

Abstract
Background:
Pediatric patients with high-risk hematologic malignancies who experience relapse after a prior allogeneic hematopoietic cell transplant (HCT) have an exceedingly poor prognosis. A second allogeneic HCT offers the potential for long-term cure but carries high risks of both subsequent relapse and HCT-related morbidity and mortality. Using haploidentical donors for HCT (haploHCT) can expand the donor pool and potentially enhance the graft-versus-leukemia effect but is accompanied by a risk of graft-versus-host disease (GVHD).
Objectives:
The goal of this protocol was to intensify the antileukemia effect of haploHCT for pediatric patients with hematologic malignancies that relapsed after prior allogeneic HCT, while limiting regimen-associated toxicities.
Study Design:
This phase II clinical trial evaluated a sub-myeloablative preparative regimen consisting of anti-thymocyte globulin (ATG), clofarabine, cytarabine, busulfan, and cyclophosphamide, in combination with plerixafor to sensitize leukemic blasts. Participants received a mobilized peripheral blood unmanipulated haploidentical donor graft with one dose of post-transplant cyclophosphamide as GVHD prophylaxis, followed by NK cell addback. Here we report the clinical outcomes and immune reconstitution of 17 participants treated on the study, and 5 additional patients treated on similar single-patient treatment plans.
Results:
Of the 22 participants analyzed, 12 (55%) had active disease at the time of HCT. The regimen provided robust immune reconstitution, with 21 participants (95%) experiencing neutrophil engraftment at a median of 14 days post HCT. In this high-risk population, the overall survival was 45% (95% CI: 24%–64%), with a 12-month event-free survival of 31% (95% confidence interval [CI]: 14%–51%) and cumulative incidence of relapse at 12 months of 50% (95% CI: 27%–69%). Four participants (18%) remain in remission at >5 years follow-up. Expected HCT-related organ-specific toxicities were observed, and 13 participants (59%) experienced acute and/or chronic GVHD.
Conclusions:
This intensified but sub-myeloablative regimen, followed by a high-dose unmanipulated haploidentical graft, post-transplant cyclophosphamide, and NK Cell infusion, resulted in adequate immune reconstitution but failed to overcome the elevated risks of relapse and treatment-related morbidity in this high-risk population.
INTRODUCTION
Pediatric patients with high-risk hematologic malignancies who experience relapse after an allogeneic hematopoietic cell transplant (HCT) have an exceedingly poor prognosis, with reported long-term survival rates of 10%–20% [1–5]. A second allogeneic HCT offers the potential for long-term survival and patients who experience remission after the first HCT have the best chance of long-term remission [1, 6–11]. Second HCT strategies in this heavily pre-treated population can fail due to either disease recurrence or high transplant-related mortality (TRM). A leading strategy to reduce TRM is using a sub-myeloablative preparative regimen, although this may be accompanied by a higher risk of relapse [12–14]. Therefore, a well-tolerated but intensive conditioning regimen that maintains substantial anti-leukemia effect and results in rapid immune recovery to protect against infection while limiting the risk of serious transplant-related complications such as graft-versus-host disease (GVHD) is ideal for a second HCT.
In the setting of second HCTs for relapsed hematologic malignancies, haploidentical donors offer several advantages. Most pediatric patients have a readily available and motivated haploidentical family donor who can proceed rapidly to HCT, and greater degrees of HLA mismatch may maximize the graft-versus-leukemia (GVL) effect [15–18]. Haploidentical HCT (haploHCT) has become a primary HCT option for patients who lack an HLA-matched donor [19–26]. To mitigate the risk of severe GVHD after a highly mismatched HCT, many haploHCT platforms use ex vivo T-cell depletion [19, 22, 27–31]. However, although this is effective in limiting GVHD, it may also dampen the GVL effect and delay T-cell immune reconstitution. Haploidentical HCT donor grafts without ex-vivo T cell depletion (also called unmanipulated) accompanied by post-transplant cyclophosphamide or other pharmacologic GVHD prophylaxis is now an established approach, with increasing experience in pediatric patients [21, 32–41].
In this phase II clinical trial, we sought to intensify the GVL effect of subsequent HCT in pediatric patients with high-risk hematologic malignancies that relapsed after the first allogeneic HCT. Our approach incorporated a high dose mobilized peripheral blood unmanipulated haploidentical graft, an intensified but sub-myeloablative preparative regimen, and natural killer cell addback to provide antiviral and antileukemic benefits. This anti-thymocyte globulin (ATG)-containing, sub-myeloablative regimen consisted of clofarabine, cytarabine, busulfan, and cyclophosphamide. Because of the anticipated high-risk population including patients with active disease, plerixafor was added to disrupt the interaction of leukemic cells with the marrow microenvironment and, potentially, to sensitize blasts to the effect of chemotherapy [42]. Our group and others have demonstrated the tolerability of adding plerixafor to an HCT conditioning regimen, and preclinical evidence supports potential mobilization and sensitization of leukemic blasts [43]. Pharmacologic GVHD prophylaxis with 1 dose of post-infusion high-dose cyclophosphamide, along with tacrolimus and mycophenolate mofetil, was based on previously reported regimens [24, 44, 45]. Herein we report the clinical outcomes and describe the immune reconstitution of the 17 participants treated on the study (NCT01621477) and of 5 additional patients who were treated on similar single-patient treatment plans.
METHODS
Participant Selection
Patients aged ≤21 years with hematologic malignancies that had relapsed or remained refractory after prior allogeneic HCT and for whom a suitable partially matched family member donor was available were enrolled on the protocol. Donors were at least single haplotype matched (≥3 of 6), but not HLA identical or 8 of 8 phenotypically matched. All participants had a new donor for second HCT (donor 2). Additional eligibility criteria included left ventricular ejection fraction >40%, glomerular filtration rate ≥70 mL/min/1.73m2, forced vital capacity ≥40% of the predicted value, Karnofsky or Lansky performance score ≥ 50, total bilirubin ≤1.5 times the upper limit of normal for age, and alanine aminotransferase ≤3 times the upper limit of normal for age. Exclusion criteria included active malignancy other than that for which transplant was indicated, active CNS leukemia, uncontrolled infection, active bronchiolitis obliterans (BOS) or cryptogenic organizing pneumonia (previously termed bronchiolitis obliterans organizing pneumonia), and pregnancy or breastfeeding. The phase II protocol was approved by the Institutional Review Board (IRB) of St. Jude Children’s Research Hospital. The study is registered at clinicaltrials.gov (NCT01621477). Written informed consent was obtained from the participant, parent, or guardian, in addition to assent from the participant when applicable. In addition to the 17 participants treated on the study, this analysis includes 5 additional patients treated on single-patient treatment plans. These patients were treated either before or after study was open to accrual, but would otherwise have met eligibility criteria
Treatment
The preparative regimen consisted of clofarabine at 40 mg/m2/day and cytarabine at 1 g/m2/day on days −9 and −8, busulfan at 0.8 mg/kg/dose every 6 hours and plerixafor at 0.24 mg/kg/day on days −7 and −6, cyclophosphamide at 15 mg/kg/day on day −5, and rabbit anti-thymocyte globulin at 1 mg/kg/day on day −4 and 3 mg/kg/day on days −3, −2, and −1. The hematopoietic progenitor cell (HPC) product was administered on day 0, and NK cell product was administered on day +6 (Figure 1A). GVHD prophylaxis included cyclophosphamide at 50 mg/kg/day on day +4 and tacrolimus and mycophenolate mofetil starting on day +11. Day +4 was chosen for cyclophosphamide administration so exposure would occur 4 days and 3 days following graft infusion for participants who would receive HPC products on Days 0 and +1 if 2 apheresis sessions were required to collect goal CD34+ cell dose. Additional supportive care and prophylaxis were administered according to institutional standard operating procedures.
Figure 1. Treatment scheme and clinical outcomes.
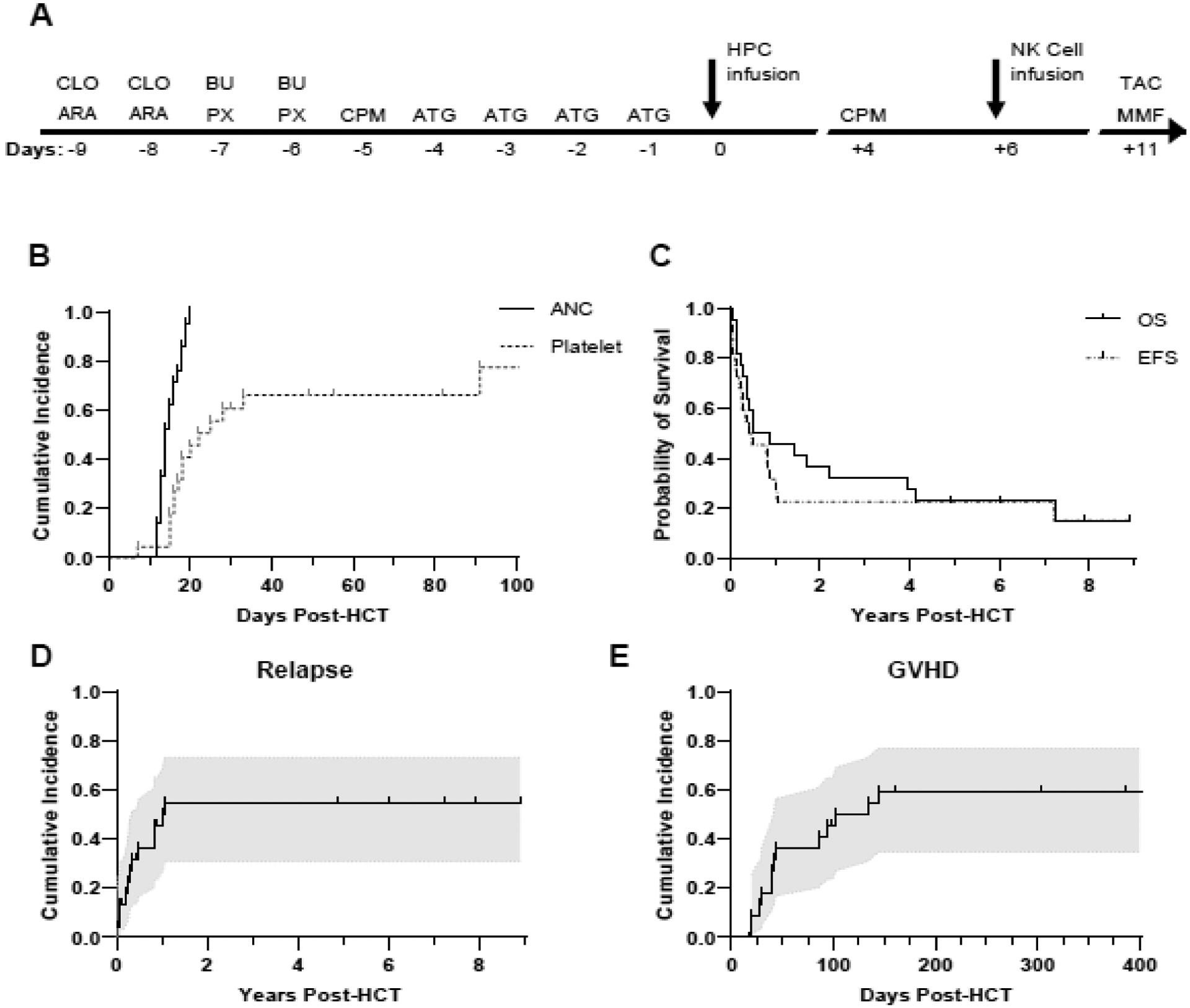
A. Protocol treatment scheme. CLO = clofarabine; ARA = cytarabine; BU = busulfan; PX = plerixafor; CPM = cyclophosphamide; ATG = anti-thymocyte globulin; HPC = hematopoietic progenitor cell; NK = natural killer; TAC = tacrolimus; MMF = mycophenolate mofetil. B. Time from HCT to first occurrence of neutrophil or platelet engraftment, censored at the time of relapse with death as a competing event. ANC = absolute neutrophil count. Median time to neutrophil engraftment was 14 days. Median time to platelet engraftment was 23.5 days. C. Overall survival (OS) and event-free survival (EFS) after HCT, censored at the time of last clinical follow-up. EFS is defined as the time from HCT to relapse of primary disease or death from any cause. Median OS was 0.72 years; median EFS was 0.47 years. D. Time from HCT to relapse of primary disease, censoring participants without relapse at the time of last clinical follow-up and accounting for death as a competing event. E. Time from HCT to first occurrence of GVHD, censoring participants without GVHD at the time of relapse or last clinical follow-up, whichever occurred first, and accounting for death as a competing event. Shaded regions indicate 95% confidence intervals.
The graft source was an unmanipulated G-CSF−mobilized peripheral blood progenitor cell product, with a target dose of ≥5 × 106 CD34+ cells/kg of recipient weight, obtained in 1 or 2 apheresis sessions. There was no maximum CD3+ cell dose. Donors underwent an additional non-mobilized leukapheresis for collection of the NK cell product on day +5. The NK cell product underwent CD3+ depletion and CD56+ enrichment with a CliniMACS cell-separation system (Miltenyi Biotec, Auburn, CA), used under a Food and Drug Administration Investigational Device Exemption [46]. Goal CD56+ cell dose > 2×106/kg (min 0.1×106/kg, max 400 ×106/kg), with maximum T cell dose of 0.05×106 CD3+CD56− cells/kg. Participants were eligible to receive additional conventional donor lymphocyte infusions (DLIs) for decreasing donor chimerism, serious viral reactivation, or evidence of residual or recurrent disease. Patients treated on single-patient treatment plans did not receive plerixafor or the NK cell infusion, but otherwise received identical treatment to patients treated on the protocol.
Post-Transplant Evaluations (Response and Toxicity Criteria)
Outcomes are censored as of August 2020. Adverse events were assessed using the CTCAE Version 3.0. Participants were followed for all NCI grade III-V adverse events from the start of the preparative regimen through the first-year post HCT, in addition to clinically significant NCI grade I or II adverse events judged to be possibly related to the protocol treatment and all infections. Acute and chronic GVHD were graded according to standard criteria [47, 48] and recorded through the time of most recent follow-up. Bone marrow evaluations were performed approximately day +21 and day +100, at 1 year post HCT, and additionally as clinically indicated because of mixed donor chimerism or concern for relapse. Complete blood counts were performed daily through engraftment, peripheral blood chimerism was assessed via variable-number tandem-repeat analysis weekly through day +100 and monthly thereafter, and lymphocyte subsets were quantified monthly. All reported chimerisms reference unsorted peripheral blood evaluations. Participants were monitored with weekly surveillance PCR for cytomegalovirus (CMV), Epstein–Barr virus (EBV), and adenovirus (ADV) through day +100.
Correlative Assays
Lymphocyte subset quantification was performed on peripheral blood using a CLIA-certified flow cytometric assay. For participants treated on the protocol, peripheral blood samples were analyzed for Vβ spectratyping as a marker of T cell receptor diversity and T cell receptor excision circles (TRECs) as a surrogate of thymopoiesis at months 2, 3, 6, and 12, using previously described methods [49]. HLA and killer immunoglobulin-like receptor (KIR) typing was performed as previously described and analyzed using the receptor–ligand model, identifying inhibitory KIR on donor NK cells and assessing the recipient’s HLA repertoire for the presence or absence of pertinent KIR ligands[50]. Detection or loss of patient-specific HLA alleles was assessed by HLA typing of post-transplant relapsed bone marrow DNA when available using sequence specific primers (SSP) (Invitrogen, Waltham, MA) [51, 52].
Statistical Analysis
Statistical analysis was performed using SAS 9.4 and figures generated using GraphPad Prism 9. Summary statistics are reported for continuous variables. Survival analyses calculations were performed using the Kaplan–Meier method and survival distributions were compared with the use of the log-rank test. Overall survival (OS) was defined as the time from HCT until death from any cause. Event-free survival (EFS) was defined as the time from HCT to relapse of primary disease or death from any cause, whichever occurred first. Relapse was defined as any evidence of hematologic, cytogenetic, or molecular recurrence of primary disease. Participants who never cleared leukemia post-HCT were noted as having relapse at 0.001 for the purpose of EFS and cumulative incidence of relapse analyses. Graft failure was defined as ANC not exceeding 500 cells/μL for 3 consecutive days post HCT with less than 5% donor 2 chimerism by day 28 (primary) or decline in ANC below 500 cells/μL accompanied by decline in donor 2 chimerism to less than 5% after prior engraftment (secondary). Surviving participants were censored at the time of last follow-up for OS and EFS. Cumulative incidence functions were estimated by the Kalbafleisch-Prentice method accounting for death as a competing risk, censoring participants without the event at the time of relapse or last follow-up, whichever occurred first, and was compared between groups with the use of Gray’s test. Cumulative incidences of neutrophil and platelet engraftment and GVHD were defined as the time from HCT until the first occurrence of the event, with neutrophil and platelet engraftment defined according to standard criteria [53]. The cumulative incidence of relapse was defined as the time from HCT until the event. Cumulative incidence of GVHD was defined as time from HCT until first diagnosis of GVHD, acute or chronic. Association of incidence of GVHD with protocol status was assessed with Fisher’s Exact test and continuous variables were compared between groups with Wilcoxon signed-rank test. Impact of NK cell dose on clinical outcomes assessed using proportional hazards regression model. Correlative assays were analyzed using 1-way ANOVA, with the Tukey–Kramer test being used for multiple comparisons between groups. TREC data was log transformed prior to statistical analysis. P values less than 0.05 were considered to indicate statistical significance.
RESULTS
Participants and Treatment
The characteristics of the 22 patients included in the analysis are presented in Table 1, with participant-specific details being noted in Table 2. All participants had received 1 prior allogeneic HCT. One participant had relapsed juvenile myelomonocytic leukemia (JMML), 1 participant had refractory acute myeloid leukemia (AML) with induction failure, 12 participants had relapsed AML, and 8 participants had relapsed acute lymphoid leukemia (ALL). Twelve participants (55%) had active disease by morphology at the time of transplant. Of the 10 participants in morphologic complete remission (marrow blast < 5%), 2 had detectable disease by flow cytometry–based minimal residual disease (MRD) assessment or RT-PCR. All participants received a graft from a haploidentical parental donor, providing a median dose of 24.65 × 106 CD34+ cells/kg (range, 4.37–115.56 × 106 CD34+ cells/kg) with a median CD3+ content of 1239.32 × 106 cells/kg (range, 186.37–3893.45 × 106 CD3+ cells/kg). The 17 participants treated on the protocol received NK cell product with a median CD56+ dose of 14.9 × 106 cells/kg and negligible CD3+ content. Natural killer (NK) cell alloreactivity was predicted in 10 of 18 evaluated recipient–donor pairs (56%) [50, 54]. Subsequent conventional DLIs were administered for mixed chimerism (UPNs 1, 9, 12, 17, and 19) or recurrent disease (UPNs 11, 12, and 16). Of participants receiving DLI for mixed chimerism, 2 corrected to 100% donor 2 after DLI. Specific indications, dosages, and response are detailed in Table S1. CD34+ boosts were administered for persistent severe leukopenia despite 100% donor 2 chimerism (UPN 7) or recurrent disease with (UPN 9) or without (UPN 12) graft failure (Table 2).
Table 1.
Patient Summary (N = 22, except as otherwise indicated)
| Age | |
| Median (range) | 6 y 11 m (11 m-19 y 8 m) |
| Sex | |
| Female | 13 (59%) |
| Male | 9 (41%) |
| Race | |
| White | 18 (82%) |
| Non-White | 4 (18%) |
| Black | 1 |
| Asian | 1 |
| Multiple | 2 |
| Ethnicity | |
| Hispanic | 1 (5%) |
| Non-Hispanic | 21 (95%) |
| Disease | |
| ALL | 8 (36%) |
| B cell | 7 |
| T cell (early T-cell precursor) | 1 |
| AML | 13 (59%) |
| JMML | 1 (5%) |
| Disease status | |
| Refractory/induction failure | 1 (5%) |
| CR1 | 1 (5%) |
| Detectable disease | 1 |
| Subsequent CR | 9 (41%) |
| Detectable disease | 1 |
| Active relapse | 11 (50%) |
| HLA match status | |
| 4 of 8 | 15 (68%) |
| 5 of 8 | 5 (23%) |
| 6 of 8 | 1 (5%) |
| 7 of 8 | 1 (5%) |
| Donor | |
| Mother | 13 (59%) |
| Father | 9 (41%) |
| CMV status (donor/patient) | |
| Negative/negative | 6 (27%) |
| Positive/positive | 11 (50%) |
| Positive/negative | 1 (5%) |
| Negative/positive | 4 (18%) |
| KIR receptor—ligand mismatch (N = 18) | |
| Yes | 10 (56%) |
| No | 8 (44%) |
| HPC product (106/kg) | |
| CD34+ median (range) | 24.65 (4.37−115.56) |
| CD3+ median (range) | 1,239.32 (186.37−3,893.45) |
| NK cell-enriched product (106/kg) (N = 17) | |
| CD56+ median (range) | 14.9 (2.69−70.73) |
| CD3+ CD56− median (range) | 0 (0−0.002) |
ALL = acute lymphoid leukemia; AML = acute myeloid leukemia; JMML = juvenile myelomonocytic leukemia; CR = complete remission; KIR = killer cell immunoglobulin-like receptor; HPC = hematopoietic progenitor cell
Table 2.
Specific Patient Characteristics
| UPN | Sex | Diagnosis | Pre-HCT Status, BM Disease Burden | Plerixafor + NK Cells | Subsequent Cell Infusions | PB Chimeris m %Donor Day +30 | Acute GVHD | Chronic GVHD | Outcome | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Organs | Grade | Organs | Grade | ||||||||
| 1 | F | AML/MDS | CR1, 3% | N | DLI (7) | 100% | Alive, CR | ||||
| 2 | F | B-ALL | CR2, 1 × 10−5* | N | 100% | Skin | I | Relapse | |||
| 3 | F | B-ALL | CR2, 2% | N | 100% | Gut, skin | II | Skin | Mild | TRM | |
| 4 | M | B-ALL | Relapse 1, 8% | N | 100% | Gut, skin | IV | TRM | |||
| 5 | F | AML | Relapse 1, Hypocellular^ | Y | 99–100% | Skin | I | Relapse | |||
| 6 | F | AML | CR2, 2% | Y | 100% | GI, oral, MSK, skin, lung | Severe | Alive,CR | |||
| 7 | M | B-ALL | Relapse 2,72% | Y | Boost(1) | 100% | Liver | IV | Relapse | ||
| 8 | F | AML | Relapse 1,16% | Y | 99–100% | Liver | III | TRM | |||
| 9 | F | B-ALL | CR2,<0.01% | Y | DLI (3) Boost (2) | 100% | TRM | ||||
| 10 | F | B-ALL | CR2,<0.01% | Y | NE | TRM | |||||
| 11 | F | AML | Relapse 1,41% | Y | DLI (2) | 100% | Relapse | ||||
| 12 | M | AML | Relapse 1,57% | Y | DLI (3) Boost (1) | 9% | Refractory | ||||
| 13 | M | AML | Relapse 1,63% | Y | 90% | Skin | I | Refractory | |||
| 14 | F | AML | Relapse 1,76% | Y | 100% | Relapse | |||||
| 15 | M | AML | CR2, 1% | Y | 100% | Skin | I | Relapse | |||
| 16 | M | AML | Relapse 1,61% | Y | DLI (1) | NE | Refractory | ||||
| 17 | M | T-ALL | CR2,<0.01% | Y | DLI (2) | 100% | Relapse | ||||
| 18 | M | AML | Relapse 1,43% | Y | 100% | Relapse | |||||
| 19 | F | AML | Ref, PIF, 12% | Y | DLI (2) | 100% | Skin, gut | III | Relapse | ||
| 20 | M | JMML | Relapse 1,0%$ | Y | 100% | Gut, liver, skin | III | Oral, skin, eye | Moderate | Alive,CR | |
| 21 | F | B-ALL | CR3,<0.01% | Y | 100% | Gut, skin | II | Lung | Severe | Alive,CR | |
| 22 | F | AML | CR3,1 × 10−3* | N | 100% | Eye, skin, gut | Moderate | TRM | |||
UPN = unique participant number; HCT = hematopoietic cell transplant; PB = peripheral blood, unsorted; GVHD = graft-versus-host disease; BM = bone marrow; F = female; M = male; AML = acute myeloid leukemia; MDS = myelodysplastic syndrome; ALL = acute lymphoid leukemia; JMML = juvenile myelomonocytic leukemia; CR = complete remission; Ref = refractory; PIF = partial induction failure; DLI = donor lymphocyte infusion; Y = yes; N = no; N/A = not available; boost = HPC product; NE = not evaluable; TRM = treatment-related mortality; MSK = musculoskeletal; mod = moderate
Detectable by PCR, negative by flow cytometry.
Leukemic blasts present
Left-shifted myeloid maturation, anemia, thrombocytopenia, and falling chimerism with no increased blasts
Outcomes
Twenty-one participants (95%) experienced neutrophil engraftment, at a median of 14 days post HCT (range 7–91 days) (Figure 1B). Fifteen participants (68%) experienced platelet engraftment, at a median of 23.5 days post HCT (range 12–20 days) (Figure 1B). Eighteen participants (82%) had >99% donor 2 chimerism on day +30, 2 were non-evaluable because of TRM or relapse, and 2 had mixed chimerism in the setting of refractory disease (Table 2). Four participants (18%) remain alive and in remission with >5 years of follow-up, with a median OS of 0.72 years (range 0.08–5.99 years) and a median EFS of 0.47 years (range 0.05 – 8.9 years) (Figure 1C). The 12-month OS and EFS are 45% (95% CI: 24%–64%) and 31% (95% CI: 14%–51%), respectively. The cumulative incidence of relapse at 12 months was 50% (95% CI: 27%–69%) (Figure 1D). Three participants had progressive disease and did not experience remission after HCT. One of 6 evaluated participants (UPN 5) had evidence of haplotype loss at the time of relapse. This participant received a maternal graft, and at the time of relapse on day +82, the paternal haplotype was not detected in a bone marrow sample. Those participants with progressive disease or subsequent relapse are all now deceased.
Regimen-Related Toxicity and GVHD
Adverse events were recorded during the 12-month monitoring period, and chronic GVHD was additionally recorded through the last clinical follow-up. Common and notable toxicities are listed in Table 3, with expected organ-specific toxicities. Fever was the most common adverse event, occurring in 19 participants (86%), and was attributed to multiple etiologies. Two participants had grade 3–4 cellular infusion reactions with HPC product infusion; there were no grade 3–4 reactions to the NK cell product. A total of 11 unique participants (50%) experienced pulmonary complications, including acute respiratory distress syndrome, idiopathic pulmonary syndrome or respiratory failure, pulmonary hemorrhage, and pleural effusion. Four participants (18%) developed hepatic veno-occlusive disease (VOD)/sinusoidal obstruction syndrome (SOS), including 1 case that was grade 5 VOD/SOS and was associated with multiorgan system failure.
Table 3.
Common and Notable Toxicities
| Toxicity | # Patients |
|---|---|
| Systemic | |
| Fever | 19 (86%) |
| Cytokine release syndrome—ATG (grade 3 or 4) | 3 (14%) |
| Cellular infusion reaction (grade 3 or 4) | 2 (10%) |
| Engraftment syndrome (grade 3 or 4) | 4 (19%) |
| Graft-versus-host disease | 13 (59%) |
| Acute | 11 (50%) |
| Grade I | 4 |
| Grade II | 2 |
| Grade III | 3 |
| Grade IV | 2 |
| Chronic | 5 (23%) |
| Mild | 1 |
| Moderate | 2 |
| Severe | 2 |
| Pulmonary | |
| ARDS/IPS/respiratory failure | 4 (18%) |
| Pulmonary hemorrhage | 4 (18%) |
| Pleural effusion | 5 (23%) |
| Hepatic/GI | |
| Hepatic VOD/SOS | 4 (18%) |
| Mucositis (grade 3 or 4) | 2 (9%) |
| Hematologic/vascular | |
| Venous thrombosis | 2 (9%) |
| Thrombotic microangiopathy | 2 (9%) |
| Cardiovascular | |
| Pericardial effusion | 2 (9%) |
| Cardiomyopathy/heart failure | 3 (14%) |
| Hypertension (grade 3 or 4) | 8 (36%) |
| Renal/genitourinary | |
| Hemorrhagic cystitis | 6 (27%) |
| Renal failure (grade 3 or 4) | 3 (14%) |
| Neurologic | |
| Seizure | 2 (9%) |
| Infectious diseases | |
| Bacteremia | 8 (36%) |
| Candidemia | 2 (9%) |
| Urinary tract infection | 3 (14%) |
| Upper respiratory tract infection | 2 (9%) |
| Enteritis | 11 (50%) |
| Viremia—by surveillance PCR | |
| ADV | 0 (0%) |
| CMV | 8 (36%) |
| EBV | 16 (73%) |
| Received rituximab treatment | 5 |
ATG = anti-thymocyte globulin; ARDS = acute respiratory distress syndrome; IPS = idiopathic pneumonia syndrome; VOD = veno-occlusive disease; SOS = sinusoidal obstruction syndrome; PCR = polymerase chain reaction; ADV = adenovirus; CMV = cytomegalovirus; EBV = Epstein–Barr virus
Renal/genitourinary complications included hemorrhagic cystitis in 6 participants (27%) and grade 3 or 4 renal failure in 3 (14%). Nine unique participants (41%) experienced cardiovascular events, including pericardial effusion, cardiomyopathy or heart failure, and hypertension. Two participants (9%) experienced seizure, 1 while receiving busulfan and 1 in the setting of clinical concern for viral encephalitis but negative infectious testing. Bacteremia was detected in 8 participants (36%) and candidemia in 2 (9%). Surveillance PCR detected CMV viremia in 8 participants (36%), with a median peak of 2.72 log10 copies/μg (range, <2.7–4.69 log10 copies/μg). None of these patients had detectable CMV viremia before HCT. EBV viremia was detected in 18 participants (82%), with a median peak of 1.44 log10 copies/μg (range, <1–4.79 log10 copies/μg). Five of these patients had detectable EBV viremia before HCT. Despite the high incidence of EBV viremia, only 5 patients required treatment with rituximab. No adenovirus viremia was detected, despite documented localized respiratory and enteric adenovirus infections (1 and 10 cases, respectively).
GVHD was diagnosed in 13 (59%) of the 22 participants, with 11 (50%) experiencing acute GVHD. Of the 14 participants alive without relapse at day +100, 5 (36%) developed chronic GVHD (Table 3), including two who developed chronic GVHD without antecedent acute GVHD (Table 2). Neither CD3+ nor CD34+ cell dose correlated with incidence of GVHD (Table S2). Chronic GVHD was mild in 1 participant, moderate in 2, and severe in 2. The cumulative incidence of acute and/or chronic GVHD at 12 months was 59% (range, 35%–77%) (Figure 1E). At last follow-up, all 4 of the long-term survivors remain in remission and 3 have experienced GVHD. One participant (UPN 1) has no evidence of GVHD and requires medical management of chemotherapy-induced cardiomyopathy, with good functional status. Another participant (UPN 6) has ventilatory-dependent BOS in addition to multifactorial dialysis-dependent chronic renal failure. Another surviving participant (UPN 21) developed BOS and requires noninvasive supplemental oxygen support. One participant (UPN 20) had chronic GVHD of the mouth, skin, and eyes that is now quiescent.
Immune Reconstitution
Quantitation of lymphocyte subsets revealed robust immune reconstitution (Figure 2A, F), with 13 of 16 evaluable participants (81%) having normal CD8+ T cell levels at 3 months and all 5 evaluable participants having normal levels at 12 months (Figure 2B). CD4+ T-cell recovery lagged, with all evaluable participants having levels below the normal limits at 3 months, 2 of 10 evaluable participants (20%) having normal levels at 6 months, and 2 of 5 evaluable participants (40%) having normal levels at 12 months (Figure 2C). B-cell recovery was intermediate, with 8 of 16 evaluable participants (50%) exhibiting normal levels by 3 months post HCT (Figure 2D). The early NK cell levels at 1 month reflected variability, with a median of 325 cells/μL (range, 10–2410 cells/μL) in participants who received NK cell product and a median of 100 cells/μL (range, 0–350 cells/μL) in patients who did not receive the NK cell product (Figure 2E). The levels of TRECs were low at 2- and 3-months post HCT relative to donors (2 months p < 0.0001, 3 months p < 0.0001), began recovering at 6 months (p = 0.0032), and were comparable to healthy donors at 12 months post HCT (p = 0.9999) (Figure 3A). This is paralleled in the Vβ spectratyping score, representing decreased clonal diversity of T cells at 2–3 months post HCT (p = 0.0020 compared to donors at 2 months, p = 0.0024 at 3 months) that improved at 6 months (p = 0.0281) and 12 months post HCT (Figure 3B).
Figure 2. Immune reconstitution post HCT.
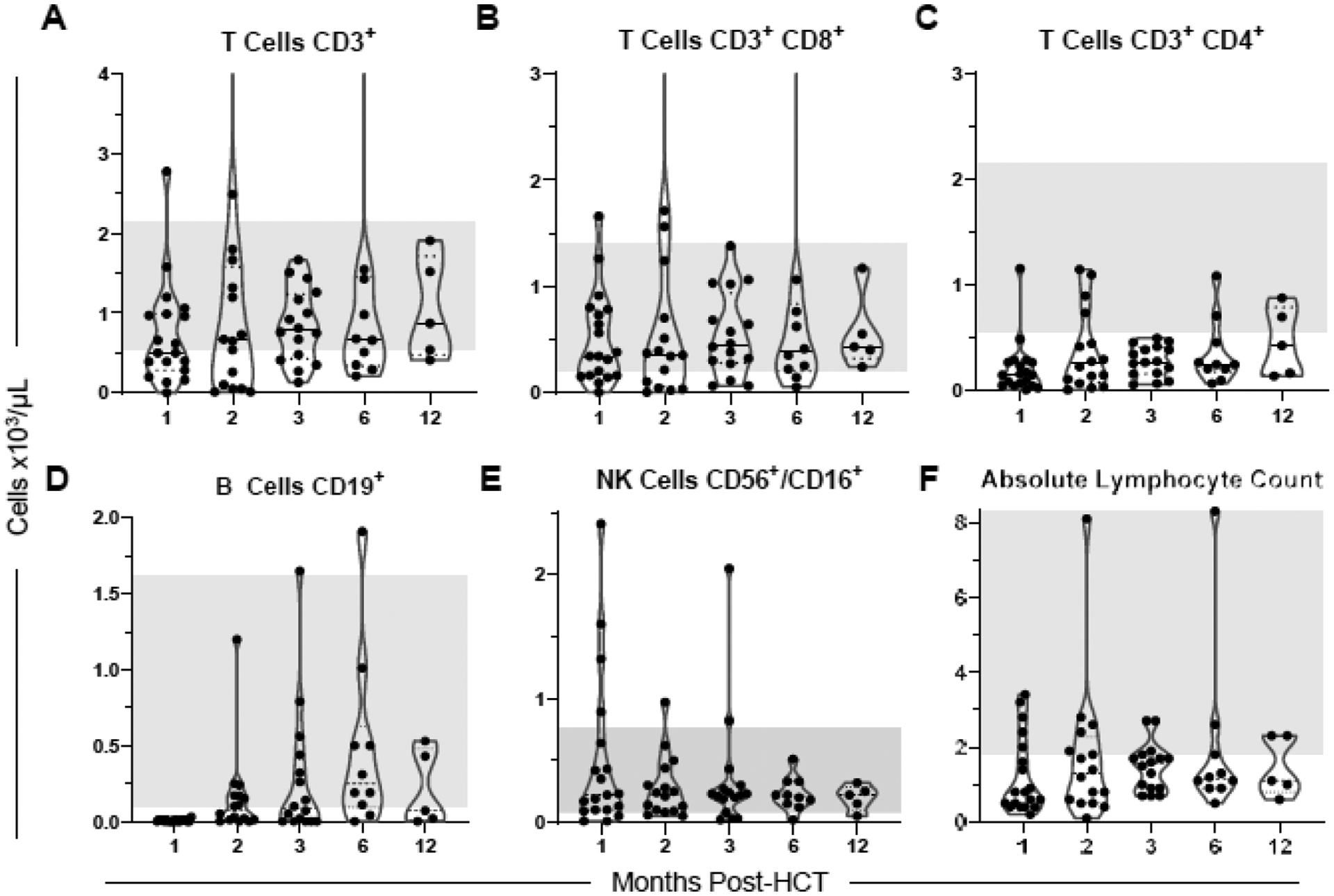
Results of monthly flow cytometry–based quantitation of lymphocyte subsets, documenting reconstitution of total CD3+ T cells (A, 2 points above axis limit: 7.61 at 2 months, 6.14 at 6 months), CD8+ T cells (B, 2 points above axis limit: 6.48 at 2 months, 4.81 at 6 months), CD4+ T cells (C), CD19+ B cells (D), CD56+/CD16+ NK cells (E), and absolute lymphocyte count (F). Shaded regions indicate normal values.
Figure 3. Characteristics of T-cell content post HCT.
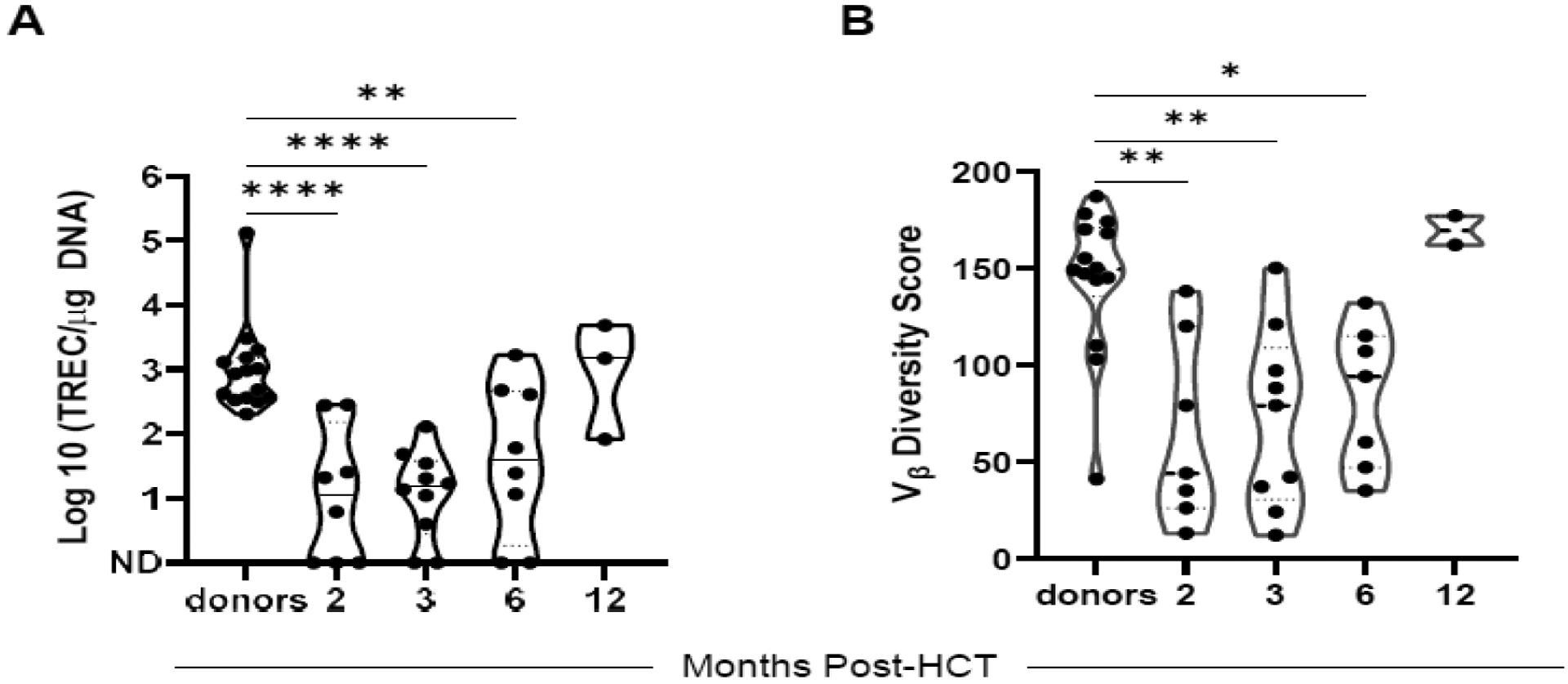
A. De novo T-cell production represented as T-cell receptor excision circles (TRECs)/μg DNA. ND = no TREC detected. An additional 2 participants had an insufficient quantity for analysis at month 2. B. Vβ spectratyping representing the diversity of the T-cell repertoire. Participants were evaluated at months 2, 3, 6, and 12 post HCT and compared to healthy donors (A, B). Results were analyzed using 1-way ANOVA with the Tukey–Kramer test for multiple comparisons. * P ≤ 0.05, ** P ≤ 0.01, **** P ≤ 0.0001
Impact of Cell Dose and Protocol Status on Clinical Outcomes
No statistically significant differences were noted in primary clinical outcomes between participants treated on protocol and those treated on single patient treatment plans, who did not receive plerixafor or NK cells (Table 4). This includes EFS (p = 0.276), OS (p = 0.383), cumulative incidence of relapse (p = 0.069), incidence of GVHD (any occurrence p = 0.3602, acute p = 1.0, chronic p = 0.9051), Of participants who experienced engraftment, those treated on single patient treatment plans had shorter median duration to ANC engraftment (12.0 vs. 15.5 days, p = 0.0018), but no difference was observed in duration to platelet engraftment based on protocol status (25.0 vs. 17.5 days, p = 0.6115). In addition, for participants who received NK cell addback, there was no statistically significant correlation between CD56+ cell dose and EFS (HR 1.004, p = 0.8184), OS (HR 1.012; p = 0.5102), or relapse risk (HR 1.008; p = 0.4926) (Table S2).
Table 4.
| SURVIVAL1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| EFS ± SE(%) | OS ± SE(%) | Relapse Cin ± SE(%) | ||||||||
| n | year 5 | p-value | year 5 | p-value | year 5 | p-value | ||||
| Protocol | 17 | 17.6±9.2 | 0.276 | 17.6±9.2 | 0.383 | 76.6±12.9 | 0.069 | |||
| Non-Protocol | 5 | 40.0±17.9 | 40.0±17.9 | 33.3±33.3 | ||||||
| GVHD2 | ||||||||||
| Any | Acute | Chronic# | ||||||||
| n | No | Yes | No | Yes | No | Yes | ||||
| Protocol | 17 | 8 | 9 | 9 | 8 | 7 | 3 | |||
| Non-Protocol | 5 | 1 | 4 | 2 | 3 | 2 | 2 | |||
| p = 0.3602 | p = 1.00 | p = 0.9051 | ||||||||
| ENGRAFTMENT3 | |||||||
|---|---|---|---|---|---|---|---|
| n | Experienced ANC >500 | Days to ANC >500 cells/μL | |||||
| Mean | Median | SD | Range | p-value | |||
| Protocol | 17 | 16 (94%) | 15.9 | 15.5 | 2.3 | 13–20 | **0.0018 |
| Non-Protocol | 5 | 5 (100%) | 12.4 | 12.0 | 0.5 | 12–13 | |
| n | Experienced Platelets >20 | Days to Platelets >20×103 cells/μL | |||||
| Mean | Median | SD | Range | p-value | |||
| Protocol | 17 | 12 (71%) | 24.0 | 17.5 | 21.9 | 7.0–91.0 | 0.6115 |
| Non-Protocol | 5 | 3 (60%) | 22.7 | 25.0 | 6.8 | 15.0–28.0 | |
Survival distributions compared using log-rank test. Cumulative incidence compared using Gray’s test.
Association of GVHD incidence with protocol status assessed with Fisher’s Exact test.
Continuous variables compared between groups with Wilcoxon signed-rank test.
of patients alive at day +100
EFS = event free survival; SE = standard error; OS = overall survival; Cin = cumulative incidence; ANC = absolute neutrophil count
DISCUSSION
In this high-risk population, increasing the dose of CD34+ and CD3+ cells by using a mobilized peripheral blood graft, adding more NK cells, and incorporating an intensified preparative regimen were not sufficient to overcome the relapse risk and were associated with treatment-related adverse effects and a high rate of GVHD. The overall survival in this cohort is comparable to the expected outcomes for pediatric patients experiencing relapse after HCT [1–5]. Despite advances in improving the tolerability of a second HCT, the key determinant of EFS and long-term success remains the ability to induce a subsequent remission [6–11]. Our cohort, which included 12 participants with active disease and 2 others in morphologic remission but with detectable MRD, for a total of 64% with evidence of disease, represented a group with an exceedingly high risk of relapse for whom no other curative therapy options were available. Of the participants with active disease, 3 did not experience complete remission after HCT. Despite attempts to optimize the GVL effect by using mismatched donors for patients with active disease [55, 56], pediatric trials with haploidentical donors for second HCTs have either confirmed that patients with active disease continue to have poor outcomes [31] or have required patients to be in remission after re-induction as a condition of enrollment [57]. Loss of the mismatched haplotype has been identified as a mechanism of relapse after a haploidentical transplant [52, 58], although this was only recently shown to be the case for the post-transplant cyclophosphamide platform [59]. Our report of a participant with haplotype loss provides additional support that this mechanism can occur after haploHCT, regardless of the platform.
This therapeutic strategy added treatment-related morbidity and GVHD without the apparent benefit of additional GVL effect, resulting in a high burden of chronic GVHD in long-term survivors. This cohort experienced higher rates of EBV reactivation than have generally been reported after post-transplant treatment with cyclophosphamide-based regimens [60, 61], although only a minority of the patients required treatment for this reactivation. This is consistent with other reports of high levels of EBV reactivation with relatively low rates of symptomatic post-transplant lymphoproliferative disorder in haploidentical regimens using both post-transplant cyclophosphamide and ATG [45]. Additionally, the number of participants with detectable EBV before HCT may have contributed to the higher-than-expected rates of reactivation. The absence of progression to post-transplant lymphoproliferative disease is consistent with other reports [62]. Other transplant complications were as expected in this heavily pre-treated population.
This sub-myeloablative conditioning regimen with a high-dose mobilized peripheral blood graft resulted in robust immune reconstitution, with recovery of lymphocyte subsets, de novo T-cell production, and clonal diversification reflecting the expected pace post HCT [60, 63]. The role of NK cells in the anti-leukemic response has been well established, particularly in AML [64]. Given the evidence that post-transplant exposure to cyclophosphamide can blunt this effect, adoptive transfer of a purified NK cell graft after receipt of post-transplant cyclophosphamide may provide additional benefit [64]. The collection, production, and infusion of an additional NK cell product was feasible in our study population. The peak NK cell numbers at 1 month and the subsequent decline probably reflect adoptively transferred NK cells. The range of this peak reflects the variability of expansion and persistence of adoptively transferred NK cells, and it accounts for non-protocol patients who did not receive the NK cell product. KIR mismatch was not required for enrollment on the protocol, and of those participants who received the NK cell product and had KIR typing available, 56% had the predicted alloreactivity. Given the high-risk character of the population, additional DLIs were frequently administered, most commonly for mixed chimerism. In one participant, serial DLIs resulted in subsequent stable chimerism and supported long-term survival with no evidence of GVHD.
This study demonstrates that additional work is needed to improve the tolerability and efficacy of a subsequent HCT for patients with relapsed hematologic malignancies after an initial HCT. Advances in immune therapies and targeted molecular therapies have provided additional salvage therapy strategies for patients with relapsed hematologic malignancies, and the ability to achieve remission in patients with post-HCT relapse will be a key factor in improving outcomes of second HCTs. Continuing to accrue experience with haploidentical platforms will enhance our ability to expand donor options and design regimens that balance optimal anti-leukemia effect with reduced toxicity.
Supplementary Material
Highlights:
Submyeloablative 2nd transplantation with unmanipulated haploidentical graft provides acceptable immune reconstitution in high-risk pediatric patients
High T cell dose and NK cell addback are feasible in the haploidentical setting
Relapse and transplant-related mortality limit success of 2nd allogeneic transplant
Acknowledgements
The authors thank Keith A. Laycock, PhD, ELS, for scientific editing of the manuscript. AT and AS acknowledge support from the American Society of Hematology (ASH) Scholar Awards. This work was supported by the National Institutes of Health (NIH)/National Cancer Institute grant P30CA021765, and the American Lebanese Syrian Associated Charities. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. We thank the referring physicians, the staff of the clinical research office for assisting with conducting the clinical study, the staff of the Human Applications Laboratory, and the staff of the Department of Bone Marrow Transplantation and Cellular Therapy for their excellent patient care. We also thank the patients who participated in this study and their caregivers, who entrusted the care of their children to us.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
BMT: Travel support from Miltenyi Biotec.
References
- 1.Sharma A, Li Y, Huang S, et al. Outcomes of pediatric patients who relapse after first HCT for acute leukemia or MDS. Bone Marrow Transplant, 56 (2021) 1866–75 [DOI] [PubMed] [Google Scholar]
- 2.Bajwa R, Schechter T, Soni S, et al. Outcome of children who experience disease relapse following allogeneic hematopoietic SCT for hematologic malignancies. Bone Marrow Transplant, 48 (2013) 661–5 [DOI] [PubMed] [Google Scholar]
- 3.Kuhlen M, Willasch AM, Dalle JH, et al. Outcome of relapse after allogeneic HSCT in children with ALL enrolled in the ALL-SCT 2003/2007 trial. Br J Haematol, 180 (2018) 82–9 [DOI] [PubMed] [Google Scholar]
- 4.Roux C, Tifratene K, Socié G, et al. Outcome after failure of allogeneic hematopoietic stem cell transplantation in children with acute leukemia: a study by the société Francophone de greffe de moelle et de thérapie cellulaire (SFGM-TC). Bone Marrow Transplant, 52 (2017) 678–82 [DOI] [PubMed] [Google Scholar]
- 5.Uden T, Bertaina A, Abrahamsson J, et al. Outcome of children relapsing after first allogeneic haematopoietic stem cell transplantation for acute myeloid leukaemia: a retrospective I-BFM analysis of 333 children. Br J Haematol, 189 (2020) 745–50 [DOI] [PubMed] [Google Scholar]
- 6.Meshinchi S, Leisenring WM, Carpenter PA, et al. Survival after second hematopoietic stem cell transplantation for recurrent pediatric acute myeloid leukemia. Biol Blood Marrow Transplant, 9 (2003) 706–13 [DOI] [PubMed] [Google Scholar]
- 7.Radich JP, Sanders JE, Buckner CD, et al. Second allogeneic marrow transplantation for patients with recurrent leukemia after initial transplant with total-body irradiation–containing regimens. J Clin Oncol, 11 (1993) 304–13 [DOI] [PubMed] [Google Scholar]
- 8.Eapen M, Giralt SA, Horowitz MM, et al. Second transplant for acute and chronic leukemia relapsing after first HLA-identical sibling transplant. Bone Marrow Transplant, 34 (2004) 721–7 [DOI] [PubMed] [Google Scholar]
- 9.Hazar V, Karasu G, Uygun V, et al. Role of a second transplantation for children with acute leukemia following posttransplantation relapse: a study by the Turkish Bone Marrow Transplantation Study Group. Leuk Lymphoma, 61 (2020) 1465–74 [DOI] [PubMed] [Google Scholar]
- 10.Naik S, Martinez C, Leung K, et al. Outcomes after Second hematopoietic stem cell transplantations in pediatric patients with relapsed hematological malignancies. Biol Blood Marrow Transplant, 21 (2015) 1266–72 [DOI] [PubMed] [Google Scholar]
- 11.Lund TC, Ahn KW, Tecca HR, et al. Outcomes after second hematopoietic cell transplantation in children and young adults with relapsed acute leukemia. Biol Blood Marrow Transplant, 25 (2019) 301–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hill BT, Bolwell BJ, Rybicki L, et al. Nonmyeloablative second transplants are associated with lower nonrelapse mortality and superior survival than myeloablative second transplants. Biol Blood Marrow Transplant, 16 (2010) 1738–46 [DOI] [PubMed] [Google Scholar]
- 13.Pulsipher MA, Boucher KM, Wall D, et al. Reduced-intensity allogeneic transplantation in pediatric patients ineligible for myeloablative therapy: results of the Pediatric Blood and Marrow Transplant Consortium Study ONC0313. Blood, 114 (2009) 1429–36 [DOI] [PubMed] [Google Scholar]
- 14.Shaw BE, Mufti GJ, Mackinnon S, et al. Outcome of second allogeneic transplants using reduced-intensity conditioning following relapse of haematological malignancy after an initial allogeneic transplant. Bone Marrow Transplant, 42 (2008) 783–9 [DOI] [PubMed] [Google Scholar]
- 15.Horowitz MM, Gale RP, Sondel PM, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood, 75 (1990) 555–62 [PubMed] [Google Scholar]
- 16.Imus PH, Blackford AL, Bettinotti M, et al. Major histocompatibility mismatch and donor choice for second allogeneic bone marrow transplantation. Biol Blood Marrow Transplant, 23 (2017) 1887–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu Y, Zhao YL, Zhang JP, et al. Unmanipulated haplo-identical donor transplantation compared with identical sibling donor had better anti-leukemia effect for refractory/relapsed acute myeloid leukemia not in remission status. Ann Hematol, 99 (2020) 2911–25 [DOI] [PubMed] [Google Scholar]
- 18.Yu S, Huang F, Wang Y, et al. Haploidentical transplantation might have superior graft-versus-leukemia effect than HLA-matched sibling transplantation for high-risk acute myeloid leukemia in first complete remission: a prospective multicentre cohort study. Leukemia, 34 (2020) 1433–43 [DOI] [PubMed] [Google Scholar]
- 19.Mamcarz E, Madden R, Qudeimat A, et al. Improved survival rate in T-cell depleted haploidentical hematopoietic cell transplantation over the last 15 years at a single institution. Bone Marrow Transplant, 55 (2020) 929–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aversa F. Haploidentical haematopoietic stem cell transplantation for acute leukaemia in adults: experience in Europe and the United States. Bone Marrow Transplant, 41 (2008) 473–81 [DOI] [PubMed] [Google Scholar]
- 21.Saglio F, Berger M, Spadea M, et al. Haploidentical HSCT with post transplantation cyclophosphamide versus unrelated donor HSCT in pediatric patients affected by acute leukemia. Bone Marrow Transplant, 56 (2021) 586–95 [DOI] [PubMed] [Google Scholar]
- 22.Bertaina A, Zecca M, Buldini B, et al. Unrelated donor vs HLA-haploidentical α/β T-cell–and B-cell–depleted HSCT in children with acute leukemia. Blood, 132 (2018) 2594–607 [DOI] [PubMed] [Google Scholar]
- 23.Luznik L, Jalla S, Engstrom LW, Iannone R, Fuchs EJ. Durable engraftment of major histocompatibility complex–incompatible cells after nonmyeloablative conditioning with fludarabine, low-dose total body irradiation, and posttransplantation cyclophosphamide. Blood, 98 (2001) 3456–64 [DOI] [PubMed] [Google Scholar]
- 24.Luznik L, O’Donnell PV, Symons HJ, et al. HLA-haploidentical bone marrow transplantation for hematologic malignancies using nonmyeloablative conditioning and high-dose, posttransplantation cyclophosphamide. Biol Blood Marrow Transplant, 14 (2008) 641–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang XJ, Liu DH, Liu KY, et al. Treatment of acute leukemia with unmanipulated HLA-mismatched/haploidentical blood and bone marrow transplantation. Biol Blood Marrow Transplant, 15 (2009) 257–65 [DOI] [PubMed] [Google Scholar]
- 26.Lu DP, Dong L, Wu T, et al. Conditioning including antithymocyte globulin followed by unmanipulated HLA-mismatched/haploidentical blood and marrow transplantation can achieve comparable outcomes with HLA-identical sibling transplantation. Blood, 107 (2006) 3065–73 [DOI] [PubMed] [Google Scholar]
- 27.Handgretinger R, Klingebiel T, Lang P, Gordon P, Niethammer D. Megadose transplantation of highly purified haploidentical stem cells: current results and future prospects. Pediatr Transplant, 7 Suppl 3 (2003) 51–5 [DOI] [PubMed] [Google Scholar]
- 28.Seif AE, Li Y, Monos DS, et al. Partially CD3+ depleted unrelated and haploidentical donor peripheral stem cell transplantation has favorable graft-versus-host disease and survival rates in pediatric hematologic malignancy. Biol Blood Marrow Transplant, 26 (2020) 493–501 [DOI] [PubMed] [Google Scholar]
- 29.Diaz MA, Zubicaray J, Molina B, et al. Haploidentical stem cell transplantation in children with hematological malignancies using αβ+ T-cell receptor and CD19+ cell depleted grafts: High CD56dim/CD56bright NK Cell Ratio Early Following Transplantation Is Associated With Lower Relapse Incidence and Better Outcome. Front Immunol, 10 (2019) 2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Locatelli F, Merli P, Pagliara D, et al. Outcome of children with acute leukemia given HLA-haploidentical HSCT after αβ T-cell and B-cell depletion. Blood, 130 (2017) 677–85 [DOI] [PubMed] [Google Scholar]
- 31.Lang P, Teltschik HM, Feuchtinger T, et al. Transplantation of CD3/CD19 depleted allografts from haploidentical family donors in paediatric leukaemia. Br J Haematol, 165 (2014) 688–98 [DOI] [PubMed] [Google Scholar]
- 32.Trujillo AM, Karduss AJ, Suarez G, et al. Haploidentical hematopoietic stem cell transplantation with post-transplantation cyclophosphamide in children with high-risk leukemia using a reduced-intensity conditioning regimen and peripheral blood as the stem cell source. Transplant Cell Ther, 27 (2021) 427.e1–.e7 [DOI] [PubMed] [Google Scholar]
- 33.Jaiswal SR, Chakrabarti A, Chatterjee S, et al. Haploidentical peripheral blood stem cell transplantation with post-transplantation cyclophosphamide in children with advanced acute leukemia with fludarabine-, busulfan-, and melphalan-based conditioning. Biol Blood Marrow Transplant, 22 (2016) 499–504 [DOI] [PubMed] [Google Scholar]
- 34.González-Llano O, González-López EE, Ramírez-Cázares AC, Marcos-Ramírez ER, Ruiz-Argüelles GJ, Gómez-Almaguer D. Haploidentical peripheral blood stem cell transplantation with posttransplant cyclophosphamide in children and adolescents with hematological malignancies. Pediatr Blood Cancer, 63 (2016) 2033–7 [DOI] [PubMed] [Google Scholar]
- 35.Berger M, Lanino E, Cesaro S, et al. Feasibility and outcome of haploidentical hematopoietic stem cell transplantation with post-transplant high-dose cyclophosphamide for children and adolescents with hematologic malignancies: an AIEOP-GITMO retrospective multicenter study. Biol Blood Marrow Transplant, 22 (2016) 902–9 [DOI] [PubMed] [Google Scholar]
- 36.Sawada A, Shimizu M, Isaka K, et al. Feasibility of HLA-haploidentical hematopoietic stem cell transplantation with post-transplantation cyclophosphamide for advanced pediatric malignancies. Pediatr Hematol Oncol, 31 (2014) 754–64 [DOI] [PubMed] [Google Scholar]
- 37.Ruggeri A, Galimard JE, Paina O, et al. Outcomes of unmanipulated haploidentical transplantation using post-transplant cyclophosphamide (PT-Cy) in pediatric patients with acute lymphoblastic leukemia. Transplant Cell Ther, 27 (2021) 424.e1–.e9 [DOI] [PubMed] [Google Scholar]
- 38.Mitchell R, Cole T, Shaw PJ, Mechinaud F, O’Brien T, Fraser C. TCR α+β+/CD19+ cell-depleted hematopoietic stem cell transplantation for pediatric patients. Pediatr Transplant, 23 (2019) e13517. [DOI] [PubMed] [Google Scholar]
- 39.Katsanis E, Sapp LN, Varner N, Koza S, Stea B, Zeng Y. Haploidentical bone marrow transplantation with post-transplant cyclophosphamide/bendamustine in pediatric and young adult patients with hematologic malignancies. Biol Blood Marrow Transplant, 24 (2018) 2034–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mo XD, Zhang XH, Xu LP, et al. Unmanipulated haploidentical hematopoietic stem cell transplantation in first complete remission can abrogate the poor outcomes of children with acute myeloid leukemia resistant to the first course of induction chemotherapy. Biol Blood Marrow Transplant, 22 (2016) 2235–42 [DOI] [PubMed] [Google Scholar]
- 41.Kobayashi S, Ito M, Sano H, et al. T-cell-replete haploidentical stem cell transplantation is highly efficacious for relapsed and refractory childhood acute leukaemia. Transfus Med, 24 (2014) 305–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burger JA, Peled A. CXCR4 antagonists: targeting the microenvironment in leukemia and other cancers. Leukemia, 23 (2009) 43–52 [DOI] [PubMed] [Google Scholar]
- 43.Maganti H, Visram A, Shorr R, Fulcher J, Sabloff M, Allan DS. Plerixafor in combination with chemotherapy and/or hematopoietic cell transplantation to treat acute leukemia: A systematic review and metanalysis of preclinical and clinical studies. Leuk Res, 97 (2020) 106442. [DOI] [PubMed] [Google Scholar]
- 44.O’Donnell PV, Luznik L, Jones RJ, et al. Nonmyeloablative bone marrow transplantation from partially HLA-mismatched related donors using posttransplantation cyclophosphamide. Biol Blood Marrow Transplant, 8 (2002) 377–86 [DOI] [PubMed] [Google Scholar]
- 45.Salas MQ, Atenafu EG, Law AD, et al. Experience Using Anti-Thymocyte Globulin With Post-Transplantation Cyclophosphamide for Graft-Versus-Host Disease Prophylaxis in Peripheral Blood Haploidentical Stem Cell Transplantation. Transplant Cell Ther, 27 (2021) 428 e1–e9 [DOI] [PubMed] [Google Scholar]
- 46.Iyengar R, Handgretinger R, Babarin-Dorner A, et al. Purification of human natural killer cells using a clinical-scale immunomagnetic method. Cytotherapy, 5 (2003) 479–84 [DOI] [PubMed] [Google Scholar]
- 47.Jagasia MH, Greinix HT, Arora M, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group report. Biol Blood Marrow Transplant, 21 (2015) 389–401 e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Przepiorka D, Weisdorf D, Martin P, et al. 1994 Consensus Conference on Acute GVHD Grading. Bone Marrow Transplant, 15 (1995) 825–8 [PubMed] [Google Scholar]
- 49.Triplett BM, Shook DR, Eldridge P, et al. Rapid memory T-cell reconstitution recapitulating CD45RA-depleted haploidentical transplant graft content in patients with hematologic malignancies. Bone Marrow Transplant, 50 (2015) 968–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leung W, Iyengar R, Turner V, et al. Determinants of antileukemia effects of allogeneic NK cells. J Immunol, 172 (2004) 644–50 [DOI] [PubMed] [Google Scholar]
- 51.Mazzi B, Clerici TD, Zanussi M, et al. Genomic typing for patient-specific human leukocyte antigen-alleles is an efficient tool for relapse detection of high-risk hematopoietic malignancies after stem cell transplantation from alternative donors. Leukemia, 22 (2008) 2119–22 [DOI] [PubMed] [Google Scholar]
- 52.Vago L, Perna SK, Zanussi M, et al. Loss of mismatched HLA in leukemia after stem-cell transplantation. N Engl J Med, 361 (2009) 478–88 [DOI] [PubMed] [Google Scholar]
- 53.Kharfan-Dabaja MA, Kumar A, Ayala E, et al. Standardizing definitions of hematopoietic recovery, graft rejection, graft failure, poor graft function, and donor chimerism in allogeneic hematopoietic cell transplantation: a report on behalf of the American Society for Transplantation and Cellular Therapy. Transplant Cell Ther, 27 (2021) 642–9 [DOI] [PubMed] [Google Scholar]
- 54.Handgretinger R, Lang P, André MC. Exploitation of natural killer cells for the treatment of acute leukemia. Blood, 127 (2016) 3341–9 [DOI] [PubMed] [Google Scholar]
- 55.Brissot E, Labopin M, Ehninger G, et al. Haploidentical versus unrelated allogeneic stem cell transplantation for relapsed/refractory acute myeloid leukemia: a report on 1578 patients from the Acute Leukemia Working Party of the EBMT. Haematologica, 104 (2019) 524–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haen SP, Groh C, Schumm M, et al. Haploidentical hematopoietic cell transplantation using in vitro T cell depleted grafts as salvage therapy in patients with disease relapse after prior allogeneic transplantation. Ann Hematol, 96 (2017) 817–27 [DOI] [PubMed] [Google Scholar]
- 57.Willasch AM, Salzmann-Manrique E, Krenn T, et al. Treatment of relapse after allogeneic stem cell transplantation in children and adolescents with ALL: the Frankfurt experience. Bone Marrow Transplant, 52 (2017) 201–8 [DOI] [PubMed] [Google Scholar]
- 58.Shyr DC, Zhang BM, Saini G, et al. HLA-haplotype loss after TCRαβ/CD19-depleted haploidentical HSCT. Bone Marrow Transplant, 56 (2021) 733–7 [DOI] [PubMed] [Google Scholar]
- 59.McCurdy SR, Iglehart BS, Batista DA, et al. Loss of the mismatched human leukocyte antigen haplotype in two acute myelogenous leukemia relapses after haploidentical bone marrow transplantation with post-transplantation cyclophosphamide. Leukemia, 30 (2016) 2102–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Massoud R, Gagelmann N, Fritzsche-Friedland U, et al. Comparison of immune reconstitution between anti-T-lymphocyte globulin and post-transplant cyclophosphamide as acute graft-versus-host disease prophylaxis in allogeneic myeloablative peripheral blood stem cell transplantation. Haematologica, (2021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Crocchiolo R, Bramanti S, Vai A, et al. Infections after T-replete haploidentical transplantation and high-dose cyclophosphamide as graft-versus-host disease prophylaxis. Transpl Infect Dis, 17 (2015) 242–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kanakry JA, Kasamon YL, Bolaños-Meade J, et al. Absence of post-transplantation lymphoproliferative disorder after allogeneic blood or marrow transplantation using post-transplantation cyclophosphamide as graft-versus-host disease prophylaxis. Biol Blood Marrow Transplant, 19 (2013) 1514–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arnold DE, MacMath D, Seif AE, et al. Immune reconstitution following TCRαβ/CD19-depleted hematopoietic cell transplantation for hematologic malignancy in pediatric patients. Transplant Cell Ther, 27 (2021) 169.e1–.e9 [DOI] [PubMed] [Google Scholar]
- 64.Van Elssen C, Ciurea SO. NK cell alloreactivity in acute myeloid leukemia in the post-transplant cyclophosphamide era. Am J Hematol, 95 (2020) 1590–8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.