

Abstract
Survival motor neuron (SMN) protein deficiency results in loss of alpha motor neurons and subsequent muscle atrophy in patients with spinal muscular atrophy (SMA). Reactive microglia have been reported in SMA mice and depleting microglia rescues the number of proprioceptive synapses, suggesting a role in SMA pathology. Here, we explore the contribution of lymphocytes on microglia reactivity in SMA mice and investigate how SMN deficiency alters the reactive profile of human induced pluripotent stem cell (iPSC)‐derived microglia. We show that microglia adopt a reactive morphology in spinal cords of SMA mice. Ablating lymphocytes did not alter the reactive morphology of SMA microglia and did not improve the survival or motor function of SMA mice, indicating limited impact of peripheral immune cells on the SMA phenotype. We found iPSC‐derived SMA microglia adopted an amoeboid morphology and displayed a reactive transcriptome profile, increased cell migration, and enhanced phagocytic activity. Importantly, cell morphology and electrophysiological properties of motor neurons were altered when they were incubated with conditioned media from SMA microglia. Together, these data reveal that SMN‐deficient microglia adopt a reactive profile and exhibit an exaggerated inflammatory response with potential impact on SMA neuropathology.
Keywords: induced pluripotent stem cell, inflammation, microglia, spinal muscular atrophy, survival motor neuron
Main Points
SMA microglia adopt a reactive morphology.
Complement gene expression, migration velocity and phagocytosis are increased in SMA microglia.
SMA microglia release factors that alter the morphology and electrophysiological properties of SMN‐deficient motor neurons in culture.
1. INTRODUCTION
Spinal muscular atrophy (SMA) is a devastating inherited neurological disease resulting in muscle weakness and subsequent atrophy. Affecting 1 in 11,000 live births worldwide, SMA was once considered one of the leading genetic causes of infant mortality (Monani, 2005; Simone et al., 2016). SMA patients have difficulty meeting developmental milestones and, in the majority of cases, rarely survive past the age of two if left untreated. SMA is characterized by deletions or mutation in the SMN1 gene, resulting in insufficient production of the survival motor neuron (SMN) protein (Lefebvre et al., 1995). Current FDA approved therapeutics, Risdiplam (Evrysdi™), Nusinersen (Spinraza®), and Onasemnogene ebeparvovec (Zolgensma®) aim to increase levels of SMN protein (Groen, 2018; Mendell et al., 2017; Mercuri et al., 2018; Messina & Sframeli, 2020). However, effectiveness and toxicity vary among patients, suggesting that further therapeutic optimization is required. Combinatorial therapy tackling multiple aspects of SMA pathogenesis is an emerging consideration.
The SMN protein is ubiquitously expressed in all tissues, and has a well‐established role in the assembly of small nuclear ribonucleoproteins (snRNPs), among many other functions (Liu et al., 1997; Massenet et al., 2002; Paushkin et al., 2002; Pellizzoni et al., 1998; Pellizzoni et al., 1999). snRNPS are essential for pre‐mRNA splicing, and have the potential to widely alter gene transcription (Yong et al., 2004). Motor neurons in the ventral horn of the spinal cord are most vulnerable to reduced levels of SMN protein (Burghes & Beattie, 2009). Furthermore, depleting SMN in motor neurons alone produces a milder disease phenotype suggesting contribution of glia in SMA disease progression (Hua et al., 2011; Park et al., 2010; Simon et al., 2017). There is growing evidence supporting SMA as a multi‐system disorder with peripheral and CNS immune cells contributing to SMA disease progression (Deguise et al., 2017; Deguise & Kothary, 2017; Martin et al., 2017; O'Meara et al., 2017; Rindt et al., 2015).
Within the CNS, glia maintain homeostasis and assist in neuronal survival through pH buffering, ion exchange, glutamate recycling, synapse structure stabilization, and clearance of debris (Hammond et al., 2019). Oligodendrocyte development and CNS myelination were normal in SMA mice suggesting oligodendrocyte function is potentially unimpaired in SMA (Ohuchi et al., 2019; O'Meara et al., 2017). Increased GFAP expression and altered astrocyte morphology has been reported in SMA mice (Rindt et al., 2015). Interestingly, selectively replacing SMN in astrocytes alone partially attenuated their pro‐inflammatory profile, improved neuromuscular circuitry, and extended life span of SMA model mice, suggesting that glia may contribute to the SMA disease phenotype (Rindt et al., 2015).
As the main innate immune cell type in the CNS, microglia influence the outcome of many neurodegenerative diseases (Alliot et al., 1999; Li & Barres, 2018; Lively & Schlichter, 2013). Microglia are highly dynamic cells that continually survey their environment for potential threats. Alterations in the pathological condition can cause a rapid response (Alliot et al., 1999; Li & Barres, 2018). Microglia function primarily through release of cytokines and trophic factors, recruitment of adaptive immune cells, as well as clearing of cellular debris and programmed elimination of neuronal cells (Hickman et al., 2018). Interactions between adaptive immune cells and antigen presenting cells are critical when responding to damage associated‐molecular patterns released after degenerating tissue and cell damage (Kigerl et al., 2014; Schetters et al., 2017; Venereau et al., 2015). The adaptive immune response is generated by the presence of lymphocytes, either T cells or B cells (Chaplin, 2010; Schenten & Medzhitov, 2011). Once mature, lymphocytes express specific antigen pattern recognition receptors that dictate highly specialized cell‐mediated responses and travel to secondary lymphoid tissues and the CNS during neuroinflammation. In SMA model mice, hypoplasia of both the spleen and thymus was observed (Deguise et al., 2017; Deguise et al., 2020; Khairallah et al., 2017). The hypoplasia coincided with an increased population of splenic B and T lymphocytes compared to controls, potentially contributing to the increased inflammatory cell interactions in SMA. Recently, SMA pre‐clinical models suggested induction of systemic inflammation (Schenten & Medzhitov, 2011; Wan et al., 2018).
Previously, microgliosis was observed in the ventral horn of severe SMA mice (Tarabal et al., 2014). Iba1, a marker for microglia, was upregulated during late stages of disease progression in SMA model mice. Interestingly, pharmacological depletion of microglia rescued proprioceptive synapses on motor neuron proximal dendrites, improving SMA mouse motor function (Vukojicic et al., 2019). However, the reactive state of microglia during SMA progression is unknown. Microglia can be neuroprotective or neurotoxic, depending on the type of stimulus and other spatiotemporal factors. Complete microglia ablation in an SMA mouse model did not prevent motor neuron death (Vukojicic et al., 2019). However, it is possible, given the spectrum of immune functions of microglia in health and disease, that completely ablating microglia may obscure differential contributions of microglia to SMA disease progression. In this study, we investigate the effect of SMN deficiency on microglia reactivity in SMA model mice and iPSC derived SMA microglia.
2. RESULTS
2.1. Microglia exhibit a reactive profile in SMA mice
Microglia migrate to and adopt a reactive profile at the site of neuronal loss in SMA model mice (Tarabal et al., 2014). What triggers this migration is unknown but there is some evidence that lymphocytes can cross the blood brain barrier during neurodegenerative conditions resulting in proliferation and activation of microglia (Brochard et al., 2009). Using flow cytometry, we evaluated the overall number of microglia (CD11b+), monocytes (CD45+), and lymphocytes (CD3+) in spinal cords extracted from unaffected and SMNΔ7 (SMN2+/+; SMN Δ7+/+; Smn−/−) mice (Figure 1a–c), which have a lifespan of about 14 days and display motor function impairment that correlates with motor neuron loss (Le et al., 2005). At postnatal day 10, near disease end‐stage in this mouse model, the number of microglia, monocytes and lymphocytes were similar between control and SMA mouse spinal cords (Figure 1a–c), suggesting no change in microglia proliferation or peripheral immune cell infiltration at disease end stage.
FIGURE 1.
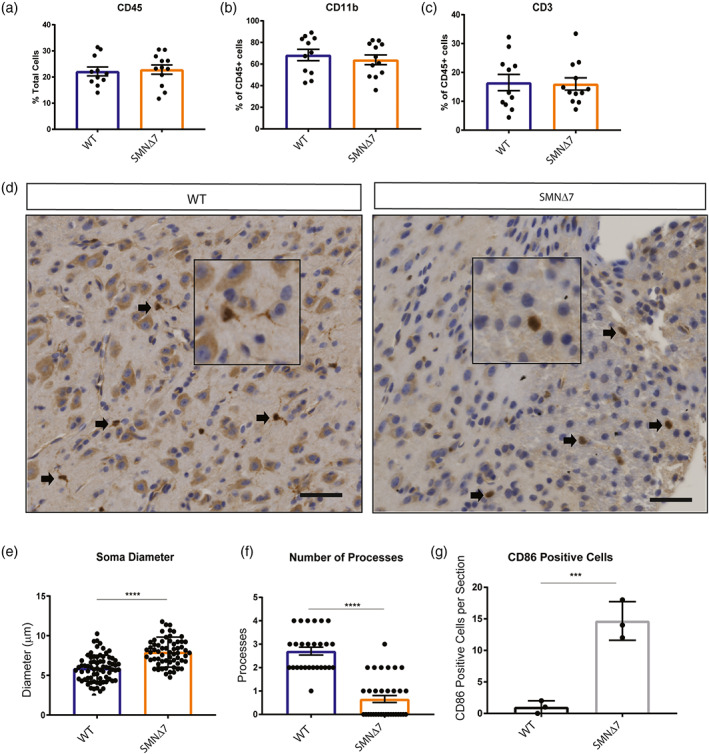
Microglia adopt a reactive morphology in SMA mouse spinal cords. Monocytes were isolated from PND10 WT and SMNΔ7 mouse spinal cords (WT n = 11, SMNΔ7 n = 12). Total (a) monocytes (CD45+), (b) microglia (CD11b+), and (c) T‐cells (CD3+) populations were quantified by flow cytometry. Error bars represent mean ± SEM. (d) Spinal cord sections from L1‐L5 of PND10 SMNΔ7 and WT mice were immuno‐stained for Iba1. DAB staining was used to visualize antibody. Scale bars = 50 μm. (e) Soma diameter, and (f) number of branches were analyzed using image J. (g) Histological quantification of the number of microglia per section co‐staining with Iba1 and CD86 (L1‐L5 spinal cord). Values represent mean ± SEM.**p = .0081, ****p = <.0001, t‐test, Iba1+ cells were evaluated from 3 wild type and 3 SMA mice
We next investigated the reactive state of microglia in the SMNΔ7 mice. Ramified quiescent microglia have a relatively small soma and elongated processes. Once activated, the microglia adopt an amoeboid morphology with an enlarged soma and retracted processes (Fujita et al., 1996; Kozlowski & Weimer, 2012). We used 7 μm thick spinal cord sections from the lumbar region (L1‐L5), where motor neuron death is prevalent in SMNΔ7 mice and determined microglia soma size and number of processes (Ling et al., 2010; Mentis et al., 2011). Soma diameter of Iba1+ microglia were significantly increased in symptomatic SMA mice compared to controls (Figure 1d,e). Moreover, SMA microglia had fewer processes compared to microglia in control mice (Figure 1d,f). Consistent with the reactive morphology, SMA microglia expressed CD86, a cell surface marker of activated microglia (Figure 1g). Together, microglia in the SMNΔ7 mouse spinal cord appear to adopt a reactive morphology with larger soma size, reduced processes, and surface expression of a classic marker of activation.
2.2. Lymphocyte ablation does not improve motor function or survival of SMA mice
Under neurodegenerative conditions, lymphocytes can infiltrate into the CNS and modulate immunological responses (Brochard et al., 2009; Mietelska‐Porowska & Wojda, 2017; Rezai‐Zadeh et al., 2009). While we saw no evidence of increased lymphocyte infiltration in the spinal cord of SMNΔ7 mice, lymphocyte‐released messengers have been reported to activate microglial and influence the progression of many neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), Alzheimer's disease (AD), and Parkinson's disease (PD) (Brochard et al., 2009; Machado‐Santos et al., 2018; Mietelska‐Porowska & Wojda, 2017; Roos et al., 2009). Here, we sought to assess if lymphocytes contribute to SMA disease pathology. T cells are typically subcategorized into two main classes, including the T helper cells (CD4+) and cytotoxic T cells (CD8+) (Pennock et al., 2013). In the SMN∆7 mouse model, overt motor neuron loss starts at post‐natal day 5 and life expectancy is approximately 14 days making it difficult to assess more subtle contributions to SMA pathology (Le et al., 2005; Monani, 2005). Due to the relatively short life span of the SMN∆7 mice it is difficult to assess underlying immune irregularities (Deguise & Kothary, 2017; Khairallah et al., 2017). To address this, we used the less severe Smn 2B/− mouse model, with an average life span of 25 days and which has previous been reported to show reactive gliosis (Bowerman et al., 2012; Cervero et al., 2018; DiDonato et al., 2001; Eshraghi et al., 2016).
To assess if CD4+ cells impact the SMA phenotype, we utilized CD4 knock‐out mice (Cd4 tm1Mak ; [Rahemtulla et al., 1991]) on the Smn 2B/− background and evaluated motor function and survival. We found no improvement in motor function, as measured by the pen test, inverted mesh grip test, and righting reflex test on Smn 2B/− ;CD4 −/− double mutant mice compared to control (Figure 2a–c). Moreover, we found no significant improvement in weight (p = .582) or survival (p = .083) of the double mutant mice (median survival 25 days) compared to controls (median survival 22 days) (Figure 2d,e). Since CD4+ T cells represent only a subset of lymphocytes, we next addressed if ablating all lymphocytes, including T cells and B cells, would alter the SMA phenotype. To do so, we used RAG1 knock‐out mice (Rag1 tm1Mom [Mombaerts et al., 1992]) on the Smn 2B/− background. The lack of RAG1 results in B and T cell deficiency due to their inability to mature (Mombaerts et al., 1992). We observed no improvement in motor function in Smn 2B/− ;Rag1 −/− double mutant mice compared to controls (p = .639;Figure 2f–h). Additionally, lymphocyte deficiency did not significantly improve the weight (p = .173) or extend the life span (p = .316) of the Smn 2B/− ;Rag1 −/− (median survival 24 days) mice compared to Smn 2B/− control (median survival 22 days) (Figure 2i,j). Next, we asked if loss of lymphocytes affected the reactive morphology of SMA microglia. Similar to the SMN∆7 mouse model, microglia from Smn 2B/− mice adopted a reactive morphology with increased soma size and reduced number of processes as observed in spinal cord sections (L1‐L5) (Figure S1a). This reactive morphology remained unchanged when Rag1 was knocked out (Figure S1b), strongly suggesting that lymphocytes do not contribute to the reactive morphology of SMA microglia and likely does not substantially contribute to the SMA disease pathology.
FIGURE 2.
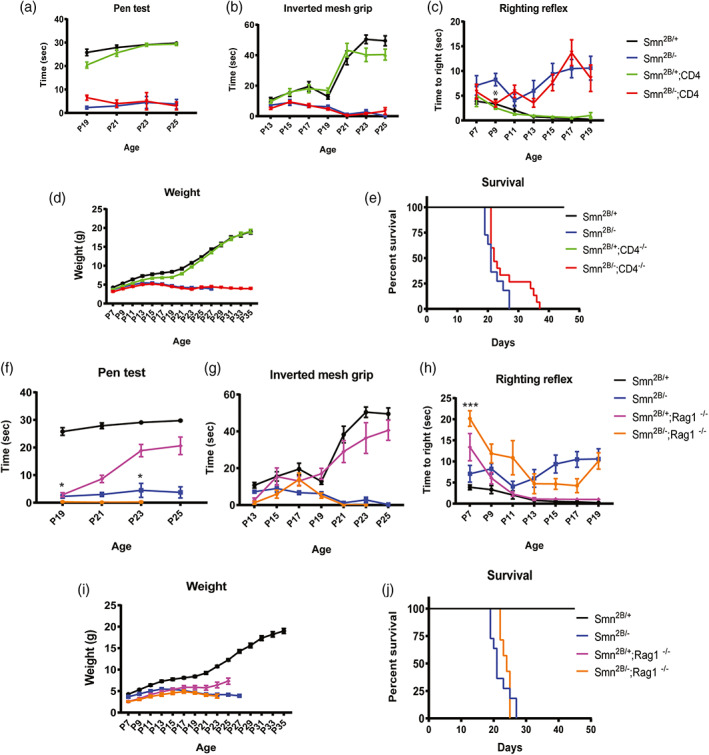
Depleting mature lymphocytes does not improve survival and motor function of Smn 2B/− mice. (a) Inverted mesh grip (starting at PND13), (b) pen test (starting at PND19), and (c) righting reflex (starting at PND7) motor function tests were assessed in Smn 2B/+ (black), Smn 2B/− (blue), Smn 2B/+ ;CD4 /− (green), Smn 2B/− ,CD4 −/− (red) mice (n 11/group) (d) Mice were weighed to assess changes in body weight. (e) Kaplan–Meier survival analysis of Smn 2B/+ (black), Smn 2B/− (blue), Smn 2B/+ ;CD4 /− (green), Smn 2B/− ,CD4 −/− (red). Motor function was assessed in Smn 2B/+ (black), Smn 2B/− (blue), Smn 2B/+ ;Rag1 −/− (pink), Smn 2B/− ;Rag1 −/− (yellow) mice (n 7/group) by (f) inverted mesh grip (starting at PND13), (g) pen test (starting at PND19), and (h) Righting reflex (starting at PND7). (i) SMA mice were weighed every other day to assess changes in body weight. (j) Kaplan–Meier survival analysis of assessed. Survival curves were analyzed using a log‐rank (Mantel–Cox) test. A P value <.05 was considered statistically significant. Significance of behavior data was determined by two‐way ANOVA. Mantel‐Cox and Gehan Breslow‐Wilcoxon analyses were performed to determine significance between groups for all significant main effects and interactions. The same group for Smn 2B/+ and Smn 2B/− control animal were used to measure the effect of CD4 and Rag1 KO on the Smn 2B/+ mutant and do not represent two different set of controls
2.3. iPSC derived SMA microglia adopt a reactive morphology
We and others have previously demonstrated that SMN deficiency can alter the development and function of non‐neuronal cells (Boyer et al., 2014; Bricceno et al., 2014; Hayhurst et al., 2012; Khayrullina et al., 2020; McCormack et al., 2021; Shafey et al., 2005; Sison et al., 2017). To determine if SMN‐deficiency contributes to microglia reactivity, we generated microglia from a human SMA patient‐derived induced pluripotent stem cell (hiPSC) line and an unaffected control (Figure S2a,b) using a modified protocol from (Haenseler et al., 2017). Given that gene expression patterns can change during iPSC differentiation, we first confirmed that SMN levels remained low in the iPSC‐derived SMA microglia (Figure S2c). We confirmed expression of microglia specific genes including TMEM119 and TREM2 and surface marker expression of Iba1, HLA molecules, CD45, and CD11b in the control and SMA microglia (Figure S2d,e). To determine if SMN deficiency was sufficient to alter microglia morphology we examined cell soma size and number of processes. Light microscope images of unaffected and SMA patient iPSC‐derived microglia were collected (Figure 3a) and individual cell morphology analyzed using ImageJ. Our morphological analysis revealed SMA microglia had a significantly fewer processes compared to controls (Figure 3a,b). However, control and SMA microglia had similar cell soma size, in contrast to what we observed in SMA mice (Figure 3a,c), suggesting the microenvironment likely contributes to some of the morphological features of SMA microglia in vivo.
FIGURE 3.
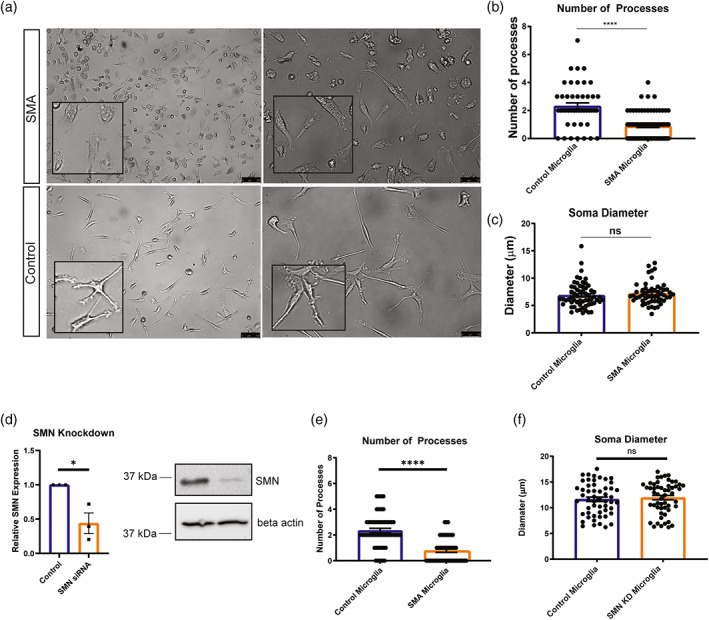
Microglia processes are retracted in iPSC derived SMA microglia. (a) Representative bright field images show control and SMA patient iPSC‐derived microglia at 10 days post differentiation. Scale bars = 100 μm (left panel) and 50 μm (right panel). (b) Number of processes (3 independent experiments were performed; a minimum of 45 microglia were counted per group). (c) Soma diameter (3 independent experiments were performed; a minimum of 50 Iba1+ microglia were counted per group) were analyzed using ImageJ. (d) Microglia derived from control cells were treated with siRNA to SMN and SMN knockdown confirmed by RT‐PCR and western blots. (e) Number of processes (3 independent experiments were performed; a minimum of 45 microglia were counted per group) following SMN knockdown in control cells. (f) Soma diameter (3 independent experiments were performed; a minimum of 50 Iba1+ microglia were counted per group) were analyzed using ImageJ following SMN knockdown in control cells. All groups were analyzed by unpaired t‐test. ****p < .0001. Values represent mean ± SEM
2.4. SMA microglia display an altered transcriptome profile
To better understand the potential roles of SMN in microglia, we performed an unbiased RNAseq analysis to assess global changes in gene expression in one human control versus one SMA patient iPSC‐derived microglia ((Figure 4a; see mRNA‐seq quality metrics Table S1). Select genes described below were validated in microglia derived from a second SMA patient iPSC line (GM23240) by qRT‐PCR. Nevertheless, we cannot completely rule out bias due to genetic background of the lines selected lines. The transcriptome analysis confirmed expression of microglia specific genes (P2RY12, CSF1, TREM2, MERTK, C1q) (Table 2). Transcriptome profiling and differential expression analysis revealed 3664 differentially expressed genes between control and SMA microglia transcripts, using a filter of log2 fold change >0.585 and false discovery rate (FDR) <0.01 (Figure 4b). A gene set enrichment analysis identified genes responsible lysosomal protein breakdown (Figure 4c), phagocytosis, cell migration and cytokine production in SMA microglia compared to control (Table 1). We found that key genes involved in cytokine signaling in innate and adaptive immune response genes, cytokine and chemokine expression (CCL2, CCR1, CCR5, CXCL10, CXCL8), and activation of toll‐like receptors (TICAM1, TIRAP, TLR1‐6) were upregulated in SMA iPSC derived microglia compared to unaffected controls (Table 2). The chemokine receptor CCR1 contributes to immune cell migration and recruitment (Chou et al., 2010; Furuichi et al., 2008) which is consistent with findings in mice. Importantly, our transcriptome analysis supports the hypothesis that SMA microglia adopt a reactive profile independent of cellular microenvironment or degenerating motor neurons.
FIGURE 4.
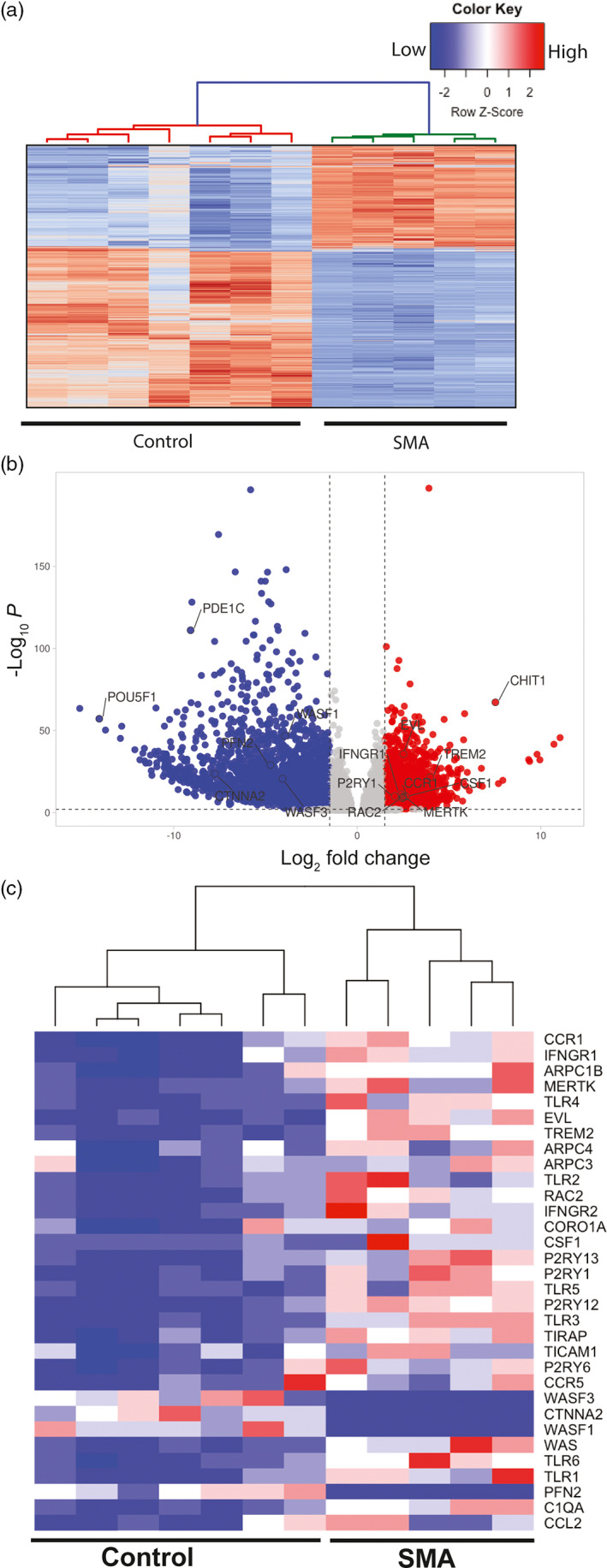
The transcriptome profile of SMA microglia is altered compared to controls. (a) Heatmap of the of the RNA‐seq data based on global mRNA expression from one SMA and one control line. Each column represents an independent differentiation (7 control and 6 SMA). Expression values were standardized and depicted on a z‐scale with red indicating high and blue indicating low expression, respectively. (b) Volcano plots illustrating fold‐change (log base 2) plotted against FDR‐adjusted p value (− log base 10) of genes differentially expressed between SMA and control microglia. A fold change of 1.5 or greater was used as the initial selection criteria. A total of 3664 transcripts were up‐ or down‐regulated in SMA microglia compared to controls. Dashed lines represent filter cutoff values for log two‐fold change of >1.5 (vertical) and FDR‐adjusted P value <.01 (horizontal). Transcripts with greater expression in SMA as compared with control are on the right of the plot (red dots). (c) Heat map of relative gene expression of select genes involved in cell migration, phagocytosis, and inflammation
TABLE 2.
Select immune related genes upregulated in SMA versus control microglia
| Gene | Log2 fold change | FDR‐adjusted p value | Confirmed in SMA line (GM23240) |
|---|---|---|---|
| CSF1 | 2.65056532 | 1.67E‐10 | ✓ |
| TREM2 | 3.98063 | 3.24E‐22 | ✓ |
| MERTK | 2.52487 | 6.73E‐10 | ✓ |
| C1Qa | 2.54305 | 1.98E‐11 | ✓ |
| CCL2 | 2.02477 | 7.95E‐4 | ✓ |
| CCR1 | 2.47138 | 1.71E‐10 | ✓ |
| CCR5 | 1.50768 | 1.132E‐3 | ✓ |
| CXCL10 | 1.90763 | 0.02237 | |
| CXCL8 | 2.44424 | 3.81E‐07 | ✓ |
| TICAM1 | 1.04313 | 2.87E‐07 | |
| TIRAP | 1.27214 | 4.00E‐18 | ✓ |
| TLR1 | 2.60297 | 2.81E‐08 | |
| TLR2 | 2.81526 | 1.06E‐10 | |
| TLR3 | 2.76389 | 2.47E‐24 | ✓ |
| TLR4 | 2.81633 | 3.02E‐13 | |
| TLR5 | 3.21768 | 7.33E‐17 | |
| TLR6 | 2.60581 | 1.51E‐10 | |
| IFNγR1 | 2.41037 | 8.06E‐12 | ✓ |
| IFNγR2 | 1.88004 | 3.39E‐10 | ✓ |
| P2RY1 | 2.04756 | 1.29E‐10 | ✓ |
| P2RY6 | 2.21671 | 1.81E‐08 | ✓ |
| P2RY12 | 3.70221 | 3.32E‐06 | ✓ |
| P2RY13 | 2.361 | 1.11E‐06 |
TABLE 1.
Pathways enriched in SMA versus control microglia
| Pathway | FDR ‐ adjusted p value |
|---|---|
| Lytic vacuole | 3.06E‐13 |
| Lysosome | 3.06E‐13 |
| Cell adhesion | 1.22E‐12 |
| Regulation of leukocyte activation | 1.23E‐11 |
| Endocytosis | 1.44E‐06 |
| Myeloid cell activation involved in immune response | 1.49E‐05 |
| Actin filament‐based process | 3.87E‐05 |
| Regulation of cytokine production | 1.12E‐4 |
Genes involved in actin dynamics and ARP2/3 pathways (ARPC1b, EVL, RAC2, WAS, WASF1, ROCK1, PFN2, CTNNA2), critical in maintaining microglia morphology and surveillance activity, were altered in SMA microglia (Table 3), supporting a previously reported role for SMN in cytoskeletal dynamics (Bricceno et al., 2014; Kolb et al., 2007; Rossoll et al., 2003; Vukojicic et al., 2019). Changes in actin filaments impact migration, chemotaxis, recruitment, as well as phagocytosis in microglia (Blanquie & Bradke, 2018; Fan et al., 2017; Pinto‐Costa & Sousa, 2020). Damaged neurons emit extracellular ATP and ADP, which act as microglia chemoattractants. Chemotaxis depends on both specific receptors and the cytoskeletal machinery, suggesting that SMA microglia may be highly stimulated by these cues (Y. Fan et al., 2017). Indeed, our analysis shows an increase in the P2Y pathway associated genes (P2RY1, P2RY12–13, P2RY6) in SMA microglia (Table 2). P2Y belongs to a purinergic G protein coupled receptors that are critical in detecting extracellular ATP levels (Pinto‐Costa & Sousa, 2020). Intriguingly, sensory synapses have been shown to be tagged with complement proteins to be phagocytosed(Vukojicic et al., 2019). Here we show that SMA microglia are primed with increased expression of complement cascade associated genes (C1qA‐C, C5, C2), as well as apoptotic cell recognizing phagocytosis receptors (CD36, FCGR3B, CD206) (Gombash et al., 2015; Menke et al., 2008; Patitucci & Ebert, 2016). In summary, microglia differentiated from SMA patient iPSCs have a unique transcriptomic profile, indicating that SMN may be a critical regulator of multiple microglial functions, particularly enriched for genes involved in cytokine release, cell migration and phagocytosis.
TABLE 3.
Select cytoskeletal‐related genes altered in SMA versus control microglia
| Gene | Log2 fold change | FDR ‐ adjusted p value | Confirmed in SMA line (GM23240) |
|---|---|---|---|
| RAC2 | 2.46119 | 2.23E‐09 | ✓ |
| WAS | 2.71399 | 2.18E‐14 | ✓ |
| ARPC1B | 1.80271 | 2.54E‐11 | ✓ |
| ARPC4 | 1.311 | 2.91E‐07 | ✓ |
| ARPC3 | 1.25563 | 2.14E‐05 | |
| EVL | 2.51318 | 2.31E‐36 | ✓ |
| CORO1A | 1.40784 | 5.47E‐4 | ✓ |
| PFN2 | −4.733528 | 9.99E‐30 | ✓ |
| CTNNA2 | −7.772883 | 2.32E‐24 | ✓ |
| WASF1 | −3.935657 | 2.30E‐47 | |
| WASF3 | −4.070919 | 2.67E‐21 |
Note: Top genes differentially expressed between SMA and control microglia. A positive fold change indicates a gene whose expression is higher in the SMA microglia, and a negative fold change indicates a gene whose expression is higher in the control microglia.
2.5. SMN deficient microglia display increased migration and phagocytic activity
Cell migration is a fundamental feature of immune cells like microglia, which is essential to their role in tissue protection, repair, and regeneration. Migration can be quantified using several metrics. Velocity is defined as the total distance that a cell migrates over a specific time, while persistence describes a cell's ability to move in a consistent direction without turning. Persistence (d/T) is measured on a scale of 0–1, with higher numbers indicating more persistence. To assess migration dynamics, both unaffected and SMA patient‐derived iPSC differentiated microglia were fluorescently labeled with cell tracker dye and migration was tracked using time‐lapse widefield fluorescent microscopy over 16 h. Velocity and persistence were calculated using ImageJ software. We found that SMA microglia displayed significantly higher velocity compared to control microglia (Figure 5a) consistent with differential expression of genes involved in actin regulation, including CORO1A, PFN2, EVL, ARPC1b, ARPC3, ARPC4, WAS, and RAC2. Transiently restoring SMN by transfecting an SMN‐GFP expression plasmid into the SMA iPSC‐derived microglia returned their velocity to levels observed in control cell (Figure 5a). We found no difference in persistence between SMA and control microglia (Figure 5b), indicating that while cell velocity was altered by SMN‐deficiency, the directionality of movement was unaffected. We next analyzed the impact of SMN deficiency on wound‐stimulated migration using a scratch wound assay (Figure 5c). Our results indicate that SMA microglia migrated into and covered a higher percentage of the wound larger area than control cells after 16 h (Figure 5d).
FIGURE 5.
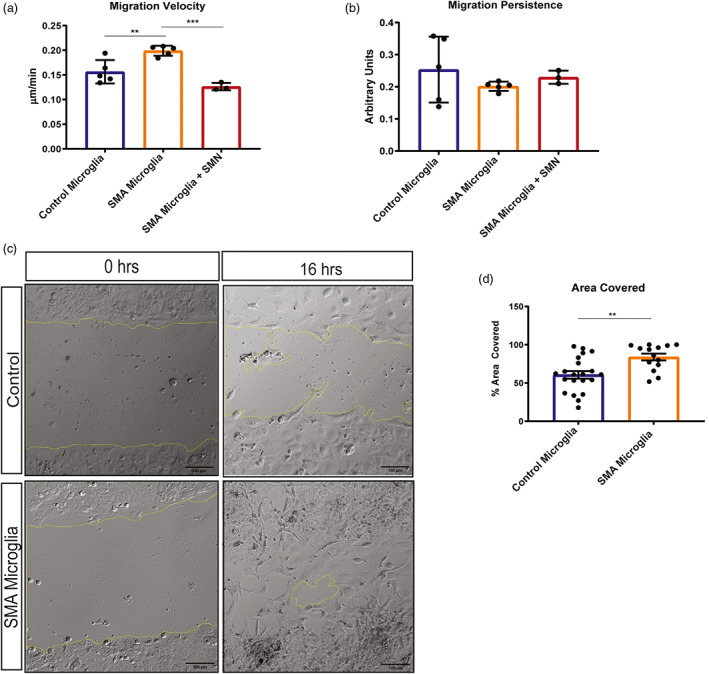
Cell velocity is increased in SMA patient iPSC derived microglia compared to control. Velocity and persistence of control (n = 5; minimum of 14 cells per group) and SMA patient iPSC derived microglia (n = 5; minimum of 14 cells per group) was performed using video analysis over 16 h, with images taken every 10 min. (a) Velocity was significantly increased, **p = .0061. SMN levels were restored in SMA microglia (n = 3; minimum of 50 cells per group) and velocity and analyzed. Transiently expressing SMN restored the cell velocity of SMA microglia to control cell levels. ****p = <.0001. (b) No change in persistence was detected in SMA versus control microglia. Restoring SMN levels did not alter persistence. All groups were analyzed by unpaired t‐test. Values represent mean ± SEM. (c) Representative photomicrographs depicting unaffected and SMA iPSC‐derived microglia plated in a monolayer following a scratch wound. A single wound was administered, and cell chamber was imaged every 10 min for 16 h and (d) total area occupied by migrating cells and, (e) total distance covered over time (rate) was quantified using ImageJ. (area covered **p = .0024; rate **p = .0051). Significance was determined by one‐way ANOVA followed by post hoc Tukey's test for multiple comparisons. Values represent mean ± SEM
Another key microglia function is removing apoptotic cells, cell debris and pathogens by phagocytosis. It was previously reported that C1q expression was increased in SMA, suggesting enhanced microglia phagocytosis of neurons in SMA model mice (Vukojicic et al., 2019). To determine if SMA microglia display greater phagocytic activity, we incubated fluorescent‐tagged beads with SMA and control microglia and measured the fluorescent intensity, indicative of bead internalization, by flow cytometry. We found that human SMA microglia have an enhanced phagocytic capacity compared to control microglia (Figure 6a).
FIGURE 6.
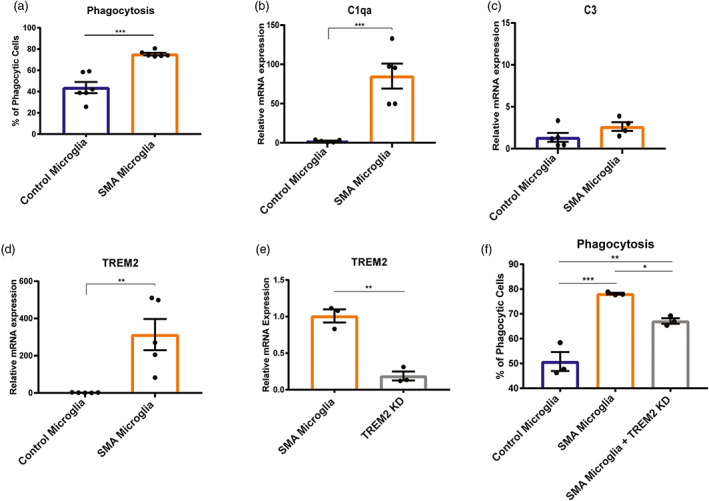
Phagocytosis is upregulated in SMA iPSC derived microglia compared to control. (a). Phagocytosis was analyzed by flow cytometry. (n = 6) ***p = .002. Gene expression analysis of (b) C1qa, (c) C3, and (d) TREM2 were determined by qRT‐PRC. (control n = 5, SMA n = 5) **p = .0058, ***p = .0008 (e) TREM2 was knocked down using siRNA (control n = 3, SMA n = 3) and gene knockdown validated by qRT‐PCR. (f) Phagocytosis of TREM2 knockdown (control n = 3, SMA n = 3) was analyzed by flow cytometry. *p = .0443, **p = .0073, ***p = .0005. All groups were analyzed by one‐way ANOVA followed by post hoc Tukey's test for multiple comparisons. Values represent mean ± SEM
The complement system and triggering receptor expressed on myeloid cells 2 (TREM2) receptor pathways, established pathways that trigger phagocytic activity in CNS immune cells (Noris & Remuzzi, 2013; Yao et al., 2019; Zabel & Kirsch, 2013), were upregulated in transcriptome of SMA microglia compared to control. Both pathways have been implicated in neurodegenerative disease such as ALS, Alzheimer's, frontotemporal dementia, and multiple sclerosis (Crehan et al., 2012; Garcia‐Reitboeck et al., 2018; Kim et al., 2017; Lian et al., 2016). Here, we sought to determine if these pathways were activated in SMA microglia. We first investigated if the complement component 1q (C1qa), a major constituent of the complement C1q protein, was differentially regulated in the human iPSC derived microglia. Indeed, C1qa expression was elevated in the SMA microglia compared to controls (Figure 6b). Expression of C3, a protein critical to complement activation (Noris & Remuzzi, 2013), was similar in SMA and control microglia suggesting that SMN‐deficiency affected expression of select complement proteins (Figure 6c). We next explored whether TREM2, a key mediator of phagocytosis in microglia, contributed to the increased phagocytic activity of SMA microglia. We found that TREM2 transcript was upregulated over 300‐fold in the SMA iPSC‐derived microglia compared to controls confirming findings from the transcriptome analysis (Figure 6d). To determine if the increased phagocytic activity of SMA microglia compared to controls was TREM2‐dependent, we knocked down TREM2 using RNAi (Figure 6e) and measured phagocytosis using a bead‐based assay. Indeed, we observed reduced phagocytic activity of SMA microglia, albeit still higher than control microglia (Figure 6f). Altogether, we showed that cell migration dynamics and phagocytosis are enhanced in SMA microglia, consistent with a role of SMN in regulating key features or microglia reactivity.
2.6. Conditioned media from SMN‐deficient microglia alters motor neuron physiology and morphology
Studies in SMA cell and mouse models have demonstrated increased excitability of SMN‐deficient motor neurons (Arumugam et al., 2017; Gogliotti et al., 2012; Mentis et al., 2011; Quinlan et al., 2019; Sun & Harrington, 2019). However, there are limited studies describing the non‐autonomous and extrinsic contributions to the enhanced excitability of SMN‐deficient neurons (Fletcher et al., 2017; Taylor et al., 2013). We next sought to determine if SMA microglia release factors that regulate motor neuron function. iPSC‐derived motor neurons were generated using an inducible neuronal line as previously described (Fernandopulle et al., 2018). Differentiated neuronal cells were validated by electrophysiology and ChAT expression by immunocytochemistry and qRT‐PCR (Figure S3b,c). Using siRNA targeting SMN, we generated SMN‐deficient motor neurons with approximately 85% reduced SMN expression compared to controls (Figure 7a), which would be comparable to severe phenotypes seen in both human and mouse models. We incubated wild type and SMN‐deficient motor neurons for 24 h in media from control and SMA microglia (Figure 7b). Individual motor neurons were randomly chosen, and electrophysiological recordings were performed under whole‐cell condition of the patch clamp technique (representative traces Figures S4 and S5). Alexa 488 was added to the internal solution to visualize the recorded cells to assess their morphology (glass capillary used for recording and to inject dye for visualization.is denoted by the white arrow, Figure 7b). The control iPSC motor neurons and SMN‐knockdown motor neurons exposed to SMA microglia media showed increased numbers of evoked action potentials in comparison to incubation in control microglia media (Figure 7c,f; Figure S5), highlighting an effect of the SMN‐depleted microglial secretome. The inward sodium and the outward rectifying potassium currents remained similar throughout the groups for control motor neurons (Figure 7d,e), however, the sodium (Figure 7g) and potassium (Figure 7h) currents were exacerbated in SMN‐knockdown motor neurons incubated with SMN microglial media. This marked difference in the current between control and SMN‐knockdown motor neurons highlights the contribution from microglial secreted signals, but also the responsiveness intrinsic to the SMN‐depleted motor neurons to those signals.
FIGURE 7.
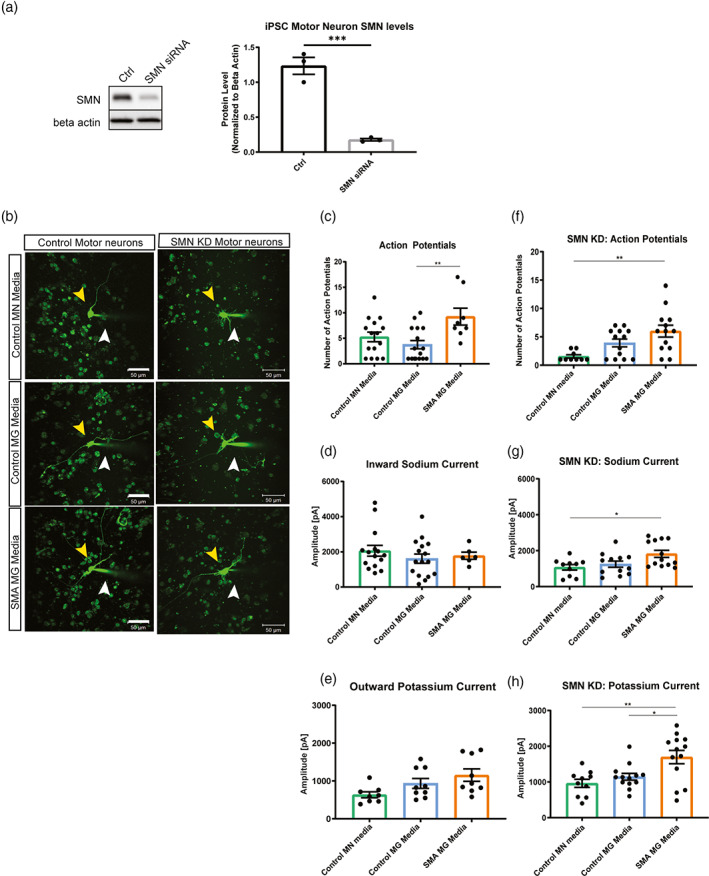
SMA microglia alter motor neuron excitability. (a) SMN knockdown in iPSC motor neurons was validated by western blot (n = 3). Analyzed by t‐test, **p = .0087 (b) control and SMN knock down motor neurons were exposed for 24 h to conditioned media. Representative images of each group were taken. Yellow arrow indicates the motor neuron that was patched. White arrow denotes glass capillary used for recording and to inject dye for visualization. Scale bar = 50 μm. (c) Number of action potentials was recorded in control motor neurons (control motor neuron media n = 15, control microglia media n = 16, and SMA iPSC derived microglia conditioned media n = 8). **p = .0053. (d) Inward sodium current was recorded in control motor neurons exposed 24 h to conditioned media (control motor neuron media n = 15, control microglia media n = 16, and SMA microglia media n = 6). (e) Outward potassium current was recorded in control motor neurons exposed for 24 h to conditioned media (control motor neuron media n = 8, control microglia media n = 9, and SMA microglia media n = 9). (f) Number of action potentials were recorded in SMN knock down motor neurons (control motor neuron media n = 10, control microglia media n = 13, and SMA microglia media n = 13). **p = .0026. (g) Inward sodium current was recorded in SMN knock down motor neurons exposed to conditioned media for 24 h (control motor neuron media n = 10, control microglia media n = 12, and SMA iPSC derived microglia conditioned media n = 13). *p = .0205 (h) outward potassium current was recorded in SMN knockdown motor neurons exposed 24 h to conditioned media (control motor neuron media n = 10, control microglia media n = 13, and SMA microglia media n = 13). *p = .0213 **p = .0034. All groups analyzed by one‐way ANOVA. Values represent mean ± SEM
Previously studies have reported aberrant growth and branching of motor neurons in models of SMA (Akten et al., 2011; Fan & Simard, 2002; McWhorter et al., 2003; Thomson et al., 2012). We next explored if conditioned media from SMA microglia impacted the morphology of motor neurons. Using Sholl analysis, we observed an increase in the number of neurite branching and enhanced extension of control and SMN‐deficient motor neurons treated with conditioned media from SMA microglia, but not conditioned from control microglia (Figure 8a,b). Together, we show that factors released by SMA microglia alter both the morphology and electrophysiological properties of motor neurons, further implicating glia in SMA pathology.
FIGURE 8.
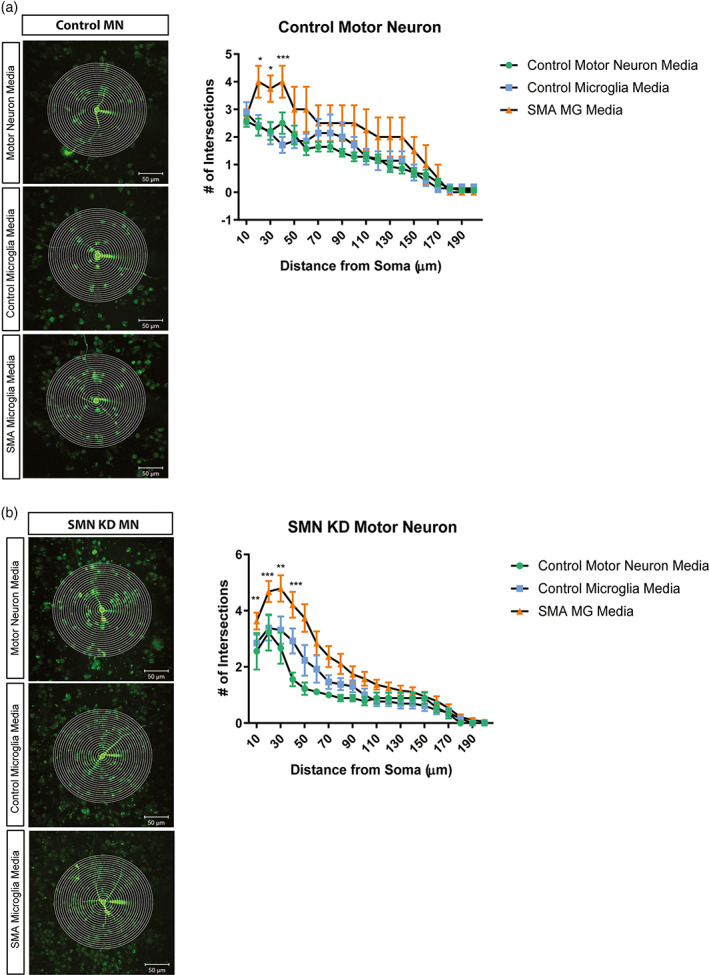
Motor neurons treated with SMA iPSC derived microglia conditioned media result in increased branching. Representative images and plot of Sholl analysis of motor neurons treated with microglia conditioned media. Total number of crossings was counted at start radius of 10 μm, with a step radius 10 μm. Branching was recorded up to 200 μm for all groups. Control motor neurons (a) and SMN deficient motor neurons (b) were treated for 24 h in control motor neuron media (green), unaffected iPSC derived microglia media (blue), and SMA iPSC derived microglia media (orange). Two‐way ANOVA was used followed by post hoc Bonferroni for multiple comparisons to analyze the Sholl plots between groups. Significance was determined when comparing unaffected iPSC derived microglia media to SMA iPSC derived microglia media. (a) *p = .0131, *p = .0108, ***p = <.001 (left to right). (b) **p = .0037, ***p = .0007, **p = .041, ***p = .0006, left to right. Values represent mean ± SEM
3. DISCUSSION
The findings in this study are consistent with previous reports that SMA microglia are reactive at the end stage of the disease in mouse models. However, it was unclear if microglia behave similarly when SMN is deficient in human cells and if peripheral immune cells contribute to the reactivity morphology of SMA microglia. In this report, we show that the inflammatory profile of microglia is minimally affected by peripheral immune cells, which are not found in abnormal numbers in the spinal cord. Using human iPSC‐derived cells, we corroborated that SMN‐deficient human microglia adopt a reactive morphology and transcriptome profile, while demonstrating enhanced migration velocity and phagocytic activity. In addition, SMN knockdown motor neurons exposed to human iPSC‐derived SMA microglial secretome shows altered electrophysiological properties coincident with enhanced branching, suggesting that microglia likely play a complex role in SMA pathology.
3.1. Lymphocytes do not play a major role in SMA disease progression
Recent studies have shown that SMN deficiency impacts the development and spleen size of three major SMA model mice although the role of these peripheral tissues on SMA pathology had not been fully characterized (Deguise & Kothary, 2017; Khairallah et al., 2017). Given the reactive profile of microglia in SMNΔ7 mice, we examined the impact of lymphocytes on the SMA pathology by genetically preventing their lymphocyte maturation using CD4 −/− and Rag1 −/− mice crossed unto the Smn 2B/− transgenic mouse background. Several recent studies have noted that the spleen and thymus were significantly smaller in the SMA mice, potentially contributing to the lack of proper lymphocyte maturation and organ development. By genetically inhibiting the production of mature lymphocytes, we rule out the possibility that developmental abnormalities, such as immature or dysfunctional T cells, could impact the immune response in SMA. We found that depleting T helper cells (CD4 −/− ) or complete lymphocyte ablation (Rag1 −/− ) did not improve the life span of these animals or their ability to perform motor function. Importantly, we show that the reactive profile of SMA microglia is unaltered by loss of lymphocytes.
3.2. Microglia in SMA: Helpful or harmful?
Although there is a growing body of evidence incriminating microglia in neurodegenerative disease pathogenesis, very little is known on how microglia regulate neuronal function and survival in disease models. As the primary CNS line of defense, microglia are particularly sensitive to immediate changes in their micro‐environment and can respond accordingly by either migrating to the site of injury, engulfing the imminent danger, or releasing factors to recruit additional aid. Microglia in SMA mouse models have been reported to adopt a pro‐inflammatory phenotype, with an increase in reactive oxygen species, release of TNFα, as well as increased NFκB activity (Ando et al., 2020). Additionally, SMN deficient microglia have an increased phagocytic behavior in SMA mice compared to controls (Vukojicic et al., 2019). Although our findings show no overall increase in microglia population (Figure 1a), morphological assessment indicates an increased soma and retracted processes, indicative of a reactive morphology in the SMNΔ7 mouse spinal cord.
To test whether this pro‐inflammatory phenotype is conserved in a human model, we differentiated microglia from unaffected and SMA patient iPSCs. We found that human IPSC‐derived microglia had increased phagocytic activity as previously reported in the mice models of SMA. Moreover, we show that SMN‐deficient microglia migrated more readily than control cells. Previously, it has been shown that the SMN protein plays a role in cytoskeletal dynamics (Bowerman et al., 2007; Chaytow et al., 2018). These changes in actin dynamics could account for the alterations in movement and phagocytosis when SMN is deficient in microglia. C1q and TREM2‐dependent phagocytosis pathways have been implicated in neurodegeneration. Similar to reports in SMA mice, we saw increased expression of C1q in our human iPSC microglia suggesting this pathway is activated when SMN‐deficient. In contrast, we saw no change in C3 expression between control and SMA microglia as reported in mice. We suspect that the C3 increase previously documented in the SMA mouse models is not a result of SMN‐deficiency in microglia, but potentially a response to injured neurons or other cells. CNS cells such as astrocytes may be contributing to the activation of microglia. In vitro co‐culture and conditioned media experiments using iPSCs could further clarify the contribution of astrocytes to microglia reactivity in SMA.
TREM2, which stimulates phagocytosis and has been implicated in astrocyte and microglia crosstalk, was highly upregulated in SMN‐deficient microglia. This suggests that SMA microglia may have an enhanced phagocytic response due to both the complement pathway and the TREM2 pathway. Intriguingly, TREM2 has not only been associated with phagocytosis, but migration. Here, we provide evidence that SMN deficient human iPSC derived microglia displayed increased migration velocity, partly consistent with TREM2 upregulation. Based on these findings, we hypothesize that SMA microglia respond quickly to damaged motor neurons, leading to enhanced phagocytosis of spinal cord motor neurons, potentially explaining the rapid deterioration in motor function and clearance of motor neurons early in SMA pathogenesis (Swoboda et al., 2005).
3.3. SMN deficient microglia release factors that influences motor neuron excitability
The complex signaling between motor neurons and microglia in SMA is not well understood. In the healthy condition, microglia help to maintain neuronal homeostasis through phagocytosis and release of inflammatory cytokines. We show that SMA microglia not only display enhanced phagocytic activity but release factors that alter motor neuron morphology and electrical properties. Cultured media from SMA patient derived microglia significantly increased the excitability of control motor neurons. Furthermore, SMN deficient motor neurons, when exposed to SMN deficient microglia conditioned media have increased generation of action potentials due in part to increase inward sodium current, as well as repolarizing outward current. These findings are consistent with altered neuronal excitability in other neurodegenerative diseases including ALS (LoRusso et al., 2019). One potential reason for the altered excitability is increased arborization in both control and SMN deficient motor neurons in the presence of media from SMA microglia. While the underlying molecular pathways remain to be elucidated, this may represent a pathological compensatory mechanism to limit ongoing damage. This is a novel finding as it shows that microglia in SMA may have conflicting effects on motor neuron physiology, which may not be fully appreciated in studies where microglia are completely ablated.
4. CONCLUSION
Overall, we have shown that loss of lymphocytes does not improve SMA disease phenotype and likely does not contribute to the microglia phenotype in SMA mice. SMN‐deficient microglia displayed an altered transcriptome profile, increased mobility and enhanced phagocytic activity, which could modify the SMA phenotype. Further studies using iPSC‐derived microglia and neuron co‐cultures will be critical in establishing the different roles of microglia in SMA disease pathology.
4.1. Methods
4.1.1. Animals
The generation and phenotypic characterization of the SMN‐deficient transgenic animals (Jackson, SMNΔ7;SMN2;Smn −/−) has been described previously (Le et al., 2005). Transgenic and wild type littermate mice were used at postnatal day 5 (PND5) and 10 (PND10). Equal numbers of male and female mice were used. Tail snips from offspring were collected at PND2 and genotyped using PCR assays. The primers used for genotyping were 5′‐CAA ACA CCT GGT ATG GTC AGT C‐3′ and 5′‐GCA CCA CTG CAC AAC AGC CTG‐3′ for the SMN2 transgene, 5′‐GCC TGC GAT GTC GGT TTC TGT GAG G‐3′ and 5′‐CCA GCG CGG ATC GGT CAG ACG‐3′ for the mouse knockout allele, and 5′‐GTG TCT GGG CTG TAG GCA TTG C‐3′ and 5′‐GGC TGT GCC TTT TGG CTT ATC TG‐3′ for the Smn gene.
All procedures complied with the standards for the care and use of animal subjects as stated in the Guide or the Care and Use of Laboratory Animals (NIH publication No. 85–23, revised 1996) and all protocols were approved by the IACUC at the Uniformed Services University of the Health Sciences. Smn 2B/2B mice on the C57BL6 background were mated with C57BL6 wild type female mice.
The Smn 2B/− ;CD4 −/− and Smn 2B/− ;Rag1 −/− mouse models were generated using a complex breeding scheme. Cd4 tm1Mak (https://www.jax.org/strain/002663) and Rag1 tm1Mom (https://www.jax.org/strain/002216) were purchased from the Jackson Laboratory. Establishment of the line was aided by genotyping as indicated by the Jackson Laboratory protocols (Protocol −28818 Generic Cd4, Protocol 27761 Standard PCR assay – Rag1). Mice were kept in stepdown facilities given their potential compromised immune system.
4.1.2. Behavioral testing
Behavioral testing was performed as previously described in (Deguise et al., 2020). Blinding was not possible given the obvious emergence of the SMA phenotype. Briefly, righting reflex followed a similar protocol as previously described in Treat‐NMD neuromuscular network (SOP MD_M.2.2.002). The pen test followed a similar protocol as previously described in Treat‐NMD neuromuscular network (SOP SMA_M.2.1.001). A maximum of 30 s on the pen was defined as a perfect score. The inverted mesh/wire grip test followed a slightly modified protocol than previously described in Treat‐NMD neuromuscular network (SOP SMA_M.2.1.002). Briefly, a mesh grip (hole of 1 mm2) was used from P13 to P19. At P21, the mesh grip was changed to a wire cage top to provide more space for the growing mice to hold on to. Termination of this test was set at 60 s.
4.1.3. Flow cytometry
To assess immune cell populations, spinal cords were extracted from wild type and SMN∆7 mice at PND10. Using an SMN∆7 breeding colony, mice pups were genotyped at PND2. Neural Tissue Dissociation Kit P (Cat No. 130‐097‐628, Miltenyi) was used for isolating a single cell suspension for analysis. Tissue was flushed with HBSS without Ca2+ and incubated with enzymes provided in the kit. The gentleMACS OctoDissociator protocol 37C_ABDK_01 was used to carry out the mechanical and enzymatic digestion (Miltenyi). Cell suspension was filtered over 70um filter and washed with cold HBSS with Ca2+. Sample was centrifuged (300 g/10 min/4°C) and pellet was re‐suspended in PBS supplemented with 0.5% BSA buffer. Afterwards magnetic myelin removal beads II (Miltenyi) were used for negative selection of myelin from the single cell suspension. Cells were blocked with TruStain fcX (Cat. 101,319, Biolegend). CD11b (1:20, Cat. 101,207, Biolegend), CD45 (1:20, Cat. 100,312, Biolegend), and CD3 (1:20, Cat No. 100312, Biolegend) were used for positive fluorescent staining of monocytes, microglia, and T‐cells, respectfully. Cells were read in cell staining buffer on BD Accuri C6 Flow Cytometer CFlow Plus software. Cells were gated on live cells, CD45+, and further gated on CD11b+ and CD3+. Unstained cells were used as a negative control. OneComp eBeads were used for positive controls (Cat. 01‐1111‐42, Invitrogen). All analysis was done using the Flow Jo software (FlowJo, LLC, Ashland, OR).
4.1.4. Immunohistochemistry
To analyze microglia morphology throughout the SMA disease progression, spinal cords (N = 8) were extracted from wild type and SMN∆7 at PND10. Mice pups were genotyped at PND2. Lumbar regions were sectioned into 7‐μm axial segments and probed with primary antibodies for Iba1 (Wako), followed by 3,3′‐Diaminobenzidine (DAB) staining to visual stain. Outcome measures including branching, image fluorescence and soma diameter was counted in ImageJ.
To analyze effects of SMN deficiency impacting cell morphology and phenotype in vitro, SMA and control cells were fixed with 4% paraformaldehyde on round coverslips. Standard immunocytochemistry (ICC) was performed. Cells were blocked for 30 min in PBS/10% Goat serum/0.1% Triton X, at 4°C. Cells were then stained for primary antibodies including Iba1 (Wako) overnight at 4°C. Appropriate secondary antibodies linked to AlexaFluor dyes (Invitrogen, Carlsbad, CA) were incubated on the fixed cells for 1 h at room temperature. Round coverslips were mounted onto slides using mounting media containing DAPI (Vector Labs, Burlingame, CA). Cells were imaged at 20× using a Leica DM4000 B LED and corresponding software (Leica, Wetzlar, Germany).
4.1.5. Cell lines
Induced pluripotent stem cells
Two SMA iPSC lines were used; CS77iSMA1‐nxx from Cedar Sinai from a 2‐year‐old male SMA Type I patient fibroblasts (homozygous deletion of SMN1 and 3 copies of SMN2) and GM23240, purchased from Coriell Institute from a 3 year old male SMA type II patient fibroblast (homozygous deletion of SMN1 and 2 copies of SMN2). Two male control iPSC lines, ATCC‐BYSO112 Human (ATCC) and GM 25256 (Coriell Institute) were used. We obtained doxycycline inducible microglia IPSCs from Dr. Michael Ward's lab (Dräger et al., 2021). Doxycycline inducible motor neuron iPSCs were also a generous donation from the Dr. Ward (Fernandopulle et al., 2018). iPSC lines were maintained in standard 6‐well tissue culture plates were coated with growth factor reduced Matrigel (Cat. 354277, Corning) diluted 1:100 in DMEM/F12 (Gibco) same day as iPSC plating. Maximum passage of 40 was used prior to experimentation. Frozen stocks of iPSCs were thawed and plated on Matrigel coated plates in E8 and E8 Flex (Gibco). iPSC lines were passaged using 0.5 mM EDTA in PBS without CaCl2 and MgCl2 (Life Technologies). Cells were maintained at 37°C, 5% CO2. The iPSCs and differentiated cells were confirmed to be mycoplasma negative.
4.1.6. Transfection
Plasmid transfection of SMN‐GFP (1 μg) on microglia was performed using Lipofectamine 3000 (Invitrogen, cat 100022052) and P3000 (Invitrogen, cat 100022058) for 48 h according to the manufacturer protocols as previously described (Khayrullina et al., 2020). The SMN‐GFP was generously donated by Dr. Greg Matera. SMN siRNA knock‐down was done using 40 pmol of pre‐validated siRNA (IDT, cat 37206943) and RNAi Max (Life Technologies, cat 56532) per manufacturer protocol for 48 h. Opti‐MEM (Gibco, cat 11058‐021) diluent was used in both protocols. Transfection was validated by Western Blot.
4.1.7. iPSC microglia differentiation
SMA and control patient iPSCs were differentiated into microglia as previous described (Haenseler et al., 2017). iPSCs were maintained in six well low adhesion plates (Thermo Scientific) and harvested using Accutase and resuspend cells at 1 × 106 cells/ml in stem cell media supplemented with BMP4 (50 ng/ml, R&D Systems), VEGF (50 ng/ml, R&D Systems), and SCF (20 ng/ml, R&D Systems). Cells were allowed to incubate at 37°C humidified incubator with 5% CO2 for 4 days to allow for the formation of embryoid bodies (EBs). EBs were transplanted to matrigel coated flasks with XVIVO15 media (Lonza) supplemented with M‐CSF (100 ng/ml, R&D Systems) and IL‐3 (25 ng/ml, R&D Systems) and incubated for a week at 37°C, 5% CO2. EBs were embedded and allowed to extent surrounding adherent stromal cells to develop structures similar to cystic yolk‐sac. After 1 week, microglia precursors could be collected from the supernatant and plated on Matrigel‐coated plates in microglia medium consisting of Advance DMEM with N2 (1:100), IL34 (100 ng/ml, R&D Systems), and GM‐CSF (10 ng/ml, R&D Systems). Microglia‐like cells were fed every 3–4 days with microglia media. Cell based assays were carried out after 7 days of incubation.
Doxycycline‐inducible microglia iPSCs were grown in StemFlex until reaching at least 50% confluency and were grown for at least 24 h without ROCK inhibitor. Cells were dissociated and centrifuged as described above and pelleted cells were resuspended in Day 0 differentiation medium containing the following: Essential 8™ Basal Medium (Gibco) as a base, 10 nM ROCK inhibitor, and 2 mg/ml Doxycycline (Clontech). Doxycycline inducible microglia iPSCs were counted and seeded onto double coated plates (Poly‐D‐Lysine‐precoated Bio plates [Corning] + Matrigel coating) with 35,000 cells/well for 6‐well plate. Media was replaced at Day 2 with differentiation media containing Advanced DMEM/F12 Medium (Gibco) as a base medium containing 1× Antibiotic‐Antimycotic (Anti‐Anti) (Gibco), 1× GlutaMAX™ (Gibco), 2 mg/ml doxycycline, 100 ng/ml Human IL34 (Peprotech) and 10 ng/ml Human GM‐CSF (Peprotech). On day 4, the medium was replaced with Microglia medium, containing Advanced DMEM/F12 as a base medium and 1× Anti‐Anti, 1× GlutaMAX, 2 mg/ml doxycycline, 100 ng/ml Human IL‐34 and 10 ng/ml Human GM‐CSF, 50 ng/ml Human M‐CSF (Peprotech) and 50 ng/ml Human TGFB1 (Peprotech). On Day 8, the media was replaced with fresh Microglia medium. Microglia were cultured for up to 12 more days in Microglia medium with full medium changes every 3–4 days. Cells were assayed on day 9.
4.1.8. Motor neuron differentiation
Motor neurons were differentiated per protocol as previously described (Fernandopulle et al., 2018).
4.1.9. Phagocytosis assay
To assess phagocytosis, we used carboxylate‐modified microspheres with an excitation emission spectrum of 580/605 (FluoSpheres, F8821, ThermoFisher Scientific). FluoSpheres were prepared in a 3% BSA, 25 mM Na2HPO4, pH 6.0 solution as previously described (Moritz et al., 2017).5 × 105 control and SMA microglia precursors were plated per well of a six‐well plate. Phagocytosis assay was carried out after a week of differentiation. Briefly, cells were washed with 1× PBS, FluoSpheres (1:67 in Optimem) were added directly to each well, and incubated for 15 min at 37°C. Unstained cells received Optimem only. Cells were then washed three times with 1× PBS and gently scraped and centrifuged to 300 g at 10 min. Pellets were resuspended in FACS buffer in polyethylene tubes and submitted to flow cytometry (BD Accuri C6, BD Biosciences, Franklin Lakes, NJ). Unstained cells were used for gating control. A total of 5000 cells were collected per group with the threshold of 1,000,000 to exclude debris. The mean fluorescence of all viable cells was measured, and data were analyzed using unpaired Student's t tests.
4.1.10. Western blot
To assess protein level, cells were lysed and combined with Laemmli sample buffer containing β‐mercaptoethanol (1:1, Biorad, Cat. 1610737). Combined samples were boiled at 95°C for 5 min. A 35 μg of samples were loaded on 4%–12% Novex WedgeWell Tris‐Glycine Gels and run according to manufacturer's instructions. Blots were transferred to PVDF membranes using Biorad Trans‐blot Turbo. Enhanced chemiluminescence was used to acquire signal.
4.1.11. qPCR
RNA was isolated using the Qiazol and chloroform extraction method, then purified with the Qiagen RNeasy kit (Qiagen, cat 74104). cDNA conversion was performed using a Veriti thermal cycler and a high‐capacity cDNA conversion kit (Applied Biosystems). Gene expression analysis was performed using pre‐validated Taqman probes (Life Technologies). Quantification occurred in a StepOnePlus qRT‐PCR machine (Applied Biosystems) using Universal Probes master mix (BioRad) per manufacturer instructions.
4.1.12. RNA sequencing
Total RNA was quantified via a fluorescence dye‐based methodology (RiboGreen) on a Spectramax Gemini XPS plate reader (Molecular Devices, Mountain View, CA). RNA integrity was assessed using automated capillary electrophoresis on a Fragment Analyzer (Advanced Analytical Technologies, Inc, Santa Clara, CA). Total RNA input of 200 ng was used for library preparation using the TruSeq Stranded mRNA Library Preparation Kit (Illumina, San Diego, CA). Sequencing libraries from 6 SMA and 7 control technical replicates were quantified by PCR using KAPA Library Quantification Kit for NGS (Kapa, Wilmington, MA) and assessed for size distribution on a Fragment Analyzer. Sequencing libraries were pooled and sequenced on a NextSeq. 500 Desktop Sequencer (Illumina) using a NextSeq. 500 High Output Kit v2 with paired‐end reads at 75 bp length. Raw sequencing data was demuxed using bcl2fastq2 Conversion Software 2.17 before alignment using TopHat Alignment v1.0 and differential expression analysis using Cufflinks Assembly & DE v1.1.0 on BaseSpace Onsite (Illumina). Functional enrichment analysis was performed using the PANTHER Classification System (Mi et al., 2019). Significantly overrepresented gene ontology biological processes at p < .01 were adjusted using a Bonferroni correction for multiple testing.
4.1.13. Migration assay
Cellvis 8‐Chamber glass preparation
To assess baseline migration, Matrigel (Corning) ECM was prepared at 1:100 concentration in DMEM/F12 media. A 400 μl of ECM was added to a chamber. The chamber glass was then incubated at room temperature for 30 min, followed by 37°C for 1 h. The chamber glass was stored in an incubator until cells were ready to be transferred to the chambers.
Cell transfer to chamber glass
Media was aspirated off cells, and 3 ml of 1:6000 Invitrogen CellTracker Green CMFDA dye in 1× PBS was added onto the dish. The CellTracker dye was allowed to incubate on the cells at room temperature for 5 min before it was aspirated. Then, 3 ml of cold 0.5 mM EDTA (MediaTech, Inc.) was added to the dish. The dish was placed at 4°C for 10 min. After, EDTA was aspirated, and 1 ml of macrophage media was added to the dish. The dish was placed at an angle, then a Corning Cell Lifter was used to scrape cells into the media. The cells in media were then transferred to a 15 ml NEST conical and vortexed. A cell count was taken with a Marienfeld hemocytometer. A total of 5000 cells were added onto each ECM component in a Cellvis 8‐Chamber glass slide.
Live cell microscopy
The chamber glass was transferred to an environmental chamber attached to an Olympus IX83 Inverted Light Microscope. The environmental chamber is INU (Incubation System for Microscopes) Tokai Hit, and the stage conditions were set to 40°C Top Heater, 39.5°C Stage Heater, and 38°C Bath Heater. DI water was used for the bath. The camera used was Hamamatsu digital camera C13440‐20CU. Excelitas Technologies X‐cite 120 LED Boost was used for fluorescent imaging. Olympus is based in Tokyo, Japan, Hamamatsu is based in Hamamatsu, Japan, Tokai Hit is based in Fujinomiya, Shizuoka Prefecture, and Excelitas Technologies is based in Waltham, MA. The chamber glass was left in the environmental chamber for 2 h to allow the microglia to spread and settle on the ECM components. Olympus CellSens software was then used to select positions on the chamber glass. Both Relief Contrast and GFP images were taken for each position. Each condition had seven positions selected, one picture was taken every 10 min, over a 16‐h time course.
Migration assay analysis
Olympus video files were opened in ImageJ with the OlympusViewer Plugin. The GFP and Relief Contrast channels were split and only the GFP channel was utilized. The TrackMate Plugin was then used for automated tracking of the macrophages, which are recognized by their contrast from the background. Each frame the cell moves, the program creates a track throughout all 97 frames. From this, the data output includes cell number, frame number, and X‐Y position. These values are saved and utilized by the Chemotaxis Tool plugin, which calculates velocity each cell based on the track generated by TrackMate, as well as persistence (d/T), which is the propensity of a cell to continue in the direction that it is already moving instead of changing directions. “d” represents the displacement of a cell (in microns) as measured from its starting and ending position. “T” is the total length (in microns) that the cell traveled during the imaging experiment. Therefore, if d/T = 1 the cell is moving in a straight line without turning. The closer the d/T value is to 1, the greater the persistence. Velocity and Persistence values for all the cells in each condition are collected and the mean is determined for each category.
Scratch wound assay
Cells were plated on the previously prepared Cellvis 8‐Chamber Glass to 100 percent confluency. Cells were serum and cytokine starved 24 h prior to running assay. Day of experiment, 1:1000 Invitrogen CellTracker Red dye was administered in cell media. The CellTracker dye was allowed to incubate on the cells at 37°C for 1 h before. After incubation, media was aspirated, and a scratch wound was made using a pipette tip. The Hamamatsu digital camera C13440‐20CU and Excelitas Technologies X‐cite 120 LED Boost was used for fluorescent imaging. The chamber glass was left in the environmental chamber for 30 min prior to selecting positions (Olympus CellSens software). Both Relief Contrast and GFP images were taken for each position. Each condition had three positions selected, one picture was taken every 10 min, over a 16‐h time course.
4.2. Electrophysiological recordings
Motor neurons were differentiated on round glass coverslips in 24 well plates at a plating density of 250,000 cells/well for 14 days. Cells were pre‐treated 24 h before recording in desired media condition and the coverslips were transferred to the recording chamber on a Zeiss 7 MP microscope system. (Zeiss, Oberkochen, Germany). Electrophysiological recordings were performed with an Axon Multicalmp 700B amplifier and 1440A Digidata unit connected to a PC running pClamp10 software (Molecular Devices, San Jose, CA). Electrodes used for recording were pulled on a horizontal pipette puller (P‐97; Sutter Instrument Company, Novato, CA) from thin wall borosilicate glass capillaries with filament (Warner Instruments, Holliston, MA) to a pipette resistance of 3–6 M Ohm when used with the solutions below. Cells were perfused in the recording chamber with an extracellular bath solution (135 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 20 mM glucose [dextrose anhydrous] and 10 mM HEPES, adjusted to pH of 7.3 with 1 M NaOH) throughout the duration of the experiment. The pipette solution contained in mM: K‐gluconate 130, KCl 15, HEPES 5, EGTA 1, Mg‐ATP 4, and Na‐GTP 0.3. The pH was adjusted to 7.3 with a 1 M KOH solution. Additionally, 15 μM Alexa Fluor 488 Hydrazide, sodium salt (Thermo Fisher Scientific) was added to the internal solution for visualization of recorded cells. Cells were recorded in whole–cell mode which was achieved by brief suction pulses after obtaining a giga Ohm seal. Cells with an access resistance over 50 M Ohm were discarded. For current voltage relationship measurements cells were held at −70 mV and the membrane potential was stepped in 5 mV increments from −50 to +45 mV for 100 ms. To gauge cell excitability, cells were recorded under current clamp conditions with a current injection to hold the membrane potential at −70 mV. A pulse protocol depolarized the cell successively by current injections in 2–5 pA steps to elicit action potentials (AP). An active voltage response was deemed an AP if it crossed 0 mV. The maximum number of APs per cell was determined. After recording electrophysiological measures, small z‐stacks at optimal z‐step size were acquired in ZEN software (Zeiss) with the infrared laser (Vision 2, Coherent, Santa Clara, CA) tuned to 800 nm and utilizing a 40×, 1.0 NA water immersion objective. Maximum intensity projections of the acquired stacks were used to determine morphology of the recorded cells.
4.3. Motor neuron morphological assessment
The number of neurites branching from the soma was determined by Sholl analysis using the Image J software (NIH, Bethesda, MD). All tracings and analysis were performed with the experimenter blinded to the condition. Images were acquired from the electrophysiology assessments for all groups. Conditions where the dye was not properly injected into the soma prior to imaging was discarded. Briefly, images were converted to grayscale and scaled to appropriate size bar. The Sholl analysis was used under the following parameters from the chosen soma center point: start radius 10 μm, step radius 10 μm. Branching was recorded up to 200 μm for all groups.
4.3.1. Statistics
All data was analyzed using the GraphPad Prism software (GraphPad, San Diego, CA). All behavior data was analyzed by two‐way ANOVA followed by Sidak's multiple comparisons test to determine significance between groups. Flow cytometry and cell culture data between a control and SMA group was analyzed with a student's t‐test. Two‐way ANOVA was used followed by post hoc Bonferroni for Multiple Comparisons to analyze the Sholl data between groups. Student t‐test was used to analyze the mean dendrite number between groups. For all statistical tests, a p value of <.05 will be considered significant.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTION
Guzal Khayrullina, Zaida A. Alipio‐Gloria, Marc‐Olivier Deguise, Sabrina Gagnon, Lucia Chehade, Matt Stinson, Natalya Belous, and Fritz W. Lischka performed and analyzed experiments. Guzal Khayrullina, Zaida A. Alipio‐Gloria, Marc‐Olivier Deguise, Jeremy Rotty, Clifton L. Dalgard, Kristen Johnson, Rashmi Kothary and Barrington G. Burnett designed experiments and interpreted data. Kristen Johnson, Rashmi Kothary and B.G.B supervised the study and provided funding. Guzal Khayrullina and Zaida A. Alipio‐Gloria wrote the manuscript, Guzal Khayrullina and Barrington G. Burnett created figures and legends, and Guzal Khayrullina, Zaida A. Alipio‐Gloria, Marc‐Olivier Deguise, Kristen Johnson, Rashmi Kothary, and Barrington G. Burnett edited the manuscript. All authors approved the manuscript.
Supporting information
Figure S1 Loss of mature lymphocytes in Smn 2B/− ;CD4 −/− mice does not correct SMA microglia reactive morphology. A. Spinal cord sections from L1‐L5 of PND 19 CD4 −/− Het (Smn 2B/+ ;CD4 −/− ), CD4 −/− SMA (Smn 2B/− ;CD4 −/− ), Rag −/− Het (Smn 2B/+ ;Rag1 −/− ), and Rag −/− SMA (Smn 2B/− ;Rag1 −/− ) were immune‐stained for Iba‐1. DAB staining was used to visualize antibody. Representative images of Iba1+ cells were taken from each group. Scale bars = 200 μm. B. Soma Size, and number of processes were analyzed using ImageScope. Values represent mean ± SEM. * < 0.05, ****p = <.0001, 150 Iba1+ cells were counted from 3 mice per group.
Figure S2 iPSC differentiated microglia were validated for CNS microglia specific markers A. Representative images shown of microglia differentiation. B. iPSC‐microglia were immune‐stained for Iba1, a marker for microglia. C. SMN protein levels in control and SMA patient iPSC lines were validated (n = 5/group). ****p = <.0001. D. Gene expression for TREM2 and TMEM119 was analyzed in iPSCs and microglia 10 days after differentiation. (n = 5/group) *p = .0112, ****p = <.0001. E. HLA‐A,B,C, CD45, and CD11b surface marker expression was validated by flow cytometry (n = 4/group). Representative histogram is shown per marker. ****p = <.0001. All groups were analyzed by unpaired t test. Bars show mean ± SEM.
Figure S3 iPSC‐derived motor neuron was validated at 10 days after differentiation. A. Representative image indicates a motor neuron from which recording was taken. Scale bar = 50 μm. Family of whole cell currents demonstrating the presence of inward sodium current and sustained outward rectifying potassium current and action potential generation. Cell was held at −70 mV and dotted line indicates 0 mV level. Scale bars show 1 nA by 20 ms and 20 mV by 100 ms, respectively. B. Differentiated cells stain positive for choline acetyltransferase (ChAT), characteristic of cholinergic neurons and late motor neuron marker. DAPI was used to visualize cell nuclei. Scale bar = 25 μm. C. ChAT gene expression was confirmed in 3 independent motor neuron differentiations by RT‐PCR. (n = 3/group) ****p = <.0001. Values represent mean ± SEM.
Figure S4 Representative traces of currents recorded from iPSC‐derived motor neurons in the presence and absence of control and microglia conditioned media.
Figure S5 Action potential patterns in induced neuronal cells across different culture conditions. Current clamp recordings for neuronal culture cells grown in control microglia medium (MG medium), SMA microglia conditioned medium (SMA medium) and SMN deficient motor neurons (knock down; “kd”), grown in control microglia medium (kd MG medium) or SMA microglia conditioned medium (kd SMA medium). Responses to 1 s depolarizing steps induced by a current injection of the strength shown under the individual traces. Dashed line indicates 0 mV, the criterion of action potential generation. The maximum number of APs that could be elicited in a given cell (row 4) is graphed in Figure 7 C & F.
Table S1 Sequencing quality metrics for mRNA‐seq transcriptome analysis of wild type and SMA microglia.
ACKNOWLEDGMENTS
We thank Drs. Martin Doughty and Thomas Flagg for fruitful discussions regarding the experimental design and interpretation of electrophysiological data. This work was supported by grants from CureSMA, National Institute of Neurological Disorders and Stroke (R01NS091575) to Barrington G. Burnett, RO1GM134104 (NIGMS) and USU startup funds to Jeremy Rotty and National Institute of Neurological Disorders and Stroke (R01NS094721) to Kristen Johnson. Rashmi Kothary was supported by a grant from Canadian Institutes of Health Research (CIHR) (grant number PJT‐156379). Marc‐Olivier Deguise was supported by a Frederick Banting and Charles Best CIHR Doctoral Research Award and Lucia Chehade by a Vanier CIHR Doctoral Research Award. Table of Contents image was created with Biorender.com. The opinions and assertions expressed herein are those of the author(s) and do not reflect the official policy or position of the Uniformed Services University or the Department of Defense.
Khayrullina, G. , Alipio‐Gloria, Z. A. , Deguise, M.‐O. , Gagnon, S. , Chehade, L. , Stinson, M. , Belous, N. , Bergman, E. M. , Lischka, F. W. , Rotty, J. , Dalgard, C. L. , Kothary, R. , Johnson, K. A. , & Burnett, B. G. (2022). Survival motor neuron protein deficiency alters microglia reactivity. Glia, 70(7), 1337–1358. 10.1002/glia.24177
Guzal Khayrullina and Zaida A. Alipio‐Gloria are co‐first authors.
Funding information Vanier CIHR Doctoral Research Award; Frederick Banting and Charles Best CIHR Doctoral Research Award; Canadian Institutes of Health Research, Grant/Award Number: PJT‐156379; USU; NIGMS, Grant/Award Numbers: RO1GM134104, R01NS091575; National Institute of Neurological Disorders and Stroke, Grant/Award Number: R01NS094721; CureSMA
Contributor Information
Rashmi Kothary, Email: rkothary@ohri.ca.
Kristen A. Johnson, Email: kajohnson@scripps.edu.
Barrington G. Burnett, Email: barrington.burnett@usuhs.edu.
DATA AVAILABILITY STATEMENT
The data supporting the findings of this study are not applicable for depositing into a public database; however, raw data are available from the corresponding author upon reasonable request.
REFERENCES
- Akten, B. , Kye, M. J. , Hao le, T. , Wertz, M. H. , Singh, S. , Nie, D. , & Sahin, M. (2011). Interaction of survival of motor neuron (SMN) and HuD proteins with mRNA cpg15 rescues motor neuron axonal deficits. Proceedings of the National Academy of Sciences of the United States of America, 108(25), 10337–10342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alliot, F. , Godin, I. , & Pessac, B. (1999). Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Research. Developmental Brain Research, 117(2), 145–152. [DOI] [PubMed] [Google Scholar]
- Ando, S. , Osanai, D. , Takahashi, K. , Nakamura, S. , Shimazawa, M. , & Hara, H. (2020). Survival motor neuron protein regulates oxidative stress and inflammatory response in microglia of the spinal cord in spinal muscular atrophy. Journal of Pharmacological Sciences, 144(4), 204–211. [DOI] [PubMed] [Google Scholar]
- Arumugam, S. , Garcera, A. , Soler, R. M. , & Tabares, L. (2017). Smn‐deficiency increases the intrinsic excitability of motoneurons. Frontiers in Cellular Neuroscience, 11, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanquie, O. , & Bradke, F. (2018). Cytoskeleton dynamics in axon regeneration. Current Opinion in Neurobiology, 51, 60–69. [DOI] [PubMed] [Google Scholar]
- Bowerman, M. , Murray, L. M. , Beauvais, A. , Pinheiro, B. , & Kothary, R. (2012). A critical SMN threshold in mice dictates onset of an intermediate spinal muscular atrophy phenotype associated with a distinct neuromuscular junction pathology. Neuromuscular Disorders, 22(3), 263–276. [DOI] [PubMed] [Google Scholar]
- Bowerman, M. , Shafey, D. , & Kothary, R. (2007). Smn depletion alters profilin II expression and leads to upregulation of the RhoA/ROCK pathway and defects in neuronal integrity. Journal of Molecular Neuroscience, 32(2), 120–131. [DOI] [PubMed] [Google Scholar]
- Boyer, J. G. , Deguise, M. O. , Murray, L. M. , Yazdani, A. , De Repentigny, Y. , Boudreau‐Lariviere, C. , & Kothary, R. (2014). Myogenic program dysregulation is contributory to disease pathogenesis in spinal muscular atrophy. Human Molecular Genetics, 23(16), 4249–4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricceno, K. V. , Martinez, T. , Leikina, E. , Duguez, S. , Partridge, T. A. , Chernomordik, L. V. , & Burnett, B. G. (2014). Survival motor neuron protein deficiency impairs myotube formation by altering myogenic gene expression and focal adhesion dynamics. Human Molecular Genetics, 23(18), 4745–4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochard, V. , Combadiere, B. , Prigent, A. , Laouar, Y. , Perrin, A. , Beray‐Berthat, V. , & Hunot, S. (2009). Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. The Journal of Clinical Investigation, 119(1), 182–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghes, A. H. , & Beattie, C. E. (2009). Spinal muscular atrophy: Why do low levels of survival motor neuron protein make motor neurons sick? Nature Reviews. Neuroscience, 10(8), 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervero, C. , Blasco, A. , Tarabal, O. , Casanovas, A. , Piedrafita, L. , Navarro, X. , & Caldero, J. (2018). Glial activation and central synapse loss, but not motoneuron degeneration, are prevented by the Sigma‐1 receptor agonist PRE‐084 in the Smn2B/− mouse model of spinal muscular atrophy. Journal of Neuropathology and Experimental Neurology, 77(7), 577–597. [DOI] [PubMed] [Google Scholar]
- Chaplin, D. D. (2010). Overview of the immune response. The Journal of Allergy and Clinical Immunology, 125(2), S3–S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaytow, H. , Huang, Y. T. , Gillingwater, T. H. , & Faller, K. M. E. (2018). The role of survival motor neuron protein (SMN) in protein homeostasis. Cellular and Molecular Life Sciences, 75(21), 3877–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou, R. C. , Kim, N. D. , Sadik, C. D. , Seung, E. , Lan, Y. , Byrne, M. H. , & Luster, A. D. (2010). Lipid‐cytokine‐chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity, 33(2), 266–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crehan, H. , Hardy, J. , & Pocock, J. (2012). Microglia, Alzheimer's disease, and complement. International Journal of Alzheimer's Disease, 2012, 983640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deguise, M. O. , Beauvais, A. , Schneider, B. L. , & Kothary, R. (2020). Blood flow to the spleen is altered in a mouse model of spinal muscular atrophy. Journal of Neuromuscular Diseases, 7(3), 315–322. [DOI] [PubMed] [Google Scholar]
- Deguise, M. O. , De Repentigny, Y. , McFall, E. , Auclair, N. , Sad, S. , & Kothary, R. (2017). Immune dysregulation may contribute to disease pathogenesis in spinal muscular atrophy mice. Human Molecular Genetics, 26(4), 801–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deguise, M. O. , De Repentigny, Y. , Tierney, A. , Beauvais, A. , Michaud, J. , Chehade, L. , & Kothary, R. (2020). Motor transmission defects with sex differences in a new mouse model of mild spinal muscular atrophy. eBioMedicine, 55, 102750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deguise, M. O. , & Kothary, R. (2017). New insights into SMA pathogenesis: Immune dysfunction and neuroinflammation. Annals of Clinical Translational Neurology, 4(7), 522–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiDonato, C. J. , Lorson, C. L. , De Repentigny, Y. , Simard, L. , Chartrand, C. , Androphy, E. J. , & Kothary, R. (2001). Regulation of murine survival motor neuron (Smn) protein levels by modifying Smn exon 7 splicing. Human Molecular Genetics, 10(23), 2727–2736. [DOI] [PubMed] [Google Scholar]
- Dräger, N. M. , Sattler, S. M. , Huang, C. T.‐L. , Teter, O. M. , Leng, K. , Hashemi, S. H. , & Kampmann, M. (2021). A CRISPRi/a platform in iPSC‐derived microglia uncovers regulators of disease states. bioRxiv, 2006, 448639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshraghi, M. , McFall, E. , Gibeault, S. , & Kothary, R. (2016). Effect of genetic background on the phenotype of the Smn2B/− mouse model of spinal muscular atrophy. Human Molecular Genetics, 25(20), 4494–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, L. , & Simard, L. R. (2002). Survival motor neuron (SMN) protein: Role in neurite outgrowth and neuromuscular maturation during neuronal differentiation and development. Human Molecular Genetics, 11(14), 1605–1614. [DOI] [PubMed] [Google Scholar]
- Fan, Y. , Xie, L. , & Chung, C. Y. (2017). Signaling pathways controlling microglia chemotaxis. Molecules and Cells, 40(3), 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandopulle, M. S. , Prestil, R. , Grunseich, C. , Wang, C. , Gan, L. , & Ward, M. E. (2018). Transcription factor‐mediated differentiation of human iPSCs into neurons. Current Protocols in Cell Biology, 79(1), e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher, E. V. , Simon, C. M. , Pagiazitis, J. G. , Chalif, J. I. , Vukojicic, A. , Drobac, E. , & Mentis, G. Z. (2017). Reduced sensory synaptic excitation impairs motor neuron function via Kv2.1 in spinal muscular atrophy. Nature Neuroscience, 20(7), 905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita, H. , Tanaka, J. , Toku, K. , Tateishi, N. , Suzuki, Y. , Matsuda, S. , & Maeda, N. (1996). Effects of GM‐CSF and ordinary supplements on the ramification of microglia in culture: A morphometrical study. Glia, 18(4), 269–281. [DOI] [PubMed] [Google Scholar]
- Furuichi, K. , Gao, J. L. , Horuk, R. , Wada, T. , Kaneko, S. , & Murphy, P. M. (2008). Chemokine receptor CCR1 regulates inflammatory cell infiltration after renal ischemia‐reperfusion injury. Journal of Immunology, 181(12), 8670–8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Reitboeck, P. , Phillips, A. , Piers, T. M. , Villegas‐Llerena, C. , Butler, M. , Mallach, A. , & Pocock, J. M. (2018). Human induced pluripotent stem cell‐derived microglia‐like cells harboring TREM2 missense mutations show specific deficits in phagocytosis. Cell Reports, 24(9), 2300–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogliotti, R. G. , Quinlan, K. A. , Barlow, C. B. , Heier, C. R. , Heckman, C. J. , & Didonato, C. J. (2012). Motor neuron rescue in spinal muscular atrophy mice demonstrates that sensory‐motor defects are a consequence, not a cause, of motor neuron dysfunction. The Journal of Neuroscience, 32(11), 3818–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gombash, S. E. , Cowley, C. J. , Fitzgerald, J. A. , Iyer, C. C. , Fried, D. , McGovern, V. L. , & Foust, K. D. (2015). SMN deficiency disrupts gastrointestinal and enteric nervous system function in mice. Human Molecular Genetics, 24(19), 5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groen, E. J. N. (2018). Future avenues for therapy development for spinal muscular atrophy. Expert Opinion on Drug Discovery, 13(10), 899–902. [DOI] [PubMed] [Google Scholar]
- Haenseler, W. , Sansom, S. N. , Buchrieser, J. , Newey, S. E. , Moore, C. S. , Nicholls, F. J. , & Cowley, S. A. (2017). A highly efficient human pluripotent stem cell microglia model displays a neuronal‐co‐culture‐specific expression profile and inflammatory response. Stem Cell Reports, 8(6), 1727–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond, T. R. , Marsh, S. E. , & Stevens, B. (2019). Immune signaling in neurodegeneration. Immunity, 50(4), 955–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayhurst, M. , Wagner, A. K. , Cerletti, M. , Wagers, A. J. , & Rubin, L. L. (2012). A cell‐autonomous defect in skeletal muscle satellite cells expressing low levels of survival of motor neuron protein. Developmental Biology, 368, 323–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman, S. , Izzy, S. , Sen, P. , Morsett, L. , & El Khoury, J. (2018). Microglia in neurodegeneration. Nature Neuroscience, 21(10), 1359–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua, Y. , Sahashi, K. , Rigo, F. , Hung, G. , Horev, G. , Bennett, C. F. , & Krainer, A. R. (2011). Peripheral SMN restoration is essential for long‐term rescue of a severe spinal muscular atrophy mouse model. Nature, 478(7367), 123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khairallah, M. T. , Astroski, J. , Custer, S. K. , Androphy, E. J. , Franklin, C. L. , & Lorson, C. L. (2017). SMN deficiency negatively impacts red pulp macrophages and spleen development in mouse models of spinal muscular atrophy. Human Molecular Genetics, 26(5), 932–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khayrullina, G. , Moritz, K. E. , Schooley, J. F. , Fatima, N. , Viollet, C. , McCormack, N. M. , & Burnett, B. G. (2020). SMN‐deficiency disrupts SERCA2 expression and intracellular Ca(2+) signaling in cardiomyocytes from SMA mice and patient‐derived iPSCs. Skeletal Muscle, 10(1), 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl, K. A. , de Rivero Vaccari, J. P. , Dietrich, W. D. , Popovich, P. G. , & Keane, R. W. (2014). Pattern recognition receptors and central nervous system repair. Experimental Neurology, 258, 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S. M. , Mun, B. R. , Lee, S. J. , Joh, Y. , Lee, H. Y. , Ji, K. Y. , & Kang, H. S. (2017). TREM2 promotes Abeta phagocytosis by upregulating C/EBPalpha‐dependent CD36 expression in microglia. Scientific Reports, 7(1), 11118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb, S. J. , Battle, D. J. , & Dreyfuss, G. (2007). Molecular functions of the SMN complex. Journal of Child Neurology, 22(8), 990–994. [DOI] [PubMed] [Google Scholar]
- Kozlowski, C. , & Weimer, R. M. (2012). An automated method to quantify microglia morphology and application to monitor activation state longitudinally in vivo. PLoS One, 7(2), e31814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le, T. T. , Pham, L. T. , Butchbach, M. E. , Zhang, H. L. , Monani, U. R. , Coovert, D. D. , & Burghes, A. H. (2005). SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full‐length SMN. Human Molecular Genetics, 14(6), 845–857. [DOI] [PubMed] [Google Scholar]
- Lefebvre, S. , Burglen, L. , Reboullet, S. , Clermont, O. , Burlet, P. , & Viollet, L. (1995). Identification and characterization of a spinal muscular atrophy‐determining gene. Cell, 80(1), 155–165. [DOI] [PubMed] [Google Scholar]
- Li, Q. , & Barres, B. A. (2018). Microglia and macrophages in brain homeostasis and disease. Nature Reviews. Immunology, 18(4), 225–242. [DOI] [PubMed] [Google Scholar]
- Lian, H. , Litvinchuk, A. , Chiang, A. C. , Aithmitti, N. , Jankowsky, J. L. , & Zheng, H. (2016). Astrocyte‐microglia Cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer's disease. The Journal of Neuroscience, 36(2), 577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling, K. K. , Lin, M. Y. , Zingg, B. , Feng, Z. , & Ko, C. P. (2010). Synaptic defects in the spinal and neuromuscular circuitry in a mouse model of spinal muscular atrophy. PLoS One, 5(11), e15457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Q. , Fischer, U. , Wang, F. , & Dreyfuss, G. (1997). The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell, 90(6), 1013–1021. [DOI] [PubMed] [Google Scholar]
- Lively, S. , & Schlichter, L. C. (2013). The microglial activation state regulates migration and roles of matrix‐dissolving enzymes for invasion. Journal of Neuroinflammation, 10, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoRusso, E. , Hickman, J. J. , & Guo, X. (2019). Ion channel dysfunction and altered motoneuron excitability in ALS. Neurological Disorders & Epilepsy Journal, 3(2), 124. [PMC free article] [PubMed] [Google Scholar]
- Machado‐Santos, J. , Saji, E. , Troscher, A. R. , Paunovic, M. , Liblau, R. , Gabriely, G. , & Lassmann, H. (2018). The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue‐resident CD8+ T lymphocytes and B cells. Brain, 141(7), 2066–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, J. E. , Nguyen, T. T. , Grunseich, C. , Nofziger, J. H. , Lee, P. R. , Fields, D. , & Foran, E. (2017). Decreased motor neuron support by SMA astrocytes due to diminished MCP1 secretion. The Journal of Neuroscience, 37(21), 5309–5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massenet, S. , Pellizzoni, L. , Paushkin, S. , Mattaj, I. W. , & Dreyfuss, G. (2002). The SMN complex is associated with snRNPs throughout their cytoplasmic assembly pathway. Molecular and Cellular Biology, 22(18), 6533–6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack, N. M. , Villalon, E. , Viollet, C. , Soltis, A. R. , Dalgard, C. L. , Lorson, C. L. , & Burnett, B. G. (2021). Survival motor neuron deficiency slows myoblast fusion through reduced myomaker and myomixer expression. Journal of Cachexia, Sarcopenia and Muscle, 12(4), 1098–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhorter, M. L. , Monani, U. R. , Burghes, A. H. , & Beattie, C. E. (2003). Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. The Journal of Cell Biology, 162(5), 919–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell, J. R. , Al‐Zaidy, S. , Shell, R. , Arnold, W. D. , Rodino‐Klapac, L. R. , Prior, T. W. , & Kaspar, B. K. (2017). Single‐dose gene‐replacement therapy for spinal muscular atrophy. The New England Journal of Medicine, 377(18), 1713–1722. [DOI] [PubMed] [Google Scholar]
- Menke, L. A. , Poll‐The, B. T. , Clur, S. A. , Bilardo, C. M. , van der Wal, A. C. , Lemmink, H. H. , & Cobben, J. M. (2008). Congenital heart defects in spinal muscular atrophy type I: A clinical report of two siblings and a review of the literature. American Journal of Medical Genetics. Part A, 146A(6), 740–744. [DOI] [PubMed] [Google Scholar]
- Mentis, G. Z. , Blivis, D. , Liu, W. , Drobac, E. , Crowder, M. E. , Kong, L. , & O'Donovan, M. J. (2011). Early functional impairment of sensory‐motor connectivity in a mouse model of spinal muscular atrophy. Neuron, 69(3), 453–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercuri, E. , Darras, B. T. , Chiriboga, C. A. , Day, J. W. , Campbell, C. , Connolly, A. M. , & Group, C. S . (2018). Nusinersen versus sham control in later‐onset spinal muscular atrophy. The New England Journal of Medicine, 378(7), 625–635. [DOI] [PubMed] [Google Scholar]
- Messina, S. , & Sframeli, M. (2020). New treatments in spinal muscular atrophy: Positive results and new challenges. Journal of Clinical Medicine, 9(7), 2222. 10.3390/jcm9072222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi, H. , Muruganujan, A. , Huang, X. , Ebert, D. , Mills, C. , Guo, X. , & Thomas, P. D. (2019). Protocol update for large‐scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nature Protocols, 14(3), 703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mietelska‐Porowska, A. , & Wojda, U. (2017). T lymphocytes and inflammatory mediators in the interplay between brain and blood in Alzheimer's disease: Potential pools of new biomarkers. Journal of Immunology Research, 2017, 4626540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombaerts, P. , Iacomini, J. , Johnson, R. S. , Herrup, K. , Tonegawa, S. , & Papaioannou, V. E. (1992). RAG‐1‐deficient mice have no mature B and T lymphocytes. Cell, 68(5), 869–877. [DOI] [PubMed] [Google Scholar]
- Monani, U. R. (2005). Spinal muscular atrophy: A deficiency in a ubiquitous protein; a motor neuron‐specific disease. Neuron, 48(6), 885–896. [DOI] [PubMed] [Google Scholar]
- Moritz, K. E. , McCormack, N. M. , Abera, M. B. , Viollet, C. , Yauger, Y. J. , Sukumar, G. , & Burnett, B. G. (2017). The role of the immunoproteasome in interferon‐gamma‐mediated microglial activation. Scientific Reports, 7(1), 9365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noris, M. , & Remuzzi, G. (2013). Overview of complement activation and regulation. Seminars in Nephrology, 33(6), 479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohuchi, K. , Funato, M. , Ando, S. , Inagaki, S. , Sato, A. , Kawase, C. , & Hara, H. (2019). Impairment of oligodendrocyte lineages in spinal muscular atrophy model systems. Neuroreport, 30(5), 350–357. [DOI] [PubMed] [Google Scholar]
- O'Meara, R. W. , Cummings, S. E. , De Repentigny, Y. , McFall, E. , Michalski, J. P. , Deguise, M. O. , & Kothary, R. (2017). Oligodendrocyte development and CNS myelination are unaffected in a mouse model of severe spinal muscular atrophy. Human Molecular Genetics, 26(2), 282–292. [DOI] [PubMed] [Google Scholar]
- Park, G. H. , Maeno‐Hikichi, Y. , Awano, T. , Landmesser, L. T. , & Monani, U. R. (2010). Reduced survival of motor neuron (SMN) protein in motor neuronal progenitors functions cell autonomously to cause spinal muscular atrophy in model mice expressing the human centromeric (SMN2) gene. The Journal of Neuroscience, 30(36), 12005–12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patitucci, T. N. , & Ebert, A. D. (2016). SMN deficiency does not induce oxidative stress in SMA iPSC‐derived astrocytes or motor neurons. Human Molecular Genetics, 25(3), 514–523. [DOI] [PubMed] [Google Scholar]
- Paushkin, S. , Gubitz, A. K. , Massenet, S. , & Dreyfuss, G. (2002). The SMN complex, an assemblyosome of ribonucleoproteins. Current Opinion in Cell Biology, 14(3), 305–312. [DOI] [PubMed] [Google Scholar]
- Pellizzoni, L. , Charroux, B. , & Dreyfuss, G. (1999). SMN mutants of spinal muscular atrophy patients are defective in binding to snRNP proteins. Proceedings of the National Academy of Sciences of the United States of America, 96(20), 11167–11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellizzoni, L. , Kataoka, N. , Charroux, B. , & Dreyfuss, G. (1998). A novel function for SMN, the spinal muscular atrophy disease gene product, in pre‐mRNA splicing. Cell, 95(5), 615–624. [DOI] [PubMed] [Google Scholar]
- Pennock, N. D. , White, J. T. , Cross, E. W. , Cheney, E. E. , Tamburini, B. A. , & Kedl, R. M. (2013). T cell responses: Naive to memory and everything in between. Advances in Physiology Education, 37(4), 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto‐Costa, R. , & Sousa, M. M. (2020). Profilin as a dual regulator of actin and microtubule dynamics. Cytoskeleton (Hoboken), 77(3–4), 76–83. [DOI] [PubMed] [Google Scholar]
- Quinlan, K. A. , Reedich, E. J. , Arnold, W. D. , Puritz, A. C. , Cavarsan, C. F. , Heckman, C. J. , & DiDonato, C. J. (2019). Hyperexcitability precedes motoneuron loss in the Smn(2B/−) mouse model of spinal muscular atrophy. Journal of Neurophysiology, 122(4), 1297–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahemtulla, A. , Fung‐Leung, W. P. , Schilham, M. W. , Kundig, T. M. , Sambhara, S. R. , & Narendran, A. (1991). Normal development and function of CD8+ cells but markedly decreased helper cell activity in mice lacking CD4. Nature, 353(6340), 180–184. [DOI] [PubMed] [Google Scholar]
- Rezai‐Zadeh, K. , Gate, D. , & Town, T. (2009). CNS infiltration of peripheral immune cells: D‐Day for neurodegenerative disease? Journal of Neuroimmune Pharmacology, 4(4), 462–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rindt, H. , Feng, Z. , Mazzasette, C. , Glascock, J. J. , Valdivia, D. , Pyles, N. , & Lorson, C. L. (2015). Astrocytes influence the severity of spinal muscular atrophy. Human Molecular Genetics, 24(14), 4094–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos, M. , Sarkozy, A. , Chierchia, G. B. , De Wilde, P. , Schmedding, E. , & Brugada, P. (2009). Malignant ventricular arrhythmia in a case of adult onset of spinal muscular atrophy (Kugelberg‐Welander disease). Journal of Cardiovascular Electrophysiology, 20(3), 342–344. [DOI] [PubMed] [Google Scholar]
- Rossoll, W. , Jablonka, S. , Andreassi, C. , Kroning, A. K. , Karle, K. , Monani, U. R. , & Sendtner, M. (2003). Smn, the spinal muscular atrophy‐determining gene product, modulates axon growth and localization of beta‐actin mRNA in growth cones of motoneurons. The Journal of Cell Biology, 163(4), 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenten, D. , & Medzhitov, R. (2011). The control of adaptive immune responses by the innate immune system. Advances in Immunology, 109, 87–124. [DOI] [PubMed] [Google Scholar]
- Schetters, S. T. T. , Gomez‐Nicola, D. , Garcia‐Vallejo, J. J. , & Van Kooyk, Y. (2017). Neuroinflammation: Microglia and T cells get ready to tango. Frontiers in Immunology, 8, 1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafey, D. , Cote, P. D. , & Kothary, R. (2005). Hypomorphic Smn knockdown C2C12 myoblasts reveal intrinsic defects in myoblast fusion and myotube morphology. Experimental Cell Research, 311(1), 49–61. [DOI] [PubMed] [Google Scholar]
- Simon, C. M. , Dai, Y. , Van Alstyne, M. , Koutsioumpa, C. , Pagiazitis, J. G. , Chalif, J. I. , & Mentis, G. Z. (2017). Converging mechanisms of p53 activation drive motor neuron degeneration in spinal muscular atrophy. Cell Reports, 21(13), 3767–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simone, C. , Ramirez, A. , Bucchia, M. , Rinchetti, P. , Rideout, H. , Papadimitriou, D. , & Corti, S. (2016). Is spinal muscular atrophy a disease of the motor neurons only: Pathogenesis and therapeutic implications? Cellular and Molecular Life Sciences, 73(5), 1003–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sison, S. L. , Patitucci, T. N. , Seminary, E. R. , Villalon, E. , Lorson, C. L. , & Ebert, A. D. (2017). Astrocyte‐produced miR‐146a as a mediator of motor neuron loss in spinal muscular atrophy. Human Molecular Genetics, 26(17), 3409–3420. [DOI] [PubMed] [Google Scholar]
- Sun, J. , & Harrington, M. A. (2019). The alteration of intrinsic excitability and synaptic transmission in lumbar spinal motor neurons and interneurons of severe spinal muscular atrophy mice. Frontiers in Cellular Neuroscience, 13, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swoboda, K. J. , Prior, T. W. , Scott, C. B. , McNaught, T. P. , Wride, M. C. , Reyna, S. P. , & Bromberg, M. B. (2005). Natural history of denervation in SMA: Relation to age, SMN2 copy number, and function. Annals of Neurology, 57(5), 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarabal, O. , Caraballo‐Miralles, V. , Cardona‐Rossinyol, A. , Correa, F. J. , Olmos, G. , Llado, J. , & Caldero, J. (2014). Mechanisms involved in spinal cord central synapse loss in a mouse model of spinal muscular atrophy. Journal of Neuropathology and Experimental Neurology, 73(6), 519–535. [DOI] [PubMed] [Google Scholar]
- Taylor, A. S. , Glascock, J. J. , Rose, F. F., Jr. , Lutz, C. , & Lorson, C. L. (2013). Restoration of SMN to Emx‐1 expressing cortical neurons is not sufficient to provide benefit to a severe mouse model of spinal muscular atrophy. Transgenic Research, 22(5), 1029–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson, S. R. , Nahon, J. E. , Mutsaers, C. A. , Thomson, D. , Hamilton, G. , Parson, S. H. , & Gillingwater, T. H. (2012). Morphological characteristics of motor neurons do not determine their relative susceptibility to degeneration in a mouse model of severe spinal muscular atrophy. PLoS One, 7(12), e52605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venereau, E. , Ceriotti, C. , & Bianchi, M. E. (2015). DAMPs from cell death to new life. Frontiers in Immunology, 6, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukojicic, A. , Delestree, N. , Fletcher, E. V. , Pagiazitis, J. G. , Sankaranarayanan, S. , Yednock, T. A. , & Mentis, G. Z. (2019). The classical complement pathway mediates microglia‐dependent remodeling of spinal motor circuits during development and in SMA. Cell Reports, 29(10), 3087–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan, B. , Feng, P. , Guan, Z. , Sheng, L. , Liu, Z. , & Hua, Y. (2018). A severe mouse model of spinal muscular atrophy develops early systemic inflammation. Human Molecular Genetics, 27(23), 4061–4076. [DOI] [PubMed] [Google Scholar]
- Yao, H. , Coppola, K. , Schweig, J. E. , Crawford, F. , Mullan, M. , & Paris, D. (2019). Distinct signaling pathways regulate TREM2 phagocytic and NFkappaB antagonistic activities. Frontiers in Cellular Neuroscience, 13, 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong, J. , Wan, L. , & Dreyfuss, G. (2004). Why do cells need an assembly machine for RNA‐protein complexes? Trends in Cell Biology, 14(5), 226–232. [DOI] [PubMed] [Google Scholar]
- Zabel, M. K. , & Kirsch, W. M. (2013). From development to dysfunction: Microglia and the complement cascade in CNS homeostasis. Ageing Research Reviews, 12(3), 749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Loss of mature lymphocytes in Smn 2B/− ;CD4 −/− mice does not correct SMA microglia reactive morphology. A. Spinal cord sections from L1‐L5 of PND 19 CD4 −/− Het (Smn 2B/+ ;CD4 −/− ), CD4 −/− SMA (Smn 2B/− ;CD4 −/− ), Rag −/− Het (Smn 2B/+ ;Rag1 −/− ), and Rag −/− SMA (Smn 2B/− ;Rag1 −/− ) were immune‐stained for Iba‐1. DAB staining was used to visualize antibody. Representative images of Iba1+ cells were taken from each group. Scale bars = 200 μm. B. Soma Size, and number of processes were analyzed using ImageScope. Values represent mean ± SEM. * < 0.05, ****p = <.0001, 150 Iba1+ cells were counted from 3 mice per group.
Figure S2 iPSC differentiated microglia were validated for CNS microglia specific markers A. Representative images shown of microglia differentiation. B. iPSC‐microglia were immune‐stained for Iba1, a marker for microglia. C. SMN protein levels in control and SMA patient iPSC lines were validated (n = 5/group). ****p = <.0001. D. Gene expression for TREM2 and TMEM119 was analyzed in iPSCs and microglia 10 days after differentiation. (n = 5/group) *p = .0112, ****p = <.0001. E. HLA‐A,B,C, CD45, and CD11b surface marker expression was validated by flow cytometry (n = 4/group). Representative histogram is shown per marker. ****p = <.0001. All groups were analyzed by unpaired t test. Bars show mean ± SEM.
Figure S3 iPSC‐derived motor neuron was validated at 10 days after differentiation. A. Representative image indicates a motor neuron from which recording was taken. Scale bar = 50 μm. Family of whole cell currents demonstrating the presence of inward sodium current and sustained outward rectifying potassium current and action potential generation. Cell was held at −70 mV and dotted line indicates 0 mV level. Scale bars show 1 nA by 20 ms and 20 mV by 100 ms, respectively. B. Differentiated cells stain positive for choline acetyltransferase (ChAT), characteristic of cholinergic neurons and late motor neuron marker. DAPI was used to visualize cell nuclei. Scale bar = 25 μm. C. ChAT gene expression was confirmed in 3 independent motor neuron differentiations by RT‐PCR. (n = 3/group) ****p = <.0001. Values represent mean ± SEM.
Figure S4 Representative traces of currents recorded from iPSC‐derived motor neurons in the presence and absence of control and microglia conditioned media.
Figure S5 Action potential patterns in induced neuronal cells across different culture conditions. Current clamp recordings for neuronal culture cells grown in control microglia medium (MG medium), SMA microglia conditioned medium (SMA medium) and SMN deficient motor neurons (knock down; “kd”), grown in control microglia medium (kd MG medium) or SMA microglia conditioned medium (kd SMA medium). Responses to 1 s depolarizing steps induced by a current injection of the strength shown under the individual traces. Dashed line indicates 0 mV, the criterion of action potential generation. The maximum number of APs that could be elicited in a given cell (row 4) is graphed in Figure 7 C & F.
Table S1 Sequencing quality metrics for mRNA‐seq transcriptome analysis of wild type and SMA microglia.
Data Availability Statement
The data supporting the findings of this study are not applicable for depositing into a public database; however, raw data are available from the corresponding author upon reasonable request.