

Abstract
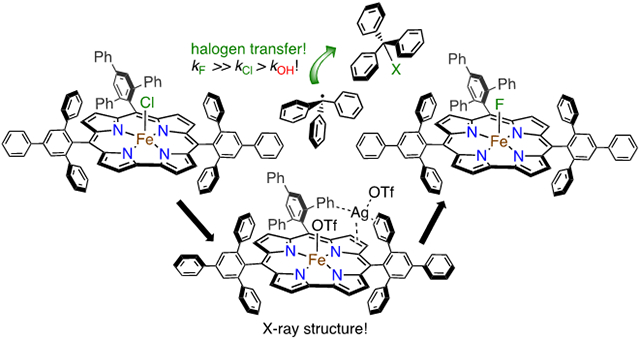
High-valent iron halide corroles were examined to determine their reactivity with carbon radicals and their ability to undergo radical rebound-like processes. Beginning with Fe(Cl)(ttppc) (1) (ttppc = 5,10,15-tris(2,4,6-triphenylphenyl)corrolato3−), the new iron corroles Fe(OTf)(ttppc) (2), Fe(OTf)(ttppc)(AgOTf) (3), and Fe(F)(ttppc) (4) were synthesized. Complexes 3 and 4 are the first iron triflate and iron fluoride corroles to be structurally characterized by single crystal X-ray diffraction. The structure of 3 reveals an AgI─pyrrole (η1 − π) interaction. The Fe(Cl)(ttppc) and Fe(F)(ttppc) complexes undergo halogen transfer to triarylmethyl radicals, and kinetic analysis of the reaction between (p-OMe-C6H4)3C• and 1 gave k = 1345 ± 34 M−1 s−1 at 23 °C and 2.18 ± 0.17 M−1 s−1 at −60 °C, and ΔH⧧ = +9.77 ± 0.29 kcal mol−1; ΔS⧧ = −14.39 ± 1.29 cal mol−1 K−1 through an Eyring analysis. Complex 4 is significantly more reactive, giving k = 1.158 x 105 ± 0.064 M−1 s−1 at 23 °C. The data point to a concerted mechanism, and show the trend X = F− > Cl− > OH− for Fe(X)(ttppc). This study provides mechanistic insights into halogen rebound for an iron porphyrinoid complex.
Graphical Abstract
Introduction.
The conversion of C-H bonds into other functional groups is of widespread importance in native biological processes and synthetic chemistry. Both heme and nonheme metalloenzymes catalyze such reactions with remarkable selectivity.1 The heme monooxygenase Cytochrome P450 (CYP) is one of the most well-studied enzymes in this class, yet much remains unknown regarding how selectivity is tuned by different CYPs.2-5 For example, hydroxylation by CYP,6 where R-H is converted to ROH, is sometimes diverted to a desaturation pathway that can result in toxic alkene metabolites,7-10 or in the decarboxylation of fatty acids, as in the case of CYP-OleT.11-15 This divergence occurs at the so-called “rebound” step, in which a transient, high-valent FeIV(OH)(porph) intermediate (protonated Compound-II (Cpd-II)) transfers •OH to the nascent carbon radical R•, or can mediate the other processes shown in Scheme 1.16
Scheme 1.

C-H Bond Activation by Cytochrome P450
Our laboratory has been interested in determining the fundamental properties of the metal center that may contribute to steering the outcome of the rebound process by studying appropriate porphyrinoid model compounds. Part of the challenge in studying this process in a direct fashion was the need to synthesize a suitable terminal Fe(OH) complex at the same redox level as Cpd-II, which was unknown at the time we began our efforts. We reported the first example of such a complex, taking advantage of a sterically encumbered corrole platform to give Fe(OH)(ttppc) (ttppc = 5,10,15-tris(2,4,6-triphenyl)phenyl corrolato3−).17, 18 We also recently described the manganese analog of this complex, Mn(OH)(ttppc), and showed that both species are capable of transferring •OH to tertiary carbon radicals.19 A detailed kinetic and thermodynamic analysis suggested that these reactions operate via a highly concerted mechanism, with C-O bond formation concomitant with M-OH bond cleavage and one-electron reduction of the metal center to the MIII oxidation state.
Given our success in studying •OH radical transfer reactions with the Fe and Mn ttppc complexes, we hypothesized that the sterically encumbered ttppc platform would allow us to examine the related halogen transfer reactions via a direct, rebound-type process. Our goals were to 1) establish that an iron corrole could mediate the direct transfer of halogens to carbon radicals, 2) determine the comparative reactivity of Fe(X) (X = Cl, F, OH) with carbon radicals, 3) define the mechanism of halogen transfer, and 4) gain insight into the general rules that guide the rebound process in heme and nonheme iron enzymes as well as related catalysts. The halogenation of C-H bonds is of considerable synthetic and biological significance. These reactions are mediated by heme and nonheme iron enzymes, including the heme enzyme chloroperoxidase, although this enzyme does not follow the C-H abstraction/rebound pathway seen for CYP.20 However, the nonheme iron halogenases (e.g. SyrB2, WelO5) carry out selective halogenation of a range of C-H bonds, and are proposed to follow a rebound mechanism in which a putative FeIII(OH)(halide) intermediate preferentially transfers the halogen atom over the hydroxyl group to the nearby carbon radical (Scheme 2).21-31 The factors that control the selectivity for nonheme iron-catalyzed halogenation remain under debate, but efforts have recently been made to reengineer nonheme Fe halogenases with specific regio- and stereoselectivities.32-35
Scheme 2.

C-H Bond Chlorination by αKG-Dependent Enzymes
From a synthetic perspective, the halogenation of C-H bonds has become an increasingly prominent target for catalysis.36-40 The development of either heme41-46 or nonheme47-54 base metal (e.g. Fe, Mn) synthetic catalysts for selective C-H halogenation has been particularly alluring because of their anticipated environmental and economic benefits, as well as their relationship to the biological systems. One prominent example in oxidative C-H fluorination involves homogenous manganese porphyrin catalysts that are postulated to operate via transfer of fluorine from an MnIV(F)2(porph) intermediate to carbon radicals.41-44 In related work, a heterogenous MOF-based catalyst with incorporated Mn porphyrin centers was shown to catalyze the C-H halogenation of inert hydrocarbons in high yield.55 A good example of the power of nonheme iron complexes to catalyze the tandem reactions of C-H cleavage and halogenation was reported with the use of a combination of a room-temperature stable nonheme iron(IV)-oxo complex and an iron(II)-halide complex to catalyze the selective halogenation of aliphatic substrates.48 In all of these cases, the rates of halogen transfer compared to other pathways, such as hydroxyl transfer, are key to the efficiencies and selectivities of these systems. However, there remains much unknown about what features of a metal catalyst influence the rates of halogen (or other group) transfer to carbon radicals.
In the present work, we report the synthesis and first crystallographic structural characterization of a fluoride-ligated iron corrole, Fe(F)(ttppc).56 Examination of the reactivity of this complex together with the previously reported Fe(Cl)(ttppc)17 has led to the first direct observations of halogen transfer (or halogen “rebound”), from an iron porphyrinoid complex to a carbon radical. Kinetic and thermodynamic analyses show that halogen transfer is significantly faster than hydroxyl transfer. To carry out these studies we also synthesized the first triflate-ligated iron corrole (Fe(OTf)(ttppc)), a versatile synthon in which the weakly bound OTf− ligand can be replaced by exogenous donors. Density functional theory calculations on the complete structures for the Fe(X)(ttppc) complexes (156 atoms) helped support the spectroscopic and structural analyses.
EXPERIMENTAL SECTION
Materials.
All chemicals were purchased from commercial sources and used without further purification unless otherwise stated. Reactions involving inert atmospheres were performed under Ar by using standard Schlenk techniques or in an N2-filled dry-box. Acetonitrile, benzonitrile, pentane and toluene were purified via a Pure-Solv solvent purification system from Innovative Technologies, Inc. Acetonitrile was further dried and purified by distilling over CaH2. Pentane and toluene were further dried and purified by distilling over sodium and benzophenone. Deuterated solvents for NMR and 57Fe (95.93%) were purchased from Cambridge Isotopes, Inc. Solvents were degassed by repeated cycles of freeze–pump–thaw and stored over 4 Å molecular sieves in an N2-filled drybox prior to use. 57FeCl2 (Fe 95.93%) was synthesized using a previously published method.17 Tris(p-tert-butylphenyl)methyl bromide and tris(p-methoxyphenyl)methyl tetrafluoroborate were synthesized following previously reported procedure.17, 57 Tris(p-methoxyphenyl)methyl chloride, tris(p-methoxyphenyl)methanol, and triphenylmethyl tetrafluoroborate were obtained from Alfa-Aesar and Sigma Aldrich, and were used as received. The triphenylmethyl radical source, Gomberg’s dimer (Ph3C=C6H5CPh2), tris(p-methoxyphenyl)methyl radical and tris(p-tert-butylphenyl)methyl radical were synthesized following literature procedures.17
Instrumentation.
UV-vis measurements were performed on a Hewlett- Packard Agilent 8453 diode-array spectrophotometer with a 3.5 mL quartz cuvette (path length = 1 cm) fitted with a septum. For all UV-vis spectroscopy measurements acquired below 23 °C, data collections were performed on a Varian Cary 60 Bio spectrophotometer. For reactions with total reaction time of <10 s, stopped-flow experiments were carried out using a HiTech SHU-61SX2 (TgK scientific Ltd.) stopped-flow spectrophotometer with a xenon light source and Kinetic Studio software. Electrospray ionization mass spectra (ESI-MS) were acquired using a Finnigan LCQ Duo ion-trap mass spectrometer equipped with an electrospray ionization source (Thermo Finnigan, San Jose, CA) in positive ion mode. Samples were introduced into the instrument at a rate of 15 μL/min using a syringe pump via a silica capillary line. The heated capillary temperature was 110 °C and the spray voltage was 4 kV. Gas Chromatography (GC-FID) was carried out on an Agilent 6850 gas chromatograph fitted with a DB–5 5% phenylmethyl siloxane capillary column (30 m x 0.32 mm x 0.25 μm) and equipped with a flame–ionization detector. Gas chromatography mass spectrometry (GC-MS) was performed using a Shimadzu GC17A/QP5050A GC/MS combination (Shimadzu Instruments, Columbia, MD). The GC17A is equipped with a low polarity (5% phenyl-, 95% methyl- siloxane) capillary column (30 m length, 0.25 mm ID, 0.25 μm film thickness, 10 m length guard column). Samples were dissolved in toluene and were injected into the instrument using an autosampler. The QP5050A is an EI quadrupole based mass spectrometer with a maximum scan range of 900 amu and an ionizing electron energy of 70 eV. 1H and 19F {1H} NMR spectra were recorded on a Bruker Advance 300 and 400 MHz NMR spectrometer. Electron paramagnetic resonance (EPR) spectra were recorded with a Bruker EMX spectrometer equipped with a Bruker ER 041 X G microwave bridge and a continuous-flow liquid helium cryostat (ESR900) coupled to an Oxford Instruments TC503 temperature controller for low temperature data collection. Mössbauer spectra were recorded on a spectrometer from SEE Co. (Edina, MN) operating in the constant acceleration mode in a transmission geometry. The sample was kept in an SVT-400 cryostat from Janis Research Co. (Wilmington, MA), using liquid N2 as a cryogen for 80 K measurements. Isomer shifts were determined relative to the centroid of the spectrum of a metallic foil of α-Fe collected at room temperature. Data analysis was performed using version F of the program WMOSS (www.wmoss.org), and quadrupole doublets were fit to Lorentzian lineshapes. Cyclic voltammetry was performed on an EG&G Princeton Applied Research potentiostat/galvanostat model 263A with a three-electrode system consisting of a glassy carbon working electrode, an Ag wire reference electrode with 0.25 M Bu4N+PF6− in C6H5CN, and a platinum wire counter electrode. Potentials were referenced using a ferrocene standard. Scans were run under an Ar atmosphere at 23 °C using Bu4N+PF6− (0.25 M) as the supporting electrolyte.
Fe(OSO2CF3)(ttppc) (2).
An amount of Fe(Cl)(ttppc)17 (15 mg, 11.5 μmol) was dissolved in toluene (5 mL) to give a dark red solution, and AgOTf (3.0 mg, 12 μmol, 1.0 equiv) was added, forming a dark red suspension. The reaction mixture was stirred in the dark at 23 °C for 1.4 h, without any noticeable color change. An amount of pentane (10 mL) was added, and the mixture was stored in the freezer at −35 °C for 12 h, during which time a fine, off-white precipitate formed which was presumed to be silver salts. The reaction mixture was then filtered, and the supernatant was dried under vacuum to give a red solid (15 mg, 93% yield). UV-vis (toluene): λmax, nm (ε x 10−4 M−1 cm−1): 421 (3.40), 532 (0.86).1H NMR (400 MHz, C6D5CD3) δ (ppm): 8.52 (d), 8.28 (d), 8.21 (t), 8.09 (t), 6.91 (t), 6.82 (t), 3.92 (br), −0.10, −7.38, −9.34, −10.91, −14.10, −52.00. Anal. Calcd for C99H67N4FeO3SF3 (2 • C7H8): C, 78.98; H, 4.49; N, 3.72. Found C, 79.40; H, 4.21; N, 3.96.
[Fe(OSO2CF3)(ttppc)(AgOTf)] (3).
An amount of Fe(Cl)(ttppc)17 (15 mg, 11.5 μmol) was dissolved in toluene (5 mL) to give a dark red solution, and AgOTf (6.5 mg, 25 μmol, 2.2 equiv) was added, forming a dark red suspension. The reaction mixture was stirred in the dark at 23 °C for 3 h, without any noticeable color change. The crude mixture was stored in the freezer at −35 °C for 12 h, during which time a fine, off-white precipitate formed which was presumed to be silver salts. The solution was then filtered, and the supernatant volume was reduced under vacuum to 1 mL. Crystals of 2 suitable for XRD were obtained by slow vapor diffusion of pentane into toluene over 3 weeks, giving dark red blocks (16 mg, 85% yield). UV-vis (toluene): λmax, nm: 358, 419, 533. ESI-MS (m/z): isotopic cluster at 1521.2, ([M – OTf]+) (calcd 1521.26). 1H NMR (400 MHz, C6D5CD3) δ (ppm): 8.50 (d), 8.28 (d), 8.20 (t), 8.09 (t), 6.92 (t), 6.81 (t), 4.07 (s,br), −0.10, −6.68, −8.11, −9.94, −11.20, −14.09, −52.20. Anal. Calcd for C93H59N4FeO6S2F6Ag: C, 66.87; H, 3.56; N, 3.35. Found C, 65.84; H, 4.14; N, 3.23.
Fe(F)(ttppc) (4).
An amount of Fe(Cl)(ttppc)17 (25 mg, 19.2 μmol) was dissolved in toluene (6 mL) to give a dark red solution, and an amount of AgOTf (0.77 mL, 19.2 μmol, from a 25 mM stock solution in CH3CN) was added, forming a dark red suspension. The reaction mixture was stirred in the dark at 23 °C for 2 h, without any noticeable color change. After which time, the resulting solution was passed through a Celite® plug to remove silver salts, and CsF (3.0 mg, 19.7 μmol) was added and the reaction mixture was stirred for 1 h at 23 °C, forming a dark red suspension. An amount of hexane (8 mL) was added, and stored in the freezer at −35 °C for 12 h, during which time a fine, white precipitate formed which was presumed to be cesium salts. The reaction mixture was then filtered, and the volume of supernatant was reduced under vacuum to give a red solid (21 mg, 87%). A crystal of 4 suitable for XRD was obtained by reducing the volume of supernatant to 1 mL, and layering hexane over 2 weeks, giving a few, very small dark red blocks (10 – 20 microns per side). However, attempts to scale-up this crystallization led to a significant amount of precipitation, and thus the routine preparation of 4 did not involve recrystallization. Isolation of 4 as a red solid, as described above, gave material which yielded highly reproducible UV-vis, 1H/19F NMR, and Mössbauer spectra. The Mössbauer spectra indicated that 4 is one species (99% of total Fe). UV-vis (toluene): λmax, nm (ε x 10−4 M−1 cm−1): 348 (2.18), 413 (3.33), 488 (1.62), 634 (0.26). ESI-MS (m/z): isotopic cluster at 1283.8 (M+) (calcd 1283.4). 1H NMR (400 MHz, C6D5CD3) δ (ppm): 20.01, 8.44 (d), 8.34 (d), 8.04 (m), 3.99 (br), −2.96, −6.27, −6.99, −8.63, −29.45.
Ph3CF.
An amount of [(C6H5)3C]+(BF4)− (50 mg) was dissolved in acetonitrile (3 mL) and potassium fluoride (26 mg, 0.45 mmol, 3 equiv) was added. The reaction mixture was stirred at 23 °C for 2 h. A color change of an orange to colorless solution was observed. A white suspension was formed after the addition of benzene (1 mL) which was presumed to be the formation of potassium salts. The supernatant was passed through Celite® plug to give a colorless solution. The filtrate was dried under vacuum to give a white solid (36 mg, 91%). White crystals of product were obtained by layering pentane into dichloromethane. GC-MS (C6H5CH3): m/z 262 (M+) (calcd 262.4). 1H NMR (400 MHz, C6D6) δ (ppm): 7.01 – 7.12 (m, 10H), 7.18 – 7.12 (m, 1H), 7.26 – 7.32 (m, 2H). 19F {1H} NMR (300 MHz, C6D6) δ (ppm): −125.6 (s) (δ = −125.4 ppm58 in CDCl3).
(p-MeO-C6H4)3CF.
An amount of [(p-MeO-C6H4)3C]+(BF4)− (32 mg) was dissolved in acetonitrile (3 mL) and potassium fluoride (13 mg, 0.23 mmol, 3 equiv) was added. The reaction mixture was stirred at 23 °C for 2 h. A color change of a dark red to colorless solution was observed. A white suspension was formed by addition of benzene (1 mL) which was presumed to be the formation of potassium salts. The supernatant was passed through Celite® plug to give a colorless solution. The filtrate was dried under vacuum to give a white solid (25 mg, 93%). 1H NMR (400 MHz, C6D6) δ (ppm): 7.06 – 7.12 (m, 6H), 6.65 – 6.72 (m, 6H), 3.46 (s, 9H). 19F {1H} NMR (300 MHz, C6D6) δ (ppm): −119.8 (s).
Reaction of Fe(Cl)(ttppc) (1) with Triphenylmethyl Radical (Ph3C•). 1H NMR Spectroscopy.
An amount of 1 (20 mg, 15 μmol) was dissolved in C6D6 (0.75 mL, 26.9 mM) and Ph3C=C6H5CPh2 (58 mg, 0.12 mmol, 8 equiv) dissolved in C6D6 (1.75 mL) was added. The reaction was stirred at 50 °C for 5 h. An aliquot of the reaction mixture (0.75 mL) was loaded into an NMR tube and a 1H NMR spectrum was collected. The aromatic region of the spectrum is dominated by peaks from the excess Ph3C=C6H5CPh2 starting material, but a multiplet at 7.34 – 7.39 ppm is well-separated and indicates formation of Ph3CCl based on comparison with an independent sample. The reaction mixture was quenched with EtOH (1 mL), and then the volatiles were removed under vacuum to give a dark red solid. Analysis by TLC of the crude solid dissolved in ethyl acetate (eluent: hexanes/ethyl acetate 6/1 v/v) showed multiple spots, but a major spot (rf = 0.34) matched Ph3CCl + Ph3COH, which ran together as seen by independent standards. Separation of the major spot by silica gel chromatography (hexanes/ethyl acetate 6/1 v/v) gave a resulting white solid which was analyzed by 1H NMR spectroscopy. Major peaks for both Ph3CCl and Ph3COH were observed, and integration against a standard (hexamethyldisiloxane) gave an average yield for the Ph3CCl of 36 ± 1% (average of 3 runs). The production of Ph3COH can be assigned to the reaction of excess Ph3C• with dioxygen in the air.
Reaction of Fe(F)(ttppc) (4) with Triphenylmethyl Radical (Ph3C•). 1H & 19F {1H} NMR Spectroscopy.
Complex 4 was made fresh, stored at −35 °C, and used within 2 – 3 days for all reactivity studies. The complex was not crystallized prior to routine use, but the purity of each batch was assessed by 1H and 19F NMR spectroscopy. The 1H NMR spectrum was consistent with 4 as the only major corrole species in solution, and the 19F {1H} NMR spectrum showed no peaks corresponding to F− impurities. An amount of 4 (0.55 mL of a 4.25 mM solution in C6D6, 2.34 μmol) was combined with Ph3C=C6H5CPh2 (0.11 mL of a 62 mM solution in C6D6, 6.8 μmol) in an NMR tube. The reaction mixture was manually mixed for 30 min at 23 °C, after which time a 19F {1H} NMR spectrum was recorded. A single peak at −125 ppm was observed, indicating the formation of Ph3CF based on comparison with an independent sample. Integration against an internal standard (CF3C(O)OC2H5, −74.85 ppm) gave an average yield of Ph3CF = 82% ± 3% (average of 2 runs).
Kinetic Study of Fe(Cl)(ttppc) (1) and Tris-(para-Methoxyphenyl)methyl Radical ((p-MeO-C6H4)3C•) at 23 °C.
Under light-limiting conditions, (p-MeO-C6H4)3C• was freshly prepared in toluene according to a literature procedure.17 The concentration of radical (0.86 −2.54 mM) was determined by UV-vis with the known extinction coefficient (ε = 570 M−1 cm−1 in toluene)17. Varying amounts of radical (27 – 79 equiv) were combined with 1 (64 μM, 0.2 mL) in toluene in a single-mix stopped-flow apparatus at 23 °C. The spectral changes showed isosbestic conversion of 1 to FeIII(ttppc) (λmax = 573 nm) over 1 – 2 s. The pseudo first-order rate constants, kobs, were obtained by nonlinear least-squares fitting of a plot of absorbance at 573 nm corresponding to the production of FeIII(ttppc). The (Abst) versus time (t) plots were fit according to the equation Abst = Absf + (Abs0 − Absf)exp(−kobst), where Abs0 and Absf are the initial and final absorbance values, respectively. The second-order rate constant (k2) was obtained from a slope of the best-fit line of a plot of kobs versus radical concentration.
Kinetic Study of Fe(Cl)(ttppc) (1) and Tris-(para-Methoxyphenyl)methyl Radical ((p-MeO-C6H4)3C•) at −60 °C.
Under light-limiting conditions, (p-MeO-C6H4)3C• was freshly prepared in toluene according to a literature procedure.17 The concentration of radical was determined by UV-vis with the known extinction coefficient (ε = 570 M−1 cm−1 in toluene)17. Varying amounts of radical (19 – 33 equiv) were combined with 1 (27 – 30 μM, 2.15 mL – 1.97 mL) in toluene at −60 °C to start the reaction. The spectral changes showed isosbestic conversion of 1 to FeIII(ttppc) (λmax = 573 nm) over 30 – 60 min. The pseudo first-order rate constants, kobs, were obtained by nonlinear least-squares fitting of a plot of absorbance at 573 nm corresponding to the production of FeIII(ttppc). The (Abst) versus time (t) plots were fit according to the equation Abst = Absf + (Abs0 − Absf)exp(−kobst), where Abs0 and Absf are the initial and final absorbance values, respectively. The second-order rate constant (k2) was obtained from a slope of the best-fit line of a plot of kobs versus radical concentration.
Reaction of Fe(Cl)(ttppc) (1) with Tris-(para-Methoxyphenyl)methyl Radical ((p-MeO-C6H4)3C•). GC-FID.
A fresh batch of (p-MeO-C6H4)3C• stock solution in toluene was prepared from (p-MeO-C6H4)3CCl following a literature procedure.17 Solid 1 (3.6 mg, 2.8 μmol) was dissolved in an aliquot of the radical stock solution (18 mM, 0.5 mL, 3.34 equiv). The reaction mixture was stirred for 30 min at 23 °C, after which time eicosane (10 μL of an 8.8 mM stock solution in toluene) was added as an internal standard for GC-FID. Analysis of the reaction mixture by GC-FID indicated formation of (p-MeO-C6H4)3CCl (RT = 26 min) based on comparison with an independent sample. The radical stock solution typically contains a small amount of unreacted (p-MeO-C6H4)3CCl as seen by GC-FID, and this background signal is subtracted from the signal for the product in order to obtain a yield for the reaction of 1 and (p-MeO-C6H4)3C• by GC-FID. Comparison with a calibration curve based on the internal standard gave an average yield of 81 ± 5% for (p-MeO-C6H4)3CCl (average of 3 runs).
Reaction of Fe(Cl)(ttppc) (1) with Tris-(para-Methoxyphenyl)methyl Radical ((p-MeO-C6H4)3C•). Mössbauer Spectroscopy.
An amount of 57Fe-1 (3 mg, 2.3 μmol) was dissolved in toluene (0.4 mL) and frozen in a Mössbauer cup in N2(l). A Mössbauer spectrum (80 K) was recorded, showing a sharp, quadrupole doublet corresponding to 57Fe-1 (≥ 99% of the fit). The solution was thawed and placed in a reaction vessel. An amount of freshly prepared (p-MeO-C6H4)3C• (1.4 mL of a stock solution (3.5 mM) in toluene, 2.1 equiv) was added to the solution of 57Fe-1, and the reaction mixture was stirred for 1 h at 23 °C, after which time the solvent was removed under vacuum to prepare a more concentrated solution for analysis by Mössbauer spectroscopy. The resulting red solid was dissolved in toluene (0.4 mL) and transferred to a Mössbauer sample holder, which was frozen in liquid N2 prior to Mössbauer spectroscopy.
Reaction of Fe(F)(ttppc) (4) with Tris-(para-Methoxyphenyl)methyl Radical ((p-MeO-C6H4)3C•). 1H & 19F {1H} NMR spectroscopy.
An amount of 4 (0.35 mL of a 4.5 mM stock solution in C6D5CD3, 1.55 μmol) was combined with a freshly prepared batch of (p-MeO-C6H4)3C• (0.5 mL of a 3.2 mM stock solution in C6D5CD3,17 1.04 equiv). The reaction mixture was manually mixed in an NMR tube for 30 min at 23 °C, after which time a 19F {1H} NMR spectrum was recorded. A single peak at −120 ppm was observed, indicating the formation of (p-MeO-C6H4)3CF based on comparison with an independent sample. Integration against an internal standard (CF3C(O)OC2H5, −74.85 ppm) gave an average yield of (p-MeO-C6H4)3CF = 93 ± 1% (average of 2 runs).
Kinetic Study of Fe(F)(ttppc) (4) and Tris-(para-Methoxyphenyl)methyl Radical ((p-MeO-C6H4)3C•) at 23 °C.
Under light-limiting conditions, (p-MeO-C6H4)3C• was freshly prepared in toluene according to a literature procedure.17 The concentration of radical (0.35 – 0.62 mM) was determined by UV-vis with the known extinction coefficient (ε = 570 M−1 cm−1 in toluene)17. Varying amounts of radical (28 – 50 equiv) were combined with 4 (25 μM, 0.2 mL) in toluene in a single-mix stopped-flow apparatus at 23 °C. The spectral changes showed isosbestic conversion of 4 to FeIII(ttppc) (λmax = 573 nm) over 0.15 s at 23 °C. The pseudo first-order rate constants, kobs, were obtained by nonlinear least-squares fitting of a plot of absorbance at 573 nm corresponding to the production of FeIII(ttppc). The (Abst) versus time (t) plots were fit according to the equation Abst = Absf + (Abs0 − Absf)exp(−kobst), where Abs0 and Absf are the initial and final absorbance values, respectively. The second-order rate constant (k2) was obtained from a slope of the best-fit line of a plot of kobs versus radical concentration.
Computational methods.
All calculations were performed with the ORCA 3.0.3 program package. Starting geometry optimizations for 1 and 4 were derived from XRD structures. Details of the steps taken to achieve the best, optimized geometries and frequencies are given in the Supporting Information. The 57Fe Mössbauer parameters for 1, 3, and 4 were determined from a single point energy calculation using TPSSh or B3LYP functional with DKH2 relativistic approximation and NORI approximation at the def2-TZVP level of theory.59 A series of grids (Grid5 and FinalGrid6) were set to a TightSCF convergence. The iron atom had an effective core potential of “CP(PPP)” and the radial integration accuracy was increased using the “SpecialGridIntAcc” command (value 7). A simulated solvent effect (COSMO) was employed for these calculations. The calculated electron density at the 57Fe nucleus (denoted ρ0) was transformed into an isomer shift (δ) according to the equation: δ = α(ρ0 − C) + β where α, β, and C are calibration constants. A published calibration curve of isomer shift with α = −0.1706325, β = 0.33226751, and C = 23615 was used for B3LYP and α = −0.172806013, β = 0.323223706, and C = 23600 was used for TPSSh.59 The calculated quadrupole splitting (ΔEQ) was used without any calibration or correction.
Results and Discussion
Fe(OSO2CF3)(ttppc) (2).
To gain access to the axially ligated fluoride complex Fe(F)(ttppc), we initially followed the method reported by Gross for the generation of Fe(F)(tpfc) (tpfc = tris(pentafluorophenyl)corrolato3−).56 This method involved metathesis of Cl− for F− by addition of AgF to the Fe(Cl)(tpfc) starting material in CH2Cl2. In this case, Fe(F)(tpfc) was only metastable, decaying within 30 min as seen by 1H NMR spectroscopy. Addition of AgF to Fe(Cl)(ttppc) did not result in a noticeable change in the UV-vis spectrum, but did show new peaks by 1H NMR. However, this species was not stable over 24 h, similar to the observed lack of stability seen for Fe(F)(tpfc). The NMR data showed the presumed Fe(F)(ttppc) was converting back to the Fe(Cl)(ttppc) starting material over this period. Thus an alternative method was sought for the preparation of Fe(F)(ttppc).
Addition of 1 equiv of AgOTf to Fe(Cl)(ttppc) (1) in toluene causes the slow precipitation of an off-white solid over 1 h which was assumed to be AgCl (Scheme 3). Analysis of the reaction mixture by UV-vis spectroscopy shows the loss of 1 and the appearance of new bands at 421 and 532 nm. This new product was moisture-sensitive, making it impractical to purify by standard chromatographic methods under aerobic conditions. After workup to remove Ag salts, a new compound was isolated as a dark red solid. This complex was identified as the desired triflate-ligated complex, Fe(OSO2CF3)(ttppc) (2) (vide infra). The UV-vis spectrum of 2 is compared with that of the starting material 1 in Figure 1. The Soret band for 1 at 407 nm is red-shifted by 14 nm in 2, and the Q-band at 637 nm is no longer visible in the spectrum for 2, but a shoulder is seen at 532 nm. However, the spectrum of 2 in the presence of the coordinating solvent CH3CN (1/1 v/v CH3CN/C6H5CH3) exhibits new bands at 360, 403, 545 and 640 nm (Figure S3), suggesting that OTf− is displaced by CH3CN in this solvent system.
Scheme 3.

Synthesis of Complex 2
Figure 1.

UV-vis spectra of 1 (blue, dashed line) and 2 (red, solid line) in toluene at 23 °C.
The 1H NMR spectrum of 2 in C6D5CD3 reveals 6 paramagnetic peaks from δ = 0 to −60 ppm that are in the same region as peaks seen for Fe(Cl)(ttppc) and Fe(OH)(ttppc) (Figure 2), and the related Fe(Cl)(tpc) (tpc = triphenylcorrolato3−).17, 60 These resonances have been assigned to the β-CH pyrrole protons for iron triphenylcorrole complexes.56, 61-63 The 19F {1H} NMR spectrum of 2 in C6D5CD3 has a broad peak at δ = −76.4 ppm (full width at half maximum (FWHM) = 692 Hz), which can be compared to the sharp resonance for AgOTf in the same solvent at −76.5 ppm (FWHM = 2.82 Hz) (Figure S4). The broad line in C6D5CD3 is consistent with coordination of the triflate ligand to the paramagnetic iron center, although tight binding of OTf − is expected to give a larger downfield shift for the OTf − peak.64, 65 In contrast, the 1H NMR spectrum of 2 in the presence of CD3CN (CD3CN/C6D5CD3 (1/1 v/v) exhibits paramagnetic peaks between 0 to −50 ppm that are shifted from neat C6D5CD3 (Figure S5). The corresponding 19F {1H} NMR spectrum exhibits a much sharper peak at δ = −77.73 ppm (FWHM = 127 Hz), consistent with displacement of the OTf− (FWHM = 23.9 Hz) axial ligand by CH3CN, as indicated by the UV-vis spectra in the same solvent (Figure S3). Electron paramagnetic resonance (EPR) spectroscopy on 2 (X-band, 20 K) showed only a baseline signal, as expected for an integer spin, S = 1 ground state (Figure S6), and as seen for the starting chloride complex 1 and Fe(OH)(ttppc).17 The zero-field Mössbauer spectrum of 2 in toluene gave well-defined doublets with δ = 0.19 mm s−1; ∣ΔEQ∣ = 3.02 mm s−1 at 125 K and δ = 0.20 mm s−1; ∣ΔEQ∣ = 3.01 mm s−1 at 80 K (Figure 3). Neutral Fe(X)(ttppc) complexes are formally in the +4 oxidation state. However, corroles can behave as non-innocent ligands, and other iron-halide corroles have been described as exhibiting electronic structures closer to FeIII(X)(corrole•+).66-69 We avoid this ambiguity by leaving out formal oxidation state assignments in the formulas for the Fe(X)(ttppc) complexes.
Figure 2.

1H NMR spectra of 2 (top) and 3 (bottom) in C6D5CD3 at 23 °C. Inset: expanded region from 6 – 9 ppm. Chemical shifts labeled in red highlight the peak at −7.4 ppm in 2 which splits into two peaks in the presence of AgOTf in 3.
Figure 3.

Zero-field 57Fe Mössbauer spectra of 2 at 125 K (top), 2 at 80 K (middle), 3 at 80 K (bottom) in toluene. Experimental data = black circles, best fit = red line.
[Fe(OSO2CF3)(ttppc)(AgOTf)] (3).
During efforts to synthesize the triflate-ligated complex 2, another species was isolated and structurally characterized by XRD. This complex was produced from the reaction of 1 with 2.2 equiv of AgOTf in toluene. Dark red crystals suitable for XRD were grown from slow vapor diffusion of pentane into toluene. The crystal structure of 3 is shown in Figure 4, revealing a five-coordinate, mononuclear iron complex with an axial OTf− ligand, and a single Ag ion in close contact with a pyrrole ring and two peripheral phenyl groups. A second OTf− anion coordinates to the AgI, completing its coordination sphere. The Ag − Cβ distances of 2.55(2) and 2.46(2) Å are similar to what has been seen for Ag − Cβ interactions (2.44, 2.48 Å) in a NiII porphyrin complex from XANES and DFT analysis.70 There are also relatively close contacts (2.55(2) – 2.62(2) Å and 2.53(2) – 2.57(2) Å) between the AgI ion and two of the carbon atoms on each of the adjacent phenyl rings, similar to other η2 − π interactions seen for the AgI ion and benzene derivatives.71 A number of AgI − π interactions involving pyrrole rings have been structurally characterized,72 but to our knowledge, the structure of 3 represents the first example of a crystallographically characterized porphyrin or corrole complex that exhibits AgI − π bonding. The Fe─O1 bond length of 1.990(19) Å is similar to FeIII(OTf)(tpp) (tpp = triphenylporphyrinato2−) (Fe-O = 1.946(6) Å – 1.996(3) Å),73-76 and the average Fe-Npyrrole distance of 1.901(2) Å is in line with other iron (ttppc) (S = 1) structures (Table S5). The iron atom in 3 is displaced by 0.374 Å from the least-squares plane defined by the 4 Npyrrole atoms toward the OTf− ligand, the same amount of displacement as seen for Fe(OH)(ttppc).17
Figure 4.

Displacement ellipsoid plot (40% probability) of 3 at 110 K. Hydrogen atoms omitted for clarity. Inset: chemical structure of 3.
The UV-vis spectrum for 3 is similar to 2 in toluene (Figure S7). The difference between both complexes was the appearance of a shoulder at 358 nm unseen for 2. The Soret band for 3 is slightly blue-shifted by 2 nm. These changes indicate that the second coordination sphere AgI ion has some influence on the absorbance bands arising from the π system of the corrole. Mass spectrometry (ESI-MS) in 1/1 (v/v) acetone/toluene gives a major peak at m/z 1521.2, corresponding to [3 – OTf]+ (Figure S8). These data show that the structure of 3, including the peripherally bound AgI ion, remains intact in solution.
The 1H NMR spectrum of 3 in C6D5CD3 reveals 7 paramagnetic peaks from δ = 0 to −50 ppm. A comparison of the spectra for 2 and 3 indicates that the outer-sphere AgI ion in 3 causes a splitting of the peak at δ = −7.39 ppm seen in 2, resulting in two new peaks at δ = −6.68 and −8.11 ppm (Figure 2). The other peaks in the spectrum for 3 (δ = 7 – 9 ppm) do not show any significant change from those seen for 2. The 19F {1H} NMR spectrum of 3 in C6D5CD3 reveals two peaks at δ = −48.96 and −76.60 ppm (Figure S4). The broad peak at −48.96 ppm (FWHM = 750 Hz) can be assigned to the triflate bound to the iron center, while the relatively sharp peak at δ = −76.6 ppm (FWHM = 91.6 Hz) can be assigned to the outer-sphere OTf− coordinated to the AgI ion.
The zero-field Mössbauer spectrum of 3 generated in situ in toluene at 80 K revealed an isomer shift close to 2 (δ = 0.22 mm s−1 versus 0.20 mm s−1) and the quadrupole splitting is slightly shifted (∣ΔEQ∣ = 3.36 mm s−1 versus 3.02 mm s−1) (Figure 3). The combined data suggests the peripheral AgI ion does exert a small influence on electronic environment around the iron center. Density functional theory (DFT) calculations were employed to corroborate the Mössbauer spectrum. The Mössbauer parameters were computed via unrestricted single point energy calculations (B3LYP/def2-TZVP) on the fixed geometry from the XRD structure for 3 with an S = 1 spin state. The calculations gave δ = 0.22 mm s−1; ∣ΔEQ∣ = 3.27 mm s−1, which were a good match to the experimental data.
Synthesis and Structural Characterization of Fe(F)(ttppc) (4).
The fluoride complex Fe(F)(ttppc) (4) was synthesized as shown in Scheme 4, starting from Fe(OTf)(ttppc) generated in situ followed by addition of CsF. A white precipitate was observed after 1 h, and UV-vis analysis of the reaction mixture showed formation of a new species (λmax = 348, 413, 488 and 634 nm). Isolation of the product was accomplished by precipitation with pentane followed by filtration to give 4 as a red solid (87%). Analysis of the isolated product by ESI-MS(+) revealed a dominant isotopic cluster centered at 1283.8 m/z, corresponding to [4]+ (Figure S9), in which the axial ligand remains intact.
Scheme 4.

Synthesis of Complex 4
The UV-vis spectrum of crystalline 4 gives λmax = 351(sh), 412, 505(sh), and 635 nm in toluene (Figure S10), and the spectrum of 4 generated in situ is nearly the same (Figure 5). The Soret band for 4 is red-shifted by 6 nm compared to the chloride complex 1 (λmax = 371, 407, 519, and 637 nm). The shoulder in the near-UV for 4 is blue-shifted compared to 1 (348 versus 371 nm), and the shoulder at 519 nm seen for 1 is not observed in the spectrum for 4. These differences are clear markers for the identity of the axial halide donor.
Figure 5.

UV-vis spectra of Fe(X)(ttppc) where X = Cl (blue, dashed line) or F (red, solid line) in toluene at 23 °C.
Dark red crystals of 4 suitable for X-ray structure determination were grown from layering hexane into toluene. The crystal structure of 4 is shown in Figure 6, revealing a five-coordinate, mononuclear iron complex with an axial F− ligand. The structure of 4 represents the first example of a crystallographically characterized iron fluoride corrole complex. The Fe─F bond is disordered over two positions that lie on opposite sides of the corrole plane, leading to a major component (ca. 72%) with an Fe-F distance of 1.972(2) Å, and a minor component with an Fe-F distance of 1.815(5) Å. These distances can be compared to ferric fluoride porphyrin (FeIII─F = 1.792(3) – 1.865(3) Å), or ferric fluoride 10-thiacorrole (Scor = 2,3,7,8,12,13,17,18-octaethyl-10-thiacorrolato3−) (FeIII–F = 1.898(2) Å).77-84 The elongated Fe-F distance seen for the more populated component may be due to intramolecular C-H---F interactions with two of the phenyl rings (C42/52(H)---F = 3.196(2)/3.201(2) Å).85-90 The average Fe−Npyrrole distance of 1.893(16) Å is in line with other iron ttppc structures (Table S5).17 The iron atom in 4 is displaced by 0.32 Å from the least-squares plane defined by the 4 Npyrrole atoms toward the F− ligand in both major and minor components, which is the same amount of displacement seen for Fe(Cl)(ttppc) (0.341 Å).17
Figure 6.

Displacement ellipsoid plot (50% probability) of 4 at 110 K. Hydrogen atoms are omitted for clarity.
Density functional theory (DFT) calculations were employed to help understand the molecular structures of 1 and 4. Geometry optimized models of 1 and 4 were tested with BP86 functional at the def2-TZVP level of theory with a ground spin state of S = 1. The BP86-optimized structure of 1 has Fe-Cl = 2.195 Å, which is 2.7% shorter than the Fe-Cl distance seen by XRD, whereas for 4 the calculated Fe-F bond length is 1.806 Å, which matches well with the minor component in the crystal structure. The calculated Fe-N, C-C and C-N distances for both complexes were a close match with XRD (Figure S12). Corresponding frequency calculations showed no imaginary frequencies, indicating that the optimized structures are at their ground state energies.
The 1H NMR spectrum of 4 reveals six paramagnetic peaks in the upfield region between δ = 0 to −35 ppm, and two downfield peaks at 3.99 and 20.01 ppm in C6D5CD3 (Figure 7). For comparison the spectrum for the Fe(Cl) complex is also shown in Figure 7, showing that the spectra are clearly sensitive to the identity of the axial ligand. Earlier NMR work on a series of selectively deuterated iron corroles with different axial halide ligands, Fe(X)(tdcc) (X = I, Br, Cl, F; tdcc = 5,10,15-tris(2,6-dichlorophenyl)corrolato3−), gave definitive assignments for the paramagnetic peaks in the 1H NMR spectra.56 Based on these assignments, the peaks at 20.01 ppm and −29.5 ppm for 4 can be attributed to the β-CH pyrrole protons on the C2 and C3 positions, respectively. The 19F NMR spectrum for 4 does not reveal any signals between −300 and +600 ppm, likely because of fast relaxation of the 19F nucleus caused by the paramagnetic iron center. The paramagnetic spin ground state for 4 was confirmed by Evans method, giving μeff = 2.63 μB, consistent with a triplet spin ground state (S = 1; μtheor = 2.71 μB) (Figure S13). The EPR (X-band, 20 K) spectrum of 4 in toluene showed only a baseline signal, as expected for an integer-spin system (Figure S14).
Figure 7.

1H NMR (400 MHz) spectrum of Fe(X)(ttppc) with X = F (top) and Cl (bottom) in C6D5CD3 at 23 °C.
The zero-field Mössbauer spectra of 1 and 4 in toluene at 80 K consist of well-defined doublets with δ = 0.17 mm s−1; ∣ΔEQ∣ = 2.76 mm s−1 for 1 and δ = 0.18 mm s−1; ∣ΔEQ∣ = 2.33 mm s−1 for 4 (Figure 8). A previous report of 1 at 4 K gave δ = 0.18 mm s−1; ∣ΔEQ∣ = 2.86 mm s−1.17 A comparison of parameters to other iron chloride corroles, Fe(Cl)(tpc) (δ = 0.19 mm s−1; ∣ΔEQ∣ = 2.93 mm s−1 at 77 K) and Fe(Cl)(tpfc) (δ = 0.18 mm s−1; ∣ΔEQ∣ = 2.93 mm s−1 at 80 K), shows that the tri(aryl) meso substituents have only a minor impact on the Mössbauer parameters.66, 91
Figure 8.

Zero-field 57Fe Mössbauer spectra of Fe(X)(ttppc) with X = Cl (top), and F (bottom) in toluene at 80 K. Experimental data = black circles, best fit = red line.
The calculated Mössbauer parameters shown in Table 1 were determined from unrestricted single point energy calculations. The calculated values for both the isomer shifts and quadrupole splittings for 1, 3 and 4 align well with the experimental data. Broken symmetry DFT calculations on Fe(X)(tpfc) (X = Cl, F), involving 3 α electrons on Fe and one β electron on the corrole (FeIII(tpfc+•) configuration), gave Mössbauer parameters of δ = 0.181 mm s−1; ∣ΔEQ∣ = 2.45 mm s−1 for X = Cl−, and δ = 0.194 mm s−1; ∣ΔEQ∣ = 2.03 mm s−1 for X = F−.91 These data suggest that both the unrestricted and broken symmetry approaches lead to similar modeling of the Mössbauer spectra.
Table 1.
Mössbauer Spectroscopic Parameters (δ [mm s–1] and ∣ΔEQ∣ [mm s–1]): Experimental Data and Calculated Valuesa
| Complex | δexp | δcalc | ∣ΔEQ∣exp | ∣ΔEQ∣calc |
|---|---|---|---|---|
| 1 a | 0.17 | 0.178 | 2.76 | 2.730 |
| 3 b | 0.22 | 0.220 | 3.36 | 3.293 |
| 4 a | 0.18 | 0.202 | 2.33 | 2.343 |
Calculated values from a BP86 geometry optimization followed by a single-point energy calculation at the B3LYP/def2TZVP level of theory. These values were confirmed with the alternate functional TPSSh (Table S6).
Structure from XRD.
Electrochemistry.
Cyclic voltammograms of the halide complexes are shown in Figure 9. For the chloride complex an irreversible reduction at Ered = −0.57 V along with a small oxidation peak at −0.06 V and a reversible oxidation at E1/2 = 0.50 V was observed with a scan rate of 50 mV s−1. As the scan rate was increased by 50 mV increments, an oxidative wave grows in at −0.54 V, giving a reversible reduction at E1/2 = −0.58 V, as shown in Figure 9 for a scan rate of 200 mV s−1. We assign this reversible wave to the first reduction for [Fe(Cl)(ttppc)]0/[Fe(Cl)(ttppc)]−. The irreversibility at slower scan rates is consistent with dissociation of the chloride ligand upon reduction of the complex. The spectrum for the fluoride complex 4 reveals a quasi-reversible reduction at E1/2 = −0.44 V and a reversible oxidation at E1/2 = +0.48 V. There are also smaller redox events centered at −0.05 V and −0.10 V which may come from minor species such as Fe(OH)(ttppc) generated by reaction of the Fe(F) complex with residual water.17 The CV data show that the identity of the halide ligand has a significant effect on the first reduction potential, but not on the first oxidation potential. The reduction wave for the fluoride complex is shifted 140 mV positive compared to the chloride complex, consistent with the greater electronegativity of F versus Cl, and indicates that complex 4 is easier to reduce than complex 1. Interestingly, the analogous hydroxide species, Fe(OH)(ttppc), appears to exhibit a reversible reduction at −0.11 V in PhCN, which is shifted even more positive by 330 mV when compared to the fluoride complex and makes the Fe(OH) complex the strongest oxidant of the series.17
Figure 9.

Cyclic voltammograms of 1 (2 mM) with a scan rate of 50 mV/s (black) and 200 mV/s (blue) (top), and 4 (3 mM) with a scan rate of 100 mV/s (black) in benzonitrile (bottom). The solution contains Bu4N+PF6− (0.25 M) supporting electrolyte.
Reaction of Fe(X)(ttppc) (X = Cl, F) with Triphenylmethyl Radical.
Initial studies began with the unsubstituted triphenylmethyl radical, delivered from the stable dimer form (Ph3C=C6H5CPh2). This dimeric starting material is in equilibrium with the monomeric radical form in solution (Ph3C=C6H5CPh2 ⇆ 2 Ph3C• (3.5% monomer in toluene at 20 °C, λmax, nm (ε = L mol−1 cm−1) = 515 (661)).92, 93 The addition of excess Ph3C=C6H5CPh2 to 1 or 4 in toluene at 23 °C (Scheme 5) resulted in the complete conversion of the starting Fe complexes to an FeIII complex characterized by λmax at 420, 575, 760 nm for 1 and 420, 573, 762 nm for 4 within 1 min by UV-vis (Figure 10).17
Scheme 5.

Reaction of Fe(X)(ttppc) (X = Cl or F) with Triphenylmethyl Radical
Figure 10.

UV-vis spectra for the reaction of Fe(X)(ttppc) where X = Cl (top) or F (bottom) with Ph3C=C6H5CPh2 in toluene at 23 °C. Spectra before (blue) and after (red) the addition of excess Ph3C=C6H5CPh2.
Analysis of the reaction mixture for the chloride complex 1 and Ph3C• showed the formation of both FeIII(ttppc) and Ph3CCl by 1H NMR spectroscopy. However, quantitation of the organic product Ph3CCl could not be obtained by NMR integration methods because the broadened peaks for Ph3CCl overlap with the starting (Ph3C=C6H5CPh2) dimer. However, running the reaction on a larger scale (20 mM) produced enough of the chlorinated Ph3CCl to isolate by chromatography. Slight warming of the reaction to 50 °C on this scale was employed to drive the reaction to completion within 5 h. Conversion of 1 to FeIII(ttppc) was confirmed by NMR spectroscopy (Figure S16), and thin-layer chromatography (TLC) revealed a major spot for the expected Ph3CCl (rf = 0.34). Partial purification by silica gel chromatography (eluent: 6/1 v/v hexane/ethyl acetate) led to isolation of Ph3CCl together with an amount of Ph3COH impurity, which co-elutes with the chlorinated product (Figure S17). A yield of 33% for Ph3CCl was obtained (average of 3 runs) by 1H NMR analysis and comparison with an internal standard.
The reaction of the iron fluoride complex 4 with Ph3C• was analyzed by both 1H and 19F {1H} NMR spectroscopies, and showed formation of FeIII(ttppc) and Ph3CF products (Figure S18 - S22). In this case, quantitation of the fluorinated product could be carried out by integration of the 19F {1H} NMR spectrum, avoiding the problem of overlap with the starting material (Ph3C=C6H5CPh2) material seen for the chloride reaction, and making it possible to obtain the yield of Ph3CF in situ. A sharp peak for Ph3CF at −125.6 ppm in the 19F {1H} NMR spectrum was integrated to give a yield of 82% (average of 2 runs).
Reaction of Fe(X)(ttppc) (X = Cl, F) with Tris-(para-Methoxyphenyl)methyl Radical.
Addition of the para-methoxy-substituted radical derivative ((p-MeO-C6H4)3C•) to 1 led to similar reactivity. Production of (p-MeO-C6H4)3CCl was confirmed by GC-FID, and a yield for (p-MeO-C6H4)3CCl of 81% (average of 3 runs) was obtained (Figure S23). Mössbauer spectroscopy on the reaction mixture confirms the formation of an iron(III) corrole (δ = 0.18 mm s−1; ∣ΔEQ∣ = 3.94 mm s−1) (Figure 11).81, 91, 94-99
Figure 11.

Zero-field 57Fe Mössbauer spectrum (C6H5CH3, 80 K) of 1 (top), and 1 + (p-MeO-C6H4)3C• (bottom). Experimental data = black circles, best fit = red line.
The reaction of the fluoride complex 4 was also examined with (p-MeO-C6H4)3C• by 1H and 19F {1H} NMR spectroscopy. Product analysis by 1H NMR spectroscopy confirms the complete conversion of the starting fluoride complex to the ferric product (Figure S24). The 19F {1H} NMR shows a sharp peak at −119.8 ppm corresponding to the fluorinated product (p-MeO-C6H4)3CF. Quantitation by integration of the 19F {1H} NMR spectrum against an internal standard gave a yield for (p-MeO-C6H4)3CF of 93% (average of 2 runs) (Figure S25).
Kinetics.
The rate of the reaction for 1 and (p-MeO-C6H4)3C• under pseudo-first-order conditions (excess radical) was examined. The addition of (p-MeO-C6H4)3C• to 1 in toluene resulted in the rapid, isosbestic conversion of 1 to FeIII(ttppc), as seen by UV-vis spectroscopy (Figure 12b). The reaction was monitored by measuring corresponding growth curve for FeIII(ttppc) at λmax = 573 nm, and the best fit led to a pseudo-first-order rate constant (kobs) (Figure S27 - S28). Measuring kobs at different radical concentrations (27 – 79 equiv at 23 °C and 19 – 33 equiv at −60 °C) led to a linear correlation between rate constant and [radical], which gave a second-order rate constant (k2) from the slope of the best-fit line (Figure 12c and Figure S28), with k2 = 1344.6 ± 34.1 M−1 s−1 at 23 °C and 2.183 ± 0.171 M−1 s−1 at −60 °C. The observed second-order rate constant is ~4 times faster than that seen for Fe(OH)(ttppc) with (p-MeO-C6H4)3C• at 23 °C.17
Figure 12.

(a) Overlay of UV–vis spectra for 1 (blue, solid line), 1 + (p-MeO-C6H4)3C• (red, dashed line), and (p-MeO-C6H4)3C• (green, dotted line) in toluene. (b) UV-vis spectra for reaction of 1 (32 μM) with (p-MeO-C6H4)3C• (62 equiv) in toluene from 0 (blue) to 2 sec (red) at 0.04 s intervals (grey) at 23 °C. Inset: Changes in absorbance versus time for the formation of FeIII(ttppc) (black dots) fit to a single exponential expression (red line). (c) Plot of kobs (23 °C) versus [(p-MeO-C6H4)3C•] with best-fit line. (d) Eyring plot of (ln(kh/kBT) versus 1/T (−70 °C to 23 °C) with best-fit line.
Temperature-dependent studies of the (p-MeO-C6H4)3C• reaction with 1 (−70 to 23 °C) gave the Eyring plot shown in Figure 12d, where k = kobs/[radical]. The plot is linear and gives activation parameters: ΔH⧧ = +9.77 ± 0.29 kcal mol−1 and ΔS⧧ = −14.39 ± 1.29 cal mol−1 K−1. These values can be compared to the activation parameters found for the tBu-substituted radical (p-tert-butyl-C6H4)3C• in reactions with Fe(OH)(ttppc) (ΔH⧧ = 13.4 kcal mol−1; ΔS⧧ = −5.6 cal mol−1 K−1) or Mn(OH)(ttppc) (ΔH⧧ = 10.9 kcal mol−1; ΔS⧧ = −15.5 cal mol−1 K−1). The faster rate of Cl• transfer versus •OH transfer can be associated with a lowering of the enthalpic contribution to the reaction barrier by 3.63 kcal mol−1, which compensates for an additional entropic penalty (−TΔS⧧(Cl) = 4.3 kcal mol−1; −TΔS⧧(OH): 1.7 kcal mol−1; T = 298 K). This results suggests that the Fe-Cl bond in 1 may be more susceptible to homolytic dissociation, i.e. has a lower bond dissociation energy, than the Fe-OH bond in Fe(OH)(ttppc).
The negative ΔS⧧ value for 1 lies in between that seen for the reactions of Fe(OH)(ttppc) with (p-tert-butyl-C6H4)3C• (ΔS⧧ = −5.6 cal mol−1 K−1) and (p-CN-C6H4)3C• (ΔS⧧ = −28.4 cal mol−1 K−1), and is consistent with a concerted mechanism, as opposed to a step-wise mechanism with outer-sphere electron-transfer (ET) as the rate-determining step.19 Early work on reactions of metalloporphyrins with sterically encumbered alkyl halides showed that a concerted, SN2-like mechanism could be distinguished from an outer-sphere ET mechanism by the observation of a negative ΔS⧧ values for the SN2 pathway, as compared to a small, positive ΔS⧧ value for an ET pathway.100
To gain further insight regarding the role of electron-transfer in these reactions, the redox potential for the [(p-MeO-C6H4)3C]+/(p-MeO-C6H4)3C•] couple was examined. This couple is reported as −0.58 V in CH3CN,101 but in order to make a direct comparison with 1, we re-measured it in PhCN. Cyclic voltammetry revealed a single, reversible wave at E1/2p-OMe = −0.60 V (Figure S30). Comparison of E1/2p-OMe with the reduction potential for 1 in PhCN (E1/2 = −0.58 V) gives ΔG°ET = −0.46 kcal mol−1 for an outer-sphere electron-transfer reaction, which is slightly exergonic. In contrast, the redox couple for the p-tBu radical derivative is E1/2p-tBu = −0.24 V (in CH3CN).101 This value predicts that ET would be largely endergonic with 1, with ΔG° = +7.84 kcal mol−1. In accordance with these thermodynamic trends, no reaction is observed for the p-tBu radical derivative with 1 (Figure S31 - S32). Taken together, these observations suggest that there is a modest amount of charge-transfer associated with the concerted transition state for halogen transfer, as implicated by computational studies for Fe(OH)(ttppc).19
The addition of (p-MeO-C6H4)3C• to 4 in toluene resulted in the rapid isosbestic conversion of 4 to FeIII(ttppc), as seen for 1 (Figure 13a). Measuring kobs at different radical concentrations led to a second-order rate constant of k2 = 115867 ± 6407 M−1 s−1 at 23 °C (Figure 13b). This second-order rate constant is ~86 times faster than that seen for Fe(Cl)(ttppc), and ~313 times faster than Fe(OH)(ttppc).17 The significantly enhanced reaction rate of the fluoride complex as compared to the chloride complex correlates with their respective redox potentials, in which 4 exhibits a reduction wave that is 0.14 V more positive than 1 (Figure 9). However, comparison with the Fe(OH)(ttppc) complex does not follow this trend; the reported value of E½ = −0.11 V for the hydroxide complex indicates it is significantly more oxidizing than either 4 or 1. It is not clear at this time as to the origins of the rate differences with the OH complex, but it suggests that the rate of hydroxyl group transfer may be influenced more by other factors, including possible entropic effects, as compared to the redox potential.
Figure 13.

(a) UV-vis spectra for reaction of 4 (12.5 μM) with (p-MeO-C6H4)3C• (31 equiv) in toluene from 0 (blue) to 0.1 s (red) at 0.0015 s intervals (grey) at 23 °C. Inset: Changes in absorbance versus time for the formation of FeIII(ttppc) (black dots) fit to a single exponential expression (red line). (b) Plot of kobs versus [(p-MeO-C6H4)3C•] with best-fit line.
Conclusions.
The synthesis and characterization of the new corrole complexes 2 – 4 were described. Complexes 2 and 4 are the first structurally characterized triflate- and fluoride-ligated iron corroles, respectively. The triflate-ligated complex represents a new synthon for accessing a wide range of iron corroles with various axial donors. The reactivity of 1 and 4 with triphenylmethyl radical derivatives shows that iron corroles are capable of mediating halogen transfer to carbon radicals via a process that mimics the rebound step seen for heme or porphyrin-catalyzed hydroxylation. The mechanism of halogen transfer appears to be in line with a concerted mechanism, as described previously for hydroxyl transfer with Fe and Mn corroles, and in which partial charge-transfer occurs in the transition state.19 From the kinetic studies, it is clear that the rate of group transfer from iron corrole to a tertiary carbon radical occurs in the following order: F > Cl > OH. These findings could be important for the future design of iron corroles, or other porphyrinoid species, as catalysts for either halogenation or hydroxylation of organic substrates. They also suggest that the redox potentials of the iron complexes are only one piece of the puzzle when predicting the reactivity of these radical transfer reactions.
Metallocorroles are promising candidates for oxidative C-H functionalization, and the sterically encumbered ttppc ligand has already shown utility in shape-selective, catalytic epoxidations.102 Our current findings suggest that base metal corroles (e.g. Fe, Mn) may be excellent candidates for the development of efficient and selective halogenation catalysts because of their ability to undergo facile, concerted transfer of an equivalent of X• (X = Cl, F) to carbon radicals. The high-valent metal-halide intermediates can be expected to exhibit relatively modest reduction potentials and good stability, helping to avoid oxidative decomposition, or the non-productive oxidation of substrates. In particular, the sterically encumbered ttppc complexes may exhibit desirable patterns of selectivity in the catalytic halogenation of C-H bonds. Future efforts in our laboratory will test these ideas.
Supplementary Material
Scheme 6.

Reaction of 4 with Tris-(para-Methoxyphenyl)methyl Radical
Synopsis:
The new triflate- and fluoride-ligated iron corroles Fe(OTf)(ttppc), Fe(OTf)(ttppc)(AgOTf), and Fe(F)(ttppc) were synthesized, and the latter two complexes were characterized by single crystal X-ray diffraction. The Fe(F)(ttppc) reacts with tertiary carbon radicals to give the fluorinated product and the one-electron reduced FeIII(ttppc), in a process that models the rebound step in halogenases and hydroxylases. Kinetic studies provided a direct comparison of reaction rates and mechanism between Fe(X)(ttppc) (X = F, Cl, OH) species.
ACKNOWLEDGMENT
The authors would like to thank the NIH (GM101153) (D.P.G.) for the financial support of this research. Part of this research project was conducted using computational resources and scientific computing services at the Maryland Advanced Research Computing Center (MARCC). The authors thank Dr. Jaime Combariza, associate research professor of Chemistry and director of MARCC, for help with calculations.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: xxx.
X-ray crystallographic data for 3 (CIF)
X-ray crystallographic data for 4 (CIF)
NMR, UV-vis, Mossbauer, EPR spectra, electrochemical and kinetics data, GC–FID, GC–MS, ESI-MS data, Tables S1 – S6, computational details.
REFERENCES
- 1.Sahu S; Goldberg DP, Activation of Dioxygen by Iron and Manganese Complexes: A Heme and Nonheme Perspective. J. Am. Chem. Soc, 2016, 138, 11410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poulos TL, Heme Enzyme Structure and Function. Chem. Rev, 2014, 114, 3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rittle J; Green MT, Cytochrome P450 Compound I: Capture, Characterization, and C-H Bond Activation Kinetics. Science, 2010, 330, 933. [DOI] [PubMed] [Google Scholar]
- 4.Denisov IG; Makris TM; Sligar SG; Schlichting I, Structure and Chemistry of Cytochrome P450. Chem. Rev, 2005, 105, 2253. [DOI] [PubMed] [Google Scholar]
- 5.Makris TM; Koenig K. v.; Schlichting I; Sligar SG, The status of high-valent metal oxo complexes in the P450 cytochromes. J. Inorg. Biochem, 2006, 100, 507. [DOI] [PubMed] [Google Scholar]
- 6.Ortiz de Montellano PR, Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes. Chem. Rev, 2010, 110, 932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rettie AE; Sheffels PR; Korzekwa KR; Gonzalez FJ; Philpot RM; Baillie TA, CYP4 Isoenzyme Specificity and the Relationship between ω-Hydroxylation and Terminal Desaturation of Valproic Acid. Biochemistry, 1995, 34, 7889. [DOI] [PubMed] [Google Scholar]
- 8.Rettie AE; Boberg M; Rettenmeier AW; Baillie TA, Cytochrome P-450-catalyzed desaturation of valproic acid in vitro. Species differences, induction effects, and mechanistic studies. J. Biol. Chem, 1988, 263, 13733. [PubMed] [Google Scholar]
- 9.Rettie AE; Rettenmeier AW; Howald WN; Baillie TA, Cytochrome P-450—catalyzed formation of delta 4-VPA, a toxic metabolite of valproic acid. Science, 1987, 235, 890. [DOI] [PubMed] [Google Scholar]
- 10.Wise CE; Grant JL; Amaya JA; Ratigan SC; Hsieh CH; Manley OM; Makris TM, Divergent mechanisms of iron-containing enzymes for hydrocarbon biosynthesis. J. Biol. Inorg. Chem, 2017, 22, 221. [DOI] [PubMed] [Google Scholar]
- 11.Matthews S; Belcher JD; Tee KL; Girvan HM; McLean KJ; Rigby SEJ; Levy CW; Leys D; Parker DA; Blankley RT; Munro AW, Catalytic Determinants of Alkene Production by the Cytochrome P450 Peroxygenase OleTJE. J. Biol. Chem, 2017, 292, 5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsieh CH; Huang X; Amaya JA; Rutland CD; Keys CL; Groves JT; Austin RN; Makris TM, The Enigmatic P450 Decarboxylase OleT Is Capable of, but Evolved To Frustrate, Oxygen Rebound Chemistry. Biochemistry, 2017, 56, 3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grant JL; Mitchell ME; Makris TM, Catalytic strategy for carbon−carbon bond scission by the cytochrome P450 OleT. Proc. Natl. Acad. Sci. U. S. A, 2016, 113, 10049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zachos I; Gaßmeyer SK; Bauer D; Sieber V; Hollmann F; Kourist R, Photobiocatalytic decarboxylation for olefin synthesis. Chem. Commun, 2015, 51, 1918. [DOI] [PubMed] [Google Scholar]
- 15.Dennig A; Kuhn M; Tassoti S; Thiessenhusen A; Gilch S; Bülter T; Haas T; Hall M; Faber K, Oxidative Decarboxylation of Short-Chain Fatty Acids to 1-Alkenes. Angew. Chem., Int. Ed. Engl, 2015, 54, 8819. [DOI] [PubMed] [Google Scholar]
- 16.Huang X; Groves JT, Beyond ferryl-mediated hydroxylation: 40 years of the rebound mechanism and C─H activation. J. Biol. Inorg. Chem, 2017, 22, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zaragoza JPT; Yosca TH; Siegler MA; Moënne-Loccoz P; Green MT; Goldberg DP, Direct Observation of Oxygen Rebound with an Iron-Hydroxide Complex. J. Am. Chem. Soc, 2017, 139, 13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaragoza JPT; Cummins DC; Mubarak MQE; Siegler MA; de Visser SP; Goldberg DP, Hydrogen Atom Abstraction by High-Valent Fe(OH) versus Mn(OH) Porphyrinoid Complexes: Mechanistic Insights from Experimental and Computational Studies. Inorg. Chem, 2019, 58, 16761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cummins DC; Alvarado JG; Zaragoza JPT; Effendy Mubarak MQ; Lin Y-T; de Visser SP; Goldberg DP, Hydroxyl Transfer to Carbon Radicals by Mn(OH) vs Fe(OH) Corrole Complexes. Inorg. Chem, 2020, 59, 16053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Timmins A; De Visser SP, A Comparative Review on the Catalytic Mechanism of Nonheme Iron Hydroxylases and Halogenases. Catalysts, 2018, 8. [Google Scholar]
- 21.Vaillancourt FH; Yin J; Walsh CT, SyrB2 in syringomycin E biosynthesis is a nonheme FeII α-ketoglutarate- and O2-dependent halogenase. Proc. Natl. Acad. Sci. U. S. A, 2005, 102, 10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mehmood R; Qi HW; Steeves AH; Kulik HJ, The Protein’s Role in Substrate Positioning and Reactivity for Biosynthetic Enzyme Complexes: The Case of SyrB2/SyrB1. ACS Catal., 2019, 9, 4930. [Google Scholar]
- 23.Menon BRK; Richmond D; Menon N, Halogenases for biosynthetic pathway engineering: Toward new routes to naturals and non-naturals. Catal. Rev, 2020, 1. [Google Scholar]
- 24.Gao S-S; Naowarojna N; Cheng R; Liu X; Liu P, Recent examples of α-ketoglutarate-dependent mononuclear non-haem iron enzymes in natural product biosyntheses. Nat. Prod. Rep, 2018, 35, 792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chatterjee S; Paine TK, Hydroxylation versus Halogenation of Aliphatic C─H Bonds by a Dioxygen-Derived Iron─Oxygen Oxidant: Functional Mimicking of Iron Halogenases. Angew. Chem., Int. Ed. Engl, 2016, 55, 7717. [DOI] [PubMed] [Google Scholar]
- 26.Wong C; Fujimori DG; Walsh CT; Drennan CL, Structural Analysis of an Open Active Site Conformation of Nonheme Iron Halogenase CytC3. J. Am. Chem. Soc, 2009, 131, 4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Srnec M; Solomon EI, Frontier Molecular Orbital Contributions to Chlorination versus Hydroxylation Selectivity in the Non-Heme Iron Halogenase SyrB2. J. Am. Chem. Soc, 2017, 139, 2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borowski T; Noack H; Radoń M; Zych K; Siegbahn PEM, Mechanism of Selective Halogenation by SyrB2: A Computational Study. J. Am. Chem. Soc, 2010, 132, 12887. [DOI] [PubMed] [Google Scholar]
- 29.Martinie RJ; Livada J; Chang W.-c.; Green MT; Krebs C; Bollinger JM; Silakov A, Experimental Correlation of Substrate Position with Reaction Outcome in the Aliphatic Halogenase, SyrB2. J. Am. Chem. Soc, 2015, 137, 6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yadav V; Rodriguez RJ; Siegler MA; Goldberg DP, Determining the Inherent Selectivity for Carbon Radical Hydroxylation versus Halogenation with FeIII(OH)(X) Complexes: Relevance to the Rebound Step in Non-heme Iron Halogenases. J. Am. Chem. Soc, 2020, 142, 7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galonić DP; Barr EW; Walsh CT; Bollinger JM; Krebs C, Two interconverting Fe(IV) intermediates in aliphatic chlorination by the halogenase CytC3. Nat. Chem. Biol, 2007, 3, 113. [DOI] [PubMed] [Google Scholar]
- 32.Agarwal V; Miles ZD; Winter JM; Eustáquio AS; El Gamal AA; Moore BS, Enzymatic Halogenation and Dehalogenation Reactions: Pervasive and Mechanistically Diverse. Chem. Rev, 2017, 117, 5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duewel S; Schmermund L; Faber T; Harms K; Srinivasan V; Meggers E; Hoebenreich S, Directed Evolution of an FeII-Dependent Halogenase for Asymmetric C(sp3)─H Chlorination. ACS Catal., 2020, 10, 1272. [Google Scholar]
- 34.Hillwig ML; Liu X, A new family of iron-dependent halogenases acts on freestanding substrates. Nat. Chem. Biol, 2014, 10, 921. [DOI] [PubMed] [Google Scholar]
- 35.Matthews ML; Neumann CS; Miles LA; Grove TL; Booker SJ; Krebs C; Walsh CT; Bollinger JM, Substrate positioning controls the partition between halogenation and hydroxylation in the aliphatic halogenase, SyrB2. Proc. Natl. Acad. Sci. U. S. A, 2009, 106, 17723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng Q; Ritter T, New Directions in C─H Fluorination. Trends Chem., 2019, 1, 461. [Google Scholar]
- 37.Szpera R; Moseley DFJ; Smith LB; Sterling AJ; Gouverneur V, The Fluorination of C─H Bonds: Developments and Perspectives. Angew. Chem., Int. Ed. Engl, 2019, 58, 14824. [DOI] [PubMed] [Google Scholar]
- 38.Champagne PA; Desroches J; Hamel J-D; Vandamme M; Paquin J-F, Monofluorination of Organic Compounds: 10 Years of Innovation. Chem. Rev, 2015, 115, 9073. [DOI] [PubMed] [Google Scholar]
- 39.Eisenstein O; Milani J; Perutz RN, Selectivity of C─H Activation and Competition between C─H and C─F Bond Activation at Fluorocarbons. Chem. Rev, 2017, 117, 8710. [DOI] [PubMed] [Google Scholar]
- 40.Tarantino G; Hammond C, Catalytic C(sp3)─F bond formation: recent achievements and pertaining challenges. Green Chem., 2020, 22, 5195. [Google Scholar]
- 41.Liu W; Groves JT, Manganese Catalyzed C─H Halogenation. Acc. Chem. Res, 2015, 48, 1727. [DOI] [PubMed] [Google Scholar]
- 42.Huang X; Liu W; Hooker JM; Groves JT, Targeted Fluorination with the Fluoride Ion by Manganese-Catalyzed Decarboxylation. Angew. Chem., Int. Ed. Engl, 2015, 54, 5241. [DOI] [PubMed] [Google Scholar]
- 43.Li G; Dilger AK; Cheng PT; Ewing WR; Groves JT, Selective C─H Halogenation with a Highly Fluorinated Manganese Porphyrin. Angew. Chem., Int. Ed. Engl, 2018, 57, 1251. [DOI] [PubMed] [Google Scholar]
- 44.Liu W; Huang X; Placzek MS; Krska SW; McQuade P; Hooker JM; Groves JT, Site-selective 18F fluorination of unactivated C─H bonds mediated by a manganese porphyrin. Chem. Sci, 2018, 9, 1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo M; Seo MS; Lee Y-M; Fukuzumi S; Nam W, Highly Reactive Manganese(IV)-Oxo Porphyrins Showing Temperature-Dependent Reversed Electronic Effect in C─H Bond Activation Reactions. J. Am. Chem. Soc, 2019, 141, 12187. [DOI] [PubMed] [Google Scholar]
- 46.Liu W; Groves JT, Manganese Porphyrins Catalyze Selective C─H Bond Halogenations. J. Am. Chem. Soc, 2010, 132, 12847. [DOI] [PubMed] [Google Scholar]
- 47.Jana RD; Sheet D; Chatterjee S; Paine TK, Aliphatic C─H Bond Halogenation by Iron(II)-α-Keto Acid Complexes and O2: Functional Mimicking of Nonheme Iron Halogenases. Inorg. Chem, 2018, 57, 8769. [DOI] [PubMed] [Google Scholar]
- 48.Rana S; Biswas JP; Sen A; Clémancey M; Blondin G; Latour J-M; Rajaraman G; Maiti D, Selective C─H halogenation over hydroxylation by non-heme iron(IV)-oxo. Chem. Sci, 2018, 9, 7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Puri M; Biswas AN; Fan R; Guo Y; Que L, Modeling Non-Heme Iron Halogenases: High-Spin Oxoiron(IV)─Halide Complexes That Halogenate C─H Bonds. J. Am. Chem. Soc, 2016, 138, 2484. [DOI] [PubMed] [Google Scholar]
- 50.Sharma N; Lee Y-M; Li X-X; Nam W; Fukuzumi S, Regioselective Oxybromination of Benzene and Its Derivatives by Bromide Anion with a Mononuclear Nonheme Mn(IV)─Oxo Complex. Inorg. Chem, 2019, 58, 14299. [DOI] [PubMed] [Google Scholar]
- 51.Rana S; Bag S; Patra T; Maiti D, Catalytic Electrophilic Halogenations and Haloalkoxylations by Non-Heme Iron Halides. Adv. Synth. Catal, 2014, 356, 2453. [Google Scholar]
- 52.Bower JK; Cypcar AD; Henriquez B; Stieber SCE; Zhang S, C(sp3)─H Fluorination with a Copper(II)/(III) Redox Couple. J. Am. Chem. Soc, 2020, 142, 8514. [DOI] [PubMed] [Google Scholar]
- 53.Kannan N; Patil AR; Sinha A, Direct C─H bond halogenation and pseudohalogenation of hydrocarbons mediated by high-valent 3d metal-oxo species. Dalton Trans., 2020, 49, 14344. [DOI] [PubMed] [Google Scholar]
- 54.Bakhoda AG; Wiese S; Greene C; Figula BC; Bertke JA; Warren TH, Radical Capture at Nickel(II) Complexes: C─C, C─N, and C─O Bond Formation. Organometallics, 2020, 39, 1710. [Google Scholar]
- 55.Lv X-L; Wang K; Wang B; Su J; Zou X; Xie Y; Li J-R; Zhou H-C, A Base-Resistant Metalloporphyrin Metal–Organic Framework for C─H Bond Halogenation. J. Am. Chem. Soc, 2017, 139, 211. [DOI] [PubMed] [Google Scholar]
- 56.Simkhovich L; Gross Z, Halogeno-Coordinated Iron Corroles. Inorg. Chem, 2004, 43, 6136. [DOI] [PubMed] [Google Scholar]
- 57.Horn M; Mayr H, Electrophilicity versus Electrofugality of Tritylium Ions in Aqueous Acetonitrile. Chem. - Eur. J, 2010, 16, 7478. [DOI] [PubMed] [Google Scholar]
- 58.Bloom S; McCann M; Lectka T, Photocatalyzed Benzylic Fluorination: Shedding “Light” on the Involvement of Electron Transfer. Org. Lett, 2014, 16, 6338. [DOI] [PubMed] [Google Scholar]
- 59.Bjornsson R; Neese F; DeBeer S, Revisiting the Mössbauer Isomer Shifts of the FeMoco Cluster of Nitrogenase and the Cofactor Charge. Inorg. Chem, 2017, 56, 1470. [DOI] [PubMed] [Google Scholar]
- 60.Steene E; Wondimagegn T; Ghosh A, Electrochemical and Electronic Absorption Spectroscopic Studies of Substituent Effects in Iron(IV) and Manganese(IV) Corroles. Do the Compounds Feature High-Valent Metal Centers or Noninnocent Corrole Ligands? Implications for Peroxidase Compound I and II Intermediates. J. Phys. Chem. B, 2001, 105, 11406. [Google Scholar]
- 61.Walker FA; Licoccia S; Paolesse R, Iron corrolates: Unambiguous chloroiron(III) (corrolate)2− π-cation radicals. J. Inorg. Biochem, 2006, 100, 810. [DOI] [PubMed] [Google Scholar]
- 62.Simkhovich L; Goldberg I; Gross Z, Iron(III) and Iron(IV) Corroles: Synthesis, Spectroscopy, Structures, and No Indications for Corrole Radicals. Inorg. Chem, 2002, 41, 5433. [DOI] [PubMed] [Google Scholar]
- 63.Pan Z; Harischandra DN; Newcomb M, Formation of stable and metastable porphyrin- and corrole-iron(IV) complexes and isomerizations to iron(III) macrocycle radical cations. J. Inorg. Biochem, 2009, 103, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gordon JB; McGale JP; Prendergast JR; Shirani-Sarmazeh Z; Siegler MA; Jameson GNL; Goldberg DP, Structures, Spectroscopic Properties, and Dioxygen Reactivity of 5- and 6-Coordinate Nonheme Iron(II) Complexes: A Combined Enzyme/Model Study of Thiol Dioxygenases. J. Am. Chem. Soc, 2018, 140, 14807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jesse KA; Filatov AS; Xie J; Anderson JS, Neocuproine as a Redox-Active Ligand Platform on Iron and Cobalt. Inorg. Chem, 2019, 58, 9057. [DOI] [PubMed] [Google Scholar]
- 66.Krzystek J; Schnegg A; Aliabadi A; Holldack K; Stoian SA; Ozarowski A; Hicks SD; Abu-Omar MM; Thomas KE; Ghosh A; Caulfield KP; Tonzetich ZJ; Telser J, Advanced Paramagnetic Resonance Studies on Manganese and Iron Corroles with a Formal d4 Electron Count. Inorg. Chem, 2020, 59, 1075. [DOI] [PubMed] [Google Scholar]
- 67.Ghosh A, Electronic Structure of Corrole Derivatives: Insights from Molecular Structures, Spectroscopy, Electrochemistry, and Quantum Chemical Calculations. Chem. Rev, 2017, 117, 3798. [DOI] [PubMed] [Google Scholar]
- 68.Ganguly S; Giles LJ; Thomas KE; Sarangi R; Ghosh A, Ligand Noninnocence in Iron Corroles: Insights from Optical and X-ray Absorption Spectroscopies and Electrochemical Redox Potentials. Chem. - Eur. J, 2017, 23, 15098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ganguly S; McCormick LJ; Conradie J; Gagnon KJ; Sarangi R; Ghosh A, Electronic Structure of Manganese Corroles Revisited: X-ray Structures, Optical and X-ray Absorption Spectroscopies, and Electrochemistry as Probes of Ligand Noninnocence. Inorg. Chem, 2018, 57, 9656. [DOI] [PubMed] [Google Scholar]
- 70.Maeda K; Takahashi T; Tomifuji R; Hirao N; Kurahashi T; Matsubara S, Ligand-controlled Behavior of Ag(I)─π Complex as σ-Lewis Acid. Chem. Lett, 2018, 47, 532. [Google Scholar]
- 71.Maier JM; Li P; Hwang J; Smith MD; Shimizu KD, Measurement of Silver─π Interactions in Solution Using Molecular Torsion Balances. J. Am. Chem. Soc, 2015, 137, 8014. [DOI] [PubMed] [Google Scholar]
- 72.Baudron SA, Ag(I)-π interactions with pyrrolic derivatives. Coord. Chem. Rev, 2019, 380, 318. [Google Scholar]
- 73.Fang M; Wilson SR; Suslick KS, A Four-Coordinate Fe(III) Porphyrin Cation. J. Am. Chem. Soc, 2008, 130, 1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gismelseed A; Bominaar EL; Bill E; Trautwein AX; Winkler H; Nasri H; Doppelt P; Mandon D; Fischer J; Weiss R, Six-coordinate quantum-mechanically weakley spin-mixed (S = 5/2, 3/2) (triflato)aquoiron(III) "picket-fence" porphyrin complex: synthesis and structural, Mössbauer, EPR, and magnetic characterization. Inorg. Chem, 1990, 29, 2741. [Google Scholar]
- 75.Gonzalez JA; Wilson LJ, Spin-state isomerism in crystalline (trifluoromethanesulfonato)(meso-tetraphenylporphinato)iron(III) [FeIII(TPP)(OSO2CF3)]. Inorg. Chem, 1994, 33, 1543. [Google Scholar]
- 76.Bhowmik S; Ghosh SK; Rath SP, Control of spins by ring deformation in a diiron(iii)bisporphyrin: reversal of ClO4− and CF33SO3− ligand field strength in the magnetochemical series. Chem. Commun, 2011, 47, 4790. [DOI] [PubMed] [Google Scholar]
- 77.Sil D; Kumar A; Rath SP, Diiron(III)─μ-Fluoro Bisporphyrins: Effect of Bridging Ligand on the Metal Spin State. Chem. - Eur. J, 2016, 22, 11214. [DOI] [PubMed] [Google Scholar]
- 78.Anzai K; Hatano K; Lee YJ; Scheidt WR, Preparation and molecular stereochemistry of fluoro(meso-tetraphenylporphinato)iron(III). Inorg. Chem, 1981, 20, 2337. [Google Scholar]
- 79.Scheidt WR; Lee YJ; Tamai S; Hatano K, Preparation and molecular stereochemistry of anionic difluoro(meso-tetraphenylporphinato)iron(III), a high-spin six-coordinate iron(III) porphyrinate with an unusually expanded core and a hydrogen-bonded "distal" imidazole. J. Am. Chem. Soc, 1983, 105, 778. [Google Scholar]
- 80.Shelly K; Bartczak T; Scheidt WR; Reed CA, Coordinated hexafluoroantimonate: x-ray crystal structure of the intermediate-spin iron(III) tetraphenylporphinato complex Fe(TPP)(FSbF5).C6H5F. Inorg. Chem, 1985, 24, 4325. [Google Scholar]
- 81.Sakow D; Baabe D; Böker B; Burghaus O; Funk M; Kleeberg C; Menzel D; Pietzonka C; Bröring M, Iron 10-Thiacorroles: Bioinspired Iron(III) Complexes with an Intermediate Spin (S=3/2) Ground State. Chem. - Eur. J, 2014, 20, 2913. [DOI] [PubMed] [Google Scholar]
- 82.Lee SC; Holm RH, Fluoride-bridged dimers: binuclear copper(II) complexes and iron(III)-copper(II) assemblies. Inorg. Chem, 1993, 32, 4745. [Google Scholar]
- 83.Meininger DJ; Muzquiz N; Arman HD; Tonzetich ZJ, A convenient procedure for the synthesis of fluoro-iron(III) complexes of common synthetic porphyrinates. J. Porphyrins Phthalocyanines, 2014, 18, 416. [Google Scholar]
- 84.Ohgo Y; Neya S; Ikeue T; Takahashi M; Takeda M; Funasaki N; Nakamura M, Molecular Structures of Five-Coordinated Halide Ligated Iron(III) Porphyrin, Porphycene, and Corrphycene Complexes. Inorg. Chem, 2002, 41, 4627. [DOI] [PubMed] [Google Scholar]
- 85.Taylor R, It Isn’t, It Is: The C─H⋯X (X = O, N, F, Cl) Interaction Really Is Significant in Crystal Packing. Cryst. Growth Des, 2016, 16, 4165. [Google Scholar]
- 86.Varadwaj PR; Varadwaj A; Marques HM; Yamashita K, Significance of hydrogen bonding and other noncovalent interactions in determining octahedral tilting in the CH3NH3PbI3 hybrid organic-inorganic halide perovskite solar cell semiconductor. Sci. Rep, 2019, 9, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hu P; Wang S; Chaturvedi A; Wei F; Zhu X; Zhang X; Li R; Li Y; Jiang H; Long Y; Kloc C, Impact of C─H⋯X (X = F, N) and π─π Interactions on Tuning the Degree of Charge Transfer in F6TNAP-Based Organic Binary Compound Single Crystals. Cryst. Growth Des, 2018, 18, 1776. [Google Scholar]
- 88.Saha BK; Saha A; Sharada D; Rather SA, F or O, Which One Is the Better Hydrogen Bond (Is It?) Acceptor in C─H⋯X─C (X− = F−, O=) Interactions? Cryst. Growth Des, 2018, 18, 1. [Google Scholar]
- 89.Crispini A; Aiello I; La Deda M; Godbert N; Ghedini M; Lelj F; Amati M, Fluorine Interactions in the 3D Packing of “Pt(IV)I2” Organometallic Molecular Materials: Structural and Computational Approaches. Cryst. Growth Des, 2017, 17, 409. [Google Scholar]
- 90.Mandal A; Patel BK; Shukla R; Chopra D, Impact of the complementary electronic nature of C─X and M─X halogens and intramolecular X⋯O interaction on supramolecular assemblies of Zn(II) complexes of o-halophenyl substituted hydrazides. CrystEngComm, 2017, 19, 1607. [Google Scholar]
- 91.Ye S; Tuttle T; Bill E; Simkhovich L; Gross Z; Thiel W; Neese F, The Electronic Structure of Iron Corroles: A Combined Experimental and Quantum Chemical Study. Chem. - Eur. J, 2008, 14, 10839. [DOI] [PubMed] [Google Scholar]
- 92.Roy MF; Marvel CS, The Dissociation of Hexa-p-alkylphenylethanes1. J. Am. Chem. Soc, 1937, 59, 2622. [Google Scholar]
- 93.Colle TH; Glaspie PS; Lewis ES, The triphenylmethyl radical: equilibrium measurements and the reaction with thiophenol. J. Org. Chem, 1978, 43, 2722. [Google Scholar]
- 94.Pangia TM; Davies CG; Prendergast JR; Gordon JB; Siegler MA; Jameson GNL; Goldberg DP, Observation of Radical Rebound in a Mononuclear Nonheme Iron Model Complex. J. Am. Chem. Soc, 2018, 140, 4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Leeladee P; Jameson GNL; Siegler MA; Kumar D; de Visser SP; Goldberg DP, Generation of a High-Valent Iron Imido Corrolazine Complex and NR Group Transfer Reactivity. Inorg. Chem, 2013, 52, 4668. [DOI] [PubMed] [Google Scholar]
- 96.Kurahashi S; Ikeue T; Sugimori T; Takahashi M; Mikuriya M; Handa M; Ikezaki A; Nakamura M, Formation and characterization of five- and six-coordinate iron(III) corrolazine complexes. J. Porphyrins Phthalocyanines, 2012, 16, 518. [Google Scholar]
- 97.Tanaka T; Ooi S; Ide Y; Ikeue T; Suzuki M; Chen PPY; Takahashi M; Osuka A, Different Antiferromagnetic Coupling between 5,5′- and 10,10′-Linked Iron(III) Corrole Dimers. Eur. J. Inorg. Chem, 2017, 1374. [Google Scholar]
- 98.Gütlich P, Bill E, and Trautwein AX, Mössbauer Spectroscopy and Transition Metal Chemistry: Fundamentals and Applications; Springer-Verlag: Berlin: 2011. [Google Scholar]
- 99.Kostka KL; Fox BG; Hendrich MP; Collins TJ; Rickard CEF; Wright LJ; Münck E, High-valent transition metal chemistry. MÖssbauer and EPR studies of high-spin (S = 2) iron(IV) and intermediate-spin (S = 3/2) Iron(III) complexes with a macrocyclic tetraamido-N ligand. J. Am. Chem. Soc, 1993, 115, 6746. [Google Scholar]
- 100.Lexa D; Saveant JM; Su Khac B; Wang DL, Single electron transfer and nucleophilic substitution. Reaction of alkyl bromides with aromatic anion radicals and low-oxidation-state iron porphyrins. J. Am. Chem. Soc, 1988, 110, 7617. [Google Scholar]
- 101.Volz H; Lotsch W, Polarographische reduktion von triarylmethylkationen. Tetrahedron Lett., 1969, 10, 2275. [Google Scholar]
- 102.Liu H-Y; Yam F; Xie Y-T; Li X-Y; Chang CK, A Bulky Bis-Pocket Manganese(V)─Oxo Corrole Complex: Observation of Oxygen Atom Transfer between Triply Bonded MnV≡O and Alkene. J. Am. Chem. Soc, 2009, 131, 12890. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.