

Abstract
Background.
Tumor infiltrating lymphocytes (TILs) confer a survival benefit among ovarian cancer patients; however, little work has been conducted in racially diverse cohorts.
Methods.
The present study investigated racial differences in the tumor immune landscape and survival of age- and stage-matched Non-Hispanic Black and Non-Hispanic White women with high-grade serous ovarian carcinoma (HGSOC) enrolled in two population-based studies (n=121 in each racial group). We measured TILs (CD3+), cytotoxic T-cells (CD3+CD8+), regulatory T-cells (CD3+FoxP3+), myeloid cells (CD11b+), and neutrophils (CD11b+CD15+) via multiplex immunofluorescence. Multivariable Cox proportional hazard regression was used to estimate the association between immune cell abundance and survival overall and by race.
Results.
Overall, higher levels of TILs, cytotoxic T-cells, myeloid cells, and neutrophils were associated with better survival in the intratumoral and peritumoral region, irrespective of tissue compartment (tumor, stroma). Improved survival was noted for T-regulatory cells in the peritumoral region and in the stroma of the intratumoral region, but no association for intratumoral T-regulatory cells. Despite similar abundance of immune cells across racial groups, associations with survival among Non-Hispanic White women were consistent with the overall findings, but among Non-Hispanic Black women, most associations were attenuated and not statistically significant.
Conclusions.
Our results add to the existing evidence that a robust immune infiltrate confers a survival advantage among women with HGSOC; however, Non-Hispanic Black women may not experience the same survival benefit as Non-Hispanic White women with HGSOC.
Impact.
This study contributes to our understanding of the immunoepidemiology of HGSOC in diverse populations.
INTRODUCTION
Ovarian cancer is the deadliest gynecological malignancy in the U.S., with only 49% of women with ovarian cancer surviving five years after diagnosis (1). Marked health disparities in ovarian cancer exist by race, where Black women have the highest mortality to incidence ratio and the worst survival of all racial/ethnic groups (1,2). Moreover, Black women with ovarian cancer have not experienced the steady improvement in survival rates over time as have White women (3). Although prognostic factors such as receipt of guideline-adherent treatment, access to care, comorbidities, and stage have been investigated as contributing causes to the disparate survival rates by race, these factors do not completely account for the disparity (4–7). Thus, additional approaches investigating biologic determinants of disparities in ovarian cancer are critically needed.
Accumulating evidence indicates that ovarian tumors are immunogenic and harbor a host of immune cells within the tumor microenvironment (8–11). Tumor infiltrating lymphocytes (TILs) are present in tumor islets of more than half of ovarian cancer patients and are associated with favorable outcomes alone or in combination with other immune cells (8,11–15). Immune cells contributing to tumor immune evasion, such as T-regulatory cells or tumor-associated macrophages, have been associated with poor tumor characteristics and worse outcomes in ovarian cancer (16–18). So far, the investigation of the ovarian tumor immune microenvironment has been conducted in study populations of primarily White women, and only one prior study in Black women investigated two immune checkpoint proteins in ovarian cancer (19). With known differences in immune responses between racial and ethnic groups (20,21), we examined the hypothesis that differences in tumor microenvironment immune cell abundance may contribute to differences in overall survival by race among Non-Hispanic Black and Non-Hispanic White women with the most common and one of the deadliest histotypes of ovarian cancer (22), high-grade serous ovarian carcinoma (HGSOC).
MATERIALS AND METHODS
Study population
This study includes two population-based case-control studies, the African-American Cancer Epidemiology Study (AACES) (23) and the North Carolina Ovarian Cancer Study (NCOCS) (24), that have been previously described (23,24). Briefly, AACES enrolled 593 African-American women diagnosed with epithelial ovarian cancer during December 2010 – December 2015 and 752 controls from eleven geographic areas in the U.S. (Alabama, metropolitan Detrioit, Georgia, Illinois, Louisiana, New Jersey, North Carolina, Ohio, South Carolina, Tennessee, and Texas). The NCOCS was conducted from 1999 through 2008 and enrolled 958 women with epithelial ovarian cancer and 1,056 controls residing in 48 counties in North Carolina. In both studies, formalin-fixed paraffin embedded tumor blocks were procured from the facility where the primary debulking surgery was completed, and a centralized expert pathology review was conducted to confirm diagnosis, histology, and grade. First-line treatment was abstracted from medical records or collected from cancer registries in AACES, and for NCOCS, medical record abstractions were completed on a subset of cases that were treated at two large hospital systems in North Carolina. Vital status and follow-up information has been updated annually. Written informed consent was obtained from AACES and NCOCS participants, and both studies were conducted in accordance with the U.S. Common Rule and approved by the relevant institutional review boards.
In the present study, we restricted to HGSOC tumors to limit contributions of disease heterogeneity by histotype (13). We also restricted to women that were treatment-naïve at the time of debulking surgery as chemotherapy has been shown to induce changes to the tumor immune landscape (25). To control for age and stage in the study design, 130 HGSOC tumors from Non-Hispanic Black women were matched to 130 Non-Hispanic White with HGSOC (1:1 match) by five-year age group and stage at diagnosis (localized/regional, distant).
Multiplex immunofluorescence
To measure immune cell abundance, multiplex immunofluorescence staining was completed using the Opal™ chemistry and multispectral microscopy Vectra system (Akoya Biosciences; Marlborough, MA). Whole tumor sections were stained for seven fluorophore-labeled markers: DAPI, pancytokeratin (PCK), CD3, CD8, FoxP3, CD11b, and CD15 (Figure 1, Panel A). DAPI and PCK are markers of tissue and cell segmentation. CD3 is a marker of overall tumor-infiltrating lymphocytes (TILs), and CD8 and FoxP3 are markers of TIL subsets, cytotoxic and regulatory T-cells, respectively. CD11b identifies myeloid cells, and CD15 in combination with CD11b identifies polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs). As the majority of the PMN-MDSCs are neutrophils, we used CD15+CD11b+ as a proxy for neutrophils. Stained slides were scanned, and image capture and analysis was performed using Akoya Biosciences Inform and Spotfire software. A pathologist selected six regions of interest (ROIs) for image analysis (Figure 1, Panel B); three from the intratumoral region and three from the peritumoral region (~90% or ~40–50% tumor content by morphology and PCK expression, respectively). The ROIs are 20X magnification with an image size of 1348 (width) x 1008 (height) pixels at a resolution of 0.5 microns per pixel. Quality control steps were completed to remove any ROIs with poor DAPI staining quality or atypical/non-specific staining that would result in inaccurate cell quantification. Because of this, nine women (and their corresponding match) were removed from subsequent analyses, resulting in a sample size of 121 Non-Hispanic Black and 121 Non-Hispanic White women with HGSOC.
Figure 1.
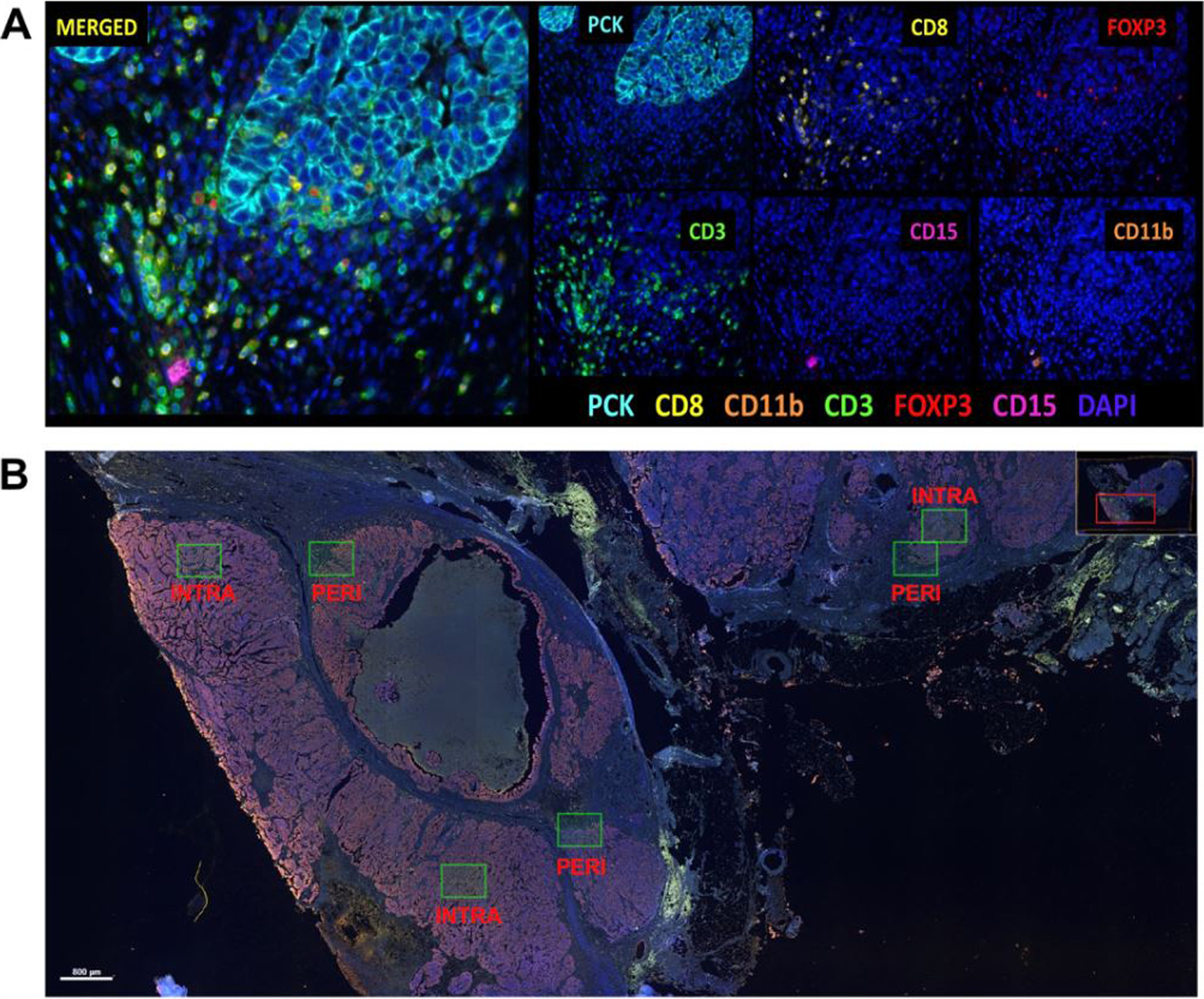
Image analysis of multiplex immunofluorescence panel. A) HGSOC tissue section stained for the seven-marker multiplex immunofluorescence panel. Each marker has a fluorophore that is identified individually (6 panels on the right) and then merged (left panel). B) Example of the ROI selection for intratumoral and peritumoral ROIs.
HGSOC: high-grade serous ovarian carcinoma, ROI: region of interest, PCK: pan-cytokeratin, CD: cell differentiation, INTRA: intratumoral, PERI: peritumoral.
Immune cell abundance
We created categories for immune cell abundance to use for downstream analyses. For the more abundant cell types (CD3+, CD3+CD8+, CD3+FoxP3+), we used a cut-off of 1% (<1%, ≥1%), while for the cell types with a larger proportion of zero positive cells (CD11b+, CD11b+CD15+), we examined these markers as presence vs. absence. This step was completed first at the ROI-level, but also at the patient-level, where the immune cell abundance measures were averaged across the intratumoral and peritumoral ROIs separately and then categorized according to the average.
Two approaches were used to collectively characterize the immune cells. First, we identified immune signatures using model-based clustering (mclust R package) (26) on the normalized immune cell abundance measures (using the scale function in R) in the tumor and stroma from the intratumoral and peritumoral ROIs. The optimal number of signatures was determined by mclust from the Bayesian Information Criterion. Second, we applied the immunoscore that has been developed and validated in colorectal cancer (27,28) to the present study. The immunoscore is determined by quantifying the density of CD3+ and CD8+ cells at the tumor center and the invasive margin and scoring these densities from low to high (low: ≤25%, intermediate: >25 to 70%, high: >70%). We used the intratumoral ROIs as a proxy for the tumor center and the peritumoral ROIs as a proxy for the invasive margin.
Statistical analyses
We assessed the concordance of percent tumor and stroma as well as immune cell positivity across the intratumoral and peritumoral ROIs using intraclass correlation coefficients (ICC). Hierarchical clustering and heatmaps of the square root transformed and standardized (according to the median and median absolute deviation to account for outliers and data skewness) abundance data in the tumor and stroma were generated separately for intratumoral and peritumoral ROIs using the ComplexHeatmap package in R and annotated according to race. We used Wilcoxon rank-sum tests and chi-square tests to examine differences in immune cell abundance by race. Due to the number of tests, we used adjusted p-values (q-values) to control the false discovery rate and correct for multiple comparisons.
Follow-up time was calculated as the time from interview to death or date of last contact. Cox proportional hazard regression models were used to estimate hazard ratios (HRs) and 95% confidence intervals (CIs) for the association of immune cell abundance with risk of all-cause mortality using a repeated measures framework where the ROIs were clustered by patient using the cluster option in coxph in R. We additionally investigated the association of the immune signatures and immunoscore with risk of all-cause mortality. We adjusted models for the matching variables, age (continuous, years) and stage (localized, regional, distant), and included race (Non-Hispanic Black, Non-Hispanic White) when modelling the overall study population. Heterogeneity in the associations by race was assessed using cross-product interaction terms for each immune marker and race. We tested the proportional hazards assumption by evaluating Schoenfeld residuals for each covariate individually and collectively. No violations of proportional hazards were observed for the models examining the clustering approaches; however, stage and CD3+FoxP3+ violated the proportional hazards assumption for the models of abundance. Therefore, we included stage as a strata term in the models to allow for different baseline hazard fuctions by stage. Since we were interested in estimating HRs for CD3+FoxP3+, we used accelerated failure time models with a Weibull distribution. The parameter estimates and standard errors (SE) were then transformed to HRs and 95% CIs using the following formulas: HR = and 95% CI = where p is the scale parameter and βCD3FoxP3 and SECD3FoxP3 are the parameter estimate and SE, respectively, for the association between CD3+FoxP3+ and risk of mortality.
Three sensitivity analyses were conducted. As self-reported race and ethnicity may not be entirely concordant with genetic ancestry (29), we used available genetic data on 232 women to estimate the proportion of intercontinental ancestry using 2,318 ancestry informative markers via the FastPop R package (30,31). We repeated the race-stratified analyses additionally adjusting for the proportion of African, Asian, and European ancestry. We also repeated the analyses adjusting for other prognostically relevant inflammatory-related exposures, including self-reported body mass index (BMI) one year prior to diagnosis (<25, 25–29, 30–35, and ≥35 kg/m2), smoking status (never, former, current smoker), and a history of diabetes (yes, no). To assess whether treatment impacted our findings, we additionally adjusted for debulking status (optimal [<1cm of residual disease] or suboptimal debulking [≥1cm of residual disease]) among the subset of women with available data (n=172; 500 intratumoral ROIs and 485 peripheral ROIs). For women with missing debulking status, we used CA-125 values after the last cycle of adjuvant chemotherapy as a proxy for debulking status (32–34) where available (<35 optimal debulking, ≥35 suboptimal debulking). While other first-line treatment-level characteristics were collected, we did not adjust for these variables as neoadjuvant chemotherapy status was used to determine study inclusion, and all but two women received adjuvant chemotherapy.
Data availability
The data that support the findings of this study are available upon reasonable request from the corresponding author.
RESULTS
Study population characteristics
The median age at diagnosis was 57.8 years and most patients had distant stage disease (78%) (Table 1). Compared to Non-Hispanic White women, Non-Hispanic Black women had a higher BMI (26.5 vs. 31.7 kg/m2, respectively; P<0.001), were more likely to have a history of diabetes (5% vs. 23%, respectively; P <0.001), and were more likely to be suboptimally debulked (10% vs. 30%, respectively; P <0.001).
Table 1.
Patient characteristics overall and by race
| Overall (N=242) | Non-Hispanic Black (N=121) | Non-Hispanic White (N=121) | ||
|---|---|---|---|---|
|
|
||||
| Patient characteristics | Mean (SD) or n (%) | Mean (SD) or n (%) | Mean (SD) or n (%) | p-value |
|
| ||||
| Age at diagnosis | ||||
| Continuous, years | 57.8 (8.9) | 57.7 (9.1) | 57.9 (8.8) | 0.9 |
| <50 years | 44 (18) | 22 (18) | 22 (18) | >0.9 |
| 50–59 years | 101 (42) | 51 (42) | 50 (41) | |
| 60–69 years | 69 (29) | 34 (28) | 35 (29) | |
| ≥70 years | 28 (12) | 14 (12) | 14 (12) | |
| Stage | 0.11 | |||
| Localized | 31 (13) | 12 (10) | 19 (16) | |
| Regional | 22 (9) | 15 (12) | 7 (6) | |
| Distant | 189 (78) | 94 (78) | 95 (79) | |
| Body mass index a | ||||
| Continuous, kg/m2 | 29.1 (8.0) | 31.7 (9.0) | 26.5 (6.0) | <0.001 |
| <25 kg/m2 | 88 (37) | 26 (22) | 62 (52) | <0.001 |
| 25–29 kg/m2 | 64 (27) | 32 (27) | 32 (27) | |
| 30–34 kg/m2 | 41 (17) | 28 (23) | 13 (11) | |
| ≥35 kg/m2 | 47 (20) | 34 (28) | 13 (11) | |
| Unknown | 2 | 1 | 1 | |
| Smoking status | 0.5 | |||
| Never smoker | 120 (50) | 56 (46) | 64 (53) | |
| Former smoker | 91 (38) | 47 (39) | 44 (36) | |
| Current smoker | 31 (13) | 18 (15) | 13 (11) | |
| Diabetes | <0.001 | |||
| No | 208 (86) | 93 (77) | 115 (95) | |
| Yes | 34 (14) | 28 (23) | 6 (5) | |
| Genetic ancestry | ||||
| African ancestry | 0.42 (0.41) | 0.80 (0.12) | 0.01 (0.02) | <0.001 |
| European ancestry | 0.57 (0.41) | 0.17 (0.12) | 0.98 (0.03) | <0.001 |
| Asian ancestry | 0.02 (0.02) | 0.03 (0.03) | 0.01 (0.02) | <0.001 |
| Unknown | 10 | 2 | 8 | |
| Debulking Status b | <0.001 | |||
| Optimal | 138 (80) | 57 (70) | 81 (90) | |
| Suboptimal | 34 (20) | 25 (30) | 9 (10) | |
| Unknown | 70 | 39 | 31 | |
| Adjuvant chemotherapy | 0.2 | |||
| No | 2 (1) | 2 (3) | 0 (0) | |
| Yes | 169 (99) | 77 (97) | 92 (100) | |
| Unknown | 71 | 42 | 29 | |
SD: standard deviation.
Calculated from self-reported height and weight one year prior to diagnosis.
Optimal debulking status is defined as no gross residual disease or <1cm of residual disease from cytoreductive surgery. Suboptimal debulking status is defined as ≥ 1cm of residual disease from cytoreductive surgery. For cases with unknown debulking status, CA-125 following adjuvant chemotherapy was used as a proxy for debulking status, where CA-125 < 35 was categorized as optimal and CA-125 ≥ 35 as suboptimal.
Consistency across ROIs
Across 242 HGSOC tumors, 706 intratumoral ROIs and 689 peritumoral ROIs were examined. The average proportion of tumor cells present across the intratumoral ROIs was 86% (14% stroma) and 43% (57% stroma) across the peritumoral ROIs. There was high consistency in the percent tumor across the intratumoral and peritumoral ROIs for each participant (ICC=0.83, 95% CI=0.81–0.86 and ICC =0.81, 95% CI=0.78–0.84, respectively). Generally, the ICCs were moderate to excellent for immune cell abundance across the ROIs (ICCs ranging from 0.42 to 0.95; Supplemental Table 1).
Immune cell abundance
Stromal CD3+ cells were the most prominent immune cell type in both the intratumoral and peritumoral ROIs (Figure 2, Panel A), averaging 6.4% CD3+ stromal cells in the intratumoral ROIs and 10.8% in the peritumoral ROIs (Supplemental Table 2). No clustering pattern in immune cell abundance by race was observed in Figure 2. Non-Hispanic Black women had a slightly lower abundance of some of the T-cells compared to Non-Hispanic White women, but only the racial differences in stromal CD3+CD8+ in the peritumoral ROIs remained statistically significant after multiple test correction (Figure 2, Panel B and Supplemental Table 2; q-value=0.02 for continuous and q-value=0.04 for categorical measurement of stromal CD3+CD8+).
Figure 2.
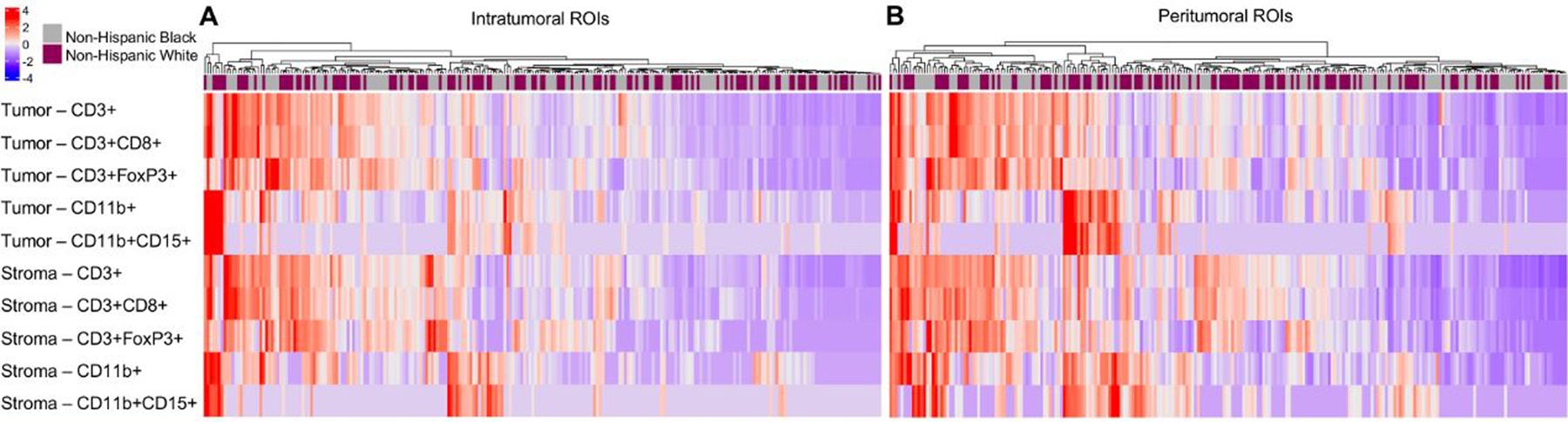
Heatmap of the average immune cell abundance in the tumor and stroma compartments in the intratumoral (Panel A) and peritumoral (Panel B) ROIs annotated by race. Immune cell abundance was standardized by the median and median absolute deviation.
Clustering analysis revealed five immune signatures, which range in immune cell involvement from low (Signature 1) to high (Signature 5). Supplemental Figure 1 provides the distribution of each immune cell in the tumor and stroma stratified by the immune signatures. About 15% of the women were in Signature 5 (Supplemental Table 2). The application of the immunoscore to HGSOC showed that a fifth of the study population (21%) had a high immunoscore (e.g., higher CD3+ and CD3+CD8+ levels in the tumor core and invasive margin). For both the immune signatures and the immunoscore, no differences were observed by race.
Survival analysis
During up to 20 years of follow-up (median, 4.3 years), there were 176 deaths (73%) overall. Higher vs. lower levels of overall TILs were associated with better outcomes in both the intratumoral and peritumoral ROIs (HR=0.68, 95% CI=0.53–0.88 and HR=0.73, 95% CI=0.55–0.97, respectively) and these associations were similar irrespective of tumor/stroma (Table 2). Similarly, a survival benefit was observed for cytotoxic T-cells in the intratumoral ROIs. In the peritumoral region, better survival was observed for higher levels of cytotoxic T-cells in the tumor (HR=0.69, 95% CI=0.52–0.92) but a weaker association was found for overall cytotoxic T-cells (HR=0.81, 95% CI=0.62–1.04) and in the stroma (HR=0.82, 95% CI=0.63–1.08). In the intratumoral ROIs, improved survival was noted for higher levels of T-regulatory cells in the stroma (HR=0.69, 95% CI=0.49–0.96), and although the magnitude of the association with survival was similar for T-regulatory cells overall and in the tumor, the associations were not statistically significant. In the peritumoral ROIs, better outcomes were observed for higher levels of T-regulatory cells overall and within the tumor and stroma. Presence of myeloid cells and neutrophils were associated with better outcomes regardless of the tissue compartment and type of ROI.
Table 2.
Hazard ratios and 95% confidence intervals for the association of immune cell abundance with risk of all-cause mortality in the total study population overall and by tissue compartments (tissue, stroma) in the intratumoral and peritumoral ROIs
| Overall | Tumor | Stroma | ||||
|---|---|---|---|---|---|---|
|
|
||||||
| Immune cell abundance | N (Event N)a | HR (95% CI)b | N (Event N)a | HR (95% CI)b | N (Event N)a | HR (95% CI)b |
|
| ||||||
| Intratumoral ROIs | ||||||
| T-cells, CD3+ | ||||||
| <1% | 354 (275) | 1.00 (Referent) | 392 (308) | 1.00 (Referent) | 296 (229) | 1.00 (Referent) |
| ≥1% | 352 (236) | 0.69 (0.53, 0.89) | 314 (203) | 0.67 (0.52, 0.86) | 410 (282) | 0.77 (0.60, 0.98) |
| Cytotoxic T-cells, CD3+CD8+ | ||||||
| <1% | 492 (377) | 1.00 (Referent) | 512 (393) | 1.00 (Referent) | 429 (326) | 1.00 (Referent) |
| ≥1% | 214 (134) | 0.60 (0.45, 0.81) | 194 (118) | 0.61 (0.44, 0.84) | 277 (185) | 0.69 (0.54, 0.90) |
| T-regulatory cells, CD3+FoxP3+c | ||||||
| <1% | 625 (462) | 1.00 (Referent) | 653 (480) | 1.00 (Referent) | 534 (404) | 1.00 (Referent) |
| ≥1% | 81 (49) | 0.79 (0.51, 1.22) | 53 (31) | 0.70 (0.42, 1.19) | 172 (107) | 0.67 (0.49, 0.94) |
| Myeloid cells, CD11b+ | ||||||
| Absent | 282 (233) | 1.00 (Referent) | 347 (286) | 1.00 (Referent) | 433 (341) | 1.00 (Referent) |
| Present | 424 (278) | 0.73 (0.59, 0.89) | 359 (225) | 0.67 (0.54, 0.82) | 273 (170) | 0.71 (0.56, 0.91) |
| Neutrophils, CD11b+CD15+ | ||||||
| Absent | 592 (446) | 1.00 (Referent) | 550 (420) | 1.00 (Referent) | 641 (478) | 1.00 (Referent) |
| Present | 114 (65) | 0.48 (0.35, 0.66) | 156 (91) | 0.58 (0.43, 0.79) | 65 (33) | 0.43 (0.28, 0.67) |
| Peritumoral ROIs | ||||||
| T-cells, CD3+ | ||||||
| <1% | 168 (130) | 1.00 (Referent) | 296 (230) | 1.00 (Referent) | 148 (115) | 1.00 (Referent) |
| ≥1% | 515 (360) | 0.74 (0.56, 0.98) | 387 (260) | 0.70 (0.55, 0.89) | 535 (375) | 0.75 (0.56, 1.01) |
| Cytotoxic T-cells, CD3+CD8+ | ||||||
| <1% | 295 (218) | 1.00 (Referent) | 446 (338) | 1.00 (Referent) | 270 (198) | 1.00 (Referent) |
| ≥1% | 388 (272) | 0.82 (0.63, 1.06) | 237 (152) | 0.70 (0.53, 0.93) | 413 (292) | 0.84 (0.64, 1.09) |
| T-regulatory cells, CD3+FoxP3+c | ||||||
| <1% | 500 (385) | 1.00 (Referent) | 591 (443) | 1.00 (Referent) | 469 (359) | 1.00 (Referent) |
| ≥1% | 183 (105) | 0.58 (0.40, 0.82) | 92 (47) | 0.65 (0.43, 0.98) | 214 (131) | 0.63 (0.46, 0.87) |
| Myeloid cells, CD11b+ | ||||||
| Absent | 167 (135) | 1.00 (Referent) | 380 (307) | 1.00 (Referent) | 194 (159) | 1.00 (Referent) |
| Present | 516 (355) | 0.77 (0.61, 0.97) | 303 (183) | 0.67 (0.54, 0.83) | 489 (331) | 0.73 (0.58, 0.91) |
| Neutrophils, CD11b+CD15+ | ||||||
| Absent | 481 (365) | 1.00 (Referent) | 551 (414) | 1.00 (Referent) | 498 (376) | 1.00 (Referent) |
| Present | 202 (125) | 0.65 (0.50, 0.84) | 132 (76) | 0.65 (0.47, 0.89) | 185 (114) | 0.64 (0.49, 0.84) |
ROI: region of interest, HR: hazard ratio, CI: confidence interval, CD: cell differentiation.
N is the number of ROIs and event N is the number of deaths; patients can be represented multiple times as they can have a maximum of three intratumoral and three peritumoral ROIs. All models are clustered by patient to take into account the repeated measures.
Estimated using a repeated measures framework where each ROI is included in the models, clustered by patient. Models are adjusted for age at diagnosis (years) and race (Non-Hispanic White, Non-Hispanic Black), and stage (localized, regional, distant) is included as a strata term.
As CD3+FoxP3+ violated the proportional hazard assumption, accelerated failure time models with a Weibull distribution were used, clustering on subject ID. The scale parameter was used to convert the parameter estimates and standard errors to HRs and 95% CIs.
Among Non-Hispanic White women, 93 deaths (77%) occurred during up to 20 years of follow-up (median, 4.5 years), and among Non-Hispanic Black women, 83 deaths (69%) occurred during up to 17 years of follow-up (median, 4.0 years). Stratifying by race revealed similar associations to the findings in the overall study population for Non-Hispanic White women; however, in Non-Hispanic Black women, the associations for TILs, cytotoxic T-cells, and T-regulatory cells were attenuated and not statistically significant (Table 3). For example, higher levels of overall CD3+ cells in the intratumoral ROIs were associated with better survival among Non-Hispanic White women (HR=0.58, 95% CI=0.41–0.82) but the association was weaker among Non-Hispanic Black women (HR=0.80, 95% CI=0.55–1.18). The associations of myeloid cells and neutrophils in the intratumoral ROIs with survival were similar among Non-Hispanic Black and White women, but the pattern of the associations among Non-Hispanic Black women being slightly weaker and not statistically significant was present for these immune cell types in the peritumoral ROIs. However, no statistically significant interactions between immune cell abundance and race were observed.
Table 3.
Hazard ratios and 95% confidence intervals for the association of immune cell abundance with risk of all-cause mortality by race and tissue compartments in the intratumoral and peritumoral ROIs
| Overall |
Tumor |
Stroma |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Non-Hispanic Black |
Non-Hispanic White |
Non-Hispanic Black |
Non-Hispanic White |
Non-Hispanic Black |
Non-Hispanic White |
|||||||
| Immune cells | N (Event N)a | HR (95% CI)b | N (Event N)a | HR (95% CI)b | N (Event N)a | HR (95% CI)b | N (Event N)a | HR (95% CI)b | N (Event N)a | HR (95% CI)b | N (Event N)a | HR (95% CI)b |
|
| ||||||||||||
| Intratumoral ROIs | ||||||||||||
| T-cells, CD3+ | ||||||||||||
| <1% | 169 (125) | 1.00 (Referent) | 185 (150) | 1.00 (Referent) | 188 (142) | 1.00 (Referent) | 204 (166) | 1.00 (Referent) | 140 (102) | 1.00 (Referent) | 156 (127) | 1.00 (Referent) |
| ≥1% | 175 (110) | 0.80 (0.55, 1.18) | 177 (126) | 0.58 (0.41, 0.82) | 156 (93) | 0.73 (0.49, 1.08) | 158 (110) | 0.59 (0.43, 0.83) | 204 (133) | 0.91 (0.64, 1.31) | 206 (149) | 0.67 (0.48, 0.93) |
| Cytotoxic T-cells, CD3+CD8+ | ||||||||||||
| <1% | 242 (177) | 1.00 (Referent) | 250 (200) | 1.00 (Referent) | 251 (184) | 1.00 (Referent) | 261 (209) | 1.00 (Referent) | 217 (156) | 1.00 (Referent) | 212 (170) | 1.00 (Referent) |
| ≥1% | 102 (58) | 0.69 (0.44, 1.08) | 112 (76) | 0.54 (0.37, 0.80) | 93 (51) | 0.63 (0.38, 1.03) | 101 (67) | 0.58 (0.37, 0.90) | 127 (79) | 0.73 (0.49, 1.09) | 150 (106) | 0.67 (0.48, 0.93) |
| T-regulatory cells, CD3+FoxP3+ c | ||||||||||||
| <1% | 293 (204) | 1.00 (Referent) | 332 (258) | 1.00 (Referent) | 311 (217) | 1.00 (Referent) | 342 (263) | 1.00 (Referent) | 259 (183) | 1.00 (Referent) | 275 (221) | 1.00 (Referent) |
| ≥1% | 51 (31) | 1.06 (0.67, 1.67) | 30 (18) | 0.56 (0.27, 1.16) | 33 (18) | 0.92 (0.51, 1.65) | 20 (13) | 0.54 (0.24, 1.21) | 85 (52) | 1.01 (0.67, 1.52) | 87 (55) | 0.51 (0.32, 0.82) |
| Myeloid cells, CD11b+ | ||||||||||||
| Absent | 138 (110) | 1.00 (Referent) | 144 (123) | 1.00 (Referent) | 172 (138) | 1.00 (Referent) | 175 (148) | 1.00 (Referent) | 210 (160) | 1.00 (Referent) | 223 (181) | 1.00 (Referent) |
| Present | 206 (125) | 0.79 (0.60, 1.04) | 218 (153) | 0.69 (0.52, 0.93) | 172 (97) | 0.67 (0.50, 0.89) | 187 (128) | 0.69 (0.51, 0.92) | 134 (75) | 0.80 (0.57, 1.12) | 139 (95) | 0.66 (0.47, 0.93) |
| Neutrophils, CD11b+CD15+ | ||||||||||||
| Absent | 289 (209) | 1.00 (Referent) | 303 (237) | 1.00 (Referent) | 273 (196) | 1.00 (Referent) | 277 (224) | 1.00 (Referent) | 317 (226) | 1.00 (Referent) | 324 (252) | 1.00 (Referent) |
| Present | 55 (26) | 0.52 (0.32, 0.83) | 59 (39) | 0.46 (0.30, 0.71) | 71 (39) | 0.66 (0.45, 0.96) | 85 (52) | 0.53 (0.33, 0.84) | 27 (9) | 0.35 (0.16, 0.76) | 38 (24) | 0.48 (0.27, 0.84) |
| Peritumoral ROIs | ||||||||||||
| T-cells, CD3+ | ||||||||||||
| <1% | 95 (72) | 1.00 (Referent) | 73 (58) | 1.00 (Referent) | 150 (114) | 1.00 (Referent) | 146 (116) | 1.00 (Referent) | 90 (67) | 1.00 (Referent) | 57 (47) | 1.00 (Referent) |
| ≥1% | 242 (157) | 0.81 (0.54, 1.22) | 273 (203) | 0.70 (0.48, 1.03) | 187 (115) | 0.77 (0.54, 1.12) | 200 (145) | 0.62 (0.45, 0.87) | 247 (162) | 0.86 (0.57, 1.31) | 289 (214) | 0.62 (0.40, 0.95) |
| Cytotoxic T-cells, CD3+CD8+ | ||||||||||||
| <1% | 164 (118) | 1.00 (Referent) | 131 (100) | 1.00 (Referent) | 227 (164) | 1.00 (Referent) | 219 (174) | 1.00 (Referent) | 150 (107) | 1.00 (Referent) | 120 (91) | 1.00 (Referent) |
| ≥1% | 173 (111) | 0.90 (0.61, 1.31) | 215 (161) | 0.77 (0.54, 1.09) | 110 (65) | 0.82 (0.53, 1.26) | 127 (87) | 0.6 (0.42, 0.87) | 187 (122) | 0.85 (0.57, 1.26) | 226 (170) | 0.85 (0.59, 1.22) |
| T-regulatory cells, CD3+FoxP3+ c | ||||||||||||
| <1% | 238 (176) | 1.00 (Referent) | 262 (209) | 1.00 (Referent) | 282 (204) | 1.00 (Referent) | 309 (239) | 1.00 (Referent) | 226 (163) | 1.00 (Referent) | 243 (196) | 1.00 (Referent) |
| ≥1% | 99 (53) | 0.74 (0.46, 1.19) | 84 (52) | 0.49 (0.29, 0.81) | 55 (25) | 0.69 (0.40, 1.21) | 37 (22) | 0.64 (0.35, 1.15) | 111 (66) | 0.83 (0.54, 1.27) | 103 (65) | 0.52 (0.33, 0.82) |
| Myeloid cells, CD11b+ | ||||||||||||
| Absent | 91 (69) | 1.00 (Referent) | 76 (66) | 1.00 (Referent) | 193 (150) | 1.00 (Referent) | 187 (157) | 1.00 (Referent) | 101 (78) | 1.00 (Referent) | 93 (81) | 1.00 (Referent) |
| Present | 246 (160) | 0.92 (0.65, 1.30) | 270 (195) | 0.66 (0.49, 0.89) | 144 (79) | 0.69 (0.50, 0.95) | 159 (104) | 0.67 (0.50, 0.89) | 236 (151) | 0.88 (0.63, 1.23) | 253 (180) | 0.63 (0.47, 0.82) |
| Neutrophils, CD11b+CD15+ | ||||||||||||
| Absent | 237 (172) | 1.00 (Referent) | 244 (193) | 1.00 (Referent) | 274 (194) | 1.00 (Referent) | 277 (220) | 1.00 (Referent) | 245 (175) | 1.00 (Referent) | 253 (201) | 1.00 (Referent) |
| Present | 100 (57) | 0.75 (0.52, 1.08) | 102 (68) | 0.59 (0.41, 0.85) | 63 (35) | 0.72 (0.47, 1.12) | 69 (41) | 0.62 (0.40, 0.97) | 92 (54) | 0.79 (0.55, 1.13) | 93 (60) | 0.56 (0.38, 0.81) |
ROI: region of interest, HR: hazard ratio, CI: confidence interval, CD: cell differentiation.
N is the number of ROIs and event N is the number of deaths; patients can be represented multiple times as they can have a maximum of three intratumoral and three peritumoral ROIs. All models are clustered by patient to take into account the repeated measures.
Estimated using a repeated measures framework where each ROI is included in the models, clustered by patient. Models are adjusted for age at diagnosis (years) and stage (localized, regional, distant) is included as a strata term.
As CD3+FoxP3+ violated the proportional hazard assumption, accelerated failure time models with a Weibull distribution were used, clustering on subject ID. The scale parameter was used to convert the parameter estimates and standard errors to HRs and 95% CIs.
The immune signature with the highest vs. lowest immune cell involvement (Signature 5 vs. 1) was associated with a lower risk of mortality (HR=0.47, 95% CI=0.25–0.88) as was a high vs. low immunoscore (HR=0.54, 95% CI=0.33–0.87). These inverse associations with survival were similar among Non-Hispanic White women, but among Non-Hispanic Black women, these associations were attenuated and not statistically significant (Table 4).
Table 4.
Adjusted hazard ratios and 95% confidence intervals for the association of immune cell abundance clustering approaches with risk of all-cause mortality overall and by race
| Overall |
Non-Hispanic Black |
Non-Hispanic White |
||||
|---|---|---|---|---|---|---|
| Clustering approaches | Cases (Deaths) | HR (95% CI)a | Cases (Deaths) | HR (95% CI)a | Cases (Deaths) | HR (95% CI)a |
|
| ||||||
| Immune signatures | ||||||
| Signature 1 | 22 (19) | 1.00 (Referent) | 13 (11) | 1.00 (Referent) | 9 (8) | 1.00 (Referent) |
| Signature 2 | 30 (24) | 1.05 (0.57, 1.92) | 15 (12) | 1.00 (0.44, 2.32) | 15 (12) | 1.19 (0.48, 2.93) |
| Signature 3 | 69 (54) | 1.09 (0.64, 1.86) | 37 (28) | 1.26 (0.62, 2.57) | 32 (26) | 0.87 (0.38, 1.99) |
| Signature 4 | 82 (55) | 0.69 (0.40, 1.17) | 32 (19) | 0.89 (0.42, 1.92) | 50 (36) | 0.52 (0.24, 1.15) |
| Signature 5 | 35 (20) | 0.49 (0.26, 0.92) | 22 (11) | 0.64 (0.28, 1.51) | 13 (9) | 0.34 (0.13, 0.90) |
| Immunoscore | ||||||
| Low | 62 (46) | 1.00 (Referent) | 33 (25) | 1.00 (Referent) | 29 (21) | 1.00 (Referent) |
| Intermediate | 126 (98) | 1.06 (0.75, 1.51) | 62 (43) | 1.14 (0.69, 1.89) | 64 (55) | 0.95 (0.57, 1.60) |
| High | 50 (28) | 0.56 (0.35, 0.90) | 24 (13) | 0.68 (0.35, 1.33) | 26 (15) | 0.45 (0.23, 0.91) |
ROI: region of interest, HR: hazard ratio, CI: confidence interval, CD: cell differentiation.
Models are adjusted for age at diagnosis (years) and stage (localized, regional, distant). The models in the overall study population are additionally adjusted for race (Non-Hispanic Black, Non-Hispanic White).
Sensitivity analysis
Among women who self-reported as Non-Hispanic White, the mean proportion of European ancestry was 97.9%, and for women who self-reported as Non-Hispanic Black, the mean proportion of African ancestry was 80.2% (Supplemental Figure 2). Additionally adjusting for ancestry proportions in the models resulted in similar findings and our conclusions remained the same (Supplemental Table 3). Likewise, our results were consistent with the main findings when adjusting for inflammatory-related exposures (Supplemental Table 4). After restricting to women with available treatment data, we observed that adjustment for debulking status resulted in slightly attenuated HRs but the conclusions remained the same (Supplemental Table 5).
DISCUSSION
Among Non-Hispanic Black and White women with HGSOC, we observed a range of immune cell abundance patterns, with a high infiltrate of T-cells and myeloid cells present in ~15% of HGSOC tumors. Higher levels of most of the studied immune cell types conferred a survival advantage irrespective of tissue and ROI location. The exception to this was cytotoxic T-cells overall and in the stroma of the peritumoral region and T-regulatory cells overall and in the tumor of the peritumoral region where a non-statistically significant inverse association with survival was present. Despite similar immune cell abundance across racial groups, the associations with survival among Non-Hispanic White women were similar to the overall findings, but among Non-Hispanic Black women, associations were attenuated and not statistically significant.
The known survival benefit of TILs and cytotoxic T-cells among ovarian cancer patients was also noted in the present study. However, we observed slightly weaker associations for cytotoxic T-cells in the peritumoral region. Investigations of the prognostic significance of T-cell populations in ovarian cancer often focus on intratumoral T-cells and do not always differentiate the tissue compartment (tumor, stroma). Similar to the present study, Sato et al. (12) and Han, et al. (35) observed no association with survival for stromal TILs or peritumoral TILs in ovarian cancer. Our data also indicated that a subset of women had tumors with an accumulation of cytotoxic T-cells in the periphery without infiltration into the tumor core, also known as immune-excluded tumors (36). As these excluded cytotoxic T-cells cannot infiltrate the tumor core to exert antitumor activity, the weaker associations with survival for peritumoral cytotoxic T-cells may be due, in part, to these immune-excluded tumors. These findings emphasize the importance of the tissue compartment of immune cell populations when investigating survival.
Despite similar abundance of T-cells in Non-Hispanic Black and White women, the survival advantage observed for high TILs, cytotoxic T-cells, and immunoscore was attenuated in Non-Hispanic Black women. However, no statistically significant differences in the associations with survival were observed by race. We speculate with a few explanations for these findings. It is possible that the small sample size could have impacted our precision and ability to detect small effect sizes among Non-Hispanic Black women. Likewise, unmeasured confounders, particularly prognostically relevant comorbidities not collected in both studies that differ in prevalence by race (e.g., heart disease, hypertension), could also bias our results. For known confounders, we used a matched design to ensure comparability across race for age and stage, and also noted consistent findings after performing sensitivity analysis adjusting for debulking status, inflammatory-related exposures, and genetic ancestry. Alternatively, our findings could be due to a higher proportion of exhausted T-cells among Non-Hispanic Black vs. White women. This could result in a lower proportion of T-cells contributing to antitumor activity, negating the survival benefit typically observed with a robust T-cell infiltrate. A recent study by Yao, et al. (37) showed a higher prevalence of an exhausted CD8+ T-cell phenotype among Black vs. White breast cancer patients, which was also associated with worse outcomes. Additionally, other prognostically relevant immune cells not measured in this study, particularly B-cells (15), may be present in the tumor microenvironment or co-localized with T-cells at different levels in Non-Hispanic Black vs. White women. BRCA1/2 mutation status has also been associated with tumor immunity (38) and differs in prevalence across race and ethnicity among ovarian cancer patients (39). Among 209 women in the present analysis with BRCA1/2 mutation status, Non-Hispanic Black women had a higher prevalence of pathogenic BRCA2 mutations compared to Non-Hispanic White women (8% vs. 0%, respectively; P=0.004), but no differences were observed in pathogenic BRCA1 mutation prevalence by race (5% vs. 9%, respectively; P=0.2). Immune cell abundance did not differ by BRCA1/2 pathogenic mutation prevealence except for slightly less neutrophils in the intratumoral region among BRCA1 pathogenic mutation carriers vs. non-carriers. However, the association of neutrophils with survival in the intratumoral region was consistent with the overall findings after adjusting for BRCA1 mutation status (HRTotal = 0.48, 95% CI=0.35–0.66, HRNon-HispanicWhite=0.45, 95% CI=0.29–0.66, HRNon-HispanicBlack=0.53, 95% CI=0.33–0.85). Thus, it is unlikely that BRCA mutation status is the cause of the observed differences by race in the present analysis. Additional work characterizing the T-cell population (e.g., exhausted, activated), investigating other potentially co-localized immune cell types, replicating these findings in a large sample size, and a full accounting of confounders is needed.
We observed a survival advantage for women with higher levels of T-regulatory cells in the invasive margin and the stroma of the tumor core, but the inverse association with survival was not statistically significant for intratumoral T-regulatory cells. T-regulatory cells promote tumor growth and progression via impairment of effector immune cell responses (40). A recent meta-analysis showed that T-regulatory cells were associated with poor outcomes for most solid tumors (41); however, the literature is inconsistent in ovarian cancer (16,42–47). Even though our findings are consistent with some of the studies in ovarian cancer (44–47), they contradict the known immunosuppressive function of T-regulatory cells and may be attributable to a few factors. The association with survival may differ according to patient or clinical characteristics as two ovarian cancer studies noted better survival for higher levels of T-regulatory cells among specific patient subgroups, women with advanced stage disease (44) and HGSOC (45). While FoxP3 is considered the most reliable marker for T-regulatory cells, some studies show that it is also expressed by other lymphoid, myeloid, and epithelial cells (48). Thus, the better prognosis could be due to staining of not only suppressive T-regulatory cells, but also effector cells that confer antitumor activity. T-regulatory cells in the tumor core have been the focus of most research without investigation of the peritumoral region or differentiating the tissue compartment of the immune cells (tumor vs. stroma). Hermans, et al. (46) also reported better outcomes among ovarian cancer patients with stromal T-regulatory cells peripherally located around the tumor center and observed that these stromal T-regulatory cells were part of lymphoid structures, which have been associated with better outcomes in ovarian cancer (15). Therefore, it is possible that the observed associations are reflective of other unmeasured, co-localized immune cells where the T-regulatory cells are a passenger but not independently associated with outcomes.
Our data show that the presence of myeloid cells, especially neutrophils, was associated with better survival. Myeloid cells include many immune cell types (e.g., macrophages, dendritic cells, neutrophils) that can have diverging impacts on outcomes. Neutrophils display both antitumor and protumorigenic activity, yet a recent meta-analysis of tumor-associated neutrophils in cancer patients observed worse overall and progression-free survival for higher levels of intratumoral neutrophils (49). As we used PMN-MDSCs as a proxy for neutrophils and PMN-MDSCs generally support tumor progression (50), it is likely that our findings reflect neutrophil populations displaying antitumor activity. Future work should deeply characterize the various myeloid subsets to better understand their prognostic impact.
This study is strengthened by the inclusion of data and biospecimens from well-established population-based studies and the use of multiplex immunofluorescence to simultaneously and objectively measure abundance and co-localization of immune cells. Despite these strengths, the limitations of our study should be considered. To address the potential of a bias due to ROI selection, we randomly selected a subset of tumors (n=19) and repeated the image analysis for the more abundant immune cell types (CD3+, CD3+CD8+) in the entire invasive margin to compare to the peritumoral ROIs. We found high correlations (r ranging from 0.81 to 0.86) for the abundance of CD3+CD8+ and CD3+, except for stromal CD3+ which was lower at 0.61. The studied immune cell types are a fraction of the immune cells in the tumor microenvironment and do not capture the complexity of immune cell interactions in tumors. While we differentiated the tissue compartment of immune cells and assessed both the intratumoral and peritumoral regions, measurements of immune cell abundance do not fully take into account the spatial contexture of the tumor microenvironment. The present study did not have cause of death or recurrence information for all participants; thus, we could not examine associations with ovarian cancer-specific or progression-free survival.
This study is the first, to our knowledge, to investigate racial differences in tumor immunity and survival of women with HGSOC. We add to the existing evidence that a robust immune infiltrate confers a survival advantage among women with HGSOC. The observed associations were consistently in the inverse direction for Non-Hispanic Black and White women but the associations were weaker among Non-Hispanic Black women. A more complete accounting of patient characteristics and a deep characterization of T-cell populations as well as other co-localized immune cell types is needed to better understand the reasons for these observed racial differences. Moreover, further investigation is warranted to replicate these findings in a large, external cohort of diverse HGSOC patients.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to acknowledge the support of the Advanced Analytical and Digital Pathology Lab in the Pathology Department and the Biostatistics and Bioinformatics Shared Resource at the H. Lee Moffitt Cancer Center and Research Institute, an NCI-designated Comprehensive Cancer Center. We would also like to thank Drs. Jill Barnholtz-Sloan and Ellen Funkhouser for their contributions to the AACES. We acknowledge the AACES interviewers, Christine Bard, LaTonda Briggs, Whitney Franz (North Carolina), and Robin Gold (Detroit). We also acknowledge the individuals responsible for facilitating case ascertainment across the study sites, including Christie McCullum-Hill (Alabama); Rana Bayakly, Vicki Bennett, Judy Andrews, and Debbie Chambers (Georgia); the Louisiana Tumor Registry; Lisa Paddock and Manisha Narang (New Jersey); Diana Slone, Yingli Wolinsky, Steven Waggoner, Anne Heugel, Nancy Fusco, Kelly Ferguson, Peter Rose, Deb Strater, Taryn Ferber, Donna White, Lynn Borzi, Eric Jenison, Nairmeen Haller, Debbie Thomas, Vivian von Gruenigen, Michele McCarroll, Joyce Neading, John Geisler, Stephanie Smiddy, David Cohn, Michele Vaughan, Luis Vaccarello, Elayna Freese, James Pavelka, Pam Plummer, William Nahhas, Ellen Cato, John Moroney, Mark Wysong, Tonia Combs, Marci Bowling, Brandon Fletcher (Ohio); Susan Bolick, Donna Acosta, Catherine Flanagan (South Carolina); Martin Whiteside (Tennessee) and Georgina Armstrong and the Texas Registry, Cancer Epidemiology and Surveillance Branch, Department of State Health Services.
FUNDING
This study was supported by K99/R00CA218681 (PI: Peres). The AACES is supported by R01CA237318 (PI: Schildkraut, Lawson) and R01CA142081 (PI: Schildkraut), and NCOCS is supported by R01CA076016 (PI: Schildkraut).
Footnotes
Conflict of interest disclosure: The authors declare no potential conflcits of interst.
REFERENCES
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin 2021;71(1):7–33 doi 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Peres LC, Schildkraut JM. Racial/ethnic disparities in ovarian cancer research. Adv Cancer Res 2020;146:1–21 doi 10.1016/bs.acr.2020.01.002. [DOI] [PubMed] [Google Scholar]
- 3.Howlader N, Noone A, Krapcho M, Miller D, Brest A, Yu M, et al. SEER Cancer Statistics Review, 1975–2017, National Cancer Institute. Bethesda, MD, https://seer.cancer.gov/csr/1975_2017/, based on November 2019 SEER data submission, posted to the SEER web site, April 2020. [Google Scholar]
- 4.Bandera EV, Lee VS, Rodriguez-Rodriguez L, Powell CB, Kushi LH. Racial/Ethnic Disparities in Ovarian Cancer Treatment and Survival. Clin Cancer Res 2016;22(23):5909–14 doi 10.1158/1078-0432.CCR-16-1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen F, Bailey CE, Alvarez RD, Shu XO, Zheng W. Adherence to treatment guidelines as a major determinant of survival disparities between black and white patients with ovarian cancer. Gynecol Oncol 2021;160(1):10–5 doi 10.1016/j.ygyno.2020.10.040. [DOI] [PubMed] [Google Scholar]
- 6.Hildebrand JS, Wallace K, Graybill WS, Kelemen LE. Racial disparities in treatment and survival from ovarian cancer. Cancer Epidemiol 2019;58:77–82 doi 10.1016/j.canep.2018.11.010. [DOI] [PubMed] [Google Scholar]
- 7.Whitmore G, Ramzan A, Sheeder J, Guntupalli SR. African American women with advanced-stage ovarian cancer have worse outcomes regardless of treatment type. Int J Gynecol Cancer 2020;30(7):1018–25 doi 10.1136/ijgc-2019-000555. [DOI] [PubMed] [Google Scholar]
- 8.Biswas S, Mandal G, Payne KK, Anadon CM, Gatenbee CD, Chaurio RA, et al. IgA transcytosis and antigen recognition govern ovarian cancer immunity. Nature 2021;591(7850):464–70 doi 10.1038/s41586-020-03144-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Payne KK, Mine JA, Biswas S, Chaurio RA, Perales-Puchalt A, Anadon CM, et al. BTN3A1 governs antitumor responses by coordinating alphabeta and gammadelta T cells. Science 2020;369(6506):942–9 doi 10.1126/science.aay2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelson BH. The impact of T-cell immunity on ovarian cancer outcomes. Immunol Rev 2008;222:101–16 doi 10.1111/j.1600-065X.2008.00614.x. [DOI] [PubMed] [Google Scholar]
- 11.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med 2003;348(3):203–13 doi 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 12.Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A 2005;102(51):18538–43 doi 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ovarian Tumor Tissue Analysis C, Goode EL, Block MS, Kalli KR, Vierkant RA, Chen W, et al. Dose-Response Association of CD8+ Tumor-Infiltrating Lymphocytes and Survival Time in High-Grade Serous Ovarian Cancer. JAMA Oncol 2017;3(12):e173290 doi 10.1001/jamaoncol.2017.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nielsen JS, Sahota RA, Milne K, Kost SE, Nesslinger NJ, Watson PH, et al. CD20+ tumor-infiltrating lymphocytes have an atypical CD27- memory phenotype and together with CD8+ T cells promote favorable prognosis in ovarian cancer. Clin Cancer Res 2012;18(12):3281–92 doi 10.1158/1078-0432.CCR-12-0234. [DOI] [PubMed] [Google Scholar]
- 15.Kroeger DR, Milne K, Nelson BH. Tumor-Infiltrating Plasma Cells Are Associated with Tertiary Lymphoid Structures, Cytolytic T-Cell Responses, and Superior Prognosis in Ovarian Cancer. Clin Cancer Res 2016;22(12):3005–15 doi 10.1158/1078-0432.CCR-15-2762. [DOI] [PubMed] [Google Scholar]
- 16.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 2004;10(9):942–9 doi 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 17.Barnett JC, Bean SM, Whitaker RS, Kondoh E, Baba T, Fujii S, et al. Ovarian cancer tumor infiltrating T-regulatory (T(reg)) cells are associated with a metastatic phenotype. Gynecol Oncol 2010;116(3):556–62 doi 10.1016/j.ygyno.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 18.Yuan X, Zhang J, Li D, Mao Y, Mo F, Du W, et al. Prognostic significance of tumor-associated macrophages in ovarian cancer: A meta-analysis. Gynecol Oncol 2017;147(1):181–7 doi 10.1016/j.ygyno.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 19.Mills AM, Peres LC, Meiss A, Ring KL, Modesitt SC, Abbott SE, et al. Targetable Immune Regulatory Molecule Expression in High-Grade Serous Ovarian Carcinomas in African American Women: A Study of PD-L1 and IDO in 112 Cases From the African American Cancer Epidemiology Study (AACES). Int J Gynecol Pathol 2019;38(2):157–70 doi 10.1097/PGP.0000000000000494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quach H, Rotival M, Pothlichet J, Loh YE, Dannemann M, Zidane N, et al. Genetic Adaptation and Neandertal Admixture Shaped the Immune System of Human Populations. Cell 2016;167(3):643–56 e17 doi 10.1016/j.cell.2016.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ye CJ, Feng T, Kwon HK, Raj T, Wilson MT, Asinovski N, et al. Intersection of population variation and autoimmunity genetics in human T cell activation. Science 2014;345(6202):1254665 doi 10.1126/science.1254665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peres LC, Cushing-Haugen KL, Kobel M, Harris HR, Berchuck A, Rossing MA, et al. Invasive Epithelial Ovarian Cancer Survival by Histotype and Disease Stage. J Natl Cancer Inst 2019;111(1):60–8 doi 10.1093/jnci/djy071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schildkraut JM, Alberg AJ, Bandera EV, Barnholtz-Sloan J, Bondy M, Cote ML, et al. A multi-center population-based case-control study of ovarian cancer in African-American women: the African American Cancer Epidemiology Study (AACES). BMC Cancer 2014;14:688 doi 10.1186/1471-2407-14-688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schildkraut JM, Moorman PG, Halabi S, Calingaert B, Marks JR, Berchuck A. Analgesic drug use and risk of ovarian cancer. Epidemiology 2006;17(1):104–7 doi 10.1097/01.ede.0000190538.55645.f8. [DOI] [PubMed] [Google Scholar]
- 25.Lo CS, Sanii S, Kroeger DR, Milne K, Talhouk A, Chiu DS, et al. Neoadjuvant Chemotherapy of Ovarian Cancer Results in Three Patterns of Tumor-Infiltrating Lymphocyte Response with Distinct Implications for Immunotherapy. Clin Cancer Res 2017;23(4):925–34 doi 10.1158/1078-0432.CCR-16-1433. [DOI] [PubMed] [Google Scholar]
- 26.Yeung KY, Fraley C, Murua A, Raftery AE, Ruzzo WL. Model-based clustering and data transformations for gene expression data. Bioinformatics 2001;17(10):977–87 doi 10.1093/bioinformatics/17.10.977. [DOI] [PubMed] [Google Scholar]
- 27.Angell HK, Bruni D, Barrett JC, Herbst R, Galon J. The Immunoscore: Colon Cancer and Beyond. Clin Cancer Res 2020;26(2):332–9 doi 10.1158/1078-0432.CCR-18-1851. [DOI] [PubMed] [Google Scholar]
- 28.Pages F, Mlecnik B, Marliot F, Bindea G, Ou FS, Bifulco C, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet 2018;391(10135):2128–39 doi 10.1016/S0140-6736(18)30789-X. [DOI] [PubMed] [Google Scholar]
- 29.Mersha TB, Abebe T. Self-reported race/ethnicity in the age of genomic research: its potential impact on understanding health disparities. Hum Genomics 2015;9:1 doi 10.1186/s40246-014-0023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amos CI, Dennis J, Wang Z, Byun J, Schumacher FR, Gayther SA, et al. The OncoArray Consortium: A Network for Understanding the Genetic Architecture of Common Cancers. Cancer Epidemiol Biomarkers Prev 2017;26(1):126–35 doi 10.1158/1055-9965.EPI-16-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y, Byun J, Cai G, Xiao X, Han Y, Cornelis O, et al. FastPop: a rapid principal component derived method to infer intercontinental ancestry using genetic data. BMC Bioinformatics 2016;17:122 doi 10.1186/s12859-016-0965-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pelissier A, Bonneau C, Chereau E, de La Motte Rouge T, Fourchotte V, Darai E, et al. CA125 kinetic parameters predict optimal cytoreduction in patients with advanced epithelial ovarian cancer treated with neoadjuvant chemotherapy. Gynecol Oncol 2014;135(3):542–6 doi 10.1016/j.ygyno.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 33.Zorn KK, Tian C, McGuire WP, Hoskins WJ, Markman M, Muggia FM, et al. The prognostic value of pretreatment CA 125 in patients with advanced ovarian carcinoma: a Gynecologic Oncology Group study. Cancer 2009;115(5):1028–35 doi 10.1002/cncr.24084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zwakman N, van de Laar R, Van Gorp T, Zusterzeel PL, Snijders MP, Ferreira I, et al. Perioperative changes in serum CA125 levels: a prognostic factor for disease-specific survival in patients with ovarian cancer. J Gynecol Oncol 2017;28(1):e7 doi 10.3802/jgo.2017.28.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han LY, Fletcher MS, Urbauer DL, Mueller P, Landen CN, Kamat AA, et al. HLA class I antigen processing machinery component expression and intratumoral T-Cell infiltrate as independent prognostic markers in ovarian carcinoma. Clin Cancer Res 2008;14(11):3372–9 doi 10.1158/1078-0432.CCR-07-4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hegde PS, Chen DS. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020;52(1):17–35 doi 10.1016/j.immuni.2019.12.011. [DOI] [PubMed] [Google Scholar]
- 37.Yao S, Cheng TD, Elkhanany A, Yan L, Omilian A, Abrams SI, et al. Breast Tumor Microenvironment in Black Women: A Distinct Signature of CD8+ T Cell Exhaustion. J Natl Cancer Inst 2021. doi 10.1093/jnci/djaa215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Przybytkowski E, Davis T, Hosny A, Eismann J, Matulonis UA, Wulf GM, et al. An immune-centric exploration of BRCA1 and BRCA2 germline mutation related breast and ovarian cancers. BMC Cancer 2020;20(1):197 doi 10.1186/s12885-020-6605-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurian AW, Ward KC, Howlader N, Deapen D, Hamilton AS, Mariotto A, et al. Genetic Testing and Results in a Population-Based Cohort of Breast Cancer Patients and Ovarian Cancer Patients. J Clin Oncol 2019;37(15):1305–15 doi 10.1200/JCO.18.01854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saleh R, Elkord E. FoxP3(+) T regulatory cells in cancer: Prognostic biomarkers and therapeutic targets. Cancer Lett 2020;490:174–85 doi 10.1016/j.canlet.2020.07.022. [DOI] [PubMed] [Google Scholar]
- 41.Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta-analysis. Sci Rep 2015;5:15179 doi 10.1038/srep15179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Polcher M, Braun M, Friedrichs N, Rudlowski C, Bercht E, Fimmers R, et al. Foxp3(+) cell infiltration and granzyme B(+)/Foxp3(+) cell ratio are associated with outcome in neoadjuvant chemotherapy-treated ovarian carcinoma. Cancer Immunol Immunother 2010;59(6):909–19 doi 10.1007/s00262-010-0817-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adams SF, Levine DA, Cadungog MG, Hammond R, Facciabene A, Olvera N, et al. Intraepithelial T cells and tumor proliferation: impact on the benefit from surgical cytoreduction in advanced serous ovarian cancer. Cancer 2009;115(13):2891–902 doi 10.1002/cncr.24317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leffers N, Gooden MJ, de Jong RA, Hoogeboom BN, ten Hoor KA, Hollema H, et al. Prognostic significance of tumor-infiltrating T-lymphocytes in primary and metastatic lesions of advanced stage ovarian cancer. Cancer Immunol Immunother 2009;58(3):449–59 doi 10.1007/s00262-008-0583-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Milne K, Kobel M, Kalloger SE, Barnes RO, Gao D, Gilks CB, et al. Systematic analysis of immune infiltrates in high-grade serous ovarian cancer reveals CD20, FoxP3 and TIA-1 as positive prognostic factors. PLoS One 2009;4(7):e6412 doi 10.1371/journal.pone.0006412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hermans C, Anz D, Engel J, Kirchner T, Endres S, Mayr D. Analysis of FoxP3+ T-regulatory cells and CD8+ T-cells in ovarian carcinoma: location and tumor infiltration patterns are key prognostic markers. PLoS One 2014;9(11):e111757 doi 10.1371/journal.pone.0111757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mhawech-Fauceglia P, Wang D, Ali L, Lele S, Huba MA, Liu S, et al. Intraepithelial T cells and tumor-associated macrophages in ovarian cancer patients. Cancer Immun 2013;13:1. [PMC free article] [PubMed] [Google Scholar]
- 48.Devaud C, Darcy PK, Kershaw MH. Foxp3 expression in T regulatory cells and other cell lineages. Cancer Immunol Immunother 2014;63(9):869–76 doi 10.1007/s00262-014-1581-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen M, Hu P, Donskov F, Wang G, Liu Q, Du J. Tumor-associated neutrophils as a new prognostic factor in cancer: a systematic review and meta-analysis. PLoS One 2014;9(6):e98259 doi 10.1371/journal.pone.0098259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou J, Nefedova Y, Lei A, Gabrilovich D. Neutrophils and PMN-MDSC: Their biological role and interaction with stromal cells. Semin Immunol 2018;35:19–28 doi 10.1016/j.smim.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available upon reasonable request from the corresponding author.