

Abstract
Alpha-tocotrienol (α-TCT) is a member of the vitamin E family. It has been reported to protect the brain against various pathologies including cerebral ischemia and neurodegeneration. However, it is still unclear if α-TCT exhibits beneficial effects during brain development. We hypothesized that treatment with α-TCT improves intracellular redox homeostasis supporting normal development of neurons. We found that primary hippocampal neurons isolated from rat feti grown in α-TCT-containing media achieved greater levels of neurite complexity compared to ethanol-treated control neurons. Neurons were treated with 1μM α-TCT for 3 weeks, and media was replaced with fresh α-TCT every week. Treatment with α-TCT increased α-TCT levels (26 pmol/mg protein) in the cells, whereas the control neurons did not contain α-TCT. α-TCT treated neurons produced adenosine triphosphate (ATP) at a higher rate and increased ATP retention at neurites, supporting formation of neurite branches. Although treatment with α-TCT alone did not change neuronal viability, neurons grown in α-TCT were more resistant to death at maturity. We further found that mRNA and protein levels of B-cell lymphoma-extra large (Bcl-xL) are increased by α-TCT treatment without inducing post-translational cleavage of Bcl-xL. Bcl-xL is known to enhance mitochondrial energy production which improves neuronal function including neurite outgrowth and neurotransmission. Therefore α-TCT-mediated Bcl-xL upregulation may be the central mechanism of neuroprotection seen in the α-TCT treated group. In summary, treatment with α-TCT upregulates Bcl-xL and increases ATP levels at neurites. This correlates with increased neurite branching during development and with protection of mature neurons against oxidative stress.
Keywords: Bcl-xL, vitamin E, mitochondria, ATP, hippocampal neuron
Graphic Abstract
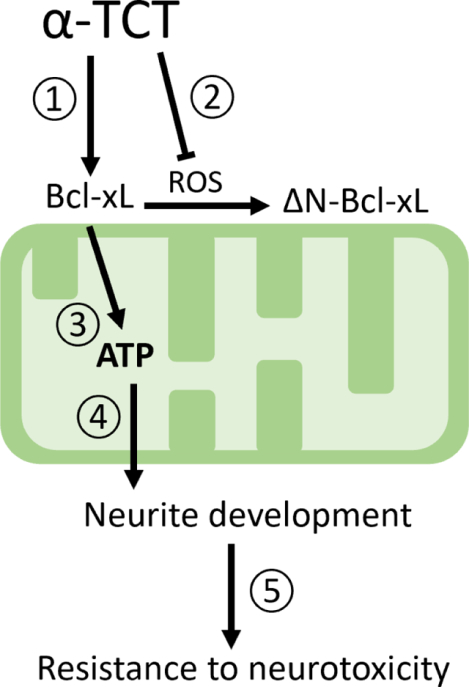
Treatment with α-TCT upregulates Bcl-xL (①) but does not cause post-translational cleavage of Bcl-xL that generates neurotoxic ΔN-Bcl-xL (②). Bcl-xL improves energy metabolism (③) at neurites advancing arborization during development of primary hippocampal neurons (④). Mature neurons with advanced neurite complexity are protected against neurotoxic insults(⑤).
1. Introduction
Neurite arborization, or branching, is an important biological process during neuronal development and plays a role in increasing neurite complexity and synaptic plasticity. Neurite arborization requires production and organization of cytoskeletal elements. Energy in the form of mitochondrial adenosine triphosphate (ATP) is necessary for the formation and maintenance of functional synapses [1]. Therefore, protecting or enhancing the function of mitochondria could be key to supporting neurite complexity during neuronal development.
B-cell lymphoma-extra large (Bcl-xL) is an anti-apoptotic member of the B-cell lymphoma-2 (Bcl-2) family proteins found in mitochondria. Bcl-xL binds to other Bcl-2 proteins such as Bcl-2-associated X (Bax), Bcl-2-antagonist/killer (Bak), and Bcl-2-associated agonist of cell death (Bad), and this interaction inhibits mitochondria-mediated apoptosis, enhancing cell survival [2–9]. In addition to this classic role as an anti-apoptotic protein, other functions of Bcl-xL have been reported by various research groups during the previous two decades. Bcl-xL is necessary for neuronal differentiation and development [10]. The cornu ammonis (CA)1-CA3 regions of the hippocampus and the upper layer of cortical neurons are particularly sensitive to Bcl-xL quantity [11], and depletion of Bcl-xL during development is highly associated with neurobehavioral deficits in mice [11]. Importantly, Bcl-xL regulates neuronal energy metabolism by enhancing ATP production via forming a multiprotein complex with the F1Fo ATP synthase [12, 13], maintaining mitochondrial inner membrane potential [14], and controlling mitochondrial morphology, fusion, fission [15, 16], and mitophagy [17, 18]. These actions improve neuronal energy metabolism supporting neurite outgrowth [19], synapse formation, and neurotransmission [8, 16, 20, 21]. Therefore, approaches that support mitochondrial function by maintaining or enhancing the abundance of Bcl-xL may favor neuronal growth and development.
Alpha-tocotrienol (α-TCT) is a fat-soluble nutrient in the vitamin E family found in plant-based food sources such as palm oil and rice bran oil. Although α-TCT is a form of vitamin E typically found less abundantly in the diet and tissues compared to alpha-tocopherol (α-TCP), supplementation of α-TCT is shown to exhibit neuroprotective properties when delivered to the brain in various models, including rodents, canines, and humans. Mice orally gavaged with 50mg/kg α-TCT for 13 weeks exhibited increased α-TCT levels (18 pmol/mg protein) in the brain [22]. Ten weeks of oral supplementation of α-TCT (61.52 mg twice/day) in canines increased α-TCT levels in the cerebral cortex (77.4 nmol/g protein), whereas no α-TCT was detected in the brain of the control group [23]. Brain tissue from human subjects supplemented with α-TCT (61.52 mg daily) for an average of 261 days contained 1.29 nmol/g of α-TCT [24]. The neuroprotective roles of α-TCT differ from those of α-TCP. Specifically, hippocampal HT4 cells treated with nM-μM α-TCT show greater protection against excitotoxicity, glutamate-induced calcium overload and cellular oxidant production, compared to the α-TCP treated group [25], indicating biological functions beyond its classic antioxidant properties. Supplementation with α-TCT (50mg/kg) in mice upregulates the expression of the multidrug resistance-associated protein 1 in the brain, enhancing the efflux of a neurotoxic byproduct of glutathione during cerebral ischemia [22]. Treatment of primary hippocampal neurons with α-TCT (1 μM) inhibits the formation of ΔN-Bcl-xL, a pro-death post-translational cleavage product of Bcl-xL, protecting neurons against excitotoxicity [7, 26]. Supplementation of the tocotrienol-rich fraction (60 mg/kg) extracted from palm oil inhibits the formation of β-amyloid in both in vitro and in vivo Alzheimer’s models [27]. α-TCT (5 μM) treatment protects axons and dendrites against hydrogen peroxide-induced degeneration in cerebellar granule cells [28]. Although α-TCT is protective when applied to in in vitro and in vivo pathology models, its role in neurons during normal physiological development is unclear.
In this study, we hypothesized that α-TCT provides benefits during normal development of the brain. To improve our understanding of the role of α-TCT in neuronal development, we examined whether treatment with α-TCT influences arborization of neurites. Specifically, the effect of chronic exposure of α-TCT on Bcl-xL expression was explored due to Bcl-xL’s known role in enhanced ATP production efficiency and reactive oxygen species (ROS) production [12, 14].
2. Methods and Materials
2.1. Culture of primary hippocampal neurons
Primary rat hippocampal neurons were prepared from rat feti (Sprague-Dawley, day 18 of gestation; Envigo, Indianapolis, IN) as described previously [19, 29–31]. Briefly, neurons (0.4 × 106 cells/ 35mm plate) were seeded on poly-l-lysin coated plates and grown in neurobasal medium supplemented with B-27, glutamine, and antibiotics (Invitrogen Gibco Life Technologies, Carlsbad, CA) for 20–22 days in vitro (DIV). Cell culture media contain 0.0916 μg/ml α-TCP with no α-TCT. α-TCT treatment: Neurons were either treated with α-TCT (1μM, ≥98% purity, Cayman chemical, Ann Arbor, MI) or a vehicle (ethanol). Neuronal media, a combination of conditioned neurobasal media with freshly prepared 1μM α-TCT or ethanol, were replaced every week for 3 weeks. Human subjects supplemented with 61.52 mg α-TCT contain α-TCT in the nanomolar range in their brain tissues [24]. Treatment with 1μM α-TCT in vitro system results in a similar range, pico to nano molar levels α-TCT in brain cells [25]. Transfection: Neurons were transfected with copGFP or lenti-Bcl-xL shRNA-GFP (Santa Cruz Biotechnology, Dallas, TX) at DIV7 and experiments were then performed 3 weeks after transfection. All protocols were approved by the Institutional Animal Care Committee (IACUC) of The University of Alabama, Tuscaloosa, AL (17-06-0324) and Yale University, New Haven, CT (2019–10388).
2.2. Sholl analysis
Sholl analysis was used to quantify neurite branches as previously described [32–34]. In brief, fluorescent micrographs of primary hippocampal neurons were opened using the Simple Neurite Tracer plugin for ImageJ (National Institutes of Health, Bethesda, MD, USA), and neurites from the soma and daughter branches from the neurites were selected using the Path Manager function. Saved traces were analyzed using Sholl analysis, a Sholl profile was created with the number of intersections at the specific radium (20, 40, 60, 80, 100, 120, 140, 160, 180, and 200 μm from soma), and micrographs were converted with pseudo-color. For the pseudo-color, different colors of neurites correspond to the number of intersections at the same radius. The largest and smallest number of intersections in the image correspond to white and black respectively, and the remaining colors are assigned based on a linear scale.
2.3. 2’,7’- dichlorodihydrofluorescein diacetate (H2DCFDA) staining
Primary hippocampal neurons were treated with 10 μM of 2’,7’-dichlorodihydrofluorescein diacetate (H2DCFDA) (ThermoFisher) solution prepared in a light protected vessel, then incubated for 30min at 37°C in the dark [35], and processed as the manufacturer’s protocol. After incubation, neurons were carefully washed with pre-warmed HBSS. Intracellular fluorescence was measured using a fluorescent microplate reader (CLARIOstar, BMG Labtech) at excitation and emission wavelengths of 470 and 515nm, respectively.
2.4. Fluo-4 staining
Primary hippocampal neurons were treated with 2.5 μM Fluo-4 (Invitrogen) solution prepared in a light protected vessel, then incubated for 30 min at 37°C as per the manufacturer’s protocol [36]. Micrographs were taken using a Zeiss Axiovert A1 microscope (Zeiss, Oberkochen, Germany). Fluo-4 fluorescence density per cell was analyzed using AxioVision 4.9.
2.5. Antioxidant capacity
Samples were deproteinated according to a published method using methanol/acetonitrile/acetone (1:1:1, v/v/v) added to samples in a ratio of 1:4 (v/v) [37]. This method allows for detection of small molecular weight antioxidants < 6 kDa in size. The antioxidant capacity of neuronal lysates was measured using the oxygen radical absorbance capacity (ORAC) assay on a FLUOstar Optima plate reader (BMG Labtech) in accordance with the method by Prior et al [38]. Samples were prepared in 7% randomly methylated beta-cyclodextrin (RMCD) buffer to liberate hydrophobic antioxidants while blocking release of hydrophilic antioxidants or phosphate buffer pH 7.4 to liberate hydrophilic antioxidants while blocking the release of hydrophobic antioxidants. The compound 2,2-azobis(2-amidino-propane) dihydrochloride was used as the peroxyl radical generator and Trolox, a water-soluble analogue of vitamin E, was used as the reference antioxidant standard. Peroxyl radicals decrease fluorescence by causing oxidation of a fluorescein probe. Changes in fluorescein fluorescence in RMCD buffer and phosphate buffer indicate hydrophobic and hydrophilic antioxidant capacity, respectively.
2.6. ATP Measurement
Total ATP: Primary hippocampal neurons were seeded on 96 well plates (0.015 × 106 neurons/well) for DIV 20–22. Neurons were treated with α-TCT, glutamate or a combination of both for 8h. Neuronal ATP production was measured by using ATPlite™ Luminescence Assay System (PerkinElmer, Waltham, MA) according to the manufacturer’s protocol. In brief, the plates were washed with sterile PBS, and cells were lysed for 5 min. Cells were then incubated with substrate (luciferin) for 10 min. The reaction between ATP, luciferase, and luciferin produced bioluminescence. ATP-induced-luminescence was measured with CLARIOstar microplate reader (BMG Labtech, Cary, NC). ATP/ADP ratio: DIV 7 primary hippocampal neurons were transfected with GW1-PercevalHR, a sensor developed by Gary Yellen lab [39, 40]. At DIV 20–22, neuronal images were taken using a Nikon C2 Laser Scanning Confocal Microscope (excitation, 488nm; emission, 530nm) at Optical Analysis Facility at University of Alabama. The ratio of fluorescence intensities was calculated F488nm/ F405nm. Pseudocolor ratiometric images were made using NIS-Elements 5.11 with Ratio View Live Ratio Graphing Module (Nikon)[41].
2.7. Oxygen consumption assay
The level of cellular oxygen consumption was measured by applying a fluorescence-based probe, MitoXpress Xtra (Agilent) as per manufacturer’s protocol. In brief, MitoXpress Xtra (10 μl) was added to neurons (90 μl media) grown in 96-well plate. Immediately after treatment, all wells were sealed by 100 μl mineral oil. Antimycin (1μM), an inhibitor of the Complex III, was used as a negative control.
2.8. Lactate dehydrogenase (LDH) assay
The level of cytotoxicity in primary neurons was assayed by measuring leakage of LDH using an in vitro toxicology assay kit (Sigma-Aldrich) as previously described [7]. In brief, the culture media and lysed cells were collected after treatment of neurons with lycopene and hydrogen peroxide for 24h. The LDH assay mixture was added to each sample. After 20 min incubation, the reaction was terminated by adding 1N HCl. LDH activity was spectrophotometrically measured with a CLARIOstar microplate reader (BMG Labtech, Cary, NC) with absorbance set at 490nm. Data were calculated by finding the activity of LDH which leaked into the medium from damaged cells / total LDH activity in the culture.
2.9. Immunoblots
Primary hippocampal neurons were scraped and lysed in the 1X cell signaling buffer (Cell signaling Technology, Danvers, MA) and protein concentration was determined using BCA protein reagents (Thermo Scientific, Rockford, IL). Samples (50–100 μg of protein/lane) were separated on a 4–12% SDS-polyacrylamide gel (Bio-Rad, Hercules, CA) and probed with anti-ΔN-Bcl-xL (1:10, Pacific Immunology, Ramona, CA), anti-Bcl-xL (1:1000, #2764, Cell signaling), anti-brain-derived neurotrophic factor (BDNF) (1:100, sc-546, Santa Cruz), anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:1000, #5174, Cell Signaling) and anti-beta actin (1:1000, #A3854, Sigma) antibodies. Anti-ΔN-Bcl-xL is custom-produced (peptide sequence: CZ DSP AVN GAT GHS SSL D. 1:100). Membranes were treated with ECL reagents (Perkin Elmer, Waltham, MA) and levels of chemiluminescence were analyzed using ImageJ software (National Institutes of Health, Bethesda, MD).
2. 10. mRNA analysis
Primary hippocampal neurons were scraped, and total RNA was isolated from the neurons using the Absolutely RNA Miniprep Kit (Agilent, Santa Clara, CA). The abundance of mRNA for Bcl-xL was quantified using real time PCR. Bcl-xL mRNA expression values were normalized using GAPDH housekeeping gene and calculated using the 2−ΔΔCT method. The following primer sets were used: Rat Bcl-xL Forward, 5-TAT TGG TGA GTC GGA TTG CA-3; Rat Bcl-xL Reverse, 5-GCT CTC GGG TGC TGT ATT GT-3; Rat GAPDH Forward, 5-ATG ACT CTA CCC ACG GCA AG-3; Rat GAPDH reverse, 5-GGA AGA TGG TGA TGG GTT TC-3.
2.11. HPLC-MS detection and quantification of vitamin E
Vitamin E was detected and quantified according to a previously published protocol [25], with modifications. Briefly, primary hippocampal neurons were washed four times with ice-cold PBS. All washes were saved and analyzed for the presence of vitamin E. Neurons were collected in PBS containing 1 mM Na2EDTA, 1 mM butylated hydroxytoluene, and 30 mM SDS. Internal standard: delta-tocotrienol (δ-TCT) was added to obtain 1 μg/mL concentration and the mixture was centrifuged at 3200 rpm for 10 min. The mixture was vortexed at 4 °C for 15 min, then 100 μL of the mixture was transferred to a new microcentrifuge tube, 140 μL of ethanol was added, and the mixture was vortexed for 2 min. Subsequently, 140 μL of n-hexane was added, and the mixture was vortexed for 3 min. The top n-hexane layer was transferred to a new microcentrifuge tube and evaporated to dryness. The remaining solid residue was resuspended in 100 μL methanol and analyzed using HPLC-MS.
To monitor the presence of vitamin E in culture media and the washes, we mixed 0.925 mL of these solutions with 0.3 mg Na2EDTA, 0.025 mL of butylated hydroxytoluene (10 mg/mL), and 0.5 mL of 0.1 M SDS and vortexed the solution at 4 °C for 15 min. We transferred 100 μL of the mixture to a new microcentrifuge tube and extracted vitamin E as described above for the cell pellet procedure. All samples were analyzed using Agilent 1260 Infinity II system, comprised of an Infinity II Binary Pump, Infinity Multisampler, Multicolumn Thermostat, Diode Array Detector (DAD, UV spectrometer), Agilent’s Instrument Control Framework, and Mass Selective Detector (MSD, mass spectrometer). A 150 × 4.6 mm, 5 μM Acclaim™ C30 column was used for analysis at room temperature. The mobile phase comprised (A) deionized ultrapure water containing 0.1% formic acid and (B) methanol containing 0.1% formic acid. The following elution gradient was applied: 0–5 min 95% B; 5–10 min 95%–100% B; 10–17 min 100% B; 17–20 min 95% B; 20–25 min 95% B. Each sample (50 μL) was injected onto the column operating at a 1.0 mL/min mobile phase flow rate. The eluate was monitored by DAD at 290 nm, and UV-VIS spectra within the range of 190–400 nm were stored for all peaks. All samples were analyzed using the MSD in electrospray positive ionization mode with a drying gas flow rate of 12 L/min, nebulizer pressure of 35 psig, a drying gas temperature of 350 °C, and capillary voltage of 4,500 V. MSD was used in single ion monitoring mode to detect α-TCT (m/z 425), δ-TCT (m/z 397).
For quantitative analysis a 5-point (0.122, 0.244, 0.480, 0.970 and 1.95 μg/mL) matrix-matched calibration curve for α-tocotrienol (Cayman Chemicals, 10 mg/ml) was prepared. Vit. E standard solutions were analyzed using above-mentioned HPLC-MS method. Areas under the α-tocotrienol peaks were measured and used to construct the calibration plot.
2. 12. Statistical analyses
Data are reported as mean ± SEM of at least three independent cultures with multiple independent experimental designs, such as independent neuronal isolations, independent performance dates, and independent plates within a culture using separately prepared reagents. Difference in the means between 2 groups was tested using student’s t-test. To compare 3 or more groups, one-way ANOVA with a Tukey’s post-hoc analysis was applied. Comparisons across multiple factors were examined using a two-way ANOVA. P<0.05 is considered statistically significant. P values are provided in figure legends.
3. Results
3.1. Primary hippocampal neurons grown in α-TCT containing media show advanced neurite arborization
The role of α-TCT during neurite development was investigated using rat primary hippocampal neurons grown in neurobasal media with or without α-TCT. Conditioned media with or without 1 μM α-TCT were replaced weekly for 3 weeks. Oral supplementation of α-TCT (61.52 mg) in humans and canines results in levels of α-TCT in the brain in the range of nmol/g protein [23, 24], whereas in vitro treatment with 1 μM α-TCT results in similar levels, pmol to nmol α-TCT in brain cells [25]. Cellular uptake of exogenously introduced α-TCT was quantified, and we consistently found that primary hippocampal neurons treated with 1 μM α-TCT increased the concentration of α-TCT in neurons (26 pmol/mg), whereas α-TCT was not detected in the vehicle treated control neurons (Figure 1A). Neurons in the control and α-TCT group were not challenged with any known toxin; thus, both groups grew normally without demonstrating cytotoxicity (Figure 1B). However, we found that primary hippocampal neurons treated with α-TCT showed increased neurite complexity. Sholl analysis was applied to quantify the number of branches within a consistent radius from the soma center, and α-TCT treated neurons had significantly increased numbers of branch points 60μm from the soma (Figure 1C). A two-way ANOVA showed that there was not a statistically significant interaction between the effects of distance and treatment (P-value=0.7599). Original GFP fluorescent micrographs were converted to pseudo-color with 16-color annotation: red indicates a higher number of branch points, whereas blue indicates a lower number of branch points (Figure 1D). Based on the Sholl analysis, treatment with α-TCT significantly increased the number of neurite branch points at 60 μm from the soma compared to the vehicle-treated control group.
Figure 1. α-TCT promotes growth and development of primary hippocampal neurons.
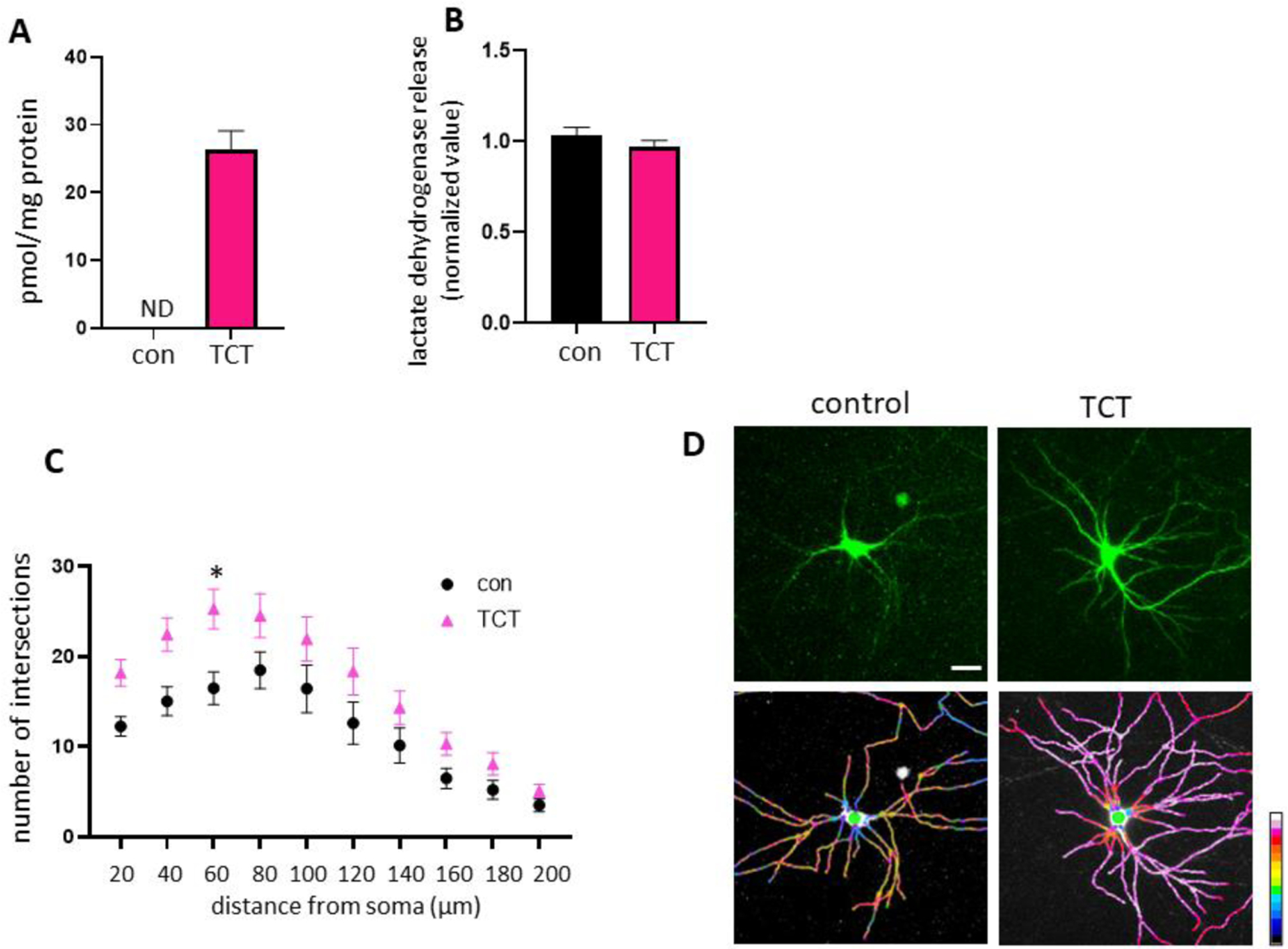
Primary hippocampal neurons were grown in neurobasal media with or without 1μM α-TCT for 3 weeks. A, Treatment with α-TCT significantly increased cellular uptake of α-TCT in primary hippocampal neurons (n=3). Primary hippocampal neurons grown in media containing α-TCT did not change lactate dehydrogenase release (n=7), indicating no significant loss of neuronal membrane integrity (B), but α-TCT treatment increased arborization of neurites (C, n=30). *p<0.05, Two-Way ANOVA followed by Bonferroni multiple comparisons test; Interactive effect was not significant. Interaction p value = 0.7599. D, Neurites treated with or without 1μM α-TCT were visualized with GFP plasmid (top panel) and pseudo-color ratiometric micrographs (bottom panel: red, more branches; blue, fewer branches). Scale bar=20μm.
α-TCT, alpha-tocotrienol; GFP, green fluorescent protein
3.2. Treatment with α-TCT enhances ATP at primary hippocampal neurites
Since neurite extension and branching require abundant energy, we tested if treatment with α-TCT supports overall ATP levels at neurites in primary hippocampal neurons by assaying bioluminescence produced via ATP, luciferase and luciferin. Primary hippocampal cultures grown in α-TCT had significantly increased ATP levels in whole neurons (Figure 2A). We further tested if α-TCT-mediated ATP increase specifically at neurites in primary hippocampal neurons. We applied a GW1-PercevalHR probe [39] to monitor local distribution of ATP in hippocampal neurites. Binding of the probe to ATP causes conformational changes leading to changes in probe fluorescence. Both control and α-TCT treated neurons demonstrated ATP-positive signals throughout the cell body and neurites. However, primary hippocampal neurons grown in α-TCT had significantly increased ATP levels in their neurites compare to controls (Figure 2B–C). Therefore, higher levels of ATP at neurites are correlated with increased growth and branching in primary hippocampal neurons in the α-TCT treated group. Since oxidative phosphorylation requires oxygen, we further measured oxygen consumption rate by applying mitoXpress Xtra. MitoXpress Xtra is a fluorescence probe quenched by oxygen, therefore higher cellular oxygen consumption leads to greater fluorescence signal. We found that treatment with α-TCT did not change the oxygen consumption rate (Figure 2D), suggesting that α-TCT treatment improves ATP production efficiency (increased ATP production per unit oxygen consumed).
Figure 2. α-TCT treatment improves energy metabolism in primary hippocampal neurons.
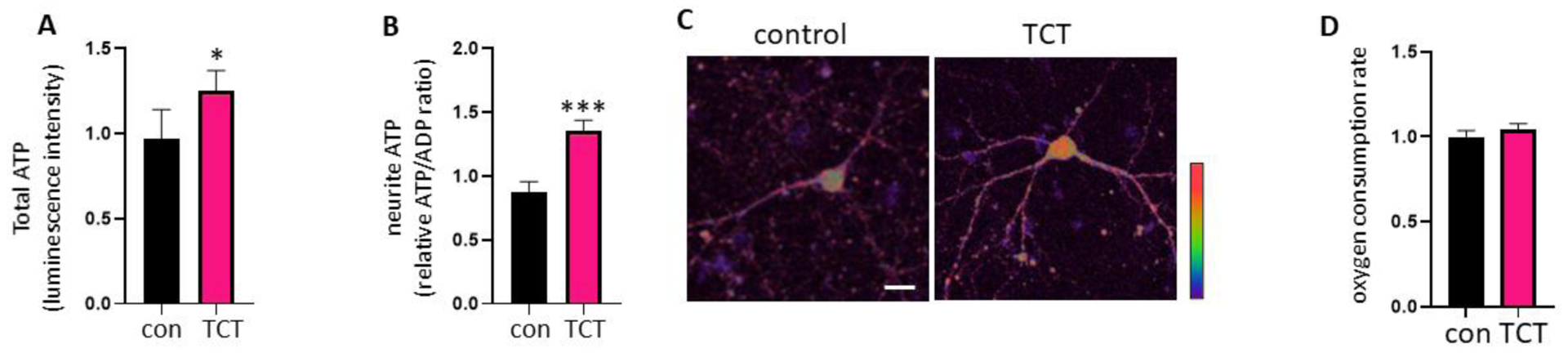
Primary hippocampal neurons were grown in neurobasal media with or without α-TCT (1μM) for 3 weeks. ATP levels in whole neurons (A, n=4) and neurites (B, n=23) were measured. *p<0.05 and ***p<0.001, two-tailed Student’s t-test. C, Pseudocolour ratiometric micrographs of neurons indicating ATP/ADP ratio using the PercevalHR sensor. Red: higher ATP/ADP; Blue: lower ATP/ADP. Scale bar=20μm. D, Treatment with α-TCT did not change MitoXpress Xtra signal, indicating no significance in neuronal oxygen consumption rate (n=4).
α-TCT, alpha-tocotrienol; GFP, green fluorescent protein; ATP, adenosine triphosphate; ADP, adenosine diphosphate
3.3. Treatment with α-TCT upregulates Bcl-xL in primary hippocampal neurons
The pro-survival mitochondrial protein Bcl-xL has been shown to regulate mitochondrial population and neuronal ATP production [12, 13, 15]. Importantly, we reported that Bcl-xL is necessary for neurite outgrowth in primary hippocampal neurons [19]. Depletion of Bcl-xL may not cause immediate neuronal death but does increase vulnerability of neurons to cytotoxicity [9, 19]. We tested if α-TCT-mediated ATP increases and enhanced neurite arborization are associated with increased levels of Bcl-xL in the cells. We have previously shown that 24h incubation with α-TCT does not influence the abundance of Bcl-xL [7]. However, primary hippocampal neurons grown with α-TCT for 3 weeks show increased mRNA and protein levels of Bcl-xL (Figure 3A–B), whereas treatment with α-TCP showed no change (Figure 3C). Post-translational cleavage of Bcl-xL is an important modification that alters the pro-survival function of Bcl-xL [42–44]. Accumulation of the cleavage product of Bcl-xL, ΔN-Bcl-xL, promotes mitochondrial leak channel activity, loss of mitochondrial inner membrane potential and alteration of ATP levels [31, 45, 46]. Thus, increased ΔN-Bcl-xL may impair energy demanding processes in neurons including neurite arborization. Indeed, primary hippocampal neurons overexpressing ΔN-Bcl-xL fail to achieve neurite complexity (Figure 3D–E). Thus, maintaining balance between Bcl-xL and ΔN-Bcl-xL is critical for neuronal survival [7, 31]. We tested if increased Bcl-xL protein promoted by α-TCT leads to increased formation of ΔN-Bcl-xL. Treatment with a α-TCT did not cause increased ΔN-Bcl-xL levels, but it did lead to an increase in full length Bcl-xL levels (Figure 3B). We were unable to detect ΔN-Bcl-xL in either control or the α-TCT treated group (Figure 3B) indicating that post-translational cleavage does not occur during prolonged α-TCT treatment. In contrast to increases in full length Bcl-xL, we have shown previously that primary hippocampal neurons lacking Bcl-xL have impaired neurite extension [19]. Consistently, primary hippocampal neurons transduced with Bcl-xL shRNA fail to achieve normal mature neuronal morphology (Figure 3F).
Figure 3. α-TCT treatment upregulates Bcl-xL in primary hippocampal neurons.
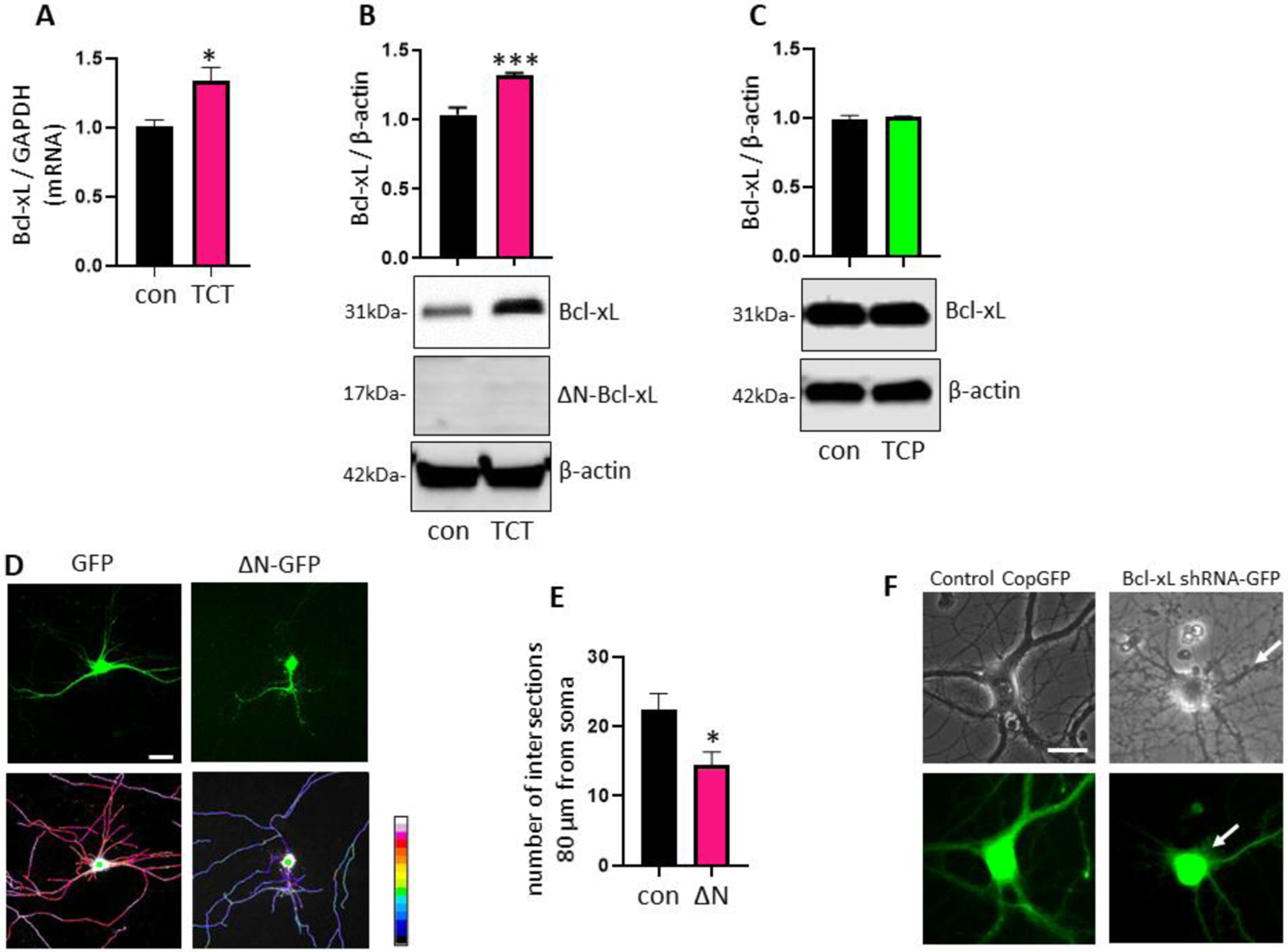
Primary hippocampal neurons were grown in neurobasal media with or without α-TCT (1μM) for 3 weeks. Treatment with α-TCT significantly increased mRNA (A, n=3) and protein (B, n=3) levels of Bcl-xL. *p<0.05 and ***p<0.001, two-tailed Student’s t-test. C, Treatment with α-TCP did not change protein levels of Bcl-xL (n=3). Primary hippocampal neurons were transduced with or without pME-HA-ΔN-Bcl-xL plasmid for 3 weeks. D, Neurites were visualized with GFP plasmid (top panel) and pseudo-color ratiometric micrographs (bottom panel: red, more branches; blue, fewer branches). Scale bar=20μm. E, Neurons overexpressing ΔN-Bcl-xL significantly decreased neurite arborization. *p<0.05, two-tailed Student’s t-test. F, Primary hippocampal neurons transduced with Bcl-xL shRNA showed thinner and less defined (arrows) neurite morphology. Top panel: phase contrast; Bottom panel: GFP (Green: GFP transduced neurons). Scale bar=20μm. α-TCT, alpha-tocotrienol; α-TCT, alpha-tocopherol; Bcl-xL, B-cell lymphoma-extra large; GFP, green fluorescent protein
As an explanation for the increase in Bcl-xL expression, we sought a growth factor that might account for increased neurite extension under antioxidant conditions. Brain-derived neurotrophic factor (BDNF), is a neurotrophic factor with known functions in neuritogenesis and neurogenesis [47], therefore we tested if Bcl-xL was required for BDNF maturation. We found that Bcl-xL is essential for maturation of BDNF because primary hippocampal neurons transduced with Bcl-xL shRNA produced pro-BDNF but failed to convert pro-BDNF to mature BDNF (Figure 4), placing pro-BDNF processing downstream of Bcl-xL activity.
Figure 4. Bcl-xL is essential for maturation of BDNF in primary hippocampal neurons.
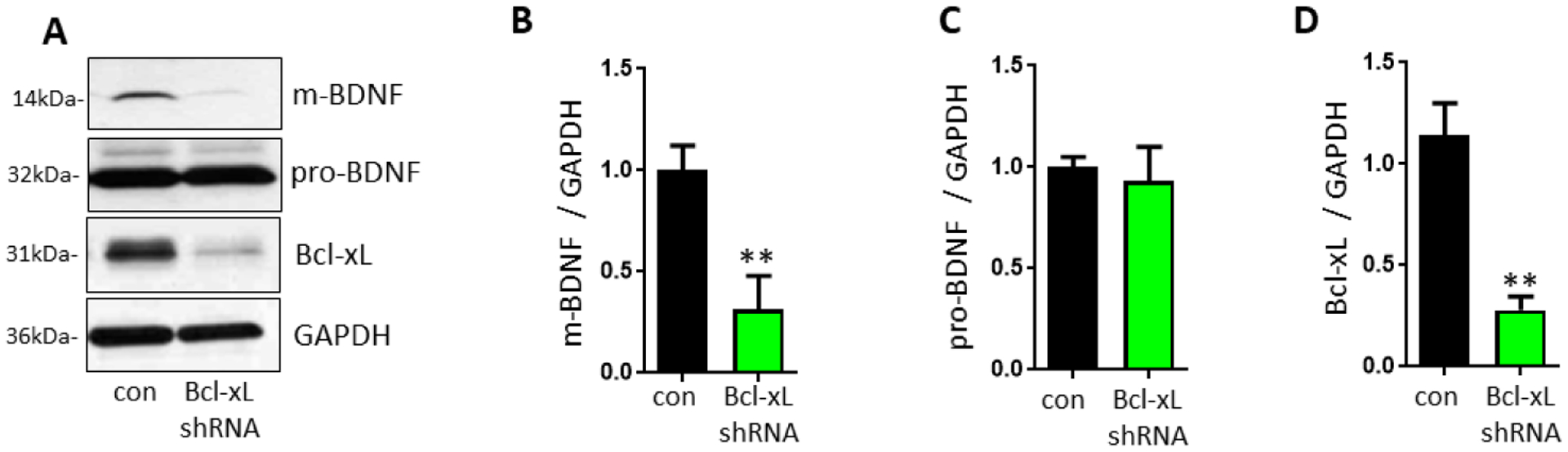
Primary hippocampal neurons were transduced with control copGFP or Bcl-xL shRNA-GFP for 3 weeks. A, Neurons lacking Bcl-xL showed decreased protein levels of mature BDNF. B-D, Immunoblot with mature-BDNF (B, n=4), pro-BDNF (C, n=4), and Bcl-xL (D, n=3). **p<0.01, two-tailed Student’s t-test. α-TCT, alpha-tocotrienol; Bcl-xL, B-cell lymphoma-extra large; BDNF, Brain-derived neurotrophic factor
3.4. Treatment with α-TCT improves redox homeostasis in primary hippocampal neurons
We recently reported that ROS causes post-translational cleavage of Bcl-xL to produce ΔN-Bcl-xL, and application of antioxidants prevents ΔN-Bcl-xL formation and protects neurons [7]. To investigate if α-TCT prevents formation of ΔN-Bcl-xL even as it upregulates full length Bcl-xL (Figure 3), we measured antioxidant capacity and oxidative stress levels in neurons treated with or without α-TCT. We measured quenching of fluorescein fluorescence via the presence of peroxyl radicals in 7% RMCD or phosphate buffer, liberating hydrophobic or hydrophilic antioxidants, respectively. We found that primary hippocampal neurons grown in neurobasal media with α-TCT showed improved lipophilic antioxidant capacity (Figure 5A). Since α-TCT is a well-studied lipophilic antioxidant, it was not surprising to see enhanced lipophilic antioxidant capacity by the addition of α-TCT. Interestingly, treatment with α-TCT also enhanced hydrophilic antioxidant capacity (Figure 5B). We further measured mitochondrial ROS levels in live primary hippocampal neurons by using mitoSOX, a red fluorescent probe for mitochondrial superoxide. Neurons grown in α-TCT containing media showed lower levels of mitoSOX fluorescence (Figure 5C).
Figure 5. α-TCT improves antioxidant capacity in primary hippocampal neurons.
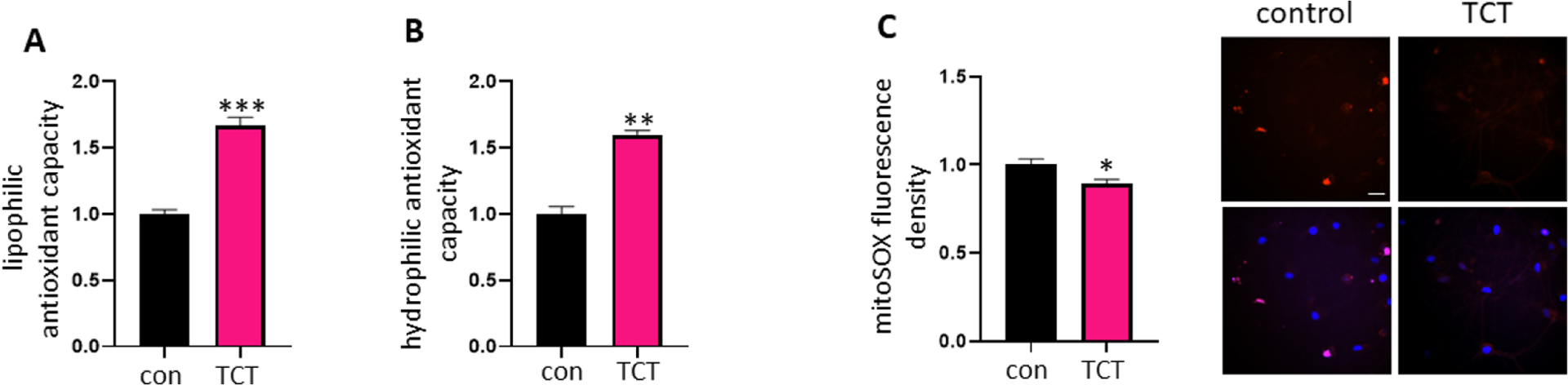
Primary hippocampal neurons were grown in neurobasal media with or without α-TCT (1μM) for 3 weeks. Quantification of intracellular lipophilic antioxidant capacity (A, n=3) and hydrophilic antioxidant capacity (B, n=3) were assayed. Treatment with α-TCT significantly increases both lipophilic and hydrophilic antioxidant capacity in neurons. **p<0.01 and ***p<0.001, two-tailed Student’s t-test. C, Treatment with α-TCT significantly decreases mitoSOX signal (n=35). p<0.05, two-tailed Student’s t-test. Scale bar=20μm. Top panel: mitoSOX; Bottom panel; DAPI merged (Red: mitoSOX; Blue: DAPI). α-TCT, alpha-tocotrienol; DAPI, 4′,6-diamidino-2-phenylindole
3.5. Primary hippocampal neurons grown in α-TCT containing media are resistant to ROS-associated damage
We subsequently tested whether primary hippocampal neurons grown in α-TCT were more resistant to neurotoxic challenge at maturity. Unlike α-TCP, α-TCT has been shown to protect neurons against glutamate-induced excitotoxicity [7, 25]. Excitotoxicity is known to promote ROS generation [48, 49]. Therefore, we first tested whether treatment with glutamate increases endogenous ROS production. Primary hippocampal neurons grown with or without α-TCT treatment were challenged with glutamate. We measured intracellular hydrogen peroxide levels using 2’,7’-dichlorofluorescein. Treatment with glutamate significantly increased the 2’,7’-dichlorofluorescein-positive fluorescent signal, indicating the generation of endogenous hydrogen peroxide. Neurons grown with α-TCT supplementation had decreased accumulation of intracellular ROS (Figure 6A). We then tested whether α-TCT prevents mitochondrial superoxide production against ROS challenge. Neurons treated with hydrogen peroxide significantly increased the mitoSOX positive signal, indicating accumulation of mitochondrial superoxide, whereas treatment with α-TCT prevented the effect of hydrogen peroxide (Figure 6B). Next, we used fluorescent microscopy with a Fluo-4 probe to detect increased intracellular calcium levels. Fluo-4 binding to calcium increases fluorescence intensity. Primary hippocampal neurons cultured with α-TCT were resistant to ROS-associated calcium surges (Figure 6C). Additionally, we measured lactate dehydrogenase release, a marker of cell membrane integrity loss. Neurons grown with α-TCT were protected against excitotoxicity (Figure 6D). Notably, 1 μM of α-TCT showed the most significant protection, whereas treatment with α-TCP was less effective in protecting neurons during the excitotoxic challenge.
Figure 6. α-TCT alleviates reactive oxygen species-mediated damage in primary hippocampal neurons.
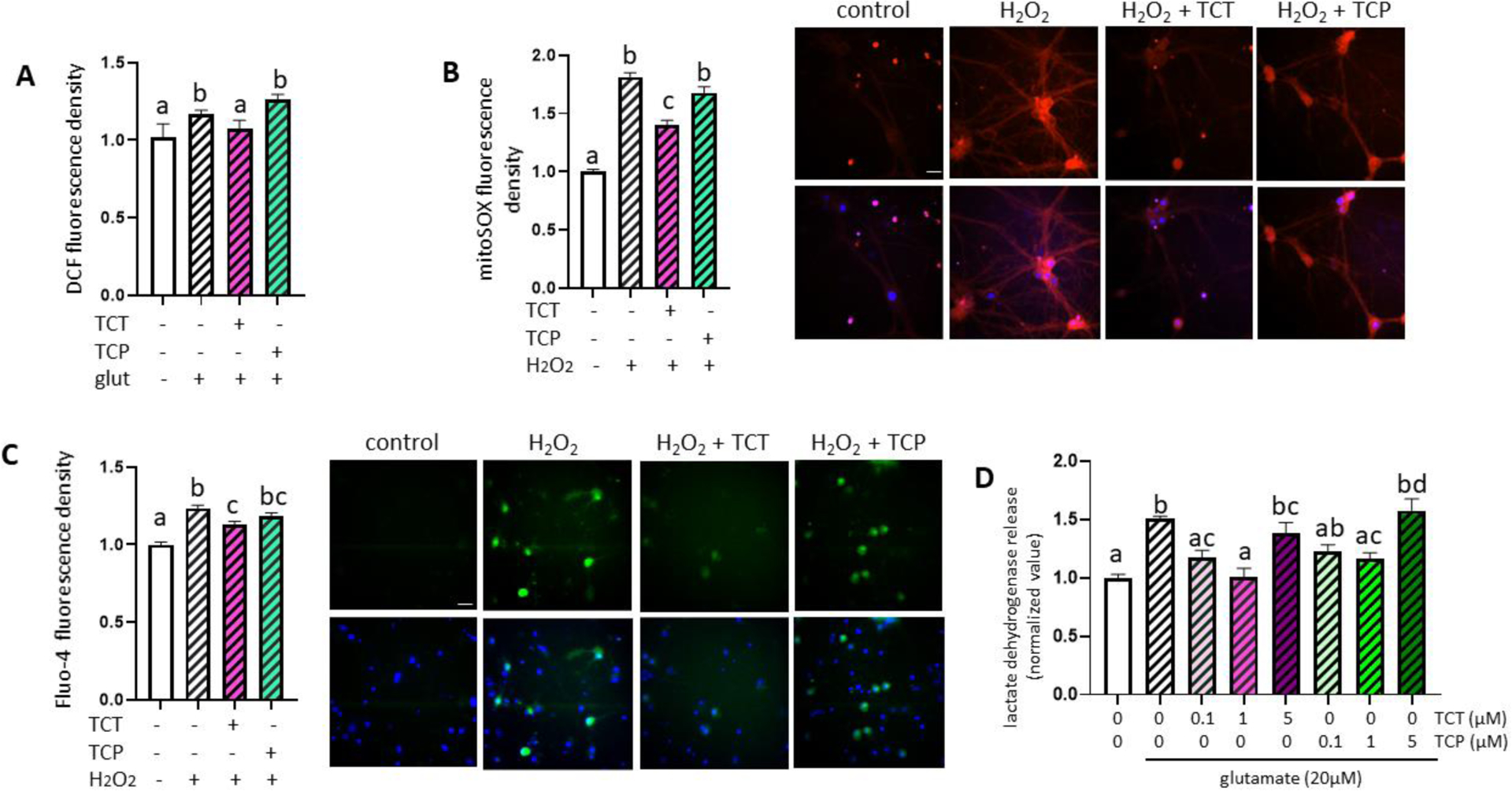
Primary hippocampal neurons were grown in neurobasal media with α-TCT (1μM) or α-TCP (1μM) for 3 weeks, then neurons were challenged with glutamate (20 μM) for 6h. Intracellular hydrogen peroxide levels were assayed by measuring 2’7’- dichlorofluoresce (DCF) (A, n=5). One-way ANOVA with a Tukey’s post-hoc analysis. Neurons grown with α-TCT (1μM) or α-TCP (1μM) were challenged with hydrogen peroxide (25 μM) for 6h, and mitochondrial oxidative stress were measured by mitoSOX staining (B, n=50). One-way ANOVA with a Tukey’s post-hoc analysis. Scale bar=20μm. Top panel: mitoSOX; Bottom panel; DAPI merged (Red: mitoSOX; Blue: DAPI). C, Intracellular calcium levels were measured by Fluo-4 (n=100). One-way ANOVA with a Tukey’s post-hoc analysis. Scale bar=20μm. Top panel: Fluo-4; Bottom panel; Hoechst merged (Green: Fluo-4; Blue: Hoechst). D, Primary hippocampal neurons were treated with α-TCT (0.1, 1, and 5μM), α-TCP (0.1, 1, and 5μM), and glutamate (20 μM) for 24h. Quantified neuronal toxicity was measured by lactate dehydrogenase (LDH) release (n=4). One-way ANOVA with a Tukey’s post-hoc analysis. α-TCT, alpha-tocotrienol; α-TCT, alpha-tocopherol; DAPI, 4′,6-diamidino-2-phenylindole
4. Discussion
In this study, we find that α-TCT supports neurite development of primary hippocampal neurons. We show that primary hippocampal neurons grown with α-TCT had increased neurite arborization related to improved neurite ATP production efficiency. Treatment with α-TCT improved neuronal energy metabolism as evidenced by an increase in ATP/ adenosine diphosphate (ADP) ratio. This indicates that α-TCT may help neurites produce local ATP more efficiently at metabolically active regions. We also suggest that a molecular target, Bcl-xL, may directly cause the increase in efficiency of ATP production. Bcl-xL is a mitochondrial protein that exhibits neuroprotective properties during various neurotoxic challenges such as excitotoxicity, oxidative stress, and hypoxia [7, 19, 31]. Bcl-xL increases mitochondrial biomass in neuronal processes and regulates mitochondrial fission [15, 16]. Bcl-xL binds directly to the β-subunit of the F1Fo ATP synthase, the key subunit that converts ADP to ATP, and this interaction between Bcl-xL and the β-subunit closes a non-selective proton leak channel in the mitochondrial inner membrane [8, 9, 12, 13, 46]. Therefore, α-TCT-mediated upregulation of Bcl-xL may improve neuronal energy metabolism by enhancing the local mitochondrial population and increasing the efficiency of mitochondrial ATP production. We previously reported that Bcl-xL is required for elongation of neurites in developing primary hippocampal neurons [19]. Bcl-xL depletion does not cause immediate toxicity, but hippocampal neurons lacking Bcl-xL fail to increase neurite complexity during development, and this leads to death at maturity [19], most likely related to lack of formation of salubrious synaptic connections.
Although Bcl-xL is a well-studied anti-apoptotic protein, it undergoes post-translational N-terminal cleavage to pro-death ΔN-Bcl-xL [44]. Accumulation of ΔN-Bcl-xL causes abnormal mitochondrial channel activity and lowers mitochondrial inner membrane potential causing neuronal death [31, 42, 43, 50]. In this study, we found that primary hippocampal neurons overexpressing ΔN-Bcl-xL failed to undergo normal neurite arborization. ΔN-Bcl-xL-mediated mitochondrial dysfunction and subsequent local energy depletion may impair normal development of hippocampal neurites. Therefore, maintaining the balance between ΔN-Bcl-xL and Bcl-xL appears to be critical for neuronal growth and survival. We tested if α-TCT-induced Bcl-xL upregulation enhances formation of ΔN-Bcl-xL. We demonstrated that primary hippocampal neurons treated with α-TCT had increased mRNA and protein levels of Bcl-xL without a concomitant increase in ΔN-Bcl-xL. We recently reported that ROS are a major contributor of ΔN-Bcl-xL production [7], and that application of α-TCT prevents accumulation of ΔN-Bcl-xL. In support of this, our current study shows that neurons grown with α-TCT had improved hydrophobic and hydrophilic antioxidant capacity, preventing generation of peroxyl radicals. Thus, improved intracellular redox status may allow neurons to maintain a favorable Bcl-xL/ΔN-Bcl-xL ratio, and an improved Bcl-xL/ΔN-Bcl-xL ratio may help neurons to maintain redox homeostasis.
Recently, dietary antioxidants and phytochemicals have been recognized as regulators of Bcl-xL quantity and may attenuate or prevent mitochondrial dysfunction associated with brain pathology [51, 52]. Although α-TCT-induced increase in Bcl-xL expression has not been reported previously, primary cortical neurons treated with α-TCT had significantly increased protein levels of Bcl-2 in the absence of neurotoxic stimuli [53]. Treatment with α-TCP, a vitamin E isoform, decreases Bax to Bcl-xL ratio during hydrogen peroxide-induced oxidative stress in an adrenal phaeochromocytoma 12 neuronal cell line, and this is correlated with retention of mitochondrial membrane potential [54]. Similarly, subcutaneous delivery of α-TCP decreased Bax to Bcl-xL ratio in rat hippocampi undergoing haloperidol-induced neurotoxicity [55]. Post et al. further showed that α-TCP prevents haloperidol-induced nuclear translocation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) and phosphorylation of nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor alpha (IkBα). Although this group did not demonstrate the direct association between NF-kB and Bcl-xL, NF-kB is reported to regulate the BCL2L1 gene encoding Bcl-xL protein [56, 57]. Involvement of TCTs in the transcriptional regulation of Bcl-xL is intensively studied in cancer research [58–60]. Although we have limited information elucidating α-TCT-mediated cell signaling pathways in the current study, these published reports implicate that α-TCT may regulate Bcl-xL gene expression.
The hydrophobic nature of α-TCT may explain its effectiveness in protecting biological membrane structure. Specifically, the mitochondrial inner membrane is essential to ROS and ATP production due to the presence of the electron transport chain. We recently showed that treatment with α-TCT blocks glutamate-induced mitochondrial superoxide generation, supporting the maintenance of mitochondrial inner membrane potential [7]. Protection of mitochondria further decreased endogenous ROS production, improved neuronal energy metabolism, and enhanced neuronal survival against oxidative stress. Based on the oxygen consumption assay, α-TCT may also prevent the wasting of oxygen atoms during oxidative phosphorylation. Bcl-xL was previously shown to enhance the efficiency of FoF1 ATP synthase by promoting ATP production without requiring high levels of oxygen [12]. Therefore, it is possible that α-TCT-mediated Bcl-xL upregulation inhibits mitochondrial ROS generation by preventing unnecessary oxygen consumption.
We demonstrate that treatment with α-TCT upregulates Bcl-xL in primary hippocampal neurons and supports neurite development by improving neuronal energy metabolism. We also found that neurons grown in α-TCT are resistant to neurotoxic stimulation at maturity. However, it is still unknown if α-TCT regulates Bcl-xL degradation. This may be attributed to limitations in the current study. Future investigation of Bcl-xL ubiquitination or mitophagy mechanisms will strengthen our understanding of the role of α-TCT in maintaining Bcl-xL protein levels in neurons. Although we primarily focused on the role of α-TCT during neurite development, acknowledging α-TCP is the major form of vitamin E in diet, it may be important to investigate whether altering the ratio of α-TCT to α-TCP may optimize neurite development but may also protect against known neurotoxic insults.
Highlights.
Alpha-tocotrienol (α-TCT) increases branching of primary hippocampal neurons.
α-TCT improves neuronal energy and upregulates B-cell lymphoma-extra large (Bcl-xL).
Primary hippocampal neurons grown in α-TCT are resistant to oxidative stress.
Sources of Support
This work was supported by the University of Alabama (RG14811, Small Grant Program; A-52248-217101-100, Crenshaw Research Fund), and National Institute of Neurological Disorders and Stroke (NIH NS045876).
Abbreviations
- ADP
Adenosine diphosphate
- ATP
Adenosine triphosphate
- Bad
Bcl-2-associated agonist of cell death
- Bak
Bcl-2-antagonist/killer
- Bax
Bcl-2-associated X
- Bcl-xL
B-cell lymphoma-extra large
- BDNF
Brain-derived neurotrophic factor
- DIV
Days in vitro
- GFP
Green fluorescent protein
- H2DCFDA
2’,7’- dichlorodihydrofluorescein diacetate
- IkBα
Kappa light polypeptide gene enhancer in B-cells inhibitor alpha
- LDH
Lactate dehydrogenase
- NF-kB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- ORAC
Oxygen radical absorbance capacity
- RMCD
Randomly methylated beta-cyclodextrin
- ROS
Reactive oxygen species
- α-TCP
Alpha-tocopherol
- α-TCT
Alpha-tocotrienol
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Declarations
All authors declare no conflict of interest.
References
- [1].Harris JJ, Jolivet R, Attwell D. Synaptic energy use and supply. Neuron. 2012;75:762–77. doi: 10.1016/j.neuron.2012.08.019. [DOI] [PubMed] [Google Scholar]
- [2].Liu X, Dai S, Zhu Y, Marrack P, Kappler JW. The structure of a bcl-xl/bim fragment complex: Implications for bim function. Immunity. 2003;19:341–52. [DOI] [PubMed] [Google Scholar]
- [3].Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, et al. Structure of bcl-xl-bak peptide complex: Recognition between regulators of apoptosis. Science. 1997;275:983–6. [DOI] [PubMed] [Google Scholar]
- [4].Tan Y, Demeter MR, Ruan H, Comb MJ. Bad ser-155 phosphorylation regulates bad/bcl-xl interaction and cell survival. J Biol Chem. 2000;275:25865–9. doi: 10.1074/jbc.M004199200. [DOI] [PubMed] [Google Scholar]
- [5].Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, et al. Proapoptotic bak is sequestered by mcl-1 and bcl-xl, but not bcl-2, until displaced by bh3-only proteins. Genes Dev. 2005;19:1294–305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Billen LP, Kokoski CL, Lovell JF, Leber B, Andrews DW. Bcl-xl inhibits membrane permeabilization by competing with bax. PLoS Biol. 2008;6:e147. doi: 10.1371/journal.pbio.0060147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Park HA, Mnatsakanyan N, Broman K, Davis AU, May J, Licznerski P, et al. Alpha-tocotrienol prevents oxidative stress-mediated post-translational cleavage of bcl-xl in primary hippocampal neurons. Int J Mol Sci. 2019;21:220. doi: 10.3390/ijms21010220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jonas EA, Porter GA, Alavian KN. Bcl-xl in neuroprotection and plasticity. Front Physiol. 2014;5:355. doi: 10.3389/fphys.2014.00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Park HA, Jonas EA. Mitochondrial membrane protein bcl-xl, a regulator of adult neuronal growth and synaptic plasticity: Multiple functions beyond apoptosis. Neural Regen Res. 2014;9:1706–7. doi: 10.4103/1673-5374.143413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chang MY, Sun W, Ochiai W, Nakashima K, Kim SY, Park CH, et al. Bcl-xl/bax proteins direct the fate of embryonic cortical precursor cells. Mol Cell Biol. 2007;27:4293–305. doi: 10.1128/MCB.00031-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nakamura A, Swahari V, Plestant C, Smith I, McCoy E, Smith S, et al. Bcl-xl is essential for the survival and function of differentiated neurons in the cortex that control complex behaviors. J Neurosci. 2016;36:5448–61. doi: 10.1523/JNEUROSCI.4247-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Alavian KN, Li H, Collis L, Bonanni L, Zeng L, Sacchetti S, et al. Bcl-xl regulates metabolic efficiency of neurons through interaction with the mitochondrial f1fo atp synthase. Nat Cell Biol. 2011;13:1224–33. doi: 10.1038/ncb2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chen R, Park HA, Mnatsakanyan N, Niu Y, Licznerski P, Wu J, et al. Parkinson’s disease protein dj-1 regulates atp synthase protein components to increase neuronal process outgrowth. Cell Death Dis. 2019;10:469. doi: 10.1038/s41419-019-1679-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chen YB, Aon MA, Hsu YT, Soane L, Teng X, McCaffery JM, et al. Bcl-xl regulates mitochondrial energetics by stabilizing the inner membrane potential. J Cell Biol. 2011;195:263–76. doi: 10.1083/jcb.201108059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Berman SB, Chen YB, Qi B, McCaffery JM, Rucker EB 3rd, Goebbels S, et al. Bcl-x l increases mitochondrial fission, fusion, and biomass in neurons. J Cell Biol. 2009;184:707–19. doi: 10.1083/jcb.200809060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Li H, Chen Y, Jones AF, Sanger RH, Collis LP, Flannery R, et al. Bcl-xl induces drp1-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 2008;105:2169–74. doi: 10.1073/pnas.0711647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the bcl-xl-beclin 1 peptide complex: Beclin 1 is a novel bh3-only protein. J Biol Chem. 2007;282:13123–32. doi: 10.1074/jbc.M700492200. [DOI] [PubMed] [Google Scholar]
- [18].Arena G, Gelmetti V, Torosantucci L, Vignone D, Lamorte G, De Rosa P, et al. Pink1 protects against cell death induced by mitochondrial depolarization, by phosphorylating bcl-xl and impairing its pro-apoptotic cleavage. Cell Death Differ. 2013;20:920–30. doi: 10.1038/cdd.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Park HA, Licznerski P, Alavian KN, Shanabrough M, Jonas EA. Bcl-xl is necessary for neurite outgrowth in hippocampal neurons. Antioxid Redox Signal. 2015;22:93–108. doi: 10.1089/ars.2013.5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li H, Alavian KN, Lazrove E, Mehta N, Jones A, Zhang P, et al. A bcl-xl-drp1 complex regulates synaptic vesicle membrane dynamics during endocytosis. Nat Cell Biol. 2013;15:773–85. doi: 10.1038/ncb2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jonas E Bcl-xl regulates synaptic plasticity. Mol Interv. 2006;6:208–22. doi: 10.1124/mi.6.4.7. [DOI] [PubMed] [Google Scholar]
- [22].Park HA, Kubicki N, Gnyawali S, Chan YC, Roy S, Khanna S, et al. Natural vitamin e alpha-tocotrienol protects against ischemic stroke by induction of multidrug resistance-associated protein 1. Stroke. 2011;42:2308–14. doi: 10.1161/STROKEAHA.110.608547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rink C, Christoforidis G, Khanna S, Peterson L, Patel Y, Abduljalil A, et al. Tocotrienol vitamin e protects against preclinical canine ischemic stroke by inducing arteriogenesis. J Cereb Blood Flow Metab. 2011;31:2218–30. doi: 10.1038/jcbfm.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Patel V, Rink C, Gordillo GM, Khanna S, Gnyawali U, Roy S, et al. Oral tocotrienols are transported to human tissues and delay the progression of the model for end-stage liver disease score in patients. J Nutr. 2012;142:513–9. doi: 10.3945/jn.111.151902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sen CK, Khanna S, Roy S, Packer L. Molecular basis of vitamin e action. Tocotrienol potently inhibits glutamate-induced pp60(c-src) kinase activation and death of ht4 neuronal cells. J Biol Chem. 2000;275:13049–55. doi: 10.1074/jbc.275.17.13049. [DOI] [PubMed] [Google Scholar]
- [26].Park HA, Jonas EA. Deltan-bcl-xl, a therapeutic target for neuroprotection. Neural Regen Res. 2017;12:1791–4. doi: 10.4103/1673-5374.219033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ibrahim NF, Yanagisawa D, Durani LW, Hamezah HS, Damanhuri HA, Wan Ngah WZ, et al. Tocotrienol-rich fraction modulates amyloid pathology and improves cognitive function in abetapp/ps1 mice. J Alzheimers Dis. 2017;55:597–612. doi: 10.3233/JAD-160685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Fukui K, Ushiki K, Takatsu H, Koike T, Urano S. Tocotrienols prevent hydrogen peroxide-induced axon and dendrite degeneration in cerebellar granule cells. Free Radic Res. 2012;46:184–93. doi: 10.3109/10715762.2011.647689. [DOI] [PubMed] [Google Scholar]
- [29].Beaudoin GM 3rd, Lee SH, Singh D, Yuan Y, Ng YG, Reichardt LF, et al. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat Protoc. 2012;7:1741–54. doi: 10.1038/nprot.2012.099. [DOI] [PubMed] [Google Scholar]
- [30].Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–15. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- [31].Park HA, Licznerski P, Mnatsakanyan N, Niu Y, Sacchetti S, Wu J, et al. Inhibition of bcl-xl prevents pro-death actions of deltan-bcl-xl at the mitochondrial inner membrane during glutamate excitotoxicity. Cell Death Differ. 2017;24:1963–74. doi: 10.1038/cdd.2017.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Longair MH, Baker DA, Armstrong JD. Simple neurite tracer: Open source software for reconstruction, visualization and analysis of neuronal processes. Bioinformatics. 2011;27:2453–4. doi: 10.1093/bioinformatics/btr390. [DOI] [PubMed] [Google Scholar]
- [33].Pool M, Thiemann J, Bar-Or A, Fournier AE. Neuritetracer: A novel imagej plugin for automated quantification of neurite outgrowth. J Neurosci Methods. 2008;168:134–9. doi: 10.1016/j.jneumeth.2007.08.029. [DOI] [PubMed] [Google Scholar]
- [34].Jansen J, Scott M, Amjad E, Stumpf A, Lackey KH, Caldwell KA, et al. Bcl-xl is required by primary hippocampal neurons during development to support local energy metabolism at neurites. Biology (Basel). 2021;10:772. doi: 10.3390/biology10080772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Duan Y, Gross RA, Sheu SS. Ca2+-dependent generation of mitochondrial reactive oxygen species serves as a signal for poly(adp-ribose) polymerase-1 activation during glutamate excitotoxicity. J Physiol. 2007;585:741–58. doi: 10.1113/jphysiol.2007.145409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Xu B, Chen S, Luo Y, Chen Z, Liu L, Zhou H, et al. Calcium signaling is involved in cadmium-induced neuronal apoptosis via induction of reactive oxygen species and activation of mapk/mtor network. PLoS One. 2011;6:e19052. doi: 10.1371/journal.pone.0019052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Crowe KM. Optimizing protein precipitation efficiency for assessing the contribution of low molecular weight compounds to serum antioxidant capacity. Clin Biochem. 2014;47:116–8. doi: 10.1016/j.clinbiochem.2014.06.021. [DOI] [PubMed] [Google Scholar]
- [38].Prior RL, Hoang H, Gu L, Wu X, Bacchiocca M, Howard L, et al. Assays for hydrophilic and lipophilic antioxidant capacity (oxygen radical absorbance capacity (orac(fl))) of plasma and other biological and food samples. J Agric Food Chem. 2003;51:3273–9. doi: 10.1021/jf0262256. [DOI] [PubMed] [Google Scholar]
- [39].Tantama M, Martinez-Francois JR, Mongeon R, Yellen G. Imaging energy status in live cells with a fluorescent biosensor of the intracellular atp-to-adp ratio. Nat Commun. 2013;4:2550. doi: 10.1038/ncomms3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Berg J, Hung YP, Yellen G. A genetically encoded fluorescent reporter of atp:Adp ratio. Nat Methods. 2009;6:161–6. doi: 10.1038/nmeth.1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Vaarmann A, Mandel M, Zeb A, Wareski P, Liiv J, Kuum M, et al. Mitochondrial biogenesis is required for axonal growth. Development. 2016;143:1981–92. doi: 10.1242/dev.128926. [DOI] [PubMed] [Google Scholar]
- [42].Ofengeim D, Chen YB, Miyawaki T, Li H, Sacchetti S, Flannery RJ, et al. N-terminally cleaved bcl-xl mediates ischemia-induced neuronal death. Nat Neurosci. 2012;15:574–80. doi: 10.1038/nn.3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Jonas EA, Hickman JA, Chachar M, Polster BM, Brandt TA, Fannjiang Y, et al. Proapoptotic n-truncated bcl-xl protein activates endogenous mitochondrial channels in living synaptic terminals. Proc Natl Acad Sci U S A. 2004;101:13590–5. doi: 10.1073/pnas.0401372101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Clem RJ, Cheng EH, Karp CL, Kirsch DG, Ueno K, Takahashi A, et al. Modulation of cell death by bcl-xl through caspase interaction. Proc Natl Acad Sci U S A. 1998;95:554–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mnatsakanyan N, Jonas EA. Atp synthase c-subunit ring as the channel of mitochondrial permeability transition: Regulator of metabolism in development and degeneration. J Mol Cell Cardiol. 2020;144:109–18. doi: 10.1016/j.yjmcc.2020.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, et al. An uncoupling channel within the c-subunit ring of the f1fo atp synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci U S A. 2014;111:10580–5. doi: 10.1073/pnas.1401591111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Cohen-Cory S, Kidane AH, Shirkey NJ, Marshak S. Brain-derived neurotrophic factor and the development of structural neuronal connectivity. Dev Neurobiol. 2010;70:271–88. doi: 10.1002/dneu.20774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Reynolds IJ, Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following nmda receptor activation. J Neurosci. 1995;15:3318–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Rego AC, Oliveira CR. Mitochondrial dysfunction and reactive oxygen species in excitotoxicity and apoptosis: Implications for the pathogenesis of neurodegenerative diseases. Neurochem Res. 2003;28:1563–74. doi: 10.1023/a:1025682611389. [DOI] [PubMed] [Google Scholar]
- [50].Hickman JA, Hardwick JM, Kaczmarek LK, Jonas EA. Bcl-xl inhibitor abt-737 reveals a dual role for bcl-xl in synaptic transmission. J Neurophysiol. 2008;99:1515–22. doi: 10.1152/jn.00598.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Park HA, Broman K, Stumpf A, Kazyak S, Jonas EA. Nutritional regulators of bcl-xl in the brain. Molecules. 2018;23:3019. doi: 10.3390/molecules23113019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Park HA, Hayden MM, Bannerman S, Jansen J, Crowe-White KM. Anti-apoptotic effects of carotenoids in neurodegeneration. Molecules. 2020;25:3453. doi: 10.3390/molecules25153453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Numakawa Y, Numakawa T, Matsumoto T, Yagasaki Y, Kumamaru E, Kunugi H, et al. Vitamin e protected cultured cortical neurons from oxidative stress-induced cell death through the activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. J Neurochem. 2006;97:1191–202. doi: 10.1111/j.1471-4159.2006.03827.x. [DOI] [PubMed] [Google Scholar]
- [54].Vlasova YA, Zakharova IO, Avrova NF. The effects of alpha-tocoferol and h2o2 on the mitochondrial membrane potential and bax/bcl-xl ratio in pc12 cells. Neurochemical Journal. 2016;10:318–22. [Google Scholar]
- [55].Post A, Rucker M, Ohl F, Uhr M, Holsboer F, Almeida OF, et al. Mechanisms underlying the protective potential of alpha-tocopherol (vitamin e) against haloperidol-associated neurotoxicity. Neuropsychopharmacology. 2002;26:397–407. doi: 10.1016/S0893-133X(01)00364-5. [DOI] [PubMed] [Google Scholar]
- [56].Chen C, Edelstein LC, Gelinas C. The rel/nf-kappab family directly activates expression of the apoptosis inhibitor bcl-x(l). Mol Cell Biol. 2000;20:2687–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Khoshnan A, Tindell C, Laux I, Bae D, Bennett B, Nel AE. The nf-kappa b cascade is important in bcl-xl expression and for the anti-apoptotic effects of the cd28 receptor in primary human cd4+ lymphocytes. J Immunol. 2000;165:1743–54. doi: 10.4049/jimmunol.165.4.1743. [DOI] [PubMed] [Google Scholar]
- [58].Husain K, Francois RA, Yamauchi T, Perez M, Sebti SM, Malafa MP. Vitamin e delta-tocotrienol augments the antitumor activity of gemcitabine and suppresses constitutive nf-kappab activation in pancreatic cancer. Mol Cancer Ther. 2011;10:2363–72. doi: 10.1158/1535-7163.MCT-11-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ahn KS, Sethi G, Krishnan K, Aggarwal BB. Gamma-tocotrienol inhibits nuclear factor-kappab signaling pathway through inhibition of receptor-interacting protein and tak1 leading to suppression of antiapoptotic gene products and potentiation of apoptosis. J Biol Chem. 2007;282:809–20. doi: 10.1074/jbc.M610028200. [DOI] [PubMed] [Google Scholar]
- [60].Prasad S, Gupta SC, Tyagi AK, Aggarwal BB. Gamma-tocotrienol suppresses growth and sensitises human colorectal tumours to capecitabine in a nude mouse xenograft model by down-regulating multiple molecules. Br J Cancer. 2016;115:814–24. doi: 10.1038/bjc.2016.257. [DOI] [PMC free article] [PubMed] [Google Scholar]