

Abstract
Objective
Antitumor viral vaccines, and more particularly poxviral vaccines, represent an active field for clinical development and translational research. To improve the efficacy and treatment outcome, new viral vectors are sought, with emphasis on their abilities to stimulate innate immunity, to display tumor antigens and to induce a specific T‐cell response.
Methods
We screened for a new poxviral backbone with improved innate and adaptive immune stimulation using IFN‐α secretion levels in infected PBMC cultures as selection criteria. Assessment of virus effectiveness was made in vitro and in vivo.
Results
The bovine pseudocowpox virus (PCPV) stood out among several poxviruses for its ability to induce significant secretion of IFN‐α. PCPV produced efficient activation of human monocytes and dendritic cells, degranulation of NK cells and reversed MDSC‐induced T‐cell suppression, without being offensive to activated T cells. A PCPV‐based vaccine, encoding the HPV16 E7 protein (PCPV‐E7), stimulated strong antigen‐specific T‐cell responses in TC1 tumor‐bearing mice. Complete regression of tumors was obtained in a CD8+ T‐cell‐dependent manner after intratumoral injection of PCPV‐E7, followed by intravenous injection of the cancer vaccine MVA‐E7. PCPV also proved active when injected repeatedly intratumorally in MC38 tumor‐bearing mice, generating tumor‐specific T‐cell responses without encoding a specific MC38 antigen. From a translational perspective, we demonstrated that PCPV‐E7 effectively stimulated IFN‐γ production by T cells from tumor‐draining lymph nodes of HPV+‐infected cancer patients.
Conclusion
We propose PCPV as a viral vector suitable for vaccination in the field of personalised cancer vaccines, in particular for heterologous prime‐boost regimens.
Keywords: adaptive immunity, IFN‐α, innate immunity, intratumoral injection, Poxviral cancer vaccine
In this study, we identified an immunostimulatory vaccine vector, pseudocowpox virus (PCPV), selected on its capacity to induce type I IFN response in primary immune cells from healthy donors and cancer patients. PCPV reactivates the ex vivo T‐cell response of cancer patients. In murine tumor models, the use of the PCPV vaccine can lead to complete tumor regression and systemic immunity.

Introduction
Cancer vaccines are regaining interest as a promising immunotherapeutic treatment modality, alone or in combination with immune checkpoint inhibition or T‐cell‐based technologies. 1 , 2 In particular, the development of methods allowing the identification of patient‐specific neoantigens has renewed interest in their use to induce personalised adaptive response against tumors. 3 Viral vectors have been employed for designing cancer vaccines thanks to their intrinsic immune activity, being able to evoke a robust adaptive response in the absence of an adjuvant. 4 , 5 The stimulation of innate immunity by the vector itself is an important attribute that can increase the potency of vaccines. 6 , 7
When delivered and exposed in the cytoplasm, nucleic acids from viral vectors act as pathogen‐associated molecular patterns (PAMPS) sensed by pattern recognition receptors (PRRs). 8 Cyclic GMP‐AMP synthase, cGAS, is the major cytosolic DNA sensor that binds dsDNA to catalyse the synthesis of a special asymmetric cyclic dinucleotide 2’3’‐cGAMP. This molecule binds and activates stimulator of interferon genes (STING) for subsequent production of type I interferons (IFNs) and other immunomodulatory genes. 9 , 10 , 11 Within the tumor microenvironment, expression of type I IFNs and IFN‐stimulated genes correlates with more favorable clinical outcome, explained by the enhanced stimulation of both innate and adaptive immune responses. 12 , 13 IFN‐α can locally induce the rapid differentiation of monocytes into highly activated dendritic cells that are particularly effective in priming adaptive immunity. 14 , 15 For instance, the combination of IFN‐α with a poxviral vaccine targeting CEA showed positive preclinical results in models of colorectal and pancreatic adenocarcinomas. 16 Additionally, type I IFNs suppress tumor growth by activating the STAT3–granzyme B pathway in tumor‐infiltrating cytotoxic T lymphocytes. 17 On the other hand, type I IFNs may upregulate PD‐L1 in tumor cells, which can lead to T‐cell exhaustion. 18
Poxviruses have been selected as a cancer vaccine vector platform for several reasons, such as safety, large gene capacity for insertion of the antigen of interest, as well as any additional immunomodulatory genes, and broad tropism that can result in a large number of cells expressing the heterologous antigen. 4
In our experience, the modified vaccinia virus Ankara (MVA), an attenuated orthopoxvirus vector, historically selected for prophylactic vaccination and gene therapy purposes, 19 has shown good antitumor properties. TG4001, a MVA vaccine encoding HPV16 proteins, was tested in HPV‐associated cervical intraepithelial neoplasia and showed significant effect on viral clearance and histological resolution in treated patients. 20 , 21 TG4050, a patient‐tailored MVA vaccine targeted at tumor‐specific antigens, 22 entered into a phase 1–2 clinical trial for patients with ovarian or head and neck cancers (ClinicalTrials.gov Identifiers NCT04183166 and NCT0839524). Poxviral strains are known to differ significantly in their ability to downmodulate immune properties in vivo. 23 Vaccinia virus (VACV) genomes indeed contain several genes involved in inhibiting the interferon pathways, probably reflecting a strong evolutionary selection for this strategy of immune evasion. 24 , 25 MVA was generated by successive passage of a VACV strain in primary chicken embryonic fibroblasts on the criteria of safety and immunogenicity, leading to a loss of approximately 10% of its original genome, but still encodes genes involved in taming the type I IFN response, such as K3 and E3. 19 , 26
We thus sought an alternative poxviral strain allowing for unrestrained IFN‐α secretion in human primary cells. A variety of poxviruses covering the main genera were subjected to a screening assay. Animal viruses were deliberately included, presuming that immune escape mechanisms prone to silence type I IFN would be restricted to their respective hosts and thus be non‐functional in humans. Among the tested viruses, the bovine parapoxvirus pseudocowpox virus (PCPV) turned out to be the best inducer of IFN‐α in human peripheral blood mononuclear cells (PBMCs). The PCPV strain TJS used in this study was isolated from a human case of Milker’s nodules 27
In this study, the performance of PCPV was tested in various immune models. In vitro, viral infection activated human NKs and APCs, and released T cells from the suppressive activity of MDSCs. PCPV allowed the ex vivo stimulation of tumor antigen‐specific T cells from cancer patients. In vivo, this PCPV vaccine induced a strong antigen‐specific T‐cell response, led to both antigen‐dependent and antigen‐independent control of tumor growth and increased survival in syngeneic tumor models. Our findings demonstrate that PCPV might thus be an improved vector for the design of efficient cancer vaccines.
Results
PCPV selection, infection and immunomodulatory properties in human immune cells
The Poxviridae family is divided into Entomopoxvirinae, infecting insects, and Chordopoxvirinae, infecting a large range of vertebrates. The latter subdivides into 18 genera including Orthopoxvirus and Parapoxvirus (according to the International Committee on Taxonomy of Viruses, ICTV). Included in this study were VACV strain Copenhagen, MVA, cowpox virus (CPXV), raccoonpox virus (RCNV), rabbitpox virus (RPV), PCPV, orf virus (ORFV), Yaba‐like disease virus (YLDV), fowlpox virus (FPV), swinepox virus (SWPV), myxoma virus (MYXV) and Cotia virus (CTV). These wild‐type viral strains were screened for the induction of IFN‐α secretion in primary human immune cells. PBMCs from healthy donors were infected at various multiplicity of infections (MOIs), and IFN‐α concentrations were measured in the cell culture supernatants after overnight incubation. The bovine Parapoxvirus PCPV turned out to be by far the best inducer of IFN‐α, leading to an up to 200‐fold increase in IFN‐α concentration in the supernatant compared with the uninfected control (Figure 1a). PCPV augmented not only the secretion of IFN‐α (4000 pg mL−1) but also the levels of IFN‐β (490 pg mL−1) and IFN‐γ (1800 pg mL−1) (Supplementary figure 1a). Within PCPV‐infected PBMCs, IFN‐α‐secreting cells were mainly plasmacytoid dendritic cells (pDCs) and monocytes (Supplementary figure 1b). At the RNA level, IFN‐α‐ and IFN‐β‐specific transcripts were clearly upregulated (Supplementary figure 1c).
Figure 1.
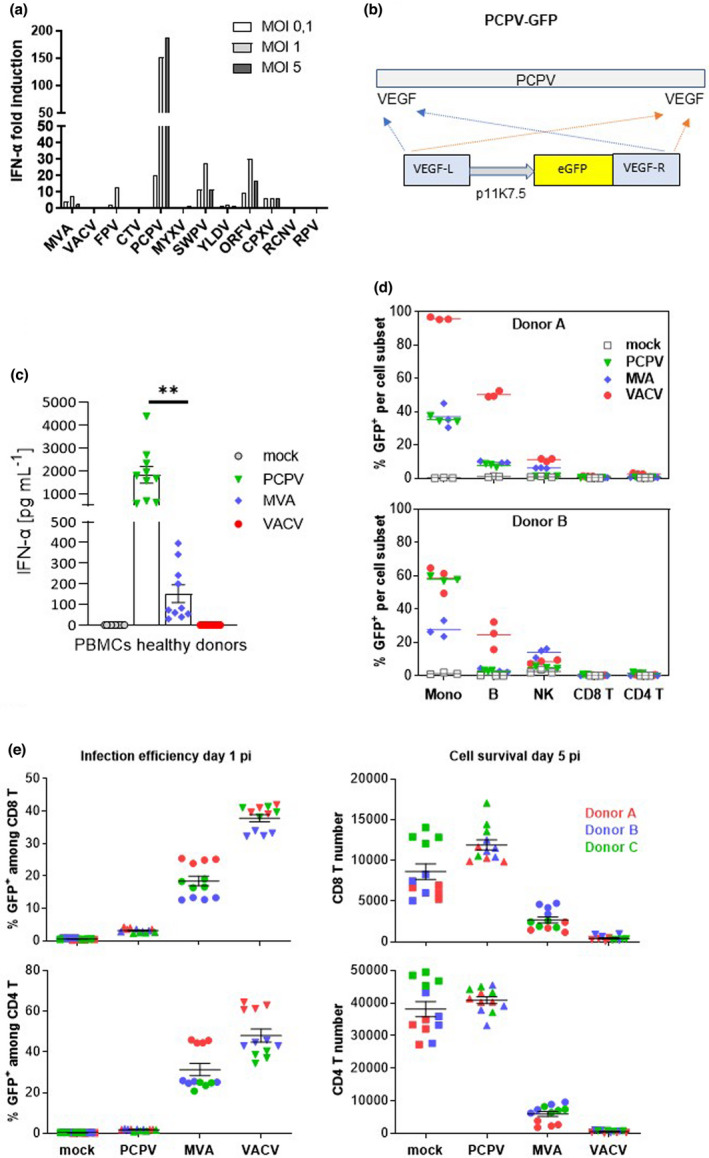
Selection and characterisation of PCPV. (a) Screening of poxviruses for IFN‐α induction in human PBMCs: immune cells were infected at MOI 0.1, 1 and 5 with each of the indicated viruses, and IFN‐α secretion was measured in the cell culture supernatant the next day. Shown is the mean fold induction of IFN‐α secretion compared with mock‐infected cells from two experiments with two donors each. (b) Schematic representation of the PCPV genome with VEGF genes located at both extremities. An eGFP expression cassette controlled by the poxvirus‐specific promoter p11K7.5 was inserted via homologous recombination in both VEGF loci. (c) PBMCs from 10 healthy donors were infected at the MOI 0.3 with PCPV, MVA and VACV viruses encoding GFP. The next day, the supernatant was harvested, and IFN‐α was quantified. Scatter plots present data from a single experiment with ten different blood donors as mean ± SEM. (d) Infection profile: PBMCs were incubated with either PCPV‐GFP, MVA‐GFP or VACV‐GFP at a MOI of 1, or left untreated (mock). The percentage of GFP‐expressing cells within live monocytes (mono), B lymphocytes (B), T lymphocytes (CD4+ T, CD8+ T) and natural killer (NK) cells was determined by flow cytometry at 1 day post‐infection. Mean of triplicate samples is indicated. Data are from two independent experiments with one PBMC donor each. (e) Activated T cells resist PCPV, but not MVA or VACV infection: PBMCs were pretreated with polymer‐bound anti‐CD3 plus anti‐CD28 and IL‐2 for 2 days and then incubated with PCPV‐GFP, MVA‐GFP or VACV‐GFP at MOI 1 in quadruplicate. The percentage of GFP+ cells (infection efficiency) and the total cell count (cell survival) were determined by flow cytometry for both activated CD8 and CD4 T‐cell subsets, at 1 or 5 days post‐infection, respectively. Scatter plots present data from a single experiment with three different blood donors as mean ± SEM.
A recombinant PCPV was engineered to encode enhanced green fluorescent protein (eGFP), as schematically depicted (Figure 1b, Supplementary figure 2a), and compared with recombinant MVA and VACV, encoding the same reporter gene in PBMCs from ten healthy blood donors (Supplementary figure 2a, b). PCPV induced significantly higher levels of secreted IFN‐α (about 2000 pg mL−1) than MVA (about 150 pg mL−1), or VACV (IFN‐α was undetectable) (Figure 1c). Further, PBMCs were incubated with these viruses at MOI 1, and the next day, GFP‐positive subpopulations were determined. PCPV‐infected monocytes were detected at high frequency, and B and NK cells to a lower extent (Figure 1d, Supplementary figure 3). CD3+CD56−CD4− cells, considered to be CD8+ T cells, and CD3+CD56−CD4+ T cells were only poorly infected with all tested viruses (Figure 1d). The overall infection profile of PCPV‐GFP under these conditions was comparable to the profiles observed with MVA‐GFP or VACV‐GFP. Next, activated T cells were expanded from PBMCs according to protocols established for CAR‐T technology, using polymer‐bound anti‐CD28 and anti‐CD3, and IL‐2, 28 and incubated with each GFP virus. Both MVA and VACV efficiently infected activated T cells, as previously described, 29 leading to the appearance of GFP+ T cells at day 1 after infection. Strikingly, PCPV hardly infected CD4+ and CD8+ T cells (Figure 1e). Five days after infection, PCPV‐treated cells were still alive, while MVA‐ and VACV‐infected cultures were dying. Importantly, the comparative innocuity of PCPV towards activated T cells distinguished this vector from other poxviral vectors.
Activation of APCs was tested in human immature monocyte‐derived dendritic cells (moDCs). These cells were infected by each GFP‐encoding virus at MOI of 0.3. At this MOI, the PCPV‐induced secretion of IFN‐α in moDCs was not at its maximum (Supplementary figure 4). The day after infection, the co‐stimulatory molecules CD86 and CD80, the activation marker CD83 and the antigen‐presenting molecule HLA‐DR were analysed. Compared to treatment with MVA, infection with PCPV upregulated CD83, CD80 and HLA‐DR, and increased CD86 expression (Figure 2a, b). In contrast, VACV did not induce maturation of moDCs, confirming earlier reports. 30 Compared to the stimulation with the TLR7/8 ligand R848, the effect induced by PCPV was either comparable (CD80) or lower (CD83, CD86 and HLA‐DR) (Figure 2a). Negative effects of virus treatment on moDC viability were negligible (Figure 2c).
Figure 2.
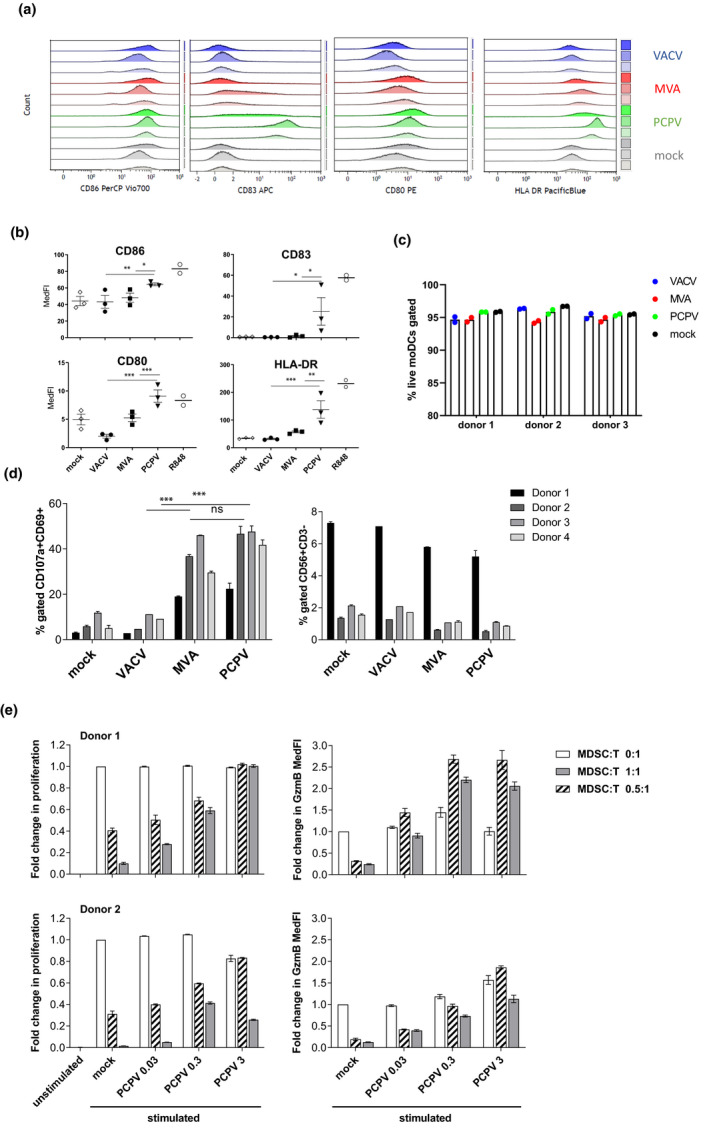
Immunomodulatory effects of PCPV in human immune cells. Infection and activation of APCs: moDCs from three healthy donors were infected at MOI 0.3 with PCPV, MVA or VACV, or treated with the TLR7/8 ligand R848 at 10−4 M. The markers CD86, HLA‐DR, CD83 and CD80 were followed by flow cytometry 16 h after infection. Gating strategy: FSC/SSC, singlets, live/ respective marker (medFI) in live cell population. (a) Median fluorescence intensities presented as staggered overlays. (b) Means of median fluorescence intensities in live cells (MedFI) ± SEM are represented. A repeated mixed model was built, if group effect was found statistically significant, and post hoc comparisons were made with the Tukey multiplicity adjustment: PCPV was able to upregulate all the four markers significantly better than MVA and VACV (CD80: P‐values < 0.001); CD83: compared to MVA, P‐value = 0.017 and VACV, P‐value = 0.015; CD86: compared to MVA, P‐value = 0.012 and VACV, P‐value = 0.003; and HLA‐DR: compared to MVA, P‐value = 0.004 and VACV, P‐value < 0.001. (c) Shown is the percentage of live moDCs at harvest time point after infection with respective virus at MOI 0.3. (d) Activation and degranulation of NK cells: PBMCs from four donors were infected at MOI 0.3 with either PCPV, MVA or VACV. After overnight incubation, K562 cells were added as targets at a ratio of 1:1. After 90 min of co‐culture, the percentage of activated, degranulating NKs was determined as CD69+CD107a+ cells within the population of NK cells (CD3−CD56+). Mean ± SD for each donor is shown. A repeated mixed model was built, if group effect was found statistically significant; post hoc comparisons were made with the Tukey multiplicity adjustment: PCPV and MVA increased the fraction of activated NK significantly better than VACV (P‐value < 0.001), but no significant differences were found between them (P‐value = 0.382). (e) MDSCs lose their suppressive function upon treatment with PCPV: the suppressive activity was tested in a proliferation assay using autologous CD8+ T as responder cells. CFSE‐labelled T cells were cultured with or without MDSCs (MDSC:T ratios were 0.5 :1, 1:1 or 0:1, respectively) for 4 days in the presence or absence of PCPV, and polymer‐bound anti‐CD3/anti‐CD28 plus IL‐2, or left unstimulated. The percentage of proliferating granzyme B+ CD8+ T cells, and the median fluorescence intensity of granzyme B in proliferating CD8+ T cells were determined. Shown is the fold change in proliferation and granzyme B expression compared with stimulated T cells cultured alone. Bars represent mean ± SEM of quadruplicate or triplicate samples for donor 1 and quadruplicate samples for donor 2. Data are from two independent experiments with one blood donor each.
The effect of PCPV on NK cells was assessed in a co‐culture assay of PBMCs with the human erythroleukaemia cell line K562 as target cell. CD69 and CD107 were used as NK cell activation and degranulation markers, respectively. 31 By measuring the percentages of total NK cells (CD3−CD56+) and of activated degranulating NK cells (CD3−CD56+CD69+CD107a+) in the co‐culture, we confirmed that both PCPV and MVA could increase the fraction of activated degranulating NK cells (Figure 2d, Supplementary figure 5a). VACV had no effect on NK cell activation. In line with this observation, PCPV infection increased the secretion of IFN‐γ in PBMC cultures (Supplementary figure 1a). Activation was associated with a slight decrease in NK cell frequency, which could be explained by cell death induced by degranulation. PCPV neither efficiently infected nor killed activated NK cells (Supplementary figure 5b). We suggest that the activation of NK cells was mediated by soluble factors secreted from PCPV‐infected cells, since the supernatant taken from MVA‐ and PCPV‐infected cell cultures induced degranulation of NK cells co‐cultured with K562 cells (Supplementary figure 5c). Secretion of IFN‐γ in these co‐cultures was best from NK cells treated with supernatant from PCPV‐infected cells (Supplementary figure 5c).
The ‘pro‐IFN’ phenotype induced by PCPV predestines this vector for near‐tumor or intratumoral applications. 13 By this route of administration, the virus itself and the induced cytokines would likely modulate the tumor‐infiltrating myeloid‐derived suppressor cells (MDSCs) geared to inhibit NK cells and tumor‐specific T cells. To test this hypothesis, MDSCs were derived from human monocytes cultured in the presence of GM‐CSF and IL‐6. 32 These cells suppressed both the proliferation of autologous activated CD8+ T cells and their production of granzyme B (Figure 2e). In the presence of increasing doses of PCPV, T cells regained their functional capacities. IP‐10, considered to be a surrogate marker for IFN response and an attractant for activated T cells, 33 was found to be increased in a dose‐dependent manner in the supernatant of infected MDSCs cultured alone (Supplementary figure 6a). Furthermore, PCPV was deleterious to MDSCs in co‐cultures since they died in a PCPV dose‐dependent manner (Supplementary figure 6b).
We then probed PCPV in blood cells from cancer patients taken before treatment onset (Supplementary figure 7). Like in PBMCs from healthy donors, PCPV induced significantly higher IFN‐α secretion than MVA. The absolute level of IFN‐α secretion, however, was lower than in PBMCs from healthy donors.
In summary, we showed that PCPV (i) efficiently induces secretion of IFN‐α in PBMCs from both healthy donors and cancer patients; (ii) infects APCs, leading to upregulation of co‐stimulatory and antigen‐presenting molecules; (iii) infects PBMCs, leading to activation and degranulation of NK cells; (iv) induces little or no toxicity towards activated T cells; and (v) impedes the suppressive activity of MDSCs on T cells.
PCPV controls tumor growth and induces an adaptive antitumor response
To assess whether these remarkable features observed in vitro translate into in vivo antitumor activity, PCPV was tested in the MC38 syngeneic model. After subcutaneous (sc) inoculation of colon adenocarcinoma cells, 1 × 107 plaque‐forming units (pfu) of either PCPV, MVA or buffer were applied at the same site at days 1, 7 and 14 during tumor expansion. In contrast to MVA, administration of PCPV induced tumor regression in half of the mice (5 of 10 mice) (Figure 3a) and increased survival rates (Figure 3b). To investigate the role of T cells in these therapeutic effects, CD8+ T cells were depleted in vivo by an anti‐CD8a antibody, the day before inoculation of MC38 cells, and the days before the second and third virus injection. Depletion of CD8+ T cells reduced the survival of PCPV‐treated animals (Figure 3c). Splenic lymphocytes from survivors were stimulated by mitomycin‐treated MC38 cells, or by a non‐related peptide as a negative control. Strikingly, ELISpot analysis showed the induction of MC38‐specific responses in these animals despite the absence of any known tumor antigen in the PCPV vector (Figure 3d).
Figure 3.
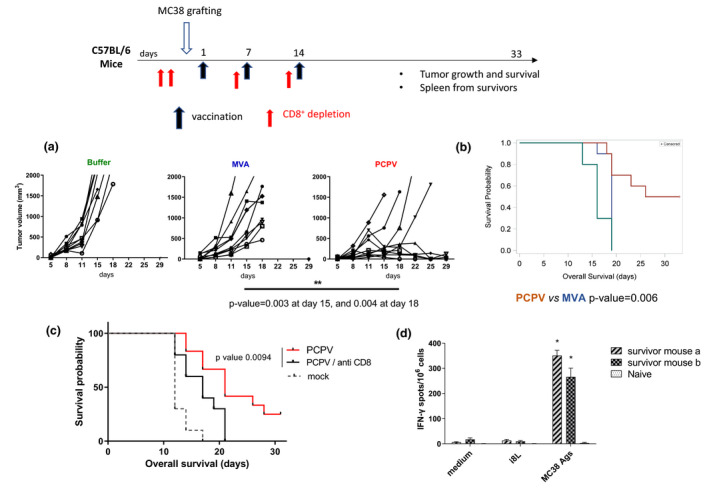
PCPV primes specific T‐cell response without encoding tumor antigen. (a) At day 0, the colon carcinoma cell line MC38 (2 × 106 cells) was injected subcutaneously in C57BL/6 mice (10 mice per group). At days 1, 7 and 14, 1 × 107 pfu of either PCPV‐E7 or MVA‐E7 (E7 being an irrelevant antigen in MC38 tumor model), or buffer was injected at the cell line injection site and later in the emerging tumors. Tumor growth evolution was statistically analysed with a repeated mixed model built on the calculated tumor diameter as response. (b) Survival: Groups were compared with a log‐rank test. (c) CD8 cell depletion reduced survival in the MC38 model (10 mice per group): 200 µg of a CD8‐depleting antibody (clone 53‐6‐7) was injected intraperitoneally at days −2, −1, 6 and 13. One of two experiments is shown. (d) IFN‐γ ELISpot on splenic lymphocytes from survivors: cells were stimulated with either culture medium, a non‐related peptide (I8L) or MC38 cells treated with mitomycin C. Means of quadruplicates with SD are shown.
Viability of MC38 cells infected with PCPV at various MOIs was assessed in vitro (Supplementary figure 8a). No signs of cytotoxicity during the 5 days of observation were observed, whereas the oncolytic vector VACV efficiently killed MC38 cells (Supplementary figure 8a). This suggests that direct tumor cell lysis was not the main mechanism involved in PCPV antitumor effects.
Assessment of the immunogenicity of recombinant PCPV‐ HPV16 E7(Δ21‐26)
A PCPV‐based vaccine encoding the non‐oncogenic form of the E7 protein, deleted for amino acids 21–26, of human papillomavirus type 16 (HPV16) was prepared. PCPV‐E7 or MVA‐E7, or the recombinant E7 protein adjuvanted with poly(I:C), was injected intravenously (iv) in naïve mice at days 1 and 8. ‘Empty’ MVA was included as control. Mice were sacrificed at day 16, spleens were pooled, and splenic lymphocytes were prepared for ELISpot analyses. PCPV‐E7‐vaccinated mice showed a significant increase in the number of E7‐specific T cells (stimulation with peptide R9F), and the response was comparable to that observed after vaccination with the adjuvanted E7 protein (Figure 4a). The effect of the latter might be underestimated since protein and adjuvant might dissociate in the bloodstream before being ingested by antigen‐presenting cells. MVA‐E7 proved to be more effective than PCPV‐E7 by a factor of 2 (P‐value = 0.0286). MVA vaccines with therapeutic efficacy in preclinical tumor models were shown previously to induce the appearance of a CD3dimCD8dim T‐cell population in the lungs upon repeated iv injection. 34 Antigen (R9F)‐specific CD8+ T cells were exclusively detected within this T‐cell population consisting of short‐lived effector cells (SLECs) and early effector cells (EECs). In the present study, we confirm the appearance of the CD3dimCD8dim T‐cell population after MVA injection (Figure 4b). This population was also observed in PCPV‐treated animals, although to a lesser extent. The proportion of gated CD3dimCD8dim cells was higher after MVA injection than after PCPV injection, respectively, 24% ± 2 (SEM) for MVA, 5% ± 0.2 (SEM) for PCPV and 1% ± 0,1 (SEM) for buffer. However, the absolute numbers of antigen (R9F)‐specific IFN‐γ‐secreting CD8+ T detected in this subpopulation were comparable after treatment with either vaccine. Thus, a significantly higher percentage of antigen‐specific CD8+ T cells was observed in the PCPV‐E7 group (Figure 4c).
Figure 4.
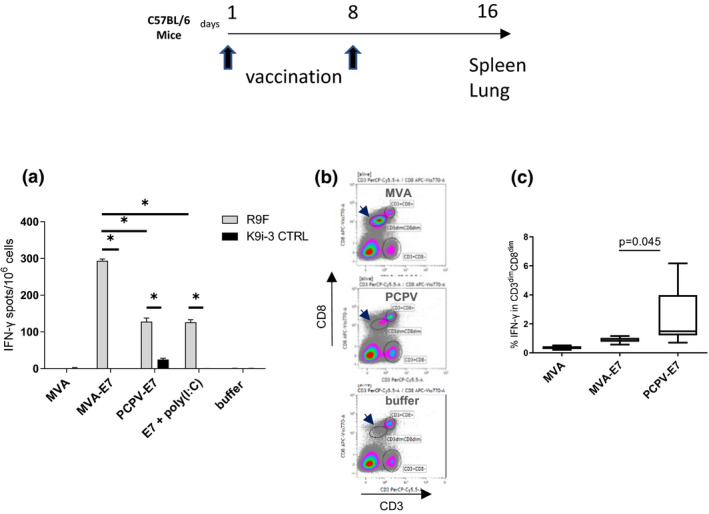
Immunogenicity of PCPV‐E7 in naïve mice. The immunogenicity of PCPV‐E7 was compared with the MVA vector and adjuvanted E7m protein. 5x105 pfu of either PCPV‐E7, MVA‐E7, empty MVA, or 50 µg of recombinant E7m protein adjuvanted with 100 µg LMW poly(I:C) was injected intravenously at days 1 and 8 in C57BL/6 mice (6 mice per group). At day 16, mice were sacrificed, and spleen and lungs were harvested. (a) ELISpot (IFN‐γ) on pooled splenic lymphocytes: antigen specificity was probed with the HPV16E7‐specific peptide R9F and the non‐related peptide K9i‐3C as a negative control. Mean of quadruplicates with SD is shown. (b) Appearance of the CD3dimCD8dim population in lung: this new T‐cell population appeared after repeated injection of MVA, but much less for PCPV. The percentage of gated CD3dimCD8dim population reached 24% ± SEM 2 (MVA), 5% ± SEM 0.2 (PCPV) and 1% ± SEM 0.1 (buffer). (c) Detection of IFN‐γ‐secreting T cells in the lungs of mice vaccinated with either empty MVA, MVA‐E7 or PCPV‐E7. Intracellular cytokine staining allowed the analysis of the percentage of R9F‐specific IFN‐γ‐secreting cells within the CD3dimCD8dim population of individual lung preparations. The CD3+CD8+ population did not show increased IFN‐γ secretion (data not shown). Groups were compared by the Wilcoxon test (equivalent to the Mann–Whitney U‐test). Results are represented as whisker plots: Min to Max.
Heterologous vaccination leads to enhanced TC1 tumor regression and protective immunity
We tested PCPV‐E7 in heterologous prime‐boost schedules with MVA‐E7 in the HPV syngeneic TC1 tumor model (Figure 5a). Injection of PCPV‐E7 into palpable tumors, followed by systemic iv injection of MVA‐E7, led to a significant increase in the survival rate (Figure 5b), often accompanied by complete tumor regression (Figure 5c). The effect of the heterologous prime‐boost strategy was significantly stronger than the homologous prime‐boost treatment with MVA‐E7. Survivors having completely rejected the tumors after heterologous vaccination were frequent: from 10 treated animals, 8 survived, and 6 completely resolved the tumor. One example is shown (Figure 5d). Survivors having completely resolved their TC1 tumor after heterologous prime‐boost treatment were rechallenged with TC1 cells on both flanks, 37 days after last virus injection. Six of six animals were completely protected from tumor growth during 36 days of observation, while all 6 animals in the control group of naïve animals developed tumors on both sides (Figure 5e). These results suggest that systemic antitumor immunity was generated.
Figure 5.
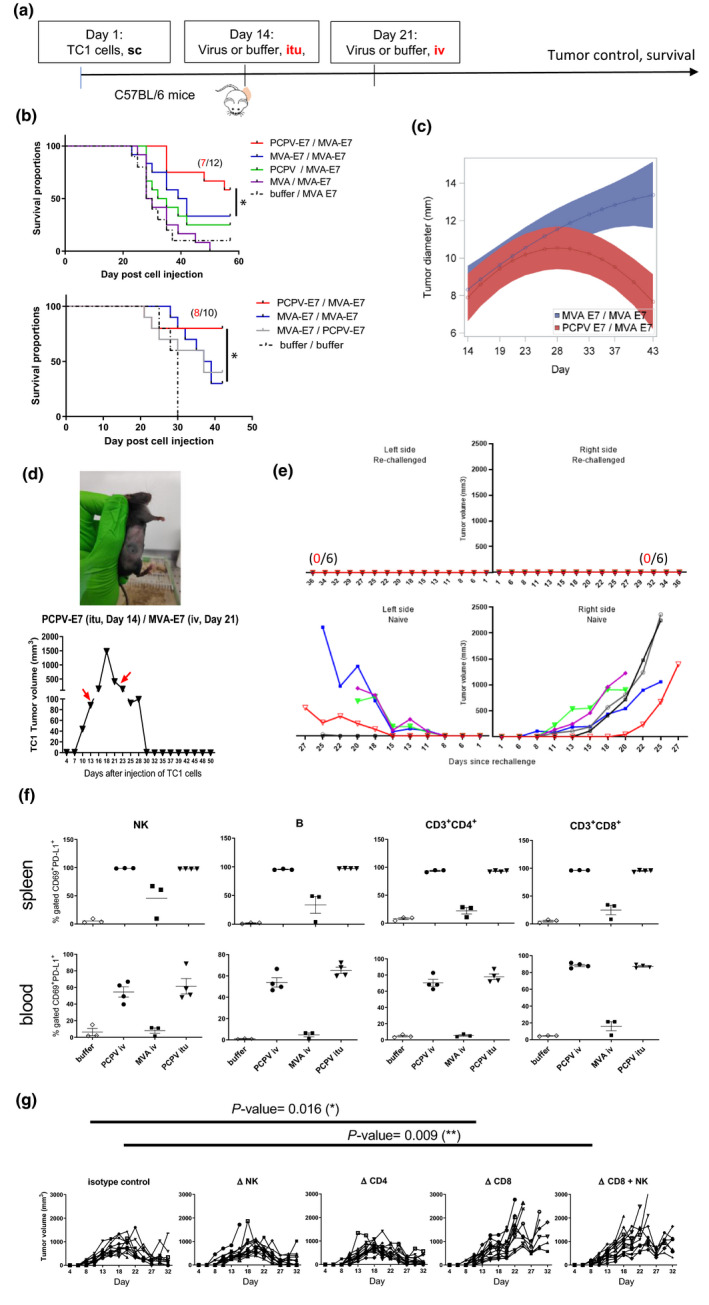
Specific antigen vaccination in the syngeneic TC1 model. (a) C57BL/6 mice (12 mice per group) were subcutaneously injected with tumor cells (5 × 105 cells). When tumors became palpable at day 14, 5 × 106 pfu of either MVA‐E7, PCPV‐E7, empty MVA or PCPV was injected into the growing tumors. One time, 106 pfu of either MVA‐E7 or PCPV‐E7 was used for the second vaccination by the intravenous route at day 21. (b) Survival data from two independent experiments are shown. Treatment effect on oversurvival was analysed using a Cox regression model for each experiment to produce a meta‐analysis. Hazard ratios and associated standard error were estimated, and meta‐analysis was performed using a fixed effect model by weighting estimation with the inverse variance. A mixed model with the treatment group, day, experiment and interactions of these terms including quadratic effect of Day was built with pooled data to estimate the differences between groups over time. The comparison between heterologous prime boost with PCPV‐E7 / MVA‐E7 vs MVA‐E7 / MVA‐E7 showed a significant difference: the P‐value was 0.0052. (c) Evolution of tumor diameter for PCPV‐E7 itu/MVA‐E7 iv (red) versus MVA‐E7 itu/MVA‐E7 iv (blue), analysed with mixed model for interaction effect day² × group, the P‐value was 0.001. (d) Example of one mouse having completely rejected a TC1 tumor after heterologous prime‐boost treatment: PCPV‐E7 was injected intratumorally at day 14 into a tumor of 90 mm3, the volume of the tumor continued to grow to 1500 mm3 until day 18, then started to regress. MVA‐E7 was injected intravenously at day 21. At day 30, tumor had completely disappeared. (e) Mice, having completely rejected TC1 tumors after heterologous prime‐boost treatment with PCPV‐E7 and MVA‐E7, were challenged with TC1 tumor cells (5 × 105), injected in both flanks. Tumor growth was monitored over 36 days (6 mice per group; upper panel: rechallenged mice, lower panel: control mice). (f) Activation status of immune cells in spleen and blood: Three or four TC1 tumor‐bearing mice were injected intravenously with 1 × 106 pfu of either MVA‐E7 or PCPV‐E7, or intratumorally with 5 × 106 pfu of PCPV‐E7. The next day, splenocytes and blood cells were analysed by flow cytometry. The percentages of CD69 and PD‐L1‐double‐positive NK, B and CD4+ or CD8+ T cells were measured. Shown are individual results per mouse and mean ± SEM per group. (g) Effect of depletion of CD8, CD4 or NK on TC1 tumor growth in mice treated with PCPV‐E7 intratumorally / and MVA‐E7 intravenously (10 mice per group). 200 µg of anti‐CD4+ (clone GK1.5) and anti‐CD8+ antibodies (clone 53‐6.7), or the isotype control for CD8 (clone 2A3), was injected intraperitoneally at days 12, 13, 20 and 28 after injection of TC1 cells. Treatment with 150 µg of the NK‐depleting antibody NK1.1 (clone PK136) was administered intraperitoneally at days 11, 13 and 15, and then twice per week. Tumor diameter evolution in individual mice is shown.
The efficacy of the heterologous regimen (PCPV‐E7/MVA‐E7) might have benefited from the absence of cross‐reactive neutralising immunity. It was reported that individuals infected with cowpox virus, closely related to VACV, did not develop immunity against PCPV and vice versa. 35 In support, we have shown that patient sera containing neutralising antibodies to MVA and VACV did not impede infection with PCPV (Supplementary table 1). Furthermore, repeated treatment of mice with PCPV did neither generate detectable levels of neutralising antibodies against PCPV (Supplementary figure 9a) nor against MVA (Supplementary figure 9b). This observation, however, could not explain why the survival rate was not improved when MVA‐E7 was injected first, and PCPV‐E7 second (Figure 5b). To assess whether a difference in the innate immune stimulation could be causal, we measured the activation status of various immune cells in TC1 tumor‐bearing animals the day after virus injection, considering CD69 and PD‐L1 as activation markers 36 , 37 (Figure 5f). All NK, B and T cells in the spleen, and 60–90% of these cell populations in the blood, were activated after PCPV treatment whatever the administration route, intravenously (iv) or intratumorally (itu). This was far larger than after MVA treatment. These data suggest that in vivo, the PCPV vaccine is a more potent immune stimulator than the MVA vaccine. This hypothesis was further supported by a trend for better efficacy of empty PCPV as a priming agent than that of empty MVA (Figure 5b). The consequence of depleting NK, CD8+ or CD4+ T cells on the therapeutic outcome was measured. Depletion of CD8+ T cells abolished tumor control, while the depletion of CD4+ T cells had no effect (Figure 5g). Treatment with the NK1.1 NK‐depleting antibody had no effect on the therapeutic outcome. Depletion of both CD8+ T cells and NK cells induced a reduction in tumor control, which was comparable to the treatment with anti‐CD8 alone. Of note, the complete loss of CD8+ and CD4+ T cells in the spleen was confirmed at the day of virus injection, that is days 14 and 21, but NK depletion proved incomplete (Supplementary figure 10). Overall, the therapeutic vaccination schedule consisting of PCPV‐E7 injection followed by MVA‐E7 led to efficient tumor control, often associated with tumor regression. This effect was abolished by depletion of CD8+ T cells. Surviving animals were protected against a tumor rechallenge. Furthermore, PCPV administration led to a much stronger systemic activation of NK, B and T cells than that observed with MVA.
After homologous treatment with PCPV‐E7/PCPV‐E7, several TC1 tumor‐carrying mice died. This contrasted with observations of good tolerability of repeated PCPV iv injection in naïve mice or MC38 tumor‐bearing animals (itu) shown above. Equally well tolerated was PCPV encoding beta‐galactosidase in a homologous prime‐boost approach in the beta‐galactosidase‐positive CT26.CL25 model (Supplementary figure 11). IV injection of PCPV‐bgal first, followed by sc injection of the same virus, led to the best survival proportions equalled only by heterologous treatment with PCPV‐bgal/MVA‐bgal (Supplementary figure 11, MVA‐bgal, schematically depicted in Supplementary figure 2b).
Safety implications of PCPV constructs
PCPV development as a cancer vaccine must be accompanied by a parallel predictive safety testing in various animal species. The biodistribution of PCPV‐GFP, injected iv at 1 × 107 pfu in naïve immunocompetent and immunodeficient BALB/c mice, was studied. The presence of viruses was analysed on days 7, 14 and 21 post‐injection in liver, spleen, brain, lung, kidneys, ovaries, lymph nodes, heart, skin and blood, by a plaque‐forming assay on permissive BT cells. While virus titres in most organs were undetectable or low, they were higher in the spleen (about 150 pfu/mg of tissue). The titres persisted between days 7 and 14 and decreased afterwards (Supplementary figure 12a). It is difficult to know whether the presence of the virus in the spleen is because of low replication or retention in this organ. All mice were well during the experiment; no decrease in weight was observed (Supplementary figure 12b).
In a dose tolerability study in C57BL/6 mice, conducted according to Good Laboratory Practices (GLP), PCPV‐E7 was administered intravenously on days 1, 4 and 8, at 0 (vehicle), 5 × 105, 5 × 106 and 1 × 107 pfu. Furthermore, the 5 × 106 pfu intermediate dose was also tested in combination with TG4001 (MVA‐HPV16‐E6‐E7), administered at 1 × 106 pfu on day 14. Animals were observed up to day 30. The following endpoints/parameters were evaluated: body weight, food and water consumption, clinical observation, haematology, clinical chemistry, organ weights and gross pathology and histopathology examination. The results are summarised in Supplementary table 2. These treatment‐related effects were not considered to be a human health risk. Both series of mouse experiments showed safety of PCPV vectors. Based on these findings, the highest non‐severely toxic dose (HNSTD) was determined as > 1 × 107 pfu.
PCPV‐E7 activates E7‐specific T cells from HPV16+‐infected cancer patients
PCPV as a therapeutic vaccine candidate was challenged in a translational setting in HPV16+‐infected HNSCC cancer patients. The goal was to test whether PCPV‐E7 could amplify HPV16E7‐specific T cells from patients’ tumor‐draining lymph nodes (TDLNs). We used the MVA‐E7 vaccine as control for some assays, encoding HPV16E7 under the control of p7.5K as is the case in TG4001, which has already been evaluated in humans. 1 , 20 , 21
We quantified the viral‐mediated cytotoxicity on TDLN cells, at different MOIs. Despite variability among patients, cytotoxicity was higher for MVA than for PCPV in all tested samples (Figure 6a). In fact, toxicity increased for MVA beyond a MOI of 1, while TDLN cells survived at similar MOI for PCPV, as well as for control or soluble E7 protein. For further experiments, PCPV was used at a MOI of 3, E7‐encoding vectors behaving similar to ‘empty’ vectors (Supplementary figure 13a).
Figure 6.
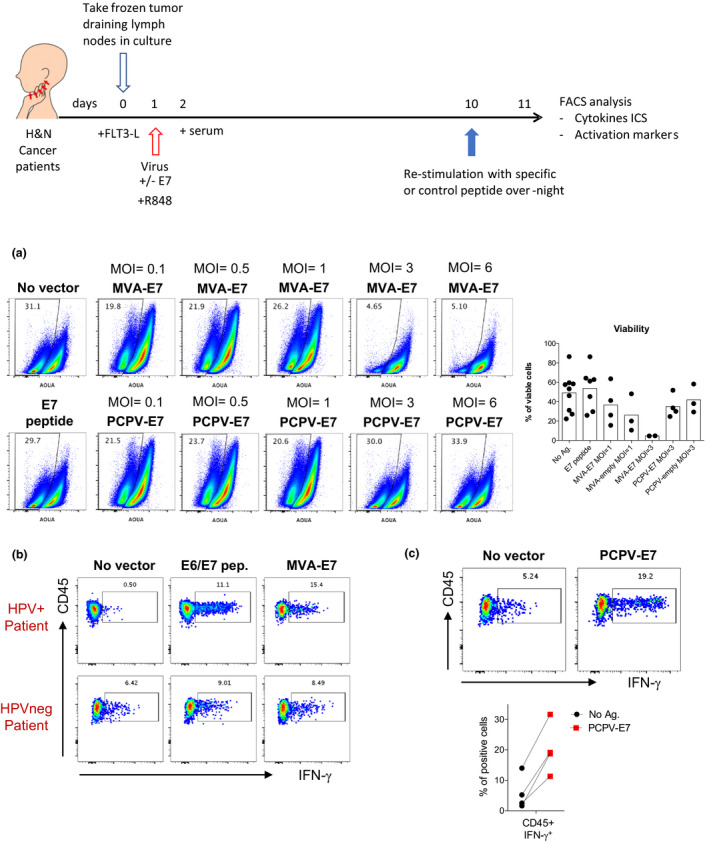
PCPV‐encoded HPV16E7 activates E7‐specific T cells from cancer patients. (a) Total suspensions of immune cells from TDLN of HNSCC patients were incubated with different MOIs of MVA‐E7 and PCPV‐E7 vectors for 10 days. Pseudocolor plots and graphics show quantification of live cells by gating on live cells in distinct conditions. (b, c) Total cell suspensions of immune cells from TDLN of HNSCC patients were submitted to an expansion protocol during 10 days in the presence of E6/E7 synthetic peptides, MVA‐E7 (MOI = 1) (b) or PCPV‐E7 (MOI = 3) (c). Cells were subsequently restimulated by synthetic E7 peptides overnight, and IFN‐γ production was evaluated by flow cytometry among live CD45+CD3+CD8–HLA‐DR+CD25+PD1high‐activated cells. In c, the graph depicts 4 independent assays.
Best culture conditions were set up to test whether the antigens encoded by the viral vectors could effectively induce specific T‐cell responses upon infection of TDLN cells. First, we confirmed that the E7 protein was correctly produced in MVA‐E7‐infected human monocytes (Supplementary figure 13b). MVA‐E7 was then used to infect TDLN cells obtained from head and neck squamous cell carcinoma (HNSCC) patients, either HPV16+ or HPV–, in an in vitro assay that we adapted from Lissina et al. 38 (Table 1). Briefly, total TDLN cells were cultured in the presence of Flt3L, and the TLR7/8 agonist R848, to induce the differentiation and maturation of endogenous APCs present in the unfractionated TDLNs. These cells were then infected with the virus vectors for 9 days and, at day 10, restimulated with the E7 peptide for 12h before analysing cytokine production and cell phenotype. As observed in Figure 6b and Supplementary figure 13 c, higher frequencies of IFN‐γ‐producing T cells were detected in HPV+‐infected than in HPV–‐infected patients, suggesting that the response was specific to the antigen encoded in the viral vector. As previously reported, the responding T cells were predominantly PD1high. 39 , 40 , 41 Our culture conditions allowed us to conclude that the viral‐encoded E7 gene is effectively translated, processed and presented by the endogenous APCs present in the TDLN sample, inducing detectable antigen‐specific T‐cell responses.
Table 1.
Clinical, biological and pathological characteristics of the HNSCC patients
| Age (years) | Gender | HPV type (by PCR) | Location | Previous treatment | pTNM (8th edition AJCC) |
|---|---|---|---|---|---|
| 52 | M | 16 | Oropharynx : Tonsil | No | pT3N0 |
| 64 | M | 16 | Oropharynx : Tonsil | No | pT2N1 |
| 60 | M | 16 | Oropharynx : Tonsil | No | pT2N1 |
| 54 | M | 16 | Oropharynx : Tonsil | No | pT1N1 |
| 62 | M | 16 | Oropharynx : Tonsil | No | pT1N2 |
| 55 | M | 16 | Oropharynx : Tonsil | No | pT2N1 |
| 71 | M | 16 | Oropharynx : Tonsil | No | pT2N1 |
| 51 | M | Negative | Oropharynx : Tonsil | No | pT2N0 |
| 51 | M | Negative | Oropharynx : Tonsil | No | pT0N1 |
The responding cells were predominantly CD3+CD8– T lymphocytes, while the CD3+CD8+ T‐cell response was inconsistently detected (Supplementary figure 13c). To determine whether our in vitro culture conditions could also sustain antigen‐specific CD3+CD8+ T‐cell responses, we tested other viral antigens in the TDLN assay. High frequencies of responding CD3+CD8+ T cells were induced by peptide antigens from cytomegalovirus (CMV), Epstein–Barr virus (EBV) and influenza A virus (FLU) (Supplementary figure 13d).
We next studied the ability of PCPV to induce T‐cell responses specific to virus‐encoded antigens. As observed for MVA, the E7 protein was well produced in human monocytes infected with the PCPV‐E7 (Supplementary figure 13b). As shown in Figure 6c, PCPV‐7 was able to induce the expansion of E7‐specific T‐cell responses, as evidenced by the higher frequencies of IFN‐γ‐producing CD3+CD8– PD1high T cells at the end of the culture than that of uninfected cells from the same patient. Interestingly, infection with the empty PCPV vector also induced a detectable response with higher frequency of IFN‐γ‐producing CD3+CD8– PD1high T cells than that of the uninfected control (Supplementary figure 13c).
Overall, these results show that PCPV was able to induce an antigen‐specific T‐cell response after infection of TDLN cells from cancer patients, confirming that the antigen encoded in the virus is correctly expressed upon infection and presented to T cells, together with the appropriate stimuli to allow activation/recall of an immune response.
Discussion
MVA has an unmatched safety and efficacy record as smallpox vaccine; its activity as a cancer vaccine, however, needs improvement. One attempt for improvement was the deletion of remaining immunomodulatory genes, for instance the IL‐18 binding protein. The resulting vector, though, did not increase antitumor activity, 42 and the massive deletion of clusters of fifteen immunomodulatory genes did not improve the immunogenicity of the resulting MVAs. 43 However, recent reports underlined that reinforcement of the type I IFN responses could be a set screw to improve antitumor immune responses. 15 , 44 , 45 These findings supported retrospectively our approach to select PCPV based on its capacity to stimulate IFN‐α secretion in human cells. It distinguishes between MVA and VACV by its higher capacity to induce secretion of IFN‐α in PBMCs not only from healthy donors but also from cancer patients. Compared with healthy donors’ PBMCs, cells from cancer patients, exposed to the same viruses, secreted less IFN‐α, suggesting the need for strong stimulation. We tried to understand the difference in IFN induction capacity between PCPV and VACV from the analysis of their genomes. Smith et al. reviewed the various immune evasion strategies of Vaccinia, highlighting the fact that about one third of the genetic coding capacity of Vaccinia is devoted to immunomodulatory proteins. 24 Among them, more than 10 viral proteins act upstream or downstream of the IFN pathway. However, the analysis of the sequence of the reference PCPV strain VR364 (NCBI entry NC_013804.1) identified fewer interferon‐related proteins. 46 The only annotated gene was ORF20, an ortholog for the VACV dsRNA‐binding protein E3L, but with only 26% sequence identity (UniProt D3IZG7 versus P21081; not shown). We found no PCPV orthologs for the soluble IFN‐receptor proteins B8R or B18R that are shared in VACV and MVA. A more extensive analysis would be necessary to understand the underlying mechanisms of the high IFN induction capacity of PCPV.
PCPV is of bovine origin but infected human primary cell subsets, as well as MVA and VACV. The main producers of IFN‐α in PCPV‐treated PBMCs were pDCs and monocytes. This pattern was also described for the coxsackievirus A21 (CVA21). 47 We could not yet demonstrate infection of pDCs by PCPV, while some studies have shown that myxoma virus 48 and Coxsackievirus may infect pDCs, the latter via the ICAM receptor. An indirect stimulation of pDCs, for example via the exchange of microvesicles containing viral nucleic acids, could be an alternative explanation, especially, since the GC‐rich genome of PCPV has been proposed to be a ligand for the TLR9 receptor, well expressed in pDCs. 49
PCPV induced not only IFN‐α but also led to the activation of human APCs and NKs, and reduced apparent suppressive effects of MDSCs. In preclinical models, intratumoral injection of PCPV‐E7 induced antigen‐specific T cells against tumor‐presented antigens / tumor cells, and vector‐encoded epitopes. In an ex vivo translational setting using tumor‐draining lymph node cells from cancer patients, we also noted activation of antigen‐specific T cells after exposure to PCPV‐E7.
In the human MDSC/CD8+ T‐cell proliferation assay, PCPV unleashed T‐cell proliferation. The underlying mechanism could be that PCPV efficiently killed suppressive MDSC cells or reprogrammed them. 50 Type I IFN response could be implied in the activation of the STAT3–granzyme B pathway in co‐cultivated T cells. 17 The effect of poxviral vectors on activated T cells was studied with the perspective of intratumoral injection of the virus, which implies an encounter with tumor‐resident and tumor antigen‐specific T cells. Inspired by Reinhard et al. 2 we might consider combination with recombinant T‐cell technologies. For these reasons, we probed the effect of our vectors on activated human T cells in vitro. We confirmed that activated T cells were readily infected by VACV and MVA 29 ; the infected cell cultures perished. This negative effect was not observed in PCPV‐treated cultures, most likely since the virus poorly infected activated human T cells. For this reason, we hypothesise that PCPV could be a better cancer vaccination vector after intratumoral injection, and of special interest for combination therapies with activated T‐cell therapies.
PCPV‐induced maturation of antigen‐presenting moDCs was superior to what was observed with orthopoxviruses and, as expected, inferior to stimulation with the TLR ligand R848. This difference might be explained by stoichiometric differences in available ligands (excess of small molecules), and virus‐induced shutdown of cellular protein synthesis. Thus, a combination with TLR ligands represents an interesting option for vaccination with PCPV, like, for example, the combination with synthetic TLR‐based neovaccines. 51 , 52 PCPV induced the activation of NK cells within PBMCs, leading to degranulation in contact with target cells, a potent innate immune response leading to cancer cell killing. 47
Vaccination of naïve mice with PCPV‐E7 allowed detection of E7‐specific splenic lymphocytes and T cells in the lung, confirming that the antigen expression levels obtained in vivo were sufficient to induce adaptive immune responses against the vector‐encoded antigen. Also in mice, treatment with PCPV strongly activated NK cells, which most likely contribute to tumor destruction, cancer antigen release and generation of tumor‐specific T‐cell responses. This indirect mechanism of antigen‐specific immune activation could explain, at least in part, the PCPV‐mediated induction of antigen‐specific immunity in the MC38 tumor model. Further, PCPV‐induced type I interferon secreted in MC38 and TC1 tumor could be involved in the activation of MHC class I‐dressed CD11b+ conventional dendritic cells to promote protective antitumor CD8+ T‐cell immunity. 15
Complete tumor regression and increased survival were observed in the syngeneic TC1 model after heterologous treatment with PCPV‐E7 and MVA‐E7. Besides the expected antigen‐specific CD8+ T‐cell response, indispensable for the observed effect, we observed stronger activation of B cells and stimulation of NK cells than after treatment with MVA, which could explain the higher efficacy when PCPV was injected first in the tumor than MVA. Repeated PCPV treatment did not induce detectable levels of PCPV‐neutralising antibodies in mice, and PCPV was not neutralised by VACV‐specific human‐neutralising antibodies. The use of non‐cross‐reactive vectors in heterologous prime‐boost regimen PCPV‐E7/MVA‐E7 might be important to maintain efficient infection and antigen expression. Further, the combination of different viruses might also help to avoid subversion of immune response towards weaker tumor antigens.
In ex vivo studies using TDLN from cancer patients, PCPV‐E7 and MVA‐E7 stimulation resulted in activation/reinvigoration of E7‐specific T cells. The higher induction of co‐stimulatory molecules in APCs, and the lower toxicity towards activated T cells could underlie the higher efficacy of PCPV‐E7. Interestingly, without encoding the specific antigen, wt PCPV induced the production of IFN‐γ by T cells, at close levels to those generated by PCPV‐E7. This could be attributed to its strong ‘Signal 2’ effects, inducing upregulation of co‐stimulatory molecules and cytokine production by the antigen‐presenting cells. In addition, our ex vivo experimental settings included an overnight restimulation using synthetic commercial E7 peptides for all tested groups. This step may reactivate residual T‐cell clonotypes that survived during cultures and are able to recognise/cross‐react the E7 peptides. However, further analysis of APCs and the characterisation of cytokine released from TDLN cells need to be performed considering the distinct subsets of immune cells that populate these tissues.
PCPV‐E7 induced a strong T‐cell response in TDLN cells of HPV+‐infected patients, which was predominantly mediated by CD3+CD8– T cells. In support, a comparison of different poxviruses coding HIV antigens showed different T‐cell predominance among vectors, and polyfunctional CD4+ T‐cell responses were as efficient as CD8+ T cell upon viral challenge in a non‐human primate model. 53 Concerning the nature of the antigen, analysis of PBMCs from HPV+‐infected oropharyngeal cancer patients detected both CD4+ and CD8+ IFN‐γ‐producing T cells upon stimulation with peptide libraries spanning the entire length of the E7 protein. 54 Moreover, analysis of TILs adoptively transferred into HPV+‐infected cervical cancer patients who underwent complete tumor regression revealed that T cells reactive to E7 were mainly CD4+PD1hi. 41 Similar results were observed for neoantigen‐specific TILs: tumor progression after adoptive transfer therapy was controlled exclusively by CD4+ T cells. 55 Finally, several teams have reported that, despite the use of MHC class I binding prediction algorithms, neoantigen peptide or RNA vaccines elicit predominant CD4+ T‐cell responses. 56 , 57 , 58 Whether these CD4+ T cells represent cytotoxic CD4+ T cells is still unclear. Along these lines, it has been suggested that CD4+ T cells could be involved in antitumoral responses. 59 , 60
Besides the vectorisation of tumor‐specific antigens, PCPV was shown to induce significant antigen‐independent activation of innate immunity, activation of APCs and NK cells and the shutdown of immunosuppressive mechanisms. These properties can be of therapeutic interest. For instance, PCPV could be used during priming to convert cold tumors into hot tumors, prior to the administration of other therapies such as immune checkpoint inhibition or vaccination, as shown for intratumoral administration of rotavirus. 61 Also, PCPV pretreatment might sensitise the tumor microenvironment to immune checkpoint treatment as described in Zemek et al. 62 PCPV hardly infects human‐activated T cells. Therefore, intratumoral injection of PCPV can be considered to be a safe strategy, sparing activated tumor‐resident T cells. In vitro observations allow hypothesis that suppressive cells such as MDSCs are readily infected and destroyed by the virus, thus allowing T cells to resume proliferation and cytotoxicity. PCPV could be used for prime vaccination of patients awaiting their personalised neoepitope vaccines, a time frame that could be profited as ‘time to prime’. Along this line, our data put forward that empty PCPV is capable of effectively restimulating HPV16E7‐specific T cells (Supplementary figure 13e). Furthermore, PCPV armed with viral epitopes (e.g. E7 or others) could be useful to reactivate virus‐specific memory T cells within the tumor, as recently proposed for cancer immunotherapy. 63
Before PCPV could become an important player in cancer vaccination in the clinic, regulatory standards must be met. The vector is currently undergoing preclinical safety testing. In a case study, infection of humans with PCPV was benign 27 and so were the frequent infections and reinfections of workers in the meat industry with the related parapoxvirus orf. 64 No fatal outcome is described in the literature.
To our knowledge, the use of PCPV as a viral vector in clinical applications has not been published so far. In the case of proving its safety, PCPV could become a powerful tool as vaccine backbone and versatile immunomodulator, proposing interesting options for combination therapies in cancer, and for prophylactic vaccination in heterologous prime‐boost regimens.
Methods
Experimental design
The primary objective of the study was to identify a poxviral backbone with improved innate and adaptive immune stimulation. The selection criterion, highest IFN‐α secretion levels in infected PBMC cultures, was met by PCPV. The expected innate immunostimulatory capacity of this virus was confirmed in various immune activation assays using cells from several donors. Human primary immune cells were obtained from healthy donors from the Etablissement Français du Sang (EFS), Strasbourg, France. Efficient IFN‐α secretion was confirmed in cohorts of healthy donor and cancer patients’ PBMCs. Immunogenicity of PCPV‐encoded antigens and antitumor activity of virus treatment were determined in mice. Mouse group sizes were based on data from previous experiments; mice were randomly distributed between groups. Then, the viability and reactivation of antigen‐specific T cells within ex vivo cultures from draining lymph nodes of cancer patients were tested. Group sizes were limited by the availability of patient material. Replication of experiments and the number of biological and technical replicates varied between experiments as described in the figure captions.
Viruses
For the selection approach, cowpox virus strain Brighton (VR‐302™) (CPV), raccoonpox virus strain Herman (VR‐838™) (RCNV), orf virus strain NZ2 (VR‐1548™) (ORFV), pseudocowpox virus strain TJS (VR‐634™) (PCPV), myxoma virus strain Lausanne (VR‐115™) (MYXV), Yaba‐like disease virus (VR‐937™) (YLDV), swinepox virus strain Kasza (VR‐363™) (SWPV), Cotia virus strain SP AN 32 (VR‐464™) (CTV) and rabbitpox virus strain Utrecht (VR1591™) (RPV) were obtained from American Type Culture Collection (ATCC, Rockville, USA) and were produced on HeLa cells except for SWPV produced on ESK‐4 cells. 65 The Fowlpox virus strain FP9 (FPV) was kindly provided by Professor Skinner (Imperial College London, UK) and was produced on chicken embryonic fibroblasts (CEFs).
MVATGN33.1 (empty MVA) and MVA‐GFP have been described previously. 66 , 67 MVA‐E7 is a research surrogate of the cancer vaccine TG4001, 20 devoid of HPV16‐E6 and IL‐2 expression cassettes present in TG4001. All VACVs were derived from the Copenhagen strain. Empty VACV (VVTG18058) and VACV‐GFP expressing the GFP gene under the control of the p11K7.5 promoter were deleted in thymidine kinase (J2R) and in the large subunit of ribonucleotide reductase (I4L) genes. 68 MVA and VACV vectors were generated and amplified on CEF as described. 67
Cell lines
The murine lung tumor cell line TC1, co‐transformed with human papillomavirus 16 (HPV16) E6/E7 and c‐Ha‐Ras, the human erythroleukaemia cell line K562, and the bovine Bos Taurus turbinate (BT) cells were obtained from the ATCC. The murine colon cancer cell line MC38 was kindly provided by Dr James W Hodge (NIH, USA). MC38 cells were cultivated in Dulbecco’s modified Eagle’s medium (DMEM, Gibco), supplemented with 2 mM L‐glutamine (Sigma‐Aldrich, Saint Louis, MO, USA), 40 mg L−1 gentamicin (Sigma‐Aldrich, Saint Louis, MO, USA) and 10% FCS. TC1 cells were maintained in the same medium, and further supplemented with 500 µg mL−1 G418 (Gibco‐Aldrich) and 200 µg mL−1 hygromycin B (Roche, Mannheim, Germany). K562 cells were cultured in ISCOVE’s Modified Dulbecco’s Medium (IDMEM) (Gibco) supplemented with 10% foetal calf serum (FCS, Gibco, Life Technologies, CA, USA). BT cells were cultivated in DMEM supplemented with 10% foetal horse serum (HyClone, Logan, USA), 2 mM L‐glutamine and 40 mg L−1 gentamicin. Regular mycoplasma testing was carried out for all cell lines (Clean Cells, Vendée, France).
Genetic engineering, production and purification of PCPV vectors
Recombinant PCPV vectors were constructed from the wild‐type strain TJS by insertion of expression cassettes by homologous recombination in the non‐essential VEGF gene. The VEGF gene is present in two copies in the PCPV genome on the left and right genome termini. The transfer plasmid used for homologous recombination at the VEGF locus was generated by inserting two sequences of about 300 bp flanking the PCPV VEGF gene in the pUC18 plasmid. The sequences upstream of the VEGF gene correspond to nucleotide position 6391 to 6089 or 138899 to 139201 of PCPV (GenBank NC_013804). The sequences downstream the VEGF gene correspond to nucleotide positions 5545–5225 or 139745–140065 of PCPV.
Generation of PCPV‐GFP (PCPTG19106)
The selection cassette (eGFP/GPT), a fusion of the gene encoding the enhanced green fluorescent protein (eGFP) and the gene encoding the E.coli xanthine‐guanine phosphoribosyl transferase (GPT), 68 was positioned under the control of the p11K7.5 vaccinia promoter and inserted in the transfer plasmid leading to pTG19106. PCPV‐GFP was generated by homologous recombination in BT cells infected with PCPV and transfected by nucleofection with pTG19106 (according to Amaxa Nucleofector Technology). Fluorescent and selective (GPT+) plaques were selected. Recombinant virus was isolated from GFP‐fluorescent plaques and submitted to additional plaque purification in BT cells. Virus structure and the absence of parental PCPV were confirmed by multiple PCRs and DNA sequencing of the expression cassette. PCPV‐GFP (PCPTG19106) was amplified in BT cells and purified.
Generation of PCPV‐E7 (PCPTG19178)
The sequence coding for HPV16 E7 protein was deleted from aa 21 to 26 to obtain a non‐oncogenic protein E7(Δ21‐26). The signal peptide of the glycoprotein precursor of rabies virus ERA strain (SR) and the membrane‐anchoring peptide derived from the rabies glycoprotein (TMR) were added, respectively, upstream and downstream of the mutated HPV16 E7 protein. 20 The sequence coding for SR‐E7(Δ21‐26)‐TMR was positioned under the control of p7.5K vaccinia promoter and inserted in the transfer plasmid leading to pTG19178. The PCPV‐E7 virus was generated by homologous recombination in BT cells infected with PCPTG19106 and transfected by nucleofection with pTG19178. Recombinant virus was isolated by selected GFP‐fluorescent negative plaques and submitted to additional plaques purification in BT cells. Virus structure and the absence of parental PCPTG19106 were confirmed by multiple PCRs and DNA sequencing of the expression cassette. The resulting virus PCPV‐E7 (PCPTG19178) was amplified in BT cells and purified according to the protocol established for MVA. Virus stocks were titrated on BT cells by plaque assay.
Protein vaccination
Non‐oncogenic recombinant HPV16 E7(Δ21‐26) protein was produced in Escherichia coli BL21(DE3) as recombinant glutathione S‐transferase (GST) fusion protein. After production, purification and thrombin cleavage, the protein was dialysed against PBS and concentrated to 1–2 mg mL–1 with Vivaspin Sartorius 5000 MWCO concentrator. Protein concentration was determined by the Bradford assay using BSA as standard. An endotoxin level of 2.6 EU per mg protein was found using the LAL chromogenic assay Endosafe PTS (Charles River, Franklin, MA, USA).
Fifty µg of recombinant protein was mixed with 100 µg of low molecular weight LMW poly(I:C) (InvivoGen, Toulouse, France) for vaccination approaches.
Luminex analysis
Cytokine and chemokine profiles in cell culture supernatants were quantified using an appropriate ProcartaPlex immunoassay (Human Inflammation Panel 20‐Plex, Human IFN‐β or IFN‐α Simplex, Invitrogen, by Thermo Fisher Scientific, Vienna, Austria). The analysis was carried out according to the manufacturer’s recommendations using a MagPix device and the ProcartaPlex Analyst 1.0 software.
Human primary cells
Human peripheral blood mononuclear cells (PBMCs) were isolated from leucocyte concentrates from healthy donors obtained from the Etablissement Français du Sang (EFS), Strasbourg, France. PBMCs were isolated by density gradient centrifugation using Ficoll–Paque PLUS (GE Healthcare, Uppsala, Sweden). Cell populations were magnetically sorted from PBMCs by positive or negative selection using an autoMACS PRO separator (Miltenyi Biotec). Monocytes for the MDSC/T assay were sorted by CD14+ positive selection (Human CD14 MicroBeads, 130‐050‐201) and CD8+ T cells by negative selection (Human CD8+ T Cell Isolation Kit, 130‐096‐495). Monocytes (Classical Monocyte Isolation Kit, 130‐117‐337), NK cells (NK cell isolation kit 130‐092‐657) and pDCs (pDC Isolation Kit II, 130‐097‐415) for all other tests were sorted by depletion. All kits were purchased from Miltenyi Biotec and used according to the manufacturer’s recommendations.
Viable frozen PBMCs from untreated cancer patients were purchased from CliniSciences, Nanterre, France.
In vitro studies with human immune cells
Infection profile in PBMCs
To define the tropism of each poxvirus for mononuclear cell subsets, PBMCs were seeded at 4 × 105 cells/well in Roswell Park Memorial Institute (RPMI)‐1640 medium (Sigma) supplemented with 10% heat‐inactivated FCS, 2 mM L‐glutamine and 40 µg mL−1 gentamycin in round‐bottom 96‐well plates and infected with recombinant PCPV‐GFP, MVA‐GFP or VACV‐GFP at a MOI of 1. The next day, cells were washed with PBS, incubated with FcR Blocking Reagent (Miltenyi Biotec) diluted 1/10 and subjected to surface staining with either anti‐lineage (CD3/19/20/56)‐APC (UCHT1, HIB19, 2H7, 5.1H11; BioLegend, San Diego, CA, USA), anti‐CD14‐PerCP‐Cy5.5 (M5E2; BD Pharmingen, San Diego, CA, USA) and anti‐CD16‐BV605 (3G8; BioLegend) or anti‐CD3‐PE‐Vio770 (REA613; Miltenyi Biotec), anti‐CD4‐APC (A161A1; BioLegend), anti‐CD56‐BV421 (5.1H11; BioLegend) and anti‐CD19‐AF700 (HIB19; BioLegend) antibodies in combination with LIVE/DEADTM near‐IR fluorescent reactive dye (Molecular Probes, Eugene, OR, USA). Samples were run on a MACSQuant 16 flow cytometer (Miltenyi Biotec) and analysed using the KALUZA 1.3 software (Beckman Coulter, Life Sciences, Villepinte, France). The percentage of infected cells (GFP+) within live monocytes (Lin–CD14+CD16‐/bright), and CD4+ T (CD3+CD56−CD4+), CD8+ T (CD3+CD56−CD4−), NK (CD3−CD56+) and B (CD3−CD56−CD19+) singlets was determined.
Activation of antigen‐presenting cells (APCs)
Immature dendritic cells (moDCs) were derived from monocytes cultured in granulocyte–macrophage colony‐stimulating factor (GM‐CSF, 20 ng mL−1) and IL‐4 (10 ng mL−1), both from Miltenyi Biotec, for 7 days. To confirm the dendritic cell phenotype, the expression of anti‐CD1a‐FITC (HI149; BD Pharmingen) was verified. For stimulation assays, moDCs were infected, and the next day, activation was measured in live cells by flow cytometry using the antibodies against CD80‐PE (REA661; Miltenyi Biotec), CD83‐APC (REA714; Miltenyi Biotec), CD86‐PerCp‐Vio700 (FM95; Miltenyi Biotec) and HLA‐/DR Pacific Blue (1243; BioLegend). To control activation, the cells were activated with the TLR7/8 ligand R848 diluted to 10−4 M (InvivoGen).
NK degranulation assay
PBMCs from three donors were infected at MOI 0.3 with PCPV, empty MVA or VACV. After o/n incubation, an equal number of K562 cells and anti‐CD107a‐APC (clone RH4A3; FastImmune, BD Biosciences, San Jose, CA, USA) were added to the culture. After 90‐min incubation at 37°C, cells were stained with Live/Dead, anti‐CD3‐FITC (clone UCHT1; BioLegend), anti‐CD56‐BV510 (NCAM162; BD Horizon) and anti‐CD69‐Alexa Fluor 700 (FN50; BD Pharmingen). Within the population of NK cells (CD3−CD56+), activated degranulating NKs were determined as CD69+CD107a+.
MDSC/T‐cell assay
To assess the effects of poxviral vectors on MDSCs, CD14+ monocytes were seeded at 5 × 105 cells mL−1 in RPMI‐1640 medium supplemented with 10% heat‐inactivated FCS, glucose (up to 4.5 g L−1; Sigma), 2 mM L‐glutamine, 40 µg mL−1 gentamycin, 1 mM sodium pyruvate (Sigma), 1× MEM‐NEAA (Gibco), 10 ng mL−1 human GM‐CSF and 10 ng mL−1 human IL‐6 (both cytokines from Miltenyi Biotec) for 6 days. Medium and cytokines were replenished every 2–3 days. Adherent MDSCs were recovered using 10 mM EDTA (Sigma‐Aldrich) in PBS or Detachin Cell Detachment Solution (Genlantis, San Diego, CA, USA) followed by gentle scraping.
MDSCs were seeded in RPMI‐1640 medium supplemented with 10% heat‐inactivated FCS, 2 mM L‐glutamine and 40 µg mL−1 gentamycin in round‐bottom 96‐well plates. 1 × 105 autologous CellTraceTM CFSE‐labelled (5 µM; Invitrogen) CD8+ T cells were either mixed with MDSC or seeded alone, and cultures were treated with PCPV at MOIs of 0.03, 0.3 and 3 in the presence of TransAct T Cell Reagent – Large Scale (Miltenyi Biotec) diluted 1/17.5 and 40 U mL−1 human IL‐2 (Miltenyi Biotec). On day 4, T cells were analysed for proliferation and granzyme B expression by flow cytometry.
Cells were stained with anti‐CD8‐APC (BW135/80; Miltenyi Biotec) in combination with LIVE/DEADTM violet fluorescent reactive dye (Molecular Probes). Intracellular staining was then performed with the Cytofix/Cytoperm Fixation/Permeabilization Kit from BD Biosciences (Pont‐de‐Claix, France) and anti‐granzyme B‐PE (REA226) or isotype control antibody (REA293) from Miltenyi Biotec. Samples were run on a MACSQuant 10 flow cytometer (Miltenyi Biotec) and analysed using the KALUZA 1.3 software. Proliferation was assessed by CFSE dilution. Single, live (LIVE/DEADlow) CD8+ leucocytes were gated as the T‐cell population. The percentage of proliferating granzyme B+ cells within the CD8+ population was determined. Granzyme B expression was quantified as median fluorescence intensity relative to isotype control staining in proliferating CD8+ cells.
Infection of activated T cells
PBMCs were first plated at 2 × 106 cells mL–1 of TexMACS medium (Miltenyi Biotec) supplemented with 20 U mL−1 human IL‐2 in 6‐well plates, and T cells were stimulated for 2 days by adding MACS GMP T Cell TransAct (final dilution 1/17.5; Miltenyi Biotec). PBMCs were then seeded at 2 × 105 and 1 × 105 cells per well in 96‐well plates for 1‐ and 5‐day cultures, respectively, and incubated with recombinant PCPV‐GFP (crude SN), MVA‐GFP or VV‐GFP at a MOI of 1 in TexMACS medium without cytokine. Half the medium was replaced at day 1 and day 4 post‐infection in 5‐day cultures. Cells were stained and analysed by flow cytometry as described for experiments studying poxvirus tropism. The percentage of GFP+ cells and the total cell count were measured for CD8 (CD3+CD4−CD19−) and CD4 (CD3+CD4+CD19−) T subsets.
Studies in vivo (naïve mice and syngeneic tumor models)
Female C57BL/6 and BALB/c mice 7 to 8 weeks of age were used for the experiments. The mice were purchased from Charles River and maintained in a temperature‐ and humidity‐controlled animal facility (21 ± 2°C; 15–70% humidity), with a 12‐h light–dark cycle and free access to water and a standard rodent chow (D04, SAFE, Villemoisson‐sur‐Orge, France). All animal procedures and experiments were approved by the local ethical committee (C2EA – 17 Comité d’éthique Com’Eth) for animal care and use and research minister.
MC38 or TC1 tumor cells were subcutaneously injected in one abdominal flank. The size of superficial tumor was assessed three times per week using a calliper, and the body weight (BW) was monitored at the same time. Tumor volume was calculated with the spheroid formula V = (LxW^2)/2 after calculation of width and length diameter in the right angles. The real body weight was calculated after estimation of the tumor weight. Ethical criteria are driven by animal well‐being and are regulated by the ethical committee and the animal welfare structure. A maximal tumor volume of 2000 mm3 and a maximum of 10% of BW loss between two measures have been considered as an ethical endpoint. Additional endpoints such as ulceration, necrosis and distension of covering tissues are recorded and would lead to terminate animals humanely when the degree of suffering cannot be justified by the scientific objective.
Depleting antibodies were purchased from BioXCell (Lebanon, NH, USA) and injected ip 69 : 200 µg of anti‐CD4+ (clone GK1.5) and anti‐CD8+ antibodies (clone 53–6.7), or the isotype control for CD8 (clone 2A3), was injected ip at days 12, 13, 20 and 28 after sc injection of TC1 cells. 150 µg of anti‐NK1.1 (clone PK136) antibody was injected ip at days 11, 13 and 15, and then twice per week. The schedule in MC38 tumor model was as follows: CD8+ cells were depleted using the anti‐CD8 (clone 53–6.7) antibody, and its isotype control (2A3). 200 µg of anti‐CD8 was injected days 2 and 1 before, and days 6 and 13 after, MC38 cell injection.
Murine ex vivo studies
ELISpot
Microwell plates (Millipore, MSIPS4W10) were coated with anti‐mouse IFN‐γ antibody (Mabtech, AN18, 3321‐3‐1000), washed and saturated with complete medium (CM).
Tests were performed with fresh splenic lymphocytes isolated from splenocyte suspension by gradient centrifugation (Lympholyte®‐M, Cedarlane, Burlington, Canada). Cells were mixed with various stimuli: R9F (RAHYNIVTF) is a H‐2Db‐restricted HPV16E7‐specific peptide, and I8L (IAYKYAQL) is a H‐2Kb HPV16E1‐specific peptide. K9i‐3 (KNGENAQAI) is a negative control peptide. As positive control, concanavalin A (ConA) was used. To stimulate with MC38 cells, 1 × 107 cells were treated with mitomycin C (50 µg mL−1) for 1 h at 37°C. The treated cells were mixed with splenocytes at ratios of 1/10 and 1/20 and included in the test. Plates were incubated for 20 h at 37°C. The following day, IFN‐γ‐positive cells were detected using biotinylated anti‐mouse IFN‐γ monoclonal antibody (Mabtech) and ExtrAvidin‐phosphatase alkaline (SIGMA), and spot‐forming units were visualised using BCIP/NBT substrate (SIGMA). Spots were counted with an ELISpot reader (CTL Immunospot Reader, S5UV). Results were expressed for each quadruplicate as the mean number of spot‐forming units (sfu) per 1 × 106 splenic lymphocytes.
ICS IFN‐γ in lungs
The lungs of immunised mice were treated individually, as described. 34 Briefly, lungs were enzymatically and mechanically dissociated (Miltenyi products: tumor dissociation kit, C‐tubes and gentleMACS). 2 × 106 cells were stimulated in 150 µL TexMACS medium (Miltenyi Biotec) in the presence of 1 µg anti‐CD28 (clone PV‐1), and either the HPV16E7‐specific peptide R9F or the control peptide I8L. After 5 h of incubation in the presence of brefeldin, cells were analysed by flow cytometry using anti‐CD3‐PerCPCy5.5 (clone 145‐2C11) and anti‐CD8a‐APCVio770 (clone 53.6‐7) antibodies. After permeabilisation (Cytofix/Cytoperm, BD Bioscience), activation was assessed by intracellular staining with anti‐IFN‐γ‐FITC (clone XMG1.2) or its isotype control.
Surface staining of mouse splenocytes and peripheral blood cells
To probe the activation status of murine immune cells or to confirm the depletion of cell population, a standard procedure was applied. C57BL/6 mice with or without tumor were killed, and peripheral blood was taken from heart immediately post‐mortem and transferred to an EDTA‐coated tube. Splenocytes were collected by mashing the spleen through a 70‐µM cell strainer. After filtration, red blood cells were lysed (BD Pharm Lyse, BD BioScience), and splenocytes and blood samples were stained with Live/Dead NearIR (Invitrogen), anti‐CD3ε‐PerCPCy5.5 (clone 145‐2C11), anti‐CD8‐V500 (clone 53‐6.7), anti‐CD4‐AF700 (clone RM4‐5) and anti‐CD19‐V450 (clone 1D3), provided by Miltenyi Biotec, and anti‐CD49b‐FITC (clone HMa2), anti‐CD69‐APC (clone H1.2F3) and anti‐PD‐L1‐PE (clone 10F.9G2), provided by BioLegend. Isotype controls for anti‐CD69 (Armenian hamster IgG) and anti‐PD‐L1 (rat IgG2b) were provided by BioLegend. Stained blood cells were treated with RBL buffer, before all samples were analysed on a MACSQuant 16 flow cytometer. Gated on live cells, CD3+CD4+, CD3+CD8+, CD3−CD49b+ (NK) and CD3−CD19+ (B) cell populations were measured. Within these populations, the proportions of PD‐L1+CD69+ double‐positive cells were quantified.
Clinical samples
Fresh tumor‐draining lymph nodes (TDLNs) were collected from patients diagnosed with head and neck squamous cell carcinoma (HNSCC) localised to tonsils having undergone standard‐of‐care primary surgical resection at the Institut Curie Hospital (Paris, France), within 3h after surgical resection from the Department of Pathology, as surgical residues. Nine patients were included in this study, which was performed in compliance with the principles of Good Clinical Practice and the Declaration of Helsinki. All patients signed a consent form mentioning that their operative specimens might be used for scientific purposes, and eight of these patients were included in the clinical trial SCANDARE NCT03017573. The study was conducted in a laboratory that operates under exploratory research principles in accordance with institutional ethical guidelines. Table 1 summarises the clinical, biological and pathological characteristics of the HNSCC patients included in this study. Patients had not received radiotherapy or chemotherapy treatment prior to surgery. Tumor staging and lymph node invasion by tumor cells were assessed macroscopically and confirmed by histopathology according to the 8th edition of the AJCC Cancer Staging Manual. 70 HPV16 status was assessed at the Pathology Department of the Institut Curie. HPV typing was conducted using total DNA isolated from formalin‐fixed tissue blocks. Real‐time polymerase chain reaction (RT‐PCR) was performed with SYBR Green and specific primers for HPV16 and HPV18 using a 7900HT Fast Real‐Time PCR System (Applied Biosystems). In case of negative results, a universal PCR for all HPV serotypes was performed, and in case of positive results, sequencing was performed to determine the serotype. Thus, HPV‐negative patients are negative for all HPV serotypes.
Samples and cell isolation
Samples were obtained within 3 h after the primary surgery, cut into small fragments and digested with 0.1 mg mL−1 Liberase TL (Roche) in the presence of 0.1 mg mL−1 DNase (Roche) for 30 min at 37°C in CO2‐independent medium (Gibco). Cells were filtered on a 40‐µm cell strainer (BD Bioscience) and frozen in 10% DMSO in FCS. 71 , 72
In vitro assay for T‐cell activation/expansion
As the frequency of tumor‐specific cells can be very limited, we used a step of in vitro expansion, adapted from Lissina et al. 38 Frozen cells either from lymph node or from tumor were thawed and incubated in AIM‐V medium (Gibco) supplemented with Flt‐3‐ligand (50 ng mL−1 R&D). After 24 h, R848 (0.5 µg mL−1; InvivoGen) and hIL‐2 (50 U mL−1; Novartis) were added, together with the different virus or control or viral antigens (1 µg mL−1; Miltenyi Biotec). At day 2, 5% of human AB serum (Corning) was added to each well and medium was subsequently replaced every 2–3 days. At day 10, cells were restimulated with the viral peptides for 12 h before flow cytometry analysis, including surface markers and intracellular cytokine staining.
Statistics
For tumor size analyses, to fit with statistical assumptions, tumor diameter (expressed in mm) was derived from tumor volume calculated with length and width (expressed in mm3) using the following formula: Diameter = 2 × (3 × Volume/4)1/3. A repeated mixed model was built using this calculated tumor diameter as response. Treatment group, Day (as continuous) and the interaction between both factors should be considered to be fixed effects, and the quadratic effect of Day was also evaluated by adding the interaction group × Day² in the mixed model. Mouse was considered to be a random effect, and a repeated measure over time was considered with a spatial power structure for variance–covariance. If some effects were found to be significant, post hoc comparisons were made with the Tukey multiplicity adjustment.
Conflicts of interest
All authors except RR, FK, CS, CLT, CaH and EP are employees of Transgene. Institute Curie employees RR, FK and EP receive funding from Transgene.
Author contribution
Rodrigo Nalio Ramos: Conceptualization; Data curation; Investigation; Writing – original draft; Writing – review & editing. Caroline Tosch: Data curation; Investigation; Methodology. Fiorella Kotsias: Data curation; Investigation; Validation; Writing – original draft; Writing – review & editing. Marie‐Christine Claudepierre: Investigation. Doris Schmitt: Investigation. Christelle Remy‐Ziller: Data curation; Investigation; Visualization. Chantal Hoffmann: Investigation; Methodology. Marine Ricordel: Investigation. Virginie Nourtier: Investigation. Isabelle Farine: Investigation. Laurence Laruelle: Investigation; Validation. Julie Hortelano: Data curation; Formal analysis; Visualization. Clementine Spring‐Giusti: Conceptualization; Writing – original draft. Christine Sedlik: Project administration. Christophe LeTourneau: Project administration. Caroline Hoffmann: Project administration. Nathalie Silvestre: Conceptualization; Supervision; Writing – original draft; Writing – review & editing. Philippe Erbs: Supervision; Writing – review & editing. Kaïdre Bendjama: Conceptualization; Supervision; Writing – original draft; Writing – review & editing. Christine Thioudellet: Conceptualization; Data curation; Investigation; Visualization; Writing – original draft; Writing – review & editing. Eric Quemeneur: Project administration; Writing – original draft; Writing – review & editing. Eliane Piaggio: Resources; Supervision; Validation; Writing – review & editing. Karola Rittner: Conceptualization; Data curation; Investigation; Project administration; Supervision; Validation; Writing – original draft; Writing – review & editing.
Supporting information
Supplementary figures 1–13
Supplementary tables 1 and 2
Acknowledgments
We acknowledge Professor James W Hodge (NIH) for providing us with the MC38 murine colon cancer cell line and the PHENOMIN‐Institut Clinique de la Souris, PHENOMIN (ICS, Illkirch, France) for conducting animal experiments. We thank our colleagues at Transgene: Johann Foloppe for providing the collection of Poxviridae used in screening approach, Patricia Kleinpeter for providing the HPV16E7m protein, and Berangere Bastien and Benoit Sansas for supervising statistical analyses. Renaud Colisson (Thermo Fisher Scientific) is acknowledged for his help in analysing Quantiplex data. We thank the SCANDARE team: from the scientific coordination unit of the Department of Drug Development and Innovation – Maud Kamal, Linda Larbi Cherif and Charlotte Lecerf; from the clinical research unit – Anne‐Sophie Plissonnier, Yasmine Yousfi, Alexia Savignoni, Amir Kadi, Frederique Berger, Gillon Veronique, Simondi Cecile, Gobillion Aline, Anne Blondel, Gwenaele Jarry, Nesrine Boutoula and Audrey Faye; from the Centre de Ressource Biologique – Odette Mariani, Thierry Bronzini, Michele Galut, Aurore Loistron and Céline Meaudre; from the pathology unit – Anne‐Vincent Salomon, Jerzy Klijanienko, Charlotte Martinat and Samira Miladi; from the Immunologie Clinique unit – Lantz Olivier, Ana Lalane, Delphine Louis, Okan Altuncu and Christina Ekwegbara; from the Direction des données Thomas Balezeau and Xose Fernandez; from the surgery unit – Nathalie Badois, Anne Chilles, Maria Lesnik, Samar Krhili, Olivier Choussy, Rabah Taouachi, Jean Baptiste Lecanu, Manuel Erminy, Antoine. Dubray Vautrin, Christian Chappey and Jules Ceccaldi; and from the Immunotherapy unit – Philemon Sirven. Rodrigo N Ramos, Fiorella Kotsias and Eliane Piaggio are supported by LabEx DCBIOL (ANR‐10‐IDEX‐0001‐02 PSL; ANR‐11‐LABX‐0043); SIRIC INCa‐DGOS‐Inserm 12554; and Center of Clinical Investigation (CIC IGR‐Curie 1428).
References
- 1. Le Tourneau C, Delord J, Cassier P et al. Phase IB/II trial of TG4001 (Tipapkinogene Sovacivec), a therapeutic HPV vaccine, and Avelumab in patients with recurrent/metastatic HPV16+ cancers. In: ESMO , editor. ESMO 2019 Annual congress. Barcelona: Annals of Oncology; 2019.
- 2. Reinhard K, Rengstl B, Oehm P et al. An RNA vaccine drives expansion and efficacy of claudin‐CAR‐T cells against solid tumors. Science 2020; 367: 446–453. [DOI] [PubMed] [Google Scholar]
- 3. Peng M, Mo Y, Wang Y et al. Neoantigen vaccine: an emerging tumor immunotherapy. Mol Cancer 2019; 18: 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu MA. Immunologic basis of vaccine vectors. Immunity 2010; 33: 504–515. [DOI] [PubMed] [Google Scholar]
- 5. Draper SJ, Heeney JL. Viruses as vaccine vectors for infectious diseases and cancer. Nat Rev Microbiol 2010; 8: 62–73. [DOI] [PubMed] [Google Scholar]
- 6. Ulmer JB, Wahren B, Liu MA. Gene‐based vaccines: recent technical and clinical advances. Trends Mol Med 2006; 12: 216–222. [DOI] [PubMed] [Google Scholar]
- 7. Zhu J, Huang X, Yang Y. Innate immune response to adenoviral vectors is mediated by both Toll‐like receptor‐dependent and ‐independent pathways. J Virol 2007; 81: 3170–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Delaloye J, Roger T, Steiner‐Tardivel QG et al. Innate immune sensing of modified vaccinia virus Ankara (MVA) is mediated by TLR2‐TLR6, MDA‐5 and the NALP3 inflammasome. PLoS Pathog 2009; 5: e1000480. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9. Dai P, Wang W, Cao H et al. Modified vaccinia virus Ankara triggers type I IFN production in murine conventional dendritic cells via a cGAS/STING‐mediated cytosolic DNA‐sensing pathway. PLoS Pathog 2014; 10: e1003989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol 2015; 15: 760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yu L, Liu P. Cytosolic DNA sensing by cGAS: regulation, function, and human diseases. Signal Transduct Target Ther 2021; 6: 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol 2015; 15: 405–414. [DOI] [PubMed] [Google Scholar]
- 13. Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer 2016; 16: 131–144. [DOI] [PubMed] [Google Scholar]
- 14. Rizza P, Moretti F, Belardelli F. Recent advances on the immunomodulatory effects of IFN‐ α: implications for cancer immunotherapy and autoimmunity. Autoimmunity 2010; 43: 204–209. [DOI] [PubMed] [Google Scholar]
- 15. Duong E, Fessenden TB, Lutz E et al. Type I interferon activates MHC class I‐dressed CD11b+ conventional dendritic cells to promote protective anti‐tumor CD8+ T cell immunity. Immunity 2022; 55: 308–323.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hance KW, Rogers CJ, Zaharoff DA, Canter D, Schlom J, Greiner JW. The antitumor and immunoadjuvant effects of IFN‐α in combination with recombinant poxvirus vaccines. Clin Cancer Res 2009; 15: 2387–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lu C, Klement JD, Ibrahim ML et al. Type I interferon suppresses tumor growth through activating the STAT3‐granzyme B pathway in tumor‐infiltrating cytotoxic T lymphocytes. J Immunother Cancer 2019; 7: 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Musella M, Manic G, De Maria R, Vitale I, Sistigu A. Type‐I‐interferons in infection and cancer: unanticipated dynamics with therapeutic implications. Oncoimmunology 2017; 6: e1314424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Volz A, Sutter G. Modified vaccinia virus Ankara: history, value in basic research, and current perspectives for vaccine development. Adv Virus Res 2017; 97: 187–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brun JL, Dalstein V, Leveque J et al. Regression of high‐grade cervical intraepithelial neoplasia with TG4001 targeted immunotherapy. Am J Obstet Gynecol 2011; 204: 169.e1–169.e8. [DOI] [PubMed] [Google Scholar]
- 21. Harper DM, Nieminen P, Donders G et al. The efficacy and safety of Tipapkinogen Sovacivec therapeutic HPV vaccine in cervical intraepithelial neoplasia grades 2 and 3: Randomized controlled phase II trial with 2.5years of follow‐up. Gynecol Oncol 2019; 153: 521–529. [DOI] [PubMed] [Google Scholar]
- 22. Malone B, Tosch C, Grellier B et al. Performance of neoantigen prediction for the design of TG4050, a patient specific neoantigen cancer vaccine. In: AACR, editor. Proceedings of the 111th Annual Meeting of the American Association for Cancer Research. Philadelphia (PA): AACR; 2020.
- 23. de Freitas LFD, Oliveira RP, Miranda MCG et al. The virulence of different vaccinia virus strains is directly proportional to their ability to downmodulate specific cell‐mediated immune compartments In Vivo . J Virol 2019; 93: e02191–e2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Smith GL, Benfield CTO, Maluquer de Motes C et al. Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. J Gen Virol 2013; 94: 2367–2392. [DOI] [PubMed] [Google Scholar]
- 25. Georgana I, Sumner RP, Towers GJ, Maluquer de Motes C. Virulent poxviruses inhibit DNA sensing by preventing STING activation. J Virol 2018; 92: e02145–e2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Antoine G, Scheiflinger F, Dorner F, Falkner FG. The complete genomic sequence of the modified vaccinia Ankara strain: comparison with other orthopoxviruses. Virology 1998; 244: 365–396. [DOI] [PubMed] [Google Scholar]
- 27. Friedman‐Kien AE, Rowe WP, Banfield WG. Milker's nodules: isolation of a poxvirus from a human case. Science 1963; 140: 1335–1336. [DOI] [PubMed] [Google Scholar]
- 28. Zhang W, Jordan KR, Schulte B, Purev E. Characterization of clinical grade CD19 chimeric antigen receptor T cells produced using automated CliniMACS Prodigy system. Drug Des Devel Ther 2018; 12: 3343–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chahroudi A, Chavan R, Kozyr N, Waller EK, Silvestri G, Feinberg MB. Vaccinia virus tropism for primary hematolymphoid cells is determined by restricted expression of a unique virus receptor. J Virol 2005; 79: 10397–10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Engelmayer J, Larsson M, Subklewe M et al. Vaccinia virus inhibits the maturation of human dendritic cells: a novel mechanism of immune evasion. J Immunol 1999; 163: 6762–6768. [PubMed] [Google Scholar]
- 31. Shabrish S, Gupta M, Madkaikar M. A modified NK cell degranulation assay applicable for routine evaluation of NK cell function. J Immunol Res 2016; e3769590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Casacuberta‐Serra S, Pares M, Golbano A, Coves E, Espejo C, Barquinero J. Myeloid‐derived suppressor cells can be efficiently generated from human hematopoietic progenitors and peripheral blood monocytes. Immunol Cell Biol 2017; 95: 538–548. [DOI] [PubMed] [Google Scholar]
- 33. Mohty AM, Grob JJ, Mohty M, Richard MA, Olive D, Gaugler B. Induction of IP‐10/CXCL10 secretion as an immunomodulatory effect of low‐dose adjuvant interferon‐α during treatment of melanoma. Immunobiology 2010; 215: 113–123. [DOI] [PubMed] [Google Scholar]
- 34. Remy‐Ziller C, Thioudellet C, Hortelano J et al. Sequential administration of MVA‐based vaccines and PD‐1/PD‐L1‐blocking antibodies confers measurable benefits on tumor growth and survival: Preclinical studies with MVA‐βGal and MVA‐MUC1 (TG4010) in a murine tumor model. Hum Vaccin Immunother 2018; 14: 140–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nomland R, Mc KA. Milkers' nodules; report of ten cases. AMA Arch Derm Syphilol 1952; 65: 663–674. [DOI] [PubMed] [Google Scholar]
- 36. Dong W, Wu X, Ma S et al. The mechanism of anti‐PD‐L1 antibody efficacy against PD‐L1‐negative tumors identifies NK cells expressing PD‐L1 as a cytolytic effector. Cancer Discov 2019; 9: 1422–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhao Y, Lee CK, Lin CH et al. PD‐L1:CD80 cis‐heterodimer triggers the co‐stimulatory receptor CD28 while repressing the inhibitory PD‐1 and CTLA‐4 pathways. Immunity 2019; 51: 1059–1073.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lissina A, Briceno O, Afonso G et al. Priming of qualitatively superior human effector CD8+ T cells using TLR8 ligand combined with FLT3 ligand. J Immunol 2016; 196: 256–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gros A, Robbins PF, Yao X et al. PD‐1 identifies the patient‐specific CD8+ tumor‐reactive repertoire infiltrating human tumors. J Clin Invest 2014; 124: 2246–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gros A, Tran E, Parkhurst MR et al. Recognition of human gastrointestinal cancer neoantigens by circulating PD‐1+ lymphocytes. J Clin Invest 2019; 129: 4992–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stevanovic S, Pasetto A, Helman SR et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science 2017; 356: 200–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Del Medico Zajac MP, Molinari P, Gravisaco MJ et al. MVAΔ008 viral vector encoding the model protein OVA induces improved immune response against the heterologous antigen and equal levels of protection in a mice tumor model than the conventional MVA. Mol Immunol 2021; 139: 115–122. [DOI] [PubMed] [Google Scholar]
- 43. Alharbi NK, Spencer AJ, Hill AV, Gilbert SC. Deletion of fifteen open reading frames from modified vaccinia virus ankara fails to improve immunogenicity. PLoS One 2015; 10: e0128626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Das K, Belnoue E, Rossi M et al. A modular self‐adjuvanting cancer vaccine combined with an oncolytic vaccine induces potent antitumor immunity. Nat Commun 2021; 12: 5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang W, Liu S, Dai P et al. Elucidating mechanisms of antitumor immunity mediated by live oncolytic vaccinia and heat‐inactivated vaccinia. J Immunother Cancer 2021; 9: e002569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hautaniemi M, Ueda N, Tuimala J, Mercer AA, Lahdenpera J, McInnes CJ. The genome of pseudocowpoxvirus: comparison of a reindeer isolate and a reference strain. J Gen Virol 2010; 91: 1560–1576. [DOI] [PubMed] [Google Scholar]
- 47. Muller LME, Holmes M, Michael JL et al. Plasmacytoid dendritic cells orchestrate innate and adaptive anti‐tumor immunity induced by oncolytic coxsackievirus A21. J Immunother Cancer 2019; 7: 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cao H, Dai P, Wang W et al. Innate immune response of human plasmacytoid dendritic cells to poxvirus infection is subverted by vaccinia E3 via its Z‐DNA/RNA binding domain. PLoS One 2012; 7: e36823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. von Buttlar H, Siegemund S, Buttner M, Alber G. Identification of Toll‐like receptor 9 as parapoxvirus ovis‐sensing receptor in plasmacytoid dendritic cells. PLoS One 2014; 9: e106188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ramos RN, Rodriguez C, Hubert M et al. CD163+ tumor‐associated macrophage accumulation in breast cancer patients reflects both local differentiation signals and systemic skewing of monocytes. Clin Transl Immunology 2020; 9: e1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lynn GM, Sedlik C, Baharom F et al. Peptide‐TLR‐7/8a conjugate vaccines chemically programmed for nanoparticle self‐assembly enhance CD8 T‐cell immunity to tumor antigens. Nat Biotechnol 2020; 38: 320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ni Q, Zhang F, Liu Y et al. A bi‐adjuvant nanovaccine that potentiates immunogenicity of neoantigen for combination immunotherapy of colorectal cancer. Sci Adv 2020; 6: eaaw6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mooij P, Balla‐Jhagjhoorsingh SS, Koopman G et al. Differential CD4+ versus CD8+ T‐cell responses elicited by different poxvirus‐based human immunodeficiency virus type 1 vaccine candidates provide comparable efficacies in primates. J Virol 2008; 82: 2975–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Masterson L, Lechner M, Loewenbein S et al. CD8+ T cell response to human papillomavirus 16 E7 is able to predict survival outcome in oropharyngeal cancer. Eur J Cancer 2016; 67: 141–151. [DOI] [PubMed] [Google Scholar]
- 55. Tran E, Turcotte S, Gros A et al. Cancer immunotherapy based on mutation‐specific CD4+ T cells in a patient with epithelial cancer. Science 2014; 344: 641–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sahin U, Derhovanessian E, Miller M et al. Personalized RNA mutanome vaccines mobilize poly‐specific therapeutic immunity against cancer. Nature 2017; 547: 222–226. [DOI] [PubMed] [Google Scholar]
- 57. Keskin DB, Anandappa AJ, Sun J et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019; 565: 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sonntag K, Hashimoto H, Eyrich M et al. Immune monitoring and TCR sequencing of CD4 T cells in a long term responsive patient with metastasized pancreatic ductal carcinoma treated with individualized, neoepitope‐derived multipeptide vaccines: a case report. J Transl Med 2018; 16: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Quezada SA, Simpson TR, Peggs KS et al. Tumor‐reactive CD4+ T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med 2010; 207: 637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Xie Y, Akpinarli A, Maris C et al. Naive tumor‐specific CD4+ T cells differentiated in vivo eradicate established melanoma. J Exp Med 2010; 207: 651–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shekarian T, Sivado E, Jallas AC et al. Repurposing rotavirus vaccines for intratumoral immunotherapy can overcome resistance to immune checkpoint blockade. Sci Transl Med 2019; 11: eaat5025. [DOI] [PubMed] [Google Scholar]
- 62. Zemek RM, De Jong E, Chin WL et al. Sensitization to immune checkpoint blockade through activation of a STAT1/NK axis in the tumor microenvironment. Sci Transl Med 2019; 11: eaav7816. [DOI] [PubMed] [Google Scholar]
- 63. Rosato PC, Wijeyesinghe S, Stolley JM et al. Virus‐specific memory T cells populate tumors and can be repurposed for tumor immunotherapy. Nat Commun 2019; 10: 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Robinson AJ, Petersen GV. Orf virus infection of workers in the meat industry. N Z Med J 1983; 96: 81–85. [PubMed] [Google Scholar]
- 65. Ricordel M, Foloppe J, Pichon C et al. Oncolytic properties of non‐vaccinia poxviruses. Oncotarget 2018; 9: 35891–35906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schaedler E, Remy‐Ziller C, Hortelano J et al. Sequential administration of a MVA‐based MUC1 cancer vaccine and the TLR9 ligand Litenimod (Li28) improves local immune defense against tumors. Vaccine 2017; 35: 577–585. [DOI] [PubMed] [Google Scholar]
- 67. Erbs P, Findeli A, Kintz J et al. Modified vaccinia virus Ankara as a vector for suicide gene therapy. Cancer Gene Ther 2008; 15: 18–28. [DOI] [PubMed] [Google Scholar]
- 68. Foloppe J, Kempf J, Futin N et al. The enhanced tumor specificity of TG6002, an armed oncolytic vaccinia virus deleted in two genes involved in nucleotide metabolism. Mol Ther Oncolytics 2019; 14: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Caudana P, Nunez NG, De La Rochere P et al. IL2/Anti‐IL2 complex combined with CTLA‐4, but not PD‐1, blockade rescues antitumor NK cell function by regulatory T‐cell modulation. Cancer Immunol Res 2019; 7: 443–457. [DOI] [PubMed] [Google Scholar]
- 70. Amin MB, Greene FL, Edge SB et al. The eight edition AJCC cancer staging manual: continuing to build a bridge from a population‐based to a more "personalized" approach to cancer staging. CA Cancer J Clin 2017; 67: 93–99. [DOI] [PubMed] [Google Scholar]
- 71. Bourdely P, Anselmi G, Vaivode K et al. Transcriptional and functional analysis of CD1c+ human dendritic cells identifies a CD163+ subset priming CD8+CD103+ T cells. Immunity 2020; 53: 335–352.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nunez NG, Tosello Boari J, Ramos RN et al. Tumor invasion in draining lymph nodes is associated with Treg accumulation in breast cancer patients. Nat Commun 2020; 11: 3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures 1–13
Supplementary tables 1 and 2