

Abstract
This study aims to push the frontier of the engineering of human cathelicidin LL-37, a critical antimicrobial innate immune peptide that wards off invading pathogens. By sequentially truncating the smallest antibacterial peptide (KR12) of LL-37 and conjugating them with fatty acids with varying chain lengths, a library of lipopeptides is generated. These peptides are subjected to antibacterial activity and hemolytic assays. Candidates (including both forms made of L and D-amino acids) with the optimal cell selectivity are subsequently fed to the second layer of in vitro filters, including salts, pH, serum, and media. These practices lead to the identification of a miniature LL-37 like peptide (D-form) with selectivity, stability and robust antimicrobial activity in vitro against both Gram-positive and negative bacteria. Proteomic studies reveal much less serum proteins binding to the D-form than the L-form peptide. C10-KR8d targets bacterial membranes to become helical, making it difficult for bacteria to develop resistance in a multiple passage experiment. In vivo, C10-KR8d is able to reduce bacterial burden of methicillin-resistant Staphylococcus aureus (MRSA) USA300 LAC in neutropenic mice. In addition, this designer peptide prevented bacterial biofilm formation in a catheter-associated mouse model. Meanwhile, C10-KR8d also recruited cytokines to the vicinity of catheters to clear infection. Thus, based on the antimicrobial region of LL-37, this study succeeds in identifying the smallest anti-infective peptide C10-KR8d with both robust antimicrobial, antibiofilm, and immune modulation activities.
Keywords: Human cathelicidin, in vitro filtering, in vivo efficacy, peptide library, proteomics, bacterial resistance
Graphical Abstract
Like plants and insects, humans also deploy host defense antimicrobial peptides (AMPs). These innate immune peptides play a critical role in warding off invading pathogenic bacteria, viruses, fungi, and parasites.1 Some of these peptides are able to eliminate the ESKAPE pathogens (Enterococcus faecium, Staphycococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) in both the planktonic and biofilm forms.2 In addition, AMPs also regulate the immune systems by neutralization of endotoxin (lipopolysaccharides), association with cell receptors, and recruitment of immune cells to clear infection.3-5 As of January 8, 2021, 141 human AMPs have been registered in the antimicrobial peptide database (original website: http://aps.unmc.edu/AP under maintenance; currently available at https://wangapd3.com) (for a review, see 6). The major families of human AMPs are defensins, cathelicidins, histatins, and lactoferricin. Of note, there are numerous human defensins, but only one cathelicidin gene. Also, some known polypeptides possess antimicrobial activity, including cytokines, neuropeptides, and β-amyloid peptides.5, 6
Unlike horse, sheep and pigs, there is only one cathelicidin gene in humans.7 Interestingly, human cathelicidin can be processed into different molecular forms. LL-37, one of the mature AMPs, is most widely studied.8-10 Another form, ALL-38, which contains one extra alanine at the N-terminus, is released in the human reproduction system, probably to protect the fertilized egg from microbial infection.11 ALL-38 is one residue shorter than FALL-39 originally predicted prior to the isolation of LL-37.7 Recently, TLN-58, an even longer form, is found from a diseased state.12 Other processed forms of human cathelicidin are also possible. For instance, an alternative form detected in fat cells has not been sequenced.13 In addition, human LL-37 can be cleaved into a variety of fragments on human skin,14 further enriching the peptide reservoir of the human cathelicidin.
The interest in identifying the antimicrobial regions of human LL-37 resulted in numerous artificial peptides. Many active fragments involve the central region of LL-37.15-24 The major antimicrobial region (residues 17-32) of LL-37 was identified by two-dimensional (2D) NMR spectroscopy using the TOCSY-trim technology.24 An N-terminally glycine-appended version of this major antibacterial peptide (FK-16) is referred to as GF-17.25 GF-17 is demonstrated to have different activities, including antibacterial,24,25 anticancer,24 antibiofilm,26 anti-HIV,27 anti-Zika,28 anti-influenza,29 anti-Ebola virus,30 and spermicidal activities.31 GF-17 eliminates both Gram-positive and negative pathogens.25,32 It is possible to convert GF-17 to narrow-spectrum antimicrobial molecules. Partial incorporation of D-amino acids into GF-17 led to GF-17d3, which is active against Escherichia coli and A. baumannii, but not other bacteria tested (supporting information in 32). It was also possible to make the peptide only inhibitory to Gram-positive pathogens such as methicillin-resistant S. aureus (MRSA).32 Alternatively via sequence truncation of GF-17, we obtained KR12 (residues 18-29 of LL-37), which is the minimal antibacterial sequence of LL-3733,34 that inhibits E. coli, but not MRSA. These results suggest different sequence requirements for cationic peptides to inhibit Gram-positive and Gram-negative pathogens.
To develop the potential medical use of LL-37, we previously succeeded in converting GF-17 into a stable, selective and potent peptide 17BIPHE2 against the ESKAPE pathogens in planktonic or biofilm forms.32, 35, 36 Others attempted to engineer new peptides based on FK-13 or KR12, the core and minimal antimicrobial peptides of LL-37 we previously identified. These range from simple amino acid substitutions to more sophisticated cyclization of KR12.38-41
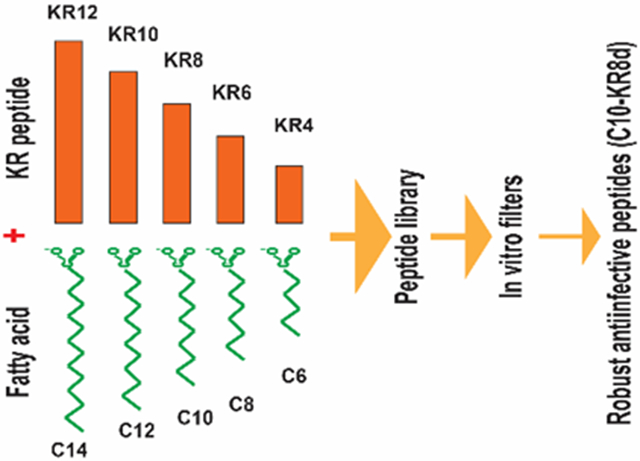
This study aimed at pushing the frontier of the LL-37 engineering by addressing two important questions. First, can we further shorten the peptide length via conjugation? Second, can we design more robust LL-37 peptides that keep antimicrobial activity under different media conditions, including pH, serum and salt? We obtained a library of new peptides by conjugating numerous short KR12 peptides with fatty acids at varying chain lengths (see TOC). Based on antibacterial and hemolytic assays, we identified outstanding candidates with excellent antimicrobial potency and high cell selectivity. This success enabled us to characterize peptide conformation, mechanism of action, and serum protein binding by mass spectrometry. Importantly, MRSA did not develop resistance to the optimal peptide C10-KR8d in a multiple passage experiment, although multiple genes are responding to it. Also, we demonstrate the in vivo efficacy of the peptide using both topical and systemic murine models. Finally, we discuss the potential of our new peptide as a promising lead for developing topical antimicrobials against antibiotic-resistant bacteria.
RESULTS
Systematic Search of Selective Peptides by a 2D Molecular Array.
To identify highly selective antimicrobial peptides, we varied the peptide length from 4 to 12 amino acids (aa) and the fatty acid chain length from C6 to C14 with a step size of 2 in both cases. Various peptide segments were generated by truncating the KR12 peptide sequence (KRIVQRIKDFLR-amide)33 from the C-terminus, two residues at a time. In each case, a fatty acid was attached to the N-terminal amine of the peptide segment by forming an amide bond. In addition, the underlined valine was replaced by a tryptophan (W) residue to facilitate peptide quantification by UV spectroscopy at 280 nm. The generated peptides were named according to the nomenclature convention for LL-37. The names of these lipopeptides are represented using a general format Cm-KRn, where Cm stands for a fatty acid with m carbons, whereas KRn means a peptide with n amino acids and starting with KR (n is an even number here). As controls, both human LL-37 and its smallest antibacterial peptide KR12 were included. Antimicrobial activities of the peptides were tested using a panel of bacteria, including E. coli, K. pneumoniae, P. aeruginosa, and S. aureus. The minimal inhibitory concentrations (MIC) of this library of peptides are listed in Table 1. LL-37 was not active against S. aureus USA300 in 100% trypic soy broth (TSB), but inhibited Gram-negative E. coli ATCC25922 (MIC 3.1 μM). Under such a condition, KR12 was not active against MRSA and weakly active against E. coli (MIC 50 μM). These results agree with the previous MIC data.42 However, most of these KR12 derived peptides gained activity against MRSA after attaching an acyl chain at the N-terminus. Table 1 shows that those with very short peptide segments or fatty acid chains were inactive against S. aureus. The active peptides contained C8 to C14 fatty acid chains with MIC in the range of 1.5-12.5 μM. We also evaluated peptide toxicity by measuring the ability of the peptide to lyse human red blood cells. The concentration that causes 50% hemolysis (HC50 in Table 1) was estimated for each peptide based on peptide dose-dependent lysis data. Some peptides conjugated with a C6 (aa4-aa10), C8 (aa4-aa8), or C10 (aa6 and aa8) fatty acid chain showed poor hemolytic activity (HC50 >200 μM in Table 1). These results rejected both the C12 (HC50 < 75 μM) and C14 (HC50 < 25 μM) fatty acid chains (blue and purple in Figure 1B) due to higher hemolysis. They also allowed us to reject the peptides with a C6 fatty acid chain owing to either high hemolytic ability or weak antibacterial activity against all the bacteria tested in Table 1. Two peptides, C10-KR8 and C8-KR10, were found to be less hemolytic. Both are active against S. aureus, E. coli, and P. aeruginosa (MIC 1.6-12.5 μM). They also inhibited clinical strains of S. aureus, including USA200, USA400, Newman, and Mu50 (1.6-6.2 μM in Table S1). However, based on cell selectivity index (CSI), i.e., the ratio of HC50 and MIC (Table 1), C10-KR8 (CSI 200) was more selective than C8-KR10 (CSI 40-80). Therefore, C10-KR8 is used for detailed study below. Its L-form (made of L-amino acids) and D-form (made of D-amino acids) have the same CSI in Table 1. The D-form is shortened into C10-KR8d.
Table 1.
Antimicrobial and hemolytic activities of KR12 derived lipopeptides 1
| Peptide | Amino acid sequence |
Acyl chain |
MIC (μM) | HC50 (μM) |
CSI 2 | |||
|---|---|---|---|---|---|---|---|---|
| EC | KP | PA | SA | |||||
| LL-37 | See legend 3 | None | 3.1-6.2 | 6.2 | >50 | >50 | 170 | ND |
| KR12 | KRIWQRIKDFLR | None | >50 | >50 | 25 | >50 | >340 | ND |
| C14-KR10 | KRIWQRIKDF | C14 | 50 | 6.2 | 12.5 | 3.1 | <25 | <8 |
| C14-KR8 | KRIWQRIK | C14 | 12.5 | >50 | 6.2 | <1.5 | <25 | <16 |
| C14-KR6 | KRIWQR | C14 | 12.5 | >50 | 6.2 | 3.1 | <25 | <8 |
| C14-KR4 | KRIW | C14 | 12.5-50 | 12.5 | 6.2 | 3.1 | <25 | <8 |
| C12-KR12 | KRIWQRIKDFLR | C12 | >50 | 12.5 | >50 | 6.2 | <25 | <4 |
| C12-KR10 | KRIWQRIKDF | C12 | 6.2 | 3.1 | 3.1 | 1.5 | <25 | <16 |
| C12-KR8 | KRIWQRIK | C12 | 6.2 | >50 | 3.1 | 1.5 | 35 | 24 |
| C12-KR6 | KRIWQR | C12 | 12.5 | >50 | 6.2 | 3.1 | 75 | 24 |
| C12-KR4 | KRIW | C12 | 12.5 | >50 | 6.2 | 3.1 | 30 | 10 |
| C10-KR12 | KRIWQRIKDFLR | C10 | >50 | 6.2-12.5 | 25 | 3.1 | <25 | <8 |
| C10-KR10 | KRIWQRIKDF | C10 | 3.1 | 6.2 | 6.2 | 1.5 | 50 | 33 |
| C10-KR8 | KRIWQRIK | C10 | 12.5 | 50 | 6.2 | 1.5-3.1 | 300 | 200 |
| C10-KR8d | KRIWQRIK | C10 | 12.5 | 25 | 6.2 | 1.5-3.1 | 300 | 200 |
| C10-KR6 | KRIWQR | C10 | 50 | >50 | 25 | 12.5 | >340 | >27 |
| C10-KR4 | KRIW | C10 | 50 | >50 | 12.5 | 12.5 | 150 | 12 |
| C8-KR12 | KRIWQRIKDFLR | C8 | 6.2 | <6.2 | 6.2 | 3.1 | <25 | <8 |
| C8-KR10 | KRIWQRIKDF | C8 | 6.2 | 25 | 6.2 | 3.1-6.2 | 250 | 40-80 |
| C8-KR10d | KRIWQRIKDF | C8 | 6.2 | 12.5 | 12.5 | 3.1 | 150 | 48 |
| C8-KR8 | KRIWQRIK | C8 | 25 | >50 | 12.5 | 6.2 | >200 | 32 |
| C8-KR6 | KRIWQR | C8 | 50 | >50 | >50 | >50 | >200 | ND |
| C8-KR4 | KRIW | C8 | >50 | >50 | >50 | >50 | >200 | ND |
| C6-KR12 | KRIWQRIKDFLR | C6 | 3.1 | 6.2 | 12.5 | <1.5 | 25 | 16 |
| C6-KR10 | KRIWQRIKDF | C6 | 50 | >50 | 50 | 25 | >200 | >8 |
| C6-KR8 | KRIWQRIK | C6 | >50 | >50 | >50 | >50 | >200 | ND |
| C6-KR6 | KRIWQR | C6 | >50 | >50 | >50 | >50 | >200 | ND |
| C6-KR4 | KRIW | C6 | >50 | >50 | >50 | >50 | >200 | ND |
All peptides are C-terminally amidated. MIC, the minimal inhibitory concentration; EC, E. coli ATCC25922; KP, Klebsiella pneumonia ATCC 13883; PA, P. aeruginosa PAO1; SA, S. aureus USA300 LAC. ND = not determined.
CSI = HC50/MICSA where HC50 is the peptide concentration that causes 50% hemolysis and MICSA is MIC against S. aureus.
The amino acid sequence of LL-37 is LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES, where KR12 is underlined. MIC and HC50 of LL-37 were taken from ref. [36].
Figure 1.

Mass spectrometry analysis of mouse serum proteins binding to the L- and D-forms of C10-KR8 immobilized to beads (see Methods). Heat map representation of the abundances of 20 significantly up-regulated (red) and down-regulated (green) proteins (PEAKS Q) bound to C10-KR8, C10-KR8d, and the peptide uncoupled control samples (left to right) after unsupervised hierarchical clustering.
Antimicrobial Robustness of the Selective Lipopeptides.
In addition to a long peptide length, another weakness of LL-37 is its loss of antimicrobial activity under certain conditions.43-45 We first compared antimicrobial activity of LL-37, KR12, and C10-KR8d in different media: Mueller Hinton Broth (MHB) and TSB. These peptides showed essentially the same MIC values (Table 2). We also evaluated the MICs in media containing varying amounts of TSB. In 50% or 100% TSB, both LL-37 and KR12 were not active against MRSA up to 50 μM, but started to gain activity at 10% TSB. In the case of E. coli ATCC 25922, both LL-37 and KR12 showed an inhibitory effect, although KR12 was less active in 100% TSB (Table 2). It appears that the anti-MRSA activity of both LL-37 and KR12 were more influenced by the concentration of TSB. However, the newly identified peptide C10-KR8d retained antibacterial activity against both MRSA and E. coli under various TSB concentrations as well as in different media. To corroborate peptide robustness, we tested antimicrobial activity of both the L- and D-forms of C10-KR8 in the presence or absence of physiological salts and at different pH values. Notably, sodium chloride did not alter the MIC values of these selective lipopeptide peptides at 100 or 200 mM (Table 3). Likewise, these peptides showed nearly the same activity at different pH conditions, from 6.8 to 8 (Table 3). However, human serum had a definitive effect on anti-MRSA activity depending on the chirality of the peptide. While the L-form rapidly lost activity in the presence of 5 or 10% human serum (MIC > 25 μM), the activity of the D-form of C10-KR8 was only slightly reduced by two fold at 10% serum, pH 7.4. To validate our finding with C10-KR8, we also compared the antibacterial activity of the L- and D-forms of C8-KR10 (octanoic acid + KRIWQRIKDF-amide) under these conditions. We obtained the same results, i.e., the D-form of C8-KR10 (C8-KR10d) is more robust than the L-form (Table 3). It appears that these D-form peptides are less likely to bind to serum proteins.
Table 2.
Effects of TSB medium percentages and different media on the antibacterial activity of C10-KR8d, KR12, and human LL-37
| Peptide | Minimal inhibitory concentration (μM) against S. aureus | |||
|---|---|---|---|---|
| 10% TSB | 50% TSB | 100% TSB | 100% MHB | |
| LL-37 | 50 | >50 | >50 | >50 |
| KR12 | 6.2 | >50 | >50 | >50 |
| C10-KR8d | 1.6 | 3.1 | 3.1 | 3.1 |
| MIC (μM) against E. coli | ||||
| LL-37 | 1.6 | 6.2 | 6.25-12.5 | 6.2 |
| KR12 | 1.6 | 3.1 | >50 | 50 |
| C10-KR8d | 1.6 | 3.1 | 6.2 | 3.1 |
Table 3.
Effects of salts, pH and human serum on peptide activity against Staphylococcus aureus USA300 LAC 1
| Peptide | Minimal inhibitory concentration (MIC, μM) | |||||||
|---|---|---|---|---|---|---|---|---|
| 0 mM | 100 mM |
200 mM |
pH=6.8 | pH=7.4 | pH=8.0 | 5% Serum |
10% Serum |
|
| C10-KR8 | 3.1 | 3.1 | 3.1 | 3.1-6.2 | 3.1 | 1.6 | >25 | >25 |
| C10-KR8d | 3.1 | 3.1 | 3.1 | 3.1 | 3.1 | 1.6 | 3.1-6.2 | 6.2 |
| C8-KR10 | 6.2 | 6.2 | 6.2 | 12.5 | 6.2 | 3.1 | >25 | >25 |
| C8-KR10d | 3.1 | 3.1 | 3.1 | 6.2 | 3.1 | 1.6 | 3.1 | 6.2 |
The MIC was normally obtained in 100% TSB at pH 7.4 (indicated by 0 mM in the table). The condition of the TSB was then altered by adjusting pH, adding NaCl or human serum.
Binding to Serum Proteins.
We reasoned that the loss of peptide activity in the presence of serum might result from binding to serum proteins. To provide evidence, we conducted a peptide binding study using both the L- and D-forms of C10-KR8. In this experiment, the peptide was attached to a commercial bead via two steps of chemical reactions described in the Methods. Our peptide immobilization to the beads allowed murine serum proteins to bind to the peptide and facilitated a thorough wash to remove unbound molecules. The bound proteins were identified by a proteomic mass spectrometry (MS) study as described in Methods. The top-20 bound proteins are provided in a heat map in Figure 1 based on protein relative abundance. The red color indicates more binding, while the green color indicates less binding. These proteins appeared to preferentially associate with the L-form of C10-KR8. Note that 16 proteins associated with the linker region of the beads without peptide coupling. Among them, five (Q8VCM7, P33622, P01942, Q61508, and P97290) overlapped with those detected in the presence of C10-KR8, but none of these 16 proteins clearly associated with C10-KR8d. Thus, the type of “baits” on the beads determines the types of the bound proteins. Table S2 provides lists of the bound serum proteins identified by MS, such as albumin, complement proteins, and numerous apolipoproteins. Table S3 calculated the intensity ratios of these apolipoproteins bound to the L and D-forms of C10-KR8. It is evident that most of the apolipoproteins, including A-I, A-IV, C-I, and C-III, showed a preferred binding to the L-form of C10-KR8. Interestingly, both apoA-II and apoE were able to bind the linker without the peptide. It appeared that the D-form peptide could enhance the binding of apoA-II, whereas the apoE binding to the linker was slightly reduced in the presence of either the L or D-form of the peptide. Therefore, unlike the D-form, the L-form of C10-KR8 is capable of binding to numerous serum proteins, including apolipoproteins (Table S2).
Peptide Stability to Proteases.
Protease degradation makes many peptides unavailable as an oral drug. Also, the cleavage of human LL-37 constitutes a virulence mechanism for bacterial infection.21 The loss of peptide activity in our antimicrobial assays in the presence of human serum could result from both protein binding and protease cleavage. Our serum protein binding above validated the preferred binding of the L-form than the D-form to serum proteins. To illustrate that protease cleavage also plays a role, we also compared peptide levels in the presence of five known proteases. Our previous study found that the major antimicrobial region of LL-37 is degraded in vitro in 4 hours.35 In line with this, the L-form of C10-KR8 was degraded within 3 h in nearly all the cases, except for S. aureus V8 protease due to a lack of acidic glutamates and aspartates in the sequence (Figure 2A, lanes 4 & 9). Nevertheless, C10-KR8d was resistant to the five proteases, including trypsin, chymotrypsin, pancreas elastase, S. aureus V8 protease, and fungal proteinase K (Figure 2B). Thus, C10-KR8d is more stable than the L-form of C10-KR8. It appeared that the protease stability of C10-KR8d is superior to 17BIPHE2, which can be cleaved by trypsin, but not chymotrypsin, S. aureus V8 protease, and fungal proteinase K.35
Figure 2.

Protease stability of the LL-37 derived C10-KR8 detected by SDS-PAGE. (A) C10-KR8 (L-form): untreated peptide (arrow); peptide treated with 2. bovine chymotrypsin; 3. porcine pancreas trypsin; 4, S. aureus V8 protease; 5. fungal proteinase K; 6, porcine pancreas elastase for 1 day; peptide treated with 7, chymotrypsin; 8. trypsin; 9, S. aureus V8 protease; 10, porcine pancreas elastase for 3 h. (B) C10-KR8d (D-form): untreated peptide (arrow); peptide treated with 2. chymotrypsin; 3. trypsin; 4, S. aureus V8 protease; 5. proteinase K; 6, porcine pancreas elastase for 1 day; peptide treated with 7, chymotrypsin; 8. trypsin; 9, S. aureus V8 protease; and 10, porcine pancreas elastase for 3 h.
C10-KR8d Targets Bacterial Membranes.
Similar MIC values for the L- and D-forms of C10-KR8d suggest membrane targeting (Table 1). To provide additional evidence for the membrane targeting of C10-KR8d, we conducted membrane permeation and depolarization experiments. Membrane permeation is indicated by the fluorescence increase of a non-membrane permeable dye propidium iodide due to binding to bacterial DNA.35 This can only occur when bacterial membranes are compromised by membrane active antibiotics such as daptomycin (Figure 3A). Similar to 17BIPHE2,35 C10-KR8d was able to permeate the membranes of S. aureus USA300 LAC. As a negative control, rifamycin, which inhibits bacterial DNA-dependent RNA synthesis, was unable to achieve the same effect. The membrane potential is essential for the normal physiology of bacteria. Some chemicals, such as Triton X-100, can destroy this potential, leading to membrane depolarization (Figure S1). We observed the same order for the depolarization ability: Triton-X100 > 17BIPHE2 > C10-KR8d > daptomycin at different concentrations, although the effect was small at 3.1 μM. The curves started to separate at 6.25 μM, but were best resolved at 12.5 μM (Figure 3B). At 25 μM, the depolarization effect of 17BIPHE2 approached that of Triton-X100 (Figure S1). We found C10-KR8d could also achieve this although at a lower level than Triton X-100. As in the case of membrane permeation (Figure 3A), C10-KR8d was also less efficient than 17BIPHE2 in membrane depolarization (Figure 3B). All these experiments support that C10-KR8d targets the membranes of MRSA.
Figure 3.

Membrane permeation and depolarization by the miniature LL-37 peptide C10-KR8d. (A) Effects of C10-KR8d, 17BIPHE2 and daptomycin on membrane integrity based on the fluorescence of a membrane non-permeable dye propidium iodide. S. aureus USA300 LAC treated with 12.5 μM of peptides and relative fluorescence intensity was recorded with time. 17BIPHE2 and daptomycin are used as positive controls, while rifamycin was included as a negative control for the assay. (B) Effects of peptides on membrane depolarization of the mid-log phase S. aureus USA300 re-suspended in PBS and glucose energized for 15 min at 37 °C. See Methods for further details. C10-KR8d, 17BIPHE2 and daptomycin (12.5 μM) were added to respective wells at post-dye equilibration pause cycle and the fluorescence was monitored for 40 minutes. The detergent Triton X-100 (0.1%) was included as a positive control.
Conformation of the Small Lipopeptides.
We also asked what conformation C10-KR8 might adopt after membrane binding. Figure 4 shows the CD spectra of both the free and bound peptides. In the 10 mM phosphate buffer (PBS), a negative band at 200 nm was observed for C10-KR8 (L-form). As anticipated, a mirror image with a positive band at ~200 nm was generated by C10-KR8d (D-form). These spectra indicate that the peptide was randomly coiled under this condition. The mirror image of the two forms of the same peptide validated the high quality of synthetic peptides. Next, we measured the CD spectra of these two forms of peptides in the presence of membrane-mimetic micelles of sodium dodecylsulfate (SDS). SDS is a useful model since we previously obtained similar helical structures for human cathelicdin LL-37 bound to SDS, dioctanoyl phosphatidylglycerol (D8PG), or lipopolysaccharides (LPS) by multidimensional NMR spectroscopy.33 For the L-form of C10-KR8 (blue), a positive band at ~195 and a negative band at 208 nm are reminiscent of the well-known CD spectra of helical proteins.11,46 The fact that the band at 222 nm was less evident implies a weak helix, probably due to a short peptide length with merely eight amino acids. Likewise, we observed a mirror image for the D-form of C10-KR8 bound to SDS micelles. These results implied that the helical structure was retained for C10-KR8 in association with bacterial membranes.
Figure 4.

Circular dichroism (CD) spectra of a highly selective lipopeptide C10-KR8 (L-form) and its D-form C10-KR8d (made of D-amino acids) (A) in PBS buffer and (B) in the presence of SDS micelles at pH 7 and 25 °C. The concentration of the peptide in all spectra was 1 mM and the peptide:SDS molar ratio was 1:60.
Resistance Development of S. aureus to the Small Lipopeptide.
Membrane targeting would make it more difficult for bacteria to develop resistance.1 To illustrate this, we conducted a multiple passage experiment.47 While the MIC of nafcillin increased by over 30 fold, no changes were observed for C10-KR8d in 14 days, indicating no resistance development of S. aureus USA300 LAC to this peptide (Figure 5).
Figure 5.

Resistance development of S. aureus USA300 LAC to C10-KR8d during 14 passages in the presence of sub-MIC of the peptide. The figure represents the average of three independent experiments.
Antimicrobial Susceptibility of the S. aureus Mutants to C10-KR8d.
The fact that MRSA did not develop resistance to C10-KR8d does not mean a lack of bacterial response. Recently, we have identified two dozens of response genes from the Nebraska Transposon Mutant Library of S. aureus USA30048 in the presence of LL-37 and its peptides.49 As C10-KR8d contains part of the core antimicrobial sequence of LL-37, we compared antimicrobial susceptibility of these transposon mutants to the new peptide designed here. The bacterial strains as well as MIC values are provided in Table 4. Among the tested strains, over half of the S. aureus mutant strains were found to be susceptible. These genes presumably contribute to the response of MRSA to C10-KR8d.49 They include the mutants for the major antimicrobial sensing system (mprF, graS, graR, and vraF) of S. aureus, potassium uptake protein (trkA), and lipoprotein signal peptidase (lspA). The major antimicrobial sensing system can modify the membrane surface with a lysine via the membrane enzyme MprF, making MRSA less susceptible to cationic antimicrobial peptides.50 The potassium uptake protein can modulate the membrane potential, again playing a role in bacterial susceptibility.51 As another example, LspA is found to be essential for resistance of Mycobacterium tuberculosis to malachite green.52 It is possible that these genes work together to reduce the likelihood of being killed by cationic AMPs. Since most of the LL-37 susceptible genes also respond to C10-KR8d, our results provide additional evidence that this newly discovered small lipopeptide acts like a miniature LL-37 peptide.
Table 4.
Antimicrobial susceptibility of the transposon mutants of S. aureus USA300 to C10-KR8d, a miniature lipoLL-37 peptide 1
| Gene ID | Name | Potential function | MIC (μM) |
Susceptible? |
|---|---|---|---|---|
| SAUSA300 | JE2 | Wild type (WT) | 3.1 | Reference |
| SAUSA300_1962 | phiPVL ORF39-like protein | 1.6-3.1 | Yes | |
| SAUSA300_1255 | mprF | Oxacillin resistance-related FmtC protein | 1.6 | Yes |
| SAUSA300_0829 | lipA | Lipoyl synthase | 3.1 | No |
| SAUSA300_1036 | RNA methyltransferase | 1.6-3.1 | Yes | |
| SAUSA300_1465 | 2-oxoisovalerate dehydrogenase, E1 component, beta subunit | 3.1 | No | |
| SAUSA300_0186 | argC | N-acetyl-gamma-glutamyl-phosphate reductase | 3.1 | No |
| SAUSA300_0988 | trkA | Potassium uptake protein | 1.6 | Yes |
| SAUSA300_1089 | lspA | lipoprotein signal peptidase | 1.6 | Yes |
| SAUSA300_1515 | ABC transporter permease | 1.6-3.1 | Yes | |
| SAUSA300_1967 | conserved hypothetical phage protein | 3.1 | No | |
| SAUSA300_1228 | thrB | homoserine kinase | 1.6-3.1 | Yes |
| SAUSA300_0394 | FAD/NAD(P)-binding Rossmann fold superfamily protein | 3.1 | No | |
| SAUSA300_0646 | graS | Sensor histidine kinase | 1.6 | Yes |
| SAUSA300_0645 | graR | DNA-binding response regulator | 1.6 | Yes |
| SAUSA300_1865 | vraR | DNA-binding response regulator | 1.6 | Yes |
| SAUSA300_0647 | vraF | ABC transporter ATP-binding protein | 1.6-3.1 | Yes |
| SAUSA300_1336 | Hypothetical protein | 3.1 | No | |
| SAUSA300_1898 | Conserved hypothetical protein | 3.1 | No | |
| SAUSA300_1037 | pheS | Phenylalanyl-tRNA synthetase subunit alpha | 1.6 | Yes |
| SAUSA300_1503 | Putative competence protein ComGB | 3.1 | No | |
| SAUSA300_1867 | Conserved hypothetical protein | 3.1 | No | |
| SAUSA300_1097 | pyrF | orotidine 5'-phosphate decarboxylase | 3.1 | No |
Mutants found previously in response to human cathelicidin LL-37 and its major antimicrobial peptides GF-17 and 17BIPHE2 [49]. No = no MIC changes of C10-KR8d against the WT and the mutant S. aureus strains measured in 50% TSB.
In Vitro and in Vivo Toxicity of the LL-37 Derived Lipopeptides.
C10-KR8 showed poor hemolysis (HC50 300 μM in Table 1). To further evaluate peptide toxicity, human HaCaT cells were treated with the peptides at multiple doses and the lethal dose where 50% cells (TC50) were dead was estimated (Figure S2). C10-KR8d was slightly more toxic than C10-KR8. To further our understanding, we also compared the toxicity of the L- and D-forms of C8-KR10. Again, C8-KR10 was slightly less toxic than C8-KR10d. These results indicate that the D-form was slightly more toxic than the L-form with TC50 between ~75 and ~100 μM. Such toxic concentrations to skin and red blood cells are much higher than the MIC values of C10-KR8d at 3.1-6.2 μM (Table 3).
We also tested peptide toxicity in mice. When C57BL/6 mice were injected with C10-KR8d intraperitoneally (i.p.) at 10 or 20 mg/kg per mouse, all mice survived during 5 days observation (Figure 6). However, all mice died in a day at a peptide dose of 40 mg/kg. This study indicates the maximal tolerated dose (MTD) is 20 mg/kg or more (but less than 40 mg/kg).
Figure 6.

Toxicity of C10-KR8d to mice for 5 days. Toxicity was assessed using female C57BL/6 mice (n=3) by intraperitoneal administration of C10-KR8d into three testing mouse groups at an increasing amount of the peptide dose of 10 (red), 20 (green), and 40 mg/kg (purple), respectively. A fourth control group was treated with vehicle (blue). See Methods for details.
Systemic in Vivo Efficacy of the Miniature LL-37 Peptides.
Since C10-KR8d has numerous drug-like properties (Table 3), we investigated its systemic efficacy in vivo. Neutropenic mice, generated by two injections of cyclophosphamide prior to infection, are widely used for this purpose since the effects of immune cells are minimized.47, 53 Based on our previous studies with other peptides and antibiotics controls, a bacterial inoculum of 2×106 colony forming units (CFU) was selected to establish the S. aureus USA300 LAC infection.47, 54 We then treated the infected mice with C10-KR8d at 5 mg/kg intraperitoneally. Compared to the untreated group, there was a significant CFU decrease in mouse lung and liver (~0.5-1 log), but not in spleen and kidney (Figure 7).
Figure 7.

Systemic efficacy of C10-KR8d in neutropenic mice infected with 2×106 CFU of S. aureus USA300 LAC. Two hours post infection, the peptide group was treated (i.p., 5 mg/kg, n = 8) with a single dose of C10-KR8d while the control infected group was untreated. The CFU difference is significant when p value < 0.05 and ns represents no significance. The statistics was determined using Mann-Whitney test in the GraphPad Prism 7.0.
Topical Antibiofilm Efficacy in a Catheter Murine Model.
Preformed biofilms associated with medical devices are notoriously difficult to get rid of by conventional antibiotics. Therefore, a preferred strategy is to prevent biofilm formation. We tested the antibiofilm capability of C10-KR8d in an established catheter model.35, 55, 56 In this experiment, catheters were inserted under the skin of the mouse flank, infected and treated by injecting the peptide into the lumen and around the catheter. After three days, mice were euthanized and the S. aureus CFUs on each catheter and its surrounding tissue were determined and presented in Figure 8A. As anticipated, we did not detect S. aureus USA300 LAC in the uninfected catheter. Our previous study confirmed that LL-23V9, an LL-23 peptide analog derived from the N-terminus of LL-37, which is inactive against S. aureus, was unable to cause any CFU decrease in mice.35 Therefore, a clear decrease in bacterial burden between the untreated and treated animal groups indicates an excellent efficacy of C10-KR8d. It appeared that the efficacy of one time treatment of C10-KR8d was superior to 17BIPHE2, which achieved a similar effect after three treatments in three days.35
Figure 8.

Biofilm inhibition capability of C10-KR8d in a catheter-associated biofilm model challenged with S. aureus USA300 LAC. (A) A single dose treatment with C10-KR8d (250 μg) into the implanted catheter lumen and surrounding tissues (50 μL/site; into lumen, four sides of the catheter) of mice (n=4). The catheter alone animal group without bacterial infection was included as a control. The CFU of each mouse represents individual points and error bars represents the deviation within the experimental group. Starred CFU differences (*) with p <0.05 were considered statistically significant within groups as determined by the Mann-Whitney test (GraphPad Prism 7). ELISA quantification of the levels of MCP-1 (B), IL-17A (C), IL-10 (D), and TNF-α (E) in the surrounding tissues of catheters embedded in mice with and without treatment with C10-KR8d.
One of the advantages of AMPs is their capability of curbing invading pathogens by multiple mechanisms, making it difficult for bacteria to develop resistance.3-9 For example, AMPs may also regulate cytokines that recruit immune cells. To get evidence, we measured the levels of cytokines in the surrounding tissues of catheters with and without C10-KR8d treatment. An up-regulation of MCP-1/CCL2 (monocyte chemoattractant protein-1) (Figure 8B) and IL-17A (Figure 8C) on day 3 after peptide treatment suggests the recruitment of monocytes35 and neutrophils,57 respectively, which could partially contribute to clearance of MRSA infection. Meanwhile, a decrease in the levels of IL-10 (Figure 8D) and TNFα (Figure 8E) could reduce inflammation to avoid potential tissue damage.4, 58
DISCUSSION
There are two major methods for drug discovery: rational design and library screening.45 The structure-based approach is widely utilized when a molecular target has been identified. Both methods have been utilized for human cathelicidin LL-37.41 While we identified the major antimicrobial peptide of human LL-37 via structural studies,24,25,33,35,36 others identified different LL-37 fragments via the library approach.15-19 This study falls into the library approach. It differs from the traditional random library in that we used a systematic approach to generating numerous peptides followed by conjugation with various fatty acids. Since some lipopeptides (e.g., daptomycin and colistin) are already in clinical use as essential antibiotics, we conjugated KR12 segments with fatty acids. Our 2D molecular array allowed us to identify small LL-37 lipopeptides with high selectivity, stability, and antimicrobial robustness. While we found two less hemolytic peptides: C10-KR8 and C8-KR10 (Table 1), a recent study found an optimal peptide when KR12 is conjugated with C8.59 It should be emphasized that our goal was not to design minimal lipopeptides (as short as two amino acids) that no longer resemble LL-37. In fact, even shorter peptides such as KRIW do not offer advantages in terms of both antimicrobial activity and cell selectivity (Table 1). In agreement, ultra-short lipopeptides may have limited cell selectivity.60, 61 Depending on the peptide sequence, there may be an optimal combination in terms of both peptide length and fatty acid chain length. Our systemic study identified a selective zone (10 carbon fatty acid × 8mer peptides or 8 carbon fatty acid × 10mer peptides) (Table 1), which may guide the construction of other selective antimicrobial lipopeptides.
C10-KR8d (made of D-amino acids), which is even more selective than C8-KR10 (Table 1), showed robust in vitro antibacterial activity under different media conditions (Table 3). In addition, the antibacterial activity of this peptide did not change in different media or with the content of TSB, whereas the antibacterial activity of both KR12 and human LL-37 were compromised in 100% TSB, especially against MRSA (Table 2). It appears that KR12 also contains the minimal LL-37 sequence required to interact with unidentified components in TSB. It is notable that C10-KR8 (made of L-amino acids) could lose activity in the presence of human serum (Table 3). Our bead binding studies revealed that many mouse plasma proteins could bind to the L-form, but very few associated with the D-form peptide. This is interesting and indicates that protein binding depends on the peptide chiral property (Table S2). Human LL-37 is known to bind to human serum protein apolipoprotein A-I, leading to a loss of its antibacterial activity in vitro.43 Indeed, our proteomic study reveals numerous murine apolipoproteins (apo), including apoA-I, apoA-IV, apoC-I, and apoC-III that preferentially bind to the L-form of C10-KR8. Since C10-KR8 associates with apoA-I similar to LL-37, our results further support that C10-KR8 retains the minimal functional core region of LL-37. Also, the tendency of the L-form of the peptide to interact with multiple serum proteins provides one possible mechanism for its loss of efficacy in vivo, since the D-form showed an antimicrobial efficacy in certain murine organs (Figures. 7 & 8). In this case, the extent of serum binding of the peptide in vitro appears to be a useful indicator for in vivo efficacy. However, one should not generalize this observation. The L- and D-forms of the database-designed DFTamP1 peptides showed similar systemic efficacy in the same mouse model, although their antibacterial activities were compromised by human serum to different extents in vitro.47 Likewise, horine has a slightly higher tendency to associate with human serum than verine. However, they are equally potent in eliminating MRSA in mice.54 It seems that serum binding may not be the sole reason for peptide activity loss. We do not exclude the possibility that, a peptide, when injected intravenously, may take advantage of the serum vehicle to reach different organs to kill pathogens.
Due to the challenging nature to develop peptides with systemic efficacy, the antimicrobial peptide field is currently focusing on topical applications. Wound healing and catheter are two commonly used models for this purpose. De Breij et al. reported a potent antibiofilm peptide SAAP-148 derived from the C-terminal region of LL-37 (residues 13-36).62 It is puzzling that the peptide was effective in mice but not in rats.63 We demonstrated the efficacy of 17BIPHE2 and its analog in preventing biofilm formation in a catheter-associated mouse model 35, 36 as well as wound healing in mice.64 While SAAP-148 consists of 24 amino acids, 17BIPHE2 is 17-residue long. Thus, our identification of a miniature LL-37 like peptide C10-KR8d here with merely eight residues further shortened the peptide length, reducing the cost of synthesis. It is remarkable that C10-KR8d (single dose treatment at 15 mg/kg) was able to achieve the same efficacy as 17BIPHE2 (three treatments in three days at 3 × 15 mg/kg) in the catheter-associated biofilm model. Our results underscore that these small LL-37 lipopeptides obtained here are potent candidates to prevent biofilm infections of antibiotic-resistant pathogens such as MRSA.
Finally, it is useful to note that S. aureus did not develop resistance to C10-KR8d in a multiple passage experiment (Figure 5). However, this evidence is insufficient for us to state that bacteria will never develop resistance to the newly designed peptide. Using the Nebraska Transposon Mutant Library of S. aureus USA300,48 we identified multiple genes that respond to the action of C10-KR8d (Table 4). MRSA became more susceptible in the absence of such genes as indicated by reduced MIC values evaluated under the same conditions. These genes, as well as unidentified genes of S. aureus, may work together as a network to compromise the effect of antimicrobials eventually. A combination of the newly designed peptide with inhibitors that target these genes may slow down the development of Staphylococcal resistance.
CONCLUSIONS
LL-37 is a human cathelicidin in the innate immune system important for fighting infections via both direct killing and immune modulation. This study advanced LL-37 engineering and identified a miniature LL-37 lipopeptide shorter than KR12 (the smallest antimicrobial peptide) via a systematic 2D molecular array. The sequence of the newly identified peptide has been shortened to the extent that its antimicrobial activity was not influenced by different media or the TSB media contents. Our proteomic studies indicate that C10-KR8 also retained some properties of LL-37. Indeed, the L-form of C10-KR8 lost activity in vitro due to association with numerous serum proteins. However, only few proteins bound to the peptide synthesized in D-amino acids (C10-KR8d). This D-peptide could tolerate physiological salts, pH, and serum. Such antimicrobial robustness may explain the systemic efficacy we observed for the D-form of the lipoLL-37 peptide in mice. Moreover, the miniature LL-37 like peptide C10-KR8d identified here showed an excellent activity in preventing biofilm formation of MRSA in a catheter-associated mouse model, thereby offering new therapeutic opportunities. In addition, our identification of the susceptible genes of S. aureus in the presence of C10-KR8d may lead to combined treatment to better combat such resistant pathogens.
METHODS
Peptide and Chemicals.
Peptides were synthesized by the established solid-phase method and purified to >95% (Genemed Synthesis, TX). The quality of each peptide was determined based on Mass Spectrometry and HPLC. Peptides stock solutions were made by solubilizing in autoclaved distilled water and their concentrations were quantitated using UV spectroscopy based on the tryptophan (W) at 280 nm. For LL-37 and KR12, which do not contain a W, they were quantified by using the Waddell method.65 Other chemicals were purchased from Sigma (MO, USA) unless specified.
Antibacterial Assays.
The antibacterial activity of peptides was evaluated using a standard broth microdilution protocol 66 with minor modifications as described.47 In brief, peptides (10 μL per well) with two fold dilutions were made in 96-well polystyrene microplates. Subsequently, the logarithmic phase bacterial cultures (i.e., optical density at 600 nm ≈ 0.5) were diluted to 0.001 OD and aliquoted 90 μL per well. The plates were incubated overnight at 37 °C. Bacterial controls were treated with water and the wells containing only the TSB media were used as blank. Positive controls include parent peptide LL-37 as well as KR12 discovered by our lab.33 Post incubation the plates were read at 630 nm using ChroMate4300 Microplate Reader (Awareness Technology, FL). The minimal inhibitory concentration (MIC) was the lowest peptide concentration that fully inhibited bacterial growth.
To study the influence of medium conditions on the antimicrobial activity of peptides against S. aureus USA300, different, pH, sodium chloride (NaCl), human sera were included into the antibacterial assays. Control experiments in TSB without any additions (pH 7.4) were set up in the same manner. In addition, antimicrobial activities of human LL-37, KR12, and C10-KR8d were compared in 10%, 50%, and 100% TSB as well as in TSB and MHB using E. coli ATCC 25922 and S. aureus USA300 LAC.
Orientational Immobilization of C10-KR8 Peptides on M370 Beads.
Dynabeads M-370 amine (2 × 109 beads/mL, 50 μL; Thermo Fisher Scientific, USA) were washed 3 times in sterile 1 mL PBS (GIBCO, USA) and reacted with a bi-functional SM(PEG)4 cross-linker (5 mg/mL in DMSO) containing N-hydroxysuccinimide (NHS) ester and maleimide functional groups. A stable covalent bond between the amino group of beads and the NHS ester of the cross-linker was formed in PBS at pH 7.2 at room temperature for 24 h with shaking at 80 rpm. The beads were then washed six times with PBS to remove non-reacted cross-linker. Next, peptide C10-KR8 (with a cysteine at the N-terminus) at 5 mg/mL was reacted with the free maleimide group in PBS for 24 h at room temperature with shaking at 80 rpm. Another round of washing was conducted to remove the non-reacted peptides. Both the L- and D-forms of the peptide were coupled in the same manner. A portion of the beads without peptide coupling was saved as a control.
For serum binding experiments, the beads were resuspended in 500 μL of PBS. A 200-microliter aliquot of the coated and control dynabead were incubated with 5% mouse plasma for 18 h at 37 °C. After binding, the beads were washed six times with PBS to remove unbound proteins. Finally, the beads were suspended in 50 μL PBS for mass spectrophotometric analysis to identify peptide-binding partners.
Mass Spectrometric Identification of Serum Proteins Bound to Peptides.
The samples were reduced by 10 mM dithiothreitol (DTT) and incubated for 30 min at 56 °C and back to room temperature. A final concentration of 50 mM iodoacetamide was added to alkylate free cysteines followed by further incubation for 20 min in the dark. Bound proteins were enzymatically digested by trypsin (4 μL of 0.5 μg/mL) overnight. Sixty microliters were transferred to a new Eppendorf tube and dried for 2 h. The samples were then dissolved in 20 μL of 2% acetonitrile with 0.1% formic acid before loading to Q-tip columns (Pepclean, C18 spin column). Column was activated by pipetting in and out (4 × times), 10 μL of 70% acetonitrile with 0.1% formic acid and equilibrating with the same volume of 2% acetonitrile with 0.1% formic acid. Binding of the samples was done by mixing more than 10 × times, followed by washing in the same buffer (2% acetonitrile with 0.1% formic acid). The bound peptides were then eluted with 20 μL of 70% acetonitrile with 0.1% formic acid. Samples were subjected to LC-MS analysis after being suspended in the desired buffer.
Extracted peptides were re-suspended in 2% acetonitrile (ACN) and 0.1% formic acid (FA) and loaded onto trap column Acclaim PepMap 100 75μm × 2 cm C18 LC Columns (Thermo Scientific™) at a flow rate of 4 μL/min and then separated with a Thermo RSLC Ultimate 3000 on a Thermo Easy-Spray PepMap RSLC C18 75 μm × 50 cm C-18 2 μm column (Thermo Scientific™) with a step gradient of 4–25% solvent B (0.1% FA in 80% ACN) from 10-37 min and 25–45% solvent B for 37–46 min at 300 nL/min and 50°C with a 70 min total run time. Eluted peptides were analyzed by a Thermo Orbitrap Fusion Lumos Tribrid (Thermo Scientific™) mass spectrometer in a data dependent acquisition mode. A survey full scan MS (from m/z 350–1800) was acquired in the Orbitrap with a resolution of 120,000. The AGC target for MS1 was set as 4 × 105 and the ion filling time set as 100 ms. The most intense ions with charge state 2-6 were isolated in 3 s cycle and fragmented using HCD fragmentation with 40 % normalized collision energy and detected at a mass resolution of 30,000 at 200 m/z. The AGC target for MS/MS was set as 5 × 104 and ion filling time set 60 ms dynamic exclusion was set for 30 s with a 10 ppm mass window. Protein identification was performed by searching MS/MS data against the Swiss-Prot mouse protein database downloaded on Aug 20, 2018. The search was set up for full tryptic peptides with a maximum of two missed cleavage sites. Acetylation of protein N-terminus and oxidized methionine were included as variable modifications and carbamidomethylation of cysteine was set as fixed modification. The precursor mass tolerance threshold was set 10 ppm for and maximum fragment mass error was 0.02 Da. Qualitative analysis was performed using PEAKS 8.5 software. The significance threshold of the ion score was calculated based on a false discovery rate of ≤ 1%.
Protease Stability of the Designed Peptides.
The peptide was incubated with various proteases (see the Figure 2 legend) in 10 mM PBS buffer (pH 7.4) at 37 °C for 24 h. Aliquots (20 μL) of the reaction solutions were taken at 3 and 24 h. The reaction was then stopped by mixing with 20 μL of 2× SDS loading buffer and boiling in a water bath for ~10 min. For the SDS-PAGE analysis, 10 μL of each sample was loaded to the well of a 5% stacking/ 18% resolving tricine gel and run at a constant current of 35 mA. The gels were stained using Coomassie Brilliant Blue.
Bacterial Membrane Permeation.
The experiment was performed as described with minor modifications.47 Serially diluted 10× peptides (10 μL each well) were created in 96-well microtiter plates. Propidium iodide (2 μL) at a fixed concentration of 20 μM were added to each well followed by 88 μL of the exponential phase S. aureus USA300 culture (a final OD600 ~0.1 in TSB media or PBS). Both daptomycin (98%, TSZChem, MA) and rifamycin (98%, Alfa Aesar, MA) were used as controls. The plate was incubated at 37 °C with continuous shaking at 100 rpm in a FLUOstar Omega (BMG LABTECH, NC) microplate reader. The samples fluorescence was read at every 5 minutes for 24 cycles with an excitation and emission wavelengths of 584 nm and 620 nm, respectively. Plots generated using average values from the experiments using GraphPad Prism 7.
Bacterial Membrane Depolarization.
The experiment was conducted as described.67 In brief, an overnight culture of S. aureus USA300 was sub-cultured in a fresh TSB medium and grown to the exponential phase. Cells were spun using centrifugation and washed 2 × with PBS, and re-suspended in twice the volume of PBS containing 25 mM glucose and incubated at 37 °C for 15 min. For membrane depolarization measurements, 500 nM (final concentration) of the dye DiBAC4 (3) bis-(1, 3- dibutylbarbituric acid) trimethine oxonol (ANASPEC, CA) was added and vortex gently. Aliquots of 90 μL of the energized bacteria solution were loaded to the wells and the plate was fed into a FLUOstar Omega microplate reader. Fluorescence was read for 20 min at excitation and emission wavelengths of 485 nm and 520 nm, respectively, to get dye normalization. Then 10 μL of peptide solutions was added and gently mixed. Fluorescence readings were recorded for 40 min, where triton X-100 (0.1%) was used as a positive control. Also, both daptomycin and 17BIPHE2 35 were included for comparison.
Circular Dichroism (CD).
CD spectra were measured on a Jasco J-815 spectropolariometer at UNMC in the far-UV region from 260–190 nm with a 1 nm interval, a 2 nm bandwidth using a digital integration time of 4 s and a scan speed of 20 nm per min. During measurements, the high-tension signal applied to the detector was also recorded and was subsequently converted to absorbance. Each spectrum represented the average of five individual scans with a corresponding reference measurement on pure solvent subtracted. The peptide (C10-KR8 or C10-KR8d) concentration was fixed at 1 mM in 10 mM PBS (pH 7) or in the presence of 60 fold SDS (molar ratio). Each sample was placed in a 0.1 mm quartz cuvette. The temperature was kept at 25 °C during measurements. Data were processed and converted to molar ellipticity ([θ]) using the Jasco Spectra Analysis software and plotted using GraphPad Prism 7.
Bacterial Resistance Development to the Peptide.
This experiment performed similar to the MIC determination with a few modifications as described.47 In short, an exponential phase S. aureus USA300 culture (i.e., optical density at 600 nm ≈ 0.5) was diluted and partitioned into a 96-well polystyrene microplate with ~105 bacteria per well (90 μL aliquots). After treatment with 10 μL of peptide or nafcillin solutions at various concentrations, microplates were incubated at 37 °C overnight and read on a ChroMate 4300 Microplate Reader at 630 nm (Awareness Technology, FL, USA). The wells with sub-MIC levels of the peptides that retained growth approximately half the growth of the control wells were again re-inoculated in fresh TSB with sub-MIC concertation of peptides or antibiotics to attain exponential phase for MIC determination. Up to 15 serial passages of the bacteria cultures were conducted. The increase in the fold change (MIC on given passage/ MIC recorded in first day of passage) used to determine the degree of drug resistance.
Antimicrobial Susceptibility of the LL-37 Susceptible S. aureus Strains to C10-KR8d.
We also tested antimicrobial susceptibility of over 20 MRSA transposon mutants we identified recently based on the transposon library screening in the presence of LL-37 peptides.49 This Nebraska Transposon Mutant Library of S. aureus USA300 consists of 1920 mutants, each with one non-essential gene disabled due to a transposon insertion.48 The correct insertion of the transposon to each Staph mutant had been verified in our previous publication.49 Antimicrobial susceptibility to the new peptide C10-KR8d was evaluated in the same manner as MIC assays. Susceptible strains could be inhibited at a peptide concentration lower than the MIC against the wild strain S. aureus JE2.
Hemolytic Assays.
Hemolytic assays of peptides were performed as described.54 Briefly, human red blood cells (hRBCs) obtained from UNMC Blood Bank. The cells were washed 3× with isotonic saline (0.9% NaCl) and diluted to 2% (v/v). Peptides at various concentrations were added to the 2% blood cells and incubated at 37°C for 1 h. Post incubation the plates were spun at 2000 rpm for 10 min, aliquots of the supernatant were carefully transferred to a fresh 96-well microplate. The amount of cells lysed is proportional to the hemoglobin released and measured at 545 nm using a ChroMate microplate reader (Awareness Technology, FL). The percent lysis was calculated by assuming 100% release when human blood cells were treated with 1% Triton X-100, and 0% release when incubated with PBS. The peptide concentration that caused 50% lysis of hRBCs is defined as HC50. The cell selectivity index (CSI) was calculated as the ratio between HC50 and the MIC of the corresponding peptide against MRSA.
Mammalian Cell Viability Assay.
Peptides assessed for potential cytotoxicity using HaCaT cells (Addexbio Technologies, CA). Briefly, cells were seeded at a density of 1×104 per well in a tissue culture 96-well plate. DMEM was supplemented with 10% fetal bovine serum (FBS) and incubated at 37 °C in a 5% CO2 atmosphere for 24 h. Culture medium was aspirated and replaced with fresh serum free media. Cells were exposed to different concentration of peptides for 1 h at 37 °C in a 5% CO2 atmosphere. Post-incubation, the cells were washed and incubated with 100 μL of DMEM media containing 20 μL of MTS reagent for 2 h at 37 °C. Absorbance was read at 492 nm using a ChroMate microplate reader (Awareness Technology, FL).
Ethics Statement.
All animal studies followed the protocols approved by the institutional animal care and use committee (IACUC #16-076-08-FC and 17-104-12-FC). On the study termination day, mice were euthanized humanely using CO2 followed by harvesting vital organs for laboratory analysis. All efforts were made to minimize animal pain and suffering. Mice were caged, feed, and environmentally adapted as described.54 All animal manipulations were performed in a class II laminar flow biological safety cabinet.
In Vivo Toxicity.
Toxicity was assessed in female C57BL/6 mice, by intraperitoneal administration of increasing doses of the antimicrobial peptide C10-KR8d (10, 20, and 40 mg/kg) and a control group without peptide treatment. The animals were then observed for 5 days (twice a day). The number of moribund/dead animals for each of the doses was noted and plotted.
In Vivo Efficacy of Peptides in Neutropenic Murine Infection Model.
Female C57BL/6 mice (6 weeks old) were purchased from Charles River. After environmental adaptation, mice were induced neutropenic by administering two doses of cyclophosphamide on day 1 (150 mg/kg) and Day 4 (100 mg/kg). On day 5, mice were infected with S. aureus USA300 (~2 × 106 CFU per mouse) via intraperitoneal injection (i.p.) as established previously.54 For the treatment groups, mice were i.p. treated 2 h post infection with peptides at a single dose of 5 mg/kg. Our previous study already established that this bacterial strain was able to disseminate to different organs two hours post infection.54 At the end of the experiments, all the animals were sacrificed according to institutional guidelines. Organs, including spleen, liver, lung, and kidney, were harvested, weighed and placed in 1 mL sterile PBS and stored on ice. Harvested organs were subsequently homogenized using an Omni Homogenizer. Proper dilutions were made to get countable colonies. The homogenates were plated onto blood agar plates and incubated overnight at 37 °C. The CFU of each murine tissue was plotted as an individual point and error bars represent the deviation within the experimental group. * indicates p<0.05, ** p <0.01, ***p <0.001 and NS, no significance (determined by Mann-Whitney test).
Catheter-associated Murine Biofilm Model.
Peptide potency was evaluated in the catheter model as described.35, 55 On the day of infection, mice were anesthetized using ketamine/xylazine (100/10 mg/kg). A small incision was made on the left flank region of the mouse and using small blunt spatula, a pouch was made for catheter insertion. A sterile catheter with a length of 1 cm was inserted into the pouch aseptically. The wound was sealed using wound closure VetBond glue. Bacterial inoculum (20 μL with 103 CFU) was injected into the lumen of the catheter. Two hours post infection, the peptide was injected at 15 mg/kg (50 μL at each of the five sites in and around the catheter). Mice were allowed for full recovery from anesthesia in an oxygen-enriched chamber. Three days after infection, untreated and treated animal groups were CO2 euthanized and the tissue around the catheter was harvested along with the catheter. A bacterial uninfected catheter group was included as a negative control. All the catheters were sonicated at 37 kHz for 15 minutes to release the bacteria in biofilms and the tissues were homogenized. Appropriate dilutions of tissue homogenates were made, plated onto the blood agar, and incubated at 37 °C overnight. The CFU of each mouse was plotted as an individual point and error bars represent the deviation within the experimental group. Degree of significance was represented as * p<0.05 ** p <0.01, ***p <0.001 and NS, no significance (determined by non-parametric, unpaired, Mann-Whitney test).
Cytokine Quantification by Flow Cytometry.
The catheter-associated tissue homogenates/plasma samples stored at −80 °C were thawed to room temperature for the cytokines quantification using the Bio legend individual mouse cytokine kit. The protocol for sample preparation and analysis is as per the manufacturer manual. The samples were in duplicates with appropriate standards. The data are represented as mean ± SD. Plots were generated using GraphPad prism 7, where * indicates p<0.05, ** p <0.01, ***p <0.001 and NS, no significance (non-parametric, unpaired, Mann-Whitney test).
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Drs. Gaelle Spagnol and Paul Sorgen for excellent assistance with CD spectra collection and processing.
Funding
This study was supported by Nebraska Research Initiative (NRI), NIH funding AI105147 to GW, and in part GM138552 to GW and JX. XW (General Hospital of Ningxia Medical University) and YZ (Hainan University) were visiting scholars supported by China Scholarship Council.
Footnotes
Supporting Information
Supporting information is provided free of charge.
The authors declare no competing financial interest.
REFERENCES
- (1).Mookherjee N, Anderson MA, Haagsman HP, Davidson DJ (2020) Antimicrobial host defence peptides: functions and clinical potential. Nat Rev Drug Discov. 19, 311–332. [DOI] [PubMed] [Google Scholar]
- (2).Rice LB (2008) Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J Infect Dis. 197, 1079–81. [DOI] [PubMed] [Google Scholar]
- (3).Nizet V, Ohtake T, Lauth X, Trowbridge J, Rudisill J, Dorschner RA, Pestonjamasp V, Piraino J, Huttner K, Gallo RL (2001) Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature. 414, 454–7. [DOI] [PubMed] [Google Scholar]
- (4).Pütsep K, Carlsson G, Boman HG, Andersson M (2002) Deficiency of antibacterial peptides in patients with morbus kostmann: an observation study. Lancet 360, 1144–1149. [DOI] [PubMed] [Google Scholar]
- (5).Scott MG, Davidson DJ, Gold MR, Bowdish D, Hancock RE (2002) The human antimicrobial peptide LL-37 is a multifunctional modulator of innate immune responses. J Immunol. 169, 3883–91. [DOI] [PubMed] [Google Scholar]
- (6).Wang G (2014) Human Antimicrobial Peptides and Proteins. Pharmaceuticals 7, 545–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Agerberth B, Gunne H, Odeberg J, Kogner P, Boman HG, Gudmundsson GH (1995) FALL-39, a putative human peptide antibiotic, is cysteine-free and expressed in bone marrow and testis. Proc Natl Acad Sci USA. 92, 195–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Dürr UH, Sudheendra US, Ramamoorthy A (2006) LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim Biophys Acta. 1758, 1408–1425. [DOI] [PubMed] [Google Scholar]
- (9).Vandamme D, Landuyt B, Luyten W, Schoofs L (2012) A comprehensive summary of LL-37, the factotum human cathelicidin peptide. Cell Immunol. 280, 22–35. [DOI] [PubMed] [Google Scholar]
- (10).Wang G, Mishra B, Epand RF, Epand RM (2014) High-quality 3D structures shine light on antibacterial, anti-biofilm and antiviral activities of human cathelicidin LL-37 and its fragments. Biochim. Biophys. Acta 1838, 2160–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Sørensen OE, Gram L, Johnsen AH, Andersson E, Bangsbøll S, Tjabringa GS, Hiemstra PS, Malm J, Egesten A, Borregaard N (2003) Processing of seminal plasma hCAP-18 to ALL-38 by gastricsin: a novel mechanism of generating antimicrobial peptides in vagina. J Biol Chem. 278, 28540–6. [DOI] [PubMed] [Google Scholar]
- (12).Murakami M, Kameda K, Tsumoto H, Tsuda T, Masuda K, Utsunomiya R, Mori H, Miura Y, Sayama K (2017) TLN-58, an additional hCAP18 processing form, found in the lesion vesicle of palmoplantar pustulosis in the skin. J. Invest Dermatol 137, 322–331. [DOI] [PubMed] [Google Scholar]
- (13).Zhang LJ, Guerrero-Juarez CF, Hata T, Bapat SP, Ramos R, Plikus MV, Gallo RL (2015) Dermal adipocytes protect against invasive Staphylococcus aureus skin infection. Science 347, 67–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Murakami M, Lopez-Garcia B, Braff M, Dorschner RA, Gallo RL (2004) Postsecretory processing generates multiple cathelicidins for enhanced topical antimicrobial defense. J Immunol. 172, 3070–7. [DOI] [PubMed] [Google Scholar]
- (15).Braff MH, Hawkins MA, Di Nardo A, Lopez-Garcia B, Howell MD, Wong C, Lin K, Streib JE, Dorschner R, Leung DY, Gallo RL (2005) Structure-function relationships among human cathelicidin peptides: dissociation of antimicrobial properties from host immunostimulatory activities. J Immunol. 174, 4271–8. [DOI] [PubMed] [Google Scholar]
- (16).Nagant C, Pitts B, Nazmi K, Vandenbranden M, Bolscher JG, Stewart PS, Dehaye JP (2012) Identification of peptides derived from the human antimicrobial peptide LL-37 active against biofilms formed by Pseudomonas aeruginosa using a library of truncated fragments. Antimicrob Agents Chemother. 56, 5698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).den Hertog AL, van Marle J, Veerman EC, Valentijn-Benz M, Nazmi K, Kalay H, Grün CH, Van't Hof W, Bolscher JG, Nieuw Amerongen AV (2006) The human cathelicidin peptide LL-37 and truncated variants induce segregation of lipids and proteins in the plasma membrane of Candida albicans. Biol Chem. 387, 1495–502. [DOI] [PubMed] [Google Scholar]
- (18).Nan YH, Bang JK, Jacob B, Park IS, Shin SY (2012) Prokaryotic selectivity and LPS-neutralizing activity of short antimicrobial peptides designed from the human antimicrobial peptide LL-37. Peptides. 35, 239–47. [DOI] [PubMed] [Google Scholar]
- (19).Nell MJ, Tjabringa GS, Wafelman AR, Verrijk R, Hiemstra PS, Drijfhout JW, Grote JJ (2006) Development of novel LL-37 derived antimicrobial peptides with LPS and LTA neutralizing and antimicrobial activities for therapeutic application. Peptides. 27, 649–60. [DOI] [PubMed] [Google Scholar]
- (20).Oren Z, Lerman JC, Gudmundsson GH, Agerberth B, Shai Y (1999) Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: relevance to the molecular basis for its non-cell-selective activity. Biochem J. 341 (Pt 3), 501–13. [PMC free article] [PubMed] [Google Scholar]
- (21).Sieprawska-Lupa M, Mydel P, Krawczyk K, Wójcik K, Puklo M, Lupa B, Suder P, Silberring J, Reed M, Pohl J, Shafer W, McAleese F, Foster T, Travis J, Potempa J (2004) Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob Agents Chemother. 48, 4673–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Sigurdardottir T, Andersson P, Davoudi M, Malmsten M, Schmidtchen A, Bodelsson M (2006) In silico identification and biological evaluation of antimicrobial peptides based on human cathelicidin LL-37. Antimicrob Agents Chemother. 50, 2983–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Turner J, Cho Y, Dinh NN, Waring AJ, Lehrer RI (1998) Activities of LL-37, a cathelin-associated antimicrobial peptide of human neutrophils. Antimicrob Agents Chemother. 42, 2206–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Li X, Li Y, Han H, Miller DW, Wang G (2006) Solution structures of human LL-37 fragments and NMR-based identification of a minimal membrane-targeting antimicrobial and anticancer region. J. Am. Chem. Soc 128, 5776–85. [DOI] [PubMed] [Google Scholar]
- (25).Wang G, Epand RF, Mishra B, Lushnikova T, Thomas VC, Bayles KW, Epand RM (2012) Decoding the functional roles of cationic side chains of the major antimicrobial region of human cathelicidin LL-37. Antimicrob. Agents Chemother 56, 845–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Mishra B, Golla RM, Lau K, Lushnikova T, Wang G (2016) Anti-Staphylococcal biofilm effects of human cathelicidin peptides. ACS Med. Chem. Lett 7, 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wang G Waston K Buckheit R Jr. (2008) Anti-human immunodeficiency virus type 1 (HIV-1) activities of antimicrobial peptides derived from human and bovine cathelicidins. Antimicrob. Agents Chemother 52, 3438–3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).He M, Zhang H, Li Y, Wang G, Tang B, Zhao J, Huang Y, Zheng J (2018) Cathelicidin-derived antimicrobial peptides inhibit Zika virus through direct inactivation and interferon pathway. Front Immun. 9, 722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Tripathi S, Wang G, White M, Rynkiewicz M, Seaton B, Hartshorn K (2015) Identifying the Critical Domain of LL-37 Involved in Mediating Neutrophil Activation in the Presence of Influenza Virus: Functional and Structural Analysis. PLoS One. 10, e0133454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Yu Y, Cooper CL, Wang G, Morwitzer MJ, Kota K, Tran JP, Bradfute SB, Liu Y, Shao J, Zhang AK, Luo LG, Reid SP, Hinrichs SH, Su K (2020) Engineered human cathelicidin antimicrobial peptides inhibit Ebola virus infection. iScience 23, 100999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kiattiburut W, Zhi R, Lee SG, Foo AC, Hickling DR, Keillor JW, Goto NK, Li W, Conlan W, Angel JB, Wang G, Tanphaichitr N (2018) Antimicrobial peptide LL-37 and its truncated forms, GI-20 and GF-17, exert spermicidal effects and microbicidal activity against Neisseria gonorrhoeae. Hum Reprod. 33, 2175–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Wang X, Mishra B, Lushnikova T, Narayana JL, Wang G (2018) Amino acid composition determines peptide activity spectrum and hot-spot-based design of merecidin. Adv Biosyst. 2, 1700259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Wang G (2008) Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J. Biol. Chem 283, 32637–32643. [DOI] [PubMed] [Google Scholar]
- (34).Mishra B, Epand RF, Epand RM, Wang G (2013) Structural location determines functional roles of the basic amino acids of KR-12, the smallest antimicrobial peptide from human cathelicidin LL-37. RSC Adv. 3, 19560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wang G, Hanke ML, Mishra B, Lushnikova T, Heim CE, Chittezham Thomas V, Bayles KW, Kielian T (2014) Transformation of Human Cathelicidin LL-37 into Selective, Stable, and Potent Antimicrobial Compounds. ACS Chem. Biol 9, 1997–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Narayana JL, Mishra B, Lushnikova T, Golla RM, Wang G (2019) Modulation of antimicrobial potency of human cathelicidin peptides against the ESKAPE pathogens and in vivo efficacy in a murine catheter-associated biofilm model. Biochim. Biophys. Acta 1861, 1592–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Saporito P, Vang Mouritzen M, Løbner-Olesen A, Jenssen H (2018) LL-37 fragments have antimicrobial activity against Staphylococcus epidermidis biofilms and wound healing potential in HaCaT cell line. J Pept Sci. 8, e3080. [DOI] [PubMed] [Google Scholar]
- (38).Rajasekaran G, Kim EY, Shin SY (2017) LL-37-derived membrane-active FK-13 analogs possessing cell selectivity, anti-biofilm activity and synergy with chloramphenicol and anti-inflammatory activity. Biochim Biophys Acta Biomembr. 1859, 722–733. [DOI] [PubMed] [Google Scholar]
- (39).Jacob B, Park IS, Bang JK, Shin SY (2013) Short KR12 analogs designed from human cathelicidin LL-37 possessing both antimicrobial and antiendotoxic activities without mammalian cell toxicity. J Pept Sci. 19, 700–7. [DOI] [PubMed] [Google Scholar]
- (40).Gunasekera S, Muhammad T, Strömstedt AA, Rosengren KJ, Göransson U (2020) Backbone Cyclization and Dimerization of LL-37-Derived Peptides Enhance Antimicrobial Activity and Proteolytic Stability. Front Microbiol. 11, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Wang G, Narayana JL, Mishra B, Zhang Y, Wang F, Wang C, Zarena D, Lushnikova T, Wang X (2019) Design of antimicrobial peptides: Progress made with human cathelicidin LL-37. Adv Exp Med Biol 1117, 215–240. [DOI] [PubMed] [Google Scholar]
- (42).Epand RF, Wang G, Berno B, Epand RM (2009) Lipid segregation explains selective toxicity of a series of fragments derived from the human cathelicidin LL-37. Antimicrob. Agents Chemother 53, 3705–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Wang Y, Agerberth B, Löthgren A, Almstedt A, Johansson J (1998) Apolipoprotein A-I binds and inhibits the human antibacterial/cytotoxic peptide LL-37. J Biol Chem. 273, 33115–8. [DOI] [PubMed] [Google Scholar]
- (44).Wang Y, Johansson J, Agerberth B, Jörnvall H, Griffiths WJ (2004) The antimicrobial peptide LL-37 binds to the human plasma protein apolipoprotein A-I. Rapid Commun Mass Spectrom. 18, 588. [DOI] [PubMed] [Google Scholar]
- (45).Mishra B, Reiling S, Zarena D, Wang G (2017) Host defense antimicrobial peptides as antibiotics: design and application strategies. Curr. Opin. Chem. Biol 38, 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Wang G, Li Y, Li X (2005) Correlation of three-dimensional structures with the antibacterial activity of a group of peptides designed based on a non-toxic bacterial membrane anchor. J. Biol. Chem 280, 5803–5811. [DOI] [PubMed] [Google Scholar]
- (47).Mishra B, Lakshmaiah Narayana J, Lushnikova T, Wang X, Wang G (2019) Low cationicity is important for systemic in vivo efficacy of database-derived peptides against drug-resistant Gram-positive pathogens. Proc Natl Acad Sci USA 116, 13517–13522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW (2013) A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. MBio. 4, e00537–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Golla RM, Mishra B, Dang X, Lakshmaiah Narayana J, Li A, Xu L, Wang G (2020) Resistome of Staphylococcus aureus in Response to Human Cathelicidin LL-37 and Its Engineered Antimicrobial Peptides. ACS Infect Dis. 6, 1866–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Falord M, Karimova G, Hiron A, Msadek T (2012) GraXSR proteins interact with the VraFG ABC transporter to form a five-component system required for cationic antimicrobial peptide sensing and resistance in Staphylococcus aureus. Antimicrob Agents Chemother 56, 1047–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Gries CM, Sadykov MR, Bulock LL, Chaudhari SS, Thomas VC, Bose JL, Bayles KW (2016) Potassium Uptake Modulates Staphylococcus aureus Metabolism. mSphere 1, e00125–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Banaei N, Kincaid EZ, Lin SY, Desmond E, Jacobs WR Jr, Ernst JD (2009) Lipoprotein processing is essential for resistance of Mycobacterium tuberculosis to malachite green. Antimicrob Agents Chemother. 53, 3799–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Radzishevsky IS, Rotem S, Bourdetsky D, Navon-Venezia S, Carmeli Y, Mor A (2007) Improved antimicrobial peptides based on acyl-lysine oligomers. Nat Biotechnol. 25, 657–9. [DOI] [PubMed] [Google Scholar]
- (54).Lakshmaiah Narayana J, Mishra B, Lushnikova T, Wu Q, Chhonker YS, Zhang Y, Zarena D, Salnikov ES, Dang X, Wang F, Murphy C, Foster KW, Gorantla S, Bechinger B, Murry DJ, Wang G (2020) Two distinct amphipathic peptide antibiotics with systemic efficacy. Proc Natl Acad Sci USA 117, 19446–19454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Heim CE, Hanke ML Kielian T (2014) A mouse model of Staphylococcus catheter-associated biofilm infection. Methods Mol Biol. 1106, 183–191. [DOI] [PubMed] [Google Scholar]
- (56).Menousek J, Mishra B, Hanke ML, Heim CE, Kielian T, Wang G (2012) Database screening and in vivo efficacy of antimicrobial peptides against methicillin-resistant Staphylococcus aureus USA300. Int. J. Antimicrob. Agents 39, 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Cho JS, Pietras EM, Garcia NC, Ramos RI, Farzam DM, Monroe HR, Magorien JE, Blauvelt A, Kolls JK, Cheung AL, Cheng G, Modlin RL, Miller LS (2010) IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J Clin Invest. 120, 1762–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Leech JM, Lacey KA, Mulcahy ME, Medina E, McLoughlin RM (2017) IL-10 Plays Opposing Roles during Staphylococcus aureus Systemic and Localized Infections. J Immunol. 198, 2352–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Kamysz E, Sikorska E, Jaśkiewicz M, Bauer M, Neubauer D, Bartoszewska S, Barańska-Rybak W, Kamysz W (2020) Lipidated Analogs of the LL-37-Derived Peptide Fragment KR12-Structural Analysis, Surface-Active Properties and Antimicrobial Activity. Int J Mol Sci. 21, 887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Makovitzki A, Avrahami D, Shai Y (2006) Ultrashort antibacterial and antifungal lipopeptides. Proc Natl Acad Sci U S A. 103, 15997–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Mishra B, Lushnikova T, Wang G (2015) Small lipopeptides possess anti-biofilm capability comparable to daptomycin and vancomycin. RSC Adv. 5, 59758–59769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).de Breij A, Riool M, Cordfunke RA, Malanovic N, de Boer L, Koning RI, Ravensbergen E, Franken M, van der Heijde T, Boekema BK, Kwakman PHS, Kamp N, El Ghalbzouri A, Lohner K, Zaat SAJ, Drijfhout JW, Nibbering PH (2018) The antimicrobial peptide SAAP-148 combats drug-resistant bacteria and biofilms. Sci Transl Med. 10, eaan4044. [DOI] [PubMed] [Google Scholar]
- (63).Dijksteel GS, Ulrich MMW, Vlig M, Nibbering PH, Cordfunke RA, Drijfhout JW, Middelkoop E, Boekema BKHL (2019) Potential factors contributing to the poor antimicrobial efficacy of SAAP-148 in a rat wound infection model. Ann Clin Microbiol Antimicrob. 18, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Su Y, Wang H, Mishra B, Lakshmaiah Narayana J, Jiang J, Reilly DA, Hollins RR, Carlson MA, Wang G, Xie J (2019) Nanofiber dressings topically delivering human cathelicidin LL-37 engineered peptides for treatment of biofilms in chronic wounds. Mol. Pharm 16, 2011–2020. [DOI] [PubMed] [Google Scholar]
- (65).Waddell WJ (1956) A simple ultraviolet spectrophotometric method for the determination of protein. J Lab Clin Med. 48, 311–4. [PubMed] [Google Scholar]
- (66).Clinical Laboratories Standards Institute (CLSI): M07-A10. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that grow aerobically; Approved Standard—Tenth Edition, 2015. [Google Scholar]
- (67).Marks LR, Clementi EA, Hakansson AP (2013) Sensitization of Staphylococcus aureus to 557 methicillin and other antibiotics in vitro and in vivo in the presence of HAMLET. PLoS One. 8, e63158. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.