

Abstract
Purpose
Several professional societies have published guidelines for the clinical interpretation of somatic variants, which specifically address diagnostic, prognostic, and therapeutic implications. While these guidelines for the clinical interpretation of variants include data types that may be used to determine the oncogenicity of a variant (e.g., population frequency, functional and in silico data or somatic frequency), they do not provide a direct, systematic, and comprehensive set of standards and rules to classify the oncogenicity of a somatic variant. This insufficient guidance leads to inconsistent classification of rare somatic variants in cancer, generates variability in their clinical interpretation and importantly impacts patient care. Therefore, it’s essential to address this unmet need.
Methods
Clinical Genome Resource (ClinGen) Somatic Cancer Clinical Domain Working Group and ClinGen Germline/Somatic Variant Subcommittee, the Cancer Genomics Consortium (CGC), and the Variant Interpretation for Cancer Consortium (VICC) used a consensus approach to develop a Standard Operating Procedure (SOP) for the classification of oncogenicity of somatic variants.
Results
This comprehensive SOP has been developed to improve consistency in somatic variant classification, and has been validated on 94 somatic variants in ten common cancer-related genes.
Conclusion
The comprehensive SOP is now available for classification of oncogenicity of a somatic variant.
Keywords: Cancer genetic testing, somatic variant, variant classification, oncogenicity, pathogenicity
Introduction
Somatic genetic variants identified in cancer are interpreted from multiple partially overlapping perspectives across the clinical oncology and molecular pathology communities. In discovery and translational research endeavors, it is important to determine if a particular variant observed in a gene of interest is oncogenic or not, as such knowledge provides the foundation for targeted cancer treatment research. Clinical applications are dominated by diagnostic, prognostic, or therapeutic implications, which in most cases depend on underlying variant oncogenicity determined through expert classifications.
The joint consensus of the Association for Molecular Pathology (AMP), the American Society of Clinical Oncology (ASCO), and College of American Pathologists (CAP) somatic variant clinical interpretation and reporting guidelines address diagnostic, prognostic, and therapeutic implications1. While the AMP/ASCO/CAP guidelines describe data types which can be used to determine oncogenicity of a variant, including population frequency, functional data, computational predictions, and somatic frequency, they do not provide a clear, systematic and comprehensive set of standards and rules to classify somatic variant oncogenicity. Multiple other ‘Levels of Evidence’ systems published by professional societies such as the European Society for Medical Oncology (ESMO) Scale of Clinical Actionability of molecular Targets (ESCAT)2 and knowledgebases such as CIViC3, OncoKB4, and MetaKB5 address the actionability of somatic cancer variants and provide rules for their curation, but omit or differ in criteria defining variant oncogenicity. This lack of structured guidance for biological classification of rare variants and the ambiguity of biological and clinical context in which they are identified leads to inconsistency in their clinical interpretation. This further enhances the variability of reporting across clinical laboratories and institutions and has wide-reaching consequences for therapeutic decisions.
This report describes the development of standards and guidelines for classification of oncogenicity of somatic sequence variants in cancer using a rigorous and comprehensive process. For the purpose of these guidelines, we define variant oncogenicity to mean the pathogenicity of the variant in the context of a neoplastic disease. We are specifically categorizing somatic variants for their potential to confer growth and survival advantages in tumor cells. These guidelines were inspired by the American College of Medical Genetics and Genomics (ACMG) and AMP germline pathogenicity guidelines6 and adapted to systematically and comprehensively categorize evidence of oncogenicity for somatic variants. Consensus was built through discussion with experts in translational cancer biology, bioinformatics, medical oncology and molecular pathology.
Based on this Standard Operating Procedure (SOP), somatic single nucleotide variants and small insertions/deletions can be assigned to one of the five categories (Oncogenic, Likely Oncogenic, Variant of Uncertain Significance (VUS), Likely Benign and Benign), aiding their further clinical interpretation. This SOP expands the classification of variants, from variants with well-established oncogenicity to those that can be classified using this SOP and were previously not amenable to clinical interpretation, which improves consistency and comprehensiveness of reporting of somatic variant oncogenicity in clinical practice.
Methods
A workgroup consisting of individuals from multiple organizations, laboratories, institutions, and countries, including members of the ClinGen Somatic Clinical Domain Working Group, ClinGen Germline/Somatic Variant Subcommittee, Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC) was formed with the goal of developing this SOP. The Working Group (WG) evaluated the literature and recommendations from other professional societies (ACMG, AMP, ASCO, CAP, American Association for Cancer Research (AACR), and ESMO) and the SOP structure was informed by the ACMG/AMP germline pathogenicity guidelines6,2,1.
Evidence of oncogenicity or benign impact is categorized as Very Strong, Strong, Moderate or Supporting. A point-based system based on Tavtigian et al. is used for combining evidence to classify oncogenicity of somatic variants7. Variants can be classified as: Oncogenic, Likely Oncogenic, Variant of Uncertain Significance (VUS), Likely Benign, or Benign. Figure 1 provides a general overview of the evidence framework.
Figure 1.
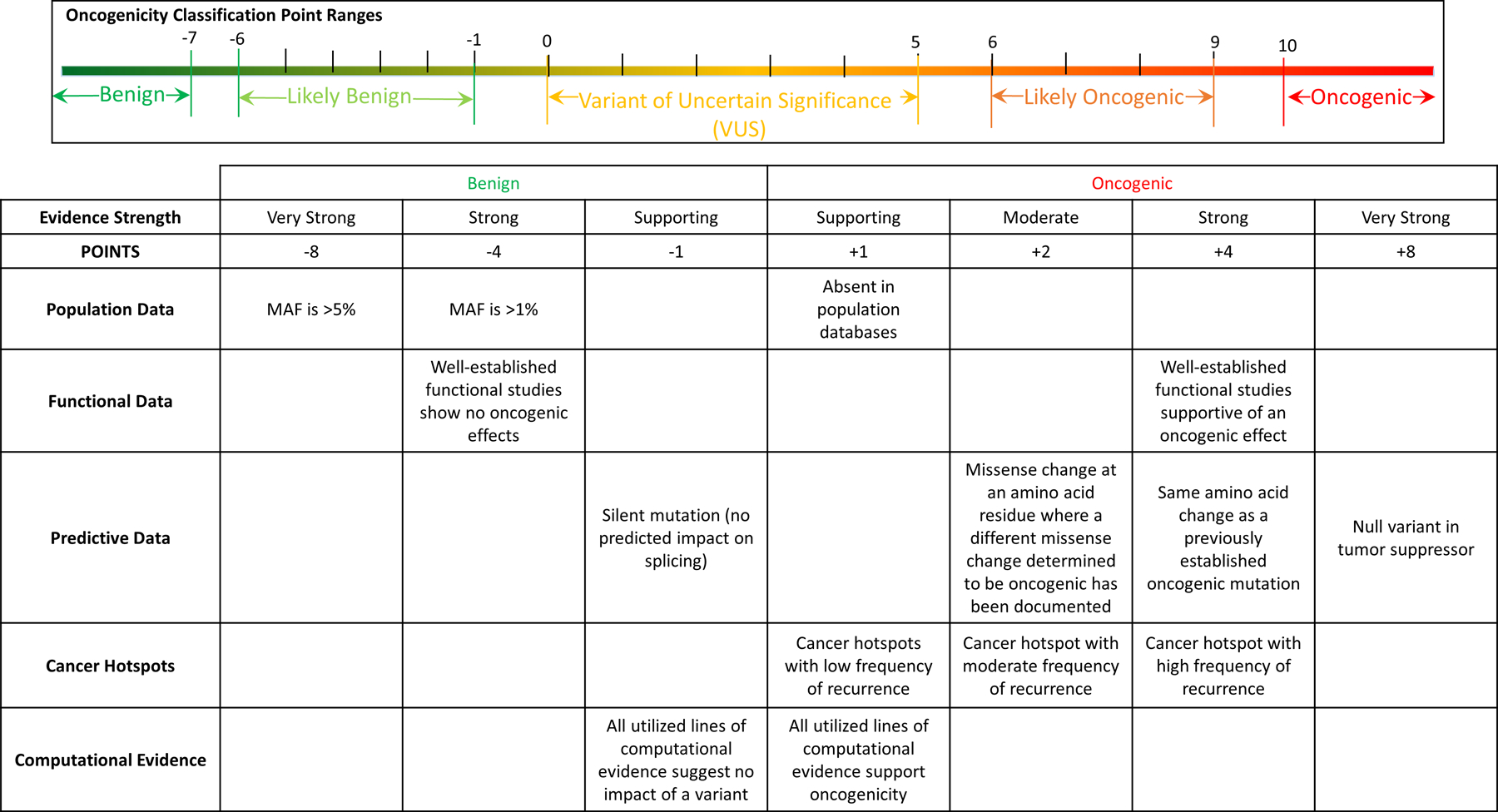
Somatic SOP Evidence Framework Overview (not all criteria are listed).
In order to test the proposed SOP we selected 9 genes covering key aspects of tumor molecular biology for initial testing. Two of these genes, KRAS and BRAF, are well-characterized oncogenes. PIK3CA is also a well-known oncogene that was selected as an example that is more challenging to interpret, in part due to the presence of hotspot variants in multiple domains8. IDH1 was selected for its neomorphic oncogenic mechanism driven by oncometabolite 2-hydroxyglutarate9. The role of EZH2 in cancer is potentially context-dependent, functioning as an oncogene or tumor suppressor gene (TSG). TERT was selected to represent non-coding oncogenic variants10,11. Finally, PTEN, TP53 and RB1 were selected to represent well-characterized TSGs. Notably, PTEN and TP53 have ClinGen germline Expert Panel guidelines available, and all variants selected for SOP evaluation in these two genes have been interpreted by a corresponding expert panel12,13. For the 9 genes, curation was completed for a set of 84 variants covering a range of classifications from benign to oncogenic. In addition, through collaboration with the ClinGen Somatic Hematologic Taskforce, oncogenicity classifications based on this SOP were performed for 10 FLT3 variants involving both tyrosine-kinase and non-tyrosine kinase domains. Determination of the oncogenicity for variants of FLT3 gene is important due to the availability of FDA-approved targeted therapeutic drugs that include highly selective tyrosine kinase inhibitors such as Midostaurin and Gilteritinib14,15. Thus, the final evaluation set consisted of 10 genes and 94 variants. All variants included in this SOP evaluation are listed in Table 1 and in supplemental table 1 using HGVS nomenclature. Each variant was evaluated independently by at least two curators; differences in evaluation between curators were reconciled via consensus agreement of classifications in regular monthly meetings of the working group. Classifications for all variants listed in Table 1 are provided in supplemental material.
Table 1.
Gene variants used to test Somatic SOP
| Gene Symbol | KRAS | BRAF | PIK3CA | IDH1 | EZH2 | TERT | FLT3 | PTEN | TP53 | RB1 |
|---|---|---|---|---|---|---|---|---|---|---|
| Transcript ID | NM_004985.5 | NM_004333.6 | NM_006218.4 | NM_005896.4 | NM_004456.4 | NM_198253.3 | NM_004119.3 | NM_000314.6 | NM_000546.5 | NM_000321.2 |
| Variant | p.Gly12Ala | p.Pro14Arg | p.Met1? | p.Phe32Val | c.-7-2A>C | c.-124C>T | p.Asp835Tyr | c.-9C>G | p.Pro47Ser | p.Ile124ArgfsTer6 |
| p.Gln61Lys | p.Ala31Gly | p.Ile391Met | c.123-4C>T | p.Asp136Val | c.-146C>T | p.Asp835Glu | p.His123Arg | p.Val73Met | p.Ser443Ter | |
| p.Ser65Ile | p.Pro403LeufsTer8 | p.Glu418Lys | p.Val71Ile | p.Asp233Gly | c.-124C>A | p.Asp835His | p.Arg130Gln | p.Cys124Ter | c.607+1G>A | |
| p.Arg164Gln | p.Asp445His | p.Glu453Lys | p.Arg132His | p.Val626Met | c.-332C>T | p.Asp835Val | p.Thr131Ile | p.Cys135Gly | c.2211+5G>T | |
| p.Asn486_Pro490del | p.Glu545Ala | p.Arg132Cys | p.Arg658Thr | c.-245T>C | p.Asp835Ile | p.Leu182Ser | p.Val173Met | c.1216-29A>G | ||
| p.Gly469Ala | p.Lys733Arg | p.Arg132Ser | p.Ala682Gly | p.Arg230Ter | p.Asn676Lys | p.Leu193= | p.Glu180Lys | p.Gln689His | ||
| p.Gly469Glu | p.His1047Gln | p.Val178Ile | p.Ala682Val | p.Trp581Ter | p.Tyr842Cys | p.Pro204Ala | p.Glu298Lys | p.Ser567Leu | ||
| p.Asp594Asn | p.His1047Arg | p.Asp220Gly | p.Arg684Cys | p.Phe691Leu | c.634+5G>C | p.Gly334Arg | p.Trp563Leu | |||
| p.Val600Lys | p.His1065Tyr | p.Glu262Lys | p.Arg690His | p.Tyr693Cys | p.Lys322Ter | p.Ala347Asp | p.Arg661Trp | |||
| p.Lys601Glu | p.Asn1068LysfsTer5 | p.Ser695Leu | p.Tyr693Asn | p.Pro354Leu | p.His365Tyr | p.Glu554Lys | ||||
| p.Tyr731Phe | p.Tyr606Cys | |||||||||
| p.Tyr731His | ||||||||||
| p.Tyr646Phe |
“p.“ denotes the variant at the protein level, “c.“ denotes the variant at the cDNA level. Note, the first 5 TERT variants are in promoter region and the negative number indicates the number of base pairs upstream of the ATG translation start site.
General Considerations
Intended Scope
This SOP is focused on the classification of oncogenicity of somatic genetic variants in tumor cells such as single nucleotide variation (SNV) and insertions/deletions including deletions covering multiple exons.
This SOP should not be used for classification of pathogenicity of germline cancer predisposition variants.
Classification of oncogenicity of variants using this SOP should be done in the context of relevant tumor type(s). Detailed discussion on this topic can be found in the special considerations section.
This SOP is not intended for classification of oncogenicity of fusions, copy number variants (with exception of deletions limited to within a single gene), or other chromosomal rearrangements.
This SOP is not intended for determining the diagnostic, prognostic or therapeutic value of variants, but may be used to support these interpretations.
This SOP is intended to be used in conjunction with AMP/ASCO/CAP style somatic guidelines for clinical actionability assessment. Figure 2 (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5707196/figure/fig2/) from Li et al. describes AMP/ASCO/CAP somatic guideline tiers. Oncogenicity can be reported alongside clinical actionability assessed by these guidelines and inform the decision-making process. From a clinical perspective, we expect the main practical benefit of this SOP to be for somatic variants which do not directly align with AMP/ASCO/CAP guideline designation as tier I. For a missense somatic variant that is not directly listed in FDA approval(s) or practice guideline(s), an SOP-based oncogenicity classification may provide a rationale to assign such variant to tier III (VUS), tier IV (benign) or potentially to tier I or tier II. This assessment might further depend on tumor type, ‘absence’ of wild-type allele in tumor DNA (for TSGs) and relevant approval and/or practice guidelines. The immediate clinical relevance of independent oncogenicity assessment arises when practice guidelines / approvals omit or do not specify variants or describe variant types instead (e.g., “EGFR Exon 19 deletion”).
Variants in genes known to be associated with hereditary cancer syndromes require special consideration. If the somatic variant is in a gene known to cause hereditary cancer, ACMG/AMP ClinGen germline gene specific expert panel guidelines provide important gene specific nuances, such as minor allele frequency cutoffs, functional evidence guidance, computational evidence guidance, etc. For somatic variants in hereditary cancer genes with VUS oncogenicity classification by this SOP but with evidence of pathogenicity in the germline context, we recommend using ClinGen gene-specific germline variant curation expert panel guidelines-based curation where evidence is applicable. If such gene-specific germline expert panel guidelines do not exist for a particular gene, the generic ACMG/AMP germline pathogenicity guidelines may be considered. This recommendation can potentially improve some variant classifications by taking advantage of segregation and other gene-specific information. The following provides an example for such a scenario. PTEN NM_000314.6:c.610C>G (p.Pro204Ala) variant is classified as VUS by this SOP as can be seen in supplemental materials; however, it is classified as Likely Pathogenic by ClinGen PTEN germline Expert Panel in part due to taking advantage of hereditary-based information (https://erepo.genome.network/evrepo/ui/classification/CA000535/MONDO:0017623/003).
Recommendations for Database Use
Population databases
Genome Aggregation Database (gnomAD https://gnomad.broadinstitute.org) aggregates population data from multiple sources, incorporating data from several key sources (e.g., Exome Aggregation Consortium (ExAC) and 1000 Genomes). We therefore recommend using gnomAD over these constituent resources. There are a number of important caveats to keep in mind in regards to population data. Insertions and deletions may be underrepresented, in part due to technical challenges associated with their detection. Moreover, due to clonal hematopoiesis, some variants in gnomAD (and similar databases) may actually be somatic and not germline. Variants likely causing clonal hematopoiesis have been reported in multiple cancer relevant genes, for example: DNMT3A, TET2, IDH2, TP53, KRAS, and SF3B1. Examining read data in the gnomAD variant report page for suspiciously low Variant Allele Frequency (VAF) may help to identify such variants. Some variants are derived from normal tissue in individuals who are known to have cancer and the non-cancer subset of gnomAD excludes such individuals.
Cancer databases
Cancer hotspots (https://www.cancerhotspots.org) provides information about statistically significant recurrently mutated positions identified in ~25,000 tumor samples16,17. Determination of statistical significance takes in account nucleotide mutability and gene-specific SNV rates. It is important to keep in mind that statistical significance in this resource is calculated for amino acid position and not for particular change at that amino acid position. This resource has good coverage for many solid tumors, however it has limited coverage for hematological, rare and pediatric malignancies. For pediatric malignancies resources, PeCanPIE (https://pecan.stjude.cloud/pie) may be considered18.
Catalogue Of Somatic Mutations In Cancer (COSMIC https://cancer.sanger.ac.uk/cosmic) provides information on frequency of somatic variants in cancer. It is important to keep in mind that data in COSMIC is not adjusted by nucleotide mutability and varying gene-specific SNV rates. COSMIC data are provided by methods with different degrees of genome coverage and therefore coverage across gene sequences may not be uniform. In some cases, the origin of the variant (germline or somatic) may not be known or available.
Medical clinical literature
When a variant cannot be found in pre-existing population or cancer databases and functional data are not available, a search of the medical literature is often performed to assess if the variant has been previously described in an oncogenic setting. This is of particular relevance for rare variants. The literature can vary in quality and scope, from case reports to seminal studies of publicly available cancer genomes (e.g. The Cancer Genome Atlas). Some of such literature could be of a clinical nature and by itself not applicable as evidence for oncogenicity classification, however it might be useful to capture such information as “clinical annotation“. Such annotations may be helpful for utilisation in the AMP/ASCO/CAP guidelines for clinical interpretation. While the clinical context in which the variant has arisen may not be relevant to the evaluation of oncogenicity, it could be described within the annotation in a relevant context, e.g. IDH1/2 variants in gliomas have prognostic significance, whereas in chondrosarcomas, IDH1/2 variants have diagnostic utility, and therapeutic implications in acute myeloid leukemia (AML). When providing information in clinical annotations for a novel variant from the medical clinical literature, it is important to report the nature of the evidence, the study type and sample size, type of data presented (i.e. genomic or transcriptomic level, tumor-only or paired tumor/normal samples), and its relevance to the tumor type.
Computational Prediction Algorithms
Number of computational tools can be useful for variant oncogenicity classification. Such tools include conservation analysis tools such as phyloP, phastCons19 http://compgen.cshl.edu/phast, also available online through https://run.opencravat.org20, splicing impact predictors such as spliceAI21 https://spliceailookup.broadinstitute.org, varSEAK https://varseak.bio, and number of in silico variant effect prediction algorithms for missense variants. In silico variant effect prediction algorithms performance varies depending on gene, variant type, variant frequency, and many other factors. Supplementary table 2 lists 19 algorithms for prediction of effect of missense variants, as described below these predictors were evaluated in context of single nucleotide missense variants classified as part of this SOP testing.
In silico variant effect prediction algorithms were evaluated for 70 single nucleotide missense variants occurring within the coding region of each gene. Supplementary table 3 contains all variant coordinates used, oncogenicity SOP results (predictions, points, and evidence codes) and scores for 19 such algorithms. For 15 of these 19 algorithms we also obtained variant effect interpretations (“pathogenic”, “deleterious”, “damaging”, “high”, etc.) based on recommended score cutoffs. Using these scores and predictions we performed Spearman correlations, receiver operator characteristic (ROC) area under the curve (AUC), positive predictive value (PPV) and negative predictive value (NPV) analyses (Supplementary Methods for details) to assess their performance relative to Oncogenicity SOP points and classifications (Supplementary Figure 1). In general, scores from the in silico predictors were positively correlated with oncogenicity SOP points (mean spearman correlation 0.376). The algorithms achieved PPV (mean of 0.915), NPV (mean of 0.664) and ROC AUC (mean of 0.825) values when evaluated against the Oncogenicity SOP classifications. The evaluation of test variants selected here should not be considered a comprehensive assessment of the performance of these algorithms, in this analysis, VEST-422, PROVEAN23, and CADD24,25 had the highest agreement with Oncogenicity SOP results. Scores from each algorithm tended to be highly correlated to the scores from other algorithms (many of which use similar assumptions and training data). Notable partial exceptions were CHASMplus General and CHASMplus Cancer Type Specific which is interesting because these are among a small list of algorithms that actually use tumor/somatic data directly in their training. Ghosh et al. provide rationale for selecting potential combinations of predictors26. Overall, this analysis suggests that while in silico predictions have value in assessing variant impact, they should only be evaluated in the context of other evidence types as recommended by this SOP and multiple algorithms should not be treated as independent supporting evidence.
Proposed Criteria for Classification of Somatic Sequence Variants in Cancer
Criteria for evidence of oncogenicity of somatic variants are provided in table 2 and criteria for evidence of benign impact of somatic variants are provided in table 3. The first letter “O” in the evidence codes in Table 2 stands for “Oncogenic“ and first two letters “SB” in Table 3 stands for “Somatic Benign“.
Table 2.
Criteria for evidence of oncogenicity of somatic variants
| Category | Evidence | Criteria | Comments/Caveats |
|---|---|---|---|
| Very Strong (8 points) | OVS1 | Null variant (nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single or multi-exon deletion) in a bona fide tumor suppressor gene. |
|
| Strong (4 points) | OS1 | Same amino acid change as a previously established oncogenic variant (using this standard) regardless of nucleotide change. Example: Val→Leu caused by either G>C or G>T in the same codon. |
|
| OS2 | Well-established in vitro or in vivo functional studies supportive of an oncogenic effect of the variant. |
|
|
| OS3 | Located in one of the hotspots in cancerhotspots.org with at least 50 samples with a somatic variant at the same amino acid position, and the same amino acid change count in cancerhotspots.org in at least 10 samples. |
|
|
| Moderate (2 points) | OM1 | Located in a critical and well-established part of a functional domain (e.g., active site of an enzyme). |
|
| OM2 | Protein length changes as a result of in-frame deletions/insertions in a known oncogene, or tumor suppressor gene or stop-loss variants in a known tumor suppressor gene. |
|
|
| OM4 | Missense variant at an amino acid residue where a different missense variant determined to be oncogenic (using this standard) has been documented. Amino acid difference from reference amino acid should be greater or at least approximately the same as for missense change determined to be oncogenic. |
|
|
| OM3 | Located in one of the hotspots in cancerhotspots.org with less than 50 samples with a somatic variant at the same amino acid position, and the same amino acid change count in cancerhotspots.org is at least 10. |
|
|
| Supporting (1 point) | OP1 | All utilized lines of computational evidence support an oncogenic effect of a variant (conservation/evolutionary, splicing impact, etc.). |
|
| OP2 | Somatic variant in a gene in a malignancy with a single genetic etiology. Example: retinoblastoma is caused by bi-allelic RB1 inactivation. |
|
|
| OP3 | Located in one of the hotspots in cancerhotspots.org and the particular amino acid change count in cancerhotspots.org is below 10. |
|
|
| OP4 | Absent from controls (or at an extremely low frequency) in Genome Aggregation Database (gnomAD). |
|
OVS1: Oncogenic Very Strong-1, OS1: Oncogenic Strong-1, OS2: Oncogenic Strong-2, OS3: Oncogenic Strong-3, OM:1 Oncogenic Moderate-1, OM2: Oncogenic Moderate-2, OM3: Oncogenic Moderate-3, OM4: Oncogenic Moderate-4, OP1: Oncogenic Supporting-1, OP2: Oncogenic Supporting-1, OP3: Oncogenic Supporting-3, OP4: Oncogenic Supporting-4
Table 3.
Criteria for evidence of benign impact of somatic variants
| Category | Evidence | Criteria | Comments/Caveats |
|---|---|---|---|
| Very strong (−8 points) | SBVS1 | Minor allele frequency is >5% in Genome Aggregation Database (gnomAD) in any of 5 general continental populations: African, East Asian, European (Non-Finnish), Latino, and South Asian. | If the somatic variant is in a gene known to cause predisposition to hereditary cancer, ACMG/AMP ClinGen germline expert panel gene specific guidelines (if they exist) must be consulted to establish a cutoff that takes disease prevalence into account. |
| Strong (−4 points) | SBS1 | Minor allele frequency is >1% in Genome Aggregation Database (gnomAD) in any of 5 general continental populations: African, East Asian, European (Non-Finnish), Latino, and South Asian. | If the somatic variant is in a gene known to cause predisposition to hereditary cancer, ACMG/AMP ClinGen germline expert panel gene specific guidelines (if they exist) must be consulted to establish a cutoff that takes disease prevalence into account. |
| SBS2 | Well-established in vitro or in vivo functional studies show no oncogenic effects. | NA | |
| Supporting (−1 point) | SBP1 | All utilized lines of computational evidence suggest no impact of a variant (conservation/evolutionary, splicing impact, etc.). | Because many in silico algorithms use the same or very similar input for their predictions, each algorithm cannot be counted as an independent criterion. Can be used only once in any evaluation of a variant. |
| SBP2 | A synonymous (silent) variant for which splicing prediction algorithms predict no impact to the splice consensus sequence nor the creation of a new splice site AND the nucleotide is not highly conserved. | NA |
SBVS1: Somatic Benign Very Strong-1, SBS1: Somatic Benign Strong-1, SBS2: Somatic Benign Strong-2, SBP1: Somatic Benign Supporting-1, SBP2: Somatic Benign Supporting-2
SOP-based examples of somatic variant classification
The following oncogenicity classifications for two variants provide illustrative examples of somatic variant classification.
Example 1: KRAS p.Arg164Gln
NM_004985.5:c.491G>A, GRCh37 (hg19) chr12:g.25362805C>T, Allele Registry ID: CA135580
An in vitro functional study by Smith et al. showed foci formation in NIH3T3 cells performed similarly to WT KRAS, however mRNA expression profiling might be indicative of a slight increase in MAPK activity27. There was an increase in nanoscale signaling complexes on the plasma membrane; however, this is not a well-established functional method, and in many instances, testing was done in cell lines with KRAS p.Arg164Gln and KRAS p.Gly12Val variants28. Based on the above evidence the SBS2 criterion was downgraded to supporting evidence level. This is a rare variant and is not listed in cancerhotspots.org. The FATHMM prediction is pathogenic (score 0.98) and CADD prediction is potentially deleterious (score 17.04), based on this evidence the OP1 criterion was satisfied. One supporting criterion in support of benign impact (−1 point) and one oncogenicity supporting criterion (1 point) were satisfied for KRAS p.Arg164Gln giving a total of 0 points which leads to a VUS oncogenicity classification for this somatic variant in neoplasms.
Example 2: BRAF p.Gly469Glu
NM_004333.6: c.1406G>A, GRCh37 (hg19) chr7:g.140481402C>T, Allele Registry ID: CA279970
Functional studies by Wan et al. and Smalley et al. indicate low BRAF kinase activity and increased signaling through CRAF29,30 (class 3 BRAF variant31). Based on this evidence the OS2 criterion was satisfied (Note: In PMID: 15035987 the amino acid (AA) numbers are shifted by one and AA 468 corresponds to AA 469.); The FATHMM prediction is pathogenic (score 0.99) and CADD prediction is deleterious (score 33), and based on this evidence the OP1 criterion was satisfied. The BRAF p.Gly469Glu AA change count in cancerhotspots.org is 4, and based on this evidence OP3 criterion was satisfied; This variant is absent in gnomAD (v.2.1.1), and based on this evidence the OP4 criterion was satisfied. One strong criterion in support of oncogenicity (4 points) and three oncogenicity supporting criteria (3 points) were satisfied for BRAF p.Gly469Glu giving a total of 7 points which leads to likely oncogenic classification of this somatic variant in neoplasms.
Criteria clarifications
In order to keep the SOP reasonably compact, criteria descriptions are concise and do not provide extensive clarifications. In order to address this, the following section provides criteria clarifications.
The OM4 criterion is intended to be used for a missense variant at an amino acid residue where a different missense variant, determined to be oncogenic (using this standard), has been documented. One of the notes for this rule stipulates that the amino acid difference from reference AA should be greater or at least approximately the same as for the missense change determined to be oncogenic. This note is intended to prevent potential misuse of this criterion in cases of AA substitutions with very similar physicochemical properties. We recommend using one of the well known AA difference metrics such as Grantham’s distance, Epstein’s coefficient of difference, or Miyata’s distance. In a generic example provided in the SOP, p.Arg156His is known to be oncogenic and p.Arg156Cys is observed. The Grantham’s distance of 180 from Arg to Cys is greater than Grantham’s distance of 29 from Arg to His, therefore this would be an appropriate use of this criterion.
Some criteria have a potential overlap which may lead to double counting. To decrease this possibility, a number of criteria have explicit exclusion(s) specified. For example, OM1 is based on variants being located in a critical and well-established part of a functional domain (e.g., active site of an enzyme). It is noted that this criterion cannot be used if OS3 is applicable because OS3 is based on a variant being in a known cancer hotspot and such hotspots are frequently located in critical functional domains.
During evaluation of OVS1 criteria we recommend considering ClinGen guidance on the interpretation of loss-of-function variants32. OVS1 can also be used for splicing variants up to −16 position at the splice acceptor site and the +5 position at the splice donor site if there is experimental evidence indicating splicing defects33. A recently created resource MutSpliceDB (https://brb.nci.nih.gov/splicing) provides a place for the genomic community to catalog RNA-seq based evidence for splice site variants effects on splicing34.
During evaluation of OS2 and SBS2 criteria we recommend considering ClinGen guidance on interpretation of functional evidence35.
Population-based criteria BA1 and SBS1 adopt most of the relevant ClinGen guidance36. General continental gnomAD populations: African, East Asian, European (Non-Finnish), Latino, and South Asian have well over 2,000 observed alleles. Finnish European population and Ashkenazi Jewish population may be used only if founder effects have been ruled out for the gene in question.
During evaluation of the following criteria: OVS1, OS2, OM1, OM2, OM4 and SBS2, underlying evidence may be insufficient to fully satisfy a criterion. In such cases curators have the option to downgrade the criterion to a lower evidence level. For example, evidence of functional impact for SBS2 may be limited or partially conflicting. In such cases, curators can downgrade evidence level to moderate or supporting.
Guidelines for Reporting Results
We endorse a stepwise, structured approach to classifying variants, in which oncogenicity classification by this guideline precedes assessment of clinical actionability, as illustrated in Figure 2. Oncogenicity classifications should be provided for all variants included in a clinical report. From the perspective of storing classifications of oncogenicity of somatic variants based on this SOP in knowledgebases, there are a few possible approaches. Ideally each met criteria code should be captured separately in addition to the overall classification, relevant tumor type(s) and comments. However, such an approach might not always be possible, especially in the short term. As an alternative, criteria codes satisfied could be captured in the comments section or overall classification statement depending on knowledgebase structure. The ClinGen Evidence Repository, which provides access to variant level evidence used and applied by ClinGen Variant Curation Expert Panels in the classification of germline variants, can be used as an example of a fully structured resource. On the other hand, ClinVar provides an example of an alternative approach. The following two links provide examples from ClinGen Evidence Repository and ClinVar for a germline TP53 variant NM_000546.5:c.537T>A (p.His179Gln) https://erepo.genome.network/evrepo/ui/classification/CA16615708/MONDO:0018875/009, https://www.ncbi.nlm.nih.gov/clinvar/variation/406578/.
Figure 2.

Stepwise process of somatic variant classification and interpretation.
Somatic variant nomenclature should follow similar guidance as outlined for germline variants in the Richards et al. publication6. A key part of this guidance is based on Human Genome Variation Society (HGVS https://varnomen.hgvs.org) nomenclature. We also strongly recommend providing a ClinGen Allele Registry ID37 for each variant (http://reg.clinicalgenome.org) in order to decrease chance of incorrect variant mapping between different genome builds and transcripts. Laboratories should adopt the MANE Select transcripts as the default reporting transcript, and additionally report variants in MANE Plus Clinical transcripts for comprehensive coverage. The MANE project (https://www.ncbi.nlm.nih.gov/refseq/MANE) provides a set of matching RefSeq and Ensembl transcripts, with the MANE Select version being well-supported by experimental data and representing the biology of the gene. Utilization of MANE transcripts allows harmonization across the community and with major genomic resources, and also decreases the chance of variant misinterpretation due to disparate transcript usage.
Discussion and Special Considerations
Curation and interpretation of oncogenic variants is a process involving several distinct stages. As indicated in the intended scope, this SOP is primarily intended to be used in conjunction with AMP/ASCO/CAP style somatic guidelines for assessing the therapeutic, diagnostic or prognostic relevance of variants. This SOP can also be integrated with other guidelines for assessing the clinical significance of cancer variants, such as ESCAT2. A more precise classification of rare and less-well characterized variants may overcome some challenges in the clinical interpretation process. In particular, this SOP might facilitate assignment of therapeutic, diagnostic or prognostic significance to variants classified as likely oncogenic or oncogenic, which might result in reassigning variants from AMP/ASCO/CAP Tier III to Tier I/II.
Functional effects of individual variants might be highly dependent on the histologic subtype and the molecular context of the tumor in which they occur. This SOP suggests that the classification of the oncogenicity of variants should be performed in context of the relevant tumor type. We recommend specifying a tumor type at the most generic level possible, for example for KRAS variants we specify “neoplasms’’ as the tumor type since KRAS variants have been observed in the majority of solid and hematological malignancies. For FLT3 variants, “hematopoietic neoplasms” is specified as the tumor type since FLT3 variants are generally limited to hematological malignancies. Some genes may have oncogenic effects and tumor suppressive effects that depend on tumor type. EZH2 is potentially one such gene. For activating EZH2 variants, “neoplasms in tissues with EZH2 expression” is specified as a tumor type. For loss of function EZH2 variants, “myeloid malignancies” is specified as a tumor type, since based on current understanding of the molecular biology of myeloid malignancies, EZH2 plays a tumor suppressive role in this context. EZH2 serves as an example of cancer relevant genes with limited understanding of the underlying molecular biology, illustrating the need to perform periodic review of variant oncogenicity curations, due in part to potential emergence of new information on the molecular biology of a gene in question.
In essentially all cases the underlying molecular mechanism of the same variant in germline and somatic context is the same. However, the implications of the same variant in germline and somatic context could be different. For example, while the BRAF p.Val600Glu variant has oncogenic effects and therapeutic implications in several malignancies, its presence in the germline results in embryonic lethality38. Germline pathogenic variants in hereditary cancer genes have lifelong implications for affected individuals, and also have potential implications to multiple family members. In sporadic malignancies, oncogenic TERT alterations are mostly copy number gains and promoter variants leading to an increase in TERT expression. In the germline context, pathogenic TERT alterations are loss-of-function events, and based on the current body of evidence, such alterations fall under the VUS umbrella in the somatic context.
Some variants in hereditary cancer genes may be present as germline, but not as somatic and vice versa. There are a few possible explanations for such observations. Variant genomic location and nucleotide composition around such locations may be susceptible to a particular type of mutational process, and that process might only be relevant in embryonic or post-natal context. Also, some variants in TSGs may have particularly strong dominant negative effects and potentially result in embryonic lethality.
Some somatic variants may not be oncogenic in a classical sense as they do not provide growth and/or survival advantage by themselves, however they may provide tumor cells with the ability to tolerate potential increases in metabolic or other stress due to other classical oncogenic variants. Also some somatic variants may provide only limited growth and/or survival advantage, but may potentially play an important role during the initial stages of neoplasm development. We consider a somatic variant’s involvement in oncogenesis as described by the Hallmarks of Cancer39 as relevant for the scope of this guideline. However, as many of these variants are part of an active and unfolding area of research, they might not be well covered by this guideline.
Clinical considerations
The current practice of precision oncology relies on individualized assessments of somatic variants and might have immediate clinical consequences for cancer patients. In order to harmonize reporting across institutions, we present this SOP for the classification of oncogenicity to be used as an element of clinical variant assessment. We are aware that many of the detected variants may fall into the VUS category. Routine clinical decisions should not be made based on variants of uncertain significance. The functional relevance of these variants should be further explored in a preclinical setting or in some instances inform therapeutic interventions in well-documented interventional or observational trials, preferably in high-volume academic centers40. The underlying rationale for inclusion of VUS in clinical trials should be based on the combination of a careful review of the respective variant, knowledge about the particular tumor type and its molecular drivers, potential therapeutic alternatives, patient medical history, performance status and additional clinical parameters. In such cases it is especially critical to provide all relevant information in a way that allows patients to make informed decisions on potential participation in clinical trials. There are a number of significant pitfalls in allowing participation in clinical trials based on VUS variants, and such trials should warrant additional vigilance by clinical trial monitoring authorities.
Clinically relevant variants detected during tumor-only sequencing can be of somatic or germline origin. In a number of instances there are significant differences in clinical implications depending on the somatic or germline variant origin. In such cases, follow-up germline testing should be considered to confirm the germline versus somatic origin41.
The ACMG/AMP germline pathogenicity guidelines provide a framework to systematically and comprehensively categorize evidence of pathogenicity or the lack thereof. However, it is a generic framework and evaluation criteria may vary due to gene specific nuances. To address such nuances, a number of ClinGen variant curation expert panels have been formed and developed gene specific guidelines, through modifications and adjustments to the underlying ACMG/AMP germline pathogenicity guidelines following a comprehensive and rigorous ClinGen process. Classifications by ClinGen variant curation expert panels are recognised by the FDA (https://clinicalgenome.org/about/fda-recognition). We envision a similar approach in the somatic space with development of gene specific somatic oncogenicity guidelines based on this SOP.
In summary, we provide a standardized and universally applicable framework and operating procedure for classification of oncogenicity of somatic variants that is an integral part of clinical variant interpretation, harmonizes reporting of variant oncogenicity and supports clinical decision making within and outside clinical trials.
Supplementary Material
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement: L.BL was an employee at QIAGEN Inc. at the time of contribution; J.D.M is a consultant for PierianDx Inc.; J.V. is an employee at Foundation Medicine Inc., Genentech Inc., Roche AG and owns stock of Roche AG; X.S.L is an employee at Congenica Ltd.
Data Availability Statement:
All data used in this article is publicly available and also listed in supplement data.
References
- 1.Li MM, Datto M, Duncavage EJ, et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn JMD 2017;19(1):4–23. doi: 10.1016/j.jmoldx.2016.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mateo J, Chakravarty D, Dienstmann R, et al. A framework to rank genomic alterations as targets for cancer precision medicine: the ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Ann Oncol Off J Eur Soc Med Oncol 2018;29(9):1895–1902. doi: 10.1093/annonc/mdy263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Griffith M, Spies NC, Krysiak K, et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat Genet 2017;49(2):170–174. doi: 10.1038/ng.3774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chakravarty D, Gao J, Phillips SM, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol 2017;2017. doi: 10.1200/PO.17.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Variant Interpretation for Cancer Consortium, Wagner AH, Walsh B, et al. A harmonized meta-knowledgebase of clinical interpretations of somatic genomic variants in cancer. Nat Genet 2020;52(4):448–457. doi: 10.1038/s41588-020-0603-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med Off J Am Coll Med Genet 2015;17(5):405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tavtigian SV, Harrison SM, Boucher KM, Biesecker LG. Fitting a naturally scaled point system to the ACMG/AMP variant classification guidelines. Hum Mutat 2020;41(10):1734–1737. doi: 10.1002/humu.24088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ligresti G, Militello L, Steelman LS, et al. PIK3CA mutations in human solid tumors: Role in sensitivity to various therapeutic approaches. Cell Cycle 2009;8(9):1352–1358. doi: 10.4161/cc.8.9.8255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2010;465(7300):966–966. doi: 10.1038/nature09132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horn S, Figl A, Rachakonda PS, et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013;339(6122):959–961. doi: 10.1126/science.1230062 [DOI] [PubMed] [Google Scholar]
- 11.Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science 2013;339(6122):957–959. doi: 10.1126/science.1229259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mester JL, Ghosh R, Pesaran T, et al. Gene‐specific criteria for PTEN variant curation: Recommendations from the ClinGen PTEN Expert Panel. Hum Mutat 2018;39(11):1581–1592. doi: 10.1002/humu.23636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fortuno C, Lee K, Olivier M, et al. Specifications of the ACMG/AMP variant interpretation guidelines for germline TP53 variants. Hum Mutat 2021;42(3):223–236. doi: 10.1002/humu.24152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med 2017;377(5):454–464. doi: 10.1056/NEJMoa1614359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perl AE, Martinelli G, Cortes JE, et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N Engl J Med 2019;381(18):1728–1740. doi: 10.1056/NEJMoa1902688 [DOI] [PubMed] [Google Scholar]
- 16.Chang MT, Asthana S, Gao SP, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol 2016;34(2):155–163. doi: 10.1038/nbt.3391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang MT, Bhattarai TS, Schram AM, et al. Accelerating Discovery of Functional Mutant Alleles in Cancer. Cancer Discov 2018;8(2):174–183. doi: 10.1158/2159-8290.CD-17-0321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McLeod C, Gout AM, Zhou X, et al. St. Jude Cloud: A Pediatric Cancer Genomic Data-Sharing Ecosystem. Cancer Discov 2021;11(5):1082–1099. doi: 10.1158/2159-8290.CD-20-1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hubisz MJ, Pollard KS, Siepel A. PHAST and RPHAST: phylogenetic analysis with space/time models. Brief Bioinform 2011;12(1):41–51. doi: 10.1093/bib/bbq072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pagel KA, Kim R, Moad K, et al. Integrated Informatics Analysis of Cancer-Related Variants. JCO Clin Cancer Inform 2020;4:310–317. doi: 10.1200/CCI.19.00132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019;176(3):535–548.e24. doi: 10.1016/j.cell.2018.12.015 [DOI] [PubMed] [Google Scholar]
- 22.Douville C, Masica DL, Stenson PD, et al. Assessing the Pathogenicity of Insertion and Deletion Variants with the Variant Effect Scoring Tool (VEST‐Indel). Hum Mutat 2016;37(1):28–35. doi: 10.1002/humu.22911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015;31(16):2745–2747. doi: 10.1093/bioinformatics/btv195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rentzsch P, Schubach M, Shendure J, Kircher M. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med 2021;13(1):31. doi: 10.1186/s13073-021-00835-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 2019;47(D1):D886–D894. doi: 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghosh R, Oak N, Plon SE. Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol 2017;18(1):225. doi: 10.1186/s13059-017-1353-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith G, Bounds R, Wolf H, Steele RJC, Carey FA, Wolf CR. Activating K-Ras mutations outwith “hotspot” codons in sporadic colorectal tumours - implications for personalised cancer medicine. Br J Cancer 2010;102(4):693–703. doi: 10.1038/sj.bjc.6605534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Šolman M, Ligabue A, Blaževitš O, et al. Specific cancer-associated mutations in the switch III region of Ras increase tumorigenicity by nanocluster augmentation. eLife 2015;4:e08905. doi: 10.7554/eLife.08905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smalley KSM, Xiao M, Villanueva J, et al. CRAF inhibition induces apoptosis in melanoma cells with non-V600E BRAF mutations. Oncogene 2009;28(1):85–94. doi: 10.1038/onc.2008.362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wan PTC, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004;116(6):855–867. doi: 10.1016/s0092-8674(04)00215-6 [DOI] [PubMed] [Google Scholar]
- 31.Yao Z, Yaeger R, Rodrik-Outmezguine VS, et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017;548(7666):234–238. doi: 10.1038/nature23291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abou Tayoun AN, Pesaran T, DiStefano MT, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat 2018;39(11):1517–1524. doi: 10.1002/humu.23626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rehder C, Bean LJH, Bick D, et al. Next-generation sequencing for constitutional variants in the clinical laboratory, 2021 revision: a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet Med Off J Am Coll Med Genet 2021;23(8):1399–1415. doi: 10.1038/s41436-021-01139-4 [DOI] [PubMed] [Google Scholar]
- 34.Palmisano A, Vural S, Zhao Y, Sonkin D. MutSpliceDB: A database of splice sites variants with RNA‐seq based evidence on effects on splicing. Hum Mutat Published online February 18, 2021:humu.24185. doi: 10.1002/humu.24185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.On behalf of the Clinical Genome Resource Sequence Variant Interpretation Working Group, Brnich SE, Abou Tayoun AN, et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med 2020;12(1):3. doi: 10.1186/s13073-019-0690-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghosh R, Harrison SM, Rehm HL, Plon SE, Biesecker LG, ClinGen Sequence Variant Interpretation Working Group. Updated recommendation for the benign stand-alone ACMG/AMP criterion. Hum Mutat 2018;39(11):1525–1530. doi: 10.1002/humu.23642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pawliczek P, Patel RY, Ashmore LR, et al. ClinGen Allele Registry links information about genetic variants. Hum Mutat 2018;39(11):1690–1701. doi: 10.1002/humu.23637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mercer K, Giblett S, Green S, et al. Expression of endogenous oncogenic V600EB-raf induces proliferation and developmental defects in mice and transformation of primary fibroblasts. Cancer Res 2005;65(24):11493–11500. doi: 10.1158/0008-5472.CAN-05-2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 40.Dickson D, Johnson J, Bergan R, Owens R, Subbiah V, Kurzrock R. The Master Observational Trial: A New Class of Master Protocol to Advance Precision Medicine. Cell 2020;180(1):9–14. doi: 10.1016/j.cell.2019.12.009 [DOI] [PubMed] [Google Scholar]
- 41.Li MM, Chao E, Esplin ED, et al. Points to consider for reporting of germline variation in patients undergoing tumor testing: a statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med Off J Am Coll Med Genet 2020;22(7):1142–1148. doi: 10.1038/s41436-020-0783-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used in this article is publicly available and also listed in supplement data.