositol trisphosphate receptor
- PI3K
Phosphoinositide 3-kinase
- PTEN
Phosphatase and Tensin Homolog
- qPCR
Quantitative polymerase chain reaction
- RhoA
Ras homolog family member A gene
- Rock2
Rho associated coiled-coil containing protein kinase 2 gene
- RyR
Ryanodine receptor
- siRNA
Small interfering RNA
- αSMA
Smooth muscle alpha-actin
- SMMHC
Smooth muscle myosin heavy chain
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
All authors have no conflict of interest to disclose.
References:
- 1.Sears MR, Greene JM, Willan AR, Wiecek EM, Taylor DR, Flannery EM, et al. A longitudinal, population-based, cohort study of childhood asthma followed to adulthood. The New England journal of medicine. 2003;349(15):1414–22. [DOI] [PubMed] [Google Scholar]
- 2.Tai A, Tran H, Roberts M, Clarke N, Gibson AM, Vidmar S, et al. Outcomes of childhood asthma to the age of 50 years. The Journal of allergy and clinical immunology. 2014;133(6):1572–8.e3. [DOI] [PubMed] [Google Scholar]
- 3.Stern DA, Morgan WJ, Halonen M, Wright AL, Martinez FD. Wheezing and bronchial hyper-responsiveness in early childhood as predictors of newly diagnosed asthma in early adulthood: a longitudinal birth-cohort study. Lancet. 2008;372(9643):1058–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGeachie MJ, Yates KP, Zhou X, Guo F, Sternberg AL, Van Natta ML, et al. Patterns of Growth and Decline in Lung Function in Persistent Childhood Asthma. The New England journal of medicine. 2016;374(19):1842–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aven L, Paez-Cortez J, Achey R, Krishnan R, Ram-Mohan S, Cruikshank WW, et al. An NT4/TrkB-dependent increase in innervation links early-life allergen exposure to persistent airway hyperreactivity. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2014;28(2):897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patel KR, Bai Y, Trieu KG, Barrios J, Ai X. Targeting acetylcholine receptor M3 prevents the progression of airway hyperreactivity in a mouse model of childhood asthma. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2017;31(10):4335–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.An SS, Bai TR, Bates JH, Black JL, Brown RH, Brusasco V, et al. Airway smooth muscle dynamics: a common pathway of airway obstruction in asthma. The European respiratory journal. 2007;29(5):834–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan F, Gao H, Zhao H, Bhatia M, Zeng Y. Roles of airway smooth muscle dysfunction in chronic obstructive pulmonary disease. Journal of translational medicine. 2018;16(1):262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dragunas G, Woest ME, Nijboer S, Bos ST, van Asselt J, de Groot AP, et al. Cholinergic neuroplasticity in asthma driven by TrkB signaling. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2020;34(6):7703–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drake MG, Scott GD, Blum ED, Lebold KM, Nie Z, Lee JJ, et al. Eosinophils increase airway sensory nerve density in mice and in human asthma. Science translational medicine. 2018;10(457). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Müller GC, Pitrez PM, Teixeira AL, Pires PS, Jones MH, Stein RT, et al. Plasma brain-derived neurotrophic factor levels are associated with clinical severity in school age children with asthma. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology. 2010;40(12):1755–9. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe T, Fajt ML, Trudeau JB, Voraphani N, Hu H, Zhou X, et al. Brain-Derived Neurotrophic Factor Expression in Asthma. Association with Severity and Type 2 Inflammatory Processes. American journal of respiratory cell and molecular biology. 2015;53(6):844–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Radzikinas K, Aven L, Jiang Z, Tran T, Paez-Cortez J, Boppidi K, et al. A Shh/miR-206/BDNF cascade coordinates innervation and formation of airway smooth muscle. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31(43):15407–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Panitch HB, Deoras KS, Wolfson MR, Shaffer TH. Maturational changes in airway smooth muscle structure-function relationships. Pediatric research. 1992;31(2):151–6. [DOI] [PubMed] [Google Scholar]
- 15.Panitch HB, Allen JL, Ryan JP, Wolfson MR, Shaffer TH. A comparison of preterm and adult airway smooth muscle mechanics. Journal of applied physiology (Bethesda, Md : 1985). 1989;66(4):1760–5. [DOI] [PubMed] [Google Scholar]
- 16.Cullen AB, Cooke PH, Driska SP, Wolfson MR, Shaffer TH. Correlation of tracheal smooth muscle function with structure and protein expression during early development. Pediatric pulmonology. 2007;42(5):421–32. [DOI] [PubMed] [Google Scholar]
- 17.Lewin G, Hurtt ME. Pre- and Postnatal Lung Development: An Updated Species Comparison. Birth defects research. 2017;109(19):1519–39. [DOI] [PubMed] [Google Scholar]
- 18.Sanderson MJ. Exploring lung physiology in health and disease with lung slices. Pulmonary pharmacology & therapeutics. 2011;24(5):452–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prakash YS. Airway smooth muscle in airway reactivity and remodeling: what have we learned? American journal of physiology Lung cellular and molecular physiology. 2013;305(12):L912–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hartman WR, Smelter DF, Sathish V, Karass M, Kim S, Aravamudan B, et al. Oxygen dose responsiveness of human fetal airway smooth muscle cells. American journal of physiology Lung cellular and molecular physiology. 2012;303(8):L711–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paez-Cortez J, Krishnan R, Arno A, Aven L, Ram-Mohan S, Patel KR, et al. A new approach for the study of lung smooth muscle phenotypes and its application in a murine model of allergic airway inflammation. PloS one. 2013;8(9):e74469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bai Y, Krishnamoorthy N, Patel KR, Rosas I, Sanderson MJ, Ai X. Cryopreserved Human Precision-Cut Lung Slices as a Bioassay for Live Tissue Banking. A Viability Study of Bronchodilation with Bitter-Taste Receptor Agonists. American journal of respiratory cell and molecular biology. 2016;54(5):656–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guedes AG, Jude JA, Paulin J, Rivero-Nava L, Kita H, Lund FE, et al. Airway responsiveness in CD38-deficient mice in allergic airway disease: studies with bone marrow chimeras. American journal of physiology Lung cellular and molecular physiology. 2015;308(5):L485–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deshpande DA, White TA, Dogan S, Walseth TF, Panettieri RA, Kannan MS. CD38/cyclic ADP-ribose signaling: role in the regulation of calcium homeostasis in airway smooth muscle. American journal of physiology Lung cellular and molecular physiology. 2005;288(5):L773–88. [DOI] [PubMed] [Google Scholar]
- 25.Deshpande DA, Guedes AGP, Graeff R, Dogan S, Subramanian S, Walseth TF, et al. CD38/cADPR Signaling Pathway in Airway Disease: Regulatory Mechanisms. Mediators of inflammation. 2018;2018:8942042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dwyer DF, Ordovas-Montanes J, Allon SJ, Buchheit KM, Vukovic M, Derakhshan T, et al. Human airway mast cells proliferate and acquire distinct inflammation-driven phenotypes during type 2 inflammation. Science immunology. 2021;6(56). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deshpande DA, Guedes AGP, Lund FE, Subramanian S, Walseth TF, Kannan MS. CD38 in the pathogenesis of allergic airway disease: Potential therapeutic targets. Pharmacology & therapeutics. 2017;172:116–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gosens R, Zaagsma J, Meurs H, Halayko AJ. Muscarinic receptor signaling in the pathophysiology of asthma and COPD. Respiratory research. 2006;7(1):73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arrighi N, Bodei S, Zani D, Michel MC, Simeone C, Cosciani Cunico S, et al. Different muscarinic receptor subtypes modulate proliferation of primary human detrusor smooth muscle cells via Akt/PI3K and map kinases. Pharmacological research. 2013;74:1–6. [DOI] [PubMed] [Google Scholar]
- 30.Jude JA, Tirumurugaan KG, Kang BN, Panettieri RA, Walseth TF, Kannan MS. Regulation of CD38 expression in human airway smooth muscle cells: role of class I phosphatidylinositol 3 kinases. American journal of respiratory cell and molecular biology. 2012;47(4):427–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cazzola M, Page CP, Rogliani P, Matera MG. β2-agonist therapy in lung disease. American journal of respiratory and critical care medicine. 2013;187(7):690–6. [DOI] [PubMed] [Google Scholar]
- 32.Ressmeyer AR, Bai Y, Delmotte P, Uy KF, Thistlethwaite P, Fraire A, et al. Human airway contraction and formoterol-induced relaxation is determined by Ca2+ oscillations and Ca2+ sensitivity. American journal of respiratory cell and molecular biology. 2010;43(2):179–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Canning BJ, Fischer A. Neural regulation of airway smooth muscle tone. Respiration physiology. 2001;125(1–2):113–27. [DOI] [PubMed] [Google Scholar]
- 34.Kistemaker LEM, Prakash YS. Airway Innervation and Plasticity in Asthma. Physiology (Bethesda, Md). 2019;34(4):283–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bai Y, Edelmann M, Sanderson MJ. The contribution of inositol 1,4,5-trisphosphate and ryanodine receptors to agonist-induced Ca(2+) signaling of airway smooth muscle cells. American journal of physiology Lung cellular and molecular physiology. 2009;297(2):L347–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krymskaya VP, Orsini MJ, Eszterhas AJ, Brodbeck KC, Benovic JL, Panettieri RA Jr., et al. Mechanisms of proliferation synergy by receptor tyrosine kinase and G protein-coupled receptor activation in human airway smooth muscle. American journal of respiratory cell and molecular biology. 2000;23(4):546–54. [DOI] [PubMed] [Google Scholar]
- 37.Billington CK, Kong KC, Bhattacharyya R, Wedegaertner PB, Panettieri RA Jr., Chan TO, et al. Cooperative regulation of p70S6 kinase by receptor tyrosine kinases and G protein-coupled receptors augments airway smooth muscle growth. Biochemistry. 2005;44(44):14595–605. [DOI] [PubMed] [Google Scholar]
- 38.Kong KC, Billington CK, Gandhi U, Panettieri RA Jr., Penn RB. Cooperative mitogenic signaling by G protein-coupled receptors and growth factors is dependent on G(q/11). FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2006;20(9):1558–60. [DOI] [PubMed] [Google Scholar]
- 39.Ethier MF, Madison JM. LY294002, but not wortmannin, increases intracellular calcium and inhibits calcium transients in bovine and human airway smooth muscle cells. Cell calcium. 2002;32(1):31–8. [DOI] [PubMed] [Google Scholar]
- 40.Tolloczko B, Turkewitsch P, Al-Chalabi M, Martin JG. LY-294002 [2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one] affects calcium signaling in airway smooth muscle cells independently of phosphoinositide 3-kinase inhibition. The Journal of pharmacology and experimental therapeutics. 2004;311(2):787–93. [DOI] [PubMed] [Google Scholar]
- 41.Doukas J, Eide L, Stebbins K, Racanelli-Layton A, Dellamary L, Martin M, et al. Aerosolized phosphoinositide 3-kinase gamma/delta inhibitor TG100–115 [3-[2,4-diamino-6-(3-hydroxyphenyl)pteridin-7-yl]phenol] as a therapeutic candidate for asthma and chronic obstructive pulmonary disease. The Journal of pharmacology and experimental therapeutics. 2009;328(3):758–65. [DOI] [PubMed] [Google Scholar]
- 42.Khindri S, Cahn A, Begg M, Montembault M, Leemereise C, Cui Y, et al. A Multicentre, Randomized, Double-Blind, Placebo-Controlled, Crossover Study To Investigate the Efficacy, Safety, Tolerability, and Pharmacokinetics of Repeat Doses of Inhaled Nemiralisib in Adults with Persistent, Uncontrolled Asthma. The Journal of pharmacology and experimental therapeutics. 2018;367(3):405–13. [DOI] [PubMed] [Google Scholar]
- 43.Wang AL, Datta S, Weiss ST, Tantisira KG. Remission of persistent childhood asthma: Early predictors of adult outcomes. The Journal of allergy and clinical immunology. 2019;143(5):1752–9.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fuchs O, Bahmer T, Rabe KF, von Mutius E. Asthma transition from childhood into adulthood. The Lancet Respiratory medicine. 2017;5(3):224–34. [DOI] [PubMed] [Google Scholar]
- 45.Billington CK, Penn RB. m3 muscarinic acetylcholine receptor regulation in the airway. American journal of respiratory cell and molecular biology. 2002;26(3):269–72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.