

SUMMARY
Circular RNAs are garnering increasing interest as potential regulatory RNAs and a format for gene expression. The characterization of circular RNA using analytical techniques commonly employed in the literature, such as gel electrophoresis can, under differing conditions, yield different results when attempting to distinguish circular RNA from linear RNA of similar molecular weights. Here we describe circular RNA migration in different conditions, analyzed by gel electrophoresis and high-performance liquid chromatography (HPLC). We characterize key parameters that affect the migration pattern of circular RNA in gel electrophoresis systems, which include gel type, electrophoresis time, sample buffer composition, and voltage. Finally, we demonstrate the utility of orthogonal analytical tests for circular RNA that take advantage of its covalently closed structure to further distinguish circular RNA from linear RNA following in vitro synthesis.
eTOC Blurb
Correctly discriminating circular RNA from its linearized form on electrophoresis systems is critical for downstream analyses; however migration differences in previous papers raised questions regarding their discrepancy. Abe et al. identify key parameters influencing circular RNA electrophoresis to resolve these differences, and recommend orthogonal methods to verify circular RNA identity.
INTRODUCTION
Circular RNAs are covalently closed, single-stranded RNA species that, naturally, arise from back splicing. Circular RNAs are prevalent in eukaryotic transcriptomes and have recently gained widespread interest for their roles in disease regulation and as therapeutic modalities for gene delivery and protein production (Chen, 2020; Wesselhoeft et al., 2018, 2019). Methods to produce synthetic circular RNA from in vitro transcription reactions using enzyme-mediated ligation or permuted autocatalytic introns have been described (Chen et al., 2017; Wesselhoeft et al., 2018, 2019). The resulting circular RNA can be identified using a range of orthogonal techniques including gel electrophoresis, RNase R digestion, oligonucleotide-guided RNase H digestion, single-hit hydrolysis, and HPLC.
Wesselhoeft et al. and Chen et al. previously reached different conclusions on the immunogenicity of synthetic circular RNAs relative to linear RNAs (Wesselhoeft et al., 2019; Chen et al., 2017; Chen et al, 2019). During efforts to resolve these conflicting conclusions, we identified differences in circular RNA migration behavior dependent on the agarose gel systems used. Wesselhoeft et al. reported on properties of synthetic circular RNAs generated using in vitro transcription reactions (Wesselhoeft et al., 2018, 2019). The authors used gel electrophoresis and HPLC methodologies to discriminate circular species and nicked linear species of the same sequence and molecular weight. As we further explored the properties of circular RNA, we identified additional analytical conditions that can influence the outcomes of such experiments. If unrecognized, investigators using certain types of electrophoresis methods could potentially mischaracterize the identity of the RNA species in question. We present our findings here to raise awareness of this issue within the circular RNA community and to describe additional tools for circular RNA identification using multiple orthogonal methods.
RESULTS
Circular RNA migrates according to molecular weight in some, but not all, agarose electrophoresis conditions
Agarose gel electrophoresis is a simple and widely used method to study the size of RNA. RNA is negatively charged and migrates toward the anode in an electric field. When applied across an agarose gel, the gel acts as a sieve to impede RNA migration based on its mass and shape. As mass is approximately related to nucleic acid length, longer RNA strands of the same structure migrate more slowly than shorter RNA. Secondary structures within RNA can influence its migration through these gels, and therefore denaturing agents are often used. We synthesized circular RNA using in vitro transcription and sought to identify different RNA species using Northern blot analysis. We designed probes to identify pre-circularization precursor RNA, circular RNA, and excised intron byproducts as shown in Figure 1A. We also designed a linear control that lacks portions of the introns needed for circularization. Standard agarose gel electrophoresis shows the precursor RNA running as expected near the 2000nt marker, which is degraded upon RNase R digestion (Figure 1B). A faster-migrating band between 1000nt and 1500nt resists RNase R digestion, indicating it is likely circular RNA. This is confirmed by Northern blot analysis, showing no signal with excised probes (probes 1 and 5) but a positive signal with the splice junction probe (probe 6).
Figure 1. Circular RNA migration using standard agarose systems.
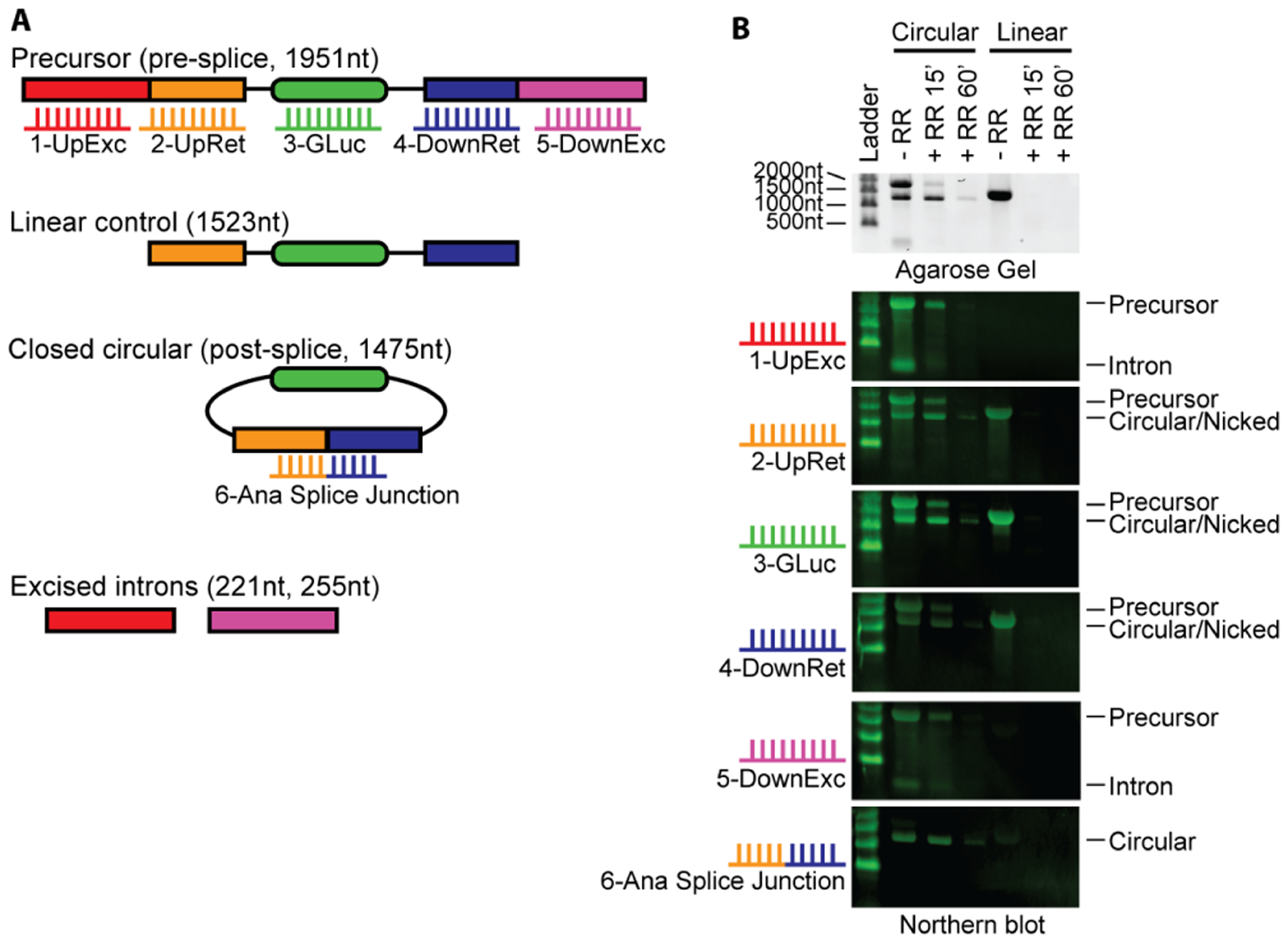
(A) Schematic and expected sizes of precursor linear RNA, linear control RNA, circular RNA, and excised introns. Linear control RNA is splicing incompetent as it lacks the full upstream and downstream introns necessary for splicing but retains remnant introns expected after splicing. Probes targeting the indicated segments of the RNA are numbered 1–6 in the indicated colors.
(B) Northern blot correctly identifies circular RNA using standard agarose electrophoresis. Circular RNA and linear control RNA were treated with RNase R for 15 minutes and 60 minutes. Samples were incubated in NorthernMax-Gly Sample Loading Dye containing glyoxal, glycerol and DMSO, and run at 50V for 80 minutes at room temperature using traditional agarose gels cast in our lab. Size markers are indicated on the left; biotinylated probes used for each Northern blot are indicated on the left and correspond with panel A. RNA species are labeled on the right. Gel and Northern blot images were cropped above the 2000nt marker for clarity.
Wesselhoeft et al. (Wesselhoeft et al., 2018, 2019) reported the use of E-Gel EX electrophoresis systems (prepackaged native agarose gel, ThermoFisher) to distinguish circular RNAs from other RNA species. They prepared samples in a denaturing buffer that was run into the native gel, and showed that the circular RNA migrates more slowly (i.e. appearing larger) than its true size under these conditions. The circular RNA migrated more slowly than the linear precursor of larger molecular weight, allowing the authors to distinguish intact circular RNA from both larger precursor RNA and equal-weight nicked circular RNA (Figure 2A, left). The RNA migration in this system is therefore different for circular and linear RNA, likely reflecting the differences in their topology. This unexpected observation was dependent on the use of E-Gel EX gels and is a useful property that was different from published reports of circular RNA electrophoresis patterns performed in different conditions (Chen et al., 2017; Zhang et al., 2014, 2016).
Figure 2. Circular RNA apparent size changes with electrophoresis conditions.
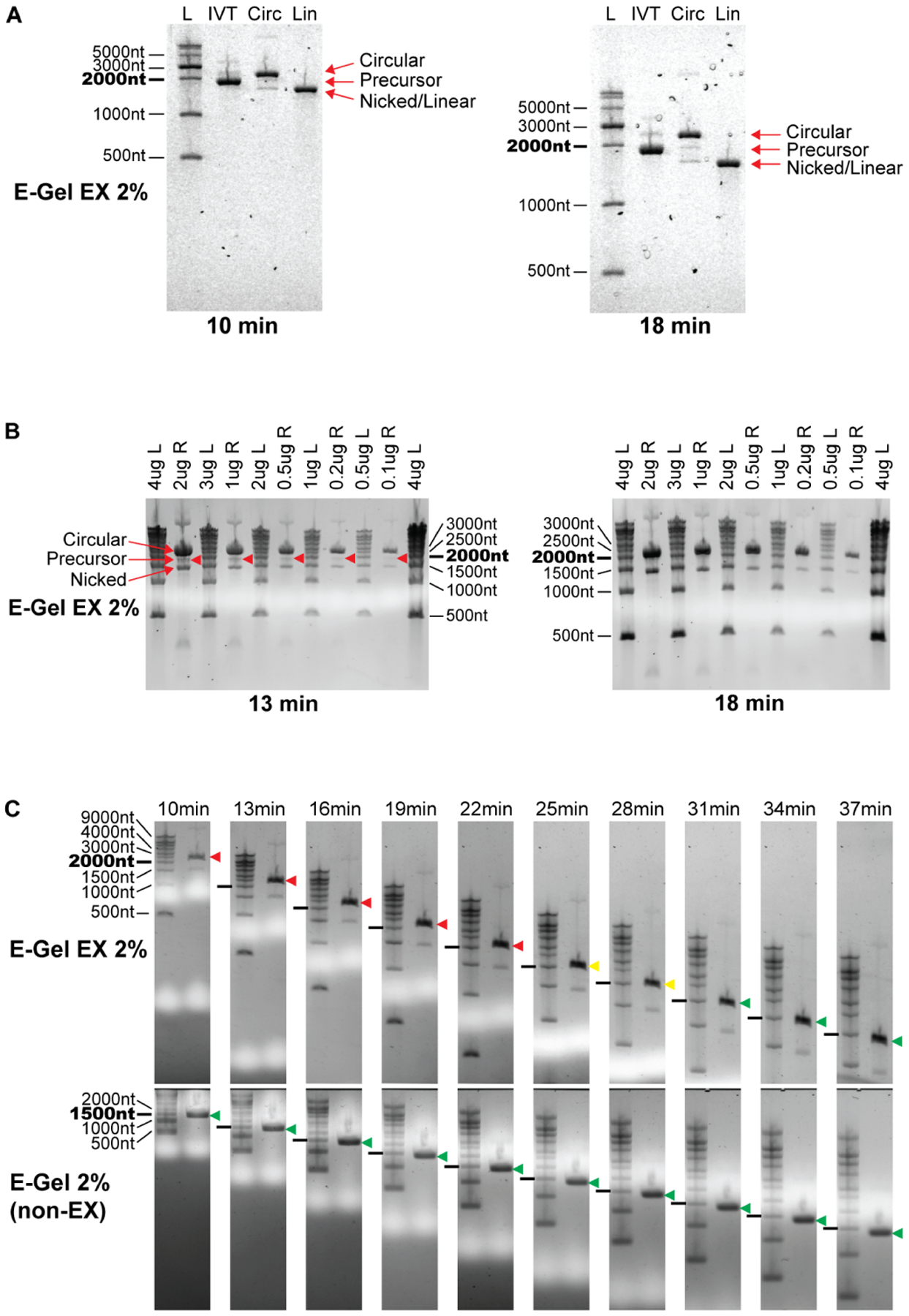
(A) Circular RNA migration on E-Gel EX systems using sample buffer containing formamide only. Circular RNA migrates slower than precursor RNA when run on E-Gel EX 2% gels using program “EX1–2%” for 10 minutes at room temperature. Extending electrophoresis duration to 18 minutes enhances separation of circular and linear splicing reaction species and maintains the same migration pattern as 10 minutes. Gel artifacts are visible as bubbles in the gel, increasing in severity with longer electrophoresis. RNA species are labeled on the right.
(B) Circular RNA migration can overlap precursor RNA when using commercially available sample buffer. Circular RNA was denatured in GLB II sample buffer and electrophoresis performed as in (A). The circular RNA band is clearly separated from the precursor band after 13 minutes of electrophoresis, however overlaps the precursor after 18 minutes. The mass of RNA loaded into the gel does not affect this migration pattern. L, Ladder; R, Circularized RNA not treated with RNaseR.
(C) Changes in circular RNA apparent size is specific to the E-Gel EX system. Top panels: E-Gel EX; bottom panels: non-EX E-Gel. RNaseR treated circularized RNA samples were denatured in GLB II buffer and subjected to the same electrophoresis conditions as in (A). Time points are indicated above gels. Circular RNA migration relative to the 2000nt RNA marker is shown by colored arrowheads (red: above 2000nt marker, yellow: with 2000nt marker, green: below 2000nt marker).
We sought to further define parameters of the E-Gel EX system that allow for this distinct migration behavior of circular RNA. We used a commercially available sample buffer containing formamide as the denaturing agent (GLBII, ThermoFisher), similar to the methods in Wesselhoeft et al, with the exception that it also contains EDTA, SDS, bromophenol blue, and xylene cyanol. When circularized RNA (without RNaseR digestion) was run on E-Gel EX 2% gels for 13 minutes using the program “EX1–2%” at ambient temperature, three predominant bands appeared: an intense upper band migrating equivalently to a 2500nt linear RNA (circular), a faint middle band migrating slightly slower than a 2000nt linear equivalent (precursor), and a clear lower band migrating just faster than a 1500nt linear equivalent (nicked) (Figure 2B, left). These results reproduced the findings of Wesselhoeft et al. (Wesselhoeft et al., 2019). When the same gel was run for an additional 5 minutes (18 minutes total) the upper band advanced from 2500nt to 2000nt, overlapping the middle band. Thus extending the run time potentially caused the circular RNA to migrate at the same position as the linear precursor (Figure 2B, right). The lower nicked RNA band was not affected by this additional time, as it migrated unchanged just below 1500nt. The amount of RNA in the well did not influence the change in apparent size, as the same migration shift was observed with different loading amounts.
A detailed analysis of purified circular RNA using the E-Gel EX system with GLBII buffer documents this migration behavior over time (Figure 2C, upper panel). We used an RNase R-treated circular RNA preparation that runs as two bands, the upper band corresponding to intact circle and lower band corresponding to a minor nicked product, prepared in GLBII buffer. Circular RNA migrated as a dominant upper band at the 2500nt marker at 10 minutes, migrated towards 2000nt at 22 minutes (indicated by red arrowheads), comigrated with the 2000nt marker at 25 minutes and 28 minutes (yellow arrowheads), and migrated below the 2000nt markers after 31 minutes (green arrowheads). This was not observed when using a sample buffer containing formamide only (Figure 2A, right), indicating that sample buffer preparation greatly influences the migration behavior of circular RNA in the E-Gel EX system. This difference was also not observed using a non-EX E-Gel, where the apparent size of circular RNA migrated according to its molecular weight at just below 1500nt throughout the electrophoresis time course (Figure 2C, lower panel).
To explore the changes in circular RNA migration pattern seen specifically with GLBII buffer on E-Gel EX systems, we tested each component of GLBII buffer separately. GLBII buffer (2x stock) contains 95% formamide, 18mM EDTA, 0.025% SDS, 0.025% bromophenol blue, and 0.025% xylene cyanol. Circularized RNA with or without RNaseR digestion was denatured 1:1 in formamide alone or with each component at the concentration in GLBII buffer (Figure 3A). The addition of EDTA caused circular RNA to migrate faster compared to formamide or formamide with SDS. EDTA also prevented nicking of circular RNA (shown by the faint nicked band) and degradation of RNA (shown by smearing below nicked RNA). This is also apparent on non-EX E-Gels (Figure 3A, bottom), where the absence of EDTA favored the accumulation of nicked RNA species when run for 31 minutes using program “EX1–2%”. Also notable in the non-EX gel is the reversed migration patterns of precursor and circular RNA, similar to that observed in non E-Gel agarose systems.
Figure 3. Sample buffer conditions affect migration pattern of circular RNA.
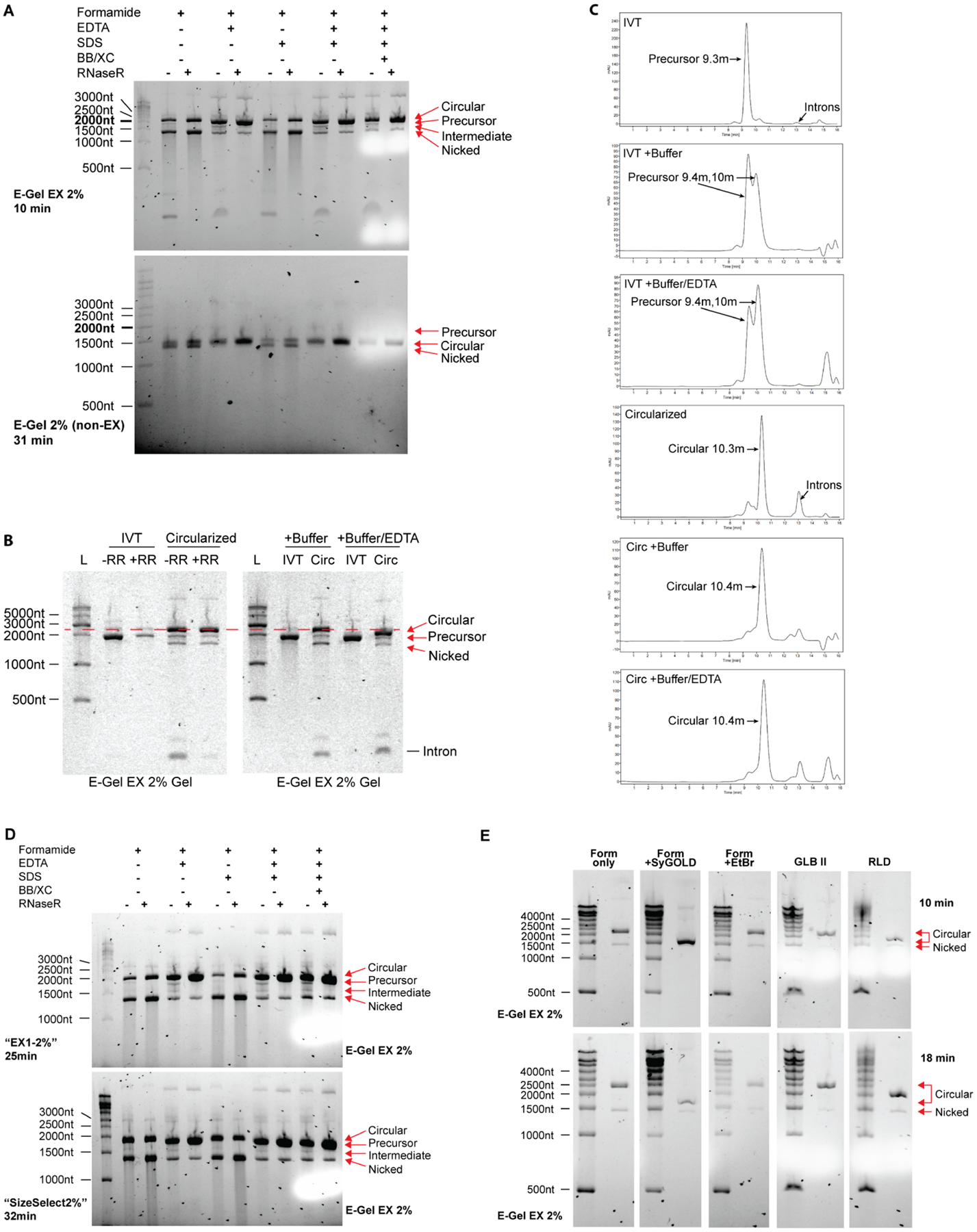
(A) EDTA decreases apparent size of circular RNA and protects circular RNA from nicking and degradation. Circularized RNA (treated with and without RNaseR) were denatured in sample buffer containing reagents depicted in the figure and run on either E-Gel EX 2% or non-EX E-Gel 2% precast gels for the indicated time using program “EX1–2%” at room temperature. Concentrations of each reagent prior to 1:1 mixing with RNA is as follows: Formamide: 95%, EDTA: 18mM, SDS: 0.025%, Bromophenol Blue (BB) and Xylene Cyanol (XC) 0.025% each. Ladder is shown on the left and was prepared in formamide only sample buffer. RNA species are labeled on the right.
(B) Buffer carryover in circular RNA preparations alters circular RNA mobility during electrophoresis in the presence of 50mM EDTA. The distance between circular RNA and linear precursor RNA narrows when 1x T4 RNA Ligase I buffer and EDTA are added to samples prior to gel loading, but not when buffer alone is added (red dotted line). A proportionally smaller shift towards lower molecular weight is visible for precursor RNA. RNA species are labeled on the right.
(C) Buffer carryover in circular RNA preparations dramatically alters precursor RNA mobility during size exclusion chromatography through the emergence of a new peak, but minimally affects circular RNA mobility. 1x T4 RNA ligase buffer I was added to samples prior to HPLC sample injection with or without 50mM EDTA. Note the lack of introns in all IVT conditions indicating that splicing is not occurring.
(D) Lower voltage causes overlap of circular RNA with precursor linear RNA. RNA was prepared in sample buffers as in (A), loaded onto two E-Gel EX 2% gels and subjected to either a high voltage program “EX1–2%” (upper) or low voltage program “SizeSelect2%” (lower) at room temperature for the time periods indicated on the left. These times are comparable with each voltage setting based on the distance of xylene cyanol migration depicted as the white areas on the right side of each gel. Ladder is shown on the left and was prepared in formamide only sample buffer. RNA species are labeled on the right.
(E) Denaturation in the presence of SYBR-GOLD I in the sample buffer abrogates the aberrant migration pattern of circular RNA seen on E-Gel EX systems. Ladder and RNaseR-treated circular RNA were denatured in the sample buffers described on top at a 1:1 ratio of sample to buffer, loaded onto E-Gel EX 2% gels, and run for 10 minutes (top panel) or 18 minutes (bottom panel) at room temperature using program “EX1–2%”. RNA species are labeled on the right. Sample buffer conditions prior to 1:1 dilution: Form only: 95% formamide; Form +SyGOLD: 95% formamide with 0.025% SYBR-GOLD I; Form +EtBr: 95% formamide with 0.025% ethidium bromide; GLB II: 95% formamide with 18mM EDTA and 0.025% each of SDS, bromophenol blue, and xylene cyanol; RLD: 95% formamide with 0.5mM EDTA and 0.025% each of SDS, ethidium bromide, bromophenol blue, and xylene cyanol.
Divalent salts, reducing agents, and other buffer components in circular RNA preparations may be present due to their use in transcription and circularization reactions. Depending on conditions used, buffer components can be carried over through transcription reaction cleanup measures, including buffer exchange filters and silica columns, which often reduce but do not eliminate these buffer components. We found that the addition of buffer containing monovalent and divalent salts did not change circular RNA migration during electrophoresis, however the addition of EDTA resulted in a shift towards smaller molecular weight for both precursor and circular RNA species (Figure 3B). This shift was proportionally more apparent for circular RNA, resulting in the near-overlap of circular and precursor RNA bands. The effects of EDTA on migration pattern were not observed when using size exclusion chromatography (Figure 3C), indicating specificity for electrophoresis-based systems.
RNA migration distance in electrophoresis systems is dependent on voltage, and we sought to determine whether voltage influenced the circular RNA migration pattern seen on E-Gel EX systems. Each E-Gel cassette is self-enclosed with prepackaged dye and running buffer, and the E-Gel device uses proprietary preset voltages that cannot be modified. We therefore used a different preset program, “SizeSelect2%” that uses a lower voltage setting based on the electrophoresis time needed to migrate visualization dyes across the cassette. We observed that an electrophoresis time of 25 minutes using program “EX1–2%” and 32 minutes using program “SizeSelect2%” produced equivalent migration of xylene cyanol on the gel (Figure 3D, white area in last two lanes equivalent to 1000nt). Circular RNA migration shifted from 2000nt in the high voltage “EX1–2%” (upper gel) to below 2000nt in the low voltage “SizeSelect2%” program (lower gel), indicating that lower voltages favor circular RNA migration closer to its expected molecular weight.. The precursor band is not distinguishable from circular RNA in the lower voltage setting, likely due to overlap with the abundant circular RNA species. Linear nicked RNA migrates consistently between the two voltage settings at just below 1500nt. The effects of EDTA are also observed with the lower voltage setting.
EX based E-Gels use SYBR-GOLD II as a detection dye, whereas standard E-Gels use SYBR-Safe. We therefore tested whether interactions between circular RNA and SYBR-GOLD II could cause the aberrant migration behavior of circular RNA. As SYBR-GOLD II is proprietary to EX E-Gels and not sold individually, we tested SYBR-GOLD I as it was commercially available with the assumption that it has similar properties to SYBR-GOLD II. We added SYBR-GOLD I to the formamide sample buffer prior to electrophoresis and observed it nearly abrogated the aberrant migration behavior of circular RNA on EX gels, with circular RNA migrating similarly to non-EX gels at just above 1500nt (Figure 3E). Addition of ethidium bromide to the formamide only sample buffer had no appreciable effect on circular RNA migration. We also tested two commercially available formamide based sample buffers: GLB II (used in previous figures) and RLD (similar to GLB II but contains 0.5mM EDTA and 0.025% ethidium bromide, ThermoFisher). GLB II sped the migration of circular RNA as seen in previous figures (circular RNA runs slightly below 2500nt), likely due to the presence of 18mM EDTA, and RLD caused a more significant shift with circular RNA migrating at 2000nt. This shift with RLD was unexpected as it contains lower EDTA than GLB II, however we cannot exclude the possibility of a combinatorial effect of each component in the RLD sample buffer. A summary of the effects on sample buffer components on circular RNA migration is provided in Table 1.
Table 1.
Effects of sample buffer reagents on circular RNA migration in E-Gel EX systems.
| Reagent | Effects on circular RNA migration |
|---|---|
| Ethylenediaminetetraacetic acid (EDTA) |
|
| Sodium Dodecyl Sulfate (SDS) |
|
| Bromophenol blue |
|
| Xylene cyanol |
|
| Sybr-GOLD |
|
| Ethidium bromide |
|
Electrophoresis generates heat in the gel and running buffer which can affect RNA stability and therefore its migration pattern. We found no significant differences in circular RNA migration when running EX E-Gels in a cold room set at 4°C for 13 minutes. This is likely due to the heat generated in the cassettes that offsets the ambient cold air (data not shown). We have noted lot-to-lot variability in EX E-Gels that can affect circular RNA migration speed but not linear RNA migration speed relative to a linear RNA ladder. Circular RNA splicing reactions run on two different lots of E-Gel EX 2% gels showed consistent linear but different circular RNA migration patterns, with a 1400nt circular RNA migrating only somewhat slower than the higher molecular weight linear precursor RNA on Gel Lot 1 (at ~2400nt), and much slower than precursor RNA on Gel Lot 2 (at ~3000nt) (Supplementary Figure 1). A trans-spliced product composed of two covalently linked linear molecules ran at around 3600nt in both gel lots. We have also observed that this reduced circular RNA migration through agarose is affected by size. The smaller circular RNA (1100nt) migrated faster than its linear precursor in Gel Lot 1 and just barely slower than its linear precursor in Gel Lot 2, suggesting that slower circular RNA migration in these gel conditions is enhanced at larger circular RNA sizes.
These results demonstrate that circular RNA band migration is dependent on multiple factors in the E-Gel EX system including electrophoresis time, voltage settings, sample buffer composition, gel batch, RNA size, and likely additional factors not analyzed here. Users wishing to exploit the slower migration of circular RNA on E-Gel EX gels may benefit from the analysis here to optimize circular RNA separation from nicked RNA of identical molecular weight. The potential for band pattern inversion when working with smaller circular RNAs should also be monitored. A summary table comparing differences between gel types is provided in Table 2.
Table 2.
Comparison of agarose-based electrophoresis systems on circular RNA migration.
| Agarose Gel | Advantages | Disadvantages |
|---|---|---|
| E-Gel EX |
|
|
| E-Gel (non-EX) |
|
|
| Self cast agarose gels |
|
|
Orthogonal methods to further confirm circular RNA identity
To further confirm the identity of circular RNA, we recommend using orthogonal methods. Here, we performed several additional analyses to verify the identity of the major product of these reactions as circular. The first method is Northern blot, which is sequence specific and can be used to detect the presence of intron fragments that are present in precursor RNA and absent in circular/nicked RNA, or exon fragments that are present in all (Chen, 2020; Jeck and Sharpless, 2014; Kristensen et al., 2019). Northern blot analysis on two electrophoresis conditions that gave contrasting migration patterns for circular RNA on E-Gel EX correctly identified circular RNA migrating above its precursor RNA in GLB II buffer and higher voltage and below its precursor RNA using RLD buffer and lower voltage settings (Figure 4A). In our experience, Northern blot resolution is diminished when using E-Gels (both EX and non-EX) and formamide, and this type of analysis has the best results with self-cast agarose gels run at lower voltages using glyoxal as a denaturing agent.
Figure 4. Orthogonal methods to confirm circular RNA identity.
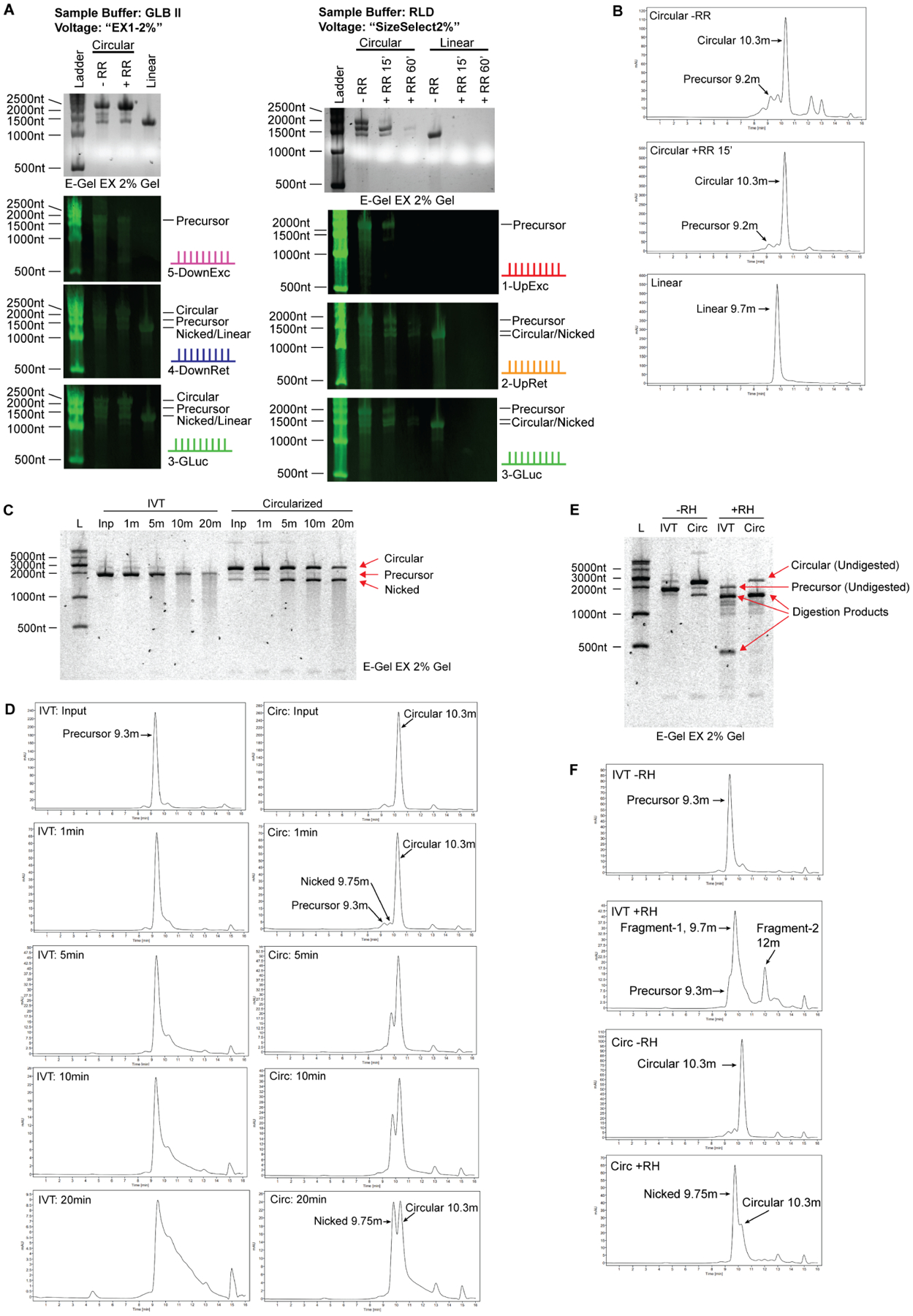
(A) Northern blot analysis of E-Gel EX system. Samples were denatured 1:1 in RLD (2xRNA Loading Dye, ThermoFisher) or GLB II (Gel Loading Buffer II, ThermoFisher), run on E-Gel EX 2% gels and subjected to different voltage programs indicated at the top. Agarose image is shown in top panel, Northern blots shown in lower panels. The probes are depicted on the right of each blot and correspond to the schematic shown in figure 1A.
(B) Size exclusion chromatography of circular RNA. Circular RNA elutes off the column with a slightly smaller molecular weight than predicated. Larger and smaller linear RNA species are on the left and right, respectively. Nicked circular RNA is visible as a minor peak at 9.75 minutes, directly before the major circular RNA peak. RNase R digestion identifies circular RNA as a single enriched peak.
(C) RNA circularity can be validated by nicking the RNA randomly using heat and divalent cations such as Mg2+. 1x T4 RNA ligase I buffer, containing magnesium, was added to RNA sample and RNA in buffer was heated at 70C for the indicated duration. IVT material, which is mostly linear, produces a smear of variable molecular weight species extending from the full-length band when randomly nicked. Circularization reaction material digested with RNase R, which is mostly circular, enriches the lower ‘nicked’ linear RNA band before producing a smear that extends from this band when randomly nicked. No smear extends from the circular RNA band. Note that the top and bottom bands are of equal molecular weight.
(D) Samples in (C) were analyzed by HPLC. Degradation of precursor RNA by heat and magnesium shows typical tailing of RNA towards lower molecular weights, consistent with the smear seen by gel electrophoresis. In contrast, degradation of circular RNA results in enrichment of a new peak corresponding to nicked RNA of equivalent molecular weight, which is followed by tailing as degradation continues. Unlike gel electrophoresis wherein intact circular RNA appears at much higher molecular weight than nicked RNA, circular RNA appears smaller than its nicked counterpart when analyzed using HPLC.
(E) Site-specific degradation using oligonucleotide-guided RNase H digestion is another method for validating RNA circularity. Site specific degradation will yield two linear products when cutting linear RNA species, and one linear product when cutting circular RNA. Predicted degradation products for precursor RNA in this experiment are 1371nt and 399nt in length, visible as the two major product bands in lane 4. Degraded circular RNA yields a single product at 1455nt. Several off-target digestion products are visible as fainter bands.
(F) Samples in (E) were analyzed by HPLC. Degradation of precursor RNA using RNase H yields two smaller fragments. Degradation of circular RNA using RNase H results in enrichment of a single fragment corresponding to nicked RNA of equivalent molecular weight, similar to the product generated by heat/magnesium nicking except that the starts and ends of the nicked molecules are expected to be homogenous.
As previously described, size exclusion chromatography can also be employed to identify circular RNA without electrophoresis (Wesselhoeft et al., 2018). Absorbance traces showed circular RNA appearing smaller than its precursor RNA, similar to non-EX agarose gel electrophoresis (Figure 4B). In contrast to both non-EX and EX agarose gel electrophoresis methods, circular RNA also appeared smaller than nicked RNA of equivalent molecular weight as previously described (Wesselhoeft et al., 2018). RNase R digestion of splicing reactions confirmed the major circular RNA peak that corresponds to the major surviving gel band. Addition of buffer components to samples prior to HPLC loading altered precursor RNA elution times and produced a second major peak (Figure 3C, middle left tracing, 10m) eluting earlier than a putative circular RNA peak (Figure 3C, middle right tracing, 10.4m) without altering free intron composition, suggesting that salt presence during sample injection may stabilize strong intron secondary structures and that dilution of sample in running buffer is not enough to compensate for this effect. Circular RNA elution times and peaks were largely unaffected, possibly because they do not contain intron structures, while free introns showed a minor shift towards a larger demonstrated molecular weight. Unlike agarose gel electrophoresis, EDTA addition did not substantially change chromatograms. Thus, care must be taken when preparing structured RNAs for native SEC-HPLC and interpreting resulting chromatograms.
Several secondary validation methods can be combined with electrophoresis and chromatography analytical steps to confirm circularity. Two exemplary methods both employ the concept of controlled degradation to introduce single cuts into the RNA backbone (nicking). Circular RNA can be nicked non-specifically by applying magnesium and heat for approximate single-hit kinetics, or specifically using oligonucleotide-guided RNase H degradation. Nicking a linear molecule results in two products, while nicking a circular RNA results in one product of equal molecular weight. Using a linear precursor RNA in vitro transcription reaction input, we saw progressive nicking of a single major band into a smear of degradation products extending from the intact band, consistent with random backbone cuts that produce fragments of variable lengths (Figure 4C). Using a majority circular RNA input, we observed depletion of the top circular RNA band and enrichment of the bottom nicked circle band as degradation progressed. Notably, a smear extended from the bottom band but not from the top band, a phenomenon that confirms the distinct identity of these two species. Analysis by HPLC reflected our gel electrophoresis observations, with a linear input progressively degrading into a smear of lower molecular weight products while a majority circular input enriches for the apparently higher molecular weight nicked RNA peak prior to tailing off into lower molecular weight products through secondary nicking events (Figure 4D).
Targeted RNA degradation using oligonucleotide-guided RNase H digestion yields a different banding pattern due to the non-random nature of the degradation site. Because the digestion site is not random as it is with divalent cation-mediated hydrolysis, two distinct bands are expected for linear RNA digestion instead of a smear of random-length products. One distinct band is expected for a nicked circular RNA, as a backbone nick anywhere within a circle will always yield a single product of uniform molecular weight regardless of whether it is targeted or random. Enzyme-negative samples showed the expected banding pattern for input materials, while RNase H-treated samples yielded the predicted banding patters for both linear and circular RNA (Figure 4E). These patterns were clearly visible by HPLC, with an apparently higher molecular weight nicked peak emerging after circular RNA digestion as expected (Figure 4F). We note that the digestion reaction was not complete, and some undigested precursor and circular RNA material survived the digestion intact. Furthermore, minority secondary digestion products were visible in both samples. Oligonucleotide design and reaction optimization are important factors to consider for targeted RNase H-mediated digestion to ensure interpretable results.
DISCUSSION
Future verification of circular RNA production and purification
The work developed here describes several simple steps to facilitate the identification of circular RNA vs. linear RNA species. First, investigators should be aware of the variation in circular RNA migration pattern seen when using the E-Gel EX system compared with standard agarose systems. Second, we encourage investigators to show molecular weight markers so that the electrophoretic migration patterns can be assessed in the context of known materials. Third, investigators can apply orthogonal detection methods such as Northern blot analysis, nicking assays, RNase R digestion, and particularly HPLC to further verify the RNA species. Some of these strategies have helped to distinguish endogenous circularized exons from host mRNA transcripts (Memczak et al., 2013; Salzman et al., 2012), while others may be most applicable to the study of synthetic circular RNAs (Zhang et al., 2014, 2016). These complementary approaches should facilitate identification of circular RNAs after in vitro synthesis.
LIMITATIONS OF THE STUDY
This paper addresses the differences in circular RNA migration behavior observed when comparing traditional agarose systems with the E-Gel EX system. The first limitation of this study includes the proprietary nature of the E-Gel EX ecosystem, where conditions such as running buffer, detection dye, and precise control of voltage settings are fixed. We addressed this limitation by modifying variables such as the sample buffer, using different preset voltage programs, and using non-EX gel cassettes. We recognize that there are other conditions not tested in this study that can influence the migration behavior of circular RNA. Second, we only tested agarose-based gel electrophoresis systems, focusing on the observations seen in E-Gel EX systems. It should be noted that this migration pattern of circular RNA migrating higher than its molecular weight has been observed in denaturing PAGE gel systems using small circular RNAs (<1000nt), however the buffer conditions were not specified (Zhang et al., 2014, 2013). We tested circular RNA sizes down to 1100nt in length (Supplementary Figure 1) and up to 2250nt (data not shown) and found consistent migration patterns, however we cannot exclude the possibility that much larger circular RNA may behave differently. Third, whether different RNA modifications affect this migration behavior is unclear, however we have found consistent migration behavior with N6-methyladenosine modification up to a level of ~10% of adenosines (Chen et al., 2019; Wesselhoeft et al., 2019, unpublished data). Fourth, this study does not address the immunogenicity of circular RNAs. Our two groups reported different strategies to overcome circRNA immunogenicity. Chen et al. reported that transfection of purified, in vitro generated circRNA into mammalian cells led to potent induction of innate immunity related to intron identity (Chen et al., 2017). In 2019, Chen et al. described that circular RNA immunogenicity can be suppressed by N6-methyladenosine modification (Chen et al., 2019). Wesselhoeft et al. reported purification methods reducing the immunogenicity of circRNAs (Wesselhoeft et al., 2019). Recently, Liu et al. reported that circRNAs made by self-splicing intron are immunogenic but circRNA made by T4 ligase have decreased immunogenicity (Liu et al., 2022). These differences remain to be addressed in future studies. Despite these limitations, we present awareness of differing circular RNA migration patterns depending on the electrophoresis system used and recommend the RNA community to use orthogonal methods in addition to electrophoretic methods to confirm circular RNA identity.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Howard Chang (howchang@stanford.edu).
Materials Availability
Plasmids and oligonucleotides generated in this study are available upon request.
Data and Code Availability
Unprocessed images have been deposited at Mendeley Data and are publically available as of the date of publication. DOIs are listed in the key resource table
This paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key Resource Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| DNase I (RNase-free) | New England Biolabs | M0303L |
| RNase R | MCLAB | RNASR-100 |
| Millennium™ RNA Markers | ThermoFisher | AM7150 |
| Bullseye Agarose | MidSci | BE-A500 |
| SYBR™ Safe DNA Gel Stain | ThermoFisher | S33102 |
| SYBR™ GOLD Nucleic Acid Gel Stain | ThermoFisher | S11494 |
| BrightStar™-Plus Positively Charged Nylon Membrane | ThermoFisher | AM10100 |
| RNA Gel Loading Dye (2X) | ThermoFisher | R0641 |
| Gel Loading Buffer II | ThermoFisher | AM8546G |
| Ultrapure™ Formamide | ThermoFisher | 15515026 |
| EDTA (0.5M), pH 8.0, RNase-Free | ThermoFisher | AM9260G |
| Ultrapure™ SDS Solution, 10% | ThermoFisher | 15553027 |
| SuperBlock™ (PBS) Blocking Buffer | ThermoFisher | 37515 |
| RiboLock RNase Inhibitor | ThermoFisher | EO0381 |
| IRDye® 800CW Streptavidin | LI-COR | 926-32230 |
| RNA ScreenTape | Agilent | 5067-5576 |
| RNA ScreenTape Sample Buffer | Agilent | 5067-5577 |
| RNA ScreenTape Ladder | Agilent | 5067-5578 |
| RNase H | New England Biolabs | M0297S |
| T4 RNA Ligase I | New England Biolabs | M0204S |
| Critical commercial assays | ||
| Platinum™ SuperFi II PCR Master Mix | ThermoFisher | 12368010 |
| NEBuilder® HiFi DNA Assembly Master Mix | New England Biolabs | E2621S |
| HiScribe T7 High Yield RNA Synthesis Kit | New England Biolabs | E2040S |
| Q5 Hot Start High-Fidelity 2x Master Mix | New England Biolabs | M0494X |
| DNA Clean & Concentrator-100 | Zymo Research | D4029 |
| Monarch® RNA Cleanup Kit | New England Biolabs | T2050L |
| NorthernMax™-Gly Kit | ThermoFisher | AM1946 |
| Pierce™ RNA 3’ End Biotinylation Kit | ThermoFisher | 20160 |
| iBlot™ 2 Transfer Stacks, nitrocellulose, mini | ThermoFisher | IB23002 |
| E-Gel™ EX Agarose Gels, 2% | ThermoFisher | G401002 |
| E-Gel™ Agarose Gels with SYBR™ Safe, 2% | ThermoFisher | G521802 |
| Deposited data | ||
| Original Image Files | This paper | DOI: 10.17632/62bwm34jvr.1 |
| Experimental models: cell lines | ||
| Experimental models: organisms/strains | ||
| Oligonucleotides | ||
| Circular precursor F: taatacgactcactatagggagaccc | This paper | N/A |
| Circular precursor R: TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTcaggaaacagctatgaccatgattacg | This paper | N/A |
| Linear control F – GLUC: CATGCATGCATGCATGGCCAGTGAATTGTAATACGACTCACTATAGGGaaaatccgttgaccttaaacgg | This paper | N/A |
| Linear control R – GLUC: aagtccgtagcgtctcg | This paper | N/A |
| 1-UpExc: ctcccgtcgagtctctgcaccttccgattagttgtaagtcatctattgtt |
This paper | BTA-NB011 |
| 2-UpRet: tgggggtggagggacttgaacccacacgaccgtttaaggtcaacggattt | This paper | BTA-NB013 |
| 3-GLUC: tcgagatccgtggtcgcgaagttgctggccacggccacgatgttgaagtc | This paper | BTA-NB019 |
| 4-DownRet: aagtccgtagcgtctcgccggtaacgcataatagccgttttgttttttgt | This paper | BTA-NB017 |
| 5-DownExc: cttactaattactacttcggcttggctcaggattgccttgtctataacta | This paper | BTA-NB012 |
| 6-ANA Splice Junction: cacgaccgtttaaggtcaacggattttaagtccgtagcgtctcgc | This paper | BTA-NB020 |
| RNase H Probe | Wesselhoeft et al., 2019 | |
| Recombinant DNA | ||
| GLuc APIE CVB3 pAC | Wesselhoeft et al., 2019 | N/A |
| circFOREIGN | Chen et al., 2019 | N/A |
| pNL1.1.PGK[Nluc/PGK] | Promega | N1441 |
| Software and algorithms | ||
| Bio-Rad Image Lab v5.2 | Bio-Rad | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
| Li-COR Image Studio Acquisition Software v3.1 | Li-COR | https://www.licor.com/bio/image-studio/ |
| Li-COR Image Studio Lite Software v5.2 | Li-COR | https://www.licor.com/bio/image-studio-lite/ |
| Other | ||
METHOD DETAILS
Plasmids, IVT Templates, and RNA synthesis
Construction of GLuc APIE CVB3 pAC has been described previously (Wesselhoeft et al., 2019) and was used as template for circular precursor and linear control PCRs. RNA was synthesized using in vitro transcription (IVT) kits (HiScribe T7 High Yield RNA Synthesis Kit). IVT templates were PCR amplified (Q5 Hot Start High-Fidelity 2x Master Mix) as described above and silica column purified prior to RNA synthesis (DNA Clean & Concentrator-100), or digested from plasmid and silica column purified (Purelink, ThermoFisher). One microgram of IVT template was used per reaction size, and reactions were incubated for two hours at 37°C with shaking at 1000rpm. The IVT template was degraded with DNaseI at two units per ten micrograms of expected RNA yield for 20 minutes at 37°C with shaking at 1000rpm. RNA was silica column purified prior to further enzymatic reactions, quantified using a Nanodrop One spectrophotometer, and verified using an Agilent TapeStation according to manufacturer’s recommendations. In some cases, uncircularized IVT product was subjected to a separate circularization step: T4 RNA ligase I buffer (New England Biolabs) was added to silica column-purified RNA at a final concentration of 1x with GTP to a final concentration of 2mM. Samples were heated at 55°C for 8m, and then silica column purified.
RNase R Digestion
Column purified RNA was digested with RNase R at a concentration of one unit per one microgram of RNA for either 15 or 60 minutes at 37°C with shaking at 1000rpm. Half the amount of RNase R was added after 7 minutes of digestion as previously described (Wesselhoeft et al., 2019). Samples were silica column purified after RNase R treatment prior to gel analysis.
Traditional Agarose Electrophoresis
Agarose gels (2%) were prepared by melting two grams of RNase-Free agarose per 100ml of TAE running buffer and poured into casting trays. In the case of Northern blots, agarose gels were prepared using 1x NorthernMax™-Gly running buffer, and RNA was denatured in glyoxal containing sample buffer by diluting 1:1 volumetrically, heating to 50°C for 30 minutes, and chilling on ice for at least three minutes. RNA was loaded into each well and run at 50V at room temperature until the bromophenol blue dye reached the edge of the gel, roughly 80 minutes. Agarose gels were post-stained with SYBR-Safe (1:10,000 dilution in 1x NorthernMax™-Gly buffer) and images were taken using a Bio-Rad Gel Doc XR and Image Lab 5.2 software using the “SYBR-Safe” settings.
E-Gel Electrophoresis System
RNA samples and markers were denatured by diluting 1:1 volumetrically with the indicated sample buffers for each figure. In all cases except for Northern blots, the 2x sample buffers contained 95% formamide. For EDTA testing, sample buffers were prepared with 95% formamide and either 18mM or 50mM EDTA (Figure 3B and 3C). For SDS testing, 0.025% SDS was included with 95% formamide. For SYBR-GOLD testing, 0.025% SYBR-GOLD was added to 95% formamide. Commercially available 2x RNA sample buffers were used where indicated (GLB II: Gel Loading Buffer II, ThermoFisher; RLD: RNA Loading Dye 2X, ThermoFisher). For carryover buffer testing, 1x RNA ligase buffer (New England Biolabs) was added to the sample prior to heating. In all cases, samples were volumetrically diluted 1:1 to a final volume of 20ul/well, heated to 70°C for at least 2 minutes, and chilled on ice for at least three minutes. Samples were loaded onto either 2% E-Gel™ EX Agarose Gels with SYBR-GOLD II™ or 2% E-Gel™ Agarose Gels with SYBR™ Safe and run on the E-Gel™ Powersnap Electrophoresis System using programs “EX1–2%” or “SizeSelect2%” for the times indicated in the figures at room temperature. Images were taken with a Bio-Rad Gel Doc™ XR and Image Lab 5.2 software using the “SYBR-Safe” settings. For Northern analysis, the E-Gel™ cassettes were opened and carefully placed onto Brightstar nylon membranes.
Northern Blot
Northern blot analysis was carried out following the NorthernMax™-Gly Kit (ThermoFisher). Briefly, RNA was prepared and separated on either standard agarose gels or E-Gels as described above. For size identification, biotinylated markers were made using the Pierce™ RNA 3’ End Biotinylation Kit and subsequently column purified. After gel electrophoresis, RNA was transferred to a Brightstar-Plus nylon membrane using the iBlot2 Dry Blotting System with the following settings: 20V for 2 min, 23V for 2 min, 25V for 3 min. In cases of transferring E-Gels, the E-Gel cassettes were opened and gels transferred onto membranes using the same settings. Membranes were gently rinsed with water and UV crosslinked with a Stratagene Stratalinker® 2400 using the “Auto Crosslink” setting. Hybridization and washing steps were performed at 42°C following the NorthernMax™-Gly Kit protocol. Hybridization probe sequences are provided in the Key Resources Table. Membranes were blocked (SuperBlock™ Blocking Buffer) for one hour at room temperature in the presence of RNase Inhibitor, subjected to IRDye® 800CW Streptavidin (Li-COR®) secondary detection reagent for one hour at room temperature and images acquired on a Li-COR® Odyssey® CLx infrared imager using Image Studio™ version 3.1. Images were exported using Image Studio Lite™ version 5.2.
RNase H Digestion
10ug of RNA was denatured at 98°C for 1 minute and then annealed with a 10-fold molar excess of DNA oligonucleotide. After reaching room temperature, RNase H buffer (New England Biolabs) was added in addition to enzyme (New England Biolabs) or water. RNase H digestion was conducted for 15 minutes at 37°C. RNA was silica column purified after digestion.
Divalent Cation Nicking
1x T4 RNA ligase I buffer (New England Biolabs), containing magnesium, was added to RNA sample and RNA in buffer was heated at 70°C for the indicated duration.
HPLC
100ng-2ug of RNA was diluted in water and run on an Agilent 1100 HPLC in 1x TE pH 6 running buffer through a 4.6×300mm size-exclusion column with particle size of 5μm and pore size of 2000Å heated to 35°C (Agilent). In some cases, 1x T4 RNA ligase buffer (New England Biolabs) was added to the sample prior to injection. In some cases, EDTA was added to the sample prior to injection to a final concentration of 50mM.
QUANTIFICATION AND STATISTICAL ANALYSIS
Significance is defined as the relative migration of circular RNA with standard RNA markers.
Supplementary Material
Highlights.
CircRNA can migrate above or below its corresponding linear RNA in native agarose gels.
Gel system, sample buffer EDTA, and detection dye affect circular RNA migration.
Orthogonal methods with Northern and HPLC confirm circular RNA identity.
ACKNOWLEDGEMENT
We thank Prof. Ling-ling Chen (SIBCB) and Prof. Sara Cherry (U. Penn) for comments. Supported by NIH 5T32AR050942-15 (B.T.A.), NIH R35-CA209919 (H.Y.C.). H.Y.C. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
H.Y.C. and R.C. are named as inventors on patents related to circular RNA held by Stanford University. D.G.A. and A.W. hold equity in Orna Therapeutics and A.W. is an employee of Orna Therapeutics. B.T.A is an employee of Eli Lilly and Company. R.C. is a consultant for Circ Bio and employee of Cartography Biosciences. H.Y.C. is a member of the Molecular Cell Advisory Board, co-founder of Accent Therapeutics, Boundless Bio, Circ Bio, Cartography Biosciences, and an advisor of 10x Genomics, Arsenal Biosciences, Chroma Medicine, and Spring Discovery.
INCLUSION AND DIVERSITY STATEMENT
While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
REFERENCES
- Chen L-L (2020). The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat. Rev. Mol. Cell Biol 21, 475–490. [DOI] [PubMed] [Google Scholar]
- Chen YG, Kim MV, Chen X, Batista PJ, Aoyama S, Wilusz JE, Iwasaki A, and Chang HY (2017). Sensing Self and Foreign Circular RNAs by Intron Identity. Mol. Cell 67, 228–238.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YG, Chen R, Ahmad S, Verma R, Kasturi SP, Amaya L, Broughton JP, Kim J, Cadena C, Pulendran B, et al. (2019). N6-Methyladenosine Modification Controls Circular RNA Immunity. Mol. Cell 76, 96–109.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeck WR, and Sharpless NE (2014). Detecting and characterizing circular RNAs. Nat. Biotechnol 32, 453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, and Kjems J (2019). The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet 20, 675–691. [DOI] [PubMed] [Google Scholar]
- Liu CX, Guo SK, Nan F, Xu YF, Yang L, and Chen LL (2022). RNA circles with minimized immunogenicity as potent PKR inhibitors. Mol. Cell 82, 420–434.e6. [DOI] [PubMed] [Google Scholar]
- Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al. (2013). Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495, 333–338. [DOI] [PubMed] [Google Scholar]
- Salzman J, Gawad C, Wang PL, Lacayo N, and Brown PO (2012). Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesselhoeft RA, Kowalski PS, and Anderson DG (2018). Engineering circular RNA for potent and stable translation in eukaryotic cells. Nat. Commun 9, 2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesselhoeft RA, Kowalski PS, Parker-Hale FC, Huang Y, Bisaria N, and Anderson DG (2019). RNA Circularization Diminishes Immunogenicity and Can Extend Translation Duration In Vivo. Mol. Cell 74, 508–520.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X-O, Wang H-B, Zhang Y, Lu X, Chen L-L, and Yang L (2014). Complementary sequence-mediated exon circularization. Cell 159, 134–147. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhang XO, Chen T, Xiang JF, Yin QF, Xing YH, Zhu S, Yang L, and Chen LL (2013). Circular Intronic Long Noncoding RNAs. Mol. Cell 51, 792–806. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yang L, and Chen LL (2016). Characterization of circular RNAs. Methods Mol. Biol 1402, 215–227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Unprocessed images have been deposited at Mendeley Data and are publically available as of the date of publication. DOIs are listed in the key resource table
This paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key Resource Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| DNase I (RNase-free) | New England Biolabs | M0303L |
| RNase R | MCLAB | RNASR-100 |
| Millennium™ RNA Markers | ThermoFisher | AM7150 |
| Bullseye Agarose | MidSci | BE-A500 |
| SYBR™ Safe DNA Gel Stain | ThermoFisher | S33102 |
| SYBR™ GOLD Nucleic Acid Gel Stain | ThermoFisher | S11494 |
| BrightStar™-Plus Positively Charged Nylon Membrane | ThermoFisher | AM10100 |
| RNA Gel Loading Dye (2X) | ThermoFisher | R0641 |
| Gel Loading Buffer II | ThermoFisher | AM8546G |
| Ultrapure™ Formamide | ThermoFisher | 15515026 |
| EDTA (0.5M), pH 8.0, RNase-Free | ThermoFisher | AM9260G |
| Ultrapure™ SDS Solution, 10% | ThermoFisher | 15553027 |
| SuperBlock™ (PBS) Blocking Buffer | ThermoFisher | 37515 |
| RiboLock RNase Inhibitor | ThermoFisher | EO0381 |
| IRDye® 800CW Streptavidin | LI-COR | 926-32230 |
| RNA ScreenTape | Agilent | 5067-5576 |
| RNA ScreenTape Sample Buffer | Agilent | 5067-5577 |
| RNA ScreenTape Ladder | Agilent | 5067-5578 |
| RNase H | New England Biolabs | M0297S |
| T4 RNA Ligase I | New England Biolabs | M0204S |
| Critical commercial assays | ||
| Platinum™ SuperFi II PCR Master Mix | ThermoFisher | 12368010 |
| NEBuilder® HiFi DNA Assembly Master Mix | New England Biolabs | E2621S |
| HiScribe T7 High Yield RNA Synthesis Kit | New England Biolabs | E2040S |
| Q5 Hot Start High-Fidelity 2x Master Mix | New England Biolabs | M0494X |
| DNA Clean & Concentrator-100 | Zymo Research | D4029 |
| Monarch® RNA Cleanup Kit | New England Biolabs | T2050L |
| NorthernMax™-Gly Kit | ThermoFisher | AM1946 |
| Pierce™ RNA 3’ End Biotinylation Kit | ThermoFisher | 20160 |
| iBlot™ 2 Transfer Stacks, nitrocellulose, mini | ThermoFisher | IB23002 |
| E-Gel™ EX Agarose Gels, 2% | ThermoFisher | G401002 |
| E-Gel™ Agarose Gels with SYBR™ Safe, 2% | ThermoFisher | G521802 |
| Deposited data | ||
| Original Image Files | This paper | DOI: 10.17632/62bwm34jvr.1 |
| Experimental models: cell lines | ||
| Experimental models: organisms/strains | ||
| Oligonucleotides | ||
| Circular precursor F: taatacgactcactatagggagaccc | This paper | N/A |
| Circular precursor R: TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTcaggaaacagctatgaccatgattacg | This paper | N/A |
| Linear control F – GLUC: CATGCATGCATGCATGGCCAGTGAATTGTAATACGACTCACTATAGGGaaaatccgttgaccttaaacgg | This paper | N/A |
| Linear control R – GLUC: aagtccgtagcgtctcg | This paper | N/A |
| 1-UpExc: ctcccgtcgagtctctgcaccttccgattagttgtaagtcatctattgtt |
This paper | BTA-NB011 |
| 2-UpRet: tgggggtggagggacttgaacccacacgaccgtttaaggtcaacggattt | This paper | BTA-NB013 |
| 3-GLUC: tcgagatccgtggtcgcgaagttgctggccacggccacgatgttgaagtc | This paper | BTA-NB019 |
| 4-DownRet: aagtccgtagcgtctcgccggtaacgcataatagccgttttgttttttgt | This paper | BTA-NB017 |
| 5-DownExc: cttactaattactacttcggcttggctcaggattgccttgtctataacta | This paper | BTA-NB012 |
| 6-ANA Splice Junction: cacgaccgtttaaggtcaacggattttaagtccgtagcgtctcgc | This paper | BTA-NB020 |
| RNase H Probe | Wesselhoeft et al., 2019 | |
| Recombinant DNA | ||
| GLuc APIE CVB3 pAC | Wesselhoeft et al., 2019 | N/A |
| circFOREIGN | Chen et al., 2019 | N/A |
| pNL1.1.PGK[Nluc/PGK] | Promega | N1441 |
| Software and algorithms | ||
| Bio-Rad Image Lab v5.2 | Bio-Rad | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
| Li-COR Image Studio Acquisition Software v3.1 | Li-COR | https://www.licor.com/bio/image-studio/ |
| Li-COR Image Studio Lite Software v5.2 | Li-COR | https://www.licor.com/bio/image-studio-lite/ |
| Other | ||