

Abstract
Astrocytes are abundant glial cells in the central nervous system (CNS) that perform diverse functions in health and disease. Astrocyte dysfunction is found in numerous diseases, including multiple sclerosis, Alzheimer disease, Parkinson disease, Huntington disease and neuropsychiatric disorders. Astrocytes regulate glutamate and ion homeostasis, cholesterol and sphingolipid metabolism and respond to environmental factors, all of which have been implicated in neurological diseases. Astrocytes also exhibit significant heterogeneity, driven by developmental programmes and stimulus-specific cellular responses controlled by CNS location, cell–cell interactions and other mechanisms. In this Review, we highlight general mechanisms of astrocyte regulation and their potential as therapeutic targets, including drugs that alter astrocyte metabolism, and therapies that target transporters and receptors on astrocytes. Emerging ideas, such as engineered probiotics and glia-to-neuron conversion therapies, are also discussed. We further propose a concise nomenclature for astrocyte subsets that we use to highlight the roles of astrocytes and specific subsets in neurological diseases.
Multiple different populations of resident non-neuronal cells, known as glial cells, have important roles in central nervous system (CNS) development, function and disease. Types of glial cell include oligodendrocytes, microglia and astrocytes1. Among these, astrocytes are the most numerous2.
Astrocytes were first described in the nineteenth century by Rudolf Virchow3. A decade later, Camillo Golgi visualized the morphology of astrocytes using the silver chromate staining technique, advancing the notion that glial cells act as a ‘glue’ of the brain4. Astrocytes were soon classified into two basic morphological subtypes: protoplasmic and fibrous5,6. Protoplasmic astrocytes are typically seen in the grey matter, and fibrous astrocytes are prevalent among the white matter of the CNS. Santiago Ramón y Cajal adopted these subtypes and further revealed the diverse morphology of astrocytes in the human cerebellum, shedding new light on their potential heterogeneity7.
Recent developments in high-throughput single-cell RNA sequencing (scRNA-seq)8–13, spatial transcriptomic methods9,12,14 and techniques for the study of astrocyte cell–cell interactions15 have provided a unique understanding of astrocyte heterogeneity and its relevance in health and disease. It is now appreciated that astrocytes comprise multiple subsets with diverse and potentially opposing roles in disease based on location, gene expression or function8,9,14,16 (BOX 1). For example, neocortical protoplasmic astrocytes display morphological heterogeneity dependent on cortical layers17, a finding that has been refined at the molecular level by scRNA-seq and spatial reconstruction analysis14. Moreover, by responding to signals in their microenvironment, astrocytes have the potential to promote or arrest inflammation and neurodegeneration, having active roles in multiple neurological diseases including multiple sclerosis (MS)8,9,15,18–28, Alzheimer disease (AD)10,29,30, Parkinson disease (PD)31,32, Huntington disease (HD)33–38 and behavioural neuropsychiatric disorders38–42 (BOXES 2,3). In this Review, we discuss recent insights into the central roles of astrocytes, their subsets and regulation, in neurological and psychiatric disorders, and their potential as targets for therapeutic intervention.
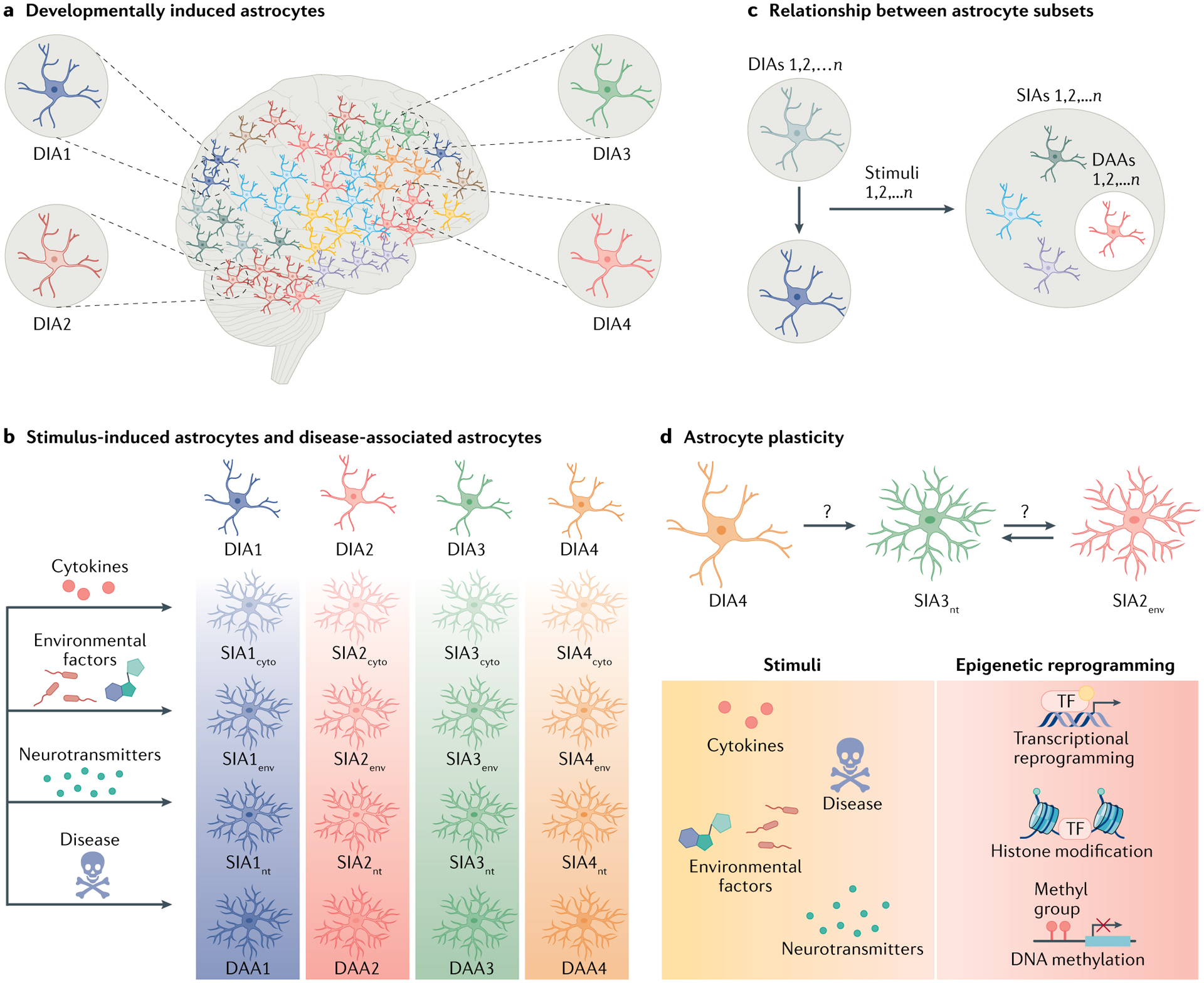
Box 1 |. Defining astrocyte subsets and plasticity.
Astrocytes are heterogeneous. Novel molecular techniques have identified astrocyte subsets as defined by their molecular signatures (based on single-cell RNA sequencing (scRNA-seq) studies) and/or function (astrocytes that promote or limit central nervous system (CNS) inflammation or neurodegeneration). These subsets likely reflect at least two sources of astrocyte heterogeneity: astrocyte subsets driven by developmental programmes (developmentally induced astrocyte (DIA) subsets); and astrocyte subsets induced in response to stimulation (stimulus-induced astrocyte (SIA) subsets).
DIAs.
Astrocyte heterogeneity emerges during neurodevelopment. As an example, regional heterogeneity of astrocytes in the spinal cord is linked to developmental transcriptional programmes driven by expression of Pax6, Nkx6.1, Reelin, Slit1 and Sema3a304–307. Similar observations have been made when studying astrocytes across cortical layers by in situ RNA profiling14. Likewise, the functional properties of astrocytes differ across brain regions, such as the dorsolateral striatum and hippocampus, when focused on electrophysiological, morphological and transcriptional attributes308. These and other data indicate that developmental programmes establish defined DIA subsets (Figure, part a). In this sense, DIA groups are similar to neuron subtypes defined by regional, electrophysiological and secretory properties.
SIAs.
Once matured, astrocytes respond to various stimuli such as neurotransmitters, cytokines, environmental chemicals, extracellular ions and microbial metabolites22,159,309. Each stimulus has the potential to induce distinct SIA subsets detectable in single-cell transcriptomic, epigenomic, metabolomic and proteomic analyses. When combined with astrocyte developmental heterogeneity, the same stimulus may induce multiple SIA subsets (Figure, parts b,c). Thus, cytokines, environmental factors and neurotransmitters can induce SIAs from various DIAs, shown here as SIA1–4cyto, SIA1–4env and SIA1–4nt, respectively, in which the number indicates the DIA subtype. In this context, disease-associated astrocytes (DAAs), which have been recently identified by single-cell/nucleus RNA-seq9,10 in CNS samples from patients affected by neurological diseases and/or preclinical models, are one example of SIAs. DAAs may result from different DIA subsets that converge on a single molecular phenotype induced by disease-related stimuli, or alternatively are the result of the expansion or activation of a single DIA subset. However, SIAs identified as DAAs may contribute to disease pathogenesis and/or participate in mechanisms of tissue repair in a manner dependent on the location and disease phase; astrocyte subsets pathogenic in one neurological disorder may limit disease pathogenesis in another. Hence, we favour the use of SIA and not DAA.
Astrocyte plasticity.
We speculate that astrocyte subsets may be interconvertible, but that their plasticity is limited by epigenetic modifications similar to those associated with the long-term modification of neuronal310 or microglial311,312 responses (Figure, part d). understanding the mechanisms that regulate astrocyte plasticity is likely to have important implications for the therapeutic targeting of astrocyte subsets that contribute to the pathogenesis of neurological disorders.
Box 2 |. Neurodegenerative diseases.
Multiple sclerosis
This chronic inflammatory disease of the central nervous system (CNS) is thought to be driven, at least during its initial phases, by an autoimmune response to myelin139. Multiple populations of immune cells, most notably t and B lymphocytes, are involved in the pathogenesis of multiple sclerosis (MS). In the late phases of MS, chronic inflammation of the CNS is thought to be driven by activation of resident glial cells, including astrocytes and microglia22,313. Current therapies target immune cell extravasation into the CNS, antibody production and B cell development314.
Alzheimer disease
Alzheimer disease (AD) is a late-onset neurodegenerative disease associated with cognitive and memory impairments. The brain in patients with AD is characterized by the presence of extracellular amyloid-β (aβ) plaques and intracellular neurofibrillary tangles (NFTs), which are composed of hyperphosphorylated tau proteins315. The first disease-modifying therapy for AD has recently been approved and promotes the clearance of aβ plaques, although it may work only in a subset of patients316,317.
Parkinson disease
This neurodegenerative disease is characterized by loss of dopaminergic neurons in the substantia nigra, which is thought to be caused by misfolded α-synuclein in Lewy bodies218. Parkinson disease (PD) is marked by motor impairments and other non-motor impairments, such as cognitive and sleep disturbances. several treatments are currently available, such as l-DOPA administration and deep brain stimulation, which help disease management, although neither cures the disease.
Huntington disease
Huntington disease (HD) is a progressive neurodegenerative disease caused by polyglutamine repeats in the huntingtin (HTT) gene, which results in misfolding of the HTT protein. HD is characterized by neurodegeneration of the striatum and cortex, as well as motor impairments. No treatments exist that alter the course of the disease, although some reduce motor symptoms.
Box 3 |. Common contributions of astrocytes to neurodegenerative diseases.
Glutamate homeostasis
Glutamate is the most prevalent excitatory neurotransmitter in the central nervous system (CNS); however, excessive glutamate levels can trigger neuronal death, a process called excitotoxicity73,193. in this context, astrocytes remove extracellular glutamate from the synaptic cleft through the high-affinity glutamate transporters excitatory amino acid transporter 1 (EAAT1) and EAAT2, which play a pivotal part in glutamate homeostasis, synaptic plasticity and neuron survival70,71. Therefore, impaired astrocytic glutamate uptake can lead to neuronal excitotoxicity, which is commonly associated with neurodegeneration73. For example, previous studies suggest that protein misfolding and aggregation (β-amyloid in alzheimer disease (AD), α-synuclein accumulation in Parkinson disease (PD) and misfolded mutant huntingtin (mHTT) in Huntington disease (HD)), hallmarks of neurodegenerative diseases, inhibit glutamate uptake in astrocytes197,198,230,318. In addition, astrocytes in experimental animal models of AD, PD and HD exhibited decreased EAAT2 expression, linked with neuronal dysfunction200,234,282,318. indeed, human astrocytes derived from patients with AD display decreased glutamate uptake concomitant with reduced EAAT2 and EAAT1 expression199. Furthermore, downregulation of EAAT2 expression correlates with disease severity in postmortem brains of patients with HD319. Overall, these studies highlight the importance and potent therapeutic value of glutamate excitotoxicity in many neurodegenerative diseases.
Ion homeostasis
Astrocytes regulate the excitability of neurons by contributing to K+ uptake and buffering in the brain. In this context, astrocytes express a wide variety of K+ channels that efficiently clear K+ ions from the extracellular fluid, a process called K+ spatial buffering320. thus, K+ channel dysfunction in astrocytes can contribute to neuronal damage, leading to neurodegeneration. For example, mHTT inclusions in astrocytes in a mouse model of HD have decreased expression of the Kir4.1 K+ channel. Selective restoration of Kir4.1 in striatal astrocytes reverts HD-like symptoms including extracellular K+ levels, abnormal motor behaviours and dysfunctional Ca2+ and glutamate signalling34,234. in preclinical PD mouse models, impairment of Kir6.1 in astrocytes aggravates neurodegeneration by increasing dopaminergic neuron damage and motor deficits68,69. Collectively, these studies demonstrate that astrocyte K+ homeostasis has a key role in neurodegeneration in both PD and HD.
Energy metabolism
Astrocytes fuel the energetic needs of neurons by supporting lactate transfer (termed the astrocyte–neuron lactate shuttle)87,88. Metabolic dysfunction of astrocytes is associated with neurodegenerative diseases including Ms93, AD90, PD89 and HD36,91,92. For instance, lactosylceramide (LacCer)-mediated sphingolipid metabolism in astrocytes contributes to experimental autoimmune encephalomyelitis (EAE) and MS pathogenesis by controlling the metabolic interactions between astrocytes and neurons93. In HD, impairment of astrocyte ascorbic acid shuttling to neurons has been linked to neurodegeneration92. Furthermore, the low level of glucose in the brain in HD promotes astrocyte metabolic reprogramming, switching from glycolysis to fatty acid oxidation, which generates reactive oxygen species and causes neuronal death246. Taken together, these findings highlight the importance of astrocyte–neuron metabolic crosstalk in neurodegeneration.
Neurotoxicity
The neurotoxic activity of astrocytes has been reported in multiple studies and is likely to involve multiple triggers, astrocyte subsets and effector mechanisms. C3+ neurotoxic astrocyte subsets induced by stimulation with the complement factor C1q, tumour necrosis factor (TNF) and IL-1α, which are produced by microglia, induce neuronal death145. Long-chain saturated lipids, synthesized by elongation of very long chain fatty acids protein 1 (ELOVL1) and secreted in lipoparticles, are the neurotoxic factors that these astrocytes secrete321. Identification of the molecular mechanisms that regulate and mediate the neurotoxic activity of these and other astrocyte subsets is likely to open novel therapeutic approaches for neurological diseases.
Astrocytes in homeostasis
Astrocytes are derived from neural progenitor cells (NPCs) in the subventricular zone (SVZ) and migrate along radial glia processes to populate all areas of the CNS43,44. Mature astrocytes participate in key processes relevant to CNS homeostasis including neurotransmitter recycling, ionic balance, modulation of synaptogenesis and synaptic transmission, and maintenance of the blood–brain barrier (BBB)45–47. Indeed, astrocytes are also one of the major components of the BBB, as astrocytic end-feet processes form a thin barrier that surrounds endothelial cells, pericytes and the basal lamina, which is called the glia limitans46–50. Interestingly, although the BBB surrounds most blood vessels in the brain, separating the CNS from the peripheral blood circulation, more than 97% of astrocytes contact the blood supply51, suggesting that astrocytes may respond to molecules produced by peripheral cells, such as the cytokines produced by circulating immune cells.
The passage of molecules and cells across the BBB is modulated by multiple cell types, including astrocytes50,52,53. Astrocytes produce the protein sonic hedgehog (SHH), which promotes anti-inflammatory signalling and contributes to the formation and maintenance of the BBB46,54. Similarly, apolipoprotein E3 (APOE3), produced by astrocytes, suppresses signalling via a cascade involving peptidyl-prolyl cis-trans isomerase A (also known as cyclophilin A (CYPA)), nuclear factor-κB (NF-κB) and metalloproteinase 9 (MMP9) in pericytes; APOE3 counteracts APOE4-associated BBB breakdown55. Conversely, astrocyte-derived vascular endothelial growth factor (VEGF), a potent angiogenic factor, promotes CNS inflammation by increasing BBB permeability and enhancing immune cell extravasation56,57. Thus, astrocytes regulate the formation, maintenance and permeability of the BBB, which is commonly disrupted in neurological diseases.
Astrocytes also have intimate roles in the regulation of synaptic function through their participation in tripartite synapses, regulating synaptic formation, transmission and plasticity39,58–60. Indeed, although astrocytes were traditionally considered to merely provide trophic and structural support for neurons, it is now clear that astrocytes engage in extensive crosstalk with neurons through multiple mechanisms involving ion channels, neurotransmitters and metabolites61–65. For example, extracellular K+ levels are crucial for the control of neuronal responses66,67. Astrocytes express K+ channels, which clear K+ ions from the extracellular environment and modulate the membrane potential and excitability of neurons. Conversely, downregulation of astrocyte inwardly rectifying K+ channel (Kir) expression results in increased extracellular K+ levels and neuronal hyperexcitability, a mechanism that is linked to neuronal dysfunction in HD33,34 and PD68,69. These findings highlight astrocyte roles in CNS homeostasis and neurodegeneration.
Tripartite synapses.
Bidirectional interactions established by astrocytes with pre- and post-synaptic nerve terminals.
Astrocytes also regulate ionic tone by recycling neurotransmitters. The high-affinity glutamate transporters excitatory amino acid transporter 1 (EAAT1, also known as GLAST) and EAAT2 (also known as GLT1) take up the neurotransmitter glutamate from the synaptic cleft for recycling70,71. The dysfunction of astrocyte glutamate transporters leads to excessive glutamate release from neurons, promoting neuronal death by excitotoxicity72, a process that has long been associated with the pathogenesis of neurodegenerative diseases73–77. Indeed, the pro-inflammatory cytokine tumour necrosis factor (TNF) boosts glutamate production78, which in turn triggers excitotoxicity and Ca2+ signalling. Ca2+ elevation in astrocytes releases glutamate, ATP and γ-aminobutyric acid (GABA)79–84. Moreover, excessive GABA and ATP release by reactive astrocytes has been linked to disease pathogenesis in AD models85,86.
Excitotoxicity.
Neuronal dysfunction and death caused by the accumulation of excess neurotransmitters, primarily glutamate, in synapses.
Finally, high extracellular K+, glutamate uptake and intracellular Ca2+ waves trigger lactate release from astrocytes. Astrocyte-produced lactate is used by neurons to fuel their energetic needs87,88. Dysfunctional astrocyte–neuron metabolic interactions are a key feature of several neurodegenerative diseases including PD89, AD90, HD36,91,92, MS93 and amyotrophic lateral sclerosis (ALS)94. Overall, these observations highlight the importance of astrocytes in the control of synaptic plasticity and CNS physiology.
Reactive astrogliosis
Pathogenic processes that target the CNS (for example, inflammation, neurodegeneration or trauma-induced injuries) are usually associated with astrogliosis, an astrocyte response characterized by functional, morphological and molecular remodelling22,95,96. Reactive astrocytes are highly heterogeneous and contribute to the pathology of neurodegenerative and neuropsychiatric diseases, but also to CNS repair. Several markers are commonly used to monitor astrocyte reactivity, but the functional investigation of molecularly defined astrocyte subtypes is still in its infancy95. The application of high-throughput sequencing techniques, including scRNA-seq and spatial transcriptomics, has enabled the identification of reactive astrocyte subsets and novel pathways involved in their regulation9,14,22.
Reactive astrocytes are now considered active players in CNS inflammation. Indeed, reactive astrocytes express a wide variety of receptors including pattern recognition receptors and cytokine receptors. Consequently, astrocytes can respond via the production of immunoregulatory molecules that may promote or limit inflammation in response to multiple cues in the CNS microenvironment97–99. In addition, newly proliferated astrocytes form a ‘glial scar’, which was initially viewed as a physical CNS barrier that interferes with axonal regrowth96,100. However, recent studies have shown that some reactive astrocytes produce molecules that support axon regeneration, such as neurotrophin-3 and brain-derived neurotrophic factor, and thus promote recovery from severe spinal cord injury (SCI)101,102. Additional reactive astrocyte subsets release a broad range of pro-inflammatory molecules that support immune cell invasion into the CNS25,50. These findings highlight the functional heterogeneity of reactive astrocytes in the context of neuroinflammation and CNS injury. Identifying the molecular mechanisms that control astrocyte subsets and function is likely to pave the way for novel therapies for neurological disorders.
Reactive astrocyte signals and pathways
Multiple signalling pathways and transcription factors modulate astrocyte responses in the context of neurodegenerative diseases, including STAT3, NFAT and NF-κB. The transcription factor NF-κB is a central regulator of inflammation and neurodegeneration103,104. In astrocytes, NF-κB signalling is triggered by pro-inflammatory mediators such as cytokines (TNF, IL-1β and IL-17)26,105, reactive oxygen species (ROS)106, Toll-like receptor (TLR) agonists107 and environmental factors26. Conversely, the downregulation of NF-κB in astrocytes has been linked to decreased CNS inflammation or injury in multiple contexts including SCI108, optic neuritis109 and experimental autoimmune encephalomyelitis (EAE, a model of multiple sclerosis)110–112, suggesting a pathogenic role for NF-κB-driven transcriptional modules in astrocytes. Of note, microbial metabolites have been shown to regulate astrocyte pathogenic functions in EAE by suppressing NF-κB signalling via suppressor of cytokine signalling 2 (SOCS2)21. NF-κB activation is also detected in preclinical models of Alzheimer disease (AD)113,114 and Huntington disease (HD)115. Protein misfolding and aggregation (that is, β-amyloid in AD, and misfolded mutant huntingtin (mHTT) in HD) triggers NF-κB activation in astrocytes, upregulating pro-inflammatory molecules during neurodegeneration.
The JAK–STAT3 pathway is a ubiquitous signalling system with newly identified roles in the control of reactive astrocytes116,117. Early studies reported that STAT3 is expressed in the brain and contributes to CNS development during astrogliogenesis118,119. In acute CNS injury, it is well established that the JAK–STAT3 pathway controls astrocyte reactivity and glial scar formation120,121. Recent studies revealed the importance of JAK–STAT3 signalling in reactive astrocytes, and thus its potential as a therapeutic target in CNS neurodegenerative diseases including MS, AD and HD116,122–125. For example, phosphorylated (activated) STAT3 has been observed in astrocytes in the white matter adjacent to active MS lesions and in EAE mice124,125. Moreover, conditional expression of a dominant mutant of the gene encoding IL-6 receptor subunit-β, a potent JAK activator in astrocytes, significantly increases EAE severity123. Furthermore, JAK–STAT3 signalling is activated in astrocytes in preclinical models of AD and HD116. Selective overexpression of suppressor of cytokine signalling 3 (SOCS3) in astrocytes suppresses JAK–STAT3 signalling, resulting in reduced astrocyte and microglia activation in the context of neurodegenerative disease models including those of AD and HD116. Interestingly, in the HD model, SOCS3 overexpression also increased mutant huntingtin (mHTT) aggregates, a hallmark of HD pathogenesis. Consistent with these findings, the conditional deletion of STAT3 in astrocytes is associated with fewer amyloid-β (Aβ) plaques, and milder deficits in spatial learning and memory, in the APP/PS1 model of AD122. However, most evidence supports a pro-inflammatory role for STAT3 signalling in reactive astrocytes. Indeed, the cell-specific inactivation of STAT3 in astrocytes decreases amyloid-β (Aβ) plaques and improves spatial learning in the amyloid precursor protein/presenilin 1 (APP/PS1) model of AD. This effect is recapitulated by the administration of a STAT3 inhibitor122. Thus, JAK–STAT3 signalling in astrocytes has important roles in both the aggravation and the alleviation of neurodegeneration.
Calcineurin (also known as protein phosphatase 3) is a serine/threonine-protein phosphatase that is abundant in the healthy brain126. Elevated intracellular Ca2+ levels activate calcineurin, which binds to and dephosphorylates NFAT, triggering NFAT nuclear translocation and transcriptional activation127. Hyperactivation of calcineurin–NFAT signalling in astrocytes is associated with CNS injury and neurological diseases128–132. For instance, calcineurin-expressing reactive astrocytes are present in patients with AD and the APP/PS1 mouse model of AD128. Similarly, NFAT activation is elevated in acute brain injury129,130, and in neurodegenerative diseases such as AD131 and PD132. Further support for the contribution of calcineurin–NFAT signalling to neuroinflammation was provided by studies based on the intracerebroventricular injection or astrocyte-specific overexpression of VIVIT, an inhibitory peptide that blocks calcineurin–NFAT signalling and ameliorates AD symptoms while reducing astrocyte and microglia activation in preclinical models133,134. Moreover, in a model of traumatic brain injury, selective blockade of calcineurin–NFAT signalling in astrocytes increases synaptic functional recovery130. Enhanced intracellular Ca2+ waves in astrocytes can also trigger the release of γ-aminobutyric acid (GABA), the major inhibitory neurotransmitter in the brain. Increased GABA uptake by neurons impairs memory and synaptic plasticity, contributing to AD pathology85. Of note, JAK–STAT and calcineurin–NFAT signalling pathways have been shown to crosstalk in other cell types135–138, suggesting that interactions between these two pathways may fulfil important roles in fine-tuning astrocyte responses in neuroinflammation and synaptic plasticity.
Astrocytes in multiple sclerosis
MS is a chronic inflammatory neurological disease thought to be driven, at least during its initial phases, by an autoimmune response to myelin139 (BOX 2). The clinical course of MS has multiple phases. Initially, approximately 85% of patients with MS present with relapsing–remitting multiple sclerosis (RRMS). Following this stage, a significant proportion of patients with RRMS develop secondary progressive multiple sclerosis (SPMS), characterized by the progressive and irreversible accumulation of neurological disability, with limited response to available therapies140. Evidence for a role of astrocytes in MS pathology was first provided in the nineteenth century, when Jean-Marie Charcot identified astrocytes as key components of MS lesions141. Studies in the EAE model further revealed diverse roles of astrocytes in CNS inflammation (FIG. 1). In the acute EAE B6 mouse model, the depletion of reactive astrocytes increases disease severity and CNS inflammation142–144. In contrast, selective depletion of reactive astrocytes during the chronic progressive phase in the NOD mouse EAE model ameliorates disease pathogenesis, limiting the recruitment and activation of microglia and monocytes19. These seemingly opposing roles of astrocytes highlight their functional heterogeneity in MS.
Fig. 1 |. Astrocyte roles in CNS inflammation.
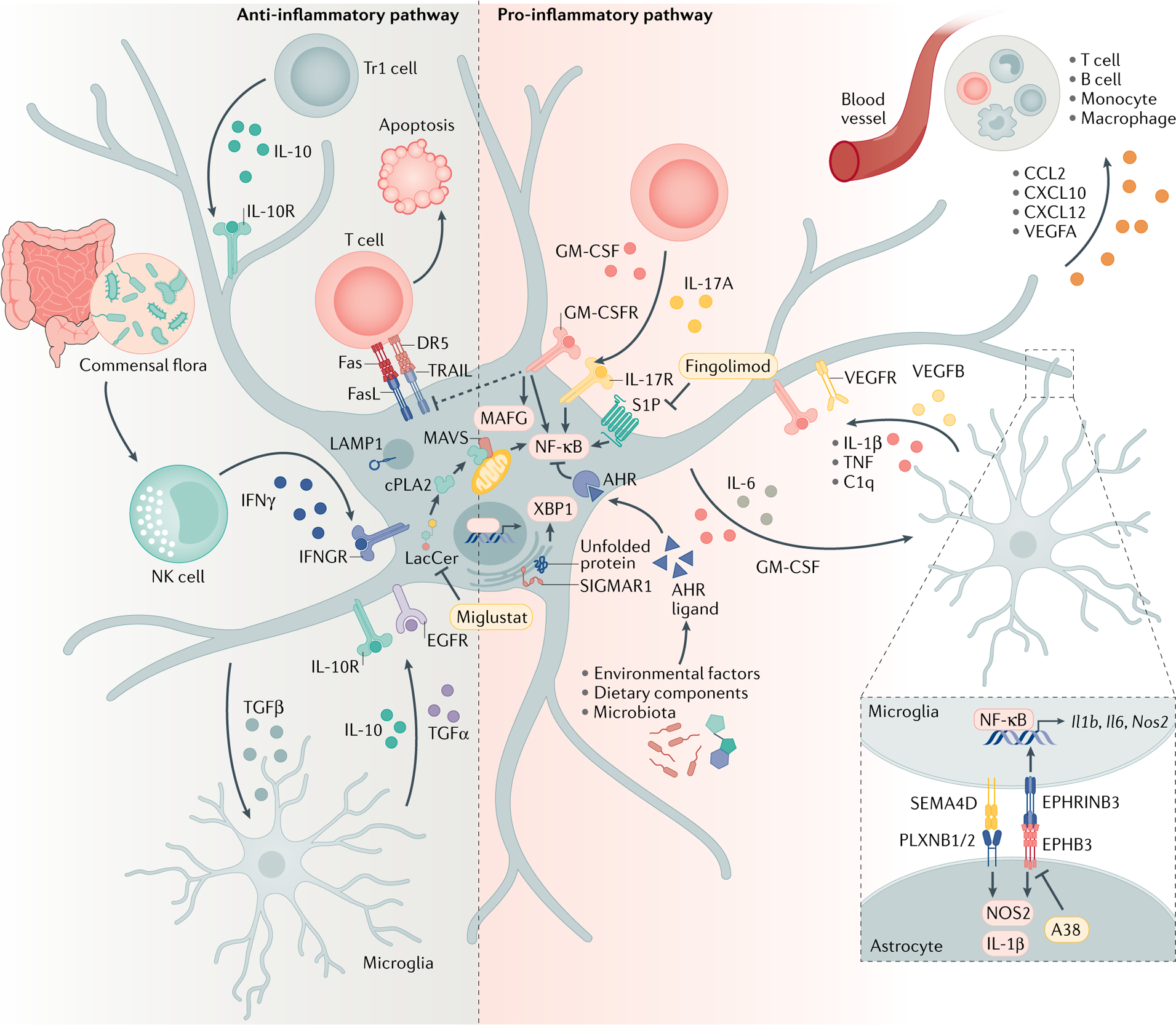
Astrocytes are active players in central nervous system (CNS) inflammation, where they have both pro-inflammatory and anti-inflammatory activities. As a result of their interactions with other cells and molecules in the CNS, astrocytes secrete various cytokines, chemokines and neuromodulatory molecules. AHR, aryl hydrocarbon receptor; CCL2, C-C motif chemokine 2; cPLA2, cytosolic phospholipase A2; CXCL10, C-X-C motif chemokine ligand 10; DR5, death receptor 5; EGFR, epidermal growth factor receptor; EPHB3, ephrin type B receptor 3; EPHNB3, ephrinB3; GM-CSF, granulocyte–macrophage colony-stimulating factor; IFNγ, interferon-γ; LacCer, lactosylceramide; LAMP1, lysosome-associated membrane glycoprotein 1; MAFG, MAF bZIP transcription factor; MAVS, mitochondrial antiviral signalling protein; NF-κB, nuclear factor-κB; NK, natural killer; NOS2, nitric oxide synthase 2; PLXNB, plexin B; SEMA4D, semaphorin4D; S1P, sphingosine 1-phosphate receptor; SIGMAR1, sigma receptor 1; TGF, transforming growth factor; TNF, tumour necrosis factor; Tr1, type 1 regulatory T cell; TRAIL, tumour necrosis factor-related apoptosis-inducing ligand; VEGF, vascular endothelial growth factor; XBP1, X-box binding protein 1.
Relapsing–remitting mutiple sclerosis.
(RRMs). The most common phenotype of multiple sclerosis, which is characterized by relapses followed by periods of partial or complete recovery. Most patients with RRMs will eventually develop secondary progressive multiple sclerosis.
Secondary progressive multiple sclerosis.
(SPMS). A phase of multiple sclerosis characterized by the progressive, irreversible accumulation of neurological disability, which shows limited response to available therapies.
Novel approaches enabled the identification of specific astrocyte subsets associated with MS pathology. Liddelow et al.145 identified a subset of neurotoxic reactive astrocytes abundant in multiple neurodegenerative diseases, including MS, and characterized by complement component 3 (C3) expression. In addition, we recently identified a subset of astrocytes, driven by the small MAF bZIP transcription factor (MAFG) and granulocyte–macrophage colony-stimulating factor (GM-CSF) signalling9, that promotes inflammation and neurodegeneration in EAE and MS. We also identified a subset of anti-inflammatory astrocytes that express the lysosome-associated membrane glycoprotein 1 (LAMP1; also known as CD107a) and tumour necrosis factor-related apoptosis-inducing ligand (TRAIL, also known as TNFSF10). These LAMP1+TRAIL+ astrocytes limit EAE pathogenesis by inducing T cell apoptosis via TRAIL–death receptor 5 (DR5) signalling8.
CNS inflammation in MS is typically initiated by BBB dysfunction and peripheral immune cell invasion, triggering the activation of astrocytes in response to pro-inflammatory cytokines and pathogen-associated molecular patterns (PAMPs)22,25,146. Indeed, reactive astrocytes produce a broad range of chemokines that attract leukocytes into perivascular spaces and the CNS parenchyma. Astrocyte-derived C-C motif chemokine 2 (CCL2) controls the onset and progression of EAE by promoting the accumulation of monocytes and T cells into the CNS26,147,148. Of note, a recent study reported that astrocytes also control the phenotype of CNS-infiltrating mononuclear phagocytes149. In addition, astrocyte-produced C-X-C motif chemokine 10 (CXCL10) promotes CNS infiltration by CD4+ T cells150. Finally, astrocytes show increased expression of CXCL12, a chemoattractant involved in the recruitment of monocytes, T cells, B cells and plasma cells, in active and chronic MS lesions151. Taken together, these findings suggest key roles for astrocytes during the initiation of neuroinflammation.
Multiple studies have further established the crosstalk between astrocytes and other cells in the CNS as a major regulatory mechanism in MS pathogenesis139. We recently described microglia–astrocyte interactions, mediated by semaphorin4D–plexinB1/2 and ephrinB3–EPHB3, that promote CNS inflammation15. Reactive astrocytes also control CNS-infiltrating T cells via the expression of pro-apoptotic molecules such as FASL and TRAIL8,152.
Conversely, astrocyte responses are regulated by T cells. IL-10 produced by regulatory T cells limits astrocyte activation and reduces neuroinflammation18. In addition, signalling triggered by GM-CSF, an important cytokine for the pathogenic activity of T cells in EAE and MS153–155, promotes the differentiation of MAFG-driven pathogenic astrocytes, which have a decreased ability to support the metabolic needs of neurons and limit oxidative stress, and simultaneously activates transcriptional programmes that promote CNS inflammation and neurodegeneration9,156. GM-CSF signalling also limits astrocyte anti-inflammatory functions by downregulating TRAIL expression, suggesting an important role for this cytokine in regulating the balance between pro- and anti-inflammatory astrocyte subsets in MS and other neurological diseases8.
Environmental factors.
Multiple environmental factors have been recently linked to astrocytes in MS pathology (FIG. 1). Importantly, astrocytes express the ligand-activated transcription factor aryl hydrocarbon receptor (AHR), which acts as a sensor for a wide range of small molecules provided by the environment, the diet, metabolic intermediates and the commensal microbiota157,158. Selective deletion of AHR in astrocytes worsens EAE, concomitant with increased expression of chemokines (CCL2, CCL20 and CXCL10) and other pro-inflammatory mediators (IL-6, IL-12, IL-23, GM-CSF and nitric oxide (NO)) in the brain21. Microbial metabolites of dietary tryptophan are an important physiological source of AHR agonists159. Indeed, some tryptophan metabolites controlled by the intestinal microbiome cross the BBB and inhibit EAE pathogenesis by suppressing astrocyte pathogenic activities; this novel gut–brain axis (GBA) provides a means through which the gut flora regulates astrocyte responses and CNS inflammation. These anti-inflammatory effects of AHR activation in astrocytes are mediated by the suppression of NF-κB signalling via SOCS2 (REF.21). A similar mechanism operates in microglia, where AHR limits NF-κB activation and also the expression of VEGFB, while increasing TGFα expression. Microglial TGFα interacts with the epidermal growth factor receptor (EGFR, also known as ERBB1), which is expressed on astrocytes, to limit their pathogenicity. In contrast, microglial VEGFB activates FLT1 signalling in astrocytes, which boosts the expression of pro-inflammatory transcriptional programmes that increase CNS pathology20. Collectively, these findings illustrate a novel mechanism by which microbial metabolites control the intrinsic pathogenic activities of microglia and astrocytes as well as their communication.
Gut–brain axis.
(GBA). Bidirectional communication between the gut microbiota and the brain.
Environmental chemicals that gain access to the CNS also have the potential to modulate astrocyte responses in CNS inflammation. By using bioinformatics, we integrated data from genomics, perturbation studies in zebrafish, mouse models and tissue samples from people with MS to evaluate the effects of common chemicals on astrocytes and identify environmental factors that modulate CNS pathology, as well as novel regulatory mechanisms and potential targets for their therapeutic modulation26. This novel platform established that the herbicide linuron boosts astrocyte pathogenic activities. Linuron boosts unfolded protein response (UPR) signalling by increasing sigma receptor 1 (SIGMAR1)-driven activation of inositol-requiring enzyme 1α (IRE1α) and X-box binding protein 1 (XBP1). Notably, the UPR is a highly conserved cellular stress response that aims to restore endoplasmic reticulum (ER) and protein homeostasis, and has been linked to several neurodegenerative diseases160–162. Indeed, IRE1α–XBP1 activation is detected in MS brain lesions, and the selective knock-down of Xbp1 or Ern1 (coding for IRE1α) in astrocytes ameliorates CNS pathology in EAE26. Moreover, a recent study reported the pathogenic role of UPR activation in astrocytes during prion-induced neurodegeneration163. Taken together, these findings suggest that SIGMAR1–IRE1α–XPB1 signalling is a common component of astrocyte pathogenic responses in neurological diseases.
Sphingolipid metabolism.
Sphingolipids are a class of lipids that are major components of the myelin sheath. Abnormal sphingolipid metabolism has been linked to MS pathology164. Indeed, sphingosine 1-phosphate (S1P) signalling is an important target in existing therapies for MS, and is associated with the control of CNS infiltration by peripheral immune cells165. In addition, the expression of S1P receptors (S1P1 and S1P3) in astrocytes is upregulated in demyelinating and chronic MS lesions. Selective knock-down of S1P signalling in astrocytes reduces EAE severity, demyelination and axonal loss166. Furthermore, S1P receptor modulation by fingolimod (FTY720), an approved therapy for MS, suppresses astrocyte pathogenic activities and ameliorates disease progression and neurodegeneration in the NOD EAE model of MS167, which recapitulates features of SPMS, supporting a role for S1P signalling in astrocytes in MS progression (FIG. 1).
The astrocyte–neuron lactate shuttle supports neuron metabolism87,88. Interestingly, sphingolipid metabolism has been recently shown to control the metabolic support of neurons by astrocytes. Levels of the ceramide-derived sphingolipid lactosylceramide (LacCer) and the enzyme β1,4-galactosyltransferase 6 (B4GALT6), which catalyses LacCer synthesis, are upregulated in the NOD EAE model and in brain samples from individuals with MS. LacCer promotes the expression of pro-inflammatory transcriptional programmes in astrocytes, and B4GALT6 inactivation in astrocytes ameliorates EAE19. Further investigations combining proteomic, metabolomic, transcriptomic and gene perturbation studies established that LacCer activates the cytosolic phospholipase A2 (cPLA2) and the mitochondrial antiviral signalling protein (MAVS). LacCer-activated cPLA2–MAVS signalling boosts NF-κB-driven pro-inflammatory transcriptional programmes in astrocytes that contribute to EAE and MS pathogenesis. In resting astrocytes, MAVS interacts with hexokinase 2 (HK2) to promote lactate production by glycolysis168. However, LacCer-induced cPLA2–MAVS interactions decrease HK2–MAVS interactions, HK2 enzymatic activity and astrocyte lactate production93. These findings identify cPLA2–MAVS–NF-κB signalling as a novel immunometabolic pathway that regulates astrocyte-driven CNS inflammation and neurodegeneration (FIG. 1).
Astrocyte–neuron lactate shuttle.
Mechanism by which lactate released by astrocytes from glycolysis is used as a metabolic substrate for neurons under normal physiological conditions.
DNA methylation.
DNA methylation is one of the best-studied epigenetic modifications in mammals. This process is controlled by DNA methyltransferases (DNMTs), which catalyse the transfer of methyl groups to DNA through a reaction that uses the methyl donor S-adenosyl methionine (SAM)169. DNA methylation represses gene expression by impairing the interactions of DNA elements with protein complexes involved in the regulation of gene transcription170. Changes in DNA methylation have been linked to MS pathogenesis171. For example, altered DNA methylation is detected in multiple immune cell types and CNS-resident cells in patients with MS172,173. Although the study of DNA methylation in astrocytes was initially focused on CNS development, recent studies highlighted the importance of DNA methylation in the control of pathogenic astrocyte activities9,174,175.
High-throughput scRNA-seq in combination with astrocyte-specific Ribotag RNA profiling identified a novel pathogenic astrocyte subset that is expanded in EAE and MS and features decreased expression of the transcription factor NRF2 and increased expression of MAFG. MAFG has dual roles in controlling NRF2 signalling, and NRF2 is a master regulator of antioxidant transcriptional responses. By forming heterodimers with NRF2, MAFG participates in NRF2-driven gene expression. However, MAFG homodimers are more abundant in CNS inflammation, and compete for MAFG–NRF2 responsive elements to suppress NRF2 signalling176. MAFG binding to its DNA responsive elements recruits the co-factor MAT2α, an enzyme that converts methionine into SAM. Together with the BACH1 and BACH2 co-factors that recruit DNA methyltransferases, MAFG–MAT2α–BACH complexes promote DNA methylation. Thus, increased MAFG expression in astrocytes promotes astrocyte pathogenic activities and CNS inflammation by downregulating NRF2 signalling. Interestingly, MAFG–MAT2α signalling in astrocytes is induced by GM-CSF, a cytokine produced by encephalitogenic T cells177,178. These observations highlight new mechanisms involved in the control of astrocyte responses in the context of CNS pathology, pointing to new therapeutic targets and challenges, as they suggest that epigenetic modifications stabilize disease-promoting astrocyte subsets.
Astrocytes in Alzheimer disease
AD is the most common neurodegenerative disease, characterized by progressive dementia with cognitive decline and impaired long-term memory179 (BOX 2). The pathological hallmark of AD is the presence of extracellular Aβ plaques and intracellular neurofibrillary tangles (NFTs), comprised of hyperphosphorylated Tau, in the brain. Aβ is a normal by-product of cellular metabolism generated by the cleavage of the transmembrane amyloid precursor protein (APP) by the aspartyl proteases β- and γ-secretase180. The accumulation of Aβ in the brain is considered an early event in AD pathogenesis. Aβ interacts with a broad range of receptors expressed by CNS-resident cells, activating signalling pathways that lead to neuroinflammation and neurodegeneration181.
Microglia and astrocytes cluster around and within Aβ plaques in CNS samples from patients with AD182. Astrocytes participate in the degradation of Aβ by secreting proteolytic enzymes such as metalloendopeptidases (NEP, ECE and IDE) and matrix metalloproteinases (MMP2 and MMP9) that cleave Aβ183. Indeed, reactive astrocytes surrounding Aβ plaques upregulate MMP2 and MMP9 in AD mouse models184,185. However, astrocytes also contribute to Aβ accumulation in the brain because they produce Aβ and release β-secretase 1 (BACE1)186–189. Cytokines such as TGFβ, IL-1β, TNF, IL-6 and interferon-γ (IFNγ) boost Aβ production by reactive astrocytes186,188,189. Moreover, pro-inflammatory cytokines induce, in astrocytes, the expression of interferon-induced transmembrane protein 3 (IFITM3), which binds to and activates γ-secretase, increasing Aβ production. Of note, IFITM3 expression is upregulated in brain samples from patients with late-onset AD, suggesting that IFITM3 may serve as a biomarker and therapeutic target in AD190.
scRNA-seq studies have analysed glial subsets in patient samples and preclinical models of AD29,30. Indeed, although multiple studies have defined microglial subsets associated with AD30,191,192, our knowledge of astrocyte heterogeneity in AD is more limited. Disease-associated astrocytes (DAAs) in AD are characterized by the upregulation of genes that encode serine protease inhibitor A3N (Serpina3n) and the lysosomal cysteine protease cathepsin B (Ctsb), both of which are involved in amyloid processing. DAAs also express pro-inflammatory genes, suggesting that this novel astrocyte subset contributes to the initial stages of AD pathogenesis10. In summary, reactive astrocytes exhibit functional diversity in AD pathology, promoting neuroprotection through Aβ degradation and clearance and also contributing to pathological processes triggered by Aβ. These findings highlight the need for a deeper characterization of astrocyte subsets in AD and the mechanisms that control them.
Glutamate homeostasis.
Glutamate is the most abundant excitatory neurotransmitter in the CNS193. However, excessive glutamate levels trigger excitotoxicity194. Astrocytes express the high-affinity glutamate transporters EAAT1 and EAAT2, which take up extracellular glutamate, thereby playing a pivotal part in glutamate homeostasis, synaptic plasticity and neuron survival70,71. Not surprisingly, deficits in glutamate transporter function have been associated with neurodegeneration in AD195. Indeed, EAAT2 knock-down in the APP/PS1 AD model exacerbated cognitive decline196. In addition, several in vitro studies suggest that Aβ directly modulates glutamate uptake in astrocytes197,198. For example, Aβ treatment inhibits the expression of EAAT2 and GLAST via a mechanism dependent on astrocytic adenosine A2A receptors197. Furthermore, human astrocytes derived from patients with AD display decreased glutamate uptake concomitant with reduced EAAT2 and GLAST expression199. Evidence that such a mechanism also occurs in vivo during neurodegeneration was recently provided by the analysis of extrasynaptic glutamate dynamics using the fluorescent glutamate indicator iGluSnFR in combination with intravital two-photon laser scanning microscopy200. These studies detected decreased EAAT2 expression in reactive astrocytes concomitant with impaired glutamate uptake in association with neuronal dysfunction in the proximity of Aβ plaques in the APP/PS1 AD model. Interestingly, ceftriaxone, an antibiotic shown to upregulate EAAT2 expression201, increased glutamate uptake via EAAT2 (REF.200) (FIG. 2), supporting the targeting of astrocytic glutamate transporters as a therapeutic approach for neurodegenerative diseases.
Fig. 2 |. Targeting astrocyte signalling in Alzheimer disease.
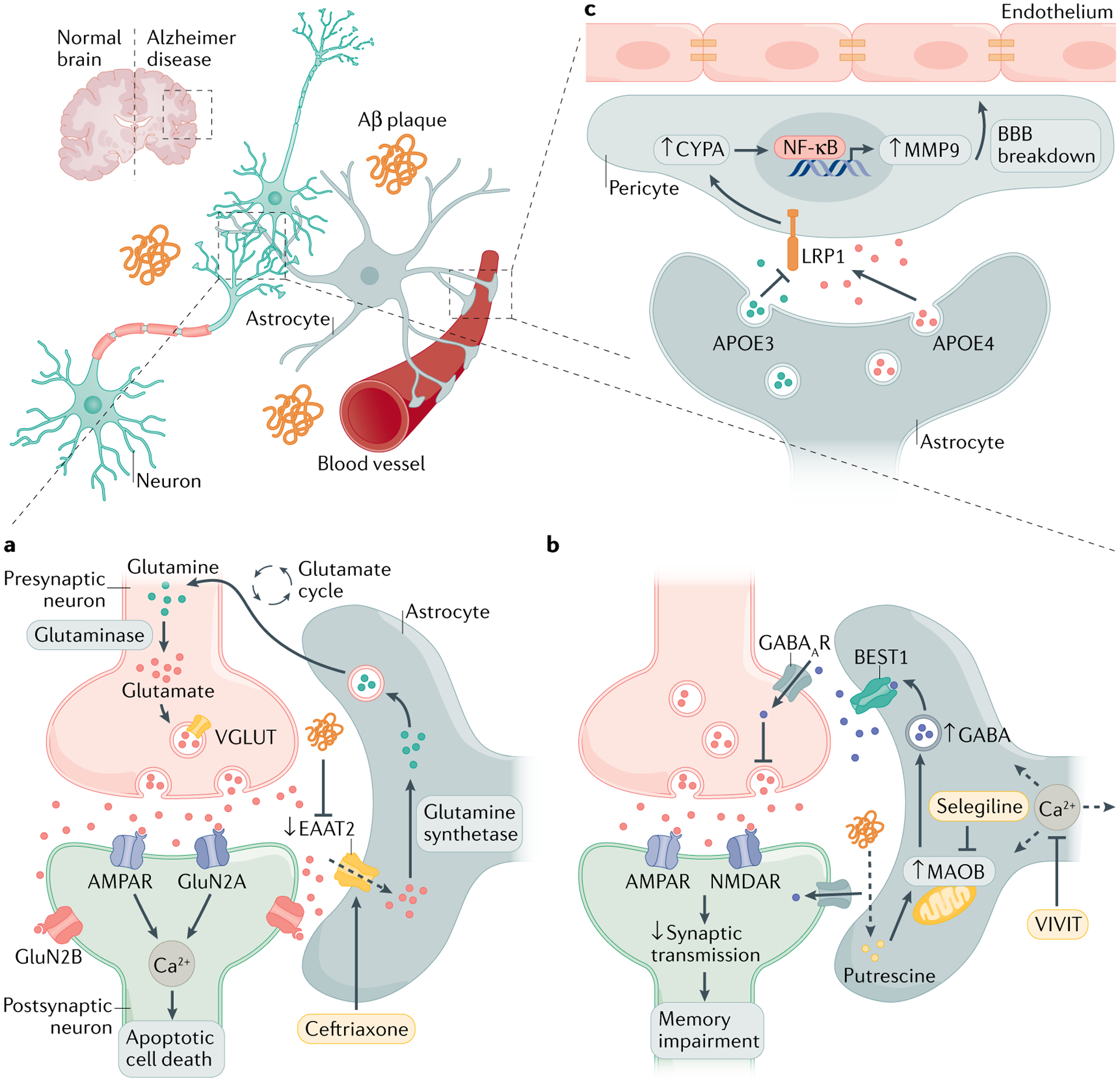
a | The accumulation of amyloid-β (Aβ) in the brain modulates glutamate uptake in astrocytes by inhibiting the glutamate transporter excitatory amino acid transporter 2 (EAAT2). This causes excessive activation of neuronally expressed glutamate receptors, which increases intracellular Ca2+ and promotes neuronal dysfunction. b | Enhanced intracellular Ca2+ waves in astrocytes increase the release of γ-aminobutyric acid (GABA), the major inhibitory neurotransmitter in the brain. Aβ plaques and putrescine trigger monoamine oxidase B (MAOB)-mediated GABA release via the Ca2+-activated anion channel bestrophin 1 (BEST1). Increased GABA uptake by neurons impairs memory and synaptic plasticity. c | Astrocytes are the main producers of apolipoprotein E (APOE), which modulates blood–brain barrier (BBB) permeability. APOE4 secretion induces cyclophilin A (CYPA)–nuclear factor-κB (NF-κB)–metalloproteinase 9 (MMP9) signalling in pericytes, which leads to BBB breakdown via the lipoprotein receptor-related protein 1 (LRP1). This APOE4-mediated pericyte activation can be negatively regulated by the production of APOE3. Strategies to rescue neuronal dysfunction mediated by glutamate dysregulation and synaptic plasticity include controlling astrocyte glutamate uptake, Ca2+ homeostasis and GABA production. AMPAR, AMPA receptor; NMDAR, N-methyl-d-aspartate receptor; VGLUT, vesicular glutamate transporter.
Calcium homeostasis.
Intracellular Ca2+ waves in astrocytes play an important part in synaptic transmission and plasticity64. Astrocytes participate in the control of long-term memory in the hippocampus via Ca2+ signalling40, suggesting a link between astrocytic Ca2+ homeostasis and AD pathology. Indeed, Aβ enhances Ca2+ signalling in astrocytes202,203, and hyperactive Ca2+ transient currents in astrocytes are detected near Aβ plaques in AD mouse models204. Conversely, intracerebroventricular injection, or astrocyte-specific overexpression, of the calcineurin–NFAT peptide inhibitor VIVIT restores glutamate dysregulation, synaptic plasticity and neuronal hyperactivity133,134, providing further support for the role of aberrant astrocyte Ca2+ signalling in AD pathogenesis.
Enhanced intracellular Ca2+ waves in astrocytes increase the release of glutamate, ATP and GABA79–84. Importantly, increased GABA levels are detected in the cerebrospinal fluid of patients with AD205. Moreover, selective blockade of GABA signalling in the APP/PS1 mouse model of AD rescues the impairment of spatial memory and hippocampal long-term potentiation (LTP)206, suggesting an important role for GABA-dependent mechanisms in AD memory impairment.
Indeed, reactive astrocytes have been reported to contribute to GABA-mediated memory loss85. When compared with homeostatic astrocytes, reactive astrocytes in close proximity to Aβ plaques release higher amounts of GABA. In APP/PS1 mice, increased monoamine oxidase B (MAOB) activity in reactive astrocytes triggers tonic GABA production via the Ca2+-activated anion channel bestrophin 1 (BEST1). The neuronal uptake of GABA via presynaptic GABA receptors results in the impairment of memory and synaptic plasticity. Interestingly, Aβ treatment induces MAOB-mediated GABA production in cultured astrocytes, suggesting that Aβ plaques are responsible for astrocytic GABA-driven memory dysfunction. Of note, the selective MAOB inhibitor selegiline limits tonic GABA release and restores impairments in spike probability, LTP and learning and memory85 (FIG. 2). These findings highlight the prominent role of astrocytic Ca2+ homeostasis in AD pathogenesis and the potential for targeting astrocyte intracellular Ca2+ signalling or GABA production as a therapeutic approach to arrest neurodegeneration in AD.
Cholesterol metabolism.
APOE participates in the transport and clearance of lipids, and the expression of certain isoforms is one of the major risk factors for AD207. Astrocytes are the main CNS producers of APOE, which transmits cholesterol to neurons via cell-surface APOE receptors. Three common APOE isoforms exist: APOE2, APOE3 and APOE4 (REF.208). A large body of evidence suggests that expression of APOE4 is the most prevalent genetic risk factor for AD, whereas APOE2 is associated with a decreased risk compared with the more common APOE3 isoform207,209,210. Subsequent studies established that APOE isoforms regulate Aβ accumulation and clearance in the brain211,212. Importantly, APOE also controls cerebrovascular integrity during neurodegeneration. For instance, individuals who carry APOE4 alleles but no signs of dementia have neurovascular dysfunction213–215. Moreover, the conditional knockout of APOE in mice causes BBB disruption216,217. Along these lines, APOE isoforms show disparate effects on BBB permeability. The inactivation of APOE4, but not of APOE2 or APOE3, results in BBB breakdown by activating CypA–NF-κB–MMP9 signalling in pericytes. This APOE4–CypA-mediated BBB breakdown induces neuronal dysfunction and degeneration. It was also shown that APOE3 secretion by astrocytes inhibits the APOE4-mediated pericyte activation responsible for BBB breakdown. Astrocyte-secreted APOE3, but not APOE4, limits CypA–NF-κB–MMP9 signalling in pericytes via lipoprotein receptor-related protein 1 (LRP1)55, thus suggesting a neuroprotective role of reactive astrocytes in APOE4-associated neurodegenerative diseases (FIG. 2). Remarkably, pathogenic DAAs upregulate Apoe expression and genes linked to Aβ metabolism and clearance in an AD mouse model10. Collectively, these findings suggest that astrocytes have important roles in APOE-dependent mechanisms of neurodegeneration and highlight the potential role of APOE as a therapeutic target in AD.
Astrocytes in Parkinson disease
PD is the second most common neurodegenerative disease; it is characterized by the loss of dopamine neurons in the substantia nigra pars compacta and abnormal aggregation of misfolded α-synuclein in Lewy bodies218 (BOX 2). The molecular mechanisms involved in the pathogenesis of PD have not been fully elucidated and there are currently limited disease-modifying strategies available. However, a large body of genome-wide association studies have identified specific genetic factors related to PD pathogenesis219. Although it is unclear whether inflammation is an initiating factor for PD, several lines of evidence suggest that pro-inflammatory glial responses contribute to PD pathogenesis32,220. For example, reactive astrocytes are detected in the substantia nigra pars compacta of patients with PD221 and preclinical models of PD222,223. In addition, nuclear receptor subfamily 4 group A member 2 (NR4A2, also known as NURR1), one of the major genetic risk factors for PD, has essential roles in the control of microglia and astrocyte neurotoxicity. NURR1 is an orphan ligand-activated transcription factor that is crucial for the development and maintenance of dopaminergic neurons. Selective downregulation of NURR1 in astrocytes results in the increased production of neurotoxic factors, including NO and ROS. These anti-inflammatory effects of NURR1 in astrocytes involve the corepressor CoREST, which cooperates with NURR1 to suppress NF-κB-driven transcriptional programmes that promote disease pathology224. Moreover, a glucagon-like peptide 1 receptor (GLP1R) agonist, a potent neuroprotective agent in neurodegenerative diseases, prevents dopaminergic neuron loss and behavioural deficits in preclinical PD mouse models by blocking the microglia-driven differentiation of C3+ neurotoxic astrocytes31. Indeed, astrocytes also contribute to PD pathology through their roles in glutamate-mediated excitotoxicity, K+ buffering and Ca2+ homeostasis, which control dopaminergic neuron death32,73,225. Collectively, these findings suggest that common mechanisms mediate the pathogenic roles of astrocytes in PD and other neurological diseases including AD and HD (BOX 3).
Astrocytes in Huntington disease
HD is a neurodegenerative disorder characterized by cognitive, motor and psychiatric symptoms that are caused by extended polyglutamine-encoding CAG repeats in the huntingtin gene (HTT)226 (BOX 2). The aggregation of misfolded mHTT is one of the hallmarks of HD and is associated with severe neurodegeneration in the striatum and the cortex226,227. Increasing evidence suggests that astrocytes express mHTT in HD and its preclinical model228–230. Indeed, selective overexpression of mHTT in mouse astrocytes results in significant motor impairment230. Conversely, the selective deletion of reactive astrocytes in HD animal models reduces neurodegeneration, suggesting that reactive astrocyte subsets contribute to HD pathogenesis231. Whether astrocytes are central drivers of neurodegeneration in HD or simply amplify it needs further investigation. However, it is now clear that mHTT accumulation leads to astrocyte dysfunction and neurodegeneration via the impairment of glutamate uptake, K+ homeostasis, Ca2+ signalling and metabolic reprogramming, all of which are mechanisms common to the pathogenesis of other neurological diseases33 (FIG. 3).
Fig. 3 |. Targeting astrocyte signalling in Huntington disease.
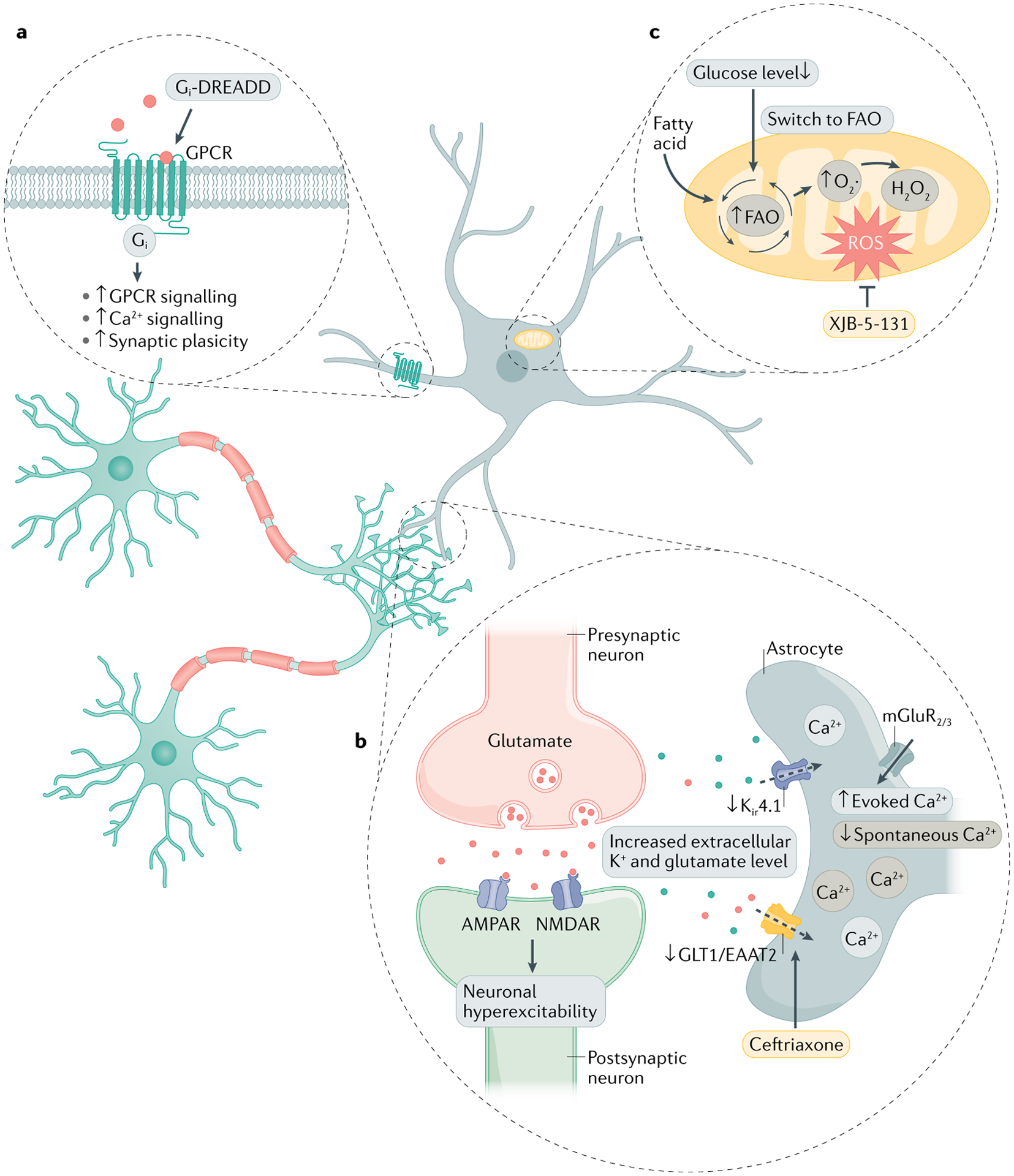
a | Selective activation of Gi–G protein-coupled receptor (GPCR) signalling pathway can recover astrocyte functional impairments and correct synaptic dysfunction. b | Mutant huntingtin protein (mHTT) inclusions in astrocytes impair the expression of excitatory amino acid transporter 2 (EAAT2) and K+ ion channel Kir4.1. Reduced glutamate transporter levels lead to robust mGlu2/3-mediated Ca2+ signalling. This gain of evoked astrocyte Ca2+ signals is followed by the loss of spontaneous Ca2+ signalling. The elevation of extracellular K+ and glutamate levels leads to neuronal hyperexcitability and the development of neurodegeneration. c | Low glucose levels in the striatum of individuals with Huntington disease (HD) triggers metabolic reprogramming in astrocytes, which switch from using glucose to fatty acids. Fatty acid oxidation (FAO) sustains energy production but also elevates levels of reactive oxygen species (ROS), which induce damage. In HD, synaptic plasticity, Ca2+ signalling and GPCR signalling is impaired in striatal astrocytes. Strategies to rescue neurodegeneration in HD include targeting astrocyte glutamate uptake, metabolism and GPCR signalling. AMPAR, AMPA receptor; NMDAR, N-methyl-d-aspartate receptor; mGluR2/3, metabotropic glutamate receptor 2/3; DREADD, designer receptor exclusively activated by designer drugs.
C3+ astrocytes have been detected in HD brain samples145. High-throughput sequencing studies have further analysed reactive astrocyte diversity in HD36,145,232, identifying additional astrocyte subsets in the cingulate cortex. These astrocyte subsets exhibit low expression of glutamate transporters (SLC1A2 and SLC1A3) and genes related to Ca2+ signalling232. Moreover, a recent study identified at least 62 differentially expressed signature genes in striatum DAAs in preclinical HD mouse models and patient samples. These DAA-associated genes were mostly related to Ca2+, glutamate and G protein-coupled receptor (GPCR) signalling, which were downregulated in HD samples. This study further identified five upstream transcriptional regulator genes (Htt, Adora2a, Mapt, Hdac4 and App), which provide candidate therapeutic targets for the control of DAAs in HD. Indeed, the selective inhibition of mHTT expression in astrocytes using zinc finger protein (ZFP) transcriptional repressors reversed the transcriptional perturbations detected in HD DAAs, highlighting the importance of mHTT in driving astrocyte responses that contribute to HD pathology36. Finally, recent scRNA-seq studies highlighted a central role for astrocyte GPCR signalling in HD pathology, as they showed that impaired synaptic plasticity, Ca2+ and GPCR signalling in striatum astrocytes could be reversed by the selective activation of Gi–GPCR signalling in astrocytes37 using designer receptors exclusively activated by designer drugs (DREADDs)233 (FIG. 3). These studies identify astrocyte GPCR signalling as a promising target to control neurodegeneration in HD. Moreover, taken together, these results point to aspects of regional astrocyte heterogeneity in HD, but also to common mechanisms of astrocyte-driven pathology across multiple neurological disorders, such as dysfunctional Ca2+ and GPCR signalling and the impairment of glutamate uptake.
Designer receptors exclusively activated by designer drugs.
(DREADDs). Widely used tool for selectively manipulating neuronal activity indirectly through g protein-coupled receptor (GPCR)-dependent signalling pathways.
Glutamate and ion homeostasis.
The glutamate transporter EAAT2 is usually downregulated in HD33, and increasing evidence suggests that this leads to astrocytic dysfunction33,230,234. For instance, mHTT expressed in astrocytes impairs EAAT2 expression by inhibiting the binding of the transcription factor SP1 to the promoter of the gene encoding EAAT2 (REF.230). Further evidence in support of dysfunctional glutamate signalling in HD astrocytes was recently provided by a study that visualized glutamate release with an intensity-based glutamate-sensing fluorescent reporter (iGluSnFR). In the R6/2 preclinical HD mouse model, striatal astrocytes exhibited reduced spontaneous Ca2+ signalling and glutamate clearance. Interestingly, EAAT2 blockade in wild-type mice induced astrocytic dysfunction similar to that detected in R6/2 mice (for example, prolonged iGluSnFR signals and enhanced mGluR2/3-mediated evoked Ca2+ signals)234, suggesting that the impairment of glutamate uptake contributes to astrocytic dysfunction in HD pathogenesis.
Furthermore, mHTT inclusions in astrocytes are associated with decreased expression of Kir4.1 in the early stages of disease in the R6/2 and Q175 HD mouse models. Kir4.1 channels control the membrane potential and excitability of neurons by promoting extracellular K+ clearance. Impaired Kir4.1 channels elevate extracellular K+ levels, leading to neuronal hyperexcitability33,34. Indeed, selective expression of Kir4.1 channels in striatal astrocytes normalizes extracellular K+ levels and HD-like symptoms, including medium spiny neuron (MSN) depolarization and abnormal motor behaviours, in preclinical models34. Further evidence to support the importance of astrocyte-mediated K+ homeostasis in HD was recently provided in a study in which restoration of Kir4.1 in striatal astrocytes partially recovered dysfunctional Ca2+ and glutamate signalling in R6/2 mice234 (FIG. 3). Collectively, these studies suggest that restoring astrocyte K+ homeostatic mechanisms may provide a therapeutic approach to control neurodegeneration in HD.
Medium spiny neuron.
(MSN). Class of inhibitory GABAergic neurons that represents ~95% of the neuronal population in the mammalian striatum.
Energy metabolism
The neuropathological hallmarks of HD include impairment of energy metabolism and oxidative stress235–237. Brain metabolism impairment in patients with HD has been detected in the striatum and cerebral cortex via positron emission tomography (PET) studies238,239, and emerging evidence suggests that dysfunctional astrocyte–neuron metabolic interactions contribute to neurodegeneration in HD91,92. For example, deficits in ascorbic acid astrocyte–neuron transport have been linked to HD symptoms in preclinical models92. Ascorbic acid is a vital antioxidant molecule in the brain240, which is taken up by neurons to inhibit glucose transport and stimulate lactate uptake. In the R6/2 mouse HD model, abnormal neuronal ascorbic acid uptake is detected in the dorsolateral striatum92,241. Moreover, in a mouse cellular model of HD, accumulation of mHTT in striatal neurons impairs ascorbic acid transporter 2 (SVCT2) translocation92, and treatment with ascorbate rescues neuronal activity and motor behaviour in R6/2 mice242,243. Thus, the impairment of neuronal ascorbic acid uptake via SVCT2 might promote neuronal metabolic failure and neurodegeneration in HD.
In addition, new evidence suggests that metabolic reprogramming in striatal astrocytes promotes neuronal death in HD. Fluorescence lifetime imaging microscopy (FLIM) revealed abnormal striatal mitochondrial dysfunction in an HdhQ(150/150) HD mouse model. In the striatum, glucose levels were low, and levels of fatty acid metabolites were elevated. Notably, this low glucose environment promotes astrocyte metabolic reprogramming, causing astrocytes to switch from glycolysis to fatty acid oxidation (FAO), which is tightly regulated to minimize the generation of ROS and neuronal hypoxia244,245. However, the accumulation of oxidized lipid bodies in the striatum of HD-like HdhQ(150/150) mice results in ROS production and neuronal toxicity. Moreover, the mitochondria-targeted electron scavenger, XJB-5-131, prevents the accumulation of lipid bodies in the brain91, providing a potential explanation for the results of a previous study in which XJB-5-131 treatment attenuated ROS production, reduced neuronal damage and recovered motor behaviours in an HD mouse model246 (FIG. 3). Taken together, these findings suggest that focusing on the link between glucose availability and striatal astrocyte metabolism may offer novel therapeutic approaches for HD.
Astrocytes in psychiatric disorders
The mechanisms described above highlight the broad contributions of astrocytes to the pathogenesis of neurodegenerative diseases. In addition, the dysregulation of astrocyte functions, such as the control of neuronal activity, release of neurotransmitters and processes associated with synaptic pruning, have all been associated with neuropsychiatric diseases. Through these and additional mechanisms, astrocytes control multiple aspects of neural circuits and behaviour42.
Synaptic pruning.
Neurodevelopmental process of eliminating neurons and synaptic connections in the brain.
The nucleus accumbens is a small forebrain area that exerts an outsized influence as a central component of the basal ganglia247. The basal ganglia consists of several GABAergic neuronal subtypes, including dopamine-responsive MSNs, which express dopamine receptors of the type I (D1R+) and type II (D2R+) classes; these receptors have opposing effects on basal ganglia outputs248. The dysfunction of dopaminergic neurotransmission in the nucleus accumbens is associated with neurodegenerative and neuropsychiatric disorders, including depression, schizophrenia and behaviours associated with fear and anxiety249–251. Hence, targeting MSNs in the nucleus accumbens is considered a potential therapeutic approach for neurological diseases.
Pioneering work from the Araque lab demonstrated that astrocytes differentially influence D1R+ and D2R+ MSN circuits in the nucleus accumbens60. Using photoactivatable Ca2+ uncaging, the authors demonstrated that astrocytes participate only in the control of homotypic MSN circuits (for example, D1R to D1R or D2R to D2R), but not heterotypic circuits (D1R to D2R). Interestingly, although astrocyte-derived Ca2+ is not crucial for the regulation of heterotypic interactions, astrocytes also participate in heterotypic MSN circuits, suggesting that additional mechanisms of astrocyte-dependent neural circuit regulation remain to be defined.
Photoactivatable Ca2+ uncaging.
Covalent attachment of a photochemical group to a biomolecule to render it inert until light irradiation releases the bond. in the case of Ca2+ uncaging, the photochemical group is attached to a Ca2+ chelator, such as EGTA.
Astrocyte Ca2+ signalling is a central mechanism in the control of neuronal function and behaviour. Khakh and collaborators38 recently demonstrated that homeostatic astrocyte Ca2+ signalling in the nucleus accumbens prevents repetitive behaviours in mice. Using an elegant combination of intravital imaging, electrophysiology and behaviour, the authors demonstrated that depletion of astrocyte Ca2+ signalling alleviates tonic inhibition of MSNs through increased activation of sodium- and chloride-dependent GABA transporter 3 (GAT3). Loss of astrocyte Ca2+ signalling led to D1R+ neuron activation via GAT3 expression, which promoted the excessive grooming commonly associated with obsessive–compulsive disorder owing to loss of GABAergic tone. Astrocyte-specific Ribotag RNA profiling252 identified Rab11a as an upstream negative regulator of GAT3 activity38. Rab11a has been reported to control GAT3 trafficking downstream of astrocyte Ca2+ influx253. Thus, a GABA-induced imbalance of astrocyte Ca2+ signalling can lead to the elevation of GAT3, mediated by the downregulation of Rab11a.
Neurons also signal to astrocytes to control behaviours associated with psychiatric diseases. Nagai et al.41 recently reported that MSN GABA release upregulates the synaptogenic cue, thrombospondin 1, in a Ca2+-dependent manner. Thrombospondin 1 induction of synapse formation potentiated corticostriatal glutamate signalling. Similar observations have been made in the context of learning and memory, during which astrocyte activation leads to the formation of hippocampal synapses, which are required for the stabilization of fear learning programmes40. A recent study proposed that defective astrocyte synapse elimination in adults leads to impaired memory formation and long-term plasticity254. These studies point to important mechanisms involved in the neuronal regulation of astrocyte synaptogenic programmes that contribute to the development of behaviours associated with anxiety, obsessive–compulsive disorder and other neuropsychiatric symptoms.
Along these lines, recent studies point to the importance of astrocyte regulation of synaptic pruning via microglia during development255,256. For instance, astrocytes promote synaptic pruning in the developing visual system via a mechanism mediated by IL-33. Microglia-dependent synaptic pruning is associated with neurodegenerative diseases such as AD, and also neuropsychiatric disorders such as schizophrenia. Pioneering work by the Barres lab demonstrated that the complement cascade participates in synaptic pruning during development257. Specifically, complement production by astrocytes was shown to trigger microglia-dependent pruning to sculpt neural circuits. Later work from Stevens and coworkers258 showed that microglial synaptic engulfment is modified by neural activity258. These insights are especially intriguing considering that the overexpression of specific complement isoforms associated with schizophrenia259 leads to dysregulated behaviour as well as dysfunctional synaptic pruning in the prefrontal cortex260. Hence, targeting dysregulated IL-33 and complement signalling may provide therapeutic avenues for disorders involving altered synapse elimination.
Astrocyte cytokine production is associated with depressive and anxiety-like behaviours261,262. For example, amygdalar adenosine signalling in astrocytes contributes to the induction of anxiety-associated phenotypes, in a region- and synapse-specific manner39. Similarly, the induction of anxiety-associated behaviours has also recently been attributed to pro-inflammatory CNS-infiltrating T cells through a metabolic programme that consists of xanthine production, which promotes adrenaline production by CD4+ T cells; adrenaline affects behaviour through direct neuronal stimulation263. These findings suggest that signalling through astrocyte-expressed GPCRs can also promote anxiety-like phenotypes. Moreover, additional Ca2+-dependent mechanisms associated with fear learning and memory40,264 may provide therapeutic targets for neuropsychiatric disease.
Beyond GPCR signalling, pro-inflammatory signals, such as those derived from pro-inflammatory T cells, can affect depressive phenotypes by acting on astrocytes. The induction of pro-inflammatory signalling pathways associated with depressive behaviours, such as the UPR26,163,265, are known to be triggered in astrocytes. These pro-inflammatory pathways can be therapeutically targeted. Indeed, NF-κB-driven pro-inflammatory signalling in astrocytes contributes to the induction of depressive behaviours266. For example, the loss of signalling by menin, a negative regulator of NF-κB267, results in increased astrocyte production of IL-1β, which promotes depressive behaviour by inducing pro-inflammatory signals in neurons268. Hence, targeting astrocyte cytokine production may offer novel therapeutic options for circuits that affect depression and related disorders.
Outlook on therapeutic strategies
Given the substantial biological insights made over the past decade, astrocytes constitute attractive therapeutic targets for neurological disorders. However, therapeutic approaches that target astrocytes, as well as other CNS-resident cells, are faced with the challenge of delivering drugs across the BBB. New developments in drug delivery suggest that this important challenge can now be properly addressed. For example, several studies have described synthetic nanoparticles that can be used for astrocyte-specific targeting269. Intraspinally administered poly(lactic-co-glycolic acid) nanoparticles are taken up by astrocytes, and persist for long periods of time in the SCI rat model270. In addition, systemically administered polyamidoamine dendrimers colocalize with astrocytes that are involved in the early progression of ischaemic injury271. Additional non-invasive molecular delivery strategies that bypass the BBB have also been developed, including receptor-mediated transcytosis, neurotropic viruses, nanoparticles and exosomes272. These exciting new approaches may facilitate the therapeutic targeting of astrocytes as described below.
Poly(lactic-co-glycolic acid) nanoparticles.
(PLGA nanoparticles). FDA-approved biodegradable polymeric nanoparticle extensively used in drug delivery systems owing to its biocompatibility and low toxicity.
Polyamidoamine dendrimers.
(PAMAM dendrimers). A class of dendrimers, hyperbranched macromolecules with numerous functional amine groups on the surface.
Metabolism.
Metabolic pathways modulate astrocyte reactivity in multiple neurological diseases and therefore may serve as viable targets for therapeutic intervention91,93,246. For example, miglustat, a FDA-approved glucosylceramide synthase (GCS) inhibitor used to treat type 1 Gaucher disease and Niemann–Pick disease type C273, ameliorates chronic progressive EAE in the NOD model by arresting immunometabolic pathways in pathogenic astrocytes93. These therapeutic effects of miglustat involve the suppression of cPLA2–MAVS signalling in astrocytes, which promotes CNS inflammation and interferes with the production of lactate involved in neuron metabolic support93. These findings also implicate other GCS inhibitors as promising candidates to modulate astrocyte responses that promote CNS inflammation and neurodegeneration. For example, venglustat, a novel CNS-penetrant oral inhibitor of GCS under investigation for the treatment of lysosomal storage diseases, may provide a useful tool to modulate astrocyte responses274.
Another metabolism-based approach to modulate astrocyte responses could be to target mitochondrial dysfunction, which has been linked with neurodegenerative diseases including MS, AD, PD and HD275,276. In HD, for example, low brain glucose levels trigger the metabolic reprogramming of striatal astrocytes, increasing ROS production and neuronal toxicity. XJB-5-131, a mitochondria-targeted antioxidant, limits ROS-induced neuronal damage mediated by astrocytes91,246, highlighting the potential of targeting astrocyte metabolism for the treatment of neurological diseases.
Physical interactions.
Cell–cell interactions play a key part in brain physiology and pathology22. For instance, astrocyte interactions with microglia control synaptic pruning, CNS inflammation and neurodegeneration15,20,145,256. Newly developed techniques allow the identification of the pathways and molecules governing CNS cell–cell interactions to systematically uncover novel therapeutic targets15,277–279. For instance, using RABID-seq, a method that combines barcoded viral tracing with scRNA-seq, we identified novel roles for axon guidance molecules (EPHB3–ephrinB3, plexinB1/2–semaphorin4D) in astrocyte–microglia interactions that promote CNS pathology in EAE and, potentially, MS. In this context, a CNS-penetrant inhibitor of EPHB3 signalling suppressed the pathogenic activities of human and mouse astrocytes in vitro and ameliorated EAE pathogenesis in acute and chronic progressive models15. These findings identify ephrinB3–EPHB3 signalling as a promising therapeutic candidate for modulating astrocyte and microglial pathogenic functions in MS, and potentially other neurological disorders as suggested by previous reports on the role of EPH signalling in the pathology of neurodegenerative diseases including AD280 and PD281.
Targeting transporters and receptors.
The glutamate transporters EAAT1 and EAAT2, which are expressed by glial cells (predominantly astrocytes), are important for neuronal survival and function70,71. Numerous studies have defined important roles for astrocytic EAAT2 in neurodegenerative diseases including AD197–200, HD33,230,234 and PD282–284. Indeed, treatment with the antibiotic ceftriaxone201, which elevates EAAT2 expression in astrocytes, restores glutamate neurotransmission and shows beneficial effects in a preclinical model of AD200, identifying EAAT2 as a promising pharmacological target for various neurodegenerative disorders.
Astrocytes also express a wide range of GPCRs, some of which can trigger intracellular Ca2+ signalling and stimulate neurotransmitter release, leading to the control of synaptic transmission285. In this context, selective activation of Gi–GPCR signalling in striatal astrocytes is reported to rescue HD-associated synaptic and behavioural phenotypes in preclinical models37. In addition, S1P receptors (which are GPCRs) promote astrocyte pathogenic responses that contribute to CNS pathology. For example, fingolimod, the first FDA-approved S1P receptor modulator, interferes with the activation of astrocytes, microglia and inflammatory monocytes, limiting disease progression in the NOD EAE model of SPMS167. Although S1P receptor modulation by fingolimod failed in the INFORMS phase III trials in primary progressive MS286; a more selective S1P receptor modulator (which targets S1P1 and S1P5), siponimod (BAF312), was recently approved for the treatment of SPMS287. Based on the emerging roles of S1P signalling and metabolism in the pathology of other neurodegenerative diseases including AD, PD and HD288, the S1P–S1P receptor axis in astrocytes constitutes an attractive candidate for therapeutic intervention in neurodegeneration.
Glia-to-neuron conversion therapy.
Astrocytes can potentially be genetically reprogrammed into functional neurons289. As astrocytes are abundant in the CNS, astrocyte reprogramming might be useful to treat neurodegeneration. Indeed, studies report promising therapeutic effects of in vivo glia-to-neuron conversion in preclinical models of major neurodegenerative diseases including AD290, PD291,292 and HD35. In HD, for example, selective inactivation of the RNA binding poly-pyrimidine tract-binding protein 1 (PTB, also known as PTBP1 (REFS293,294)) in astrocytes located in the substantia nigra and striatum resulted in the conversion of astrocytes into dopaminergic neurons, restoring dopamine levels and ameliorating motor behaviour in an animal model of PD291,292. Similarly, selective co-expression of the neuronal transcription factors neurogenic differentiation factor 1 (NEUROD1) and homeobox protein DLX2 in striatal astrocytes resulted in their conversion into GABAergic neurons and improved motor function and survival in an HD mouse model35. However, it should be noted that glia-to-neuron conversion is controversial. Using lineage tracing methods, it has recently been suggested that glia do not become neurons, but rather that endogenous neurons are labelled295,296. Overall, these studies may underscore the potential of novel regenerative medicine therapeutic interventions that target astrocytes for the treatment of neurodegenerative diseases.
Engineered probiotics.
The gut microbiota and its metabolites modulate CNS function and have been linked to several neurodegenerative diseases and neuropsychiatric disorders158,159,266,297–300. For example, in α-synuclein-overexpressing mice, a model of PD, short-chain fatty acids promote aspects of disease pathology298. Recent studies revealed that the GBA also controls astrocyte function in CNS inflammation8,20,21. For instance, the microbial tryptophan metabolite 3-indoxyl sulfate activates AHR signalling in astrocytes21 and microglia20, limiting CNS inflammation and neurodegeneration. The microbiome also educates peripheral immune cells, which then reach the CNS to control resident astrocytes, as we recently reported for the control of anti-inflammatory astrocytes by microbiome-modulated natural killer cells8. Collectively, these findings suggest that probiotics may be used to manipulate astrocyte and other CNS-resident cells in neurological diseases. In this context, recent advances in synthetic biology have led to the development of engineered probiotics that target specific molecular pathways for the treatment of inflammation, metabolic dysfunction, infection and cancer. For example, we recently developed self-tunable engineered yeast probiotics that target purinergic signalling to suppress intestinal inflammation301. Although there are no currently FDA-approved therapeutic probiotics, several engineered bacterial strains are now being evaluated in clinical trials302,303. Therefore, engineered probiotics may provide a novel approach for the therapeutic targeting of the GBA.
Conclusions and future perspectives
Recent technological developments have enabled the identification of novel astrocyte functions in health and disease, highlighting their potential as therapeutic targets. These new technologies, which include specific transgenic lines, intravital imaging, optogenetic and chemogenetic actuators, in situ sequencing, scRNA-seq and methods for the study of cell–cell interactions, have identified local, microbial and environmental factors that control diverse astrocyte subsets and hence have prominent roles in neurodegeneration. Some of these factors also offer well-defined molecular targets for therapeutic intervention in neurological diseases. However, linking transcriptionally defined astrocyte subsets in real time with neuronal activity, behaviour and disease hallmarks remains one of the great challenges in the field. Indeed, deciphering cell interactions between astrocytes and other cells may yield a greater understanding of astrocyte regulation in homeostasis, dysregulation in disease and new therapeutic targets15,277–279.
One central challenge for the therapeutic targeting of astrocytes is the existence of functional subsets with well-defined roles in health and disease that display marked differences between CNS regions, diseases or disease states (BOX 1). Interestingly, although disease-specific astrocyte triggers and responses have been identified, some astrocyte-related mechanisms of disease pathogenesis are common to multiple neurological disorders. Cross-disease mechanisms include neurotoxicity and deficits in the control of extracellular glutamate levels, recycling of K+ ions or lactate shuttling (BOX 3). The pathways regulating these and other common dysregulated astrocyte activities may offer targets to manage astrocyte-driven pathology across multiple neurological diseases. However, several fundamental questions should be addressed to enable the therapeutic targeting of astrocytes. How plastic and/or developmentally defined are astrocyte subsets? Moreover, which astrocyte subsets are shared between neurological diseases? How are they spatially distributed in the CNS? How can we compare astrocyte subsets across neurological disorders to identify common mechanisms of pathogenesis and therapeutic targets? Which experimental models are most appropriate for the functional interrogation of these subsets? And, most importantly, how do we therapeutically target specific astrocyte subsets of interest? These and other challenges emphasize the key roles of astrocytes, and their interactions, in health and disease, as well as their potential as viable targets for the treatment of neurological and neuropsychiatric disorders.
Acknowledgements
Research in the Quintana lab is supported by grants NS102807, ES02530, ES029136, AI126880 and AI149699 from the NIH; RG4111A1 from the National Multiple Sclerosis Society (to F.J.Q.), and PA-1604-08459 from the International Progressive MS Alliance. H.-G.L. was supported by a Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2021R1A6A3A14039088). M.A.W. was supported by the NIH (1K99NS114111, F32NS101790), a training grant from the NIH and Dana-Farber Cancer Institute (T32CA207201), a travelling neuroscience fellowship from the Program in Interdisciplinary Neuroscience at Brigham and Women’s Hospital, and the Women’s Brain Initiative at Brigham and Women’s Hospital.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Allen NJ & Lyons DA Glia as architects of central nervous system formation and function. Science 362, 181–185 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Freeman MR Specification and morphogenesis of astrocytes. Science 330, 774–778 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Virchow R Gesammelte Abhandlungen zur Wissenschaftlichen Medizin (Meidinger Sohn & Co., 1856). [Google Scholar]
- 4.Golgi C Contribuzione alla fina Anatomia Degli Organi Centrali del Sistema Nervosos (Tipi Fava e Garagnani, 1871). [Google Scholar]
- 5.Kölliker A Handbuch der Gewebelehre des Menschen (Wilhelm Engelmann, 1889). [Google Scholar]
- 6.Andriezen WL The neuroglia elements in the human brain. BMJ 2, 227–230 (1893). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcia-Marin V, Garcia-Lopez P & Freire M Cajal’s contributions to glia research. Trends Neurosci. 30, 479–487 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Sanmarco LM et al. Gut-licensed IFNγ+ NK cells drive LAMP1+TRAIL+ anti-inflammatory astrocytes. Nature 590, 473–479 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wheeler MA et al. MAFG-driven astrocytes promote CNS inflammation. Nature 578, 593–599 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper was the first to identify heterogeneity among astrocytes in neurological disease in an unsupervised manner using scRNA-seq and by validating a disease-associated astrocyte subset.
- 10.Habib N et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci 23, 701–706 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; Detailed analysis of disease-associated astrocytes in AD.
- 11.Zeisel A et al. Molecular architecture of the mouse nervous system. Cell 174, 999–1014.e1022 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeisel A et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Saunders A et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030.e1016 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bayraktar OA et al. Astrocyte layers in the mammalian cerebral cortex revealed by a single-cell in situ transcriptomic map. Nat. Neurosci 23, 500–509 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clark IC et al. Barcoded viral tracing of single-cell interactions in central nervous system inflammation. Science 372, eabf1230 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes the development of RABID-seq as a method for the study of astrocyte interactions in vivo using molecular barcoding and scRNA-seq. Astrocyte interactions can be mapped at single-cell resolution.
- 16.Liddelow SA & Barres BA Reactive astrocytes: production, function, and therapeutic potential. Immunity 46, 957–967 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Lanjakornsiripan D et al. Layer-specific morphological and molecular differences in neocortical astrocytes and their dependence on neuronal layers. Nat. Commun 9, 1623 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mayo L et al. IL-10-dependent Tr1 cells attenuate astrocyte activation and ameliorate chronic central nervous system inflammation. Brain 139, 1939–1957 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayo L et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat. Med 20, 1147–1156 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rothhammer V et al. Microglial control of astrocytes in response to microbial metabolites. Nature 557, 724–728 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work demonstrates for the first time that astrocyte–microglia interactions during CNS inflammation are regulated by the gut commensal flora.
- 21.Rothhammer V et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med 22, 586–597 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; First report of the control of astrocyte transcriptional programmes in CNS inflammation by the GBA via specific microbial metabolites.
- 22.Linnerbauer M, Wheeler MA & Quintana FJ Astrocyte crosstalk in CNS inflammation. Neuron 108, 608–622 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giovannoni F & Quintana FJ The role of astrocytes in CNS inflammation. Trends Immunol. 41, 805–819 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wheeler MA & Quintana FJ Regulation of astrocyte functions in multiple sclerosis. Cold Spring Harb. Perspect. Med 9, a029009 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rothhammer V & Quintana FJ Control of autoimmune CNS inflammation by astrocytes. Semin. Immunopathol 37, 625–638 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wheeler MA et al. Environmental control of astrocyte pathogenic activities in CNS inflammation. Cell 176, 581–596.e518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Itoh N et al. Cell-specific and region-specific transcriptomics in the multiple sclerosis model: focus on astrocytes. Proc. Natl Acad. Sci. USA 115, E302–E309 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spence RD et al. Estrogen mediates neuroprotection and anti-inflammatory effects during EAE through ERalpha signaling on astrocytes but not through ERbeta signaling on astrocytes or neurons. J. Neurosci 33, 10924–10933 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mathys H et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou Y et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med 26, 131–142 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yun SP et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med 24, 931–938 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Booth HDE, Hirst WD & Wade-Martins R The role of astrocyte dysfunction in Parkinson’s disease pathogenesis. Trends Neurosci. 40, 358–370 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khakh BS et al. Unravelling and exploiting astrocyte dysfunction in Huntington’s disease. Trends Neurosci. 40, 422–437 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tong X et al. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat. Neurosci 17, 694–703 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; A molecularly defined validation of a disease-associated astrocyte subset in a HD model.
- 35.Wu Z et al. Gene therapy conversion of striatal astrocytes into GABAergic neurons in mouse models of Huntington’s disease. Nat. Commun 11, 1105 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diaz-Castro B, Gangwani MR, Yu X, Coppola G & Khakh BS Astrocyte molecular signatures in Huntington’s disease. Sci. Transl. Med 11, eaaw8546 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Yu X et al. Context-specific striatal astrocyte molecular responses are phenotypically exploitable. Neuron 108, 1146–1162 e1110 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu X et al. Reducing astrocyte calcium signaling in vivo alters striatal microcircuits and causes repetitive behavior. Neuron 99, 1170–1187 e1179 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; Comprehensive report demonstrating the role of astrocyte calcium signalling in controlling behaviour and neural activity in the striatum.
- 39.Martin-Fernandez M et al. Synapse-specific astrocyte gating of amygdala-related behavior. Nat. Neurosci 20, 1540–1548 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adamsky A et al. Astrocytic activation generates de novo neuronal potentiation and memory enhancement. Cell 174, 59–71.e14 (2018). [DOI] [PubMed] [Google Scholar]; First report of bona fide astrocyte control over memory by potentiating synaptic transmission in the hippocampus.
- 41.Nagai J et al. Hyperactivity with disrupted attention by activation of an astrocyte synaptogenic cue. Cell 177, 1280–1292.e1220 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagai J et al. Behaviorally consequential astrocytic regulation of neural circuits. Neuron 109, 576–596 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Molofsky AV & Deneen B Astrocyte development: a guide for the perplexed. Glia 63, 1320–1329 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Ge WP, Miyawaki A, Gage FH, Jan YN & Jan LY Local generation of glia is a major astrocyte source in postnatal cortex. Nature 484, 376–380 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Volterra A & Meldolesi J Astrocytes, from brain glue to communication elements: the revolution continues. Nat. Rev. Neurosci 6, 626–640 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Alvarez JI, Katayama T & Prat A Glial influence on the blood brain barrier. Glia 61, 1939–1958 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abbott NJ, Ronnback L & Hansson E Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci 7, 41–53 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Mastorakos P & McGavern D The anatomy and immunology of vasculature in the central nervous system. Sci. Immunol 4, eaav0492 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Obermeier B, Daneman R & Ransohoff RM Development, maintenance and disruption of the blood-brain barrier. Nat. Med 19, 1584–1596 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sofroniew MV Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci 16, 249–263 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Foo LC et al. Development of a method for the purification and culture of rodent astrocytes. Neuron 71, 799–811 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Owens T, Bechmann I & Engelhardt B Perivascular spaces and the two steps to neuroinflammation. J. Neuropathol. Exp. Neurol 67, 1113–1121 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Engelhardt B & Coisne C Fluids and barriers of the CNS establish immune privilege by confining immune surveillance to a two-walled castle moat surrounding the CNS castle. Fluids Barriers CNS 8, 4 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alvarez JI et al. The hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 334, 1727–1731 (2011). [DOI] [PubMed] [Google Scholar]
- 55.Bell RD et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485, 512–516 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Argaw AT et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Invest 122, 2454–2468 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Argaw AT, Gurfein BT, Zhang Y, Zameer A & John GR VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc. Natl Acad. Sci. USA 106, 1977–1982 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Araque A, Parpura V, Sanzgiri RP & Haydon PG Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 22, 208–215 (1999). [DOI] [PubMed] [Google Scholar]
- 59.Chung WS, Allen NJ & Eroglu C Astrocytes control synapse formation, function, and elimination. Cold Spring Harb. Perspect. Biol 7, a020370 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martin R, Bajo-Graneras R, Moratalla R, Perea G & Araque A Circuit-specific signaling in astrocyte-neuron networks in basal ganglia pathways. Science 349, 730–734 (2015). [DOI] [PubMed] [Google Scholar]; This study validates functional astrocyte heterogeneity in the context of striatal microcircuit regulation and implicates defined pathways that controlled astrocyte–neuron communication.
- 61.Khakh BS & Sofroniew MV Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci 18, 942–952 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shigetomi E, Patel S & Khakh BS Probing the complexities of astrocyte calcium signaling. Trends Cell Biol. 26, 300–312 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Araque A et al. Gliotransmitters travel in time and space. Neuron 81, 728–739 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Volterra A, Liaudet N & Savtchouk I Astrocyte Ca2+ signalling: an unexpected complexity. Nat. Rev. Neurosci 15, 327–335 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Halassa MM & Haydon PG Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu. Rev. Physiol 72, 335–355 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Somjen GG Ion regulation in the brain: implications for pathophysiology. Neuroscientist 8, 254–267 (2002). [DOI] [PubMed] [Google Scholar]
- 67.Silver IA, Deas J & Erecinska M Ion homeostasis in brain cells: differences in intracellular ion responses to energy limitation between cultured neurons and glial cells. Neuroscience 78, 589–601 (1997). [DOI] [PubMed] [Google Scholar]
- 68.Hu ZL et al. Kir6.1/K-ATP channel on astrocytes protects against dopaminergic neurodegeneration in the MPTP mouse model of Parkinson’s disease via promoting mitophagy. Brain Behav. Immun 81, 509–522 (2019). [DOI] [PubMed] [Google Scholar]
- 69.Chen MM, Hu ZL, Ding JH, Du RH & Hu G Astrocytic Kir6.1 deletion aggravates neurodegeneration in the lipopolysaccharide-induced mouse model of Parkinson’s disease via astrocyte-neuron cross talk through complement C3-C3R signaling. Brain Behav. Immun 95, 310–320 (2021). [DOI] [PubMed] [Google Scholar]
- 70.Duan S, Anderson CM, Stein BA & Swanson RA Glutamate induces rapid upregulation of astrocyte glutamate transport and cell-surface expression of GLAST. J. Neurosci 19, 10193–10200 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Anderson CM & Swanson RA Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia 32, 1–14 (2000). [PubMed] [Google Scholar]
- 72.Schousboe A & Waagepetersen HS Role of astrocytes in glutamate homeostasis: implications for excitotoxicity. Neurotox. Res 8, 221–225 (2005). [DOI] [PubMed] [Google Scholar]
- 73.Dong X-X, Wang Y & Qin Z-H Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin 30, 379–387 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maragakis NJ & Rothstein JD Glutamate transporters: animal models to neurologic disease. Neurobiol. Dis 15, 461–473 (2004). [DOI] [PubMed] [Google Scholar]
- 75.Soni N, Reddy BV & Kumar P GLT-1 transporter: an effective pharmacological target for various neurological disorders. Pharmacol. Biochem. Behav 127, 70–81 (2014). [DOI] [PubMed] [Google Scholar]
- 76.Doble A The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacol. Ther 81, 163–221 (1999). [DOI] [PubMed] [Google Scholar]
- 77.Pitt D, Werner P & Raine CS Glutamate excitotoxicity in a model of multiple sclerosis. Nat. Med 6, 67–70 (2000). [DOI] [PubMed] [Google Scholar]
- 78.Santello M & Volterra A TNFalpha in synaptic function: switching gears. Trends Neurosci. 35, 638–647 (2012). [DOI] [PubMed] [Google Scholar]
- 79.Fiacco TA & McCarthy KD Astrocyte calcium elevations: properties, propagation, and effects on brain signaling. Glia 54, 676–690 (2006). [DOI] [PubMed] [Google Scholar]
- 80.Zorec R et al. Astroglial excitability and gliotransmission: an appraisal of Ca2+ as a signalling route. ASN Neuro 4, e00080 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee S et al. Channel-mediated tonic GABA release from glia. Science 330, 790–796 (2010). [DOI] [PubMed] [Google Scholar]
- 82.Parpura V et al. Glutamate-mediated astrocyte-neuron signalling. Nature 369, 744–747 (1994). [DOI] [PubMed] [Google Scholar]
- 83.Queiroz G, Gebicke-Haerter PJ, Schobert A, Starke K & von Kugelgen I Release of ATP from cultured rat astrocytes elicited by glutamate receptor activation. Neuroscience 78, 1203–1208 (1997). [DOI] [PubMed] [Google Scholar]
- 84.Stout CE, Costantin JL, Naus CC & Charles AC Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J. Biol. Chem 277, 10482–10488 (2002). [DOI] [PubMed] [Google Scholar]
- 85.Jo S et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med 20, 886–896 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wu Z, Guo Z, Gearing M & Chen G Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s [corrected] disease model. Nat. Commun 5, 4159 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bélanger M, Allaman I & Magistretti JP Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 14, 724–738 (2011). [DOI] [PubMed] [Google Scholar]
- 88.Magistretti PJ & Allaman I Lactate in the brain: from metabolic end-product to signalling molecule. Nat. Rev. Neurosci 19, 235–249 (2018). [DOI] [PubMed] [Google Scholar]
- 89.Sonninen TM et al. Metabolic alterations in Parkinson’s disease astrocytes. Sci. Rep 10, 14474 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Merlini M, Meyer EP, Ulmann-Schuler A & Nitsch RM Vascular beta-amyloid and early astrocyte alterations impair cerebrovascular function and cerebral metabolism in transgenic arcAbeta mice. Acta Neuropathol. 122, 293–311 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Polyzos AA et al. Metabolic reprogramming in astrocytes distinguishes region-specific neuronal susceptibility in huntington mice. Cell Metab. 29, 1258–1273 e1211 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Acuña AI et al. A failure in energy metabolism and antioxidant uptake precede symptoms of Huntington’s disease in mice. Nature Commun. 4, 2917 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chao CC et al. Metabolic control of astrocyte pathogenic activity via cPLA2-MAVS. Cell 179, 1483–1498.e1422 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ferraiuolo L et al. Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain 134, 2627–2641 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Escartin C et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci 24, 312–325 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; A recent consensus statement defining the hallmarks and open questions that surround astrocyte heterogeneity and reactivity.
- 96.Anderson MA, Ao Y & Sofroniew MV Heterogeneity of reactive astrocytes. Neurosci. Lett 565, 23–29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Safieh-Garabedian B, Mayasi Y & Saade NE Targeting neuroinflammation for therapeutic intervention in neurodegenerative pathologies: a role for the peptide analogue of thymulin (PAT). Expert Opin. Ther. Targets 16, 1065–1073 (2012). [DOI] [PubMed] [Google Scholar]
- 98.Farina C, Aloisi F & Meinl E Astrocytes are active players in cerebral innate immunity. Trends Immunol. 28, 138–145 (2007). [DOI] [PubMed] [Google Scholar]
- 99.Colombo E & Farina C Astrocytes: key regulators of neuroinflammation. Trends Immunol. 37, 608–620 (2016). [DOI] [PubMed] [Google Scholar]
- 100.Silver J & Miller JH Regeneration beyond the glial scar. Nat. Rev. Neurosci 5, 146–156 (2004). [DOI] [PubMed] [Google Scholar]
- 101.Anderson MA et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; Seminal study describing molecular mechanisms by which astrocytes control regeneration following SCI.
- 102.Anderson MA et al. Required growth facilitators propel axon regeneration across complete spinal cord injury. Nature 561, 396–400 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tak PP & Firestein GS NF-kappaB: a key role in inflammatory diseases. J. Clin. Invest 107, 7–11 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mattson MP & Camandola S NF-κB in neuronal plasticity and neurodegenerative disorders. J. Clin. Invest 107, 247–254 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kang Z et al. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity 32, 414–425 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu J & Du L PERK pathway is involved in oxygen-glucose-serum deprivation-induced NF-kB activation via ROS generation in spinal cord astrocytes. Biochem. Biophys. Res. Commun 467, 197–203 (2015). [DOI] [PubMed] [Google Scholar]
- 107.Kawai T & Akira S Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med 13, 460–469 (2007). [DOI] [PubMed] [Google Scholar]
- 108.Brambilla R et al. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J. Exp. Med 202, 145–156 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Brambilla R et al. Transgenic inhibition of astroglial NF-κB protects from optic nerve damage and retinal ganglion cell loss in experimental optic neuritis. J. Neuroinflammation 9, 213 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Brambilla R et al. Transgenic inhibition of astroglial NF-kappa B improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. J. Immunol 182, 2628–2640 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Brambilla R et al. Astrocytes play a key role in EAE pathophysiology by orchestrating in the CNS the inflammatory response of resident and peripheral immune cells and by suppressing remyelination. Glia 62, 452–467 (2014). [DOI] [PubMed] [Google Scholar]
- 112.van Loo G et al. Inhibition of transcription factor NF-kappaB in the central nervous system ameliorates autoimmune encephalomyelitis in mice. Nat. Immunol 7, 954–961 (2006). [DOI] [PubMed] [Google Scholar]
- 113.Carrero I et al. Oligomers of β-amyloid protein (Aβ1–42) induce the activation of cyclooxygenase-2 in astrocytes via an interaction with interleukin-1β, tumour necrosis factor-α, and a nuclear factor κB mechanism in the rat brain. Exp. Neurol 236, 215–227 (2012). [DOI] [PubMed] [Google Scholar]
- 114.Lian H et al. NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 85, 101–115 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hsiao HY, Chen YC, Chen HM, Tu PH & Chern Y A critical role of astrocyte-mediated nuclear factor-κB-dependent inflammation in Huntington’s disease. Hum. Mol. Genet 22, 1826–1842 (2013). [DOI] [PubMed] [Google Scholar]
- 116.Ben Haim L et al. The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer’s and Huntington’s diseases. J. Neurosci 35, 2817–2829 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ceyzeriat K, Abjean L, Carrillo-de Sauvage MA, Ben Haim L & Escartin C The complex STATes of astrocyte reactivity: how are they controlled by the JAK-STAT3 pathway? Neuroscience 330, 205–218 (2016). [DOI] [PubMed] [Google Scholar]
- 118.Zhong Z, Wen Z & Darnell JE Stat3 and Stat4: members of the family of signal transducers and activators of transcription. Proc. Natl Acad. Sci. USA 91, 4806–4810 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.He F et al. A positive autoregulatory loop of Jak-STAT signaling controls the onset of astrogliogenesis. Nat. Neurosci 8, 616–625 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shibata N et al. Activation of signal transducer and activator of transcription-3 in the spinal cord of sporadic amyotrophic lateral sclerosis patients. Neurodegener. Dis 6, 118–126 (2009). [DOI] [PubMed] [Google Scholar]
- 121.Shibata N et al. Activation of STAT3 and inhibitory effects of pioglitazone on STAT3 activity in a mouse model of SOD1-mutated amyotrophic lateral sclerosis. Neuropathology 30, 353–360 (2010). [DOI] [PubMed] [Google Scholar]
- 122.Reichenbach N et al. Inhibition of Stat3-mediated astrogliosis ameliorates pathology in an Alzheimer’s disease model. EMBO Mol. Med 11, e9665 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Haroon F et al. Gp130-dependent astrocytic survival is critical for the control of autoimmune central nervous system inflammation. J. Immunol 186, 6521–6531 (2011). [DOI] [PubMed] [Google Scholar]
- 124.Lu HC et al. STAT3 signaling in myeloid cells promotes pathogenic myelin-specific T cell differentiation and autoimmune demyelination. Proc. Natl Acad. Sci. USA 117, 5430–5441 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lu JQ, Power C, Blevins G, Giuliani F & Yong VW The regulation of reactive changes around multiple sclerosis lesions by phosphorylated signal transducer and activator of transcription. J. Neuropathol. Exp. Neurol 72, 1135–1144 (2013). [DOI] [PubMed] [Google Scholar]
- 126.Klee CB, Crouch TH & Krinks MH Calcineurin: a calcium- and calmodulin-binding protein of the nervous system. Proc. Natl Acad. Sci. USA 76, 6270–6273 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hogan PG Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 17, 2205–2232 (2003). [DOI] [PubMed] [Google Scholar]
- 128.Furman JL & Norris CM Calcineurin and glial signaling: neuroinflammation and beyond. J. Neuroinflammation 11, 158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Serrano-Perez MC et al. Response of transcription factor NFATc3 to excitotoxic and traumatic brain insults: identification of a subpopulation of reactive astrocytes. Glia 59, 94–107 (2011). [DOI] [PubMed] [Google Scholar]
- 130.Furman JL et al. Blockade of astrocytic calcineurin/NFAT signaling helps to normalize hippocampal synaptic function and plasticity in a rat model of traumatic brain injury. J. Neurosci 36, 1502–1515 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Abdul HM, Furman JL, Sama MA, Mathis DM & Norris CM NFATs and Alzheimer’s disease. Mol. Cell Pharmacol 2, 7–14 (2010). [PMC free article] [PubMed] [Google Scholar]
- 132.Caraveo G et al. Calcineurin determines toxic versus beneficial responses to α-synuclein. Proc. Natl Acad. Sci. USA 111, E3544–E3552 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Furman JL et al. Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer’s disease. J. Neurosci 32, 16129–16140 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sompol P et al. Calcineurin/NFAT signaling in activated astrocytes drives network hyperexcitability in Aβ-bearing mice. J. Neurosci 37, 6132–6148 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Choe JY, Park KY, Park SH, Lee SI & Kim SK Regulatory effect of calcineurin inhibitor, tacrolimus, on IL-6/sIL-6R-mediated RANKL expression through JAK2-STAT3-SOCS3 signaling pathway in fibroblast-like synoviocytes. Arthritis Res. Ther 15, R26 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hirano K et al. Differential effects of calcineurin inhibitors, tacrolimus and cyclosporin a, on interferon-induced antiviral protein in human hepatocyte cells. Liver Transpl. 14, 292–298 (2008). [DOI] [PubMed] [Google Scholar]
- 137.Manukyan I, Galatioto J, Mascareno E, Bhaduri S & Siddiqui MA Cross-talk between calcineurin/NFAT and Jak/STAT signalling induces cardioprotective alphaB-crystallin gene expression in response to hypertrophic stimuli. J. Cell Mol. Med 14, 1707–1716 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mencarelli A et al. Calcineurin B in CD4+ T cells prevents autoimmune colitis by negatively regulating the JAK/STAT pathway. Front. Immunol 9, 261 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Filippi M et al. Multiple sclerosis. Nat. Rev. Dis. Prim 4, 43 (2018). [DOI] [PubMed] [Google Scholar]
- 140.Faissner S, Plemel JR, Gold R & Yong VW Progressive multiple sclerosis: from pathophysiology to therapeutic strategies. Nat. Rev. Drug Discov 18, 905–922 (2019). [DOI] [PubMed] [Google Scholar]
- 141.JM C Histologie de la sclérose en plaque [Histology of multiple sclerosis]. Gaz. des. Hôpitaux 41, 554–555 (1868). [Google Scholar]
- 142.Liedtke W, Edelmann W, Chiu FC, Kucherlapati R & Raine CS Experimental autoimmune encephalomyelitis in mice lacking glial fibrillary acidic protein is characterized by a more severe clinical course and an infiltrative central nervous system lesion. Am. J. Pathol 152, 251–259 (1998). [PMC free article] [PubMed] [Google Scholar]
- 143.Voskuhl RR et al. Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J. Neurosci 29, 11511–11522 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Toft-Hansen H, Fuchtbauer L & Owens T Inhibition of reactive astrocytosis in established experimental autoimmune encephalomyelitis favors infiltration by myeloid cells over T cells and enhances severity of disease. Glia 59, 166–176 (2011). [DOI] [PubMed] [Google Scholar]
- 145.Liddelow SA et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Farez MF et al. Toll-like receptor 2 and poly(ADP-ribose) polymerase 1 promote central nervous system neuroinflammation in progressive EAE. Nat. Immunol 10, 958–964 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Moreno M et al. Conditional ablation of astroglial CCL2 suppresses CNS accumulation of M1 macrophages and preserves axons in mice with MOG peptide EAE. J. Neurosci 34, 8175–8185 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kim RY et al. Astrocyte CCL2 sustains immune cell infiltration in chronic experimental autoimmune encephalomyelitis. J. Neuroimmunol 274, 53–61 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Locatelli G et al. Mononuclear phagocytes locally specify and adapt their phenotype in a multiple sclerosis model. Nat. Neurosci 21, 1196–1208 (2018). [DOI] [PubMed] [Google Scholar]
- 150.Mills Ko E et al. Deletion of astroglial CXCL10 delays clinical onset but does not affect progressive axon loss in a murine autoimmune multiple sclerosis model. J. Neuroinflammation 11, 105 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Krumbholz M et al. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up-regulation is differentially linked to CNS immune cell recruitment. Brain 129, 200–211 (2006). [DOI] [PubMed] [Google Scholar]
- 152.Wang X, Haroon F, Karray S, Martina D & Schluter D Astrocytic Fas ligand expression is required to induce T-cell apoptosis and recovery from experimental autoimmune encephalomyelitis. Eur. J. Immunol 43, 115–124 (2013). [DOI] [PubMed] [Google Scholar]
- 153.Becher B, Tugues S & Greter M GM-CSF: from growth factor to central mediator of tissue inflammation. Immunity 45, 963–973 (2016). [DOI] [PubMed] [Google Scholar]
- 154.Croxford AL et al. The cytokine GM-CSF drives the inflammatory signature of CCR2+monocytes and licenses autoimmunity. Immun. 43, 502–514 (2015). [DOI] [PubMed] [Google Scholar]
- 155.Komuczki J et al. Fate-mapping of GM-CSF expression identifies a discrete subset of inflammation-driving T helper cells regulated by cytokines IL-23 and IL-1β. Immunity 50, 1289–1304 (2019). [DOI] [PubMed] [Google Scholar]
- 156.Wicks IP & Roberts AW Targeting GM-CSF in inflammatory diseases. Nat. Rev. Rheumatol 12, 37–48 (2016). [DOI] [PubMed] [Google Scholar]
- 157.Gutiérrez-Vázquez C & Quintana FJ Regulation of the immune response by the aryl hydrocarbon receptor. Immunity 48, 19–33 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wheeler MA, Rothhammer V & Quintana FJ Control of immune-mediated pathology via the aryl hydrocarbon receptor. J. Biol. Chem 292, 12383–12389 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rothhammer V & Quintana FJ The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol 19, 184–197 (2019). [DOI] [PubMed] [Google Scholar]
- 160.Ron D & Walter P Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol 8, 519–529 (2007). [DOI] [PubMed] [Google Scholar]
- 161.Scheper W & Hoozemans JJM The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta Neuropathol. 130, 315–331 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hetz C, Zhang K & Kaufman RJ Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol 21, 421–438 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Smith HL et al. Astrocyte unfolded protein response induces a specific reactivity state that causes non-cell-autonomous neuronal degeneration. Neuron 105, 855–866.e855 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Alaamery M et al. Role of sphingolipid metabolism in neurodegeneration. J. Neurochem 158, 25–35 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Brinkmann V et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov 9, 883–897 (2010). [DOI] [PubMed] [Google Scholar]
- 166.Choi JW et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc. Natl Acad. Sci. USA 108, 751–756 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rothhammer V et al. Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proc. Natl Acad. Sci. USA 114, 2012–2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zhang W et al. Lactate is a natural suppressor of RLR signaling by targeting MAVS. Cell 178, 176–189. e115 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Janke R, Dodson AE & Rine J Metabolism and epigenetics. Annu. Rev. Cell Dev. Biol 31, 473–496 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Jones PA Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet 13, 484–492 (2012). [DOI] [PubMed] [Google Scholar]
- 171.Huynh JL & Casaccia P Epigenetic mechanisms in multiple sclerosis: implications for pathogenesis and treatment. Lancet Neurol. 12, 195–206 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Huynh JL et al. Epigenome-wide differences in pathology-free regions of multiple sclerosis-affected brains. Nat. Neurosci 17, 121–130 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Li X, Xiao B & Chen X-S DNA methylation: a new player in multiple sclerosis. Mol. Neurobiol 54, 4049–4059 (2017). [DOI] [PubMed] [Google Scholar]
- 174.Fan G DNA methylation controls the timing of astrogliogenesis through regulation of JAK-STAT signaling. Development 132, 3345–3356 (2005). [DOI] [PubMed] [Google Scholar]
- 175.Hatada I et al. Astrocyte-specific genes are generally demethylated in neural precursor cells prior to astrocytic differentiation. PLoS ONE 3, e3189 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Katsuoka F & Yamamoto M Small Maf proteins (MafF, MafG, MafK): history, structure and function. Gene 586, 197–205 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.El-Behi M et al. The encephalitogenicity of TH17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat. Immunol 12, 568–575 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Codarri L et al. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol 12, 560–567 (2011). [DOI] [PubMed] [Google Scholar]
- 179.Long JM & Holtzman DM Alzheimer disease: an update on pathobiology and treatment strategies. Cell 179, 312–339 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Polanco JC et al. Amyloid-beta and tau complexity — towards improved biomarkers and targeted therapies. Nat. Rev. Neurol 14, 22–39 (2018). [DOI] [PubMed] [Google Scholar]
- 181.Heneka MT et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Itagaki S, McGeer PL, Akiyama H, Zhu S & Selkoe D Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J. Neuroimmunol 24, 173–182 (1989). [DOI] [PubMed] [Google Scholar]
- 183.Nalivaeva NN, Beckett C, Belyaev ND & Turner AJ Are amyloid-degrading enzymes viable therapeutic targets in Alzheimer’s disease? J. Neurochem 120, 167–185 (2012). [DOI] [PubMed] [Google Scholar]
- 184.Yan P et al. Matrix metalloproteinase-9 degrades amyloid-beta fibrils in vitro and compact plaques in situ. J. Biol. Chem 281, 24566–24574 (2006). [DOI] [PubMed] [Google Scholar]
- 185.Yin K-J et al. Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. J. Neurosci 26, 10939–10948 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lesne S et al. Transforming growth factor-beta 1 potentiates amyloid-beta generation in astrocytes and in transgenic mice. J. Biol. Chem 278, 18408–18418 (2003). [DOI] [PubMed] [Google Scholar]
- 187.Leuba G et al. Neuronal and nonneuronal quantitative BACE immunocytochemical expression in the entorhinohippocampal and frontal regions in Alzheimer’s disease. Dement. Geriatr. Cogn. Disord 19, 171–183 (2005). [DOI] [PubMed] [Google Scholar]
- 188.Blasko I et al. Costimulatory effects of interferon-gamma and interleukin-1beta or tumor necrosis factor alpha on the synthesis of Aβ1–40 and Aβ1–42 by human astrocytes. Neurobiol. Dis 7, 682–689 (2000). [DOI] [PubMed] [Google Scholar]
- 189.Zhao J, O’Connor T & Vassar R The contribution of activated astrocytes to Aβ production: implications for Alzheimer’s disease pathogenesis. J. Neuroinflammation 8, 150 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hur JY et al. The innate immunity protein IFITM3 modulates γ-secretase in Alzheimer’s disease. Nature 586, 735–740 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Keren-Shaul H et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290 e1217 (2017). [DOI] [PubMed] [Google Scholar]
- 192.Krasemann S et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581.e569 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Meldrum BS Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J. Nutr 130, 1007S–1015S (2000). [DOI] [PubMed] [Google Scholar]
- 194.Hynd MR, Scott HL & Dodd PR Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int 45, 583–595 (2004). [DOI] [PubMed] [Google Scholar]
- 195.Masliah E, Alford M, DeTeresa R, Mallory M & Hansen L Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann. Neurol 40, 759–766 (1996). [DOI] [PubMed] [Google Scholar]
- 196.Mookherjee P et al. GLT-1 loss accelerates cognitive deficit onset in an Alzheimer’s disease animal model. J. Alzheimer’s Dis 26, 447–455 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Matos M et al. Astrocytic adenosine A2A receptors control the amyloid-beta peptide-induced decrease of glutamate uptake. J. Alzheimers Dis 31, 555–567 (2012). [DOI] [PubMed] [Google Scholar]
- 198.Huang S et al. Astrocytic glutamatergic transporters are involved in Aβ-induced synaptic dysfunction. Brain Res. 1678, 129–137 (2018). [DOI] [PubMed] [Google Scholar]
- 199.Liang Z, Valla J, Sefidvash-Hockley S, Rogers J & Li R Effects of estrogen treatment on glutamate uptake in cultured human astrocytes derived from cortex of Alzheimer’s disease patients. J. Neurochem 80, 807–814 (2002). [DOI] [PubMed] [Google Scholar]
- 200.Hefendehl JK et al. Mapping synaptic glutamate transporter dysfunction in vivo to regions surrounding Aβ plaques by iGluSnFR two-photon imaging. Nat. Commun 7, 13441 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Rothstein JD et al. β-Lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 433, 73–77 (2005). [DOI] [PubMed] [Google Scholar]
- 202.Abramov AY, Canevari L & Duchen MR Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J. Neurosci 23, 5088–5095 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Lee L, Kosuri P & Arancio O Picomolar amyloid-β peptides enhance spontaneous astrocyte calcium transients. J. Alzheimers Dis 38, 49–62 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Kuchibhotla KV, Lattarulo CR, Hyman BT & Bacskai BJ Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 323, 1211–1215 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Samakashvili S et al. Analysis of chiral amino acids in cerebrospinal fluid samples linked to different stages of Alzheimer disease. Electrophoresis 32, 2757–2764 (2011). [DOI] [PubMed] [Google Scholar]
- 206.Yoshiike Y et al. GABAA receptor-mediated acceleration of aging-associated memory decline in APP/PS1 mice and its pharmacological treatment by picrotoxin. PLoS ONE 3, e3029 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Yamazaki Y, Zhao N, Caulfield TR, Liu CC & Bu G Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol 15, 501–518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Mahley RW, Weisgraber KH & Huang Y Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J. Lipid Res 50, S183–S188 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kim J, Basak JM & Holtzman DM The role of apolipoprotein E in Alzheimer’s disease. Neuron 63, 287–303 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Verghese PB, Castellano JM & Holtzman DM Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol 10, 241–252 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Holtzman DM, Herz J & Bu G Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med 2, a006312 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Kanekiyo T, Xu H & Bu G ApoE and Aβ in Alzheimer’s disease: accidental encounters or partners? Neuron 81, 740–754 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Thambisetty M, Beason-Held L, An Y, Kraut MA & Resnick SM APOE ε4 genotype and longitudinal changes in cerebral blood flow in normal aging. Arch. Neurol 67, 93–98 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Sheline YI et al. APOE4 Allele Disrupts Resting State fMRI connectivity in the absence of amyloid plaques or decreased CSF A 42. J. Neurosci 30, 17035–17040 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Reiman EM et al. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl Acad. Sci. USA 101, 284–289 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Methia N et al. ApoE deficiency compromises the blood brain barrier especially after injury. Mol. Med 7, 810–815 (2001). [PMC free article] [PubMed] [Google Scholar]
- 217.Hafezi-Moghadam A, Thomas KL & Wagner DD ApoE deficiency leads to a progressive age-dependent blood-brain barrier leakage. Am. J. Physiol. Cell Physiol 292, C1256–C1262 (2007). [DOI] [PubMed] [Google Scholar]
- 218.Poewe W et al. Parkinson disease. Nat. Rev. Dis. Prim 3, 17013 (2017). [DOI] [PubMed] [Google Scholar]
- 219.Nalls MA et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Hirsch EC & Hunot S Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 8, 382–397 (2009). [DOI] [PubMed] [Google Scholar]
- 221.Damier P, Hirsch EC, Zhang P, Agid Y & Javoy-Agid F Glutathione peroxidase, glial cells and Parkinson’s disease. Neuroscience 52, 1–6 (1993). [DOI] [PubMed] [Google Scholar]
- 222.Ciesielska A et al. The impact of age and gender on the striatal astrocytes activation in murine model of Parkinson’s disease. Inflamm. Res 58, 747–753 (2009). [DOI] [PubMed] [Google Scholar]
- 223.Morales I, Sanchez A, Rodriguez-Sabate C & Rodriguez M The astrocytic response to the dopaminergic denervation of the striatum. J. Neurochem 139, 81–95 (2016). [DOI] [PubMed] [Google Scholar]
- 224.Saijo K et al. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 137, 47–59 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Chan CS, Gertler TS & Surmeier DJ Calcium homeostasis, selective vulnerability and Parkinson’s disease. Trends Neurosci. 32, 249–256 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Bates GP et al. Huntington disease. Nat. Rev. Dis. Prim 1, 15005 (2015). [DOI] [PubMed] [Google Scholar]
- 227.Ghosh R & Tabrizi SJ Clinical features of Huntington’s disease. Adv. Exp. Med. Biol 1049, 1–28 (2018). [DOI] [PubMed] [Google Scholar]
- 228.Hebb MO, Denovan-Wright EM & Robertson HA Expression of the Huntington’s disease gene is regulated in astrocytes in the arcuate nucleus of the hypothalamus of postpartum rats. FASEB J. 13, 1099–1106 (1999). [DOI] [PubMed] [Google Scholar]
- 229.Shin JY et al. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J. Cell Biol 171, 1001–1012 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Bradford J et al. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc. Natl Acad. Sci. USA 106, 22480–22485 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Wood TE et al. Mutant huntingtin reduction in astrocytes slows disease progression in the BACHD conditional Huntington’s disease mouse model. Hum. Mol. Genet 28, 487–500 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Al-Dalahmah O et al. Single-nucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathol. Commun 8, 19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Roth BL DREADDs for neuroscientists. Neuron 89, 683–694 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Jiang R, Diaz-Castro B, Looger LL & Khakh BS Dysfunctional calcium and glutamate signaling in striatal astrocytes from Huntington’s disease model mice. J. Neurosci 36, 3453–3470 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Mochel F & Haller RG Energy deficit in Huntington disease: why it matters. J. Clin. Invest 121, 493–499 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Manoharan S et al. The role of reactive oxygen species in the pathogenesis of Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease: a mini review. Oxid. Med. Cell Longev 2016, 8590578 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Kumar A & Ratan RR Oxidative stress and Huntington’s disease: the good, the bad, and the ugly. J. Huntingtons Dis 5, 217–237 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Leenders KL, Frackowiak RS, Quinn N & Marsden CD Brain energy metabolism and dopaminergic function in Huntington’s disease measured in vivo using positron emission tomography. Mov. Disord 1, 69–77 (1986). [DOI] [PubMed] [Google Scholar]
- 239.Kuwert T et al. Cortical and subcortical glucose consumption measured by PET in patients with Huntington’s disease. Brain 113, 1405–1423 (1990). [DOI] [PubMed] [Google Scholar]
- 240.May JM, Qu ZC & Mendiratta S Protection and recycling of α-tocopherol in human erythrocytes by intracellular ascorbic acid. Arch. Biochem. Biophys 349, 281–289 (1998). [DOI] [PubMed] [Google Scholar]
- 241.Rebec GV, Barton SJ & Ennis MD Dysregulation of ascorbate release in the striatum of behaving mice expressing the Huntington’s disease gene. J. Neurosci 22, RC202 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Rebec GV, Barton SJ, Marseilles AM & Collins K Ascorbate treatment attenuates the Huntington behavioral phenotype in mice. Neuroreport 14, 1263–1265 (2003). [DOI] [PubMed] [Google Scholar]
- 243.Rebec GV, Conroy SK & Barton SJ Hyperactive striatal neurons in symptomatic Huntington R6/2 mice: variations with behavioral state and repeated ascorbate treatment. Neuroscience 137, 327–336 (2006). [DOI] [PubMed] [Google Scholar]
- 244.Schonfeld P & Reiser G Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood Flow. Metab 33, 1493–1499 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Barber CN & Raben DM Lipid metabolism crosstalk in the brain: glia and neurons. Front.Cell. Neurosci 13, 212 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Polyzos A et al. Mitochondrial targeting of XJB-5-131 attenuates or improves pathophysiology in HdhQ150 animals with well-developed disease phenotypes. Hum. Mol. Genet 25, 1792–1802 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Bostan AC & Strick PL The basal ganglia and the cerebellum: nodes in an integrated network. Nat. Rev. Neurosci 19, 338–350 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Kreitzer AC & Malenka RC Striatal plasticity and basal ganglia circuit function. Neuron 60, 543–554 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Liu C, Goel P & Kaeser PS Spatial and temporal scales of dopamine transmission. Nat. Rev. Neurosci 22, 345–358 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Horga G & Abi-Dargham A An integrative framework for perceptual disturbances in psychosis. Nat. Rev. Neurosci 20, 763–778 (2019). [DOI] [PubMed] [Google Scholar]
- 251.Russo SJ & Nestler EJ The brain reward circuitry in mood disorders. Nat. Rev. Neurosci 14, 609–625 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Sanz E et al. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc. Natl Acad. Sci. USA 106, 13939–13944 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Zhang YV, Ormerod KG & Littleton JT Astrocyte Ca2+ influx negatively regulates neuronal activity. eNeuro 4, ENEURO.0340–16.2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Lee JH et al. Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis. Nature 590, 612–617 (2021). [DOI] [PubMed] [Google Scholar]; Groundbreaking study reporting that astrocytes, not microglia, are primarily responsible for activity-induced synaptic pruning controlling memory in the adult hippocampus.
- 255.Nguyen PT et al. Microglial remodeling of the extracellular matrix promotes synapse plasticity. Cell 182, 388–403.e315 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Vainchtein ID et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 359, 1269–1273 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Stevens B et al. The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178 (2007). [DOI] [PubMed] [Google Scholar]
- 258.Schafer DP et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Sekar A et al. Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Yilmaz M et al. Overexpression of schizophrenia susceptibility factor human complement C4A promotes excessive synaptic loss and behavioral changes in mice. Nat. Neurosci 24, 214–224 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Pfau ML, Menard C & Russo SJ Inflammatory mediators in mood disorders: therapeutic opportunities. Annu. Rev. Pharmacol. Toxicol 58, 411–428 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Hodes GE, Kana V, Menard C, Merad M & Russo SJ Neuroimmune mechanisms of depression. Nat. Neurosci 18, 1386–1393 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Fan KQ et al. Stress-induced metabolic disorder in peripheral CD4+ T cells leads to anxiety-like behavior. Cell 179, 864–879.e819 (2019). [DOI] [PubMed] [Google Scholar]
- 264.Kol A et al. Astrocytes contribute to remote memory formation by modulating hippocampal-cortical communication during learning. Nat. Neurosci 23, 1229–1239 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Ii Timberlake M & Dwivedi Y Linking unfolded protein response to inflammation and depression: potential pathologic and therapeutic implications. Mol. Psychiatry 24, 987–994 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Cruz-Pereira JS et al. Depression’s unholy trinity: dysregulated stress, immunity, and the microbiome. Annu. Rev. Psychol 71, 49–78 (2020). [DOI] [PubMed] [Google Scholar]
- 267.Leng L et al. Menin deficiency leads to depressive-like behaviors in mice by modulating astrocyte-mediated neuroinflammation. Neuron 100, 551–563.e557 (2018). [DOI] [PubMed] [Google Scholar]
- 268.DiSabato DJ et al. Interleukin-1 receptor on hippocampal neurons drives social withdrawal and cognitive deficits after chronic social stress. Mol. Psychiatry 26, 4770–4782 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Zhang F, Lin YA, Kannan S & Kannan RM Targeting specific cells in the brain with nanomedicines for CNS therapies. J. Control. Rel 240, 212–226 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Wang YC et al. Sustained intraspinal delivery of neurotrophic factor encapsulated in biodegradable nanoparticles following contusive spinal cord injury. Biomaterials 29, 4546–4553 (2008). [DOI] [PubMed] [Google Scholar]
- 271.Nance E et al. Systemic dendrimer-drug treatment of ischemia-induced neonatal white matter injury. J. Control. Rel 214, 112–120 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Terstappen GC, Meyer AH, Bell RD & Zhang W Strategies for delivering therapeutics across the blood-brain barrier. Nat. Rev. Drug Discov 20, 362–383 (2021). [DOI] [PubMed] [Google Scholar]
- 273.Venier RE & Igdoura SA Miglustat as a therapeutic agent: prospects and caveats. J. Med. Genet 49, 591–597 (2012). [DOI] [PubMed] [Google Scholar]
- 274.Peterschmitt MJ et al. Pharmacokinetics, pharmacodynamics, safety, and tolerability of oral venglustat in healthy volunteers. Clin. Pharmacol. Drug Dev 10, 86–98 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Arun S, Liu L & Donmez G Mitochondrial biology and neurological diseases. Curr. Neuropharmacol 14, 143–154 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Witte ME, Mahad DJ, Lassmann H & van Horssen J Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol. Med 20, 179–187 (2014). [DOI] [PubMed] [Google Scholar]
- 277.Giladi A et al. Dissecting cellular crosstalk by sequencing physically interacting cells. Nat. Biotechnol 38, 629–637 (2020). [DOI] [PubMed] [Google Scholar]
- 278.Pasqual G et al. Monitoring T cell-dendritic cell interactions in vivo by intercellular enzymatic labelling. Nature 553, 496–500 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Turczyk BM et al. Spatial sequencing: a perspective. J. Biomol. Tech 31, 44–46 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Cisse M et al. Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature 469, 47–52 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Nkiliza A et al. RNA-binding disturbances as a continuum from spinocerebellar ataxia type 2 to Parkinson disease. Neurobiol. Dis 96, 312–322 (2016). [DOI] [PubMed] [Google Scholar]
- 282.Chung EK, Chen LW, Chan YS & Yung KK Downregulation of glial glutamate transporters after dopamine denervation in the striatum of 6-hydroxydopamine-lesioned rats. J. Comp. Neurol 511, 421–437 (2008). [DOI] [PubMed] [Google Scholar]
- 283.Chotibut T et al. Ceftriaxone reduces L-dopa-induced dyskinesia severity in 6-hydroxydopamine Parkinson’s disease model. Mov. Disord 32, 1547–1556 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Zhang Y et al. Regulation of glutamate transporter trafficking by Nedd4–2 in a Parkinson’s disease model. Cell Death Dis. 8, e2574 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Khakh BS & McCarthy KD Astrocyte calcium signaling: from observations to functions and the challenges therein. Cold Spring Harb. Perspect. Biol 7, a020404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Lublin F et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet 387, 1075–1084 (2016). [DOI] [PubMed] [Google Scholar]
- 287.Kappos L et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet 391, 1263–1273 (2018). [DOI] [PubMed] [Google Scholar]
- 288.Grassi S et al. Sphingosine 1-phosphate receptors and metabolic enzymes as druggable targets for brain diseases. Front. Pharmacol 10, 807 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Wei ZD & Shetty AK Treating Parkinson’s disease by astrocyte reprogramming: progress and challenges. Sci. Adv 7, eabg3198 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Guo Z et al. In vivo direct reprogramming of reactive glial cells into functional neurons after brain injury and in an Alzheimer’s disease model. Cell Stem Cell 14, 188–202 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Qian H et al. Reversing a model of Parkinson’s disease with in situ converted nigral neurons. Nature 582, 550–556 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Zhou H et al. Glia-to-neuron conversion by CRISPR-CasRx alleviates symptoms of neurological disease in mice. Cell 181, 590–603.e516 (2020). [DOI] [PubMed] [Google Scholar]
- 293.Xue Y et al. Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits. Cell 152, 82–96 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Xue Y et al. Sequential regulatory loops as key gatekeepers for neuronal reprogramming in human cells. Nat. Neurosci 19, 807–815 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Wang LL et al. Revisiting astrocyte to neuron conversion with lineage tracing in vivo. Cell 184, 5465–5481 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Blackshaw S et al. Ptbp1 deletion does not induce glia-to-neuron conversion in adult mouse retina and brain. bioRxiv 10.1101/2021.10.04.462784 (2021). [DOI] [Google Scholar]
- 297.Needham BD, Kaddurah-Daouk R & Mazmanian SK Gut microbial molecules in behavioural and neurodegenerative conditions. Nat. Rev. Neurosci 21, 717–731 (2020). [DOI] [PubMed] [Google Scholar]
- 298.Sampson TR et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167, 1469–1480.e1412 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Aho VTE et al. Gut microbiota in Parkinson’s disease: temporal stability and relations to disease progression. EBioMedicine 44, 691–707 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Yang D et al. The role of the gut microbiota in the pathogenesis of Parkinson’s disease. Front. Neurol 10, 1155 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Scott BM et al. Self-tunable engineered yeast probiotics for the treatment of inflammatory bowel disease. Nat. Med 27, 1212–1222 (2021). [DOI] [PubMed] [Google Scholar]
- 302.Charbonneau MR, Isabella VM, Li N & Kurtz CB Developing a new class of engineered live bacterial therapeutics to treat human diseases. Nat. Commun 11, 1738 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Aggarwal N, Breedon AME, Davis CM, Hwang IY & Chang MW Engineering probiotics for therapeutic applications: recent examples and translational outlook. Curr. Opin. Biotechnol 65, 171–179 (2020). [DOI] [PubMed] [Google Scholar]
- 304.Deneen B et al. The transcription factor NFIA controls the onset of gliogenesis in the developing spinal cord. Neuron 52, 953–968 (2006). [DOI] [PubMed] [Google Scholar]
- 305.Hochstim C, Deneen B, Lukaszewicz A, Zhou Q & Anderson DJ Identification of positionally distinct astrocyte subtypes whose identities are specified by a homeodomain code. Cell 133, 510–522 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; An early report of transcriptional programmes that regionally specify the establishment of astrocyte subsets.
- 306.Molofsky AV et al. Astrocyte-encoded positional cues maintain sensorimotor circuit integrity. Nature 509, 189–194 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work shows that spinal cord astrocytes exhibit dorsal–ventral heterogeneity, which dictates the patterning of sensory and motor neuron projections to the spinal cord.
- 307.Tsai HH et al. Regional astrocyte allocation regulates CNS synaptogenesis and repair. Science 337, 358–362 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; Functional validation of regionally defined astrocyte subsets that contribute to the establishment of CNS domains.
- 308.Chai H et al. Neural circuit-specialized astrocytes: transcriptomic, proteomic, morphological, and functional evidence. Neuron 95, 531–549.e539 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Khakh BS & Deneen B The emerging nature of astrocyte diversity. Annu. Rev. Neurosci 42, 187–207 (2019). [DOI] [PubMed] [Google Scholar]
- 310.Mews P et al. Alcohol metabolism contributes to brain histone acetylation. Nature 574, 717–721 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Ayata P et al. Epigenetic regulation of brain region-specific microglia clearance activity. Nat. Neurosci 21, 1049–1060 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Wendeln A-C et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556, 332–338 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Baecher-Allan C, Kaskow BJ & Weiner HL Multiple sclerosis: mechanisms and immunotherapy. Neuron 97, 742–768 (2018). [DOI] [PubMed] [Google Scholar]
- 314.Reich DS, Lucchinetti CF & Calabresi PA Multiple Sclerosis. N. Engl. J. Med 378, 169–180 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Selkoe DJ & Hardy J The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med 8, 595–608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Sevigny J et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 537, 50–56 (2016). [DOI] [PubMed] [Google Scholar]
- 317.Selkoe DJ Alzheimer disease and aducanumab: adjusting our approach. Nat. Rev. Neurol 15, 365–366 (2019). [DOI] [PubMed] [Google Scholar]
- 318.Gu XL et al. Astrocytic expression of Parkinson’s disease-related A53T α-synuclein causes neurodegeneration in mice. Mol. Brain 3, 12 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Arzberger T, Krampfl K, Leimgruber S & Weindl A Changes of NMDA receptor subunit (NR1, NR2B) and glutamate transporter (GLT1) mRNA expression in Huntington’s disease–an in situ hybridization study. J. Neuropathol. Exp. Neurol 56, 440–454 (1997). [DOI] [PubMed] [Google Scholar]
- 320.Kofuji P & Newman EA Potassium buffering in the central nervous system. Neuroscience 129, 1045–1056 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Guttenplan KAW et al. Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature 599, 102–107 (2021). [DOI] [PubMed] [Google Scholar]; A recent study reporting that long-chain saturated lipids are one mechanism of astrocyte-induced neurodegeneration.