

Abstract
G protein-coupled receptors (GPCRs) that couple to the Gi family of G proteins are key regulators of cell and tissue physiology. Our previous work has revealed new roles for Gαi in regulating the migration of neutrophils and fibrosarcoma cells downstream of activated chemoattractant receptors. We used an intact cell proximity-based labeling approach using BioID2 coupled to tandem mass tag (TMT)-based quantitative proteomics to identify proteins that selectively interacted with the GTP-bound form of Gαi1. Multiple targets were identified and validated for selective biotinylation by a constitutively active BioID2-tagged Gαi1 mutant, suggesting a network of interactions for activated Gαi proteins in intact cells. We showed that active Gαi1 stimulated one candidate protein, PDZ-RhoGEF (PRG) and that despite over 85% sequence identity, Gαi2 poorly activated PRG. We also demonstrated that active Gαi likely regulates polarized myosin light chain kinase phosphorylation through activation of PRG in primary human neutrophils, suggesting functional relevance for this interaction. Identification and characterization of new targets regulated by Gαi both individually, and in networks, provide insights that will aid in the investigation of the functional roles of Gi-coupled GPCRs in multiple biological processes. .
Keywords: GPCR signaling, Gαi proteins, Proximity labeling, Mass spectrometry, Interactome, PDZ-RhoGEF, Cell-migration
Summary:
Proximity labeling approach was used to identify signaling networks and signaling mechanisms downstream of Gi-coupled receptors.
Introduction
G protein-coupled receptors (GPCRs) are a major class of cell surface receptors that regulate multiple physiological and pathophysiological processes in response to various ligands. Activated GPCRs bind to heterotrimeric G proteins consisting of Gα subunits and Gβγ constitutive heterodimers and catalyze the exchange of GTP for GDP on the Gα subunits. Subsequent conformational changes in the Gα subunit cause it to dissociate from the receptor and Gβγ subunits. Both Gα and Gβγ subunits then transduce signals from receptors to downstream effector proteins, including second messenger generating enzymes and ion channels [1–4].
The α-subunits that define the basic properties of heterotrimeric G proteins are divided into four families, Gαs, Gαi/o, Gαq/11, and Gα12/13 [1]. The Gαi/o family is an abundant and ubiquitous class of G protein subunits consisting of various isoforms including Gαi1, Gαi2, Gαi3, and Gαo [5]. Gαi/o coupled GPCRs comprise nearly one hundred receptors for a wide variety of ligands including, opioids, cannabinoids, prostaglandins, histamine, somatostatins, chemokines, and neurotransmitters such as acetylcholine, adrenaline, serotonin, and dopamine [5]. Gαi/o subunit activity is classically associated with adenylate cyclase (AC) inhibition [6].
Gαi/o-coupled chemokine and chemoattractant GPCRs regulate directional cell migration and adhesion, which is involved in tissue formation, wound healing, immune responses, and cancer cell invasion and metastasis [7–9]. Gβγ subunits released from Gi heterotrimers are central mediators of chemokine driven chemotaxis, whereas Gαi has been proposed to function passively through the GDP-GTP exchange-dependent cycling of free and bound Gβγ subunits [10]. Identification of signaling mechanisms specifically downstream of Gαi subunits has been hampered by the fact that perturbations that inhibit Gαi signaling also inactivate Gβγ signaling. For example, modification of Gαi by pertussis toxin (PTX) blocks interactions between the Gαi-βγ heterotrimer and GPCRs, thereby inhibiting both Gα and Gβγ signaling [11]. Similarly, knockout (KO) of specific G protein α subunits, either in mice or with specific short inhibitory RNAs in cell culture, prevents signaling by both Gα and its associated Gβγ subunits [12]. Using a small-molecule Gβγ activator developed in our laboratory, we identified a role for active Gαi (Gαi-GTP) in regulating neutrophil and HT1080 fibrosarcoma cell migration [13, 14]. In these studies, we showed that Gβγ promotes cell adhesion and Gαi-GTP promotes de-adhesion, processes that must be coordinated in order for cells to move [14]. Gαi-GTP regulation of adhesion is independent of cAMP signaling [13]; however, a direct effector regulated by Gαi was not identified.
Methods previously used to identify new effectors of Gαi beyond AC include the yeast-two hybrid system and immunoprecipitation (IP) followed by mass spectrometry (MS) [15–17]. While these methods have successfully identified interacting proteins, they have limitations. IP-MS methods recover only strong interaction partners that survive cell lysis and repeated detergent washes. GPCR-dependent signal transduction processes often involve transient protein-protein interactions that are lost after cell disruption. The yeast two-hybrid systems lack appropriate cellular context, and only fragments of proteins are used to identify binding interactions. Cell context is critical for optimizing interactions between signal transduction components through compartmentalization and interactions with membrane surfaces. Thus, it is likely that multiple G protein interactions may have been missed by these traditional approaches.
To circumvent these challenges, we adopted a proximity labeling approach using BioID2, a promiscuous biotin ligase enzyme, coupled to affinity purification and MS [18, 19]. Our goal was to capture Gαi subunit interactions with potential signal transduction partners and complexes in intact cells of interest. Using this approach, we identified multiple known binding partners of Gαi, including Gβγ subunits and AC. Multiple classes of proteins involved in diverse cellular processes, including cell migration and amino acid transport, were identified as potential interaction partners of active Gαi1. One such protein was the RH family RhoGEF, PDZ-RhoGEF (PRG), also known as ARHGEF11. The role of PRG in regulation of cell migration downstream of Gi-coupled chemoattractant receptors is well characterized, but it is thought to be mediated primarily by additional coupling of these receptors to Gα12/13 subunits [20, 21]. We demonstrated that PRG was an effector of active Gαi1 and Gαi3 but was poorly activated by Gαi2, a highly homologous (~85% identical) Gαi family member. We demonstrated that PRG was activated downstream of Gi-coupled receptors and showed the involvement of Gi in the regulation of PRG in human neutrophils. PRG is relatively ubiquitously expressed [22]; thus its identification as a new Gαi-GTP target has implications for regulation of Rho in various tissues and cell types by Gi-coupled GPCRs.
Results
Rational Design of BioID2 Fused Gαi1
To identify proteins that selectively interact with the active form of Gαi1 using proximity labeling, we fused the promiscuous biotin ligase, BioID2 [19], to Gαi1 (BioID2-Gαi1) and constitutively active Gαi1-Q204L (BioID2-Gαi1-QL). We inserted BioID2 as an internal tag in the αb-αc loop of Gαi1, which has been shown to tolerate GFP insertion, allowing the N and C termini to interact with membranes and receptors [23] (Fig. 1A). In the absence of receptor-dependent activation in cells, Gαi1 is primarily GDP-bound and inactive (referred to as Gαi1), whereas a Q204L mutation in Gαi1 renders it GTPase-deficient, and therefore constitutively GTP-bound and active (referred to as Gαi1-QL) [24]. Comparison of active to inactive Gαi1 allowed us to search for targets that interact selectively with the activated Gαi1. As a control for general promiscuous labeling of proteins because of abundance or simply due to co-residence at the plasma membrane (PM), a PM-targeted BioID2 fused to the C-terminal PM targeting motif of KRas (BioID2-CaaX) was used. Thus, three experimental groups, BioID2-Gαi1, BioID2-Gαi1-QL, and BioID2-CaaX, were used to screen for potential targets that selectively interact with Gαi1-GTP (Fig. 1A, left).
Figure 1. Principle and Experimental Workflow of Proximity Labeling of the Gαi1 Interactome.
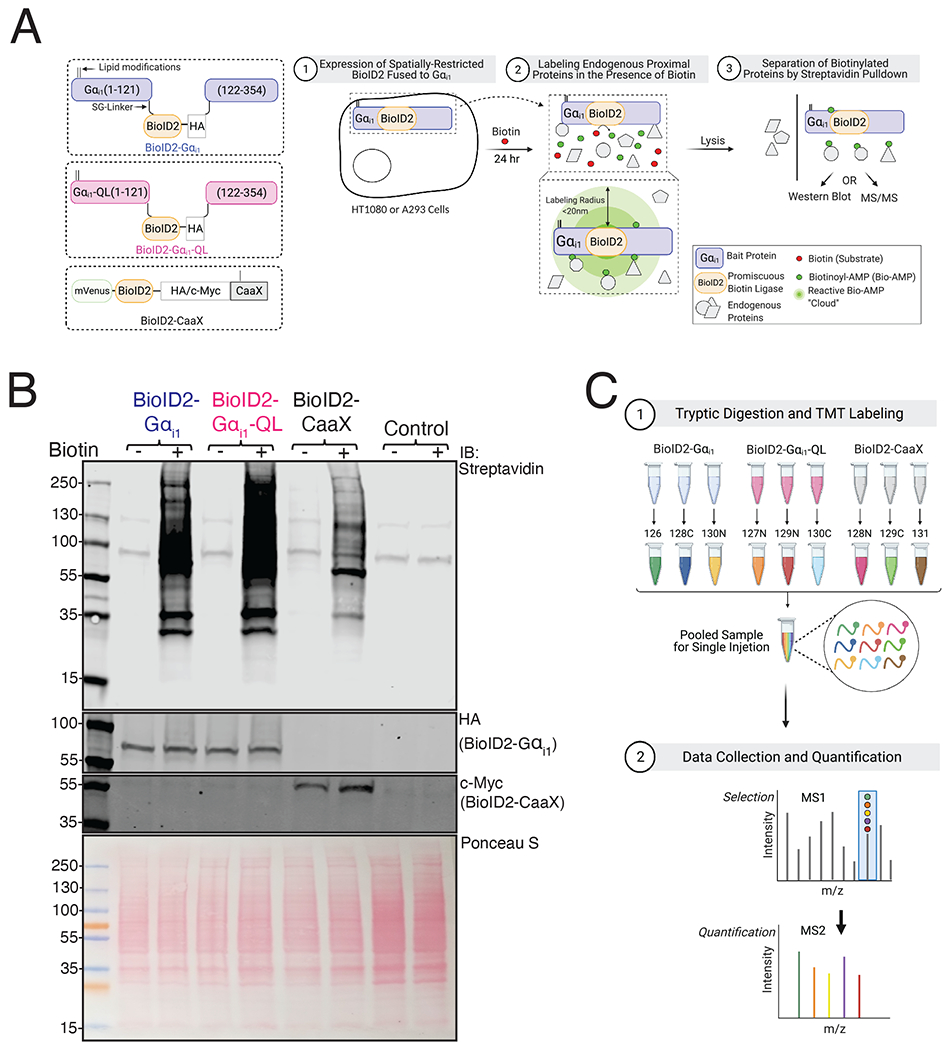
(A) Left: Schematic of BioID2 fusion constructs. BioID2 was inserted between residues Ala121-Glu122 in the αb-αc loop (the first loop of the helical domain) of human Gαi1, flanked by SG-linkers. Palmitoylation and myristylation sites on the Gαi1 subunit and the farnesylation sites on CaaX moiety are labeled as lipid modifications. Right: Schematic of principle and experimental workflow of proximity-based labeling using BioID2. HT1080 cells were used for mass spectrometry experiments and A293 cells were used for pull-down western blot. Cells were transfected with the indicated constructs and labeled for 24 hr in the presence of biotin. Gαi1 fused BioID2 biotinylates proteins in proximity (< 20 nm) in an unbiased manner to identify candidate interacting proteins of Gαi1. (B) A293 cells were transfected with indicated constructs and labeled with biotin for 24 hr. Top: Biotinylated proteins in whole-cell lysates were detected on a streptavidin Western blot. The two bands at 130 and ~90 kDa correspond to endogenously biotinylated proteins in control lanes. Middle: Cell lysates were immunoblotted with Gαi1/2 antisera to detect BioID2-Gαi1 and BioID2-Gαi1-QL and with Myc antibody to detect BioID2-CaaX. Bottom: Ponceau S-stained blot showing total protein loading. Western blots are representative of three independent experiments that yielded similar results. (C) Schematic of sample processing and mass spectrometry analysis. Samples pulled down using streptavidin beads were digested with trypsin and labelled with a TMT tag. Biological triplicate samples of lysates of cells expressing BioID2-Gαi1, BioID2-Gαi1-QL and BioID2-CaaX were pooled and resolved by LC-MS and the data was analyzed using proteome discover.
BioID2 Fused Gαi1 Localizes Predominantly to the PM and Gαi1-QL Inhibits cAMP Accumulation
To characterize the functionality of BioID2 fused Gαi1 proteins, we examined their localization in A293 cells. All three proteins, BioID2-Gαi1, BioID2-Gαi1-QL, and BioID2-CaaX, localized predominantly to the PM (fig. S1A). Next, we evaluated the ability of Gαi1-QL to inhibit AC by measuring inhibition of forskolin (Fsk)-stimulated cyclic-AMP (cAMP) production using a cAMP biosensor (cAMP-Glo™). As expected, both untagged and BioID2 tagged Gαi1-QL reduced the rate and extent of cAMP generated upon Fsk addition (fig. S1B). Conversely, the Gαi1-GDP counterparts did not affect Fsk stimulated cAMP accumulation. Western blot confirmed similar abundance of these proteins (fig. S1C). These experiments established that Gαi1-QL fused with BioID2 can localize to the PM, inhibit AC, and thus behave similarly to the untagged counterpart.
BioID2 Fused with Gαi1 Biotinylates Endogenous Proteins
We tested the biotin labeling efficiency of BioID2 fused Gαi1 proteins in A293 cells. Cells transiently transfected with the indicated cDNA clones were incubated with biotin and lysates were probed with fluorescently tagged streptavidin (Fig. 1A, right). Multiple proteins were biotinylated in BioID2-Gαi1, BioID2-Gαi1-QL, and BioID2-CaaX samples, and biotinylation was dependent on BioID2 and biotin (Fig. 1B). There were differences in the total biotinylation pattern amongst the three experimental groups, suggesting that the three fusion proteins labeled endogenous proteins differentially.
Proximity Labeling-Coupled MS in HT1080 Fibrosarcoma Cells
A goal was to identify proteins regulated by Gαi that could be involved in cell migration downstream of chemokine or chemoattractant receptors. HT1080 fibrosarcoma cells express FPR1 receptors, adhere and migrate on fibronectin-coated surfaces, and are comparatively easy to grow and transfect relative to neutrophil-like cells. Roles for Gαi1 and Gβγ in cell adhesion and migration have been previously established in these cells [14, 25]. For these reasons, we chose HT1080 cells for the proximity labeling experiments to increase the probability of identifying effectors of Gαi1 relevant to cell migration.
In HT1080 cells transfected with BioID2-Gαi1, BioID2-Gαi1-QL, or BioID2-CaaX, BioID2 fused Gαi1 subunits were expressed at levels similar to endogenous Gαi (fig. S1D). To perform quantitative comparison of biotinylated proteins after purification with streptavidin beads, each sample was labeled with a unique isobaric tandem mass tag (TMT). This allowed triplicate samples for each group to be pooled and analyzed in a single MS run so that relative protein abundances could be compared (Fig. 1C).
MS Data Analysis Identifies Multiple Known and Candidate Interacting Proteins
We detected several proteins known to interact with Gαi including, Gβ and γ subunit isoforms, which were selectively enriched in the BioID2-Gαi1 samples relative to BioID2-Gαi1-QL, as expected (Fig. 2A). Several isoforms of AC were detected, but there was no statistically significant difference between the BioID2-Gαi1-QL and BioID2-Gαi1 samples. Ric8A was also equally labeled by BioID2-Gαi1-QL and BioID2-Gαi1. Gαi-GTP effectors, GPRIN1 and RASA3 [16, 26], were highly enriched in BioID2-Gαi1-QL samples relative to BioID2-Gαi1 samples (Fig. 2A). Multiple receptors were also identified, but most were not significantly enriched in either the BioID2-Gαi1-QL or BioID2-Gαi1 samples (Data File S1).
Figure 2. Proximity Labeling Proteomics Results.
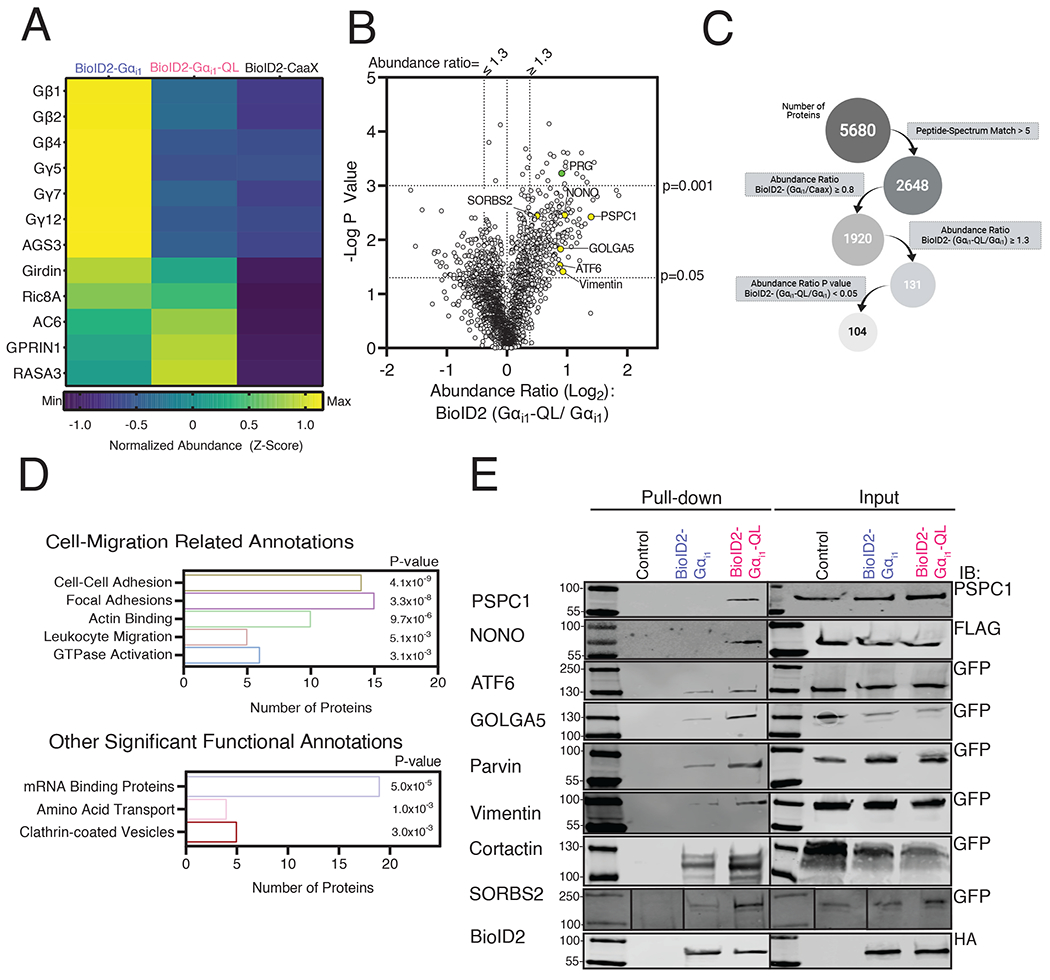
(A) Heat map showing the relative changes in abundance of known binding partners of Gαi that were identified in the mass spectrometry analysis. (B) Volcano plot of all high confidence proteins identified for which the BioID2-Gαi1/BioID2-CaaX ratio was greater than 0.8. PRG is indicated by the green dot, and the candidate proteins investigated in Fig. 2E are indicated by the yellow dots. (C) Schematic showing filtering criteria for selection of proteins enriched in BioID2-Gαi1-QL samples relative to BioID2-Gαi1. (D) Representative classes from GO analysis of proteins from (B) that met the final criteria in (C). P values were generated with the DAVID software. (E) Cells were transfected with the indicated epitope or GFP tagged proteins and BioID2-Gαi1 or BioID2-Gαi1-QL and labeled with biotin for 24 hr. Streptavidin pulldowns were performed with cell lysates and subjected to Western blotting with the indicated antibodies. Western blots are representative of experiments performed twice that yielded similar results.
Overall, ~5000 proteins were isolated and identified (Fig. 2B). We selected proteins with a minimum of 5 peptides assigned to each protein to ensure robustness of the data. We also filtered the data to include only proteins for which the ratio of normalized abundance for the BioID2-Gαi1 and BioID2-CaaX roughly equivalent or greater (BioID2-Gαi1/ BioID2-CaaX > 0.8). The rationale behind this criterion was that because BioID2-CaaX labels proteins at the PM based on proximity within the compartment, proteins labeled to a similar extent by BioID2-CaaX and BioID2-Gαi1 likely colocalized with BioID2-Gαi1 at the PM. Proteins labeled to a greater extent by BioID2-Gαi1 than BioID2-CaaX could be PM resident proteins that selectively interact with Gαi1 in its inactive GDP-bound state but could also be proteins labeled by BioID2-Gαi1 in other compartments or cytosolic proteins that interact with Gαi1 at the PM.
To identify proteins that selectively interact with Gαi1-QL, we further filtered the data and included only those proteins with BioID2-Gαi1-QL/BioID2-Gαi1 normalized abundance ratio ≥ 1.3 and a P-value < 0.05 (Fig. 2C). This resulted in a list of 104 candidate proteins (Fig. 2C, fig. S2, Data File S2). These 104 Gαi1-QL enriched proteins were analyzed using DAVID gene ontology software to identify classes of proteins involved in different cellular processes [27]. Several enriched targets regulate various aspects of cell migration (Fig. 2D top, Data File S3). These data suggest that active Gαi may regulate cell migration through a protein interaction network rather than just a single target. Other classes of proteins identified with high confidence were mRNA binding proteins, amino acid transporters, and proteins involved in clathrin-mediated endocytosis (Fig. 2D, bottom, Data File S3).
Most of these proteins have not been previously identified as targets of Gαi. To further validate selective enrichment of potential Gαi binding proteins with BioID2-Gαi1-QL, we tested a subset of enriched proteins based on the availability of epitope or fluorescent protein (FP)-tagged cDNA clones in a proximity labeling-coupled biotinylation Western blot assay. These proteins included PSPC1 (paraspeckle component 1), p54 (nuclear RNA-binding protein, 54-kD or NONO), ATF6 (Activating Transcription Factor 6), SORBS (Sorbin and SH3 domain-containing protein 2 or ArgBP2), GOLGA5 (Golgin A5) and Vimentin (Fig. 2B, highlighted in yellow). We also included two proteins of interest that did not quite reach statistical significance, parvin and cortactin.
Each protein-coding cDNA was individually co-transfected with BioID2-Gαi1-QL, BioID2-Gαi1, or control plasmid in A293 cells (Fig. 2E). Cells were treated with biotin and subjected to streptavidin pull-down. Of the 12 proteins tested, 8 showed enriched labeling by BioID2-Gαi1-QL relative to BioID2-Gαi1 (Fig. 2E), confirming preferential interaction between the active form of Gαi1 and the candidate proteins. Co-transfection of these cDNAs with BioID2 tagged Gαi1 cDNAs did not increase expression of the proteins, suggesting that increased biotin labeling was not due to an increase in expression. These data support the idea that many of the other proteins among the 104 proteins enriched in the BioID2-Gαi1-QL samples were in close proximity to the active form of Gαi1.
Overall, the data suggest that Gαi regulates multiple classes of cellular processes through mechanisms that involve coordinated network interactions with a variety of protein targets.
PRG Selectively Interacts with Active Gαi1
One protein of interest relevant to cell migration and significantly enriched in BioID2-Gαi1-QL relative to BioID2-Gαi1 samples was PDZ-RhoGEF (PRG and also known as ARHGEF11), a Rho guanine nucleotide exchange factor (Fig. 2B, green dot and 3A). PRG biotin labeling by BioID2-Gαi1 and BioID2-CaaX was similar based on the MS quantification, suggesting that labeling by inactive BioID2-Gαi1 was primarily due to membrane proximity (Fig. 3A). We decided to pursue PRG for several reasons. PRG was strongly enriched in the BioID2-Gαi1-QL samples, and PRG has a role in regulation of neutrophil migration downstream of Gi-coupled chemoattractant receptors [20, 21]. PRG localizes to the rear of migrating neutrophils and activates Rho and myosin-dependent tail retraction during migration in response to chemoattractants [21]. The abundance of endogenous PRG was similar in HT1080 cells transfected with BioID2-Gαi1, BioID2-Gαi1-QL, or BioID2-CaaX (Fig. 3B), showing that the high PRG abundance in BioID2-Gαi1-QL samples was not simply due to increased PRG expression.
Figure 3. BioID2-Gαi1-QL Interacts with PRG in Cells.
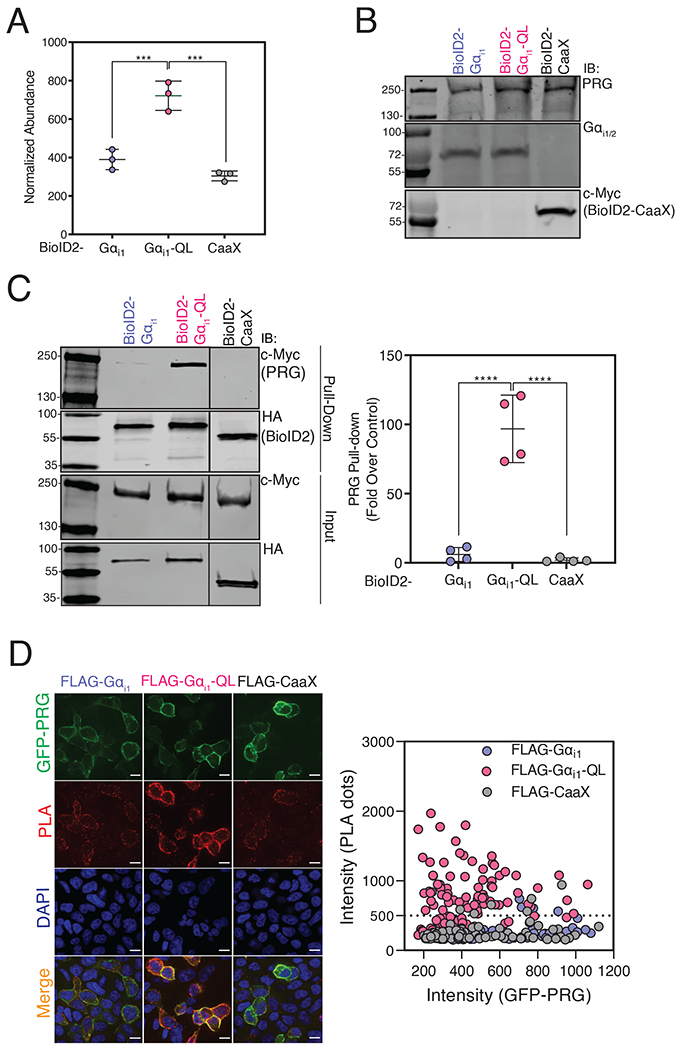
(A) Normalized abundance of PRG was quantified by MS in cells expressing BioID2-Gαi1, BioID2-Gαi1-QL, or BioID2-CaaX. The data represent the mean ± SD of three independent experiments. (****P<0.0001, one-way ANOVA with Tukey’s multiple comparisons test). (B) HT1080 cells were transfected with the indicated constructs and whole cell lysates were resolved by Western blot. (C) A293 cells were transfected with PRG and BioID2-Gαi1, BioID2-Gαi1-QL, or BioID2-CaaX and labeled with biotin for 24 hr. Cell lysates were subjected to streptavidin pulldowns. Left: Representative Western blots of PRG in streptavidin pull-downs from cells expressing BioID2-Gαi1, BioID2-Gαi1-QL, or BioID2-CaaX. Right: Quantitation (shown as mean ± SD) of three independent experiments normalized to total PRG. ***P < 0.001, ****<0.0001, one-way ANOVA with Tukey’s multiple comparisons test). (D) PLAs were performed in cells transfected with GFP-PRG and APEX2-FLAG-tagged Gαi1-WT, Gαi1-QL, or CaaX. Left: Representative images from three randomly selected fields show GFP-PRG (green), PLA reaction (red), merge (orange) and DAPI (blue). Scale bar, 10μm. Right: The intensity of the PLA signal (y-axis) was plotted against GFP-PRG expression (x-axis). For each experiment, ~100 cells per condition were analyzed, and the data are shown from one of three independent experiments that yielded similar results (See fig. S5).
To independently validate PRG enrichment in BioID2-Gαi1-QL samples by MS, we labeled A293 cells co-expressing PRG with BioID2-Gαi1, BioID2-Gαi1-QL, or BioID2-CaaX with biotin and performed streptavidin affinity pull-downs. PRG was highly enriched in streptavidin pulldowns from BioID2-Gαi1-QL-transfected cells compared to those transfected with BioID2-Gαi1 or BioID2-CaaX (Fig. 3C). Next, we used a proximity ligation assay (PLA) to test for interactions in A293 cells. A robust PLA signal in cells co-transfected with GFP-PRG and APEX-FLAG-Gαi1-QL. The low PLA signal between GFP-PRG and the controls APEX-FLAG-Gαi1-WT or APEX-CaaX may be due to colocalization at the PM, resulting in background bystander proximity labeling (Fig. 3D). These data further support selective interactions between Gαi1-GTP and PRG (Fig. 3D, fig. S5B).
Gαi1 Activates the RhoGEF Activity of PRG
These findings prompted us to investigate whether Gαi1 could activate PRG. An SRE luciferase (SRE-Luc) reporter for Rho activation [28] was used to study PRG activation in A293 cells co-transfected with PRG and Gαi1-WT or Gαi1-QL. Strong synergistic activation of SRE-Luc was observed upon PRG and Gαi1-QL co-transfection, but not upon transfection of either component alone (Fig. 4A, left), suggesting that Gαi1-QL activates PRG to stimulate Rho activation. Activation of PRG depended on the Gαi1 activation state because Gαi1-WT did not significantly increase PRG activity (Fig. 4A, left). PRG activation by Gαi1-QL was concentration-dependent (Fig. 4A middle). Gαi1-WT and Gαi1-QL were expressed at comparable levels, and PRG expression was similar in all three conditions (Fig. 4A, right).
Figure 4. Gαi1-GTP Activates PRG Activity.
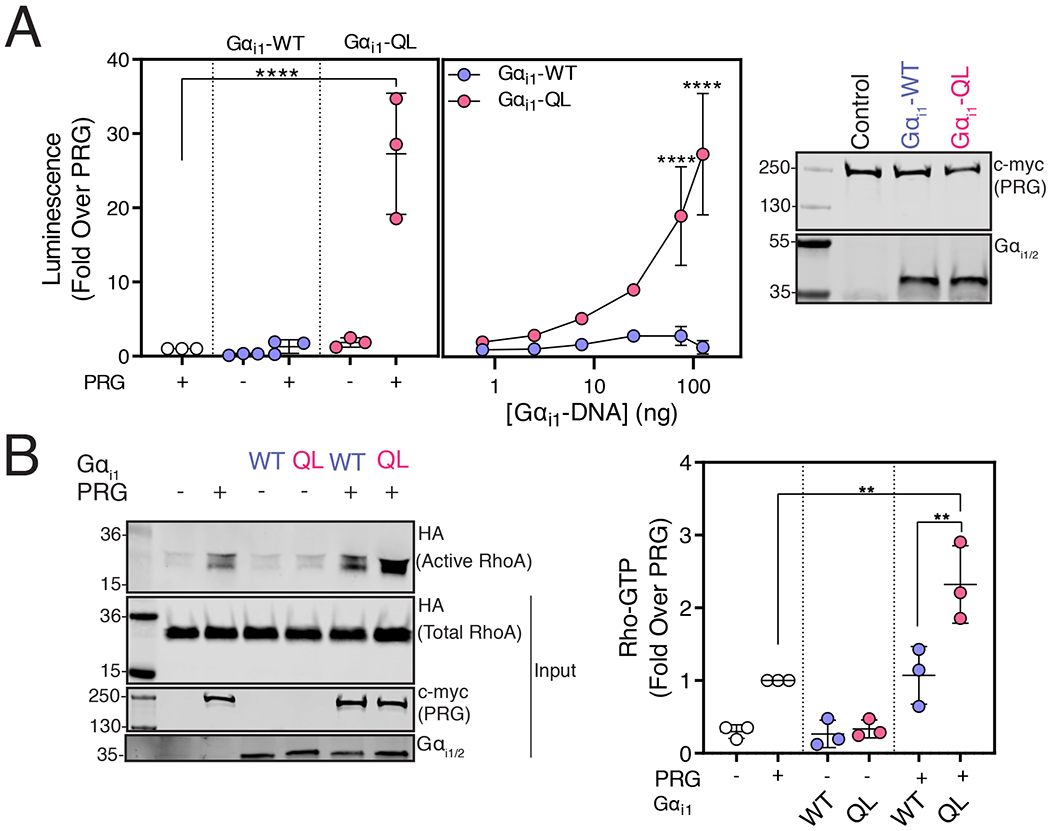
(A) A293 cells were transfected with cDNAs encoding SRE-Luciferase, PRG, and either Gαi1 or Gαi1-QL for 20 hr. Left and middle: Luminescence was measured in serum-starved cells 10 min after the addition of One-Glo™ reagent. The data represent the mean ± SD of three independent experiments. Right: Representative Western blots showing relative expression of various cDNA constructs in A293 cells. Data are representative of three independent experiments that yielded similar results. (B) A293 cells were transfected with the indicated cDNA constructs and cell lysates were incubated with GST-Rhotekin beads. Left: Representative Western blots showing bound RhoA-GTP and relative expression of transfected constructs. Right: Quantification (shown as mean ± SD) of three independent experiments, normalized to total RhoA. (**P < 0.01, ****<0.0001, Panel A left and B: one-way ANOVA with Tukey’s multiple comparisons test, Panel A middle: two-way ANOVA with Tukey’s multiple comparisons test).
To validate activation of PRG by active Gαi1, we measured active RhoA (RhoA-GTP) with a Rhotekin pull-down assay, in which RhoA binds to Rhotekin-Glutathione S-transferase (GST) in an activation-dependent manner. PRG expression led to increased RhoA-GTP levels in Rhotekin-GST pulldowns compared to control conditions, and this enhancement was further significantly increased with co-expression of Gαi1-QL but not of Gαi1-WT (Fig. 4B). These assays establish that GTP-bound Gαi1 activates the RhoGEF activity of PRG.
PRG-dependent SRE-Luc Activation is Specific to Gαi/o Proteins
Activation of PRG by Gαi1 was unexpected given that PRG is reported to be regulated by Gα12/13 [22, 29]. We tested whether PRG as measured by the SRE-Luc reporter was activated by other families of Gα subunits. Transfection of both WT and QL versions of Gαq, Gα12, Gα13 increased the activity of the SRE-Luc reporter compared to control conditions, likely due to activation of endogenous RhoGEFs in A293 cells (fig. S3A), although for reasons that are not clear, activation was equal for the WT and QL forms of these proteins. Under the conditions of this assay, Gαq, Gα12, Gα13 co-expression with PRG did not increase PRG-dependent SRE-Luc activation.
Gαi1 and Gαi3 Strongly Activate PRG, but Gαi2 is a Poor Activator
We tested Gαi family member isoforms for their ability to activate PRG. GαoA-QL, GαoB-QL showed a small increase in PRG activation whereas Gαz did not activate PRG (fig. S3B). The Gαi isoforms Gαi1, Gαi2, and Gαi3 are highly homologous, with greater than 85% amino acid sequence identity [6]. Gαi1-QL and Gαi3-QL strongly activated PRG, whereas Gαi2-QL did not (Fig. 5A). Gαi1 and Gαi2 were expressed at approximately similar levels (Fig. 5B), and Gαi3 expression could not be compared because the antibody does not recognize Gαi3. Gαi-QL versions of all three subtypes showed similar inhibition of Fsk-stimulated cAMP production (Fig. 5C, fig. S5A). To corroborate this finding, we labeled A293 cells co-expressing PRG with BioID2-tagged WT and QL versions of the Gαi isoforms in A293 cells with biotin and streptavidin affinity pull-downs were performed. PRG was highly enriched in the streptavidin pulldowns from BioID2-Gαi1-QL-expressing cells (Fig. 5D). Enrichment was lower in pulldowns from BioID2-Gαi3-QL-expressing cells and very low in those from BioID2-Gαi2-QL-expressing cells (Fig. 5D). Effector selectivity amongst these three highly related Gαi isoforms has not been previously reported.
Figure 5. PRG Activation is Gαi Isoform Specific.
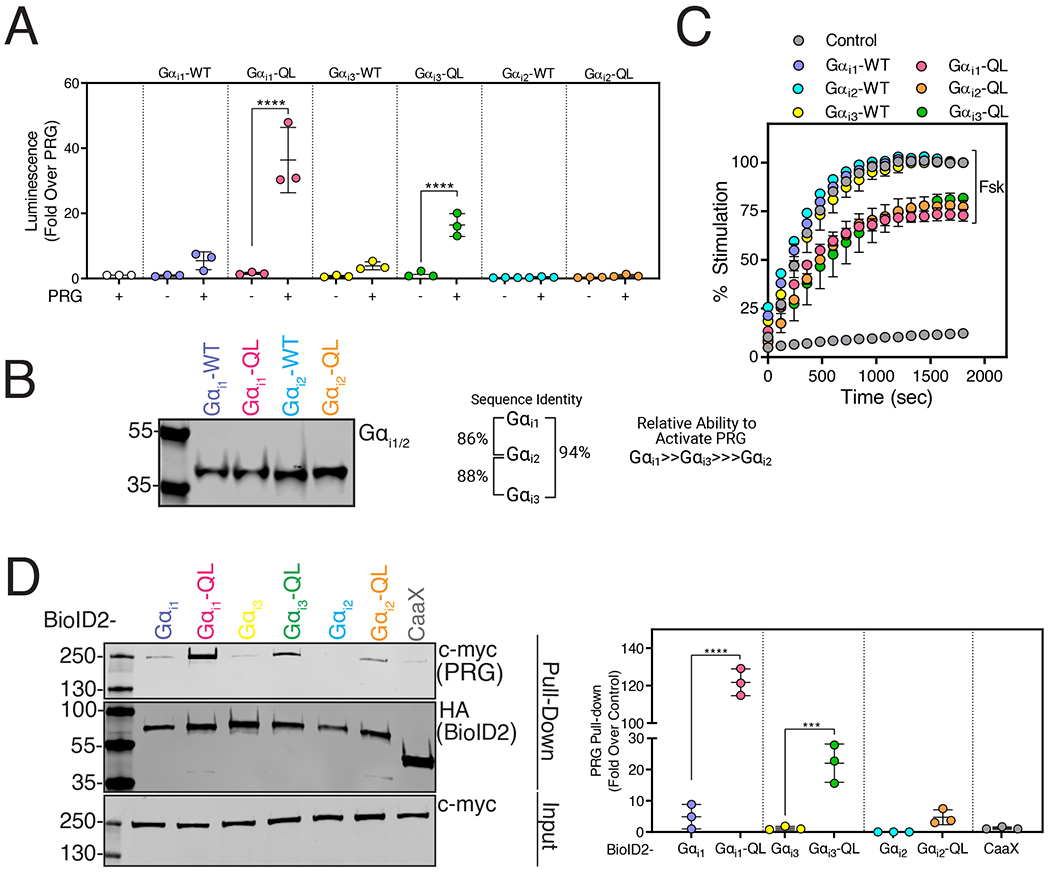
(A) A293 cells were transfected with cDNAs encoding SRE-Luciferase, PRG, and either Gαi1-QL, Gαi2-QL, or Gαi3-QL. Luminescence was measured in serum-starved cells 10 min after the addition of One-Glo™ reagent. The data represent the mean ± SD of three independent experiments. (B) Western blot showing relative expression of Gαi1 and Gαi2 constructs in A293 cells. Data are representative of three independent experiments that yielded similar results. (C) Cells were transfected with cAMP Glosensor™ and WT and QL versions of Gαi1, Gαi2, or Gαi3 for 24 hr. Luminescence was measured for 30 min (x-axis) after Forskolin (Fsk) stimulation and represented as % stimulation (y-axis) relative to the maximum signal in the respective WT group with 1 μM Fsk treatment. Data shown as mean ±SD are representative of three independent experiments (See fig. S5A for replicate experiments). (D) A293 cells were transfected with PRG and BioID2-Gαi1-QL, BioID2-Gαi3-QL, or BioID2-Gαi2-QL and labeled with biotin for 24 hr. Cell lysates were subjected to streptavidin pulldowns. Left: Representative Western blots of PRG in streptavidin pull-downs as well as of expression of BioID2-Gαi1, Gαi1-QL and BioID2-CaaX. Right: Quantitation shown as mean ± SD of three independent experiments normalized to total PRG. (***P <0.001, ****<0.0001, one-way ANOVA with Tukey’s multiple comparisons test).
Gαi1-QL is Specific for PRG Mediated RhoA Activation and Does Not Require Gα12/13
As mentioned above, Gα12/13 is reported to bind to PRG [22, 29], leading us to investigate whether Gα12/13 was required for Gαi1-QL mediated PRG activation. Gαi1-QL co-transfected with PRG robustly increased the activity of the SRE-Luc reporter, and knockout of Gα12/13 by CRISPR/Cas9-mediated gene editing did not affect PRG activation (fig. S4A, left). Gα13 KO was confirmed by Western blotting (fig. S4A, right). These findings demonstrate that Gαi1-GTP can activate RhoGEF activity of PRG in the absence of Gα12 or Gα13.
Next, we tested the ability of Gαi1-QL to activate other members of the DH-PH family of RhoGEFs in the SRE-Luc reporter assay (Fig. S4B, left). We co-transfected Gαi1-QL with GFP-p115 RhoGEF (Lsc), GFP-LARG, GFP-AKAP13 (Proto-Lbc, ARHGEF13), and GFP-PRG. Gαi1-QL robustly increased SRE-Luc reporter activity when co-transfected with PRG. Gαi1-QL also activated p115RhoGEF, although to a much lesser extent as compared to PRG activation. Gαi1-QL did not activate LARG or AKAP13 to a statistically significant extent. Western blotting demonstrated expression of the RhoGEFs at varying levels (fig. S4B, right).
The Formyl Peptide Receptor 1 (FPR1) Activates PRG through Gαi1
To determine whether PRG could be activated by Gαi downstream of a Gi-coupled receptor, we assayed PRG activation with the SRE-Luc reporter. In A293 cells stably expressing FPR1 (A293-FPR1), application of fMLF activated SRE-Luc reporter activity in a concentration-dependent fashion only when PRG and Gαi1-WT were co-transfected and not when PRG or Gαi1-WT were transfected alone (Fig. 6A). In contrast, fMLF did not increase reporter activity in cells co-transfected with Gαi2-WT and PRG (Fig. 6A), confirming that Gαi2-QL poorly activates PRG. Application of the Gαi blocker PTX significantly inhibited the fMLF-dependent increase in PRG activation in cells expressing FPR1, Gαi1 and PRG (Fig. 6B), thus providing further evidence that FPR1 activation of PRG depends on Gαi.
Figure 6. Activation of PRG Downstream of Gαi-coupled Receptor FPR1.
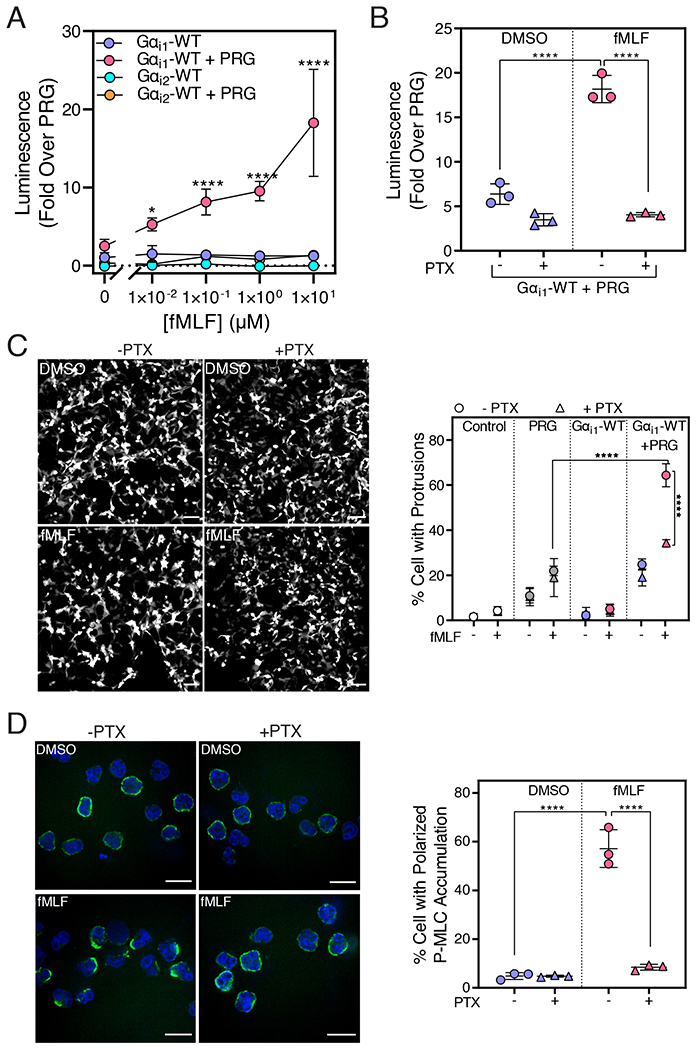
(A) A293 cells stably expressing FPR1 (A293-FPR1) were transfected with cDNAs encoding SRE-Luc, PRG, and Gαi1-WT or Gαi2-WT and incubated for 12 hr in serum free media containing fMLF (0.01, 0.1, 1 and 10 μM) or DMSO. Data from three independent experiments were plotted as mean ± SD. (B) A293-FPR1 cells were transfected and incubated for 12 hours in serum-free media containing PTX (100 ng/mL) and fMLF (10 μM). Luminescence was measured 10 min after the addition of the One-Glo™ reagent. The data combined data (mean ± SD) of three independent experiments. (C) A293-FPR1 cells were transfected with PRG, Gαi1-WT, and YFP for 36 hr. 24 hr after transfection, cells were treated with PTX (100 ng/mL) for 12 hr. Cells were stimulated with 100 nM fMLF and live cell video microscopy was performed for 40 min. Left: Representative images of A293-FPR1 cells expressing PRG + Gαi1-WT and treated with fMLF or DMSO are shown. Scale bar, 100 μm. Right: Quantitative analysis of the percentage of cells with dynamic protrusions shown as mean ± SD from three independent experiments. For each experiment, >500 cells per condition were analyzed in a blinded manner. (D) Human neutrophils were pretreated or not with PTX (500 ng/mL) for 2 hr, allowed to adhere to the fibronectin-coated surface for 15 min, and stimulated with 10 nM fMLF for 5 min. The cells were stained with a P-MLC antibody and DAPI and imaged with confocal microscopy. Left: Representative images from three randomly selected fields of view are shown. Right: Total number of cells and cells with asymmetric P-MLC localization were counted in a field and the % cells with polarized P-MLC localization (mean ± SD) from three independent experiments were plotted. (****p < 0.0001, Panel A: Two-way ANOVA with Tukey’s multiple comparisons test, Panel B, C, D: One-way ANOVA with Tukey’s multiple comparison test). Scale bar, 10 μm.
RhoA is a major regulator of cytoskeletal rearrangement and can induce peripheral protrusions [30, 31]. As an alternate measure of RhoA activation, we examined fMLF-stimulated dynamic protrusion formation in FPR1-A293 cells (Fig. 6C, left, Movies S1–4). The percentage of cells that formed protrusions in cells transfected with PRG alone was slightly but significantly higher than in cells transfected with vector control and was not affected by PTX treatment (Fig. 6C, right). Co-expression of Gαi1-WT with PRG significantly increased the percentage of cells with dynamic protrusions only when cells were stimulated with fMLF, and the activation was inhibited by PTX. Together, these data support the hypothesis that PRG RhoGEF activity can be activated downstream of Gi-coupled GPCRs through Gαi1.
Evidence for involvement of Gαi in fMLF-dependent PRG Activation in Human Neutrophils
To understand the physiological relevance of the Gαi-dependent mechanism for Rho regulation, we examined the role of Gi signaling in human neutrophils. Of the multiple G protein activated RhoGEFs expressed in neutrophils, PRG mediates Rho-dependent polarized accumulation of phosphorylated (at Ser19)-myosin light chain (P-MLC) at the trailing edge of migrating neutrophils [20, 21]. This effect has been proposed to result from FPR1-dependent Gα13 activation [20] in part because PTX treatment of the neutrophil-like cell line HL60 only partially inhibits fMLF-dependent Rho activation and asymmetric localization of P-MLC [20]. To determine if Gαi activates endogenous PRG in human neutrophils, we examined polarized P-MLC staining in human neutrophils after stimulation with a physiologically relevant concentration of fMLF. In DMSO treated neutrophils, P-MLC was uniformly distributed at the surface of cells. Stimulation with 10 nM fMLF promoted strong polarized P-MLC accumulation (Fig. 6D, left). In cells pre-treated with PTX, fMLF failed to promote polarized accumulation of P-MLC (Fig. 6D, right). Because PRG mediates the Rho-dependent polarization of P-MLC, this finding suggests that at physiological concentrations of fMLF, PRG activation in human neutrophils occurs through a Gαi-dependent mechanism.
Discussion
In this study, we used an unbiased approach to identify effectors of Gαi and focused on identifying proteins involved in chemoattractant-dependent cell migration. The proximity labeling method used in this study has advantages over previously used methods [15–17], including allowing detection of transient complex formation in the context of an intact cell. One potential drawback is that apart from detecting direct or indirect interactions, proximity-based methods can also identify the proteins that do not interact but are located within 20 nm, perhaps in the same cellular compartment. However, the ratiometric enrichment strategy employed here comparing constitutively active Gαi to inactive Gαi largely circumvented this issue. GTP binding to Gα subunits lead to conformational changes that drive new protein-protein interactions. In principle, selective enrichment in the GTP-bound state could result from a few processes: (i) GTP-selective protein-protein interactions causing an increase in proximity, (ii) GTP driven changes in Gαi subunit compartmentalization within the membrane or cell, or (iii) Gαi-GTP driven changes in selective protein expression. There is some evidence that Gαi activation leads to changes in compartmentalization within the PM but not for larger-scale changes in subcellular distribution [32]. There is overwhelming evidence that G protein α subunit activation results in conformational changes that drive new protein interactions [3]. G protein targets may not be easily identified by traditional methods due to their often low abundance, cell context specificity, and often transient interactions. Thus, proximity labeling with proper controls is a viable approach for investigating Gαi subunit interactions in intact cells and may represent a general approach for identification of signaling partners and networks downstream of G proteins.
Multiple known Gαi binding partners were identified in the proximity labeling MS experiments. Gβ subunits, Gγ subunits, and GPSM1 (AGS3) [33] were enriched in inactive Gαi1 samples, whereas GPRIN1[26], Rasa3 [16], and GIV [34] were enriched in Gαi1-QL samples. Many AC isoforms were identified but somewhat surprisingly were equally enriched in both Gαi1 and Gαi1-QL samples. It has been suggested that Gαi-AC complexes can be formed regardless of the activation state of Gi and that AC activation results from conformational rearrangement of the prebound G protein heterotrimer [35–37]. GPCRs were detected, but the overall labeling efficiency was low for many of them, leading to lower confidence in identification for some of the receptors (Data File S1), and most were not significantly enriched with either Gαi1 or Gαi1-QL. This finding could be because of their low abundance or lack of preference of the inactive receptors for Gαi1 or Gαi1-QL. Overall, many bona fide Gαi targets were identified in the MS screen, validating the method, suggesting that additional Gαi effectors are likely to be identified.
Proteins enriched in the Gαi-QL samples were grouped into unique functional categories, including classes related to cell migration (Fig 2C, Data File S2). The identification of multiple proteins suggests that the role of Gαi in cell migration may involve a network of protein-protein interactions similar to the role of Gβγ in cell migration. These potential interactions remain to be independently investigated, but many are likely “true” interaction partners. Indeed, the biotinylation validation assays with a subset of BioID2-Gαi1-QL enriched proteins, selected based on availability of the cDNA clones in DNA repositories, (Fig. 2E) support the idea that many of these proteins selectively interact with activated Gαi. In a complex process such as chemoattractant-dependent cell migration, Gαi is likely to play more than one role.
From the proteins identified in this study, we focused on PRG, a RhoGEF with well-established roles in cell migration, and demonstrated that Gαi1 strongly activated PRG in a GTP and concentration-dependent manner in intact cells. We also demonstrated Gαi-dependent PRG regulation downstream of the Gi-coupled chemoattractant receptor, FPR1, in A293 cells. In differentiated HL60 neutrophil-like cells, activation of FPR1 leads to PRG and Rho-dependent accumulation of P-MLC at the trailing edge, where it is responsible for tail retraction as the cell moves forward [20, 21], a process that has been proposed to be mediated by Gα13. In our experiments, stimulation of primary human neutrophils with a physiological concentration fMLF promoted polarized P-MLC accumulation that was inhibited by PTX, suggesting this phenotype depends on Gαi signaling. In the previous work performed with 100 nM fMLF, PTX had a partial effect on P-MLC polarization, and fMLF dependent RhoA activation was partially inhibited by PTX [20]. In contrast to this study, we used primary neutrophils and a physiological concentration of fMLF (10 nM). Our data suggest that at a low physiological concentration of chemoattractant, Gαi regulation of PRG is critical for tail retraction during cell migration, and we propose that Gα13 may perform a more dominant role in migrating neutrophils at higher concentrations of chemotactic ligand.
It is accepted that Gα13 activates RH-RhoGEFs such as PRG, and convincing evidence shows that PRG activity is regulated downstream of Gα12/13 [22, 38]. Strong data has demonstrated G13 binding to PRG and clear regulation of PRG by G12/13 coupled GRCRs in physiological settings [39], but we did not observe activation of PRG by Gα12 or Gα13 in our SRE based co-transfection experiments. Gα12 and Gα13 apparently activated endogenous RhoGEFs in our assays because they promoted SRE activation without PRG co-transfection, but surprisingly the activation was not different between WT and constitutively active forms. The reasons for these discrepancies are unclear, but demonstration of Gα12/13-dependent regulation of PRG in cell-based assays similar to those used in our studies are limited [40, 41]. Nevertheless, we observed a clear activation of PRG by Gαi that was strongly dependent on the activation state of Gαi, lending confidence to our results. Additionally, there is precedence for regulation of PRG by G proteins other than G12/13. Gαs-mediated activation of Cdc42 has been reported to require PRG [42].
From our studies we cannot conclude whether the mechanism for Gαi-dependent regulation of PDZ-RhoGEF involves direct protein-protein interactions, or if a higher order complex is involved. The strong stimulation of RhoGEF activity of PRG by Gαi suggests direct interactions, but further in vitro reconstitution experiments with purified components will be required to demonstrate direct PRG regulation by Gαi.
Of the three highly homologous Gαi isoforms, Gαi1 and Gαi3 activated PRG, but Gαi2 was a poor activator [13]. Gαi1-QL shares 86% amino acid identity with Gαi2 [43, 44], and the three Gαi isoforms inhibit AC with similar potency and efficacy [6]. These three Gαi isoforms have been studied for nearly three decades [43], and no molecular differences with respect to effector regulation have been demonstrated. Mouse neutrophils express Gαi2 and Gαi3 at similar amounts, and neutrophils from Gαi3 KO mice showed reduced ability to migrate toward a chemotactic stimulus, whereas Gαi2 KO resulted in loss of their ability to arrest [45, 46]. Therefore, this divergent role could be attributed to differential effector regulation by different Gαi subtypes.
PRG regulates various biological processes, including neurite retraction [47], cell migration, and proliferation of mouse embryonic fibroblasts [48]. Thus, these findings have broad implications for signaling by Gi-coupled receptors. Overall, identification of signaling pathways and networks regulated by Gi-coupled GPCRs has the potential to affect our understanding of the biology regulated by these ubiquitous and pharmacologically important receptors. Identification of proteins from multiple functional families indicates that Gi proteins likely play a central role in several biological processes through signaling networks that do not result in adenylate cyclase inhibition. Fuller characterization of individual candidate interactors or networks is warranted to validate and understand the roles of these interactions in Gi-coupled GPCR biology. Ultimately, discovery of new GPCR biology will lead to a greater understanding of disease pathologies, identification of new therapeutic targets, and development of innovative therapeutic strategies.
Materials and Methods
Plasmid cDNA Constructs
BioID2 fused N-terminally with c-myc tag and C-terminally with mVenus followed by CaaX PM targeting motif (KKKKKKSKTKCVIM, derived from the C terminus of KRas), was a gift from Dr. Sundeep Malik, University of Rochester. C-terminally c-myc tagged full length PRG cDNA construct in mammalian expression vector was a gift from Dr. John Tesmer, Purdue University. A293-FPR1 stable cell lines were generated by Dr. Jesi To, University of Michigan, and A293-Gα12/13 CRISPR cells were a gift from Dr. Graeme Milligan, University of Glasgow, UK. The following plasmids were obtained from Addgene. MCS-BioID2-HA (Kyle Roux, Plasmid #74224) [19], pCDNA3-HA PSPC1(Yuh-Shan Jou, Plasmid #101764) [49], FLAG-p54 (Benjamin Blencowe, Plasmid #35379) [50], pEGFP-ATF6-(S1P-) (Ron Prywes, Plasmid #32956) [51], GFP-nArgBP2 (Guoping Feng, Plasmid #74514) [52], GFP-Golgin-84-TEV (Ayano Satoh, #42108), mEmerald-Parvin-C-14 (Michael Davidson, Plasmid #54214), EGFP-Vimentin-7 (Michael Davidson, Plasmid #56439), pGFP-Cortactin (Kenneth Yamada, Plasmid #50728). All Gα clones in pcDNA3.1+ were obtained from the cDNA Resource Center. Gαi1-FLAG-APEX2, Gαi1-QL-FLAG-APEX2, Lyn-FLAG-APEX2, and EGFP-BioID2-HA-CaaX were synthesized by GenScript. The sequences of the clones are available upon request.
Design and Cloning of cDNA Constructs
BioID2-HA was inserted between Ala121 and Glu122 of human Gαi1-WT and Gαi1-QL with the linker sequence SGGGGS flanking BioID2-HA on either side. The final clone was organized as follows: Gαi1(1-121)-Linker-BioID2-HA-Linker-Gαi1(122-355). GFP-PRG was generated from PRG amplification from FL-c-myc-PRG and insertion into the pEGFP-N1 vector.
Cell Culture
A293 and HT1080 cells were obtained from American Type Culture Collection (ATCC). A293, A293-FPR1, and HT1080 cells were grown in DMEM (10013CV, Corning) supplemented with 10% fetal bovine serum (FBS) (10437028, Gibco) and 100 units of penicillin/streptomycin (P/S) (15140122, Gibco) at 37 °C with 5% CO2. Media was supplemented with 100 μg/mL Geneticin (G418) (G8168, Sigma) to select A293-FPR1 cells. Trypsin-EDTA (25200056, Gibco) was used for cell passaging.
Reagents
The following primary and secondary antibodies were utilized: Gαi1/2 (anti-sera) [53], PDZ-RhoGEF (ab110059, abcam), HA (3724, Cell Signaling), FLAG (F1804, Sigma), P-MLC (3671, Cell Signaling), c-myc (13-2500, Invitrogen), Streptavidin-IRDye800 (925-32230, LI-COR), GFP (A11122, Invitrogen). Primary antibodies were made in 3% BSA and 0.1% Sodium azide and the blots were incubated in primary antibody overnight at 4 °C except 1 hr incubation at RT for streptavidin-IRDye800. Secondary antibody goat anti-rabbit DyLight™ 800 (SA535571, Invitrogen), goat anti-mouse IRDye 800CW (926-32210, LICOR) at 1:10,000 dilution and goat anti-rabbit Alexa Fluor 488 (A11034, Invitrogen) at 1:1000 dilution.
Proximity Labeling using BioID2 Followed by Western Blotting
A293 cells were plated in a 6-well plate at a density of 0.35 × 106 cells per well. 24 hr after plating, media was replaced with 2 mL DMEM supplemented with 50 μM Biotin (B4501, Sigma) (prepared as previously described [54] and 10% FBS. The cells were then transfected with 1 μg of BioID2 clone (BioID2-Gαi1, BioID2-Gαi1-QL or BioID2-CaaX) and 100 ng of yellow fluorescent protein (YFP) cDNAs in each well using 1:3 DNA: Lipofectamine 2000 (11668019, Invitrogen) ratio. 24 hr after concurrent transfection and biotin labeling, 300 μL 1× Laemmli buffer was added per well, and the lysates were collected, boiled for 10 min at 95 °C, 40 μL of was resolved on 4-20% Mini-protean TGX™ Gel (4561094, Bio-Rad) and detected by western blot. Anti-HA (1:2000), anti-PDZ-RhoGEF (1:1000), anti-c-myc (1:2000), Streptavidin-IRDye800 (1:3000) were utilized.
A293 cells were plated in a 10 cm dish at a density of 2.0 × 106 cells per dish. The next day, media was replaced with 10 mL DMEM supplemented with 50 μM Biotin and 10% FBS. Thereafter, the cells were transfected with 3 μg of BioID2-Gαi1, BioID2-Gαi1-QL, or BioID2-CaaX and 3 μg of protein of interest (HA-PSPC1, FLAG-p54-HA, GFP-ATF6, GFP-ArgBP2, Golgin A5-GFP, Parvin-GFP, Vimentin-GFP, Cortactin-GFP) in each dish using 1:3 DNA: Lipofectamine 2000 ratio. 24 hr after transfection and labeling, the cells were harvested by centrifugation at 4000× g for 10 min and lysed in 500 μL ice-cold lysis buffer (modRIPA buffer: 50 mM Tris, 150 mM NaCl, 0.1% SDS, 0.5% Sodium deoxycholate, 1% Triton X-100, final pH 7.5) supplemented with 1× protease inhibitor (PI) cocktail (P8849, Sigma), 1 mM Phenylmethylsulfonyl fluoride (PMSF) (786-055, G-Biosciences) for 10 min on ice. The lysates were further incubated with 125 units of Benzonase (E1014-25KU, Sigma) in an end-over-end rotator at 4 °C for 20 min. 0.3% SDS was added to lysates and incubated for an additional 10 min at 4 °C. Lysates were centrifuged at 15000× g for 10 min, and the supernatant was transferred to fresh tubes and total protein concentration was equalized using Pierce 660-nm protein assay reagent (22660, Thermo Fisher Scientific). 5% of equalized lysates were taken out before pulldown to analyze the biotinylation of inputs by western blot analyses. The remaining lysates were incubated with 100 μL Pierce™ streptavidin magnetic beads slurry (88817, Thermo Fisher Scientific) per sample in an end-over-end rotator at 4 °C for 18 hr to capture biotinylated proteins. Following streptavidin pull-down, beads were washed twice with ice-cold modRIPA, and once with ice-cold 1× PBS. 30 μL 1× Laemmli buffer was added to the beads, boiled for 10 min at 95 °C, and the supernatant was loaded on the SDS-PAGE followed by western blot analyses. Antibody dilutions were anti-HA (1:2000) and anti-c-myc (1:2000).
Proximity Labeling using BioID2 for Mass Spectrometry Based Proteomic Analysis.
Low passage HT1080 cells (passage number up to 15) were used for proximity labeling experiments. HT1080 cells were plated into 175 cm2 flasks at a density of 5.5 × 106 cells per flask. The next day, media was replaced with 35 mL DMEM containing 50 μM biotin and 10% FBS. Subsequently, the cells were transfected with 8 μg of BioID2 and 4 μg of YFP cDNAs in each flask. 0.6 μL of Viromer® Red (VR-01LB-00, Lipocalyx, Germany) reagent was used per 2 μg of cDNA for transfection, resulting in ~80-85% transfection efficiency. 24 hr after labeling and transfection, the labeling medium was decanted, cells were washed twice with 1×PBS, and harvested at 4000× g for 10 min. This step was repeated twice using 1×PBS to recover the maximum number of cells. The supernatant was aspirated, and pellets were snap-frozen and stored at −80°C until further use.
All stock solutions used for streptavidin pull-down were freshly prepared, except lysis buffer. Low protein binding tubes (022431081, Eppendorf) were used for sample preparation. Frozen pellets were lysed in 1 mL of ice-cold lysis solution (composition described above) for 10 min on ice, incubated with 125 units of Benzonase with end-over-end rotation at 4 °C for 20 min. 0.3% SDS was added to lysates and incubated for another 10 min at 4 °C. Lysates were centrifuged at 15,000× g for 15 min, the supernatant was transferred to fresh tubes, and total protein concentration was equalized using Pierce 660-nm protein assay reagent. 5% of equalized lysates were saved before pull-down to analyze the biotinylation of inputs by western blot analysis. The remaining equalized lysates were incubated with 500 μL Pierce™ streptavidin magnetic beads slurry per sample, in an end-over-end overnight 4°C for 18 hr. Subsequently, the beads were washed twice with modRIPA, once with four different solutions: 1 M KCl, 0.1 M Na2CO3, 2% SDS (made in 50 mM Tris pH 7.5), and 2 M Urea (made in 10 mM Tris pH 8.0). Finally, the beads were washed twice with 1× PBS and were snap-frozen and stored at −80 °C until further processing for mass spectrometry.
Protein Digestion and TMT Labeling
On-bead digestion followed by LC-MS/MS analysis was performed at the mass spectrometry-based Proteomics Resource Facility of the Department of Pathology at the University of Michigan. Samples were reduced (10 mM DTT in 0.1 M TEAB at 45°C for 30 min), alkylated (55 mM 2-chloroacetamide at room temperature (RT) for 30 min in the dark, and subsequently digested using 1:25 trypsin (V5113, Promega): protein at 37° C with constant mixing using a thermomixer. 0.2% TFA was added to stop the proteolysis, and peptides were desalted using a Sep-Pak C18 cartridge (WAT036945, Waters Corp). The desalted peptides were dried in a Vacufuge and reconstituted in 100 μL of 0.1 M TEAB. A TMT10plex™ isobaric labeling kit (0090110, Thermo Fisher Scientific) was used to label each sample per manufacturer’s protocol. The samples were labeled with TMT 10-plex reagents at RT for 1 hr. The reaction was quenched by adding 8 μL of 5% hydroxylamine for 15 min, combined, and subsequently dried. An offline fractionation of the combined sample into 8 fractions was performed using a high pH reversed-phase peptide fractionation kit, as per manufacturer’s protocol (84868, Pierce). Fractions were dried and reconstituted in 12 μL of 0.1% formic acid/2% acetonitrile for LC-MS/MS analysis. Sample-to-TMT channel information is provided in table S1.
Liquid Chromatography-Mass Spectrometry Analysis
An Orbitrap Fusion (Thermo Fisher Scientific) and RSLC Ultimate 3000 nano-UPLC (Dionex) was used to acquire the data. To achieve superior quantitation accuracy, we employed multinotch-MS3 [55]. 2 μL of each fraction was resolved on a nano-capillary reverse phase column (PepMap RSLC C18 column, 75 μm i.d. × 50 cm; Thermo Scientific) at the flowrate of 300 nL/min using 0.1% formic acid/acetonitrile gradient system (2-22% acetonitrile in 110 min;22-40% acetonitrile in 25 min; 6 min wash at 90% followed by 25 min re-equilibration) and directly sprayed onto the Orbitrap Fusion using EasySpray source (Thermo Fisher Scientific). Mass spectrometer was set to collect one MS1 scan (Orbitrap; 120K resolution; AGC target 2x105; max IT 50 ms) followed by data-dependent, “Top Speed” (3 seconds) MS2 scans (collision-induced dissociation; ion trap; NCD 35; AGC 5x103; max IT 100 ms). For multinotch-MS3, the top 10 precursors from each MS2 were fragmented by HCD followed by Orbitrap analysis (NCE 55; 60K resolution; AGC 5x104; max IT 120 ms, 100-500 m/z scan range).
Proteome Discoverer (v2.4; Thermo Fisher) was used for data analysis. Tandem MS spectra were searched against SwissProt human protein database using the following search parameters: MS1 and MS2 tolerance were set to 10 ppm and 0.6 Da, respectively; carbamidomethylation of cysteines (57.02146 Da) and TMT labeling of lysine and N-termini of peptides (229.16293 Da) were considered static modifications; oxidation of methionine (15.9949 Da) and deamidation of asparagine and glutamine (0.98401 Da) were considered variable. Proteins and peptides that passed ≤1% false discovery rate threshold were retrained for subsequent analysis. Quantitation was performed using TMT reporter ion in MS3 spectra with an average signal-to-noise ratio of 10 and <50% isolation interference.
Normalization and Sorting Criteria
Only a small fraction of all the proteins labeled by Gαi1 BioID2, are expected to have increased enrichment in BioID2-Gαi1-QL samples relative to the BioID2-Gαi1 samples. Most of the proteins are expected to be equally enriched across samples as the majority of the labeling is based on proximity rather than Gαi1-QL-specific interactions. Therefore, to quantitatively compare the samples across groups, we summed the total TMT signal for each sample to obtain a normalization factor used to normalize the values for each protein across experimental groups.
Normalized abundance ratio and p-values were used for the subsequent analysis. Proteins constituting the active Gαi1 interactome fulfilled all the following criteria: PSM>5, Abundance ratio BioID2-Gαi1/BioID2-CaaX ≥ 0.8 and BioID2-Gαi1-QL/BioID2-Gαi1 ≥ 1.3, Abundance ratio P-value BioID2-Gαi1-QL/BioID2-CaaX <0.05.
Gene ontology analysis.
Gene ontology (Go) analysis was performed with using the DAVID Bioinformatics resource at https://david.ncifcrf.gov. Proteins selected based on the criteria in Figure 2 C were submitted based on gene identifiers to the analysis server and analyzed by functional annotation clustering.
Immunofluorescence Staining
A293 cells (2 × 104 cells/well) were plated on a poly-D-lysine coated 8-well chamber μ-slide (80826, Ibidi) and transfected with BioID2 clones (200 ng cDNA/well) the following day using 1:3 DNA: Lipofectamine 2000 ratio. 24 hr post-transfection, cells were fixed with 4% (w/v) paraformaldehyde (PFA) (15710, Electron microscopy sciences) for 10 min at RT and washed with phosphate-buffered saline (PBS, BP3994, Fisher). Subsequently, cells were blocked and permeabilized with 10% normal goat serum in 1× PBS containing 0.1% (v/v) Triton X100 (1× PBS-T) for 1 hr at RT. Primary anti-HA antibody HA (3724, Cell Signaling) was used at 1:500 dilution in 2% goat serum in 1×PBS-T overnight at 4°C. The next day, cells were washed three times with 1× PBS-T and incubated with secondary antibody (goat anti-rabbit Alexa Fluor 488) at a dilution of 1:1000 in 1× PBS-T for 1 hr at RT. The nuclei were stained with DAPI for 15 min and washed once with 1× PBS-T and 1× PBS. Cells were imaged on a LEICA DMi8 microscope in confocal mode with a 63× oil lens using 405 nm excitation for DAPI and 488 nm for Alexa Fluor 488 secondary antibody. Acquisition parameters were kept constant for all the conditions of an experiment.
Glosensor cAMP Reporter Assay
A293 cells (4 × 104 cells/well) were plated per well in a 96-well plate (655983, Greiner). The following amounts of DNA were used per well: 50 ng of pGloSensor™-20F cAMP Plasmid (E1171, Promega), 125 ng of untagged Gαi1-WT, Gαi1-QL or BioID2 fused Gαi clones or empty vector (control, pCDNA3.1+). Reverse transfection was performed using 1:3 DNA: Lipofectamine 2000 ratio. 24 hr after transfection, cells were washed once with 1× PBS, and 75 μL of 2 mM D-luciferin (LUCK-1G, Goldbio) in Leibovitz’s L-15 medium (21083-027, Gibco) was added for 2 hr at 37 °C, 5% CO2 incubator. Cells were treated with vehicle or 1 μM forskolin (Fsk) (11018, Cayman Chemicals) and luminescence was measured using a Varioskan™ LUX multimode microplate reader for 30 min.
Proximity Ligation Assay (PLA)
In situ PLAs were performed using Duolink™ Kit (DUO92101, Sigma) as per manufacturer’s protocol with some modifications. 2 × 104 cells were plated on 14 mm coverslips in a 35 mm dish (D11030H, Matsunami) and the following day, 100ng of Gαi1-FLAG-APEX2, Gαi1-QL-FLAG-APEX2 or Lyn-FLAG-APEX2 with 25 ng EGFP-PRG were transfected and the cDNAs were allowed to express for 48 hr with media change after 24 hr. The cells were then washed twice with 1× PBS and fixed with 4% PFA, and 4% sucrose made in 1× PBS for 10 min in the dark at RT. The cells were permeabilized and blocked using freshly prepared 5% goat serum, 1% BSA, 0.2% Triton X100 in TBS. Subsequently, rabbit anti-GFP (1:750) (A11122, Invitrogen) and mouse anti-FLAG-(1:750) (F1804, Sigma) antibodies were diluted in Duolink® antibody diluent and incubated overnight at 4 °C in a humidified chamber. A total 40 μL reaction mixture including PLA probe binding, ligation, amplification steps in a humidified chamber. The dilution factors for all the reagents were kept used as per manufacturer’s instructions. 2 mL of either buffer-A or B were used per wash as directed in the manual. After final washes, all the aqueous media was removed, 80 μL of mounting media was added to the cells. Random fields were imaged on the LEICA DMi8 microscope in confocal mode with a 63× oil lens, using 405 nm excitation for DAPI, 488 nm for GFP-PRG and 568 nm for PLA dots. Acquisition parameters were kept constant for all the conditions of an experiment. The intensity of PLA dots and GFP-PRG was measured for ≥ 100 cells per condition, using ImageJ, and are represented on the X and Y-axis, respectively.
Luciferase Reporter Assay
A293 cells were plated, 4 × 104 cells per well, in a 96-well plate (655983, Greiner). The following amounts of DNA were used per well: 25 ng of SRE luciferase reporter (E134A, Promega), 2.5 ng of c-myc-PRG and 125 ng of each Gα or Gα-QL subunit. For the assay to determine the concentration dependence for activation, 0.75, 2.5, 7.5, 25, 75, 125 ng Gαi1 or Gαi1-QL. pcDNA3.1 were used. Empty vector was used to keep the total amount of DNA constant in each well. Transfection was performed using 1:3 DNA: Lipofectamine 2000 (Invitrogen) ratio. Reverse transfection was performed, meaning cells were plated and transfected at the same time. 12 hr after transfection, media was replaced with 75 μL of serum-free media for another 12 hr, and 75 μL (1:1 volume) of One-Glo™ reagent (E6110, Promega) was added to each well, incubated for 10 min at RT. The luminescence signal was measured using Varioskan™LUX multimode microplate reader (Thermo Scientific™). A293-FPR1 cells were transfected and treated with 100 ng/mL PTX (P7208, Sigma) for 12 hr and subsequently, fMLF (0.01, 0.1, 1, 10 μM) was added in serum-free media with or without PTX, for the next 12 hr.
Rhotekin Pull-Down Assay
Active RhoA levels were measured using the RhoA Pull-down Activation Assay Biochem Kit (BK036-S, Cytoskeleton Inc.) using GST Rhotekin beads. The levels of the GTP-RhoA associated with GST-Rhotekin-RBD were quantified by western blot analysis. Briefly, A293 cells were plated in a 6-well plate at a density of 3.5 x 105 cells per well and transfected with 1 μg Gαi1, 100 ng c-myc-PRG and, 250 ng RhoA-HA per well using 1:3 DNA: Lipofectamine 2000 ratio. 20 hr after transfection, cells were cultured in serum-free media for 4 hr. Cells in each well were then lysed with 300 μL of RhoA lysis buffer with 1×PI (included with the kit), and lysates were equalized for total protein amount. Samples from two wells were pooled for each experimental group (total 600 μL, ~600 μg protein per experimental group). The lysates were incubated with 50 μg of GST-Rhotekin bound beads in an end-over-end rotator for 1 hr at 4 °C. Beads were washed twice with wash buffer (included with the kit), eluted in 40 μL 1× Laemmli sample buffer, and analyzed by western blot using an anti-HA (1:2000), anti-c-myc antibody (1:2000), and anti-Gαi1/2 antisera (1:3000). Band intensities were quantified using Image Studio Lite (version 5.2)
FPR1-A293 Cell Protrusions Assay
FPR-1-A293 cells (2 × 104 /well) were plated in an 8-well chamber slide coated with poly-D-lysine. The following plasmids were transfected per well: 100 ng YFP, 4 ng PRG, 125 ng Gαi1 or empty vector to bring the total amount of cDNA per well equal. Plasmids were transfected using 1:3 DNA: Lipofectamine 2000 ratio. 24 hr after transfection, the media was changed to fresh media, with or without PTX (100ng/mL) (P7208, Sigma) for 24 hr more. Subsequently, the cells were washed once with 1× PBS and placed in HBSS + HEPES (10mM) pH 7.3. The cells were imaged every 20 seconds for 40 min, and formyl-methionyl-leucyl-phenylalanine fMLF (F3506, Sigma) or vehicle was added 5 min after the video initiation. The videos were taken at 10× magnification on the LEICA DMi8 microscope using a 488 nm excitation filter. To quantify the % cells with protrusions, the total number of cells in first frame of a video were counted in ImageJ by ‘analyze particles’ option, and cell with the protrusions were manually counted from the videos, in a blinded manner.
Human Neutrophil Isolation
Human peripheral blood was obtained from the Platelet Pharmacology and Physiology Core at the University of Michigan. The core maintains a blanket IRB for basic science studies, which does not require HIPAA information, and enrolls healthy subjects that follow the protection of human subject standards. De-identified samples were used in the study. Neutrophils were isolated from human peripheral blood as described previously [56]. Freshly isolated blood was carefully layered on top of 1-step polymorphs (AN221725, Accurate chemicals and scientific corporation) (1:1 Blood and Polymorphs) and centrifuged at 1000× g for 45 min and buffy coat was transferred to fresh tubes. Red blood cells were lysed using 0.1× PBS hypotonic solution for 45 sec, and immediately 4× PBS was added. The tubes were centrifuged at 400× g for 10 min, and pelleted cells were resuspended in modified Hanks’ balanced salt solution (mHBSS). Neutrophil preparations were at least 95% pure, as confirmed by nuclear morphology.
Immunostaining of Human Neutrophils
Each well of an 8-well chamber μ-slide was coated with 5 μg of fibronectin (F1141, Sigma) overnight at 4°C. Freshly isolated human neutrophils were preincubated with either vehicle or 500 ng/mL PTX for 2 hr at 37 °C with gentle rotation before plating on the fibronectin-coated wells. 2 × 105 cells per well were allowed to adhere to the surface for 15 min at 37°C in 5% CO2. The cells were stimulated with vehicle or fMLF (10 nM) for 5 min at 37 °C in 5% CO2 incubator and then fixed with 4% PFA and 5% sucrose in ddH2O for 15 min at RT, and blocked using 10% goat serum, 3% Fatty acid-free BSA, 0.05% Saponin in 0.2% PBST for 1 hr at RT. Subsequently, the cells were incubated with 1:100 P-MLC primary antibody prepared in 2% goat serum, 0.05% saponin in 0.1% PBST. The following day, the cells were washed with 0.05% saponin in 0.1% PBST for 10 min, three times, and incubated with anti-rabbit 488 secondary for 1 hr at RT and subsequently stained with DAPI. The cells were imaged in confocal mode with a 63× oil lens, and acquisition parameters were kept constant for all the experimental conditions. Three random fields were acquired per experiment, and images from three independent experiments were analyzed by counting the number of cells with asymmetric P-MLC staining and the total cells to determine % cells with asymmetric P-MLC distribution. Representative images were captured with a 100× oil lens.
Western Blotting
Samples were resolved on 4-20% Mini-protean TGX™ Gels (4561094, Bio-Rad), were transferred to a nitrocellulose membrane (66485, Pall Corporation), and stained with Ponceau S (141194, Sigma). Membranes were blocked with 5% non-fat milk powder in TBST (0.1% Tween-20 in Tris-buffered saline) at RT for 1 hr with constant shaking. Membranes were probed with primary antibodies for 1 or 2 hr at RT or overnight at 4 °C. The membranes were washed with TBST, incubating with secondary antibody for 1 hr at RT, washed with TBST, and imaged using an Odyssey Infrared Imaging System (Li-Cor Biosciences).
Statistical Analysis
All the experiments were performed at least three times, except Figure 2E, which was repeated twice. Data shown are expressed as mean ± SD or as a representative experiment with replicate data in the figure or the supplemental figures. Statistical significance between various conditions was assessed by determining P values using the Student’s t-test, one-way or two-way ANOVA with Tukey’s multiple comparisons test. (**P < 0.01, *** < 0.001, ****<0.0001).Western blot images were scanned using Licor and quantified using Image Studio Lite (Version 5.2). All data were analyzed using GraphPad Prism 7.0 (GraphPad; La Jolla, CA), and schematic representations of the figures were created with BioRender.com and Adobe illustrator.
Supplementary Material
Acknowledgments:
We would like to thank U of M Pathology core, especially Venkatesha Barsur, Alexey Nesvizhskii, and Kevin Conlon for running mass spec samples as well as for advice on the data analysis. Srilakshmi Yalavarthi and Gautam Sule for providing isolated neutrophils for this study. We would like to thank Sundeep Malik for BioID2-CaaX cDNA, advice and scientific discussions and John Tesmer for full length PDZ-RhoGEF cDNA mammalian expression construct used in this study.
Funding:
This work is supported by NIH R35GM127303 (A.V.S.), Pharmacology Centennial Fund and Benedict and Diana Lucchesi Pre-doctoral Fellowship, Department of Pharmacology, University of Michigan (N.R.C.), NIH R01AI152517 (C.A.P.).
Abbreviations
- AC
Adenylate Cyclase
- cAMP
3′,5′-Cyclic Adenosine Monophosphate
- fMLF
N-Formylmethionine-Leucyl-Phenylalanine
- FPR1
Formyl Peptide Receptor1
- Fsk
Forskolin
- IP
Immunoprecipitation
- KO
Knockout
- MS
Mass Spectrometry
- PI
Protease Inhibitor
- PLA
Proximity Ligation Assay
- PM
Plasma Membrane
- PRG
PDZ-RhoGEF
- PTX
Pertussis Toxin
- TMT
Tandem Mass Tag
- WCL
Whole Cell Lysates
- WT
Wildtype
- YFP
Yellow Fluorescent Protein
Footnotes
Competing interests:
The authors declare that they have no competing interests.
Data and Materials Availability:
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [57] partner repository with the dataset identifier PXD027905. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes:
- 1.Hepler JR and Gilman AG, G proteins. Trends Biochem.Sci, 1992. 17: p. 383–387. [DOI] [PubMed] [Google Scholar]
- 2.Gilman AG, G proteins: transducers of receptor-generated signals. Annu Rev Biochem, 1987. 56: p. 615–649. [DOI] [PubMed] [Google Scholar]
- 3.Oldham WM and Hamm E, Structural basis of function in heterotrimeric G proteins. Quarterly Reviews of Biophysics, 2006. 39(02): p. 117–166. [DOI] [PubMed] [Google Scholar]
- 4.Smrcka AV, G protein βγ subunits: Central mediators of G protein-coupled receptor signaling, in Cell. Mol. Life Sci 2008. p. 2191–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wettschureck N and Offermanns S, Mammalian G proteins and their cell type specific functions. Physiol Rev, 2005. 85(4): p. 1159–204. [DOI] [PubMed] [Google Scholar]
- 6.Taussig R, Iniguez-Lluhi JA, and Gilman AG, Inhibition of adenylyl cyclase by Gi alpha. Science, 1993. 261(5118): p. 218–21. [DOI] [PubMed] [Google Scholar]
- 7.Murphy PM, The molecular biology of leukocyte chemoattractant receptors. Annu Rev Immunol, 1994. 12: p. 593–633. [DOI] [PubMed] [Google Scholar]
- 8.Bendall L, Chemokines and their receptors in disease. Histol Histopathol, 2005. 20(3): p. 907–26. [DOI] [PubMed] [Google Scholar]
- 9.Le Y, et al. , Chemokines and chemokine receptors: their manifold roles in homeostasis and disease. Cell Mol Immunol, 2004. 1(2): p. 95–104. [PubMed] [Google Scholar]
- 10.Neptune ER, Iiri T, and Bourne HR, Galphai is not required for chemotaxis mediated by Gi-coupled receptors. J Biol Chem, 1999. 274(5): p. 2824–8. [DOI] [PubMed] [Google Scholar]
- 11.Katada T, The inhibitory G protein G(i) identified as pertussis toxin-catalyzed ADP-ribosylation. Biol Pharm Bull, 2012. 35(12): p. 2103–11. [DOI] [PubMed] [Google Scholar]
- 12.Smrcka AV and Fisher I, G-protein betagamma subunits as multi-functional scaffolds and transducers in G-protein-coupled receptor signaling. Cell Mol Life Sci, 2019. 76(22): p. 4447–4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Surve CR, et al. , Dynamic regulation of neutrophil polarity and migration by the heterotrimeric G protein subunits Galphai-GTP and Gbetagamma. Sci Signal, 2016. 9(416): p. ra22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.To JY and Smrcka AV, Activated heterotrimeric G protein alphai subunits inhibit Rap-dependent cell adhesion and promote cell migration. J Biol Chem, 2018. 293(5): p. 1570–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tall GG, Krumins AM, and Gilman AG, Mammalian Ric-8A (synembryn) is a heterotrimeric Galpha protein guanine nucleotide exchange factor. J Biol Chem, 2003. 278(10): p. 8356–62. [DOI] [PubMed] [Google Scholar]
- 16.Nafisi H, et al. , GAP1(IP4BP)/RASA3 mediates Galphai-induced inhibition of mitogen-activated protein kinase. J Biol Chem, 2008. 283(51): p. 35908–17. [DOI] [PubMed] [Google Scholar]
- 17.Solis GP, et al. , Golgi-Resident Galphao Promotes Protrusive Membrane Dynamics. Cell, 2017. 170(6): p. 1258. [DOI] [PubMed] [Google Scholar]
- 18.Roux KJ, et al. , A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol, 2012. 196(6): p. 801–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim DI, et al. , An improved smaller biotin ligase for BioID proximity labeling. Mol Biol Cell, 2016. 27(8): p. 1188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu J, et al. , Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell, 2003. 114(2): p. 201–14. [DOI] [PubMed] [Google Scholar]
- 21.Wong K, Van Keymeulen A, and Bourne HR, PDZRhoGEF and myosin II localize RhoA activity to the back of polarizing neutrophil-like cells. J Cell Biol, 2007. 179(6): p. 1141–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fukuhara S, et al. , A novel PDZ domain containing guanine nucleotide exchange factor links heterotrimeric G proteins to Rho. J Biol Chem, 1999. 274(9): p. 5868–79. [DOI] [PubMed] [Google Scholar]
- 23.Gibson SK and Gilman AG, Gialpha and Gbeta subunits both define selectivity of G protein activation by alpha2-adrenergic receptors. Proc Natl Acad Sci U S A, 2006. 103(1): p. 212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coleman DE, et al. , Crystallization and preliminary crystallographic studies of Gi alpha 1 and mutants of Gi alpha 1 in the GTP and GDP-bound states. J Mol Biol, 1994. 238(4): p. 630–4. [DOI] [PubMed] [Google Scholar]
- 25.Ahmed SM, et al. , G protein betagamma subunits regulate cell adhesion through Rap1a and its effector Radil. J Biol Chem, 2010. 285(9): p. 6538–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen LT, Gilman AG, and Kozasa T, A candidate target for G protein action in brain. J Biol Chem, 1999. 274(38): p. 26931–8. [DOI] [PubMed] [Google Scholar]
- 27.Huang da W, Sherman BT, and Lempicki RA, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc, 2009. 4(1): p. 44–57. [DOI] [PubMed] [Google Scholar]
- 28.Hill CS, Wynne J, and Treisman R, The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell, 1995. 81(7): p. 1159–70. [DOI] [PubMed] [Google Scholar]
- 29.Chen Z, et al. , Recognition of the activated states of Galpha13 by the rgRGS domain of PDZRhoGEF. Structure, 2008. 16(10): p. 1532–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pertz O, et al. , Spatiotemporal dynamics of RhoA activity in migrating cells. Nature, 2006. 440(7087): p. 1069–72. [DOI] [PubMed] [Google Scholar]
- 31.Etienne-Manneville S and Hall A, Rho GTPases in cell biology. Nature, 2002. 420(6916): p. 629–35. [DOI] [PubMed] [Google Scholar]
- 32.Huang C, et al. , Persistent membrane association of activated and depalmitoylated G protein α subunits. Proceedings of the National Academy of Sciences, 1999. 96: p. 412–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takesono A, et al. , Receptor-independent activators of heterotrimeric G-protein signaling pathways. J Biol Chem, 1999. 274(47): p. 33202–5. [DOI] [PubMed] [Google Scholar]
- 34.Le-Niculescu H, et al. , Identification and characterization of GIV, a novel Galpha i/s-interacting protein found on COPI, endoplasmic reticulum-Golgi transport vesicles. J Biol Chem, 2005. 280(23): p. 22012–20. [DOI] [PubMed] [Google Scholar]
- 35.Navarro G, et al. , Evidence for functional pre-coupled complexes of receptor heteromers and adenylyl cyclase. (2041-1723 (Electronic)). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferré S, The GPCR heterotetramer: challenging classical pharmacology. (1873-3735 (Electronic)). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rebois RV, et al. , Heterotrimeric G proteins form stable complexes with adenylyl cyclase and Kir3.1 channels in living cells. (0021-9533 (Print)). [DOI] [PubMed] [Google Scholar]
- 38.Jackson M, et al. , Modulation of the neuronal glutamate transporter EAAT4 by two interacting proteins. Nature, 2001. 410(6824): p. 89–93. [DOI] [PubMed] [Google Scholar]
- 39.Struckhoff AP, et al. , PDZ-RhoGEF is essential for CXCR4-driven breast tumor cell motility through spatial regulation of RhoA. J Cell Sci, 2013. 126(Pt 19): p. 4514–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wells CD, et al. , Mechanisms for reversible regulation between G13 and Rho exchange factors. J Biol Chem, 2002. 277(2): p. 1174–81. [DOI] [PubMed] [Google Scholar]
- 41.Carter AM, Gutowski S, and Sternweis PC, Regulated localization is sufficient for hormonal control of regulator of G protein signaling homology Rho guanine nucleotide exchange factors (RH-RhoGEFs). J Biol Chem, 2014. 289(28): p. 19737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Castillo-Kauil A, et al. , Galphas directly drives PDZ-RhoGEF signaling to Cdc42. J Biol Chem, 2020. 295(50): p. 16920–16928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Itoh H, et al. , Presence of three distinct molecular species of Gi protein alpha subunit. Structure of rat cDNAs and human genomic DNAs. J Biol Chem, 1988. 263(14): p. 6656–64. [PubMed] [Google Scholar]
- 44.Ko EM, Leem YE, and Choi HT, Purification and characterization of laccase isozymes from the white-rot basidiomycete Ganoderma lucidum. Appl Microbiol Biotechnol, 2001. 57(1-2): p. 98–102. [DOI] [PubMed] [Google Scholar]
- 45.Kuwano Y, et al. , Galphai2 and Galphai3 Differentially Regulate Arrest from Flow and Chemotaxis in Mouse Neutrophils. J Immunol, 2016. 196(9): p. 3828–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zarbock A, et al. , Galphai2 is required for chemokine-induced neutrophil arrest. Blood, 2007. 110(10): p. 3773–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Togashi H, et al. , Functions of a rho-specific guanine nucleotide exchange factor in neurite retraction. Possible role of a proline-rich motif of KIAA0380 in localization. J Biol Chem, 2000. 275(38): p. 29570–8. [DOI] [PubMed] [Google Scholar]
- 48.Mikelis CM, et al. , PDZ-RhoGEF and LARG are essential for embryonic development and provide a link between thrombin and LPA receptors and Rho activation. J Biol Chem, 2013. 288(17): p. 12232–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yeh HW, et al. , PSPC1 mediates TGF-beta1 autocrine signalling and Smad2/3 target switching to promote EMT, stemness and metastasis. Nat Cell Biol, 2018. 20(4): p. 479–491. [DOI] [PubMed] [Google Scholar]
- 50.Rosonina E, et al. , Role for PSF in mediating transcriptional activator-dependent stimulation of pre-mRNA processing in vivo. Mol Cell Biol, 2005. 25(15): p. 6734–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen X, Shen J, and Prywes R, The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J Biol Chem, 2002. 277(15): p. 13045–52. [DOI] [PubMed] [Google Scholar]
- 52.Zhang Q, et al. , Impaired Dendritic Development and Memory in Sorbs2 Knock-Out Mice. J Neurosci, 2016. 36(7): p. 2247–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mumby SM and Gilman AG, [19] Synthetic peptide antisera with determined specificity for G protein α or β subunits, in Methods in Enzymology. 1991, Academic Press. p. 215–233. [DOI] [PubMed] [Google Scholar]
- 54.Hesketh GG, et al. , Parallel Exploration of Interaction Space by BioID and Affinity Purification Coupled to Mass Spectrometry. Methods Mol Biol, 2017. 1550: p. 115–136. [DOI] [PubMed] [Google Scholar]
- 55.McAlister GC, et al. , MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem, 2014. 86(14): p. 7150–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cash JN, et al. , Discovery of Small Molecules That Target the Phosphatidylinositol (3,4,5) Trisphosphate (PIP3)-Dependent Rac Exchanger 1 (P-Rex1) PIP3-Binding Site and Inhibit P-Rex1-Dependent Functions in Neutrophils. Mol Pharmacol, 2020. 97(3): p. 226–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Perez-Riverol Y, et al. , The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res, 2019. 47(D1): p. D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [57] partner repository with the dataset identifier PXD027905. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.