

Abstract
Objectives:
Dysregulated chondrocyte metabolism is closely associated with the pathogenesis of osteoarthritis (OA). Suppressing chondrocyte catabolism to restore cartilage homeostasis has been extensively explored, whereas far less effort has been invested toward enhancing chondrocyte anabolism. This study aimed to repurpose clinically approved drugs as potential stimulators of chondrocyte anabolism in treating OA.
Methods:
Screening of a Food and Drug Administration-approved drug library; Assays for examining the chondroprotective effects of Digoxin in vitro; Assays for defining the therapeutic effects of Digoxin using a surgically-induced OA model; A propensity-score matched cohort study using The Health Improvement Network to examine the relationship between Digoxin use and the risk of joint OA-associated replacement among patients with atrial fibrillation; Identification and characterization of the binding of Digoxin to low density lipoprotein receptor-related protein 4 (LRP4); Various assays, including use of CRISPR-Cas9 genome editing to delete LRP4 in human chondrocytes, for examining the dependence on LRP4 of Digoxin regulation of chondrocytes.
Results:
Serial screenings led to the identification of Ouabain and Digoxin as stimulators of chondrocyte differentiation and anabolism. Ouabain and Digoxin protected against OA and relieved OA-associated pain. The cohort study of 56,794 patients revealed that Digoxin use was associated with reduced risk of OA-associated joint replacement. LRP4 was isolated as a novel target of Digoxin, and deletion of LRP4 abolished Digoxin’s regulations of chondrocytes.
Conclusions:
These findings not only provide new insights into the understanding of Digoxin’s chondroprotective action and underlying mechanisms, but also present new evidence for repurposing Digoxin for OA.
Keywords: Digoxin, Ouabain, Low density lipoprotein receptor-related protein 4, Chondrocyte anabolism, Osteoarthritis
INTRODUCTION
Osteoarthritis (OA) is the most common joint disease and the leading cause of disability in older adults.1, 2 OA incurs significant financial burden with the medical cost of the disease estimated to account for between 1% and 2.5% of the gross domestic product of various high-income countries.3 Although the complex pathogenesis of OA is not fully understood, the progressive degeneration of articular cartilage is considered the major hallmark.4, 5 Articular cartilage is commonly known as a physiologically non-self-renewing avascular tissue comprised predominately of extracellular matrix (ECM) maintained through feedback from restricted population of chondrocytes, which is composed mainly of type II collagen and aggrecan.6, 7 It is increasingly understood that OA is an active dynamic alteration arising from an imbalance between the repair and destruction of joint tissues, and not a passive degeneration or so-called wear-and-tear disease as commonly described. Therefore, in addition to the well-known phenomena of increased cartilage-degrading metalloproteinases and cartilage erosion, attention should be also paid to synthesis of matrix molecules and ECM remodeling in OA.8, 9 Beyond that, articular cartilage has been shown to contain a population of stem cells or progenitor cells, similar to those found in many other adult tissues, that are thought to be involved in the maintenance of tissue homeostasis and in ECM remodeling in OA.10, 11
To date, there is no safe treatment available that can halt OA progression. The main goals of the current disease management are pain control and functional improvement with avoidance of therapeutic toxicity.12 Identifying disease-modifying treatment for OA remain an urgent unmet clinical need. Considering the huge costs in terms of time and money associated with drug development, identification of new uses for old drugs is desirable.13–15 Taking that view, we screened a drug library composed of Food and Drug Administration (FDA)-approved drugs by use of mesenchymal stem cells and chondrocytes. After preliminary validation, we found that cardenolides, represented by Ouabain and Digoxin, may have potential therapeutic effects against OA.
Cardenolides, one of the two sub-groups of cardiac glycosides, are a class of natural biologically active steroids derived from plants and have been used for the treatment of heart disease for centuries.16 Ouabain and Digoxin, two FDA-approved cardenolides, are used to increase the contractile force of the heart and decrease its rate of contraction by inhibiting the cellular Na+/K+-ATPase. Besides the well-known effect of Ouabain and Digoxin on the cardiovascular system, compelling evidence has indicated that they also participate in the regulation of inflammation. For instance, each has been found to inhibit the expressions of pro-inflammatory cytokines such as interleukin 6 (IL-6) and IL-17 under different pathological conditions.17–20 Additionally, previous research reported that Digoxin and Ouabain enhanced the functional properties of tissue-engineered cartilage in vitro.21 However, no studies have investigated the effects of cardenolides on OA. In the present study, we performed comprehensive assays with cell and animal models as well as a large population-based cohort study to demonstrate the potential clinical use of Digoxin in treating OA. Additionally, we also identified low-density lipoprotein receptor-related protein 4 (LRP4) as a new binding target of Digoxin and demonstrated that LRP4 was required for Digoxin regulation of chondrocytes.
RESULTS
Isolation of Digoxin as a potential chondroprotective drug
To isolate the small molecule drugs that may induce mRNA expression of type II collagen (Col2a1), the major component of cartilage ECM, a drug library containing 1046 FDA-approved drugs was firstly screened. Briefly, C3H10T1/2 mesenchymal stem cells were treated with drugs for 24 h individually, followed by thousands of real-time qPCR assays for examining the expression of type II collagen in response to individual drug treatment. 20 drugs that increased the expression of Col2a1 potently were identified after the 1st round screening (Supplementary Fig. 1A). These 20 drug candidates were subjected to the 2nd round screening by treating the C28I2 chondrocytes for 24 h separately. There were only 3 candidates, particularly Ouabain, that could induce the expression of COL2A1 dramatically and dose-dependently (Supplementary Fig. 1B, C). To further evaluate their potential anabolic effects, we treated C28I2 cells with these three drugs then examined the expressions of various genes known to associated with chondrocyte differentiation and metabolism. Among three drugs analyzed, only Ouabain could robustly induce the expressions of anabolic marker genes, including COL2A1, aggrecan (ACAN) and cartilage oligomeric matrix protein (COMP). Intriguingly, it strongly inhibited the expression of RUNX Family Transcription Factor 2 (RUNX2), a marker gene of chondrocyte hypertrophy and OA (Supplementary Fig. 1D–F). As Ouabain belongs to the family of cardenolides, we also treated cells with three other cardenolides: Lanatoside C, Cymarin and Digoxin. All three cardenolides could induce the expression of COL2A1, but only Digoxin could significantly increase the expressions of ACAN and COMP, two additional markers of anabolism (Supplementary Fig. 1G). Therefore, we ended up using Ouabain and Digoxin as representatives of cardenolides to conduct further experiments.
Digoxin induces chondrogenesis and regulates chondrocyte metabolism
Given the stimulatory effect of Ouabain and Digoxin on the expression of Col2a1 in C3H10T1/2 mesenchymal stem cells, we first sought to determine whether these two drugs could induce chondrogenesis. We treated micromass cultured C3H10T1/2 mesenchymal stem cells with either drug for 7 or 14 days. As shown in Fig. 1A, B, alcian blue staining validated the enhanced chondrocyte differentiation in both Ouabain and Digoxin treated groups compared to the PBS group. Moreover, we also examined the transcriptional levels of chondrogenic marker genes. After treatment with either drug, the mRNA expressions of Col2a1, Comp, Acan, SRY-Box Transcription Factor 5 (Sox5), SRY-Box Transcription Factor 6 (Sox6) and SRY-Box Transcription Factor 9 (Sox9) were all significantly upregulated (Fig. 1C).
Fig. 1. Ouabain and Digoxin enhance chondrogenesis and stimulate chondrocyte anabolism.
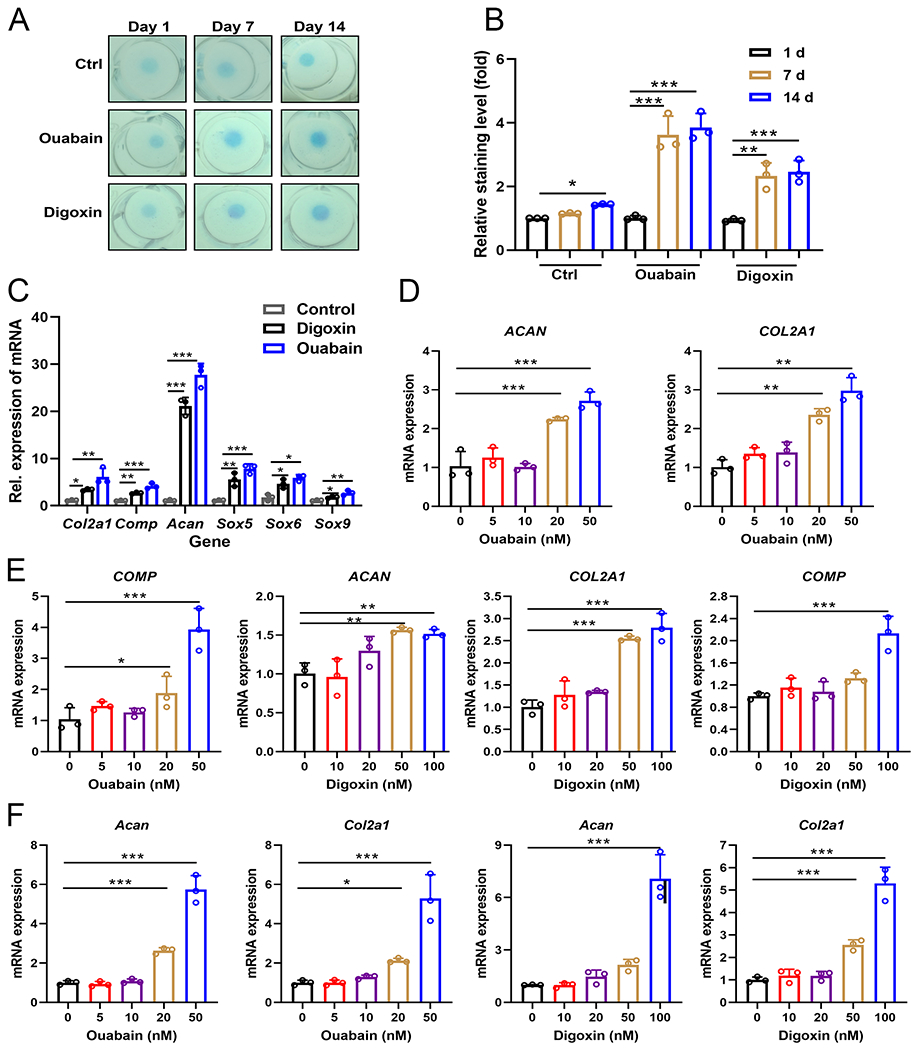
(A) C3H10T1/2 mesenchymal stem cells were incubated in the absence or presence of 50 nM Ouabain or 100 nM Digoxin for 1, 7, 14 days, followed by Alcian blue staining. (B) Quantification of (A). (C) C3H10T1/2 cells were incubated in the absence or presence of 50 nM Ouabain or 100 nM Digoxin for 2 or 7 days, qRT-PCR was performed to examine the expression of Col2a1, Comp, Acan, Sox5, Sox6 and Sox9. (D, E) mRNA levels of COL2A1, ACAN and COMP in human C28I2 chondrocytes treated with a series of Ouabain or Digoxin for 24 h, assayed by qRT-PCR analysis. (F) mRNA levels of Col2a1 and Acan in murine primary chondrocytes treated with various concentrations of Ouabain or Digoxin for 24 h, assayed by qRT-PCR analysis. (G) mRNA levels of COL2A1, ACAN and COMP in human primary normal and OA chondrocytes treated with or without Ouabain or Digoxin for 24 h, assayed by qRT-PCR analysis. The values are mean±SEM of at least 3 independent experiments; *p<0.05, **p<0.01, ***p<0.001 versus control group.
Next we tested their ability to promote anabolism in both human and mouse chondrocytes. We found that both Ouabain and Digoxin could induce the expressions of COL2A1 and ACAN in human C28I2 chondrocytes. In addition, the expression of COMP, which encodes a protein that is present in small amounts but plays a key role in matrix composition,22, 23 was also enhanced by the drugs (Fig. 1D, E). We next explored whether Ouabain and Digoxin could upregulate anabolic marker gene expressions in mouse and human primary chondrocytes. As shown in Fig. 1F, G, both Ouabain and Digoxin could stimulate chondrocyte anabolism in a dose dependent manner. In addition, human OA chondrocytes appeared to respond better than normal chondrocytes to drug treatment (Fig. 1G).
Given that these two drugs were previously reported to inhibit the expressions of pro-inflammatory cytokines under different pathological conditions,17–20 we next determined whether Ouabain and Digoxin could also inhibit pro-inflammatory cytokine activated catabolism in chondrocytes.24 C28I2 chondrocytes were incubated with pro-inflammatory cytokines (TNFα or IL-1β, known to play crucial roles in the pathogenesis of OA) for 24 h, and both Ouabain and Digoxin significantly reduced the mRNA expressions of catabolic markers, ADAM metallopeptidase with thrombospondin type 1 motif 4 (ADAMTS4) and matrix metallopeptidase 13 (MMP13)25 (Supplementary Fig. 2A, 3A). Consistently, the protein levels of ADAMTS4, MMP13 and iNOS were also downregulated in Ouabain or Digoxin treated groups (Supplementary Fig. 2D, 3D). Mouse primary chondrocytes were also treated as described above, as shown in Supplementary Fig. 2B, C and 3B, C, real-time PCR revealed significant downregulation of the expressions of catabolic marker genes, mitochondrially encoded cytochrome c oxidase II (Cox2), nitric oxide synthase 2 (Nos2), Mmp13, matrix metallopeptidase 3 (Mmp3) and ADAM metallopeptidase with thrombospondin type 1 motif 5 (Adamts5), in the drug treatment groups. The decreased protein levels of iNOS, MMP13, ADAMST5 validated the occurrence of drugs-inhibited chondrocytes catabolism (Supplementary Fig. 2E, 3E).
Digoxin protects against OA in a surgically induced model in vivo
To examine the effect of Ouabain and Digoxin on OA progression, we performed destabilization of the medial meniscus (DMM) surgery26 in 12-week-old male C57BL/6J mice with or without Ouabain or Digoxin administration commencing 3 days after surgery (Fig. 2A). We carried out histological analyses to assess knee joint damage 12 weeks after DMM surgery. As expected, cartilage degeneration was severe at 12 weeks after DMM surgery, evidenced by markedly increased Osteoarthritis Research Society International (OARSI) scores. The administration of both Ouabain and Digoxin (50 nM and 100 nM respectively for intra-articular injection) caused partial but significant reduction in cartilage degradation (Fig. 2B, C). By immunohistochemical (IHC) staining, a significant decrease in type II collagen in cartilage was observed 12 weeks after DMM surgery, which was greatly inhibited by Ouabain or Digoxin use (Fig. 2D). However, the levels of the markers of chondrocytes catabolism (cleaved aggrecan, MMP13 and ADAMTS5) was increased after DMM surgery, and Ouabain or Digoxin administration inhibited these changes (Fig. 2E). As illustrated in previous studies, during the evolution of OA, the subchondral bone undergoes marked changes in its composition and structural organization.27 Micro-computerized tomography (μCT) analysis performed in this study also showed significant difference in knee joint structure of mice in different treatment groups. Increased osteophyte formation was observed in mice 12 weeks after DMM surgery, and this effect was significantly inhibited by treatment with Ouabain or Digoxin (Fig 2F–I). The volume of calcified meniscus and synovial tissue was also quantified and exhibited similar trends to those observed in osteophytes (Fig. 2F, J, K). We further analyzed changes in subchondral bone mass and found that subchondral bone mass was significantly increased 12 weeks after DMM surgery and Ouabain or Digoxin had no significant effect on DMM-induced subchondral bone mass increase (Supplementary Fig. 4A–E). Collectively, these results suggest that Ouabain and Digoxin can limit OA development in the injury-induced OA mouse model.
Fig. 2. Ouabain and Digoxin protect against OA in a surgically induced model in vivo.
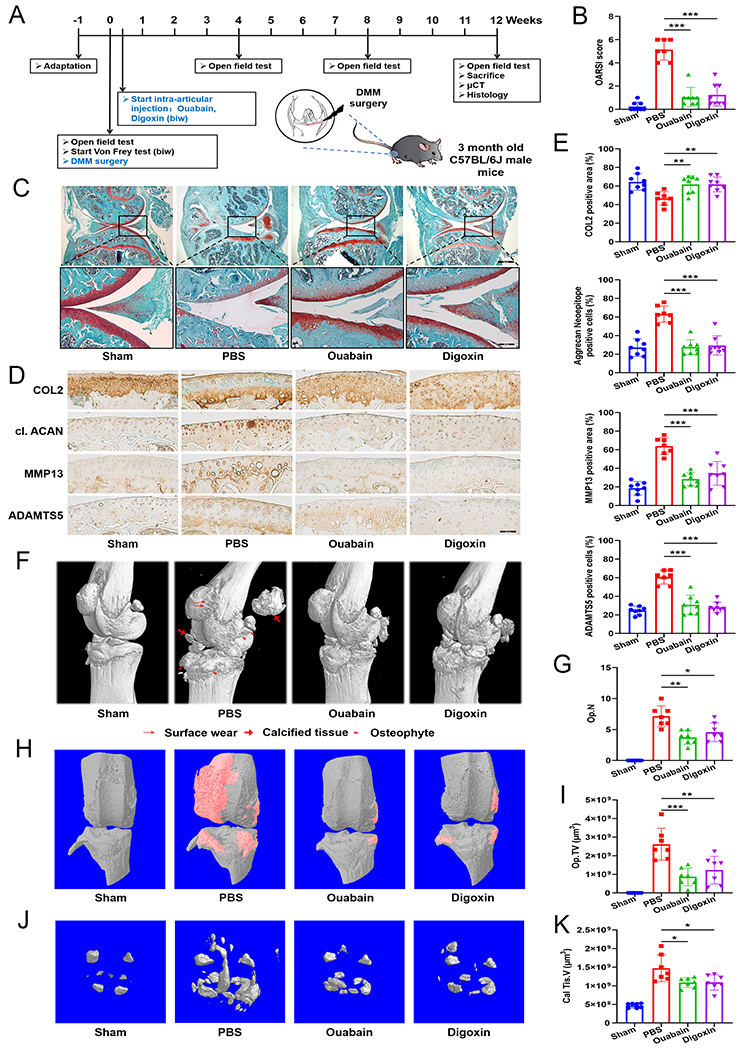
(A) Experimental flow chart. DMM surgery was performed in 12-week-old male C57BL/6J mice. Ouabain or Digoxin was administered 3 days after DMM surgery (n=8; a mouse in the PBS control group died of unknown causes 10 weeks after surgery). (B) The severity of OA-like phenotype 12 weeks after surgery was analyzed by grading histological sections using the Osteoarthritis Research Society International (OARSI) score system. (C) Representative images of Safranin O/Fast Green stained sections of knee joints from mice treated with or without Ouabain or Digoxin for 12 weeks. Scale bar=800 μm (top panel) and 200 μm (bottom panel). (D, E) Representative images of immunohistochemical staining for type II collagen, aggrecan neoepitope, MMP13 and ADAMTS5 in knee joint sections of mice treated with or without Ouabain or Digoxin for 12 weeks. Scale bar=100 μm. Positive staining for type II collagen, aggrecan neoepitope, MMP13 and ADAMTS5 were quantified. (F) Three-dimensional mirco-CT images of pathological structural changes in the mouse knee 12 weeks after surgery. (G) Osteophyte number (Op.N) and (I) size (Op.TV) in the knee of mice after DMM surgery. (H) Three-dimensional mirco-CT images of osteophyte formation between the groups. The region marked in red shows osteophyte. (J) Three-dimensional mirco-CT images of calcified meniscus and synovial tissue between the groups. (K) The volume of calcified meniscus and synovial tissue (Cal Tis.V) was quantified. *p<0.05, **p<0.01, ***p<0.001 versus PBS control group.
We also performed a series of tests to determine if Ouabain and Digoxin could lower OA pain sensitivity. The results of von Frey tests showed significantly reduced paw withdrawal response thresholds after DMM surgery in mice. The administration of Ouabain and Digoxin could significantly increase paw withdrawal response threshold, reflecting reduced pain sensitivity in mice (Fig. 3A). We also tested spontaneous activity of the mice in response to DMM surgery and cardenolide treatment by performing an open field test. We found that travel distance, maximum walking speed, active time and absolute turn angle decreased over time after DMM surgery. Treatment with Ouabain or Digoxin could significantly reverse reduced spontaneous activity caused by DMM surgery (Fig. 3B–F). Taken together, Ouabain and Digoxin were associated with reduced mechanical pain sensitivity and enhanced spontaneous ambulatory activity relative to untreated controls.
Fig. 3. Effect of Ouabain and Digoxin on pain-related behaviors in the DMM-induced OA mice model.
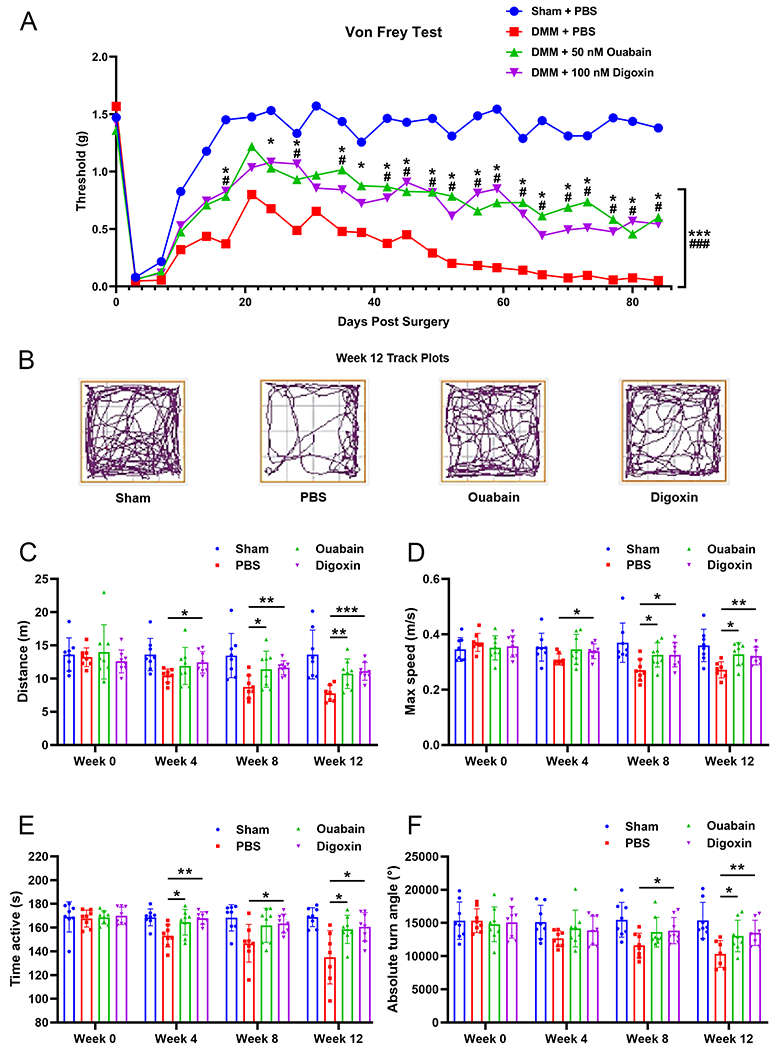
(A) Mechanical sensitivity was measured using von Frey filaments twice a week after DMM surgery. Statistical analysis was conducted using two-way analysis of variance (ANOVA) and multiple t tests. P values were compared between Ouabain (*) or Digoxin (#) group and PBS control group. (B) Representative track plots show decreased spontaneous activity of mice in open field tests after DMM surgery. Changes in spontaneous activity, including (C) travel distance, (D) max speed, (E) active time and (F) absolute turn angle were evaluated 4, 8 and 12 weeks after DMM surgery. * or #p<0.05, ** or ##p<0.01, *** or ###p<0.001 versus PBS control group.
Digoxin use is associated with reduced risk of knee or hip OA-associated joint replacement among patients with atrial fibrillation
To determine whether use of Digoxin associates with OA in human patients, we performed a sequential propensity-score matched cohort study using data from The Health Improvement Network (THIN) in the UK. The flow charts depict the selection process of individuals are shown in Supplementary Fig. 5. The baseline characteristics of each propensity-score matched cohort are shown in Supplementary Table 2. After propensity-score matching, 56,794 patients were included in the analysis (n=28,397 for each group). The mean age was 74.1 (standard deviation [SD]: 9.8) years among the Digoxin cohort and 75.1 (SD: 9.7) years among the non-user cohort; the female proportions were 49.1% and 51.5%, respectively. Overall, the characteristics among the propensity-score matched cohorts were well-balanced, with all standardized differences < 0.1. Digoxin initiators had a lower risk of knee or hip OA-associated joint replacement than non-users (Fig. 4). As shown in Table 1, 739 cases of knee or hip OA-associated joint replacement occurred among 28,397 Digoxin users (5.8 per 1000 person-years) and 854 cases occurred among 28,397 non-users (6.8 per 1000 person-years). The rate difference (RD) in knee or hip OA-associated joint replacement between Digoxin initiators and non-users was −0.9 (95% confidence interval [CI]: −1.5 to −0.3) per 1000 person-years and the HR was 0.85 (95% CI: 0.77 to 0.93). Results from the sensitivity analyses (i.e., excluding participants who had knee or hip replacement within three months after the index date, excluding participants who had extreme propensity-scores, and missing data imputation analysis) did not change substantially (Table 1)
Fig. 4.
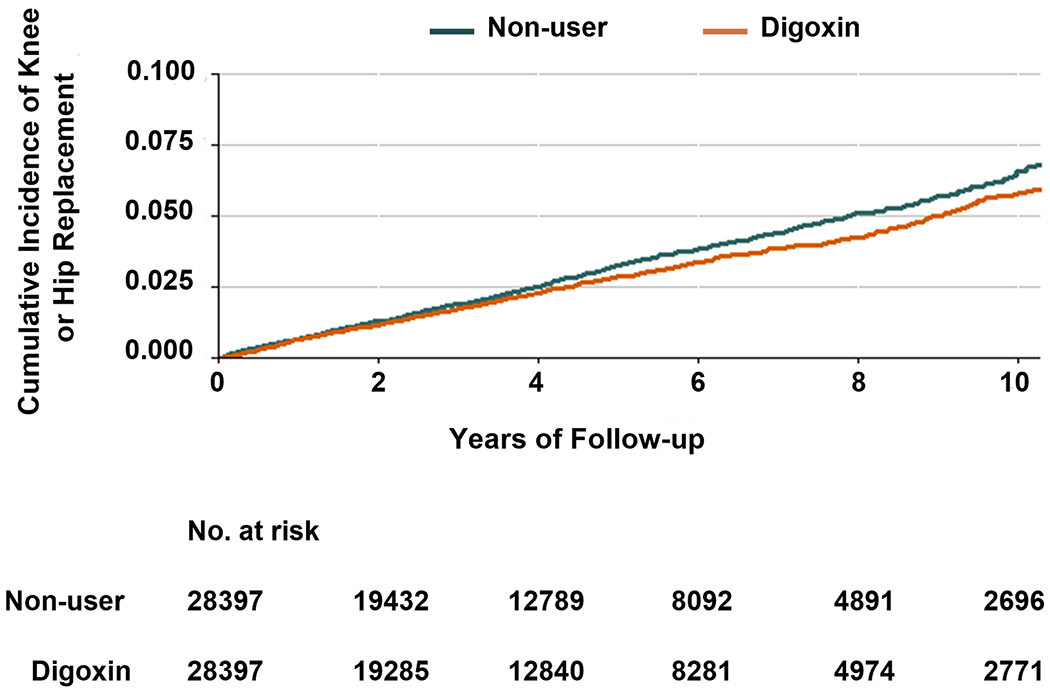
Cumulative incidence of knee or hip OA-associated joint replacement in 28397 Digoxin users and 28397 non-users, matched by propensity-score.
Table 1.
Association between Digoxin and Risk of Knee or Hip Replacement due to Osteoarthritis among Patients with Atrial Fibrillation
| Digoxin (n=28,397) | Non-user (n=28,397) | |
|---|---|---|
| Event (n) | 739 | 854 |
| Mean follow-up (years) | 4.46 | 4.45 |
| Rate of event, /1000 person-years | 5.83 | 6.75 |
| RD (95% CI), /1000 person-years | −0.9 (−1.5 to −0.3) | 0.0 (reference) |
| HR (95% CI) | 0.85 (0.77 to 0.93) | 1.00 (reference) |
| Three-month lag, HR (95% CI) a | 0.89 (0.80 to 0.98) | 1.00 (reference) |
| PS trimming, HR (95% CI) b | 0.86 (0.78 to 0.95) | 1.00 (reference) |
| Missing data imputation, HR (95% CI) c | 0.85 (0.77 to 0.95) | 1.00 (reference) |
HR, hazard ratio; RD, rate difference; 95% CI, 95% confidence interval.
This analysis introduced a three-month exposure lag period to exclude patients with knee or hip replacement within three months after treat date.
Asymmetric trimming was used to exclude participants whose propensity score was below the 2.5th percentile of the propensity score of the Digoxin cohort and above the 97.5th percentile of the propensity-score of the comparator cohort.
Imputation analysis was performed to deal with missing data. Specifically, missing values of the variables (i.e., body mass index, smoking, drinking status, or Townsend Deprivation Index) were imputed by a sequential regression method based on a set of covariates as predictors.
Digoxin regulates chondrocyte metabolism through ERK1/2, AKT and NF-κB signaling pathways
It is widely accepted that the extracellular signaling-regulated kinases (ERK) and AKT signaling pathways play an essential role in chondrocyte anabolism.28, 29 To investigate whether Ouabain and Digoxin activated anabolism through ERK1/2 and AKT signaling pathways, we treated chondrocytes with Ouabain or Digoxin at different concentrations or different time points, and then performed Western blot analysis for total and phosphorylated ERK1/2 and AKT. As shown in Supplementary Fig 6A, B, Ouabain and Digoxin could induce the phosphorylation of ERK1/2 and AKT in a dose and time dependent manner. Notably, Ouabain and Digoxin mediated activation of ERK1/2 and AKT signaling pathways and expressions of anabolic marker genes, including COL2A1, ACAN and COMP, were abolished by U0126 (ERK inhibitor) and wortmannin (AKT inhibitor) respectively (Supplementary Fig. 6C–F). These results revealed that Ouabain and Digoxin activated chondrocyte anabolism through ERK1/2 and AKT signaling pathways.
Pro-inflammatory cytokines TNFα and IL-1β play key roles by activating p38 mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinase (JNK) signaling and the transcription factor nuclear factor kappa B (NF-κB), in the pathogenesis of OA.30 Ouabain and Digoxin could inhibit both TNFα and IL-1β-induced chondrocytes catabolism promoted us to determine whether these two drugs could also affect these signaling pathways. Both Ouabain and Digoxin had no effect on the activation of p38 MAPK and JNK activated by TNFα, while dramatically inhibit NF-κB phosphorylation and transcriptional activity (Supplementary Fig. 7A–E). As illustrated in Supplementary Fig. 7F, p65 translocation from the cytoplasmic to the nuclear compartment upon stimulation with TNFα was almost abolished in the presence of Ouabain or Digoxin.
LRP4 is a novel target of Digoxin
Cardenolides bind to Na+/K+-ATPase and inhibit its activity. Accordingly, we sought to investigate whether Ouabain and Digoxin mediated chondrocyte metabolism through Na+/K+-ATPase. We thus suppressed the activity of Na+/K+-ATPase by treating C28I2 cells with Na+/K+-ATPase activity inhibitor Istaroxime hydrochloride. To our surprise, the inhibition of Na+/K+-ATPase activity did not affect either drug enhancement of anabolism or inhibition of catabolism (Supplementary Fig. 8A, B). This finding indicates that Ouabain and Digoxin may have targets other than Na+/K+-ATPase in mediation of chondrocyte homeostasis.
To address this issue, we performed the drug affinity responsive target stability (DARTS) assay and separated proteins by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). A band with the molecular weight of around 200 kDa was shown to be clearly protected by Ouabain (Fig. 5A). This band was excised for the unbiased protein identification by mass spectrometry (Fig. 5B), which identified LRP4 as a potential candidate. LRP4 is known to antagonize LRP5/6 signaling and mediate bone homeostasis.31 To confirm whether LRP4 is the target of Ouabain and Digoxin, we employed DARTS assay using a series of concentrations of protease to digest cell lysate with or without Ouabain or Digoxin incubation, and found that both drugs could protect LRP4 against enzymatic digestion (Fig. 5C).
Fig. 5. LRP4 is the target of Ouabain and Digoxin.
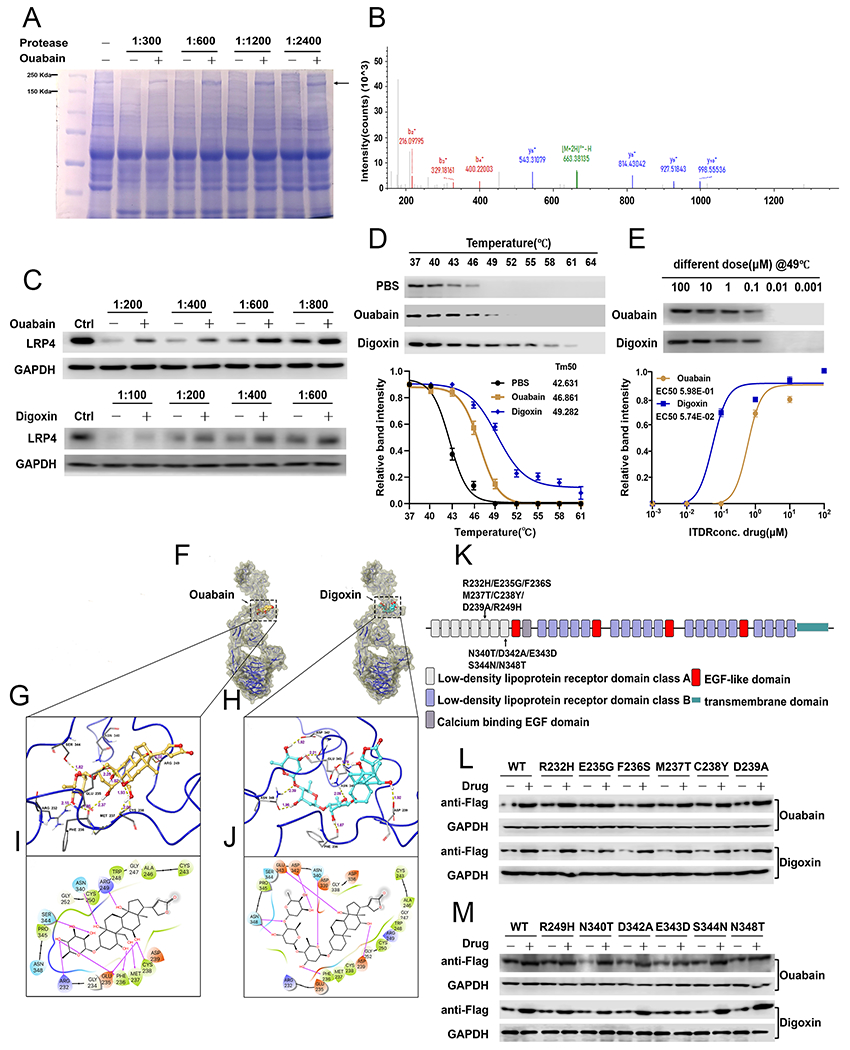
(A) Coomassie blue staining of DARTS assay. The band with molecular weight around 200 kDa was protected by Ouabain. (B) LRP4 adapted image from mass spectrometry. (C) C28I2 cells were digested with several dosages of protease with or without various concentrations of drugs, as indicated, then the level of LRP4 was assayed using Western blot. (D) C28I2 cell lysate was denatured under various temperatures and the protein level of LRP4 in control, Ouabain-treated and Digoxin-treated groups were assayed using Western blot and densitometry analysis curve. (E) Isothermal dose response with serial concentrations of drug. Protein level of LRP4 was measured via Western blot with associated curve. (F) Overview of IFD-predicted binding positions of Ouabain and Digoxin in LRP4 aa 146-737 monomer. LRP4 is shown by ribbons along with yellow surface of 70% transparency. Ouabain and Digoxin are shown by CPK representation with the following color scheme: carbon-faded orange (Ouabain) or teal (Digoxin), oxygen-red, polar hydrogen-white. Non-polar hydrogen atoms are not shown. (G, H) IFD-predicted docked LRP4-Ouabain complex (G) and LRP4-Digoxin complex (H) with the ligand depicted in ball and stick and the important interacting residues depicted as sticks. Hydrogen bonds are represented by dotted yellow lines and the distance of hydrogen bonds are measured in Å. (I, J) The 2D interaction diagrams of Ouabain (I) and Digoxin (J) docked with LRP4 aa 146-737. The amino acids within 4 Å to the ligand are shown as colored bubbles, where polar residues are cyan, hydrophobic residues are green, positively charged residues are purple, and negatively charged residues are red. Hydrogen bonds are shown by magenta arrows. (K) Schematic view of LRP4 receptor domain organizations and localization of mutations tested. (L, M) DARTS assay for serial point mutants of LRP4. C28I2 cells were transfected with the plasmid expressing various Flag-tagged LRP4 mutants, as indicated. Mutants of LRP4 were detected by Flag antibody. The values are mean±SEM of at least 3 independent experiments.
To further confirm the interaction between LRP4 and Ouabain and Digoxin, we performed the cellular thermal shift assay (CETSA),32, 33 which shows the change in thermal denaturation temperature for a target protein in the presence of various drug dosages. Both Ouabain and Digoxin prevented the denaturation of LRP4 at various temperatures, especially at 49°C, compared to control group (Fig. 5D). The melting curve showed a robust change of melting temperature (Tm) in the presence of Ouabain or Digoxin with Tm for control, Ouabain and Digoxin conditions 42.63, 46.86 and 49.28°C respectively (Fig. 5D). We also performed CETSA at 49°C with different concentrations of drugs and demonstrated that Ouabain and Digoxin prevented LRP4 denaturation in a dose-dependent manner with the EC50 of 5.98E-01 and 5.74E-02 respectively (Fig. 5E).
To further unravel the associations of LRP4 with Ouabain and Digoxin, we then employed molecular docking simulations. The structures of Ouabain and Digoxin were docked with three monomers of homology modelled human LRP4, respectively. Glide XP docking showed that both Ouabain and Digoxin had relatively lower binding free energy when they were docked into LRP4 monomer aa 146-737 compared to the other two monomers, which was reflected by the more negative values of docking scores (Supplementary Fig. 9), suggesting that Ouabain and Digoxin may have better affinity to the region from residue 146 to 737 of LRP4. Induced-fit docking (IFD) was then performed for Ouabain and Digoxin with LRP4 monomer aa146-737 to get optimal binding simulation. From IFD simulation, both Ouabain and Digoxin were predicted to majorly interact with LRP4 with docking scores of −12.496 kcal/mol and −10.149 kcal/mol (Fig. 5F). The binding pocket for Ouabain and Digoxin was lined by residues 232-239, 243-252, and 340-348 of LRP4. With abundant hydroxyl groups in the structure, Ouabain and Digoxin could form multiple hydrogen bonding interactions with LRP4, which may contribute to the good binding affinity. All the hydroxyl groups of Ouabain are predicted to be involved in hydrogen bonding interactions with LRP4, which bonded with residues Arg232, Glu235, Phe236, Met237, Cys238, Arg249, Asn340, and Ser344, respectively. Several hydroxyl groups of Digoxin served as hydrogen bond donors interacting with the side chains of Phe236, Asp239, Asp342, Glu343, and Asn348 of LRP4. Additionally, Digoxin was exhibited to be involved in hydrogen bonds as acceptor of LRP4 at residues Asn340 and Asn348 (Fig. 5G–J).
We next mutated the sites of LRP4 mentioned above and performed DARTS to determine key site(s) for the binding between LRP4 and drugs (Fig. 5K). Results showed that Ouabain largely lost, while Digoxin totally lost, its protective effect on the Glu-343 mutant of LRP4 (Fig. 5L, M), demonstrating that Glu-343 is the critical amino acid for the drugs targeting to LRP4.
LRP4 is downregulated in OA and its deficiency abolishes Digoxin regulation of chondrocyte metabolism
To explore the relationship of LRP4 and OA, we examined the expression of LRP4 in human normal and OA patients’ cartilage. LRP4 protein level was reduced in human OA cartilage compared with non-arthritic controls (Fig. 6A–C). Furthermore, in line with the results in human cartilage, the protein expression of LRP4 was decreased after murine DMM surgery, whereas intriguingly, the administration of Ouabain and Digoxin prevented OA-associated LRP4 down-regulation (Fig. 6D, E).
Fig. 6. LRP4 is down-regulated in OA cartilage and required for Ouabain and Digoxin regulation of chondrocyte anabolism.
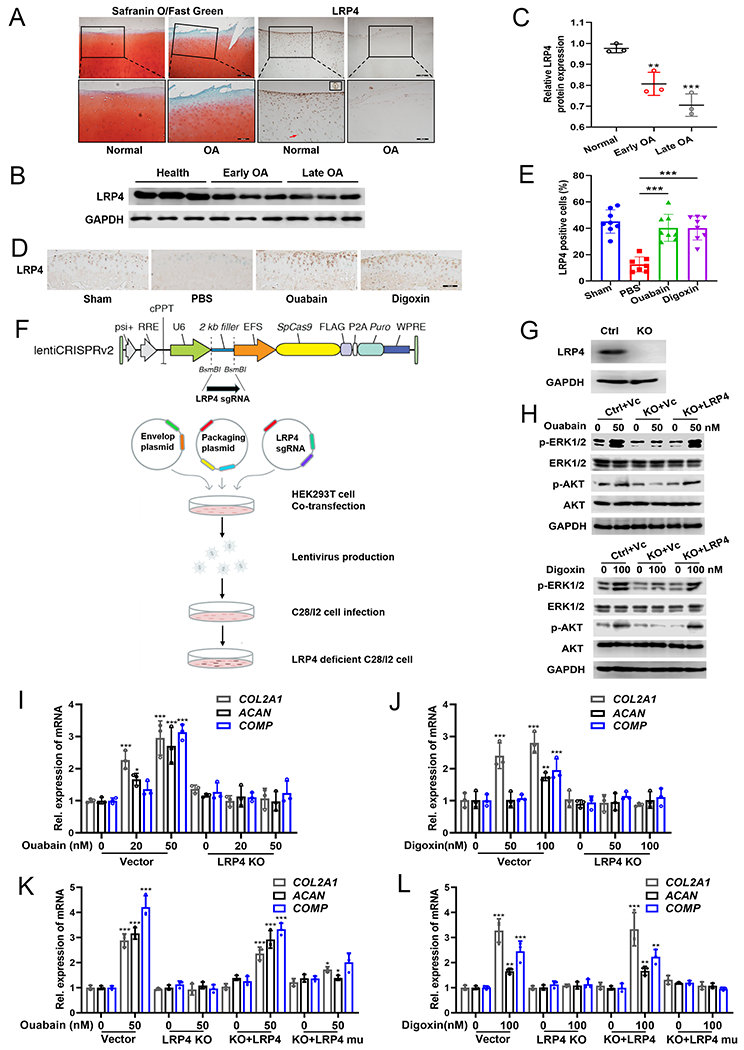
(A) Human non-arthritic and OA cartilage stained with Safranin O/Fast Green (left) and detected LRP4 by immunohistochemical staining (right). The red arrow and the inserted image indicate the expression of LRP4 in a single chondrocyte. Scale bar=400 μm (top panel) and 200 μm (bottom panel). (B) Expression of LRP4 was measured in healthy, early OA and late OA patients via Western blot. (C) Densitometry analysis of immunoblotting results shown in (B). (D, E) Immunohistochemical staining of LRP4 and quantification of LRP4 positive cells in knee joint sections of C57BL/6J mice treated with or without cardenolides for 12 weeks after DMM surgery. Scale bar=100 μm; n≥7. (F) Workflow of generating LRP4 knockout C28I2 cells using CRISPR/Cas9 technology. (G) Western blot confirmation of LRP4 knockout in C28I2 cells. (H) Immunoblotting of ERK1/2 and AKT signaling activation in control and LRP4 knockout C28I2 cells with or without re-expression of LRP4 treated with 50 nM Ouabain or 100 nM Digoxin for 30 min. (I, J) mRNA levels of COL2A1, ACAN and COMP in control or LRP4 knockout C28I2 cells treated with a series of Ouabain or Digoxin concentrations for 24 h, assayed by qRT-PCR analysis. (K, L) Control and LRP4 knockout C28I2 cells with or without re-expression of wildtype or Glu343 site mutated LRP4 were treated with 50 nM Ouabain or 100 nM Digoxin for 24 h, mRNA levels of COL2A1, ACAN and COMP were detected by qRT-PCR. LRP4 mu: Glu343 site mutation of LRP4. The values are mean±SEM of at least 3 independent experiments; *p<0.05, **p<0.01, ***p<0.001 versus control group.
To define the role of LRP4 in mediating Ouabain and Digoxin’s effects on chondrocyte metabolism, we efficiently deleted the LRP4 gene using CRISPR-Cas9 genome editing strategy (Fig. 6F, G). Notably, the activation of ERK1/2, AKT signaling pathways induced by Ouabain and Digoxin were abolished in LRP4 knockout chondrocytes, leaving expression of the anabolic marker genes, such as COL2A1, ACAN and COMP, unchanged after drug treatment (Fig. 6H, I, J). Furthermore, Ouabain and Digoxin mediated inhibition of NF-κB activation was lost in LRP4 knockout cells, resulting in increased expression of catabolic genes, including ADAMTS4 and MMP13 (Supplementary Fig. 10A–D).
We next re-expressed wildtype or Glu343 mutated LRP4 in LRP4 knockout C28I2 chondrocytes. Transfection of wildtype LRP4 expression plasmid reinstated Ouabain and Digoxin induced activation of ERK1/2, AKT signaling pathways and chondrocyte anabolism (Fig. 6H, K, L) and also restored the anti-NF-κB effect of Ouabain and Digoxin and the inhibition of chondrocyte catabolism (Supplementary Fig. 10A, B, E, F). However, expression of LRP4 containing a Glu343 site mutation in LRP4 knockout C28I2 cells did not restore the regulatory effects of Ouabain and Digoxin upon chondrocyte metabolism (Fig. 6K, L, Supplementary Fig. 10E, F). Taken together, these findings indicated that Ouabain and Digoxin facilitation of anabolism and inhibition of catabolism in chondrocytes is LRP4-dependent and Glu343 is critical for drug-mediated regulation of chondrocyte metabolism.
DISCUSSION
We have made several key observations in the studies presented here. (1) After performance of three rounds of RT-qPCR screening of an FDA-approved drug library, we identified Ouabain and Digoxin as candidates with potential to promote the anabolism of chondrocyte ECM. (2) We found that the administration of Ouabain or Digoxin limited OA development and relieved OA-associated pain sensitivity in mice, and our large population-based cohort study provides clinical evidence that Digoxin use was associated with a reduced risk of knee or hip OA-associated joint replacement among patients with atrial fibrillation. (3) Through combined use of DARTS, proteomics, CETSA, IFD and generation of point mutations, we identified LRP4 as a previously unrecognized target of Digoxin and found that Digoxin’s regulation of chondrocytes depends on the presence of LRP4.
No previous studies have directly investigated the effects of cardenolides use on OA. Though indirect evidence concerning the utility of these compounds in facilitating cartilage homeostasis has been provided through in vitro study which found that Ouabain and Digoxin could increase the functional properties of bovine articular chondrocytes. This promotion may be related to the amount of collagen cross-linking and increased Ca2+ oscillations.21 In the current study, we have implemented a murine surgically–induced OA model to provide in vivo evidence that both Ouabain and Digoxin can limit OA development and relieve OA-associated pain. Consistently, the anabolism of ECM of chondrocytes is promoted, while the catabolism is inhibited, by Ouabain and Digoxin. To explore the mechanism of this dual protective effect, we studied multiple pathways closely related to OA by using specific inhibitors of each pathway. The results confirmed that activation of AKT and ERK pathways are involved in Ouabain and Digoxin-mediated promotion of ECM anabolism, while these cardenolides exert inhibitory effects on NF-κB pathway activation lending to reduced chondrocyte catabolism.
Digoxin is known as the only safe inotropic drug for oral use that improves hemodynamics.35 This led us to hypothesize that records related to Digoxin might be accessible in general practitioner based medical records databases. Herein, we performed a sequential propensity-score matched cohort study via leveraging the data from THIN. THIN contains health information on approximately 17 million patients from 790 general practices in the UK. THIN data reflects a routine medical practice environment and have been shown to be valid for use in clinical and epidemiological research studies.36–38 Excitingly, we found that Digoxin use was associated with reduced risk of joint replacement surgery due to OA in a large population with atrial fibrillation. Despite the inevitable limitations of this database-based study (confounding factors, information authenticity, and lack of knee or hip images), this study has significant implications for the clinical prospects of Digoxin against OA.
Most of the biological activities of cardenolides are based on their ability to inhibit the membrane-bound Na+/K+-ATPase.16 Inhibition of Na+/K+-ATPase has been shown to induce cell proliferation, autophagy and even apoptosis, not only in cardiac myocytes but also other several cell lines.39, 40 Under the intervention condition in this study, both Ouabain and Digoxin treatment didn’t show any toxicity (Supplementary Fig. 11). Neither the promoting effect of Ouabain and Digoxin on cartilage ECM anabolism nor the inhibitory effect on ECM catabolism was affected by inhibiting the activity of Na+/K+-ATPase with Istaroxime hydrochloride in chondrocytes. This suggested that Ouabain and Digoxin may regulate cartilage ECM homeostasis through a completely different target that has not yet been discovered. Therefore, through combined use of DARTS, proteomics, CETSA, IFD and point mutations, we identified LRP4 as the target and Glu-343 as the critical amino acid involved in the interaction with Digoxin. By knockout and re-expression of LRP4 in chondrocytes, we demonstrated that LRP4 is indispensable in the process of both Ouabain and Digoxin promoting ECM synthesis by regulating ERK and AKT pathways and inhibiting ECM degradation by regulating NF-κB pathway (Supplementary Fig. 12).
LRP4 is a member of the low-density lipoprotein receptor family. At neuromuscular junction, neuronal agrin binds LRP4 in a complex with the muscle-specific kinase (MuSK) to form neuromuscular synapse.41 In recent decades, increasing reports describing mutations of members of the LRP family and their relation to bone has reinforced the important role of LRP family members in the pathogenesis of devastating diseases such as osteoporosis, rheumatoid arthritis and OA.31 LRP4 was reported to induce gene expressions of ECM proteins, as well as production of total proteoglycans in ATDC5 chondrocyte cells, whereas LRP4 knockdown had opposite effects and reduced mRNA expression of SOX9, a master regulator for chondrogenesis.42 LRP4 was also reported to up-regulate SOX9 expression in primary bovine chondrocytes, and LRP4 was indispensable for the promotion of chondrocyte differentiation and cartilage formation mediated by agrin.43 These findings are consistent with our current report of reduced LRP4 expression in OA cartilage and that the dependence of the chondroprotective effect of Ouabain and Digoxin on LRP4. However, the specific mechanism by which LRP4 affects chondrocyte function warranties further investigations. Despite the lack of current knowledge regarding LRP4 in chondrocytes, the roles of LRP4 and other members of the LRP family in bone has long been studied. LRP4 and family-member LRP5/6 have opposite effects on bone mass regulation31, 44 In OA specifically, in addition to the lack of reports on LRP4, the role of LRP5 and LRP6 is also controversial.45–48 Therefore, although LRP4 has been found to regulate Wnt and BMP pathways in other physiological and pathological processes,49–51 further studies are needed to determine whether LRP4 can affect the development of OA by regulating these pathways and thereby mediating the interaction with other LRPs. In our study, both Ouabain and Digoxin regulated the expression of the proteins related to AKT, ERK and NF-κB pathways in chondrocytes. This provides a new clue for the mechanism of LRP4 in chondrocyte function and the development of OA.
This study carries significance from several perspectives: This study identifies Digoxin as a stimulator of chondrocyte differentiation and anabolism, and uncovers a new strategy for enhancing chondrocyte function and cartilage integrity. Thus, Digoxin may be used for treating various chondrocyte-associated diseases and conditions, particularly OA. Due to the unique mechanism of action (e.g., targeting LRP4 to enhance chondrocyte function), Digoxin may be effective for OA patients who fail to respond to current anti-inflammatory and anti-catabolic treatments. This study identifies LRP4 as a new target of Digoxin, thus advancing our understanding of Digoxin’s action and underlying mechanisms, and also providing a solid foundation for future discoveries related to this Digoxin/LRP4 interaction in various conditions.
Limitations of this study should be also acknowledged. Although previous studies have used joint replacement as a proxy for end-stage OA and joint replacement has been generally accepted as a clinically relevant “hard” outcome in cohort studies of OA,52–54 knee images and functional data were not available in THIN; thus, we were unable to examine the association between Digoxin and the risk of progression of structural lesions and functional deterioration of OA. Although we conducted a 1:1 propensity-score matched cohort study to control for many potential confounders, this may limit generalizability of our findings to patients with OA whose characteristics differ from OA patients included in the current study. Although we have demonstrated that digoxin and ouabain can relieve OA-related pain in vivo, we failed to delve deeply into the mechanisms. The weak correlation between pain and cartilage loss, the complex classification of pain and the conduction involved,55, 56 give us reason to believe that further research, richer tools and extended time points are need to uncover a full story. Further, additional in vivo characterization and pharmaceutic kinetic assays of the drug in OA, including dose dependence, frequency and route of delivery, as well as treatment duration, warrant further investigations with preclinical animal models and human clinical trials.
In summary, functional studies, including various cell-based assays, in vivo animal model, and analysis of a large cohort of human patients, support the concept of using Digoxin as novel treatment for OA patients in clinic. Additionally, combined use of various approaches isolates LRP4 as the new target of Digoxin responsible for its chondroprotective action. Thus, this study not only provides new insights into the understanding of Digoxin’s action and underlying mechanisms, but may also broaden the clinical application of Digoxin.
Supplementary Material
KEY MESSAGES.
What is already known about this subject?
Cardenolides, including Digoxin, have been used in treating heart disease for centuries, and have been reported to enhance chondrocyte function in vitro, but how they regulate chondrocytes and whether they are therapeutic against OA remains unknown.
What does this study add?
This study identifies Digoxin as a new chondroprotective factor that protects against OA and reduces OA-associated pain in a surgically-induced mouse OA model.
Digoxin use is associated with reduced risk of knee or hip OA-associated joint replacement among patients with atrial fibrillation.
This study isolates LRP4 as a new target of Digoxin, thus advancing our understanding of Digoxin’s action and underlying mechanisms and providing a solid foundation for future discoveries relating to the Digoxin/LRP4 interaction in various conditions.
How might this impact on clinical practice or future developments?
The chondroprotective effects of Digoxin on OA support the concept that cardenolides, particularly Digoxin, may be a new option to treat patients with OA in clinics.
Acknowledgement:
The authors would like to acknowledge all lab members for insightful discussions. We thank Dr. Mary Goldring for providing us with human C28I2 chondrocytes. We are also grateful to Dr. Tanaji T. Talele (St. John’s University, New York, NY, USA) for providing the computing resources for docking analysis. We also thank NYU Proteomics Laboratory and microCT core for technique support.
Funding:
This work is supported partly by NIH research grants R01AR062207, R01AR061484, R01AR076900, R01NS103931 and a DOD research grant W81XWH-16-1-0482.
Footnotes
Competing interests: None declared.
Patient and public involvement: Patients and/or the public were not involved in the design, or conduct, or reporting or dissemination plans of this research.
Patient consent for publication: Not required.
Ethics approval: All animal procedures were carried out in accordance with institutional guidelines and approved by the Institutional Animal Care and Use Committee of New York University. Human subjects research was performed according to the Institutional Review Boards at New York University Medical Center (IRB Study Number i11-01488). This study was approved by the THIN Scientific Review Committee (20SRC003). THIN is a registered trademark of Cegedim SA in the UK and other countries. Reference made to the THIN database is intended to be descriptive of the data asset licensed by IQVIA. This work uses de-identified data provided by patients as part of their routine primary care.
Data availability statement:
No data are available from THIN. While we are unable to share the data sets, code lists are available on request. The rest of the data relevant to the study are included in the article or uploaded as supplementary information.
REFERENCES
- 1.Hunter DJ, Bierma-Zeinstra S. Osteoarthritis. The Lancet 2019;393:1745–59. [DOI] [PubMed] [Google Scholar]
- 2.Katz JN, Arant KR, Loeser RF. Diagnosis and treatment of hip and knee osteoarthritis: a review. JAMA 2021;325:568–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hunter DJ, Schofield D, Callander E. The individual and socioeconomic impact of osteoarthritis. Nat Rev Rheumatol 2014;10:437–41. [DOI] [PubMed] [Google Scholar]
- 4.Martel-Pelletier J, Barr AJ, Cicuttini FM, et al. Osteoarthritis. Nat Rev Dis Primers 2016;2:16072. [DOI] [PubMed] [Google Scholar]
- 5.Loeser RF, Collins JA, Diekman BO. Ageing and the pathogenesis of osteoarthritis. Nature Reviews Rheumatology 2016;12:412–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rahmati M, Nalesso G, Mobasheri A, et al. Aging and osteoarthritis: central role of the extracellular matrix. Ageing Res Rev 2017;40:20–30. [DOI] [PubMed] [Google Scholar]
- 7.Poole AR, Kobayashi M, Yasuda T, et al. Type II collagen degradation and its regulation in articular cartilage in osteoarthritis. Ann Rheum Dis 2002;61 Suppl 2:i78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loeser RF, Goldring SR, Scanzello CR, et al. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum 2012;64:1697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldring MB, Otero M, Tsuchimochi K, et al. Defining the roles of inflammatory and anabolic cytokines in cartilage metabolism. Ann Rheum Dis 2008;67 Suppl 3:i75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang Y, Tuan RS. Origin and function of cartilage stem/progenitor cells in osteoarthritis. Nature Reviews Rheumatology 2015;11:206–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grogan SP, Miyaki S, Asahara H, et al. Mesenchymal progenitor cell markers in human articular cartilage: normal distribution and changes in osteoarthritis. Arthritis Res Ther 2009;11:R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Niu J. Editorial: shifting gears in osteoarthritis research toward symptomatic osteoarthritis. Arthritis Rheumatol 2016;68:1797–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chong CR, Sullivan DJ. New uses for old drugs. Nature 2007;448:645–6. [DOI] [PubMed] [Google Scholar]
- 14.Sachs RE, Ginsburg PB, Goldman DP. Encouraging new uses for old drugs. JAMA 2017;318:2421–2. [DOI] [PubMed] [Google Scholar]
- 15.Pantziarka P, Pirmohamed M, Mirza N. New uses for old drugs. BMJ 2018;361:k2701. [DOI] [PubMed] [Google Scholar]
- 16.El-Seedi HR, Khalifa S, Taher EA, et al. Cardenolides: insights from chemical structure and pharmacological utility. Pharmacol Res 2019;141:123–75. [DOI] [PubMed] [Google Scholar]
- 17.Cavalcante-Silva L, Lima ÉA, Carvalho D, et al. Much more than a cardiotonic steroid: modulation of inflammation by ouabain. Front Physiol 2017;8:895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pirkmajer S, Bezjak K, Matkovič U, et al. Ouabain suppresses IL-6/STAT3 signaling and promotes cytokine secretion in cultured skeletal muscle cells. Front Physiol 2020;11:566584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee J, Baek S, Lee J, et al. Digoxin ameliorates autoimmune arthritis via suppression of Th17 differentiation. Int Immunopharmacol 2015;26:103–11. [DOI] [PubMed] [Google Scholar]
- 20.Wei Z, Wang Y, Zhang K, et al. Inhibiting the Th17/IL-17A-related inflammatory responses with digoxin confers protection against experimental abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol 2014;34:2429–38. [DOI] [PubMed] [Google Scholar]
- 21.Makris EA, Huang BJ, Hu JC, et al. Digoxin and adenosine triphosphate enhance the functional properties of tissue-engineered cartilage. Tissue Eng Part A 2015;21:884–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin EA, Liu CJ. The role of ADAMTSs in arthritis. Protein Cell 2010;1:33–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu CJ, Zhang Y, Xu K, et al. Transcriptional activation of cartilage oligomeric matrix protein by Sox9, Sox5, and Sox6 transcription factors and CBP/p300 coactivators. Front Biosci 2007;12:3899–910. [DOI] [PubMed] [Google Scholar]
- 24.Liu-Bryan R, Terkeltaub R. Emerging regulators of the inflammatory process in osteoarthritis. Nat Rev Rheumatol 2015;11:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Houard X, Goldring MB, Berenbaum F. Homeostatic mechanisms in articular cartilage and role of inflammation in osteoarthritis. Curr Rheumatol Rep 2013;15:375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fang H, Beier F. Mouse models of osteoarthritis: modelling risk factors and assessing outcomes. Nat Rev Rheumatol 2014;10:413–21. [DOI] [PubMed] [Google Scholar]
- 27.Goldring SR, Goldring MB. Changes in the osteochondral unit during osteoarthritis: structure, function and cartilage-bone crosstalk. Nat Rev Rheumatol 2016;12:632–44. [DOI] [PubMed] [Google Scholar]
- 28.Sun K, Luo J, Guo J, et al. The PI3K/AKT/mTOR signaling pathway in osteoarthritis: a narrative review. Osteoarthritis Cartilage 2020;28:400–9. [DOI] [PubMed] [Google Scholar]
- 29.Duan L, Ma B, Liang Y, et al. Cytokine networking of chondrocyte dedifferentiation in vitro and its implications for cell-based cartilage therapy. Am J Transl Res 2015;7:194–208. [PMC free article] [PubMed] [Google Scholar]
- 30.Wojdasiewicz P, Poniatowski AA, Szukiewicz D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm 2014;2014:561459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lara-Castillo N, Johnson ML. LRP receptor family member associated bone disease. Rev Endocr Metab Disord 2015;16:141–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinez MD, Jafari R, Ignatushchenko M, et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013;341:84–7. [DOI] [PubMed] [Google Scholar]
- 33.Jafari R, Almqvist H, Axelsson H, et al. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc 2014;9:2100–22. [DOI] [PubMed] [Google Scholar]
- 34.Latourte A, Kloppenburg M, Richette P. Emerging pharmaceutical therapies for osteoarthritis. Nat Rev Rheumatol 2020;16:673–88. [DOI] [PubMed] [Google Scholar]
- 35.Kjeldsen K, Nørgaard A, Gheorghiade M. Myocardial Na,K-ATPase: the molecular basis for the hemodynamic effect of digoxin therapy in congestive heart failure. Cardiovasc Res 2002;55:710–3. [DOI] [PubMed] [Google Scholar]
- 36.Lewis JD, Schinnar R, Bilker WB, et al. Validation studies of the health improvement network (THIN) database for pharmacoepidemiology research. Pharmacoepidemiol Drug Saf 2007;16:393–401. [DOI] [PubMed] [Google Scholar]
- 37.Zeng C, Dubreuil M, Larochelle MR, et al. Association of tramadol with all-cause mortality among patients with osteoarthritis. JAMA 2019;321:969–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zeng C, Bennell K, Yang Z, et al. Risk of venous thromboembolism in knee, hip and hand osteoarthritis: a general population-based cohort study. Ann Rheum Dis 2020;79:1616–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurauchi Y, Noma K, Hisatsune A, et al. Na(+), K(+)-ATPase inhibition induces neuronal cell death in rat hippocampal slice cultures: Association with GLAST and glial cell abnormalities. J Pharmacol Sci 2018;138:167–75. [DOI] [PubMed] [Google Scholar]
- 40.Felippe GC, Ribeiro SA, Ignácio DSC, et al. Na/K pump and beyond: Na/K-ATPase as a modulator of apoptosis and autophagy. Molecules 2017;22: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim N, Stiegler AL, Cameron TO, et al. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell 2008;135:334–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Asai N, Ohkawara B, Ito M, et al. LRP4 induces extracellular matrix productions and facilitates chondrocyte differentiation. Biochem Biophys Res Commun 2014;451:302–7. [DOI] [PubMed] [Google Scholar]
- 43.Eldridge S, Nalesso G, Ismail H, et al. Agrin mediates chondrocyte homeostasis and requires both LRP4 and α-dystroglycan to enhance cartilage formation in vitro and in vivo. Ann Rheum Dis 2016;75:1228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiong L, Jung JU, Wu H, et al. Lrp4 in osteoblasts suppresses bone formation and promotes osteoclastogenesis and bone resorption. Proc Natl Acad Sci U S A 2015;112:3487–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kerkhof JM, Uitterlinden AG, Valdes AM, et al. Radiographic osteoarthritis at three joint sites and FRZB, LRP5, and LRP6 polymorphisms in two population-based cohorts. Osteoarthritis Cartilage 2008;16:1141–9. [DOI] [PubMed] [Google Scholar]
- 46.Lodewyckx L, Luyten FP, Lories RJ. Genetic deletion of low-density lipoprotein receptor-related protein 5 increases cartilage degradation in instability-induced osteoarthritis. Rheumatology (Oxford) 2012;51:1973–8. [DOI] [PubMed] [Google Scholar]
- 47.Joiner DM, Less KD, Van Wieren EM, et al. Heterozygosity for an inactivating mutation in low-density lipoprotein-related receptor 6 (Lrp6) increases osteoarthritis severity in mice after ligament and meniscus injury. Osteoarthritis Cartilage 2013;21:1576–85. [DOI] [PubMed] [Google Scholar]
- 48.Shin Y, Huh YH, Kim K, et al. Low-density lipoprotein receptor-related protein 5 governs Wnt-mediated osteoarthritic cartilage destruction. Arthritis Res Ther 2014;16:R37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Y, Pawlik B, Elcioglu N, et al. LRP4 mutations alter Wnt/beta-catenin signaling and cause limb and kidney malformations in Cenani-Lenz syndrome. Am J Hum Genet 2010;86:696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ahn Y, Sims C, Murray MJ, et al. Multiple modes of Lrp4 function in modulation of Wnt/β-catenin signaling during tooth development. Development 2017;144:2824–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kumar J, Swanberg M, Mcguigan F, et al. LRP4 association to bone properties and fracture and interaction with genes in the Wnt- and BMP signaling pathways. Bone 2011;49:343–8. [DOI] [PubMed] [Google Scholar]
- 52.Neogi T, Li S, Peloquin C, et al. Effect of bisphosphonates on knee replacement surgery. Ann Rheum Dis 2018;77:92–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ballal P, Peloquin C, Boer CG, et al. Warfarin use and risk of knee and hip replacements. Ann Rheum Dis 2021;80:605–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu D, Jordan KP, Snell K, et al. Development and validation of prediction models to estimate risk of primary total hip and knee replacements using data from the UK: two prospective open cohorts using the UK Clinical Practice Research Datalink. Ann Rheum Dis 2019;78:91–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bacon K, Lavalley MP, Jafarzadeh SR, et al. Does cartilage loss cause pain in osteoarthritis and if so, how much? Ann Rheum Dis 2020;79:1105–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Conaghan PG, Cook AD, Hamilton JA, et al. Therapeutic options for targeting inflammatory osteoarthritis pain. Nat Rev Rheumatol 2019;15:355–63. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No data are available from THIN. While we are unable to share the data sets, code lists are available on request. The rest of the data relevant to the study are included in the article or uploaded as supplementary information.