

Abstract
Since loss of the NF2 tumor suppressor gene results in p21-activated kinase (Pak) activation, PAK inhibitors hold promise for the treatment of NF2-deficient tumors. To test this possibility, we asked if loss of Pak2, a highly expressed group I PAK member, affects the development of malignant mesothelioma in Nf2;Cdkn2a-deficient (NC) mice and the growth properties of NC mesothelioma cells in culture. In vivo, deletion of Pak2 resulted in a markedly decreased incidence and delayed onset of both pleural and peritoneal malignant mesotheliomas in NC mice. In vitro, Pak2 deletion decreased malignant mesothelioma cell viability, migration, clonogenicity, and spheroid formation. RNA-seq analysis demonstrated downregulated expression of Hedgehog and Wnt pathway genes in NC;Pak2−/− mesothelioma cells versus NC;Pak2+/+ mesothelioma cells. Targeting of the Hedgehog signaling component Gli1 or its target gene Myc inhibited cell viability and spheroid formation in NC;P+/+ mesothelioma cells. Kinome profiling uncovered kinase changes indicative of EMT in NC;Pak2−/− mesothelioma cells, suggesting that Pak2-deficient malignant mesotheliomas can adapt by reprogramming their kinome in the absence of Pak activity. The identification of such compensatory pathways offers opportunities for rational combination therapies to circumvent resistance to anti-PAK drugs.
Keywords: Conditional knockout mice, malignant mesothelioma, Nf2 and Cdkn2a tumor suppressor genes, PAK, Hedgehog and Wnt signaling, cancer stem cells
Introduction
Malignant mesotheliomas are medically unresponsive cancers that arise mainly from the serosal membranes lining the pleural and peritoneal cavities. Most (85%) mesotheliomas arise in the pleura, but about 10%–20% of cases develop from the peritoneal surfaces (1). Malignant mesotheliomas often occur after chronic exposure of mesothelial cells to asbestos fibers (2). Malignant mesothelioma causes more than 3,000 deaths annually in the U.S., and a significant increase in mesothelioma incidence is predicted in certain developing countries where asbestos usage is increasing at an alarming rate and where protection of workers is minimal.
Although pleural and peritoneal mesotheliomas exhibit some genomic differences, both are characterized by frequent somatic loss/inactivation of certain tumor suppressor genes, prominent among them being BAP1, NF2, and CDKN2A/B (3–14). NF2 mutations and loss of expression of the NF2 product, Merlin, have been reported in up to ~55% of mesothelioma cell lines (6). Among pleural mesotheliomas characterized in The Cancer Genome Atlas (TCGA) database, monoallelic deletions of NF2 were observed in ~35% of samples and biallelic inactivation in another 40% of tumors, with many of the latter harboring mutations of one allele (12). In peritoneal mesothelioma, mutations and allelic losses of NF2 have been observed in 21%–35% of cases (11,13,14). Underscoring the relevance of NF2 inactivation to mesothelioma pathogenesis, heterozygous Nf2 knockout mice treated with asbestos develop mesothelioma at a significantly higher frequency and markedly accelerated rate than their wild-type counterparts (7,15). Moreover, in one of these studies, 9/9 mesothelioma cell lines established from neoplastic ascites from asbestos-exposed Nf2+/− mice exhibited loss of the wild-type Nf2 allele, and expression of Merlin was consistently absent in these cells (7).
Merlin has been implicated in various tumor-related signaling pathways, prominent among them being p21-activated kinase (PAK) and Hippo signaling. Merlin regulates the protein kinases Mst1 and Mst2 (mammalian sterile 20-like 1 and −2; a.k.a. serine/threonine protein kinase Stk4 and Stk3) and the serine/threonine kinases Lats1 and −2 (large tumor suppressor 1 and −2). Merlin and each of these kinases are components of the highly conserved Hippo signaling pathway, which regulates organ size in Drosophila and mammals. Combined Mst1/2 deficiency in the liver results in loss of inhibitory phosphorylation of the downstream oncoprotein Yap1 and development of hepatocellular carcinoma (16). Nf2-deficient phenotypes in multiple tissues were suppressed by heterozygous deletion of Yap1, suggesting that Yap is a major effector of Merlin in growth regulation. The Hippo tumor-suppressive signaling pathway has also been connected with Merlin-deficient mesothelioma (17,18). Other work has shown that Merlin suppresses tumorigenesis by activating upstream components of the Hippo pathway by inhibiting the E3 ubiquitin ligase CRL4(DCAF1) (19).
Merlin is regulated by phosphorylation, with hypophosphorylated Merlin being the growth-inhibitory, functionally active tumor suppressor form, whereas hyperphosphorylated Merlin is growth-permissive (20,21). PAK directly phosphorylates Merlin at serine residue 518, a site that regulates Merlin activity and localization (22,23). Such phosphorylation of Merlin weakens its head-to-tail self-association and its association with the cytoskeleton (24). Furthermore, functional analysis of serine 518 phosphorylation has demonstrated that expression of a phospho-mimic mutant (Merlin S518D) caused striking changes in cell proliferation and shape, stimulating the creation of filopodia (25). These results strongly suggest that Merlin’s growth and motility suppression functions are attenuated following phosphorylation by PAK. Moreover, it is noteworthy that many mesotheliomas that lack NF2 mutations nevertheless show functional inactivation of Merlin via constitutive phosphorylation of Ser518, in some cases as a result of inhibition of a Merlin phosphatase (26).
PAKs are serine/threonine protein kinases that are binding partners for the small GTPases Cdc42 and Rac, and they represent one of the most highly conserved effector proteins for these enzymes (27). Mammalian tissues contain six PAK isoforms: group I (Pak1, −2, and −3) and group 2 (Pak4, −5, and −6). These two groups differ substantially in form, function and tissue expression, with only group I PAKs appearing to be involved in Merlin signaling (28). Group I PAKs, in particular Pak1, have oncogenic properties when expressed at high levels. In most settings, expression of PAK1 stimulates cell proliferation, survival, and motility (28,29). Group I PAKs are frequently activated in human malignant mesotheliomas, and genetic or pharmacologic inhibition of PAKs is sufficient to inhibit mesothelioma cell proliferation and survival (30). Importantly, Merlin is more than just a target for PAK; it is also a negative regulator of this kinase. Merlin binds to and inactivates Pak1, and loss of Merlin results in the activation of Pak1 (24). These results suggest that the growth and motility abnormalities of NF2-null cells might in part be attributed to the activation of PAK and its downstream targets. These data also suggest that inhibitors of PAK might be useful in switching off such signaling pathways.
We hypothesized that loss of PAK activity would counteract some aspects of NF2/Merlin loss-of-function by compromising key downstream oncogenic signaling pathways in mesothelial cells. To test this possibility, we crossed Nf2f/f;Cdkn2af/f mice to mice with conditional deletion of Pak2, which encodes the most highly expressed group I PAK isoform in most tissues. Cohorts of these animals were then injected in either the pleural or peritoneal cavities with adeno-Cre virus to delete floxed alleles in the mesothelial lining, with the goal being to determine if Pak2 loss diminishes tumor onset and/or progression at one or both sites where mesothelioma typically originates. We show that loss of Pak2 indeed delays mesothelioma tumorigenesis, though studies employing multiplexed kinase inhibitor beads and mass spectrometry (MIB/MS) and RNA-seq technologies revealed that these mesothelioma cells ultimately reprogram their kinome in the absence of Pak2 activity. The identification of such secondary pathways offers opportunities for the rational design of combination therapies to circumvent resistance to anti-PAK drugs.
Materials and Methods
Mouse strains
LucR;Nf2f/f;Cdknaf/f mice (31), a kind gift of Dr. Anton Berns, and Pak2f/f mice (32), were maintained in a mixed FVB/N × 129/Sv background. All studies were performed in accordance with protocol #18-03 approved by the Fox Chase Cancer Center (FCCC) Institutional Animal Care and Use Committee (IACUC). The specific primer sequences for the genotyping are listed in Supplementary Table S1. All PCR were at Ta = 58°C for 30 cycles.
Adeno-Cre injections, tumor collection, and mesothelioma cell line generation
For studies involving intrathoracic (IT) and intraperitoneal (i.p.) injections of Ad5CMVCre (adeno-Cre) virus, animals with Nf2f/f;Cdknaf/f;Pak2+/+ and Nf2f/f;Cdknaf/f;Pak2f/f genotypes were divided into two cohorts with equivalent numbers of male and female mice in each group. At 8–10 weeks of age, mice were injected either IT or i.p. with adeno-Cre virus (50 μl of 3–6 × 1010 PFU) in PBS. Expression of adeno-Cre results in the removal of the floxed exons in the Nf2, Cdkn2a, and Pak2 loci, generating the following genotypes in infected cells: Nf2Δ/Δ;Cdkn2aΔ/Δ and Nf2Δ/Δ;Cdkn2aΔ/Δ;Pak2Δ/Δ, and the resulting malignant mesotheliomas are referred to as NC;P+/+ and NC;P−/− tumors hereafter. All mice were monitored daily for tumor formation for up to 12 months. Mice were sacrificed by CO2 inhalation upon signs of pain/distress or illness as judged by lethargic behavior, weight loss or bloating, difficulty in breathing, hunched posture, rough hair coat, dehydration or detectable tumor volume approaching 10% of overall body weight. Tissues of all organs of both cavities were collected from sacrificed mice, and tumor specimens were subjected to histopathological assessment by an experienced animal pathologist (K.Q.C.) of FCCC’s Histopathology Facility. Portions of tumors were saved in RNAlater Solution (Thermo Fisher Scientific, Waltham, MA) and frozen at −80°C for subsequent study. When possible, a portion of the fresh primary tumor was also chopped into fine pieces and incubated with 0.2% collagenase for 2 h and then cultured in DMEM supplemented with 10% FCS (GIBCO) and penicillin/streptomycin (GIBCO). The minced tissues were grown at 37°C in 5% CO2 for 2–3 d. Cells that attached to culture dishes and grew out were passaged to establish cell lines. Once the pathologist confirmed by H&E and IHC that the corresponding primary tumor was diagnosed as a mesothelioma, we validated that the derived cell line expressed mesothelioma markers, e.g., WT1 and mesothelin, by semi-quantitative PCR and/or immunoblotting. All mesothelioma cell lines were tested for mycoplasma regularly using LookOut® Mycoplasma PCR Detection Kit (Millipore Sigma, # MP0035-1KT) up until the last experiments were performed. Thawing of the cell lines for experiments was typically at passage 3, and all experiments were conducted using the earliest passages possible (p4–p5), with no experiments performed beyond passage 6.
Detection of tumors and statistical considerations
Survival curves for NC;P+/+ versus NC;P−/− mice were compared using one-sided, log-rank tests. Kaplan-Meier plots were used to display time to tumor detection among the two separate cohorts for both IT and i.p. injection studies.
Tumor histopathology/immunohistochemistry
Histopathology and immunohistochemistry of malignant mesothelioma tissues were performed according to our usual method (33).
Preparation of luciferin and in vivo bioluminescent imaging (BLI)
LucR;Nf2f/f;Cdknaf/f mice were crossed to Pak2f/f mice to generate offspring with different Pak2 genotypes (+/+, +/f, f/f), which at 8–10 weeks of age were injected IT with adeno-Cre virus. Beginning at 6–7 weeks after virus injections, littermates with different genotypes were injected i.p. with 150 mg of filtered D-Luciferin, Firefly, potassium salt (Caliper Life Sciences) in PBS per kg mouse body weight 10 min before imaging. The Cre recombinase removes a floxed polyadenylation acid sequence before the ORF of the luciferase reporter transgene (LucR) permitting luciferase expression for monitoring tumor progression. BLI scans were acquired using an IVIS Spectrum Imaging System (Caliper Life Sciences) as described (34) to assess the presence and relative size of tumors, as indicated by the intensity of luminescent signals detected. The same mice were imaged weekly until tumors began to form. The experiment was repeated four times.
Preparation of lentivirus expressing shRNA against Pak2 and infection of NC;P+/+ mesothelioma cells
A Tet-pLKO-puro plasmid was a gift from Dmitri Wiederschain (Addgene, plasmid # 21915). The shRNAs targeting mouse Pak2 were created by the cloning of annealed forward and reverse oligos synthesized based on information provided at the Broad Institute’s website (https://portals.broadinstitute.org/gpp/public/). Lentivirus was produced by cotransfecting into HEK293T cells separately Tet-inducible shPak2 #70, shPak2 #85, and shGFP with psPAX2 and pMD2.G plasmids using lipofectamine 2000 (Thermo Fisher Scientific). After 24 h and 48 h, virus particles were collected, filtered and then used to infect cell lines for the in vivo experiment described below. For a second set of experiments, lentivirus was produced by transfecting 293T cells with the three separate pLKO shPak2 or with control shGFP. Lentivirus particles harboring the three different shPak2 constructs or control shGFP were used to infect peritoneal NC;P+/+ MM cell lines. Cells were seeded in 6-well plates and infected with virus particles containing polybrene (sc-134220; Santa Cruz) at a concentration of 2 μg/ml, centrifuged at 2,000 rpm for 2 h, and incubated in a humidified 5% CO2 incubator at 37°C for 24 h. The following day, supernatant was removed and cells were selected with puromycin (4 μg/mL) and then used for immunoblotting and cell viability, clonogenicity, and spheroids assays. The clone numbers and target sequences for shPak2 are shown in Supplementary Table S1.
In vivo model to assess lung tumor burden of mesothelioma cells following doxycycline-inducible shRNA knockdown of Pak2
In this experiment, we used tumor cell line (MM87) derived from an asbestos-induced mesothelioma seen in a Nf2+/−;Cdkn2a+/− mouse (35). Stable MM87 cells expressing tet-inducible lentiviruses against Pak2 (shPak2 #70 and #85) were injected into the tail vein of three NSG mice (0.5 × 106 cells/mouse), followed by i.p. administration of doxycycline beginning after d 7 and then every 2 d thereafter after. Animals were sacrificed on d 21, and then the lungs were harvested for histopathological assessment of tumor colonization using an Aperio ScanScope CS2 scanner (Aperio, Vista, CA) (see Supplementary Materials and Methods for details).
Preparation of lentivirus expressing Pak2 and infection of NC;P−/− mesothelioma cell lines
Preparation and infection of NC;P−/− mesothelioma cells with untagged Pak2, HA-tagged Pak2, and control pLenti were performed using the same protocol described above (see Supplementary Materials and Methods for details). After selection was completed, the cells were used for immunoblotting as well as cell viability and clonogenicity assays.
Immunoblot analysis and antibodies
For immunoblotting, protein lysates were prepared as previously described (35). Lysates (30-50 μg/sample) were loaded on gels, transferred to nitrocellulose membranes (1620115, Bio-Rad, Hercules, CA), and probed with primary antibodies listed in Supplementary Table S2. Anti-rabbit secondary antibody (45-000-682, 1:5000) and anti-mouse secondary antibody (45-000-679, 1:5000-50000 depending on the primary antibody) were from Fisher Scientific (Waltham, MA). Immunoblots were imaged using Immobilon Western Chemiluminescent HRP Substrate (ECL) (WBKLS0500, MilliporeSigma, Ontario Canada).
Cell fractionation studies
Cell fractionation of NC;P−/− and NC;P+/+ peritoneal mesothelioma cell lines was performed using a Protease Inhibitor Cocktail (#11836170001, MilliporeSigma), as described in the Supplementary Materials and Methods. For immunoblotting, protein lysates were prepared as previously described (35).
Migration assay
In vitro migration of NC;P−/− and NC;P+/+ mesothelioma cell lines was determined using a Transwell Multiwell Plate with Polyester Membrane Inserts (Corning, Edison, NJ), following the manufacturer’s protocol. The migrating cells on the underside of the membrane were fixed and stained with a Diff-Quik stain kit (Siemens).
Cell viability and clonogenic assays
NC;P+/+ cell lines with stable knockdown of Pak2 were trypsinized, counted, and seeded in 96-well plates at 500 cells/well. In a different experiment NC;P+/+ and NC;P−/− cell lines were seeded in 96-well plates (2,500 cells/well), and 24 h after seeding, the cells were treated with the indicated concentrations of GANT61 (in medium containing 2.5% FBS) and either JQ1 or DMSO (the latter two with 10% FBS) for 72–96 h. Cell viability was determined was assessed using a CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS) (G3582; Promega, Madison, WI), according to the manufacturer’s instructions. The same protocol was used to perform a cell viability assay involving the stable overexpression of Pak2 in NC;P−/− cells.
Clonogenic assays were performed by seeding NC;P+/+ cell lines with stable knockdown of Pak2 in triplicate in 10-cm Petri dishes at a density of 10,000 cells/dish. Colonies were stained and counted after 2 weeks, using a Diff-Quik stain kit (Siemens). Each experiment was performed in triplicate. For quantification of the clonogenic assay, each stained 10-cm dish was incubated in a shaker with 6 ml of 0.01N HCL for 15 min. Then 100 μl of solution was transferred in 96-well plates, and the absorbance was determined at 630 nm. The same protocol was used to perform a clonogenic assay for stable overexpression of Pak2 in NC;P−/− cells.
Spheroid formation assay
Spheroid formation in NC;P−/− and NC;P+/+ mesothelioma cell lines were performed using ultra-low attachment 6-well plates (5,000 cells/well, Corning #3471) in serum-free DMEM/F12 medium supplemented with B27, EGF (10 ng/mL), mouse basic FGF (10 ng/mL) and penicillin/streptomycin. Spheroids were photographed after 10 d of cell culture. Mesothelioma cell lines were incubated with or without drug (GANT61, a Gli1 inhibitor, or JQ1, an inhibitor of bromodomain proteins, one of which (BRD4) regulates Myc transcription) for 10 d, and then spheroids were photographed.
RNA-seq and gene set enrichment analyses performed on peritoneal primary mesotheliomas
RNA was isolated from NC;P+/+ and NC;P−/− primary peritoneal mesotheliomas using TRIzol (Thermo Fisher Scientific) and purified using RNeasy columns (Qiagen, Germantown, MD) following the manufacturers’ instructions. The RNA quality was determined using a Bioanalyzer (Agilent). RNA-seq analysis was performed on a set of tumors (5 primary NC;P−/− mesotheliomas and 3 primary NC;P+/+ mesotheliomas) by Novogene (Sacramento, CA), using its Illumina NovoSeq 6000 platform. Raw sequence counts for each gene were produced with HTseq (https://htseq.readthedocs.io), and differentially expressed genes were determined using DESeq2 (36). For functional enrichment analysis, genes identified as differentially expressed with nominal p-value < 0.5 were ranked by fold-change and mapped to human genes using R biomaRt (37), and these genes were analyzed using the GSEAPreranked method of Gene Set Enrichment Analysis (GSEA) (38). Further details are shown in the extended Supplementary Materials and Methods. Raw sequencing data have been deposited in the GEO repository with the dataset Series record GSE192660.
Semi-quantitative reverse transcriptase PCR analysis
For semiquantitative reverse transcriptase PCR (RT-PCR) analysis, 1 μg of total mRNA was retrotranscribed into cDNA from a subset of NC;P−/− and NC;P+/+ primary peritoneal tumors and cell lines. Normal mesothelial cell cultures (NMC) were used as controls. Reverse transcription was performed using SuperScript II Reverse Transcriptase (Thermo Fisher Scientific) following the manufacturer’s protocol. RT-PCR analysis was performed to confirm the diagnosis of malignant mesothelioma and to validate RNA sequencing experiments. PCR was performed using REDTaq (Sigma) with the annealing temperature at 58°C. PCR products were run on 2% ethidium bromide gels. The primer sequences are shown in Supplementary Tables S1 (mesothelioma marker genes) and S3 (differentially expressed genes).
MIBs preparation and chromatography
To gain an unbiased and more comprehensive view of Merlin signaling, kinome reprogramming analysis was performed using MIB/MS (39). In brief, cells were lysed on ice in buffer containing 50 mM HEPES (pH 7.5), 0.5% Triton X-100, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10 mM sodium fluoride, 2.5 mM sodium orthovanadate, 1X protease inhibitor cocktail (Roche), and 1% each of phosphatase inhibitor cocktails 2 and 3 (Sigma). Particulate was removed by centrifuging lysates at 21,000 g for 15 min at 4°C and filtering through 0.45 μm syringe filters. Protein concentrations were determined by BCA analysis (Thermo Fisher Scientific). Endogenous kinases were isolated by flowing lysates over kinase inhibitor-conjugated Sepharose beads (purvalanol B, VI16832, PP58 and CTx-0294885 beads) in 10 ml gravity-flow columns. After 2×10-ml column washes in high-salt buffer and 1×10-ml wash in low-salt buffer (50 mM HEPES, pH 7.5), 0.5% Triton X-100, 1 mM EDTA, 1 mM EGTA, 10 mM sodium fluoride, and 1M NaCl or 150 mM NaCl, respectively, retained kinases were eluted from the column by boiling in 2 × 500 μl of 0.5% SDS, 0.1 M TrisHCl (pH 6.8), and 1% 2-mercaptoethanol. Eluted peptides were reduced by incubation with 5 mM DTT at 65°C for 25 min, alkylated with 20 mM iodoacetamide at room temperature for 30 min in the dark, and alkylation was quenched with DTT for 10 min. Samples were concentrated to ~100 μl with Millipore 10-kD cutoff spin concentrators. Detergent was removed by chloroform/methanol extraction, and the protein pellet was resuspended in 50 mM ammonium bicarbonate and digested with sequencing-grade modified trypsin (Promega) overnight at 37°C. Peptides were cleaned with PepClean C18 spin columns (Thermo Fisher), dried in a speed-vac, resuspended in 50 μl of 0.1% formic acid, extracted with ethyl acetate (10:1 ethyl acetate:H2O), washed five times in 1 mL ethyl acetate, and then dried in a speed vac. Nano-LC-MS/MS and data processing for the kinome profiling analysis were performed as described in detail elsewhere (40). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD030571.
Results
Inactivation of Pak2 results in decreased incidence and delayed onset and progression of pleural and peritoneal mesotheliomas in Nf2-deficient mice
Locotemporal expression of Cre recombinase by either IT or i.p. injection of adeno-Cre virus was used to induce mesothelial cell-specific homozygous deletions of Nf2, Cdkn2a, with or without excision of Pak2, in homozygous compound CKO mice. Examples of genotyping of Nf2f/f;Cdknaf/f;Pak2+/+ and Nf2f/f;Cdknaf/f;Pak2f/f mice and immunoblotting demonstrating loss of expression of conditionally knocked out genes in malignant mesotheliomas arising after injection of adeno-Cre virus are depicted in Fig. 1A and 1B, respectively. The IT-injected mice were followed for up to 1 year, and by the end of the study, 16/19 NC;P+/+ animals (84%) developed pleural mesothelioma, with a median survival of 26 weeks. In contrast, only 8/15 (53%) NC;P−/− mice developed pleural mesothelioma, and the median survival was prolonged to 34 weeks. Among animals injected i.p. with adeno-Cre virus, 18/22 (82%) NC;P+/+ mice developed peritoneal mesothelioma (median survival: 24 weeks) versus only 10/23 (43%) NC;P−/− mice (median survival: 35 weeks). Malignant mesothelioma incidence and Kaplan-Meier survival curves of IT- and i.p.-injected mice that developed mesothelioma are shown in Figure 1C and 1D, respectively. In both the IT and the i.p. studies, the incidence of mesothelioma was much lower in NC;P−/− mice than in the NC;P+/+ cohort. Statistical differences in survivals of NC;P+/+ versus NC;P−/− mice with mesothelioma were highly significant in both IT and i.p. studies. Mesotheliomas from NC;P+/+ and NC;P−/− mice showed similar histopathology, with the vast majority of tumors in both the pleural and peritoneal models being sarcomatoid (Supplemental Figure S1), which is consistent with our previous studies of mesotheliomas in conditional NC mice (33). No purely epithelioid tumors were observed. Several were biphasic, with a predominance of spindle cells.
Figure 1.
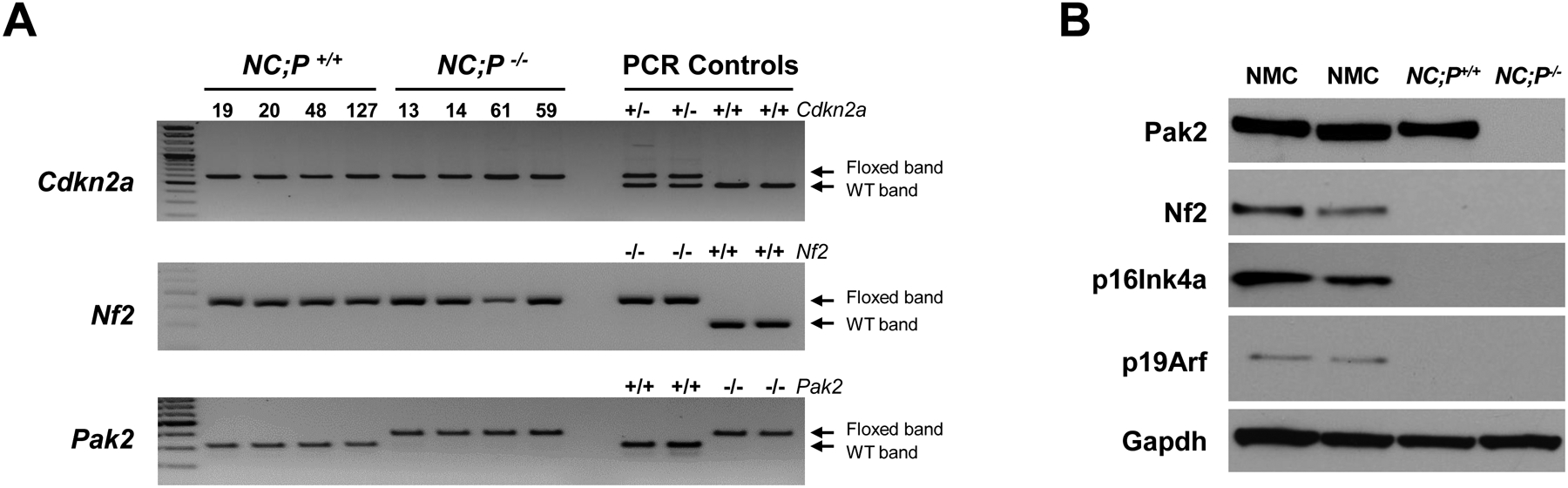
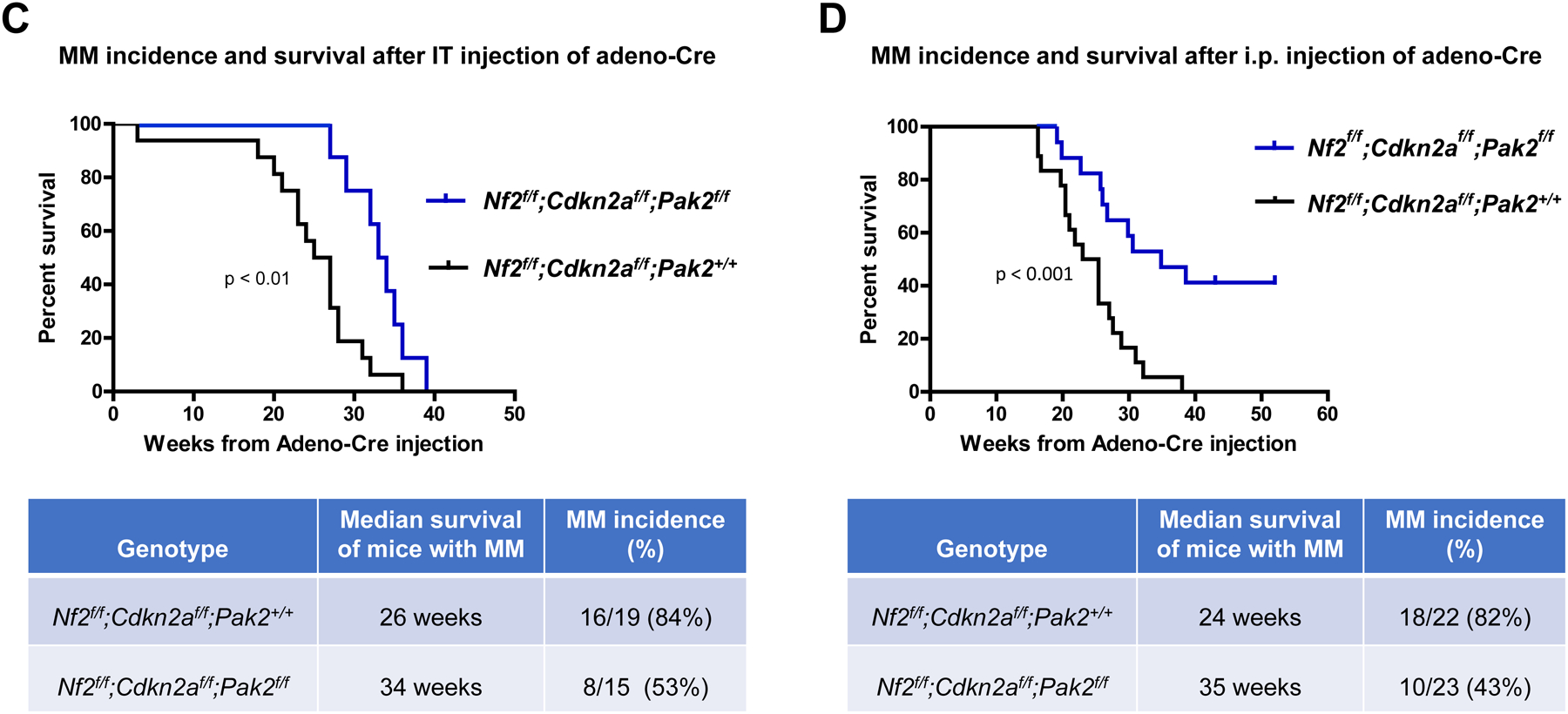
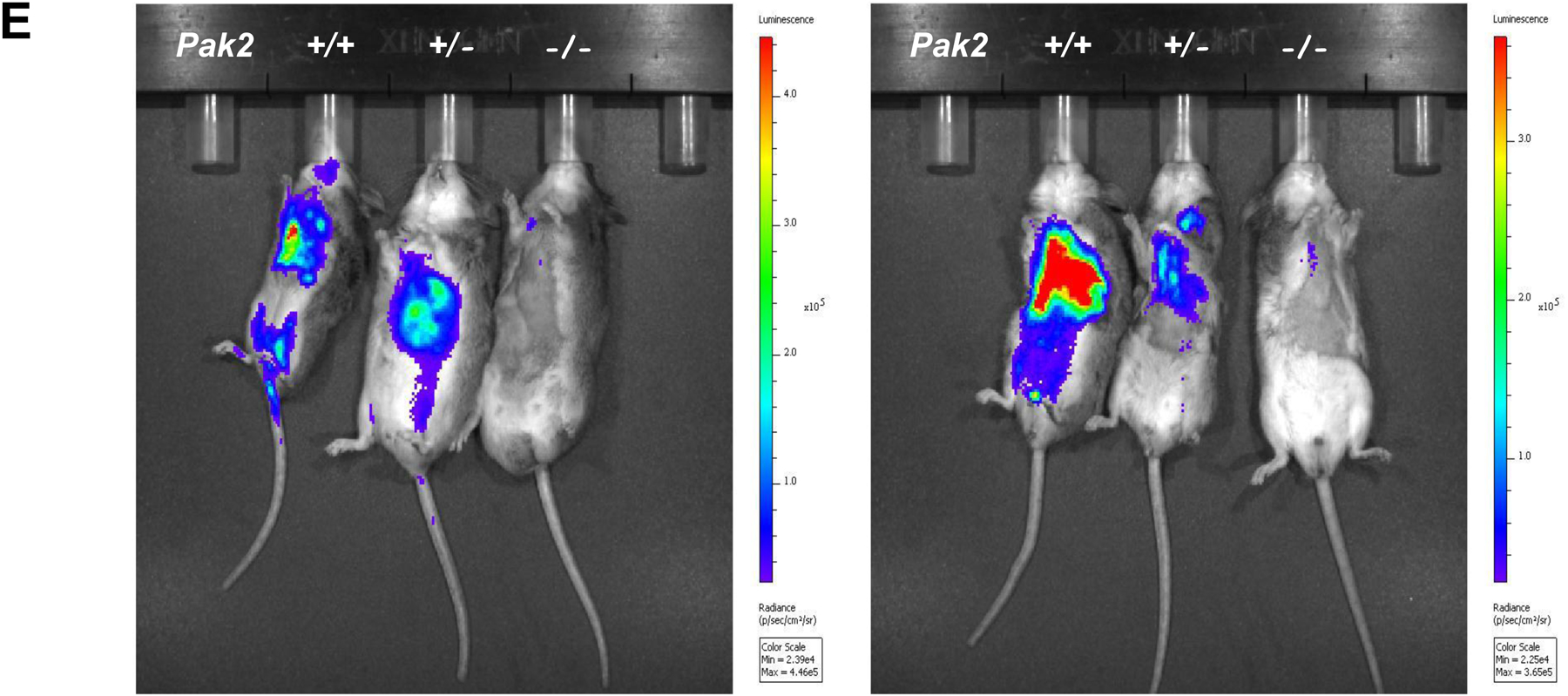
Genotyping and characterization of mesothelioma development in, Nf2f/f;Cdknaf/f;Pak2+/+ and Nf2f/f;Cdknaf/f;Pak2f/f mice injected intrathoracically or intraperitoneally with adeno-Cre virus. (A) Genotyping of tail DNA from four representative Nf2f/f;Cdknaf/f;Pak2+/+ mice and four Nf2f/f;Cdknaf/f;Pak2f/f mice that developed peritoneal mesothelioma. PCR controls for floxed and wild type alleles of Cdkn2a, Nf2 and Pak2 were from tail DNA of heterozygous and homozygous conditional knockout mice. (B) Immunoblotting of two early passage cell lines derived from NC;P−/− and NC;P+/+ peritoneal mesotheliomas demonstrating loss of expression of conditionally knocked out genes in tumor cells. NMC, normal mesothelial cells. (C, D) Malignant mesothelioma progression, incidence and Kaplan-Meier survival curves of cohorts of Nf2f/f;Cdknaf/f;Pak2+/+ and Nf2f/f;Cdknaf/f;Pak2f/f mice injected intrathoracically (C) or intraperitoneally (D) with adeno-Cre virus and succumbing to mesothelioma. Abbreviations: IT, intrathoracic (injection); i.p., intraperitoneal (injection); MM, malignant mesothelioma. (E) Bioluminescent imaging reveals delayed mesothelioma progression in mice with Nf2-null mesothelial lining and excision of one or both alleles of Pak2. ffLucR;Nf2f/f;Cdkn2af/f mice were crossed to Pak2f/f mice to generate offspring having wild type (+/+) Pak2 or with one or both floxed (+/f or f/f, respectively) Pak2 alleles. Mice were injected IT with adeno-Cre virus to excise floxed alleles of thoracic mesothelial lining cells. Infection with adeno-Cre virus also removes a floxed polyadenylation sequence before the ORF of a luciferase reporter transgene (LucR). The latter permits luciferase expression to monitor tumor progression, using D-luciferin as a substrate and bioluminescent imaging with an IVIS Imaging System. Shown is bioluminescent imaging on two sets of ffLucR;NC littermates with three different Pak2 genotypes. Mice were injected with D-luciferin 6 months (left panel) or 7 months (right) after IT injection of adeno-Cre virus; mice with excision of Pak2 show delayed tumor progression as indicated by reduced intensity of luminescent signals. Experiment was repeated four times with similar results.
Generally, the pleural mesotheliomas were solitary and more massive than the tumors observed in the peritoneum. In pleural mesotheliomas, there was no obvious overall tumor size difference between Pak2 WT and Pak2 KO tumors. Moreover, we were able to generate tumor cell lines from a high percentage of the pleural tumors, regardless of the Pak2 status. Mesotheliomas in the peritoneum typically presented as multiple, diffuse, small nodules that often caused intestinal obstructions. Notably, Pak2 loss in peritoneal mesotheliomas resulted in diminished tumor size and multiplicity. Also, while ~80% of Pak2 WT tumors formed cell lines, <20% of Pak2 KO tumors did so.
In vivo bioluminescent imaging confirms that loss of Pak2 delays malignant mesothelioma progression in Nf2-deficient mice
BLI scanning revealed intense luminescent signals in NC;P+/+ mice beginning at about 6 months, with less signal observed in NC;P+/− (heterozygous loss of Pak2) littermates (Figure 1E). BLI scanning was repeated with four sets of littermates with different Pak2 genotypes, and in each instance tumor progression, as indicated by diminished intensity of the luminescent signals observed, was consistently delayed in mice with homozygous excision of Pak2.
Knockdown of Pak2 diminishes metastatic colonization of Nf2-deficient malignant mesothelioma cells in the lung
Malignant mesothelioma cells (MM87), which were previously derived from an asbestos exposed Nf2+/−;Cdkn2a+/− mouse, were infected with tet-inducible lentiviruses against Pak2. Two clones that demonstrated marked knockdown of Pak2 (shPak2 #85 and shPak2 #70 in Figure 2A) were expanded and injected individually into the tail vein of NSG mice. The mice were injected with doxycycline i.p. after 7 d, when the tumors have become established, and then every 2 d thereafter; all animals were sacrificed on d 21, and lungs were examined histopathologically for tumor colonization (Figure 2B). H&E staining revealed that tumor colonization of the lungs by MM87 cells was consistently diminished in the cell clones expressing shRNA against Pak2 versus control MM87 cells infected with lentivirus expressing shRNA against GFP (Figure 2C), and the differences were statistically significant (p < 0.05).
Figure 2.
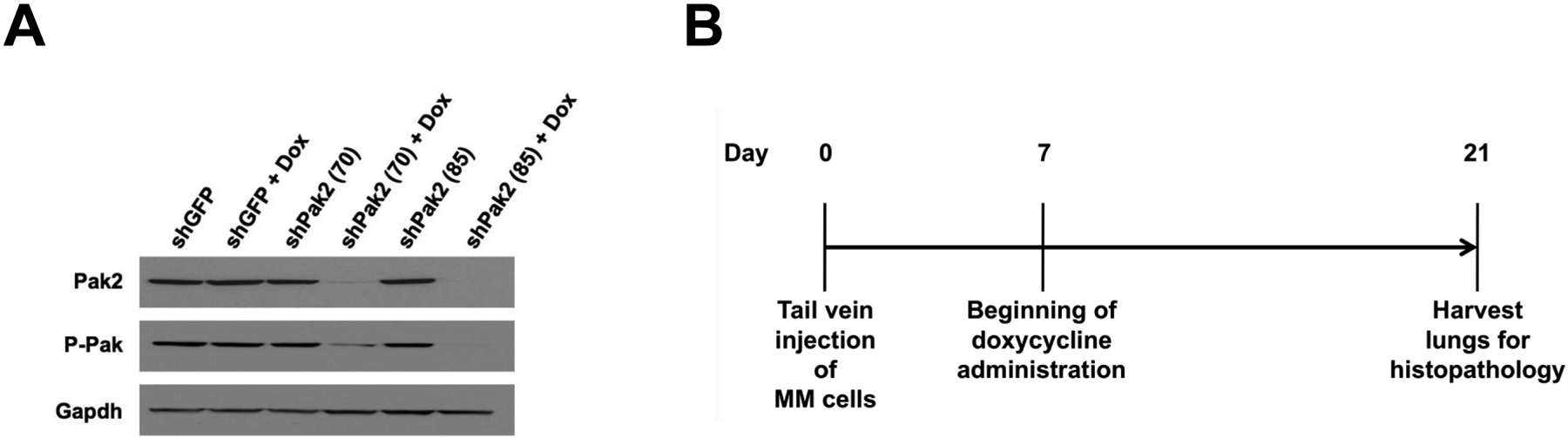
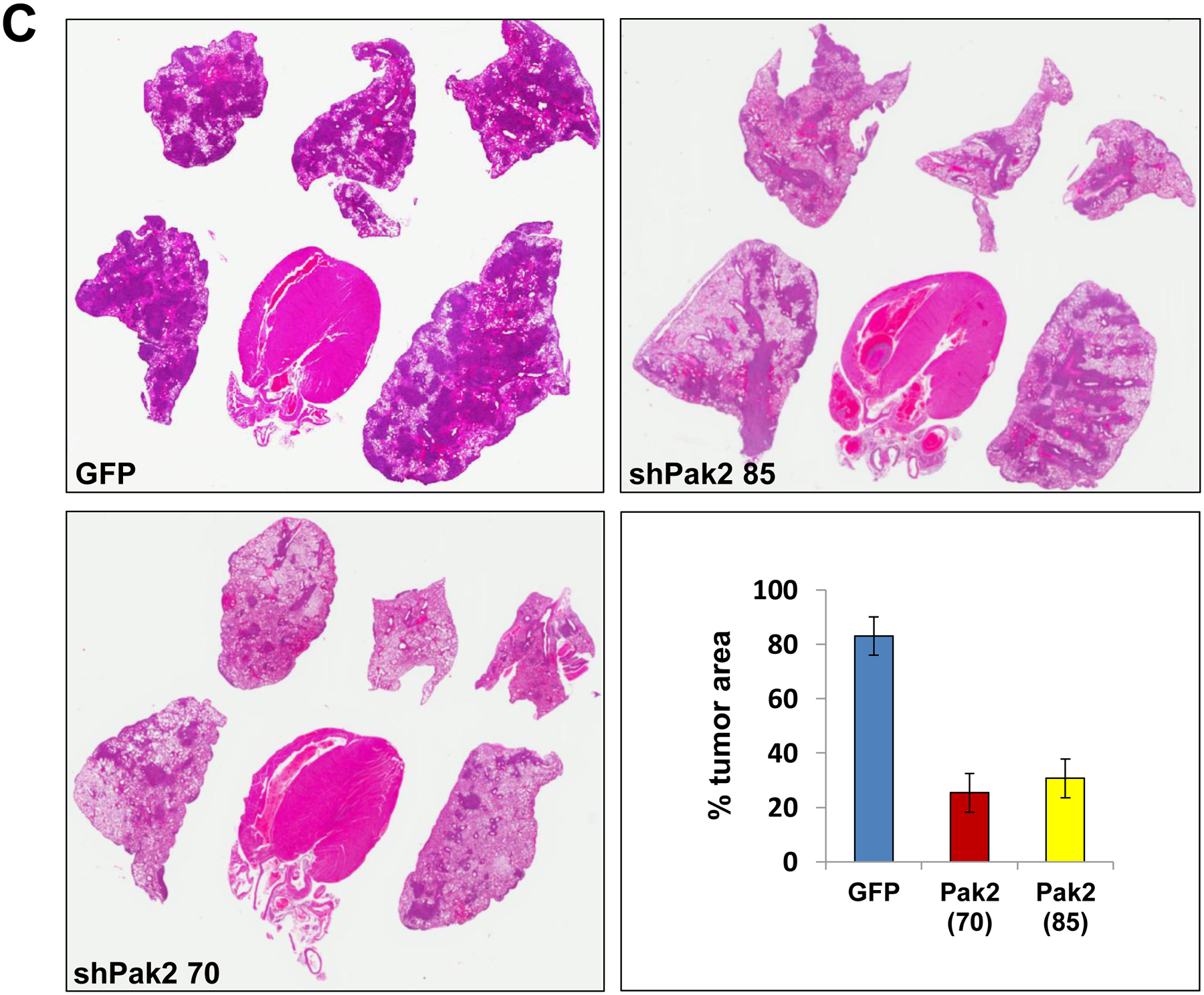
Knockdown of Pak2 diminishes metastatic colonization of Nf2-deficient mesothelioma cells in the lung. Cell line MM87, which was derived from asbestos-induced mesothelioma from a Nf2+/−;Cdkn2a+/− mouse, was infected with tet-inducible lentiviruses against Pak2. (A) Immunoblot demonstrating expression of Pak2 after knockdown with shRNA. Two clones with robust knockdown of Pak2 (shPak2 #70 and shPak2 #85) and one clone infected with lentivirus against GFP, used as a control, were selected for tail vein injections into NSG mice. (B) Malignant mesothelioma clones were each injected into the tail vein of three different NGS mice, followed by injection with doxycycline 7 d later and every 2 d thereafter; all animals were sacrificed on d 21, and lungs were collected for histopathological assessment of tumor burden. (C) H&E staining illustrating representative tumor colonization (intensely stained areas) of lungs by MM87 cells expressing shGFP, shPak2 #70 or shPak2 #85. Tumor burden was quantified using an Aperio ScanScope CS2 scanner, as described in the Supplemental Materials and Methods.
Deletion of Pak2 results in decreased tumor cell migration
We compared cell migration of pleural mesothelioma cells from NC;P+/+ mice versus mesothelioma cells from NC;P−/− mice. Twenty-two hours after seeding, the cells were evaluated for migratory ability using a transwell migration assay, as described in the Materials and Methods. As shown in Supplemental Figure S2, a pleural NC;P−/− mesothelioma cell line tested showed markedly less migration than a NC;P+/+ mesothelioma cell line.
Pak2 loss results in diminished mesothelioma cell viability, clonogenicity, and spheroid formation
The NC;P+/+ peritoneal mesothelioma cell lines tested formed large spheroids when seeded in ultra-low attachment 6-well plates in stem cell media, whereas the NC;P−/− peritoneal mesothelioma lines did not (Fig. 3A). Stable knockdown of Pak2 with lentivirus expressing three separate shRNA (shPak2 #12, #70, and #85), which was confirmed by immunoblotting (Fig. 3B), was shown to inhibit cell viability in two different NC;P+/+ mesothelioma cell lines, as assessed by MTS assay (Fig. 3C). Furthermore, stable knockdown of Pak2 caused a marked reduction in spheroid formation in NC;P+/+ cell lines (Fig. 3D). Clonogenicity was also decreased by stable Pak2 knockdown in NC;P+/+ cells (Fig. 3E). Reciprocally, re-expression of Pak2 in NC;P−/− cell lines resulted in increases in both cell viability and clonogenicity (Supplemental Figure S3).
Figure 3.
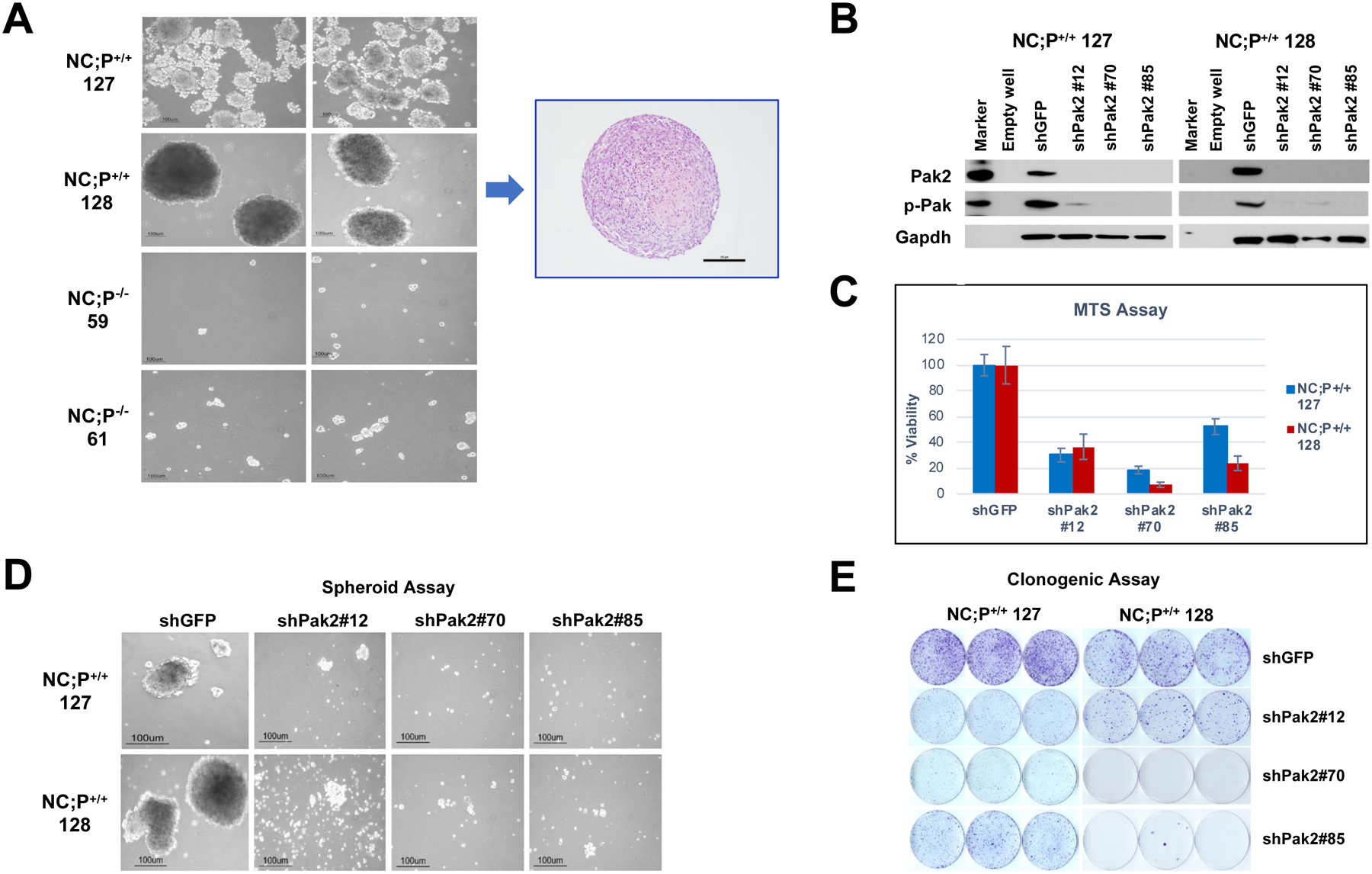
Deletion of Pak2 results in decreased peritoneal malignant mesothelioma cell viability, clonogenicity, and spheroid formation. (A) NC;P−/− mesothelioma cell lines exhibit greatly deceased spheroid formation. Mesothelioma cells were seeded in ultra-low attachment 6-well plates (5,000 cells/well) in stem cell medium, and colonies were photographed after 10 d. H&E staining of a section of typical spheroid from mesothelioma cell line NC;P+/+ 128 is also shown (A). In other experiments shown, two different NC;P+/+ mesothelioma cell lines (127, 128) were infected with lentivirus expressing shGFP (control) and three separate shRNA (#12, #70, #85) against Pak2. After 4 days, the selection with puromycin was completed, and the cells were collected for immunoblotting and seeded for the experiments described below. (B) Immunoblot analysis demonstrating knockdown of Pak2 in the two NC;P+/+ mesothelioma cell lines 4 d after selection with puromycin. Cells with stable knockdown of Pak2 inhibited mesothelioma cell viability (C), spheroid formation (D), and clonogenicity (E). The MTS assay revealed a significant reduction in cell viability 96 h after seeding with each of the three shPak2 lentiviruses compared to the shGFP lentivirus. The clonogenic assay demonstrated a marked reduction in colony formation 2 weeks after seeding with each of the three shPak2, and knock down of Pak2 similarly caused a striking decrease in spheroid formation after 10 d of growth in vitro.
RNA-seq analysis reveals that Pak2 loss in Cdkn2a;Nf2-null primary mesotheliomas leads to diminished expression of genes involved in oncogenic pathways
RNA-seq analysis was performed on 3 primary NC;P+/+ peritoneal mesotheliomas and 5 primary NC;P−/− peritoneal mesotheliomas to identify genes that are differentially expressed due to loss of Pak2, one of the main downstream effectors of Nf2. Most differentially expressed genes in the NC;P−/− tumors were downregulated. Many of the downregulated genes are involved in muscle contraction and cardiac epithelial-mesenchymal transition (EMT) pathways (Supplemental Figure S4), whereas others are involved in Wnt (Wif1, Axin2, Gli1, Gpc3, Sfrp5, Fzd4/8/9, Lrp4, Wnt9b) and Hedgehog (Gli1) signaling. Other downregulated genes involved kinase and other cancer-related pathways (Igf2, Fgfr3/4, Notch3, Hey1, Tgfb2, and Ifi44l), stem cell markers (Sox8/11), and integrins (Itga7/8). Fewer upregulated genes were observed in the NC;P−/− mesotheliomas; these included a Wnt inhibitor gene (Dkk2), a gene encoding a kinase (Styk1), and a transcription repressor gene (Foxg1). A heatmap of genes differentially expressed in NC;P−/− mesotheliomas versus NC;P+/+ mesotheliomas is shown in Figure 4, including genes involved in Wnt and Hedgehog signaling (Figure 4A) and various other pathways (Figure 4B). Multiple cancer-related genes validated by semi-quantitative RT-PCR analysis are shown in Figure 4C along with several mesothelial markers such as Wt1 and Msln (mesothelin). Immunoblot analysis of Nf2/Merlin, Pak2, p16Ink4a, and p19Arf in 3 NC;P−/− and 3 NC;P+/+ primary peritoneal mesotheliomas corresponding to a subset of the tumors used for the RNA-seq analysis are shown in Figure 4D.
Figure 4.
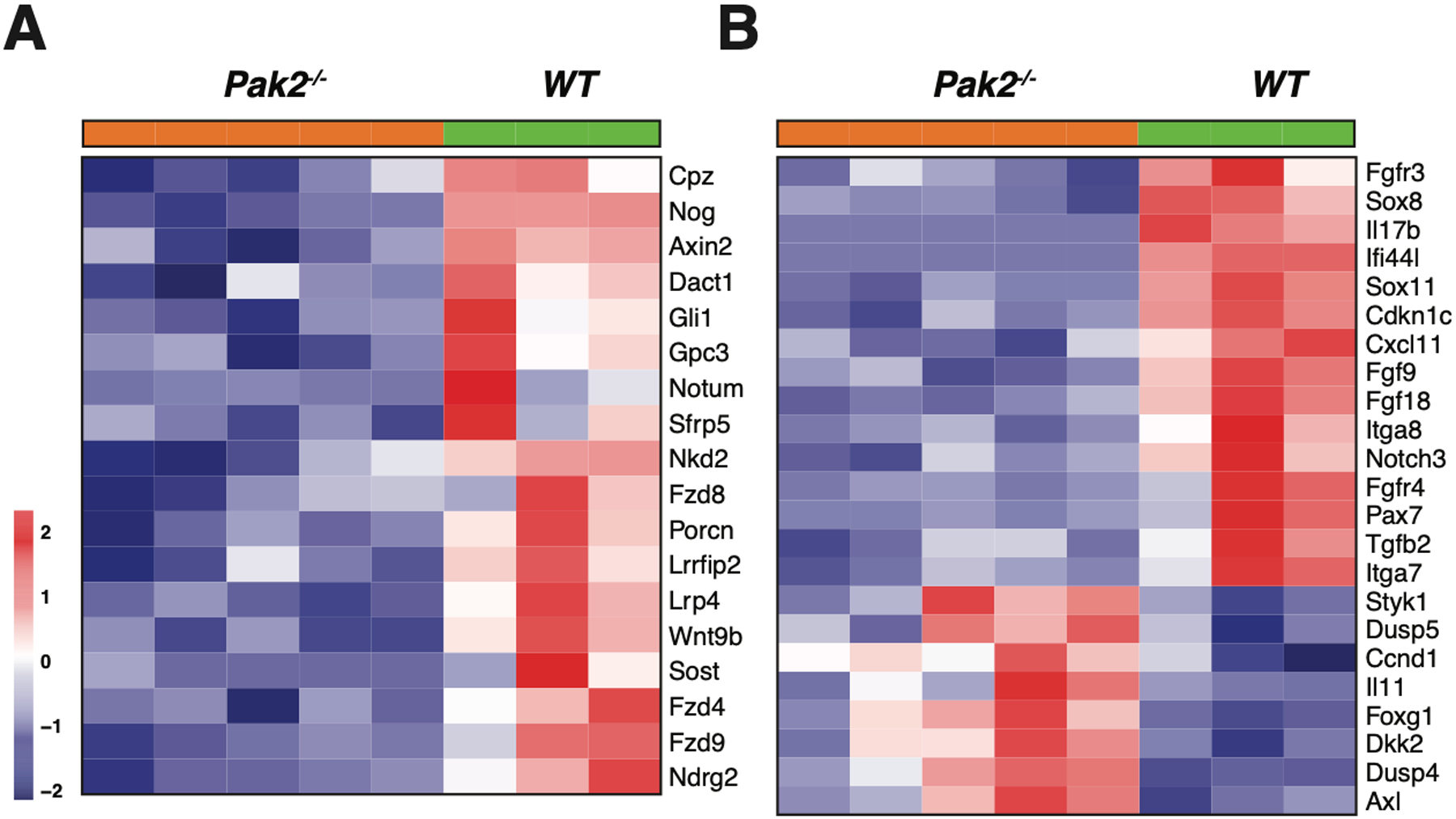

Heatmaps showing expression patterns of genes in NC;P+/+ and NC;P−/− primary malignant mesotheliomas. Heatmaps depict differentially expressed genes observed in peritoneal malignant mesotheliomas from 3 NC;P+/+ mice versus 5 peritoneal malignant mesotheliomas from NC;P−/− mice. (A) Genes involved in Wnt and Hedgehog signaling. (B) Differentially expressed genes involved in multiple pathways/functions, including Akt and Notch signaling, cardiac EMT, cell cycling, cancer stem cell (CSC) markers, and integrins. (C) Validation of a number of differentially expressed genes by semi-quantitative RT-PCR analysis. Left, Down-regulated genes in NC;P−/− primary malignant mesotheliomas include genes involved in Hedgehog signaling (Gli1, Gli2), CSC properties and metastasis (Sox8, Sox11), Wnt signaling (Fzd4, Fzd9, Axin2), and Akt signaling (Igf2, Fgfr4). Right, Validation of genes involved in Notch signaling (Notch3, Hey1) and genes involved in tumor reprogramming in NC;Pak2−/− mesothelioma cells. Note that NC;Pak2−/− cells show downregulation of IfI44L, a tumor suppressor gene that has been implicated in metastasis and drug resistance by regulating met/Src signaling (49), and upregulation of Dkk2 and Styk1, genes involved in cell proliferation and metastasis. Controls include malignant mesothelioma marker genes Msln (mesothelin), Wt1, and cytokeratin 18 (Krt18). Note that in addition to the mutant deleted Pak2 allele seen in NC;P−/− malignant mesotheliomas, there is also a weak wild type allele due to contaminating stroma in these tumor samples. NMC, normal mesothelial cells; *, wild type Pak2 allele; **, mutant Pak2 allele created after adeno-Cre excision of floxed allele. (D) Immunoblot analysis confirming the loss of expression of Pak2 (as well as of Nf2, p16Ink4a and p19Arf) in three NC;P−/− primary malignant mesotheliomas, whereas Pak2 expression is retained in the three NC;P+/+ tumors. NMC, normal mesothelial cells.
Pak2 loss in malignant mesothelioma results in downregulated Hedgehog and Wnt signaling and diminished cancer stem cell (CSC) properties
Semi-quantitative PCR analysis was also performed on early passage NC;P−/− and NC;P+/+ peritoneal mesothelioma cell lines. These studies confirmed that NC;P−/− cells exhibited down regulation of genes involved in Hedgehog signaling (Gli1, Gli2), CSCs (Sox2, Nanog, Nes), cell migration/invasion (Adam8, Mmp9), and Wnt signaling (Lrp4, Lrg6, Wnt10b, Stmn2) (Fig. 5A). Immunoblotting of NC;P−/− and NC;P+/+ cell lines demonstrated loss of expression of Pak2, Nf2, p16Ink4a, p19Arf and Myc, an activator and transcriptional target of Hedgehog signaling, in NC;P−/− cells (Fig. 5B). Cell fractionation studies of the NC;P−/− and NC;P+/+ peritoneal mesothelioma cell lines were performed, and immunoblot analysis revealed decreased nuclear expression of Gli1 and Myc in NC;P−/− cells (Fig. 5C).
Figure 5.
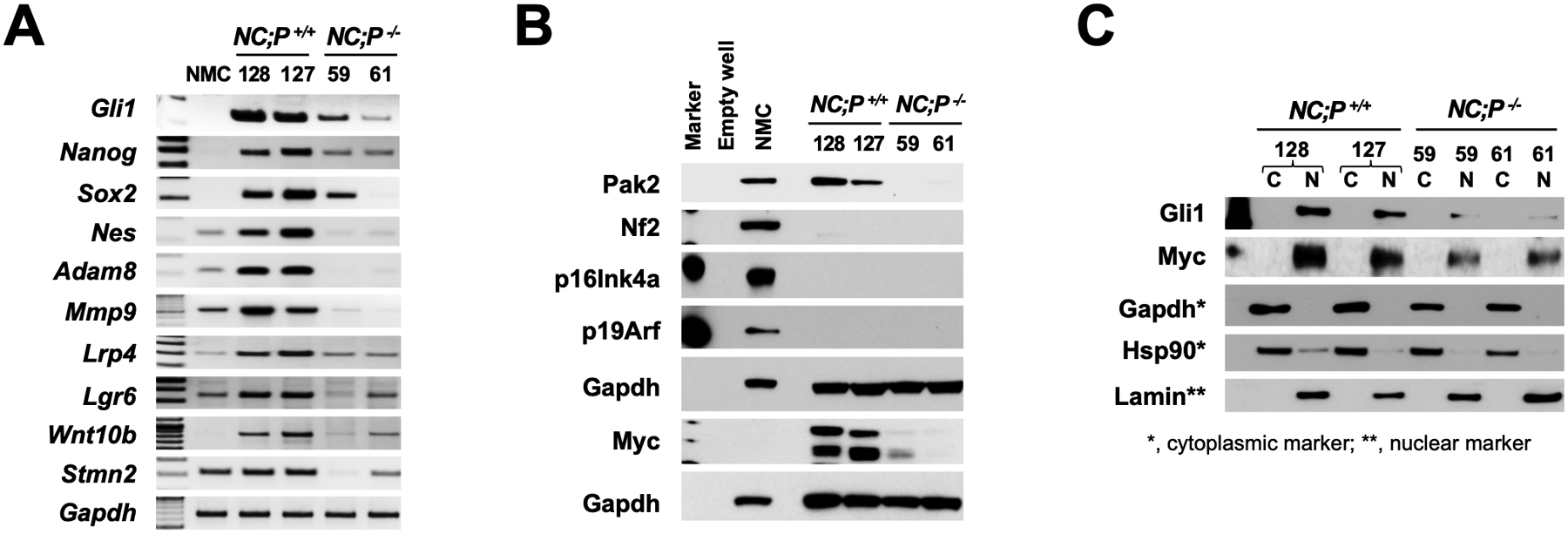
Loss of Pak2 in peritoneal mesothelioma cell lines results in down regulation of Hedgehog and Wnt signaling as well as CSC and metastasis markers. (A) Semi-quantitative PCR analysis of early passage (p. ≤8) peritoneal NC;P−/− and NC;P+/+ mesothelioma cell lines demonstrating down-regulation of several genes involved in Hedgehog signaling (Gli1), CSCs (Nanog, Nes, Sox2), migration/invasion (Adam8, Mmp9), Wnt signaling (Lrp4, Lrg6, Wnt10b, Stmn2). (B) Immunoblot analysis of NC;P−/− and NC;P+/+ mesothelioma cell lines demonstrating loss of expression of Pak2, Nf2, p16Ink4a, and p19Arf in all cell lines and loss of expression of Pak2 and the oncoprotein Myc specifically in NC;P−/− mesothelioma cells. (C) Cell fractionation analysis of peritoneal NC;P−/− and NC;P+/+ mesothelioma cells; immunoblotting demonstrates decreased expression of nuclear localized Gli1 and Myc proteins in NC;P−/− cell lines 59 and 61. C, cytoplasmic lysate; N, nuclear lysate.
Assessment of kinome reprogramming in Pak2-null pleural mesothelioma cells using MIB/MS technology
Given that the development of drug resistance is a significant factor in aggressive tumors such as malignant mesothelioma, we also used a technological approach, MIB/MS, that globally measures kinase signaling at the proteomic level (39,40) to assess dynamic reprogramming of the kinome in response to genetic inhibition of Pak2 in malignant mesothelioma. This technology was used to compare the kinase expression of an early passage NC;P+/+ pleural mesothelioma cell line with that of an early passage NC;P−/− pleural mesothelioma line, with studies performed in triplicate. Kinome activity, principal component analysis (PCA), identities of up- and down-regulated kinases, and heat-map are depicted in Figure 6A–D. The kinome profiling revealed that as compared to NC;P+/+ mesothelioma cells, NC;P−/− mesothelioma cells had multiple kinase changes suggestive of EMT. Prominent among the changes associated with Pak2 loss were up regulation of Pdgfrα, Pdgfrβ, Limk1, Fyn, Jak1 and Mkk6 along with down regulation of Ddr1. Immunoblot confirmation of expression changes in selected kinases is shown in Figure 6E, with upregulated expression of Pdgfrα, Pdgfrβ, and phospho-Stat3 being consistently observed in multiple NC;P−/− cell lines versus NC;P+/+ lines (Fig. 6F).
Figure 6.
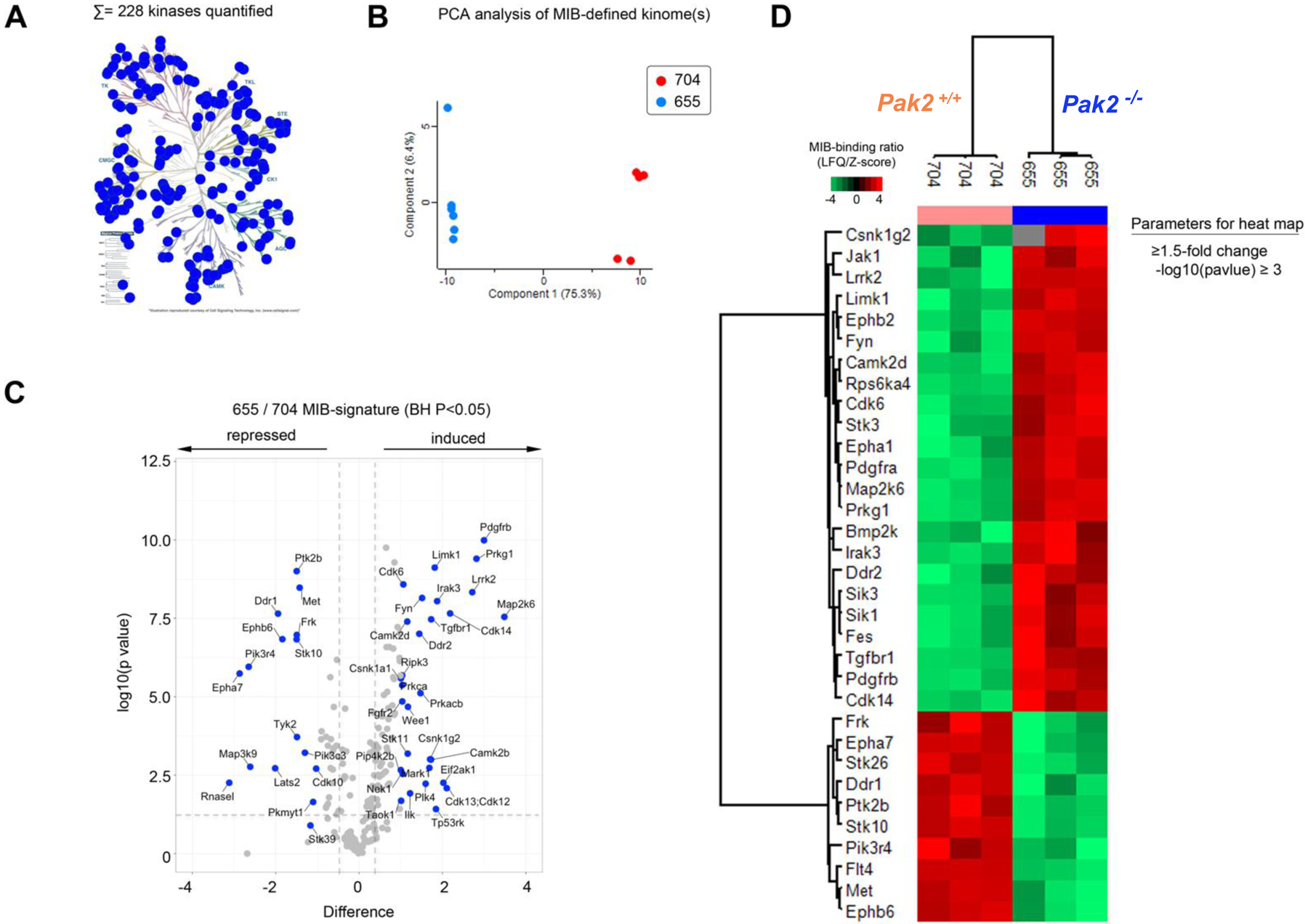
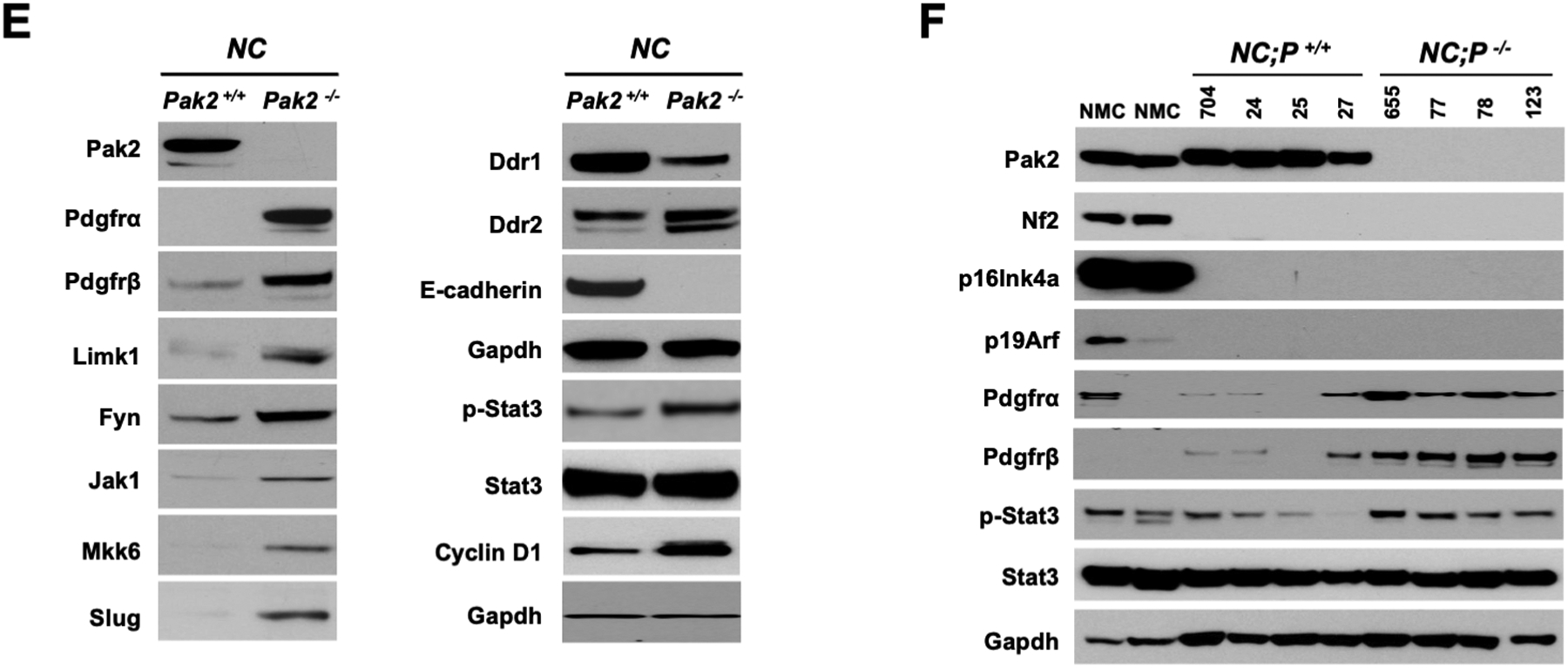
Kinome profiling of NC;P−/− pleural mesothelioma cells and NC;P+/+ mesothelioma cells. Early passage pleural mesothelioma cell cultures were derived from tumors observed in Nf2f/f;Cdkn2af/f and Nf2f/f;Cdkn2af/f;Pak2f/f mice injected IT with adeno-Cre virus. (A) Kinome activity as measure by MIBs. (B) PCA analysis, showing differential kinome profiles. (C) Volcano plot depicts kinases exhibiting induced or repressed MIB binding in cells with knockout of Pak2. (D) Heat-map depicts statistical changes in kinase levels between one NC;P−/− mesothelioma cell line and one NC;P+/+ mesothelioma cell line, with experiment performed in triplicate. (E) Immunoblot confirmation of expression changes in selected kinases in these two cell lines. (F) Immunoblot analysis confirming consistently up-regulated expression of Pdgfrα, Pdgfrβ, and p-Stat3 in NC;P−/− versus NC;P+/+ mesothelioma cell lines.
Targeting of Gli1 or Myc inhibits cell viability and spheroid formation in NC;P+/+ mesothelioma cells
Early passage NC;P−/− and NC;P+/+ peritoneal mesothelioma cell lines were seeded in 96-well plates and treated with various concentrations of GANT61, a Gli1/2 inhibitor, and JQ1, an inhibitor of bromodomain proteins, one of which (BRD4) regulates Myc transcription. MTS assays demonstrated substantial decreases in cell viability in NC;P+/+ cells compared to NC;P−/− cells at higher concentrations of GANT61 and JQ1 (Fig. 7A). Moreover, both GANT61 and JQ1 radically inhibited the formation of spheroids in NC;P+/+ cells, whereas little effect was observed with either drug in NC;P−/− cells (Fig. 7B). Semi-quantitative PCR analysis (Fig. 7C) of the NC;P+/+ cells demonstrated that GANT61 decreased the expression of Gli1 mRNA expression at a concentration of 5 μM, which was also the concentration that effectively inhibited spheroid formation (Fig. 7B). Immunoblot analysis demonstrated the JQ1 diminished the expression of Myc at 39 and 78 nM (Fig. 7D), doses that were also sufficient to markedly inhibit spheroid formation. However, we acknowledge that JQ1 has activity against several other targets besides BRD4, including other BET proteins, and we did not examine directly whether the effects we observed are mediated via BRD4.
Figure 7.

Targeting of Gli1 or Myc each inhibits cell viability and spheroid formation in NC;P+/+ mesothelioma cells. (A) Early passage NC;P−/− and NC;P+/+ peritoneal mesothelioma cell lines were seeded in 96-well plates (2,500 cells/well) and treated with indicated concentrations of GANT61 or JQ1 for 72 h in medium supplemented with 2.5% FBS for GANT61 treatment and 10% FBS for JQ1. MTS assays show significant decreases in cell viability in NC;P+/+ cells compared to NC;P−/− cells at increasing concentrations of GANT61 and JQ1. IC50 values for the two NC;P+/+ cell lines were determined using GraphPad Prism. IC50’s were not determined for the NC;P−/− cell lines, as these cells were very insensitive to GANT61 and JQ1 due to their low expression of Gli1 and Myc, respectively. (B) Morphological appearance of spheroids in presence or absence of GANT61 and JQ1. NC;P−/− and NC;P+/+ cells were seeded in 6-well plates (5,000 cells/well) in stem cell medium and treated with GANT61 (2.5 and 5 μM) or JQ1 (39 and 78 nM), and cells were photographed after 10 d. Both GANT61 (at 5 μM) and JQ1 (both at 39 and 78 nM) markedly inhibited spheroid formation in NC;P+/+ cells compared to cells cultured in medium containing vehicle (DMSO). In contrast, neither drug had much effect on NC;P−/− cells. (C) Semi-quantitative PCR analysis was performed on NC;P+/+ cell lines treated as above, and after treatment with drugs for 72 h. RNA was extracted with TRIzol and semi-quantitative PCR was performed. (D) Immunoblot analysis of NC;P+/+ cells treated with different concentrations of JQ1. Cells were seeded in 6-well plates, treated with JQ1 for 72 h, lysed, and subjected to immunoblot analysis, which demonstrates inhibition of Myc expression by the drug treatment.
Discussion
Losses of NF2 and CDKN2A are thought to play a critical role in malignant mesothelioma pathogenesis. In fact, in human peritoneal mesothelioma, homozygous deletions in CDKN2A and hemizygous loss of NF2 as detected by fluorescence in situ hybridization has been reported to confer a poor clinical outcome, whereas loss of BAP1 was not associated with clinical outcome (11). In conditional knockout mice, Nf2f/f mice and Cdkn2af/f mice injected IT with adeno-Cre virus resulted in very few pleural mesotheliomas, whereas Nf2f/f;Cdkn2af/f mice injected with adeno-Cre virus resulted in aggressive thoracic tumor growth (31,33). In the present study, Nf2f/f;Cdkn2af/f;Pak2f/f mice acquire loss of only a single Group I PAK gene in the mesothelial lining following injection with adeno-Cre virus; yet, this was sufficient to significantly decrease the incidence and delay the onset and progression of both pleural and peritoneal mesotheliomas when compared to that of NC mice retaining Pak2.
Group I PAKs are frequently activated in human malignant mesotheliomas, and genetic or pharmacologic inhibition of PAKs is sufficient to inhibit mesothelioma cell proliferation and survival (30). Pak1 is frequently overexpressed in human breast, ovarian, bladder and brain cancers, often secondary to amplification of its 11q13.5–14 chromosomal locus (41). Moreover, breast and bladder carcinoma cells bearing such amplification were shown to be highly sensitive to PAK inhibition by small molecule inhibitors or RNAi, suggesting that these cancer cells are “addicted” to Pak1 overexpression. The role of Pak2 in human neoplasia is less well studied, but it has been linked to a variety of human cancers (27). The pathways that mediate the oncogenic effects of PAK overexpression are not fully known, but include activation of the Erk pathway (via phosphorylation of Raf and Mek), inactivation of NF2/Merlin, stabilization of β-catenin, and possibly scaffolding interactions that link Pdk1 with Akt (27). Loss of PAK function is associated with decreased cell proliferation and motility, with concomitant loss of activity of these signaling pathways (42,43). Consistent with these data, we demonstrate that loss of Pak2 results in diminished mesothelioma cell migration and downregulated expression of two genes, Adam8 and Mmp9, that have been implicated in cell migration and invasion. Additionally, other in vitro studies revealed that loss of Pak2 in mesothelioma cells results in decreased cell viability, clonogenicity, and spheroid formation.
Pak1 and Pak2, while similar in sequence and structure, nevertheless have a number of distinct functions. For example, although both isoforms are broadly expressed, deletion of the Pak2 gene in mice leads to early embryonic lethality, whereas deletion of Pak1 has little effect on development, longevity, or fertility (27). Further, cardiovascular toxicity of anti-PAK small molecule inhibitors are related to blockade of PAK2 rather than PAK1 (44). The mechanistic bases for these differences are not yet well understood, but may be related to differences in intracellular locations and/or intrinsic differences in substrate preferences (27). These distinguishing features need to be kept in mind when considering potential therapeutic interventions.
Our RNA-seq analysis demonstrated that loss of Pak2 counteracts some aspects of Nf2 loss-of-function in mesothelioma cells by downregulating the expression of multiple genes involved in oncogenic pathways, particularly genes encoding proteins involved in Hedgehog (Gli1/2) and Wnt signaling. In response to activation of Hedgehog signaling, Gli proteins are processed into transcriptional activators that induce expression of target genes, including Myc, Nanog, Sox2 and Wnt, all genes that are downregulated in NC;P−/− versus NC;P+/+ cells. Notably, the CSC marker genes Sox2, Nanog and Nes (nestin), a new putative marker of CSCs (45), were all upregulated in NC;P+/+ cells, which correlates with the prominent spheroid formation properties observed in these cells. Germane to this, we have previously reported cooperativity between Nf2 and Cdkn2a losses to drive the development of highly aggressive mesotheliomas characterized by large tumor spheroids in ascites, enhanced tumor spreading capability, and the presence of a CSC population (35).
Collectively, the findings presented here point to a positive role for group I PAKs in tumorigenesis mediated by several central signaling pathways, Hedgehog and Wnt being prominent among them. Our findings further suggest that inhibitors of PAK might be useful in switching off such signaling pathways. Furthermore, these data help establish a framework for understanding malignant mesothelioma adaptation and permit the design of rational combinations of targeted agents, as clinical or near-clinical inhibitors for many protein kinases already exist.
We found that targeting of Gli1 or Myc, two activators and effectors of Hedgehog signaling, inhibited cell viability and spheroid formation in NC;P+/+ mesothelioma cells but had minimal effect on NC cells with loss of Pak2. Similarly, in studies of primary colorectal cancer organoid cultures, treatment with GANT61 inhibited expression of stem cell markers such as Myc and Nanog, which was thought to occur via decreased transcription of GLI1 (46). GLI1 is a transcriptional effector at the terminus of the Hedgehog pathway, and in certain cancers, aberrant activation of the Hedgehog pathway has been shown to promote dedifferentiation to a more stem-like phenotype (47).
The RNA-seq profiling presented here also demonstrated that numerous genes that are downregulated in NC;P−/− mesothelioma cells are involved in muscle contraction and cardiac EMT pathways. These data are consistent with previous work showing a significant role for Group I PAKs in myoblast and cardiac muscle function. For example, the Rac1-Pak2 pathway has been shown to be indispensable for zebrafish heart regeneration (48). In addition, Group I PAKs have been shown to promote skeletal myoblast differentiation during postnatal development and regeneration in mice, and adult mice conditionally lacking both Pak1 and Pak2 in the skeletal muscle lineage developed an age-related myopathy, with muscles exhibiting centrally-nucleated myofibers, fibrosis, and signs of degeneration (49). As in the RNA-seq studies, kinome profiling assayed by MIB/MS technology also uncovered kinase changes indicative of EMT in NC;P−/− mesothelioma cells, including up regulation of Pdgfrα, Pdgfrβ and Fyn kinases, as well as up regulation of the EMT transcription regulator Slug. Recent work has clarified the EMT cell plasticity program as a set of dynamic transitional states between the epithelial and mesenchymal phenotypes, with EMT and its intermediary states serving as critical drivers of organ fibrosis and tumor progression (50). The upregulation of these genes in NC;P−/− mesothelioma cells may represent an adaptive survival mechanism that can be rationally targeted to prevent the emergence of resistance to PAK small molecule inhibitors.
Collectively, the findings presented here suggest that Nf2-deficient mesothelioma cells with loss of Pak2 ultimately adapt by reprogramming their kinome and transcriptome in the absence of PAK activity. While PAKs are vital to oncogenic signaling in NF2-null mesothelioma cells, targeted genetic inactivation of Pak2 in Nf2-null mesothelioma cells resulted in downregulated expression of genes involved in oncogenic pathways. Eventually, however, malignant mesotheliomas develop in NC;P−/− mice apparently by compensatory activation of oncogenic pathways involving other kinases such as Styk1, Pdgfrα/β, Jak/Stat and Fyn, as well as by gene expression changes such as downregulation of the tumor suppressor gene If44il and upregulation of Dkk2. Such information can inform the design of rational combination therapies for malignant mesothelioma using molecularly targeted inhibitors. The identification of such secondary pathways that could be co-targeted to prevent resistance to anti-PAK drugs thus sets the stage for future preclinical studies with novel therapeutics.
Supplementary Material
Implications.
We provide evidence supporting a role for PAK inhibitors in treating NF2-deficient tumors. NF2-deficient tumors lacking Pak2 eventually adapt by kinome reprogramming, presenting opportunities for combination therapies to bypass anti-PAK drug resistance.
Acknowledgments
This work was supported by NCI grants CA148805 (to J.R. Testa and J. Chernoff) and CA06927 (to FCCC) and an appropriation from the Commonwealth of Pennsylvania to FCCC. Other support was provided by the Local #14 Mesothelioma Fund of the International Association of Heat and Frost Insulators and Allied Workers. The authors thank Michael Slifker and Dr. Alison M. Kurimchak for compiling and depositing datasets in the GEO repository and ProteomeXchange, respectively. The following FCCC core services assisted this project: Laboratory Animal, Transgenic Mouse, Genomics, Cell Culture, Histopathology, and Biostatistics and Bioinformatics Facilities.
Footnotes
Conflict of Interest Statement
JRT has provided legal consultation regarding genetic aspects of mesothelioma. The remaining authors have no potential conflicts of interest with regard to the publication of this work.
References
- 1.Kindler HL. Peritoneal mesothelioma: the site of origin matters. Am Soc Clin Oncol Educ Book 2013;33:182–8. [DOI] [PubMed] [Google Scholar]
- 2.Britton M The epidemiology of mesothelioma. Semin Oncol 2002;29:18–25. [DOI] [PubMed] [Google Scholar]
- 3.Cheng JQ, Jhanwar SC, Klein WM, Bell DW, Lee W-C, Altomare DA, et al. p16 alterations and deletion mapping of 9p21-p22 in malignant mesothelioma. Cancer Res 1994;54:5547–51. [PubMed] [Google Scholar]
- 4.Sekido Y, Pass HI, Bader S, Mew DJY, Christman MF, Gazdar AF, et al. Neurofibromatosis type 2 (NF2) gene is somatically mutated in mesothelioma but not in lung cancer. Cancer Res 1995;55:1227–31. [PubMed] [Google Scholar]
- 5.Bianchi AB, Mitsunaga S-I, Cheng JQ, Klein WM, Jhanwar SC, Seizinger B, et al. High frequency of inactivating mutations in the neurofibromatosis type 2 gene (NF2) in primary malignant mesotheliomas. Proc Natl Acad Sci U S A 1995;92:10854–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng JQ, Lee WC, Klein MA, Cheng GZ, Jhanwar SC, Testa JR. Frequent mutations of NF2 and allelic loss from chromosome band 22q12 in malignant mesothelioma: evidence for a two-hit mechanism of NF2 inactivation. Genes Chromosomes Cancer 1999;24:238–42. [PubMed] [Google Scholar]
- 7.Altomare DA, Vaslet CA, Skele KL, De Rienzo A, Devarajan K, Jhanwar SC, et al. A mouse model recapitulating molecular features of human mesothelioma. Cancer Res 2005;65:8090–5. [DOI] [PubMed] [Google Scholar]
- 8.Bott M, Brevet M, Taylor BS, Shimizu S, Ito T, Wang L, et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat Genet 2011;43:668–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Testa JR, Cheung M, Pei J, Below JE, Tan Y, Sementino E, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet 2011;43:1022–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nasu M, Emi M, Pastorino S, Tanji M, Powers A, Luk H, et al. High incidence of somatic BAP1 alterations in sporadic malignant mesothelioma. J Thorac Oncol 2015;10:565–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singhi AD, Krasinskas AM, Choudry HA, Bartlett DL, Pingpank JF, Zeh HJ, et al. The prognostic significance of BAP1, NF2, and CDKN2A in malignant peritoneal mesothelioma. Mod Pathol 2016;29:14–24. [DOI] [PubMed] [Google Scholar]
- 12.Hmeljak J, Sanchez-Vega F, Hoadley KA, Shih J, Stewart C, Heiman D, et al. Integrative molecular characterization of malignant pleural mesothelioma. Cancer Discov 2018;8:1548–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hung YP, Dong F, Torre M, Crum CP, Bueno R, Chirieac LR. Molecular characterization of diffuse malignant peritoneal mesothelioma. Mod Pathol 2020;33:2269–79. [DOI] [PubMed] [Google Scholar]
- 14.Offin M, Yang SR, Egger J, Jayakumaran G, Spencer RS, Lopardo J, et al. Molecular characterization of peritoneal mesotheliomas. J Thorac Oncol 2021;Oct 11;S1556–0864(21)03215–9. doi: 10.1016/j.jtho.2021.09.012. Online ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fleury-Feith J, Lecomte C, Renier A, Matrat M, Kheuang L, Abramowski V, et al. Hemizygosity of Nf2 is associated with increased susceptibility to asbestos-induced peritoneal tumours. Oncogene 2003;22:3799–805. [DOI] [PubMed] [Google Scholar]
- 16.Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 2009;16:425–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murakami H, Mizuno T, Taniguchi T, Fujii M, Ishiguro F, Fukui T, et al. LATS2 is a tumor suppressor gene of malignant mesothelioma. Cancer Res 2011;71:873–83. [DOI] [PubMed] [Google Scholar]
- 18.Mizuno T, Murakami H, Fujii M, Ishiguro F, Tanaka I, Kondo Y, et al. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene 2012;31:5117–22. [DOI] [PubMed] [Google Scholar]
- 19.Li W, Cooper J, Zhou L, Yang C, Erdjument-Bromage H, Zagzag D, et al. Merlin/NF2 loss-driven tumorigenesis linked to CRL4(DCAF1)-mediated inhibition of the hippo pathway kinases Lats1 and 2 in the nucleus. Cancer Cell 2014;26:48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shaw RJ, McClatchey AI, Jacks T. Localization and functional domains of the neurofibromatosis type II tumor suppressor, merlin. Cell Growth Diff 1998;9:287–96. [PubMed] [Google Scholar]
- 21.Morrison H, Sherman LS, Legg J, Banine F, Isacke C, Haipek CA, et al. The NF2 tumor suppressor gene product, merlin, mediates contact inhibition of growth through interactions with CD44. Genes Dev 2001;15:968–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiao GH, Beeser A, Chernoff J, Testa JR. p21-activated kinase links Rac/Cdc42 signaling to merlin. J Biol Chem 2002;277:883–6. [DOI] [PubMed] [Google Scholar]
- 23.Kissil JL, Johnson KC, Eckman MS, Jacks T. Merlin phosphorylation by p21-activated kinase 2 and effects of phosphorylation on merlin localization. J Biol Chem 2002;277:10394–9. [DOI] [PubMed] [Google Scholar]
- 24.Kissil JL, Wilker EW, Johnson KC, Eckman MS, Yaffe MB, Jacks T. Merlin, the product of the Nf2 tumor suppressor gene, is an inhibitor of the p21-activated kinase, Pak1. Mol Cell 2003;12:841–9. [DOI] [PubMed] [Google Scholar]
- 25.Rong R, Surace EI, Haipek CA, Gutmann DH, Ye K. Serine 518 phosphorylation modulates merlin intramolecular association and binding to critical effectors important for NF2 growth suppression. Oncogene 2004;23:8447–54. [DOI] [PubMed] [Google Scholar]
- 26.Thurneysen C, Opitz I, Kurtz S, Weder W, Stahel RA, Felley-Bosco E. Functional inactivation of NF2/merlin in human mesothelioma. Lung Cancer 2009;64:140–7. [DOI] [PubMed] [Google Scholar]
- 27.Radu M, Semenova G, Kosoff R, Chernoff J. PAK signalling during the development and progression of cancer. Nat Rev Cancer 2014;14:13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hofmann C, Shepelev M, Chernoff J. The genetics of Pak. J Cell Sci 2004;117:4343–54. [DOI] [PubMed] [Google Scholar]
- 29.Bokoch GM. Biology of the p21-activated kinases. Annu Rev Biochem 2003;72:743–81. [DOI] [PubMed] [Google Scholar]
- 30.Menges CW, Sementino E, Talarchek J, Xu J, Chernoff J, Peterson JR, et al. Group I p21-activated kinases (PAKs) promote tumor cell proliferation and survival through the AKT1 and Raf-MAPK pathways. Mol Cancer Res 2012;10:1178–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jongsma J, van Montfort E, Vooijs M, Zevenhoven J, Krimpenfort P, van der Valk M, et al. A conditional mouse model for malignant mesothelioma. Cancer Cell 2008;12:261–71. [DOI] [PubMed] [Google Scholar]
- 32.Radu M, Lyle K, Hoeflich KP, Villamar-Cruz O, Koeppen H, Chernoff J. p21-activated kinase 2 regulates endothelial development and function through the Bmk1/Erk5 pathway. Mol Cell Biol 2015;35:3990–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kukuyan AM, Sementino E, Kadariya Y, Menges CW, Cheung M, Tan Y, et al. Inactivation of Bap1 cooperates with losses of Nf2 and Cdkn2a to drive the development of pleural malignant mesothelioma in conditional mouse models. Cancer Res 2019;79:4113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Connolly DC, Hensley HH. Xenograft and transgenic mouse models of epithelial ovarian cancer and non-invasive imaging modalities to monitor ovarian tumor growth in situ: applications in evaluating novel therapeutic agents. Curr Protoc Pharmacol 2009;45:14.2.1–2.6. [PMC free article] [PubMed] [Google Scholar]
- 35.Menges CW, Kadariya Y, Altomare D, Talarchek J, Neumann-Domer E, Wu Y, et al. Tumor suppressor alterations cooperate to drive aggressive mesotheliomas with enriched cancer stem cells via a p53-miR-34a-c-Met axis. Cancer Res 2014;74:1261–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 2009;4:1184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurimchak AM, Shelton C, Duncan KE, Johnson KJ, Brown J, O’Brien S, et al. Resistance to BET bromodomain inhibitors is mediated by kinome reprogramming in ovarian cancer. Cell Rep 2016;16:1273–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurimchak AM, Kumar V, Herrera-Montávez C, Johnson KJ, Srivastava N, Devarajan K, et al. Kinome profiling of primary endometrial tumors using multiplexed inhibitor beads and mass spectrometry identifies SRPK1 as candidate therapeutic target. Mol Cell Proteomics 2020;19:2068–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dummler B, Ohshiro K, Kumar R, Field J. Pak protein kinases and their role in cancer. Cancer Metastasis Rev 2009;28:51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sells MA, Boyd JT, Chernoff J. p21-activated kinase 1 (Pak1) regulates cell motility in mammalian fibroblasts. J Cell Biol 1999;145:837–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Allen JD, Jaffer ZM, Park SJ, Burgin S, Hofmann C, Sells MA, et al. p21-activated kinase regulates mast cell degranulation via effects on calcium mobilization and cytoskeletal dynamics. Blood 2009;113:2695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rudolph J, Murray LJ, Ndubaku CO, O’Brien T, Blackwood E, Wang W, et al. Chemically diverse group I p21-activated kinase (PAK) inhibitors impart acute cardiovascular toxicity with a narrow therapeutic window. J Med Chem 2016;59:5520–41. [DOI] [PubMed] [Google Scholar]
- 45.Neradil J, Veselska R. Nestin as a marker of cancer stem cells. Cancer Sci 2015;106:803–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Usui T, Sakurai M, Umata K, Elbadawy M, Ohama T, Yamawaki H, et al. Hedgehog signals mediate anti-cancer drug resistance in three-dimensional primary colorectal cancer organoid culture. Int J Mol Sci 2018;19:1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Avery JT, Zhang R, Boohaker RJ. GLI1: A Therapeutic target for cancer. Front Oncol 2021;11:673154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peng X, He Q, Li G, Ma J, Zhong TP. Rac1-PAK2 pathway Is essential for zebrafish heart regeneration. Biochem Biophys Res Commun 2016;472:637–42. [DOI] [PubMed] [Google Scholar]
- 49.Joseph GA, Hung M, Goel AJ, Hong M, Rieder MK, Beckmann ND, et al. Late-onset megaconial myopathy in mice lacking group I Paks. Skelet Muscle 2019;9:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nieto MA, Huang RY-J, Jackson RA, Thiery JP. EMT: 2016. Cell 2016;166:21–45. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.