

Abstract
PURPOSE:
To evaluate retinal function in a family presenting with Jalili syndrome due to a previously unreported variant in CNNM4.
METHODS:
A family of three sisters with a novel CNNM4 variant, c.482T>C p.(Leu161Pro), and ten visually normal, age-similar controls participated in this study. The subjects underwent detailed dental examinations and comprehensive ophthalmological examinations that included color vision testing, retinal imaging, and electroretinography. Full-field light- and dark-adapted luminance thresholds were obtained, in addition to light- and dark-adapted measures of the pupillary light reflex (PLR; pupil constriction elicited by a flash of light) across a range of stimulus luminance.
RESULTS:
Clinical findings of cone dysfunction and amelogenesis imperfecta were observed, consistent with Jalili syndrome. Light-adapted ERGs were non-detectable in CNNM4 subjects, whereas dark-adapted ERGs were generally normal. Full-field luminance thresholds were normal under dark-adapted conditions and were elevated, but measurable, under light-adapted conditions. The CNNM4 subjects had large PLRs under dark-adapted conditions and responses near the lower limit of normal, or slightly subnormal, under light-adapted conditions.
CONCLUSION:
CNNM4 variants can result in Jalili syndrome with cone dystrophy and generally preserved rod function. The PLR may be a useful measure for evaluating cone function in these individuals, as robust cone-mediated PLRs were recordable despite non-detectable light-adapted ERGs.
Keywords: Jalili syndrome, CNNM4, pupillary light reflex, electrophysiology, photopic full-field stimulus threshold
INTRODUCTION
Jalili syndrome is a rare autosomal recessive disorder due to mutations in the CNNM4 gene (Parry et al. 2009; Polok et al. 2009). Affected patients typically exhibit visual dysfunction apparent early in life due to cone-rod degeneration, as well as amelogenesis imperfecta (AI), characterized by small discolored teeth which break easily due to defects in enamel formation (Luder et al. 2013). Since it was first recognized in an extended Arab family in the Gaza Strip (Jalili and Smith 1988), Jalili syndrome has been reported throughout the world, especially the eastern Mediterranean, Kosovo, and Africa, with fewer reports in the Americas (Daneshmandpour et al. 2019).
CNNM4 encodes a transmembrane protein critical for divalent metal ion transport and is expressed in the retina and developing teeth (Parry et al. 2009; Luder et al. 2013). Over 24 different mutations, including nonsense, splice-site, and missense variants in CNNM4 have been implicated in Jalili syndrome (Daneshmandpour et al. 2019). All patients exhibit AI and photoreceptor degeneration, usually with nystagmus and a history of decreased vision and photoaversion, but there is considerable interfamilial and intrafamilial phenotypic variability (Gerth-Kahlert et al. 2015; Topçu et al. 2016). High refractive errors (both hyperopia and myopia) as well as emmetropia have been reported, as have posterior segment findings that range from severe chorioretinal and optic nerve atrophy to normal fundi (Daneshmandpour et al. 2019). Additionally, there are reports of Jalili syndrome associated with other systemic findings, including intellectual disability in a Saudi Arabian family with a novel mutation in CNNM4 (Torres et al. 2015), myopathy with muscular overgrowth in a family with a previously unreported missense mutation in CNNM4 (Wawrocka et al. 2017), and situs inversus with keratoconus (Purwar et al. 2015).
Here we describe a novel mutation in CNNM4 in a Palestinian family with Jalili syndrome. Given the previous reports of phenotypic variability in this syndrome, we undertook a detailed investigation of visual dysfunction in this family using multimodal imaging, full-field and multi-focal electroretinography, and psychophysical luminance threshold measurements. Additionally, we used pupillometry, as our group (Collison et al. 2015, 2016, 2019) and others (Kawasaki et al. 2012; Ner et al. 2019; Krishnan et al. 2020) have employed in inherited retinal diseases, to reveal preserved cone-mediated function (Park et al. 2020) as well as the function of intrinsically photosensitive retinal ganglion cells (Kardon et al. 2009; Park et al. 2011; Kelbsch et al. 2019) in patients who have inherited retinal degenerations. To our knowledge this is the first report of Jalili syndrome due to a homozygous L161P variant in CNNM4, as well as the first report of pupillometry in Jalili syndrome, highlighting the genotypic and phenotypic spectrum of this disorder.
METHODS
Subjects
The research was approved by an institutional review board at the University of Illinois at Chicago. The tenets of the Declaration of Helsinki were followed and all subjects’ guardians provided written informed consent prior to participating. Three female subjects who are sisters (ages 5, 14 and 15 years) with molecularly confirmed variants in CNNM4 participated. The clinical characteristics of these patients are provided in Table 1. Ten visually-normal control subjects (5 male and 5 female; ages 24 to 41 years) with no history of eye disease also participated in the study. The control subjects had Early Treatment of Diabetic Retinopathy Study (ETDRS) best-corrected visual acuity (BCVA) of 0 log MAR or better and normal contrast sensitivity assessed with the Pelli-Robson chart.
Table 1.
| Case 1 (IV.2) | Case 2 (IV.3) | Case 3 (IV.6) | |
|---|---|---|---|
| Age | 15 | 14 | 5 |
| Onset | Early childhood | Early childhood | Early childhood |
| Presentation | Nystagmus | Nystagmus Exotropia |
Nystagmus Esotropia |
| BCVA | OD: 20/100 OS: 20/200 |
OD: 20/200 OS: 20/200 |
OD: 20/200 OS: 20/400 |
| Refraction | OD: −3.50 +2.75 ×095 OS: −4.50 +2.25 ×075 |
OD: plano +2.00 ×100 OS: −0.50 +2.75 ×070 |
OD: +1.50 +1.00 ×105 OS: +1.25 +1.00 ×080 |
| Color vision | Dyschromatopsia | Dyschromatopsia | n/a |
| Optic disc | Normal | Normal | Small disc |
| Macula | Foveal hypopigmentation | Foveal hyperpigmentation | Normal |
| Peripheral retina | Normal | Normal | Normal |
| ERG | Cone extinguished Rod normal |
Cone extinguished Rod normal |
n/a |
| Pupillometry | Cone: partially affected Rod: normal Melanopsin: normal |
Cone: partially affected Rod: normal Melanopsin: normal |
n/a |
Genetic testing
A sample from the proband underwent whole exome sequencing (GeneDx, Gaithersburg, MD). A sample from the subject’s mother and father were also submitted for variant segregation analysis. The proband was homozygous for a variant in CNNM4, c.482T>C p.(Leu161Pro) and each parent was heterozygous for the same variant. Additional family members underwent targeted sequencing of the CNNM4 gene (GeneDx). Variants in CNNM4 were evaluated for pathogenicity using established American College of Medical Genetics (ACMG) criteria (Richards et al. 2015).
Imaging
All patients underwent ophthalmic clinical evaluation, including BCVA using the Snellen chart and guidelines, refraction, color vision testing by monocular Farnsworth D15, slit-lamp biomiocroscopy, dilated eye exam, fundus color and autofluorescence imaging (Optos, Dunfermlime, Scotland), spectral-domain optical coherence tomography (Heidelberg Engineering, Heidelberg, Germany), and clinical evaluation with a medical geneticist. Oral health records were reviewed, including panoramic and dental radiographs.
ERG measurement
ERGs were recorded using conventional techniques that conformed to ISCEV standards (McCulloch et al. 2015) and are described in detail elsewhere (McAnany et al. 2020; Park et al. 2020). In brief, responses were recorded using DTL corneal electrodes that were referenced to the ear and grounded at the forehead. Subjects adapted to a uniform achromatic field (30 cd/m2) and light-adapted responses were elicited by LED-generated achromatic 3.0 cd-s-m−2 flashes and flicker (31.25 Hz). Subjects were then dark-adapted for 20 minutes and responses were elicited by LED-generated achromatic flashes of 0.01 and 3.0 cd-s-m-2. All stimuli were generated by and presented in a ColorDome ganzfeld and responses were acquired with an Espion E3 electrophysiology system (Diagnosys, LLC, Lowell, MA). A minimum of 3 responses for each stimulus were obtained and averaged for analysis.
Pupil measurement
Full-field light- and dark-adapted pupillometry was performed using methods that have been described in detail elsewhere (Collison et al. 2019; Park et al. 2020). In brief, protocols were used to target rod-, cone-, and melanopsin-pathway function. Rod- and melanopsin-pathway measurements were performed following 10 min of dark-adaptation using 1 sec, short wavelength (465 nm) light. Cone-pathway measurements were performed following two minutes of light adaptation to a 6 cd/m2 short-wavelength, blue (465 nm), rod suppressing field. Stimuli for the cone-pathway measurements consisted of 1 sec, long wavelength (642 nm) flashes that were presented against the rod suppressing field (the 6 cd/m2 short wavelength field was presented continuously: before, during, and after flash presentation). Under dark-adapted (rod pathway) conditions, stimulus luminance ranged from 0.0001 cd/m2 to 450 cd/m2, whereas under light-adapted (cone-pathway) conditions, stimulus luminance ranged from 0.1 cd/m2 to 450 cd/m2. For visually-normal subjects tested under conditions identical to those used in the present report, PLR sensitivity is similar for short and long wavelength stimuli under photopic conditions, whereas PLR sensitivity is approximately 2.5 log units greater for the same stimuli under scotopic conditions (Collison et al. 2019). These previous findings are expected based on the rod spectral sensitivity function for photopically matched stimuli and suggest that our photopic conditions target cone-pathway function, whereas our scotopic conditions target rod pathway function.
All stimuli were generated by and presented in a ColorDome ganzfeld (Diagnosys, LLC, Lowell, MA). Pupil responses were recorded using an infra-red videography system (Arrington Research, Inc. Scottsdale, AZ). A minimum of two responses for each stimulus luminance were obtained and averaged for analysis.
For analysis, pupil diameter was normalized by the mean baseline pupil size during the 1 sec preceding the flash, which helps to minimize the effects of inter-subject differences in baseline pupil size. The transient PLR amplitude was defined as the difference between the normalized baseline diameter and the maximum pupil constriction following stimulus onset. The transient PLR response was plotted as a function of log stimulus luminance and the data were fit with a Naka-Rushton function (McCulloch et al. 2019) to derive Pmax (the maximum saturated PLR response) and s (the PLR semi-saturation constant):
| (1) |
where L is the stimulus luminance and n is the slope of the function. The value of n was set to 0.43 (mean of the control subjects) and Pmax and s were determined by minimizing the mean squared error between the data and the fit (1/s is a measure of pupil response sensitivity).
A measure of the sustained pupil response (post-illumination pupil response; PIPR) was also obtained. The PIPR is a sustained pupillary constriction that is optimally elicited by a high luminance, short-wavelength stimulus (Gamlin et al. 2007; Park et al. 2011; Kelbsch et al. 2019). This response component is thought to be mediated by activation of ipRGC-containing melanopsin cells (Dacey et al. 2005; Gamlin et al. 2007). A 1- sec, 450 cd/m2 short wavelength flash was used to elicit the PIPR and the response was defined as the median pupil size from 5 to 7 seconds following the flash (6 – 8 sec after the 1 sec flash onset).
Full-field threshold measurement
Full-field luminance thresholds were measured using flashes of long-wavelength (642 nm) and short-wavelength (465 nm) light. Light-adapted measures were performed against a 30 cd/m2 achromatic field, whereas dark-adapted measures were performed in the dark, following 30 minutes of dark adaptation. The full-field stimulus test (FST) protocol provided by Diagnosys, LLC, and described in detail elsewhere (Roman et al. 2005) was used to measure thresholds. A subset of the controls (N = 6) participated in the dark- and light-adapted FST measurements. In the figure below, 0 dB is equal to 0.01 cd·s·m−2.
RESULTS
Genetic testing/pedigree
An extensive family history and pedigree was recorded as shown in Figure 1. The proband (Figure 1A, IV.3) is a 14 year old girl of Palestinian parents who presented with poor central vision, exotropia, nystagmus, and photoaversion. Examination was notable for Snellen BCVA at distance of 20/200 in the right eye and 20/200 in the left eye. The anterior segment was unremarkable, but the fundus exam was notable for a bull’s eye atrophic lesion of the macula with perifoveal pigmentation and a normal peripheral retina (findings summarized in Table 1). SD-OCT revealed diffuse loss of the outer retinal layers, and fundus autofluorescence revealed a bull’s eye pattern of hypo and hyper-autofluorescence. Farnsworth D15 color vision testing revealed numerous axis errors.
Figure 1:
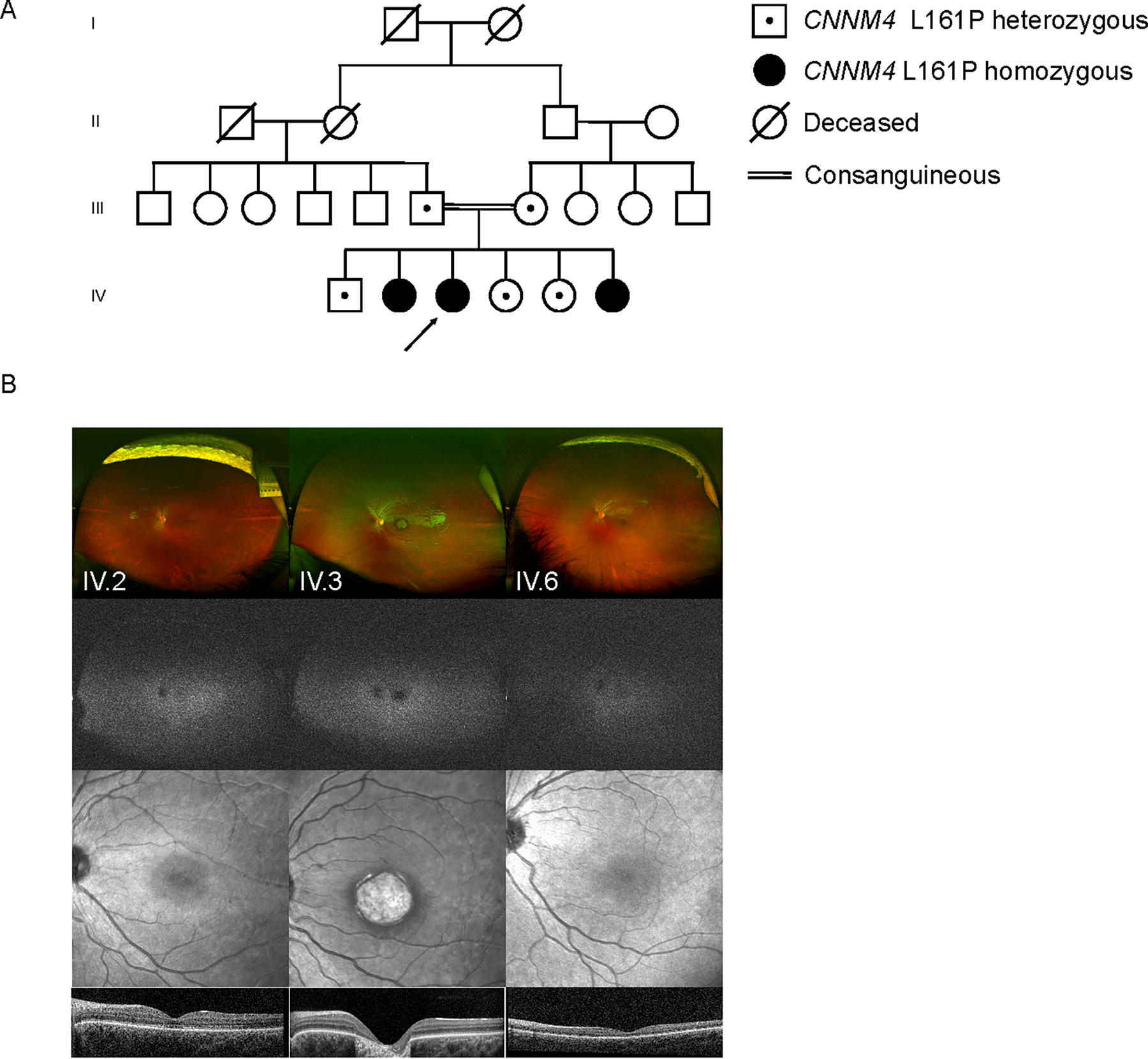
A four-generation pedigree of the family is shown (A). Ultra-widefield scanning laser ophthalmoscope (SLO) images (B, first row), ultra-widefield fundus autofluorescence (FAF) images (B, second row), near-IR reflectance images (B, third row), and optical coherence tomography (OCT) B-scans (B, fourth row) are shown for the three affected sibs (IV.2, IV.3 and IV.6) in the pedigree.
The intraoral examination revealed developmentally age-appropriate permanent dentition with generalized dental defect characterized with thin enamel, yellow-brown tooth discoloration and evidence of post-eruptive enamel breakdown. The clinical features were consistent with hypoplastic-hypomineralized type AI. Anterior open bite and spacing between teeth were also evident (Figure 2A). Radiographically, the enamel was thin, the pulp chambers of the second molars were enlarged and the unerupted third molars showed hyperplastic dental follicles. The patient had undergone restorative treatment, including stainless steel crowns on upper molars, dental restorations on lower molars and endodontic therapy (Figure 2B).
Figure 2:
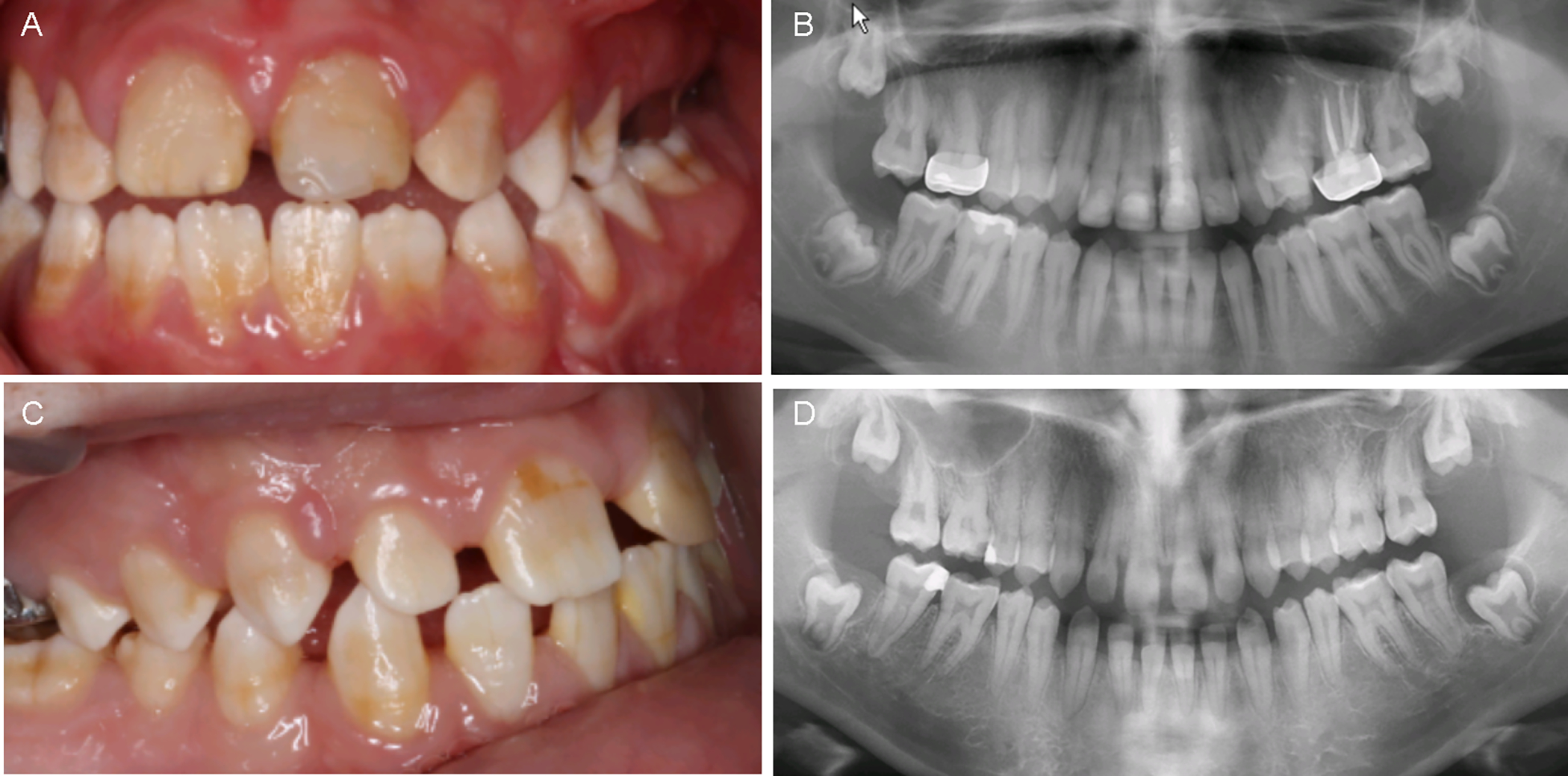
Intraoral photographs (A, C) and panoramic radiographs (B, D) of the proband (IV.3, top row) and her older sister (IV.2, bottom row).
The proband’s older sister (IV.2) also presented with similar complaints of poor central vision and photoaversion. Examination was notable for horizontal nystagmus, with Snellen BCVA at distance of 20/100 in the right eye and 20/200 in the left eye. Anterior segment was unremarkable, and fundus exam was notable for subtle hypopigmentation of the macula and a normal peripheral retina (findings summarized in Table 1). SD-OCT revealed segmental loss of reflectivity of the ellipsoid zone in the fovea, and fundus autofluorescence revealed a bull’s eye pattern of hypo and hyper-autofluorescence. Farnsworth D15 color vision testing revealed numerous axis errors.
The intraoral examination showed age-appropriate permanent dentition with spacing between the teeth. All teeth presented with thin, yellow-brown discolored enamel consistent with hypoplastic-hypomineralized type AI (Figure 2C). Radiographically, the enamel was thin, the mandibular second molars had taurodontism and the unerupted third molars exhibited hyperplastic dental follicles. The patient had undergone restorative treatment, including stainless steel crowns on upper molars, dental restorations on lower molars and endodontic therapy (Figure 2D).
The proband’s youngest sibling (IV.6) presented with complaints of blurred vision, photoaversion, and color confusion. As in her older affected sisters, examination was notable for horizontal nystagmus and decreased vision, with Snellen BCVA at distance of 20/200 in the right eye and 20/400 in the left eye. Esotropia was present and the anterior segment was likewise unremarkable. Interestingly, fundus examination was notable for absence of frank retinal atrophy or pigmentary changes in the macula or in the periphery. The optic disc appeared slightly smaller than normal, but no dysplastic features were present. SD-OCT revealed subtle interruption of the subfoveal ellipsoid zone and preserved outer retinal lamination. Fundus autofluorescence was unremarkable.
The intraoral examination showed primary dentition with present mandibular teeth and missing all maxillary teeth with the exception of the second primary molars. The patient had undergone previously a complete oral rehabilitation under general anesthesia which included provision of stainless-steel crowns on all molars, composite resin crowns on the lower anterior teeth and extraction of upper teeth. According to the dental records, the oral rehabilitation was necessitated due to presence of generalized enamel defect consistent with AI, which had led to tooth structure loss. Of note is that the patient had a history two teeth developing dental abscesses in an absence of dental caries.
The proband (IV.3) had previously been evaluated by a medical geneticist for evaluation of short stature, achromatopsia, horizontal nystagmus, dental enamel abnormalities, and joint hypermobility. Whole exome sequencing revealed she was homozygous for c.482T>C (p.L161P) in exon 1 of the CNNM4, a finding which was initially identified as a variant of unknown significance (VUS) using established ACMG criteria (Richards et al. 2015). Her affected sisters (IV.2 and IV.6) were also found to be homozygous for the same variant, while both parents and the three unaffected siblings were found to be heterozygotes.
Clinical electrophysiology
Figure 3 shows the dark-adapted (top row) and light-adapted (bottom row) ERGs for the proband and her older affected sister (IV.2, green trace, and IV.3, blue trace) compared to a visually-normal control subject (black trace). The gray boxes mark the normal ranges. (Subject IV.6 was deemed too young to participate in electrophysiological and pupillometric assessments). Panel A shows that the low luminance (0.01 cd-s-m−2) responses were well within the normal range. Panel B shows that the moderate luminance (3.0 cd-s-m−2) responses had a- and b-wave amplitudes that were within normal ranges. The implicit time of the b-wave, however, was somewhat delayed for both subjects. Panels C and D show that the light-adapted single flash and flicker responses were undetectable for both CNNM4 syndrome subjects. A multifocal electroretinogram (mfERG) was also obtained for both subjects and demonstrated non-detectable amplitudes (data not shown).
Figure 3:

ERG waveforms under dark-adapted conditions for a dim flash (0.01 cd-s-m−2) and a moderate luminance flash (3.0 cd-s-m−2) are shown in the top row (A and B, respectively). The bottom row shows ERGs under light-adapted conditions for a moderate luminance flash (3.0 cd-s-m−2) and flicker (C and D, respectively). Case 1 (IV.2) is shown in green and the proband (Case 2, IV.3) is shown in blue. Black traces represent a normal control and the gray regions highlight the normal ranges.
PLR data
Figure 4 shows normalized pupil size under three conditions that are intended to target the rod- (Figure 4A) cone- (Figure 4B) and melanopsin- (Figure 4C) pathways. Responses from the proband and her older affected sister (IV.2, green trace, and IV.3, blue trace) are shown along with the ranges of normal (gray regions). Figure 4A shows that the rod-mediated pupil response (dark-adapted, 1-sec, 0.001 cd/m2 flash) was large, falling near the upper limit of normal for both subjects. In contrast, Figure 4B shows that the cone-mediated pupil response (light-adapted, 1-sec, 10 cd/m2 flash) was attenuated, but measurable, for both subjects. Figure 4C shows that the melanopsin-mediated PIPR (dark-adapted, 1-sec, 450 cd/m2 flash) was large, falling near the upper limit of normal for both subjects. These panels are intended to illustrate pupil size over time for the three conditions; pupil sizes for the patients and controls are quantified in the right panels.
Figure 4:

Normalized pupil size is shown over time for cases 1 (IV.2, green) and 2 (IV.3, blue). Panel A shows the rod-mediated PLR, panel B shows the cone-mediated PLR, and panel C shows the melanopsin-mediated PLR. The gray regions mark the normal range. PLR amplitude is plotted as a function of log stimulus luminance measured under dark-adapted conditions with a short-wavelength stimulus (D) and light-adapted conditions with a long-wavelength stimulus (E). The range of pupil responses for the control group are shown by the shaded areas. Data for each subject are shown by the color-coded symbols.
Figures 4D–E plot normalized PLR amplitude as a function of log stimulus luminance for the two CNNM4 syndrome subjects and the range of normal (shaded areas). The solid lines are fits of Eq. 1 to the patient data. Under dark-adapted conditions (D), the two subjects had large PLRs. In fact, they had pupil responses that somewhat exceeded the range of normal at the lowest stimulus luminance level. Under light-adapted conditions (E), the PLRs tended to be slightly attenuated throughout the stimulus luminance range.
Full-field threshold data
Figure 5 shows full-field luminance thresholds obtained with long- and short-wavelength stimuli under dark-adapted (left) and light-adapted (right) conditions. The boxes show the range of normal and the horizontal bars mark the mean of each group. Under dark-adapted conditions, both subjects had normal thresholds for the long- and short-wavelength stimuli. In contrast, the subjects had elevated, but measurable, thresholds for both stimuli under light-adapted conditions. Thresholds were elevated above the normal range by approximately 10 dB for the long-wavelength stimulus and by approximately 5 dB for the short-wavelength stimulus for both subjects.
Figure 5:

Log luminance threshold is shown under dark-adapted conditions (left) and light-adapted conditions (right) for long-wavelength stimuli (left data sets) and short wavelength stimuli (right data sets). The red and blue boxes represent the ranges of normal for long- and short wavelength stimuli, respectively.
DISCUSSION
It has previously been reported that Jalili syndrome exhibits phenotypic variability with respect to the degree of retinal degeneration (Gerth-Kahlert et al. 2015; Topçu et al. 2016). To our knowledge, this is the first report of a family with molecularly confirmed variants in CNNM4 presenting with the classic dental features of Jalili syndrome, AI, but with retinal dystrophy of the cone pathway only. However, pupillometry reveals that not only are rod responses preserved, but that there is in fact some partially preserved cone-mediated responses.
The CNNM4 variant c.482T>C p.(Leu161Pro), in this family has not been previously reported as pathogenic. This variant is not observed in controls in a large population (Consortium et al. 2016) and segregates with the disease in multiple family members (as homozygous in 3 affected relatives and as heterozygous in 5 unaffected relatives). There are no known reports of patients presenting with both a cone dystrophy and AI due to variants in an alternative gene. Thus, we believe the p.(Leu161Pro) variant to be responsible for Jalili syndrome in this family.
The function of CNNM4 in the retina has been examined previously. An in vitro study demonstrated that CNNM4 interacts with IQCB1 (Li et al. 2018), which is required for mouse photoreceptor outer segment formation (Ronquillo et al. 2016). IQCB1 directly interacts with CEP290 (Schäfer et al. 2008), calmodulin, and is complexed with RPGR (Otto et al. 2005). Mutations in IQCB1 lead to diseases featuring widespread retinal degeneration, including Senior-Loken syndrome and non-syndromic Leber congenital amaurosis (Estrada-Cuzcano et al. 2011). Mutations in CNNM4 are expected to affect Mg2+ homeostasis and lead to hypomagnesemia (Yamazaki et al. 2013). While further work is needed to clarify the specific pathway leading to photoreceptor degeneration, it was proposed that a mutation in CNNM4 may lead to non-sense mediated decay, apoptosis, and/or have deleterious consequences on IQCB1 function, leading to retinal dystrophy (Li et al. 2018).
AI is the dental manifestation of Jalili syndrome and its second most prominent feature (Parry et al. 2009). AI is an umbrella term that describes a group of heterogenous hereditary conditions associated with abnormal dental enamel development (Wright et al. 2015). AI typically affects both primary and permanent dentitions and can present in isolation or in conjunction with syndromic disorders (Wright et al. 2015). The enamel development (amelogenesis) is a complex process regulated by functionally diverse genes and multifaceted molecular pathways (Parry et al. 2009; Wright et al. 2015). Disturbances in the secretory stages of amelogenesis may present phenotypically with hypoplastic (thin) enamel, while errors during the stages of deposition and maturation of the enamel matrix formation may result in hypomineralized and/or hypomature (brittle, discolored) enamel (Parry et al. 2009; Wright et al. 2015). While AI can be associated with thousands of different genes and their products, the mutation of the CNNM4 in Jalili syndrome is thought to dysregulate the transport of metal ions, specifically magnesium, leading to the development of enamel structure with deficient thickness and reduced mineral content (significantly less calcium and abnormally increased magnesium) (Luder et al. 2013). In CNNM4 (−/−) mice, there is deficient dental enamel (Yamazaki et al. 2013). All three siblings in this study showed similar anomalous tooth structure demonstrated by thin enamel and discoloration consistent with hypoplastic/ hypomineralized type AI. Anterior open bite, taurodontism and risk of spontaneous dental abscesses were also dental findings with known AI association. The dental manifestations described in this study are supported by previous literature (Jalili and Smith 1988; Luder et al. 2013; Wright et al. 2015).
A prior series discussed the natural history of Jalili syndrome and indicated progressive visual dysfunction over 10 years (Hirji et al. 2018). However, all the patients in that study presented with at least some rod pathway abnormalities on initial ERG, a finding not present in the two subjects in this study. The initial report of Jalili syndrome noted that all patients exhibited diminished visual acuity and nystagmus, and that cones initially are affected with later rod involvement (Jalili and Smith 1988). Thus, at this time, it is unclear whether the three affected subjects in this study will have progressive visual deterioration either with, or without, later rod dysfunction.
This study applied a battery of assays to measure retinal function in this family. Interestingly, despite the structural differences evident on clinical exam and imaging, the electrophysiology and pupillometry were remarkably similar in both sisters. Both subjects had no measurable light-adapted ERG, and the dark-adapted ERG findings of delayed b-wave implicit times were consistent with loss of cone pathway input. Despite the profoundly attenuated cone-pathway ERG findings, the cone-mediated PLR ranged from normal to mildly reduced. The PLR, much like the ERG, can be measured under dark- and light-adapted conditions to target rod- and cone-pathway function. It has recently been shown that a cone-mediated PLR can be recorded in individuals who have severe cone dystrophy and no measurable light-adapted full-field ERG (Park et al. 2020). Close further examination with pupillometry could help detect progression of cone dysfunction or early signs of rod pathway dysfunction in these patients.
In addition to measuring photoreceptor dysfunction with electrophysiology and pupillometry, this study utilized luminance thresholds to assay the rod and cone-photoreceptor pathways. Much like the rod-mediated PLRs, dark-adapted luminance thresholds were normal, but light-adapted thresholds were elevated. Thus, the three full-field measures of cone-pathway function provided considerably different results: light-adapted ERGs were non-detectable, light-adapted luminance thresholds were moderately elevated, and light-adapted PLRs were nearly normal. To our knowledge, this is the first report combining these three photopic measurements to characterize a photoreceptor dysfunction in a molecularly confirmed cone dystrophy.
Differences in the cone-pathway-mediated measures may be attributed to differences in the spatial summation characteristics of these tests. That is, the ffERG is a mass electrical response pooled across the retina that requires a large area of intact cone function to produce a measurable response. In contrast, the full-field stimulus test is thought to be mediated by the retinal area with greatest sensitivity (Roman et al. 2005). Likewise, the critical area of spatial summation is thought to be relatively small for the cone-mediated PLR (less than approximately 16o of visual angle) (Park and McAnany 2015). Of note, inferring retinal function based on the light-adapted ERGs alone would suggest that the two subjects in this study lack cone-pathway function. This, however, is clearly not the case: the subjects had measurable, although not necessarily normal, cone-mediated PLRs and luminance thresholds.
Although previous work in visually-normal individuals has shown that the 6 cd/m2 blue adapting field was sufficient to suppress rod-pathway activity (Collison et al. 2019), there may be rod contributions to the light-adapted PLRs in our patients who have cone dysfunction. Measuring PLRs against adapting fields of high photopic luminance could help resolve this question. Light-adapted luminance threshold measurements were similar for photopically matched long- and short-wavelength stimuli, indicating that these measures were likely cone-pathway mediated. Thus, the PLR data support the luminance threshold data, both indicating residual cone-pathway function despite the undetectable photopic ERGs. We note that photopic ERGs were only measured with a 3.0 cd-s-m−2 flash and it is possible that a higher luminance flash could elicit measurable photopic flash and flicker responses. Future work using high luminance flashes under photopic conditions are needed to address this question. In addition, future measurements in this family and other subjects with cone-pathway dysfunction may reveal how these distinct functional measurements can be selectively utilized to monitor disease progression and, potentially, response to treatment.
ACKNOWLEDGMENTS
We thank Lydia DeJonge, DDS, for providing dental photographs.
FUNDING
Supported by National Eye Institute P30EY001792 (UIC core support); unrestricted funds from Research to Prevent Blindness to the University of Illinois at Chicago
Footnotes
DECLARATION OF INTEREST
The authors have no financial declarations of interest.
REFERENCES
- Collison FT, Park JC, Fishman GA, McAnany JJ, Stone EM. Full-Field Pupillary Light Responses, Luminance Thresholds, and Light Discomfort Thresholds in CEP290 Leber Congenital Amaurosis PatientsFull-Field Measures in CEP290 Leber Congenital Amaurosis. Invest Ophth Vis Sci. 2015;56(12):7130–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collison FT, Park JC, Fishman GA, Stone EM, McAnany JJ. Two-color pupillometry in enhanced S-cone syndrome caused by NR2E3 mutations. Doc Ophthalmol. 2016;132(3):157–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collison FT, Park JC, Fishman GA, Stone EM, McAnany JJ. Two-color pupillometry in KCNV2 retinopathy. Doc Ophthalmol. 2019;139(1):11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium EA, Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacey DM, Liao H-W, Peterson BB, Robinson FR, Smith VC, Pokorny J, et al. Melanopsin-expressing ganglion cells in primate retina signal colour and irradiance and project to the LGN. Nature. 2005;433(7027):749–54. [DOI] [PubMed] [Google Scholar]
- Daneshmandpour Y, Darvish H, Pashazadeh F, Emamalizadeh B. Features, genetics and their correlation in Jalili syndrome: a systematic review. J Med Genet. 2019;56(6):jmedgenet-2018–105716. [DOI] [PubMed] [Google Scholar]
- Estrada-Cuzcano A, Koenekoop RK, Coppieters F, Kohl S, Lopez I, Collin RWJ, et al. IQCB1 Mutations in Patients with Leber Congenital Amaurosis. Invest Ophth Vis Sci. 2011;52(2):834–9. [DOI] [PubMed] [Google Scholar]
- Gamlin PDR, McDougal DH, Pokorny J, Smith VC, Yau K-W, Dacey DM. Human and macaque pupil responses driven by melanopsin-containing retinal ganglion cells. Vision Res. 2007;47(7):946–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerth-Kahlert C, Seebauer B, Dold S, Hanson JVM, Wildberger H, Spörri A, et al. Intra-familial phenotype variability in patients with Jalili syndrome. Eye. 2015;29(5):712–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirji N, Bradley PD, Li S, Vincent A, Pennesi ME, Thomas AS, et al. Jalili Syndrome: Cross-sectional and Longitudinal Features of Seven Patients With Cone-Rod Dystrophy and Amelogenesis Imperfecta. Am J Ophthalmol. 2018;188:123–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalili IK, Smith NJ. A progressive cone-rod dystrophy and amelogenesis imperfecta: a new syndrome. J Med Genet. 1988;25(11):738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardon R, Anderson SC, Damarjian TG, Grace EM, Stone E, Kawasaki A. Chromatic Pupil Responses Preferential Activation of the Melanopsin-mediated versus Outer Photoreceptor-mediated Pupil Light Reflex. Ophthalmology. 2009;116(8):1564–73. [DOI] [PubMed] [Google Scholar]
- Kawasaki A, Crippa SV, Kardon R, Leon L, Hamel C. Characterization of Pupil Responses to Blue and Red Light Stimuli in Autosomal Dominant Retinitis Pigmentosa due to NR2E3 MutationAutosomal Dominant Retinitis Pigmentosa. Invest Ophth Vis Sci. 2012;53(9):5562–9. [DOI] [PubMed] [Google Scholar]
- Kelbsch C, Strasser T, Chen Y, Feigl B, Gamlin PD, Kardon R, et al. Standards in Pupillography. Front Neurol. 2019;10:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan AK, Jacobson SG, Roman AJ, Iyer BS, Garafalo AV, Héon E, et al. Transient pupillary light reflex in CEP290- or NPHP5-associated Leber congenital amaurosis: Latency as a potential outcome measure of cone function. Vision Res. 2020;168:53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Xi Q, Zhang X, Yu D, Li L, Jiang Z, et al. Identification of a mutation in CNNM4 by whole exome sequencing in an Amish family and functional link between CNNM4 and IQCB1. Mol Genet Genomics. 2018;293(3):699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luder HU, Gerth-Kahlert C, Ostertag-Benzinger S, Schorderet DF. Dental Phenotype in Jalili Syndrome Due to a c.1312 dupC Homozygous Mutation in the CNNM4 Gene. Plos One. 2013;8(10):e78529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAnany JJ, Park JC, Fishman GA, Collison FT. Full-Field Electroretinography, Pupillometry, and Luminance Thresholds in X-Linked Retinoschisis. Invest Ophth Vis Sci. 2020;61(6):53–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch DL, Kondo M, Hamilton R, Lachapelle P, Messias AMV, Robson AG, et al. ISCEV extended protocol for the stimulus–response series for light-adapted full-field ERG. Doc Ophthalmol. 2019;138(3):205–15. [DOI] [PubMed] [Google Scholar]
- McCulloch DL, Marmor MF, Brigell MG, Hamilton R, Holder GE, Tzekov R, et al. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol. 2015;130(1):1–12. [DOI] [PubMed] [Google Scholar]
- Ner DB, Sher I, Hamburg A, Mhajna MO, Chibel R, Derazne E, et al. Chromatic pupilloperimetry for objective diagnosis of Best vitelliform macular dystrophy. Clin Ophthalmol. 2019;13:465–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto EA, Loeys B, Khanna H, Hellemans J, Sudbrak R, Fan S, et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat Genet. 2005;37(3):282–8. [DOI] [PubMed] [Google Scholar]
- Park JC, Collison FT, Fishman GA, McAnany JJ. Electrophysiological and Pupillometric Abnormalities in PROM1 Cone–Rod Dystrophy. Transl Vis Sci Technology. 2020;9(9):26–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JC, McAnany JJ. Effect of stimulus size and luminance on the rod-, cone-, and melanopsin-mediated pupillary light reflex. J Vision. 2015;15(3):13–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JC, Moura AL, Raza AS, Rhee DW, Kardon RH, Hood DC. Toward a Clinical Protocol for Assessing Rod, Cone, and Melanopsin Contributions to the Human Pupil Response. Invest Ophth Vis Sci. 2011;52(9):6624–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry DA, Mighell AJ, El-Sayed W, Shore RC, Jalili IK, Dollfus H, et al. Mutations in CNNM4 Cause Jalili Syndrome, Consisting of Autosomal-Recessive Cone-Rod Dystrophy and Amelogenesis Imperfecta. Am J Hum Genetics. 2009;84(2):266–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polok B, Escher P, Ambresin A, Chouery E, Bolay S, Meunier I, et al. Mutations in CNNM4 Cause Recessive Cone-Rod Dystrophy with Amelogenesis Imperfecta. Am J Hum Genetics. 2009;84(2):259–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purwar P, Sareen S, Bhartiya K, Inayatullah SRS, Bansal M, Chahal V, et al. Jalili syndrome presenting with situs inversus totalis and keratoconus: the first case in the Indian subcontinent. Oral Surg Oral Medicine Oral Pathology Oral Radiology [Internet]. 2015. Nov 1;120(5):e210–8. Available from: 10.1016/j.oooo.2015.04.002 [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman AJ, Schwartz SB, Aleman TS, Cideciyan AV, Chico JD, Windsor EAM, et al. Quantifying rod photoreceptor-mediated vision in retinal degenerations: dark-adapted thresholds as outcome measures. Exp Eye Res. 2005;80(2):259–72. [DOI] [PubMed] [Google Scholar]
- Ronquillo CC, Hanke-Gogokhia C, Revelo MP, Frederick JM, Jiang L, Baehr W. Ciliopathy-associated IQCB1/NPHP5 protein is required for mouse photoreceptor outer segment formation. Faseb J. 2016;30(10):3400–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer T, Pütz M, Lienkamp S, Ganner A, Bergbreiter A, Ramachandran H, et al. Genetic and physical interaction between the NPHP5 and NPHP6 gene products. Hum Mol Genet. 2008;17(23):3655–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topçu V, Alp MY, Alp CK, Bakır A, Geylan D, Yılmazoğlu MÖ. A new familial case of Jalili syndrome caused by a novel mutation in CNNM4. Ophthalmic Genet. 2016;38(2):1–6. [DOI] [PubMed] [Google Scholar]
- Torres LTL, Schorderet D, Valmaggia C, Todorova M. A novel mutation in CNNM4 (G492C) associated with Jalili Syndrome. Acta Ophthalmologica [Internet]. 2015. Oct 1;93(S255). Available from: 10.1111/j.1755-3768.2015.0606 [DOI] [Google Scholar]
- Wawrocka A, Walczak-Sztulpa J, Badura-Stronka M, Owecki M, Kopczynski P, Mrukwa-Kominek E, et al. Co-occurrence of Jalili syndrome and muscular overgrowth. American Journal of Medical Genetics Part A [Internet]. 2017. Aug 1;173(8):2280–3. Available from: 10.1002/ajmg.a.38318 [DOI] [PubMed] [Google Scholar]
- Wright JT, Carrion IA, Morris C. The Molecular Basis of Hereditary Enamel Defects in Humans. J Dent Res. 2015;94(1):52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki D, Funato Y, Miura J, Sato S, Toyosawa S, Furutani K, et al. Basolateral Mg2+ Extrusion via CNNM4 Mediates Transcellular Mg2+ Transport across Epithelia: A Mouse Model. Plos Genet. 2013;9(12):e1003983. [DOI] [PMC free article] [PubMed] [Google Scholar]