

Abstract
Atopic dermatitis is a chronic inflammatory skin disease. Patients with atopic dermatitis experience inflammatory lesions associated with intense itch and pain, which lead to sleep disturbance and poor mental health and quality of life. We review the molecular mechanisms underlying itch and pain symptoms in atopic dermatitis and discuss the current clinical development of treatments for moderate‐to‐severe atopic dermatitis. The molecular pathology of atopic dermatitis includes aberrant immune activation involving significant cross‐talk among the skin and immune and neuronal cells. Exogenous and endogenous triggers modulate stimulation of mediators including cytokine/chemokine expression/release by the skin and immune cells, which causes inflammation, skin barrier disruption, activation and growth of sensory neurons, itch and pain. These complex interactions among cell types are mediated primarily by cytokines, but also involve chemokines, neurotransmitters, lipids, proteases, antimicrobial peptides, agonists of ion channels or various G protein–coupled receptors. Patients with atopic dermatitis have a cytokine profile characterised by abnormal levels of interleukins 4, 12, 13, 18, 22, 31 and 33; thymic stromal lymphopoietin; and interferon gamma. Cytokine receptors mainly signal through the Janus kinase/signal transducer and activator of transcription pathway. Among emerging novel therapeutics, several Janus kinase inhibitors are being developed for topical or systemic treatment of moderate‐to‐severe atopic dermatitis because of their potential to modulate cytokine expression and release. Janus kinase inhibitors lead to changes in gene expression that have favourable effects on local and systemic cytokine release, and probably other mediators, thus successfully modulating molecular mechanisms responsible for itch and pain in atopic dermatitis.
Keywords: atopic dermatitis, cytokines, interleukins, neuroimmunology
In this article, we review molecules that mediate the symptoms of atopic dermatitis (AD), including cytokines that signal through the Janus kinase (JAK)/Signal transducer and activator of transcription (STAT) pathway and interact with the immune and nervous systems. We describe JAK inhibitors that are in development or approved to treat AD.

Introduction
Atopic dermatitis (AD) is a chronic, relapsing, inflammatory skin disease that typically begins during childhood 1 ; however, late‐onset or adult AD can also occur. AD prevalence is dependent on age and ranges from 2% to 15%, with about 20% of cases being moderate to severe. 2
Pruritus and pain are hallmark symptoms of AD and have an adverse impact on quality of life. 3 Patient‐reported data have established that itch and pain are the most burdensome and impactful symptoms of AD. 3 Itch and pain contribute to impairment in work productivity and social dysfunction. 4 Severe AD is associated with an increased risk of death, in particular from infection, possibly because of cutaneous or extracutaneous infection or immunosuppressive treatments. 5
The pathophysiology of AD involves aberrant interactions among various cell types in the skin, and immune and nervous systems that are mediated by a host of immune‐related molecules including toll‐like receptors (TLRs) and cytokines, neurotransmitters and their cognate receptors and various other molecules involved in signal transduction that lead to itch and pain. 6 , 7 Understanding these pathways provides insight into rational therapeutic approaches to achieve disease and symptom control and potentially offers insight into preventive options when used as maintenance therapy. Cytokines are of particular interest because they are involved in cross‐talk between the immune and sensory systems. Several key cytokines signal through a family of Janus kinase/signal transducer and activator of transcription (JAK/STAT) molecules, making these an important class of drug targets. 8
Dysregulation of adaptive/innate responses in AD
Although the development of AD is not well understood, it likely involves initial interactions between the skin microbiome and the host innate immune response that further develops with the involvement of adaptive immunity. Patients with AD have abnormal innate and adaptive immune responses. 9
Signalling through TLRs expressed on cells in the skin plays a key role in host innate immune response to pathogens. Thirteen TLRs have been identified, each reactive to components of specific types of pathogens. For example, TLR2 detects various components of gram‐positive bacteria. 6 Keratinocytes express several TLRs, including TLR2, TLR3 and TLR5, which, upon activation by specific pathogens, leads to an innate proinflammatory response including production of the cytokines, tumor necrosis factor α (TNF‐α) and IL‐6. 6 Dendritic cells (DCs) in the skin express most TLRs, and plasmacytoid DCs, which infiltrate the skin on injury, express TLR7 and TLR9 that recognise nucleic acids derived from bacteria, viruses and damaged cells. 6 TLRs on DCs signal through myeloid differentiation primary response 88 (MyD88), activating protein 1 (AP‐1) and NFκB, resulting in increased expression of genes encoding proinflammatory cytokines and subsequent AD. 6 TLR‐mediated innate immune activation in DCs also results in recruitment of macrophages and T cells, and AD subsequently occurs when excess inflammation is unresolved. 6 Thus, TLRs are at the border of innate and adaptive immunity. 6
The pathophysiology of AD is characterised by a prominent, abnormal Th2 immune response. Langerhans cells in the epidermis recognise foreign antigens and prime T cells as part of the host defence. 6 Activation of TLR2 and production of IL‐4 induce keratinocytes to produce thymic stromal lymphopoietin (TSLP), which is a key regulator of the Th2 response. TLR2 and TSLP link the innate and Th2‐skewed adaptive immune responses in patients with AD, leading to worsening and persistence of the disease. 6 TSLP also activates T cells, DCs and mast cells. 10 TSLP promotes itch via the TSLP receptor by activating cutaneous sensory neurons that express transient receptor potential ankyrin 1 (TRPA1) ion channels. 11 The long‐term and short‐term effects of TSLP on itch may be distinct, with acute effects involving activation of sensory neurons and chronic effects involving recruitment of various immune cells including macrophages and T cells. 11
Deviant Th1, Th17 and Th22 responses are also involved to varying degrees in certain AD endotypes (e.g. paediatric and Asian subtypes). 12 Th1‐mediated dysregulation has been implicated in the chronic form of AD. 2 Abnormal TLR2 activation may also induce Th1 responses and TSLP production by keratinocytes, driving AD pathology by linking the innate immune response to pathogens at the skin barrier with abnormal Th2 adaptive immune responses. 6 Th2/Th17 expression is especially prominent in African American patients with AD (Th1, Th17 and Th22). 13
Some cytokines and receptors involved in the innate and adaptive immune response modulate the sensation of itch and pain in AD via TRP ion channels. 14 In mast cell–deficient mice with transgenic expression of IL‐13 in skin, IL‐13 increases expression of TRPA1 ion channels in dorsal root ganglia (DRGs) and mast cells and induces itch. 15 Recruitment of T cells to AD lesions, production of IL‐31 and subsequent activation of sensory neurons may drive and exacerbate itch. 16 Sensory neurons that express transient receptor potential vanilloid (TRPV) 1‐4 17 , 18 and IL‐31 receptor also express TRPV1 and TRPA1 ion channels, thus linking T cell–mediated and sensory nerve–mediated itch. 19 Neurons activated by cytokines, in turn, release neurotransmitters and neuropeptides that affect innate and adaptive immune cells. 20 Thus, cytokines released from skin and immune cells have a direct impact on neurons via TRP ion channels, the activation of which produces itch.
Cytokines in AD: role in itch, pain and skin barrier dysfunction
Early events in AD
Key cytokines implicated and upregulated in AD include IL‐4, IL‐13, IL‐31 and others. 2 Although AD is primarily a disease of Th2 dysregulation, with IL‐4, IL‐13, IL‐31 and TSLP being key players, cytokines such as interferon (IFN)‐γ (Th1), IL‐17, IL‐22 (Th17) and IL‐33 are also involved. 21 Key cytokines that play a role in the early pathophysiology of itch, pain and disruption of the skin barrier in patients with AD are described below and summarised in Table 1.
Table 1.
Key cytokines involved in atopic dermatitis
| Cytokine | Cellular origin/stimulus | Cytokine receptor | Cell type(s) expressing the receptor | Overall function/AD MOA, itch, pain | References |
|---|---|---|---|---|---|
| IL‐4 | ILC2, CD4+ T cells, eosinophils, basophils | IL‐4Rα, γC | Sensory neurons | Development of Th2 cells; ↓EDC molecules; ↑itch, IL‐31, MHC class II; activate sensory neurons | 22, 24, 40, 41, 114, 141 |
| IL‐13 | Mast cells, basophils, eosinophils, ILC2, CD4+ T cells | IL‐4Rα, IL‐13rα1 | Sensory neurons | Induce Th2 response; ↑IL‐31, TRPA1, branching, itch; ↓filaggrin; activate sensory neurons | 2, 24, 40 |
| IL‐22 | Mast cells | IL‐10 family | ↓Filaggrin, claudin‐1; ↑GRP, TSLP, IL‐33, itch | 29, 30 | |
| IL‐31 | T cells, DCs, Th2 cells, skin cells, eosinophils, basophils, mast cells | IL‐31R, OSMR | DRGs, sensory nerves, leukocytes, keratinocytes | ↑NKB, itch, BNP, branching; ↓filaggrin; stimulate sensory neurons via TRPA1 | 19, 32, 35, 40, 80 |
| TSLP | Keratinocytes/in response to proteases and bacteria | TSLPR, IL‐7Rα | Sensory nerves, ILC2 | ↑IL‐31, IL‐33, itch; activate sensory neurons via TRPA1, T cells, DCs, mast cells; stimulate ILC2; link innate and adaptive immune responses | 2, 10, 11, 39, 50, 142 |
| IL‐33 | Keratinocytes, macrophages, DCs | IL‐33R, ST2 | Astrocytes, mast cells, ILC2 | ↑IL‐4, IL‐13, IL‐33, TNF‐α, CXCL8, PGD2, itch | 44, 49, 50 |
| IL‐17 | CD4+ T cells | IL‐17RA | Skin cells, ILC2 | ↑GM‐CSF | 48, 50, 54 |
| IL‐25 | Keratinocytes | IL‐17RB | ILC2 | ↑IL‐13 | 49, 50 |
| IL‐18 | Keratinocytes, macrophages, DCs | IL‐18R | Th1 cells | ↑IFN‐γ, ↑IL‐4, chemokines, inflammation, skin barrier dysfunction, skin pain | 52 |
| GM‐CSF | Keratinocytes, T cells, B cells, NK cells, monocytes/macrophages, fibroblasts, mast cells | GM‐CSFRα | Keratinocytes | Induces Th2 response | 54 |
↓, decreased; ↑, increased; AD, atopic dermatitis; BNP, brain natriuretic peptide; DC, dendritic cell; DRG, dorsal root ganglion; EDC, epidermal differentiation complex; GM‐CSF, granulocyte‐macrophage colony‐stimulating factor; GM‐CSFR, granulocyte‐macrophage colony‐stimulating factor receptor; GRP, gastrin‐releasing peptide; IL, interleukin; ILC2, type 2 innate lymphoid cells; MOA, mechanism of action; NKB, neurokinin B; OSMR, oncostatin M receptor; PGD2, prostaglandin D2; ST2, stromal cell line; TNF, tumor necrosis factor; TRPA, transient receptor potential ankyrin; TSLP, thymic stromal lymphopoietin; TSLPR, thymic stromal lymphopoietin receptor.
IL‐4
IL‐4 is produced by mast cells, type 2 innate lymphoid cells (ILC2), CD4+ T cells, eosinophils and basophils and facilitates Th2 cell development. 22 In AD lesions, IL‐4 downregulates multiple genes involved in innate defence, including genes in the epidermal differentiation complex critical for epidermal barrier function. 21 IL‐4 activates/sensitises IL‐4Ra on sensory nerve endings and induces JAK‐STAT signalling, thereby contributing to neuroinflammation and itch. 23 , 24
IL‐13
IL‐13 is produced by basophils, mast cells and eosinophils. 25 IL‐13 along with IL‐4 are major players in induction of the Th2 response and blocking expression of the barrier protein filaggrin, leading to skin barrier dysfunction. 2 Sensory neurons are activated by IL‐4 and IL‐13, which sensitise small‐diameter sensory neurons to other inducers of itch, including histamine, chloroquine, TSLP, protease‐activated receptor 2 (PAR‐2), leukotrienes and IL‐31. 24 , 26 , 27 IL‐13Ra1 is primarily responsible for the IL‐13–mediated role in itch, whereas IL‐13Ra2 is probably involved in neuroinflammation; its role in itch or pain is not yet clear. 28
IL‐22
IL‐22, which is increased in patients with AD, is produced by mast cells 29 and downregulates expression of filaggrin and the tight junction protein claudin‐1. 30 High levels of IL‐22 are also produced by circulating CD4+ and CD8+ T cells in patients with prurigo nodularis, which often presents as a manifestation of AD in African Americans. 31 IL‐22 also upregulates expression of the pruritogenic peptide gastrin‐releasing peptide (GRP) in sensory neurons and expression of TSLP and IL‐33 in epithelial cells and increases itch and AD‐like disease in mice. 30 Whether IL‐22–induced keratinocyte activation leads to the release of itch mediators is not yet clear.
IL‐31 and oncostatin M
T cell–derived pruritogenic IL‐31 is upregulated in AD, and DRGs express high levels of the IL‐31 receptor, which signals through extracellular signal‐regulated kinase to induce itch. 16 , 19 , 32 Other cell types that express IL‐31 include keratinocytes, eosinophils, basophils, mast cells, DCs and monocytes. 16 , 33 IL‐31 induces the expression of neurokinin B, which is required for IL‐31–mediated itch in DRG neurons. 34 IL‐31 downregulates filaggrin expression. 35 Consistent with a role for IL‐31 in itch, a phase 2 trial of nemolizumab, a humanised antibody targeting IL‐31 receptor A, demonstrated significant itch reduction in patients with moderate‐to‐severe AD. 36 In addition, promising results have been seen with nemolizumab in prurigo nodularis. 37
Oncostatin M (OSM), which is produced by dermal T cells and monocytes, is a highly upregulated cytokine in AD and is associated with chronic itch. OSM is a non‐canonical inducer of itch. Rather than activating sensory neurons, OSM sensitises neurons by increasing neural excitability and responses to pruritogens. The OSM receptor (OSMR; shared with the IL‐31 receptor) is expressed by natriuretic polypeptide B–positive itch‐selective neurons. OSMR knockout in DRG neurons decreases OSM‐mediated itch in mice, and pharmacologic inhibition of OSMR decreases itching in experimental inflammatory dermatitis in mice. 38
Thymic stromal lymphopoietin
In response to proteases and bacteria on the skin surface, keratinocytes produce cytokines including TSLP, 2 , 39 which promotes itch by directly activating sensory neurons. 40 TSLP stimulates ILC2, which bridges the innate and adaptive immune response in AD and secretes type 2 cytokines including IL‐4, IL‐5 and IL‐13. 41 Protease PAR‐2 activation stimulates release of TSLP from keratinocytes, which activates TSLP receptor (TSLPR) on sensory nerve endings, thereby inducing pruritus. 11 Although clinical trials of antibodies against TSLP/TSLPR have been successful in treating asthma, 42 but not yet in AD, 43 the beneficial role of TSLP/TSLPR blockage in treating itch and AD cannot be fully excluded.
IL‐33
IL‐33 is produced by keratinocytes, endothelial cells and immune cells; is upregulated in AD; and is an early ‘danger alarmin’ that activates the innate immune system. 44 IL‐33 enhances expression levels of IL‐4 and IL‐13, which in turn stimulate synthesis of the itch‐producing IL‐31. 2 , 44 In addition, IL‐33 stimulates basophils to activate ILC2 via IL‐4, and both cytokines can also induce itch. Conversely, itch mediators such as IL‐31, substance P and brain natriuretic peptide (BNP) can also stimulate release of cytokines and chemokines that are involved in inflammation, barrier dysfunction, itch and pain. 16 , 40 , 45 , 46 , 47
IL‐17
IL‐17 is produced by CD4+ T cells, regulates the expression of chemokines by keratinocytes, exacerbates AD and is found to be significantly increased in the skin of African Americans with lesional AD. 13 , 48 Whether IL‐17 cytokine family members are involved in pain or itch in patients with AD is not clear yet.
IL‐25
Stimulated keratinocytes secrete IL‐25, which is upregulated in AD lesions. 49 , 50 IL‐25 and IL‐33 induce secretion of type 2 cytokines (IL‐5, IL‐9 and IL‐13) from ILC2s, further perpetuating the pathophysiology of AD, including skin barrier dysfunction. 50 Because IL‐25 and IL‐33 are involved in eosinophil recruitment, both cytokines may be involved in eosinophil‐associated itch in patients with AD. 51
IL‐18
IL‐18 is expressed by keratinocytes, is increased in patients with AD 52 and contributes to inflammation and skin barrier dysfunction. IL‐18 increases IFN‐γ production and subsequent chemokine secretion by keratinocytes. 53 Whether IL‐18 is associated with TSLP‐mediated itch from keratinocytes or pain is not known.
Granulocyte‐macrophage colony‐stimulating factor
Granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) is produced by keratinocytes in patients with AD at higher levels than keratinocytes in healthy controls. 54 GM‐CSF plays a role in inducing the Th2 immune response in patients with AD, and its production may be mediated by the proinflammatory PAR‐2 and induced by IL‐17. 54 Thus, in the early stages of AD pathology, multiple cytokines produced by various immune and skin cells not only mediate the inflammatory symptoms of AD but also drive itch, pain and skin barrier dysfunction (Figure 1). Thus, current literature indicates that GM‐CSF is primarily indirectly involved in itch or pain in AD.
Figure 1.
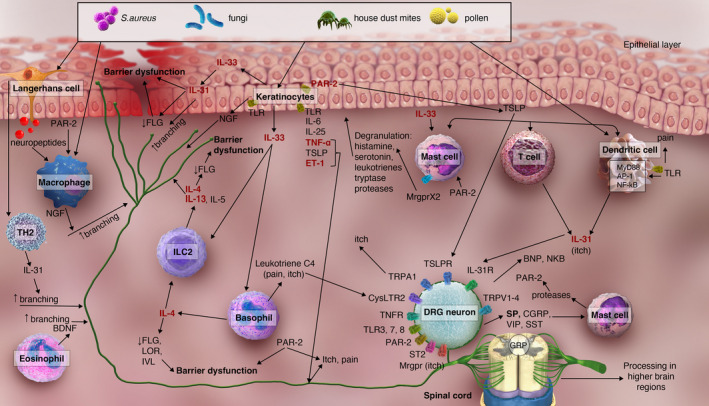
The cytokine network in atopic dermatitis with effects by and on various cell types, with a focus on neuroinflammation, itch and pain. Cytokines in red/bold text are upregulated in atopic dermatitis (AP‐1, activating protein 1; BDNF, brain‐derived neurotrophic factor; BNP, brain natriuretic peptide; CGRP, calcitonin gene‐related peptide; CysLTR2, leukotriene receptor; DRG, dorsal root ganglion; ET‐1, endothelin 1; FLG, filaggrin; GRP, gastrin‐releasing peptide; IL, interleukin; IL‐31R, IL‐31 receptor; ILC2, innate lymphoid cell 2; IVL, involucrin; LOR, loricrin; Mrgpr, Mas‐related G protein–coupled receptor; MrgprX2, Mas‐related G protein–coupled receptor X2; NGF, nerve growth factor; NKB, neurokinin B; PAR‐2, proteinase‐activated receptor 2; S. aureus, Staphylococcus aureus; SP, substance P; SST, somatostatin; ST2, IL‐33 receptor; TLR, toll‐like receptor; TNF, tumor necrosis factor; TNFR, TNF receptor; TRPA1, transient receptor potential ankyrin 1; TRPV4, transient receptor potential vanilloid 4; TSLP, thymic stromal lymphopoietin; TSLPR, thymic stromal lymphopoietin receptor; VIP, vasoactive intestinal peptide).
Cellular mediators of itch and pain in AD
Different cell types mediate longer‐term events in patients with AD and perpetuate itch and pain. Specialised cells such as Schwann cells in the peripheral nervous system and astrocytes or oligodendrocytes in the central nervous system (CNS) communicate with immune cells to mediate chronic itch and pain. 55 , 56 IL‐33 binds to its receptor expressed on astrocytes and induces production of proinflammatory cytokines via JAK2/STAT3 signalling, thus exacerbating itch. 57 Intrathecal administration of the astrocyte inhibitor, l‐α‐aminoadipate, attenuates chronic itch. 58
Glial cells also play a role in neuropathic pain. In the peripheral nervous system, satellite glial cells surround DRG cell bodies, are activated after nerve injury and play a role in the initiation and maintenance of neuropathic pain. 59 Afferent pain signals lead to secretion of adenosine triphosphate (ATP), which induces satellite glia to produce matrix metalloproteinase 9 (MMP‐9), nerve growth factor (NGF) and ATP, which in turn activate DRG neurons and result in peripheral sensitisation. 60 Schwann cells, which myelinate peripheral axons, are activated when TLR2 recognises pathogens and subsequently produce chemokines and cytokines including proinflammatory TNF‐α and IL‐6. Activation of TRPA1 in Schwann cells maintains pain sensation. 59 In the CNS, activated astrocytes produce proinflammatory molecules including CXCL1 and CCL2 that sensitise dorsal horn neurons, 60 and intrathecal injection of TNF‐α–activated astrocytes induces pain. 61 Factors secreted upon afferent pain signalling such as ATP, calcitonin gene‐related peptide (CGRP) and MMP activate microglia. 60 Microglial activation is typically early and transient compared with astrocytic activation, and spinal cord microglia produce proinflammatory cytokines that also sensitise dorsal horn neurons to pain. 57
Keratinocytes secrete TSLP and endothelin‐1 (ET‐1), thus activating nerves, inducing itch and disrupting the skin barrier function. Keratinocytes also secrete NGF, which leads to hyperinnervation, increased sensitivity and barrier dysfunction. 46 , 62 Proinflammatory IL‐33 secreted by keratinocytes stimulates mast cells to produce numerous factors including TNF‐α and prostaglandin D2. 51 Mast cells also undergo degranulation in response to sensory neuron‐derived substance P through the neurokinin receptor and Mas‐related G protein–coupled receptor (Mrgpr)X2. 62 Macrophages express NGF and can respond to neuropeptides and thus are part of the bidirectional cross‐talk between the immune and nervous systems. 62 Activated basophils show increased production of leukotriene C4, which directly activates sensory neurons via the cysteinyl leukotriene receptor 2 (CysLTR2) that acts via TRPV1 and TRPA1, leading to mast cell–dependent and basophil‐dependent acute itch (i.e. itch flares). 63 , 64 Thus, multiple cell types, including but not limited to immune and nerve cells, play roles in chronic stages of the pathology of AD (Figure 1).
Neuroinflammation and the role of cytokines in increasing itch sensitivity via neurotrophism
Neuroinflammation includes trafficking of leukocytes into the CNS and peripheral nervous system (PNS), roles for satellite glial cells and secretion of proinflammatory molecules, leading to skin barrier dysfunction, which in turn induces or aggravates itch and pain. 7 , 65
Several cytokines increase sensitivity to stimuli by inducing branching and growth of sensory afferents (i.e. neurotrophism). Growth of sensory nerves including dermal neuropeptide‐positive fibres occurs in response to IL‐31 66 and IL‐13. 15 IL‐31 may also increase sensitivity to subthreshold stimuli and induce itch. 21 In mice with transgenic expression of IL‐22 in the skin, GRP‐positive neurons in the DRG that innervated the skin were increased compared with wild‐type mice, and these mice showed intense scratching. 30 Abnormal plasticity of peripheral sensory neurons contributes to the sensory perception of pain and itch and is present in patients with chronic pruritus, including those with AD; however, this was not because of hyperinnervation. 7 , 67
Other molecules involved in itch and pain
PAR‐2
Sensory nerve cells and keratinocytes express PAR‐2, which is cleaved by proteases released by degranulated mast cells. 68 These effects are mediated by substance P and CGRP that are expressed by DRG neurons. Tryptase and PAR‐2 are increased in the skin of patients with AD, 69 and PAR‐2 overexpression in keratinocytes is sufficient to induce lesions resembling AD. 70 PAR‐2 activates TRPV4 channels in DRG neurons 71 and NFκB in keratinocytes. 72 Thus, following release of proteases by mast cells, cleaved PAR‐2 induces CGRP‐ and substance P–dependent neurogenic inflammation, itch and pain in patients with AD in a manner that also involves signalling via NFκB. 68 , 71 , 72 Exogenous trigger factors such as allergens (house dust mite allergens) and endogenous trigger factors can activate PAR‐2, thereby contributing to itch and/or pain. 70 These exogenous and endogenous factors are poorly understood.
Ion channels
Neuronal TRP calcium ion channels modulate neuroimmune interactions and mediate itch and pain. 19 TRPA1 and TRPV1 are major mediators of IL‐31–induced itch, 19 and a TRPV1 antagonist attenuates AD and itching. 73 TRP channels can be activated by bacteria such as Staphylococcus aureus, thereby inducing itch and pain in patients with AD. 74 Chemokines are involved in itch and pain owing to activation of chemokine receptors, which are G protein–coupled receptors (GPCRs). 75 The chemokines CXCL1 and CXCL2 modulate TRPV1 and reduce TRPV1 desensitisation in DRGs. 75 IL‐13 activates TRPA1, leading to itch. TRPA1 is increased in dermal sensory fibres, DRGs, mast cells and keratinocytes and is involved in pain sensation. 15 TRPV3 is expressed in keratinocytes and is overactive in AD model mice. Blocking TRPV3 attenuates itch and AD. 76 Other ion channels involved in cutaneous itch and pain include acid‐sensing ion channels, 77 potassium channels 78 as well as NaV1.7, NaV1.8 and NaV1.9 sodium channels. 79 Thus, ion channels play a role in itch and pain signalling.
Neurotransmitters
Multiple neurotransmitters play a role in itch and pain signalling. Neuropeptide Y reduces IL‐31–mediated itch via presynaptic inhibition of afferent neurons. 80 Substance P and NGF promote itch. Stimulated nerve fibres, which secrete substance P, vasoactive intestinal peptide and somatostatin, activate mast cells, which regulate nerve function. This positive feedback loop is mast cell dependent and TRPA1 dependent. 15 Substance P is increased in AD and induces production of IFN‐γ, IL‐4, IL‐10 and TNF‐α, and treatments targeting substance P may be useful for AD. 81 TNF‐α is also increased in AD 46 and potentiates TSLP‐dependent calcium ion flow in DRGs, increasing the sensation of itch and indicating synergism between these cytokines. 46 In addition to direct induction of itch, IL‐31 increases the secretion of BNP in DRGs and the skin, leading to increased secretion of other cytokines, chemokines and proteases in the skin and thus coordinating signalling that evokes itch. 45 ET‐1 induces secretion of BNP and activates sensory neurons, various immune cells and keratinocytes. 46 BNP induces itching, and inhibition of the BNP receptor, natriuretic peptide receptor 1, attenuates itch. 82 Neuronal sensory fibres and eosinophils express brain‐derived neurotrophic factor (BDNF), the levels of which are higher in the skin of patients with AD than in patients with normal skin. 83 Eosinophil‐derived BDNF appears to play a role in inducing branching of DRG neurons. 83 BDNF promotes neurite outgrowth via JAK/STAT signalling. Thus, various secreted molecules work together to facilitate itch and afferent sensitivity in patients with AD.
G protein–coupled receptors
Several GPCRs also mediate itch and pain, including Mrgpr family members and PAR‐2, as mentioned previously. 68 , 72 Neuropeptide Y, 80 mu opioids, 84 prostaglandin D2 85 and substance P signal through GPCRs. 86 The G protein–coupled oestrogen receptor modulates the effects of a TNF‐α–induced protein called A20 that is increased by formononetin, which attenuates AD. 87 TRPA1, which is involved in itch and pain sensation, is activated downstream of GPCRs. 15 Activation of GPCRs in sensory satellite glia directly modulates neuronal activity and induces analgesia. 65 Thus, GPCRs are crucially involved in itch and pain signalling in AD.
Itch, pain and the afferent nervous system
The afferent nervous pathways for itch and pain are distinct but also overlap and involve both the PNS and CNS. 88 , 89 Different pruritogens activate specific groups of afferent neurons that express their cognate receptor (e.g. substance P binds MrgprX, and the PAR‐2 agonist peptide binds PAR‐2). 90
In the PNS, itch is mediated by C‐fibres and Aδ fibres, and pain is mediated by Aβ, Aδ and C‐fibres. 91 Other cells are also involved in itch and pain such as Merkel cells, which are present in the basal portion of the epidermis and transmit touch and pressure sensations to sensory fibres. 91 Itch‐specific small‐diameter sensory neurons in mice that directly detect pruritogens express MrgprA3, MrgprC11 and MrgprD. 89 However, neurons that respond to both pain and itch have also been identified. 89 RNA‐sequencing analysis has led to identification of multiple subtypes of sensory neurons, including multiple itch pathways, one of which involves IL‐31 and leukotrienes. 92 Differences have been identified among mouse, human and non‐human primate DRGs. 93 , 94 , 95 A subset of itch‐selective DRGs was identified with BNP expression, 96 highlighting the differences among species.
In the CNS, itch circuits and pain circuits are present in the spinal cord and brain. 47 Peripheral itch‐specific sensory neurons project to GRP receptor‐positive neurons in the spinal cord. 89 A common population of dorsal horn spinal cord neurons responds to pain and itch stimuli. 19 The sensation of itch or pain may depend on population coding in which the combination of fibres that are activated determines the quality (itch or pain) of the sensation 88 , 97 and/or on higher brain processing of the signals downstream of the primary sensory neurons. 19 The hippocampus, amygdala and hypothalamus are activated by both pain and itch sensations. 89
More than half of patients with AD report skin pain. 98 In patients with AD, pain is modulated by IL‐12 and IL‐18 (which are upregulated in AD), 52 prostaglandins, leukotrienes and MMPs. 99 , 100 Additional pain mediators include bradykinin, ATP, P2X purinoceptor 3 agonists, proteases, TLR agonists, cytokines (IL‐1, IL‐6, IL‐8, IL‐17, IL‐33, TSLP and TNF‐α), neurotrophins including NGF, ET‐1, CGRP, MrgprX1, MrgprX2 and MrgprD and ion channels (TRPVs, TRPA1, acid‐sensing ion channels, potassium ions and sodium ions). 60 , 77 , 101 , 102 , 103 , 104 , 105 , 106 The activity of many of these molecules is mediated by various populations of primary afferent fibres (e.g. peptidergic and nonpeptidergic) that synapse in different layers of the dorsal horn of the spinal cord. Interneurons in the dorsal horn modulate the signal travelling from spinal cord projection neurons to higher brain regions. 107
Another mechanism of chronic itch and pain in skin involves descending central inhibition in which neural pathways from the brain inhibit pain or itch sensation of a normally nonpainful or non‐itch‐invoking stimulus. This system is impaired in patients with chronic itch. 7 , 67 Impaired descending central inhibition involves the spinal cord, brain stem and cortex and leads to central sensitisation, producing chronic pain and chronic itch 7 induced by stimuli such as touch, clothing or warmth that normally do not induce these sensations. 108 Thus, itch and pain in patients with AD result from aberrant cytokine expression, subsequent effects on the CNS and PNS and impaired neurologic function.
Neuroimmune cross‐talk controlling itch and pain in AD
The immune system and nervous system show extensive bidirectional cross‐talk. The peripheral sensory and autonomic nervous system, which innervates the skin, has cross‐talk with mast cells, DCs, Th2 cells and other immune cells. 20 Activated neurons release neurotransmitters and neuropeptides that affect innate and adaptive immune cells. 20 In turn, immune cells release cytokines that activate peripheral nerves that mediate itch. 20 Neuropeptide Y, which is expressed by dorsal horn interneurons and acts through the inhibitory Y2 receptor expressed on the central terminals of primary afferents, decreases IL‐31‐mediated itch. 80 IL‐31 and TSLP are also neuroimmune links. 11 , 19 Mast cells, eosinophils and basophils interact with peripheral nerves to mediate skin inflammation, pain and itch in patients with AD via IL‐4, IL‐13, IL‐31, OSM, neuropeptides and substance P. 25 , 38 Thus, the bidirectional communication between the nervous system and immune system can work to amplify the pathologic state in patients with AD (Figure 1).
Therapeutic approaches in AD
JAK signalling
Multiple cytokines implicated in AD signal through JAK and various STATs (Figure 2). 8 For example, IL‐4, IL‐13, IL‐31, TSLP, IL‐5, IL‐22 and IFN‐γ signal through JAK1. 109 , 110 , 111 , 112 As reviewed above, JAK signalling also plays a role in cell signalling pathways in peripheral nerves that contribute to itch and pain, thus providing a rationale for the use of JAK inhibitors (JAKi) to treat AD (Figure 3). IL‐4 and IL‐13, signalling through all members of the JAK family and STAT6 homodimers, decrease expression of genes involved in keratinocyte differentiation 111 , 113 and that of the antimicrobial genes human beta‐defensins (HBD)‐2 and HBD‐3. 114 IL‐4 also affects MHC class II expression, and IL‐4 and IL‐13 affect Th2 cell differentiation (via changes in GATA‐3 expression) and IgE class switching. 114
Figure 2.
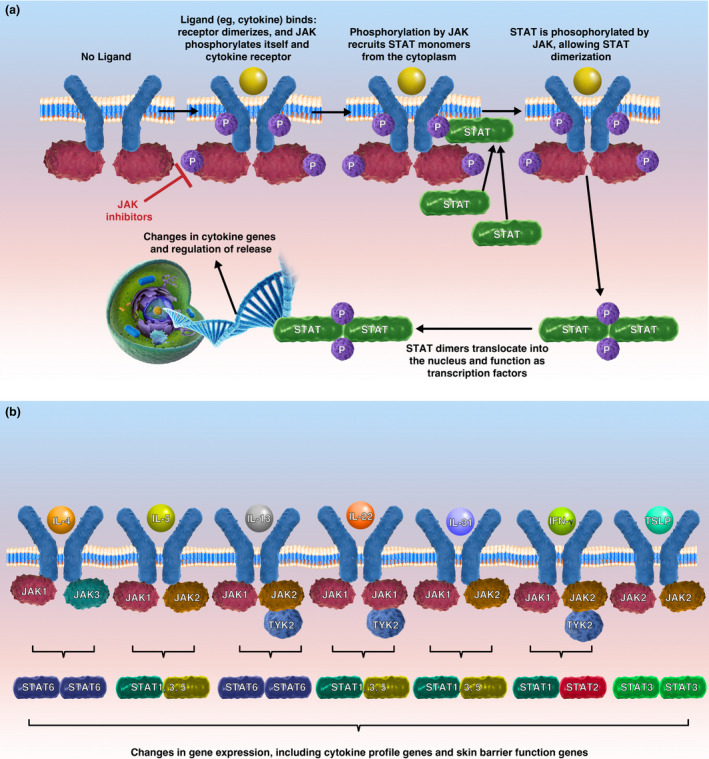
JAK/STAT signalling. (a) Upon ligand binding (e.g. cytokines), the cytokine receptor dimerises, and JAK phosphorylates itself and the intracellular region of the cytokine receptor. Phosphorylation of the receptor recruits STAT monomers from the cytoplasm. JAK phosphorylates STAT, which then forms a dimer. STAT dimers translocate into the nucleus where they act as transcription factors and impact gene expression, including expression of cytokine genes involved in inflammation, epidermal skin barrier, itch and pain. JAK inhibitors interact with the P‐loop of the JAK kinase domain, 148 and hence inhibit events downstream of JAK phosphorylation (JAK, Janus kinase; P, phosphorylation; STAT, signal transducer and activator of transcription). (b) Key cytokines in atopic dermatitis and the JAK and STAT molecules that mediate signalling (IL, interleukin; IFN, interferon; TSLP, thymic stromal lymphopoietin).
Figure 3.
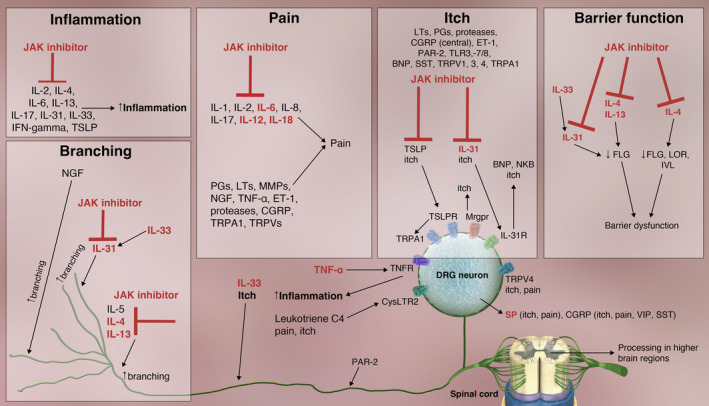
Effects of JAK inhibitors on neuroimmune circuits and barrier dysfunction in atopic dermatitis. Cytokines in red/bold text are upregulated in atopic dermatitis (BNP, brain natriuretic peptide; CGRP, calcitonin gene‐related peptide; CysLTR2, cysteinyl leukotriene receptor; DRG, dorsal root ganglion; ET‐1, endothelin 1; FLG, filaggrin; IFN, interferon; IL, interleukin; IL‐31R, IL‐31 receptor; IVL, involucrin; JAK, Janus kinase; LOR, loricrin; LT, leukotriene; MMP, matrix metalloproteinase; MrgprX1, Mas‐related G protein–coupled receptor X1; MrgprX2; Mas‐related G protein–coupled receptor X2; MrgprD, Mas‐related G protein–coupled receptor‐D; NGF, nerve growth factor; NKB, neurokinin B; PAR‐2, proteinase‐activated receptor 2; PG, prostaglandin; SP, substance P; SST, somatostatin; TLR, toll‐like receptor; TNF, tumor necrosis factor; TNFR, tumor necrosis factor receptor; TRPA1, transient receptor potential ankyrin 1; TRPV, transient receptor potential vanilloid; TSLP, thymic stromal lymphopoietin; TSLPR, thymic stromal lymphopoietin receptor; VIP, vasoactive intestinal peptide).
IL‐4 signals through JAK1 and JAK3 and STAT6 homodimers, resulting in downregulation of the expression of several genes in the epidermal differentiation complex including filaggrin (FLG), loricrin (LOR) and involucrin (IVL). 21 , 114 The use of JAKi may mitigate these effects. For example, the JAK1/3 inhibitor, tofacitinib, increases expression of DSC1, FLG and KRT1. 115
IL‐31 signals through JAK1 and JAK2 109 and induces changes in pro‐opiomelanocortin, which regulates itch in AD, 116 and downregulates filaggrin expression, resulting in disrupted barrier function. 35 IL‐31 also leads to increased secretion of other cytokines, chemokines and proteases in the skin to coordinate signalling that leads to itch. 45
IL‐33 does not signal through JAK, but induces secretion of IL‐31, TNF‐α and CXCL8. 117 , 118 Enhanced JAK signalling via elevated proinflammatory cytokines and subsequent changes in gene expression downstream of JAK signalling propagate this dysregulated immune response and exacerbate the abnormal cytokine profile.
In addition to direct effects, JAKi can indirectly suppress cytokines such as IL‐17 that do not signal through JAK/STAT by inhibiting other JAK/STAT‐dependent cytokines such as IL‐23. 8 Favourable alterations in gene expression by JAKi are also expected to lead to improvements in barrier function, pain and itch. Decreased expression of genes in the epidermal differentiation complex by IL‐4 decreases epidermal barrier function, 114 and JAKi are expected to help restore barrier function by blocking these decreases (Figure 3). Descending central inhibitory pathways are abnormal in patients with AD and in those with rheumatoid arthritis. JAKi, which are used to treat patients with rheumatoid arthritis, attenuate central pain processing that is overactive owing to chronic inflammation. 119 Thus, the use of JAKi in patients with AD is expected to restore the cytokine profile to a more normal state, improve barrier function and itch and decrease pain by affecting CNS processing.
Preventive and other approaches
The disease course of AD is often relapsing and remitting. 120 The goals of treatment include prevention of flares to help control pain and itch and to repair and maintain a functional skin barrier. 120 , 121 Preventive treatments for AD include various topical therapies as well as systemic options including phototherapy. 122 Topical treatment options include topical steroids, calcineurin inhibitors, phosphodiesterase‐4 inhibitors, JAK/STAT inhibitors and wet wrap therapy with topical steroids. 2 , 120 Occasionally, antibiotics or antiseptics are used to treat infected lesions. 121
Clinical overview and differentiation
Patient‐reported outcomes are important for selecting therapy for AD. 3 , 123 Patients with uncontrolled disease report higher burdens of itch, psychological comorbidities and sleep deprivation. 124 Inadequate disease control is common among those with moderate‐to‐severe AD, highlighting the need for more effective treatments. 1 , 124 Several inhibitors of the JAK/STAT signalling pathway, both oral and topical, have been developed or are currently under development (Table 2). 2 In patients with inadequate disease control, systemic options that target multiple cytokines in AD may afford more effective disease control than topical interventions. Here, we explore the rationale for emerging systemic treatment options for AD by considering all signs and symptoms.
Table 2.
Current and potentially future treatment options for atopic dermatitis
| Drug | Target(s) | Mode of administration | Development status | Comment | References |
|---|---|---|---|---|---|
| Baricitinib | JAK1/2 | Oral | Approved in the European Union for moderate‐to‐severe AD | Increase in number of patients achieving EASI‐50 at 16 weeks; treatment effect seen at 4 weeks | 12, 131 |
| Abrocitinib | JAK1 | Oral | Approved in several countries for moderate‐to‐severe AD | Improvements in EASI, IGA, EASI‐75, EASI‐90 and PP‐NRS | 132, 133, 134, 137, 138, 139 |
| Upadacitinib | JAK1 | Oral | 3 phase 3 trials completed; approved in the European Union and United States | About half of patients achieved EASI‐75 and IGA 0/1 | 12, 135, 136 |
| ASN002 | JAK1, 2, 3, TYK and SYK | Oral | Phase 1b | Improvement in itch starting on day 8; 83–100% of patients achieved EASI‐50 at the 2 highest doses | 12 |
| Delgocitinib | All JAKs | Topical | Approved in Japan for AD; FDA Fast‐Track Designation for chronic hand eczema | Significant improvements in modified EASI | 125 |
| Tofacitinib | JAK1/3 | Topical | Phase 2a | Off‐label; significant changes in EASI PGA, BSA and pruritus | 143 |
| Ruxolitinib | JAK1/2 | Topical | Approved for mild‐to‐moderate AD in the United States | Off‐label; IGA treatment success achieved; improvements in itch | 126, 144 |
| Roflumilast | PDE4 | Topical | Phase 2a | No significant changes in SCORAD, TEWL or pruritus | 145 |
| Tapinarof | AHR | Topical | Phase 2b | Week 12 IGA responses, EASI scores, pruritus and POEM improved; %BSA reduced | 146 |
| Dupilumab | IL‐4Rα | Subcutaneous injectable | FDA approved | ≤ 50% decrease in PP‐NRS at week 16; itch improvement as early as 1–3 days | 141 |
| Tralokinumab | IL‐13 | Subcutaneous injectable | Approved in the European Union and United States for moderate‐to‐severe AD | Improvements in IGA, pruritus, sleep interference, DLQI, POEM and SCORAD | 129, 130, 147 |
| Nemolizumab | IL‐31 | Subcutaneous injectable | Phase 2 | Significant changes in pruritus at week 12; 70–80% change in pruritus score after 64 weeks | 12, 36 |
| Tezepelumab | TSLP | Subcutaneous injectable | Phase 2 | Non‐statistically significant improvement in EASI‐50 for tezepelumab group vs placebo | 12, 43 |
| GBR 830 | OX40 | Intravenous infusion | Phase 2 | Increase in number of patients achieving EASI‐50 | 12 |
| Etokimab | IL‐33 | Phase 2a | All patients achieved EASI‐50 or better; however, primary endpoint not met in a phase 2b trial | 12 | |
| Fezakinumab | IL‐22 | Intravenous infusion | Phase 2a | Improvements in SCORAD and IGA in patients with severe, but not moderate, AD; improvements in %BSA | 12 |
%BSA, percent of body surface area; AD, atopic dermatitis; AHR, aryl hydrocarbon receptor; DLQI, Dermatology Life Quality Index; EASI, Eczema Area and Severity Index; EASI‐50, −75 and −90, 50%, 75% and 90%, respectively, improvement in EASI score from baseline; FDA, Food and Drug Administration; IGA, Investigator’s Global Assessment; IL, interleukin; JAK, Janus kinase; PDE, phosphodiesterase; POEM, Patient‐Oriented Eczema Measure; PP‐NRS, Peak Pruritus Numerical Rating Scale; SCORAD, SCORing Atopic Dermatitis; SYK, spleen tyrosine kinase; TEWL, transepidermal water loss; TSLP, thymic stromal lymphopoietin; TYK, leukocyte receptor tyrosine kinase.
Topical treatment options
The topical pan‐JAKi, delgocitinib (JTE‐052), improves barrier function by blocking IL‐4/IL‐13/JAK/STAT3/6–mediated signalling and subsequent chemokine expression by keratinocytes. 113 Delgocitinib is effective in improving the Eczema Area and Severity Index (EASI) score. 125 The topical JAKi ruxolitinib is approved for the treatment of mild‐to‐moderate AD. 126 The topical aryl hydrocarbon receptor (AHR) mediates keratinocyte differentiation. Tapinarof, an AHR modulator, increases FLG and LOR expression and also induces IL‐24, which activates JAK/STAT and decreases FLG and LOR expression. Data support a new strategy for treating AD using both AHR modulators and JAKi. 127
Systemic treatment options
Currently, approved biologic therapies for AD include dupilumab, a monoclonal antibody that targets IL‐4Rα, which is activated by IL‐4 and IL‐13, 128 and tralokinumab, which targets IL‐13. 129 , 130 Treatment with dupilumab leads to favourable changes in gene expression in patients with AD, including effects on inflammatory mediators, barrier‐related genes, chemokines and cytokines. 128 These changes parallel clinical improvements in patients receiving dupilumab. 128 Other injectable systemic therapies are currently in clinical trials for moderate‐to‐severe AD targeting IL‐13 alone or IL‐31. 16 , 36
Baricitinib, which is approved in the European Union for moderate‐to‐severe AD, is an orally available JAK1/2 inhibitor. 12 A phase 3 trial showed that at week 16, the proportion of patients with moderate‐to‐severe AD who achieved a 75% improvement in the EASI score from baseline (EASI‐75) and a validated Investigator's Global Assessment for Atopic Dermatitis response (vIGA‐AD) was significantly higher in the baricitinib 2‐mg group than the placebo group. 131 Upadacitinib and abrocitinib are orally available selective JAK1 inhibitors. 12 Abrocitinib is approved in several countries for the treatment of patients with moderate‐to‐severe AD in who are candidates for systemic therapy. 132 , 133 , 134 Upadacitinib is approved by the European Medicines Agency and the US Food and Drug Administration (FDA). Three phase 3 trials with upadacitinib showed that at week 16, the proportions of patients who had achieved EASI‐75 and a vIGA‐AD response were significantly higher in the upadacitinib 15‐ or 30‐mg groups (as monotherapy or with topical corticosteroids) than in the placebo group. 135 , 136 In patients with moderate‐to‐severe AD, abrocitinib showed acceptable safety and good efficacy, including a higher proportion of patients in the 100‐ and 200‐mg abrocitinib groups achieving EASI‐75, an Investigator’s Global Assessment response, 137 , 138 , 139 and a Peak Pruritus Numerical Rating Scale response 138 compared with placebo at week 12. ASN002 is an orally available inhibitor of all four JAK family members and spleen tyrosine kinase inhibitor (SYK), and showed rapid improvement in itching and EASI‐50 scores. 2
Thus, emerging clinical data show that JAKi are an effective systemic option for patients with moderate‐to‐severe AD, providing rapid improvement in itch symptoms, and overall, JAKi have a favourable safety profile. 140 These data are consistent with a pivotal role for the JAK/STAT signalling pathway in mediating changes in gene expression that inhibit molecular mechanisms of host immune dysregulation and itch and pain response in patients with AD.
Conclusions and future perspectives
The main symptoms of AD are itch and pain, which significantly affect patients’ quality of life. Primary afferent sensory nerves play a key role by transmitting itch and pain to the CNS; in addition, axon reflex mechanisms of those nerves release neuromediators in the skin of patients with AD, thereby aggravating inflammation, itch and pain. Recent evidence indicates that these neuroimmune circuits are induced by and also change the cytokine profile in AD, thus modulating itch and pain in those patients. Thus, cytokines affect one another and also mediate the cross‐talk between the immune and afferent nervous system via neuronal cytokine receptors. Many cytokine receptors signal through JAK, and several JAKi are in clinical development for the treatment of patients with AD. JAKi modulate gene expression to restore normal cytokine profiles, which results in downstream effects on neuroimmune cross‐talk that reduce itch and pain. Other sensations such as burning and hypersensitivity (i.e. alloknesis) caused by neurotrophic effects require more detailed investigation. Important remaining questions are as follows: (1) which cytokines are the most important in itch and/or pain; (2) which JAK/STAT pathways are the most critical to control itch and pain in AD; and (3) which JAKi and biologics are the most significant to successfully and sustainably suppress cytokine‐mediated itch and pain in AD? Answering these questions will lead to optimal control of these debilitating symptoms in patients with AD.
Conflict of interest
SGK is an advisory board member/consultant for Pfizer Inc., AbbVie, Celldex Therapeutics, Galderma, Incyte, Regeneron Pharmaceuticals and Kiniksa Pharmaceuticals and has served as an investigator for Pfizer Inc., Galderma, Kiniksa Pharmaceuticals and Sanofi. LM is an advisory board member/consultant for Pfizer Inc., AbbVie, Bayer, Galderma, LEO Pharma, Lilly, Novartis and Sanofi; has served as an investigator for Pfizer Inc., Abbvie, Almirall, Dermira, Galderma, Kiniksa Pharmaceuticals, Lilly, Novartis, Sanofi and Trevi; and has received grants from Beiersdorf, Clarins and Johnson & Johnson. CC is an employee and shareholder of Pfizer Inc. MS is an advisory board member/consultant/speaker for Pfizer Inc., AbbVie, Algorithm, Almirall, Avon, Bayer, Baiersdorf, Celgene, Chugai, Galderma, GSK, Novartis, Incyte, Kiniksa Pharmaceuticals, LEO Pharma, Eli Lilly, Janssen, Johnson & Johnson, L’Oreal, Maruho, MenloTx, Mitsubishi, Novartis, Pierre Fabre, Qatar Pharm, Regeneron, Sanofi, Toray, Vertex and Zymogenetics; and has received grant funding from Pfizer Inc., AbbVie, Almirall, Avon, BMS, Chugai, Galderma, Janssen, Johnson & Johnson, Pierre Fabre, Novartis, L’Oreal, Maruho, Sanofi, Toray and Zymogenetics.
Author contributions
Shawn G Kwatra: Conceptualization; Writing – original draft; Writing – review & editing. Laurent Misery: Writing – review & editing. Claire Clibborn: Writing – review & editing. Martin Steinhoff: Conceptualization; Writing – original draft; Writing – review & editing.
Acknowledgments
MS is supported by the National Priorities Research Program (NPRP11S‐0117‐180326) of the Qatar National Research Fund, Member of Qatar Foundation and the Internal Research Grand Competition (IRGC‐04‐SI‐17‐151) of the MRC Fund, Hamad Medical Corporation, Qatar (to MS). SGK is supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under award number K23AR077073. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Editorial/medical writing support under the guidance of the authors was provided by Dr Kristine De La Torre and Dr Jerome Sah at ApotheCom, San Francisco, CA, and was funded by Pfizer Inc., New York, NY, in accordance with Good Publication Practice (GPP3) guidelines (Ann Intern Med 2015; 163: 461–464). This review was supported by Pfizer Inc.
References
- 1. Egeberg A, Griffiths CEM, Williams HC, Andersen YMF, Thyssen JP. Clinical characteristics, symptoms and burden of psoriasis and atopic dermatitis in adults. Br J Dermatol 2020; 183: 128–138. [DOI] [PubMed] [Google Scholar]
- 2. Munera‐Campos M, Carrascosa JM. Innovation in atopic dermatitis: from pathogenesis to treatment. Actas Dermosifiliogr 2020; 111: 205–221. [DOI] [PubMed] [Google Scholar]
- 3. Newton L, DeLozier AM, Griffiths PC et al. Exploring content and psychometric validity of newly developed assessment tools for itch and skin pain in atopic dermatitis. J Patient Rep Outcomes 2019; 3: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Whang KA, Khanna R, Williams KA, Mahadevan V, Semenov Y, Kwatra SG. Health‐related QOL and economic burden of chronic pruritus. J Invest Dermatol 2021; 141: 754–760.e751. [DOI] [PubMed] [Google Scholar]
- 5. Silverwood RJ, Mansfield KE, Mulick A et al. Atopic eczema in adulthood and mortality: UK population‐based cohort study, 1998‐2016. J Allergy Clin Immunol 2021; 147: 1753–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sun L, Liu W, Zhang LJ. The role of Toll‐like receptors in skin host defense, psoriasis, and atopic dermatitis. J Immunol Res 2019; 2019: 1824624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ji RR. Neuroimmune interactions in itch: do chronic itch, chronic pain, and chronic cough share similar mechanisms? Pulm Pharmacol Ther 2015; 35: 81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Damsky W, King BA. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol 2017; 76: 736–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Skabytska Y, Kaesler S, Volz T, Biedermann T. The role of innate immune signaling in the pathogenesis of atopic dermatitis and consequences for treatments. Semin Immunopathol 2016; 38: 29–43. [DOI] [PubMed] [Google Scholar]
- 10. Ziegler SF, Roan F, Bell BD, Stoklasek TA, Kitajima M, Han H. The biology of thymic stromal lymphopoietin (TSLP). Adv Pharmacol 2013; 66: 129–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wilson S, Thé L, Batia L et al. The epithelial cell‐derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell 2013; 155: 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Renert‐Yuval Y, Guttman‐Yassky E. New treatments for atopic dermatitis targeting beyond IL‐4/IL‐13 cytokines. Ann Allergy Asthma Immunol 2020; 124: 28–35. [DOI] [PubMed] [Google Scholar]
- 13. Wongvibulsin S, Sutaria N, Kannan S et al. Transcriptomic analysis of atopic dermatitis in African Americans is characterized by Th2/Th17‐centered cutaneous immune activation. Sci Rep 2021; 11: 11175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Meng J, Li Y, Fischer MJM, Steinhoff M, Chen W, Wang J. Th2 modulation of transient receptor potential channels: an unmet therapeutic intervention for atopic dermatitis. Front Immunol 2021; 12: 696784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Oh M‐H, Oh SY, Lu J et al. TRPA1‐dependent pruritus in IL‐13‐induced chronic atopic dermatitis. J Immunol 2013; 191: 5371–5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Datsi A, Steinhoff M, Ahmad F, Alam M, Buddenkotte J. Interleukin‐31: the ‘itchy’ cytokine in inflammation and therapy. Allergy 2021; 76: 2982–2997. [DOI] [PubMed] [Google Scholar]
- 17. Vandewauw I, Owsianik G, Voets T. Systematic and quantitative mRNA expression analysis of TRP channel genes at the single trigeminal and dorsal root ganglion level in mouse. BMC Neurosci 2013; 14: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cheng W, Yang F, Liu S et al. Heteromeric heat‐sensitive transient receptor potential channels exhibit distinct temperature and chemical response. J Biol Chem 2012; 287: 7279–7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cevikbas F, Wang X, Akiyama T et al. A sensory neuron‐expressed IL‐31 receptor mediates T helper cell‐dependent itch: involvement of TRPV1 and TRPA1. J Allergy Clin Immunol 2014; 133: 448–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Voisin T, Bouvier A, Chiu IM. Neuro‐immune interactions in allergic diseases: novel targets for therapeutics. Int Immunol 2017; 29: 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim J, Kim BE, Leung DYM. Pathophysiology of atopic dermatitis: clinical implications. Allergy Asthma Proc 2019; 40: 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gadani SP, Cronk JC, Norris GT, Kipnis J. IL‐4 in the brain: a cytokine to remember. J Immunol 2012; 189: 4213–4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Campion M, Smith L, Gatault S, Métais C, Buddenkotte J, Steinhoff M. Interleukin‐4 and interleukin‐13 evoke scratching behaviour in mice. Exp Dermatol 2019; 28: 1501–1504. [DOI] [PubMed] [Google Scholar]
- 24. Oetjen LK, Mack MR, Feng J et al. Sensory neurons co‐opt classical immune signaling pathways to mediate chronic itch. Cell 2017; 171: 217–228.e213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nakashima C, Ishida Y, Kitoh A, Otsuka A, Kabashima K. Interaction of peripheral nerves and mast cells, eosinophils, and basophils in the development of pruritus. Exp Dermatol 2019; 28: 1405–1411. [DOI] [PubMed] [Google Scholar]
- 26. Liang G, Barker T, Xie Z, Charles N, Rivera J, Druey KM. Naive T cells sense the cysteine protease allergen papain through protease‐activated receptor 2 and propel TH2 immunity. J Allergy Clin Immunol 2012; 129: 1377–1386.e1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Voisin T, Perner C, Messou MA et al. The CysLT(2)R receptor mediates leukotriene C(4)‐driven acute and chronic itch. Proc Natl Acad Sci USA 2021; 118: e2022087118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xiao S, Lu Z, Steinhoff M et al. Innate immune regulates cutaneous sensory IL‐13 receptor alpha 2 to promote atopic dermatitis. Brain Behav Immun 2021; 98: 28–39. [DOI] [PubMed] [Google Scholar]
- 29. Mashiko S, Bouguermouh S, Rubio M, Baba N, Bissonnette R, Sarfati M. Human mast cells are major IL‐22 producers in patients with psoriasis and atopic dermatitis. J Allergy Clin Immunol 2015; 136: 351–359.e351. [DOI] [PubMed] [Google Scholar]
- 30. Lou H, Lu J, Choi EB et al. Expression of IL‐22 in the skin causes Th2‐biased immunity, epidermal barrier dysfunction, and pruritus via stimulating epithelial Th2 cytokines and the GRP pathway. J Immunol 2017; 198: 2543–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Belzberg M, Alphonse MP, Brown I et al. Prurigo nodularis is characterized by systemic and cutaneous T helper 22 immune polarization. J Invest Dermatol 2021; 141: 2208–2218.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sonkoly E, Muller A, Lauerma AI et al. IL‐31: a new link between T cells and pruritus in atopic skin inflammation. J Allergy Clin Immunol 2006; 117: 411–417. [DOI] [PubMed] [Google Scholar]
- 33. Roh YS, Choi J, Sutaria N, Belzberg M, Kwatra MM, Kwatra SG. IL‐31 inhibition as a therapeutic approach for the management of chronic pruritic dermatoses. Drugs 2021; 81: 895–905. [DOI] [PubMed] [Google Scholar]
- 34. Sakata D, Uruno T, Matsubara K et al. Selective role of neurokinin B in IL‐31‐induced itch response in mice. J Allergy Clin Immunol 2019; 144: 1130–1133.e1138. [DOI] [PubMed] [Google Scholar]
- 35. Huth S, Schmitt L, Marquardt Y et al. Effects of a ceramide‐containing water‐in‐oil ointment on skin barrier function and allergen penetration in an IL‐31 treated 3D model of the disrupted skin barrier. Exp Dermatol 2018; 27: 1009–1014. [DOI] [PubMed] [Google Scholar]
- 36. Ruzicka T, Hanifin JM, Furue M et al. Anti‐interleukin‐31 receptor A antibody for atopic dermatitis. N Engl J Med 2017; 376: 826–835. [DOI] [PubMed] [Google Scholar]
- 37. Ständer S, Yosipovitch G, Legat FJ et al. Trial of nemolizumab in moderate‐to‐severe prurigo nodularis. N Engl J Med 2020; 382: 706–716. [DOI] [PubMed] [Google Scholar]
- 38. Tseng PY, Hoon MA. Oncostatin M can sensitize sensory neurons in inflammatory pruritus. Sci Transl Med 2021; 13: eabe3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nakajima S, Kabata H, Kabashima K, Asano K. Anti‐TSLP antibodies: targeting a master regulator of type 2 immune responses. Allergol Int 2020; 69: 197–203. [DOI] [PubMed] [Google Scholar]
- 40. Erickson S, Heul AV, Kim BS. New and emerging treatments for inflammatory itch. Ann Allergy Asthma Immunol 2021; 126: 13–20. [DOI] [PubMed] [Google Scholar]
- 41. Eberl G, Colonna M, Di Santo JP, McKenzie AN. Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science 2015; 348: aaa6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Corren J, Parnes JR, Wang L et al. Tezepelumab in adults with uncontrolled asthma. N Engl J Med 2017; 377: 936–946. [DOI] [PubMed] [Google Scholar]
- 43. Simpson EL, Parnes JR, She D et al. Tezepelumab, an anti‐thymic stromal lymphopoietin monoclonal antibody, in the treatment of moderate to severe atopic dermatitis: a randomized phase 2a clinical trial. J Am Acad Dermatol 2019; 80: 1013–1021. [DOI] [PubMed] [Google Scholar]
- 44. Imai Y. Interleukin‐33 in atopic dermatitis. J Dermatol Sci 2019; 96: 2–7. [DOI] [PubMed] [Google Scholar]
- 45. Meng J, Moriyama M, Feld M et al. New mechanism underlying IL‐31‐induced atopic dermatitis. J Allergy Clin Immunol 2018; 141: 1677–1689.e1678. [DOI] [PubMed] [Google Scholar]
- 46. Meng J, Wang J, Buddenkotte J, Buhl T, Steinhoff M. Role of SNAREs in atopic dermatitis‐related cytokine secretion and skin‐nerve communication. J Invest Dermatol 2019; 139: 2324–2333. [DOI] [PubMed] [Google Scholar]
- 47. Steinhoff M, Schmelz M, Szabó IL, Oaklander AL. Clinical presentation, management, and pathophysiology of neuropathic itch. Lancet Neurol 2018; 17: 709–720. [DOI] [PubMed] [Google Scholar]
- 48. Hassan Z, Luvsannyam E, Patel D, Nukala S, Puvvada SR, Hamid P. Review of prominent cytokines as superior therapeutic targets for moderate‐to‐severe atopic dermatitis. Cureus 2020; 12: e9901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Klonowska J, Gleń J, Nowicki RJ, Trzeciak M. New cytokines in the pathogenesis of atopic dermatitis‐new therapeutic targets. Int J Mol Sci 2018; 19: 3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Salimi M, Barlow JL, Saunders SP et al. A role for IL‐25 and IL‐33‐driven type‐2 innate lymphoid cells in atopic dermatitis. J Exp Med 2013; 210: 2939–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cevikbas F, Steinhoff M. IL‐33: a novel danger signal system in atopic dermatitis. J Invest Dermatol 2012; 132: 1326–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zedan K, Rasheed Z, Farouk Y et al. Immunoglobulin e, interleukin‐18 and interleukin‐12 in patients with atopic dermatitis: correlation with disease activity. J Clin Diagn Res 2015; 9: WC01–WC05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jiang Y, Tsoi LC, Billi AC et al. Cytokinocytes: the diverse contribution of keratinocytes to immune responses in skin. JCI Insight 2020; 5: e142067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Asahina R, Maeda S. A review of the roles of keratinocyte‐derived cytokines and chemokines in the pathogenesis of atopic dermatitis in humans and dogs. Vet Dermatol 2017; 28: 16‐e15. [DOI] [PubMed] [Google Scholar]
- 55. Lowy DB, Makker PGS, Moalem‐Taylor G. Cutaneous neuroimmune interactions in peripheral neuropathic pain states. Front Immunol 2021; 12: 660203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Donnelly CR, Andriessen AS, Chen G et al. Central nervous system targets: glial cell mechanisms in chronic pain. Neurotherapeutics 2020; 17: 846–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ji RR, Donnelly CR, Nedergaard M. Astrocytes in chronic pain and itch. Nat Rev Neurosci 2019; 20: 667–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liu T, Han Q, Chen G et al. Toll‐like receptor 4 contributes to chronic itch, alloknesis, and spinal astrocyte activation in male mice. Pain 2016; 157: 806–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wei Z, Fei Y, Su W, Chen G. Emerging role of Schwann cells in neuropathic pain: receptors, glial mediators and myelination. Front Cell Neurosci 2019; 13: 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Xu M, Bennett DLH, Querol LA et al. Pain and the immune system: emerging concepts of IgG‐mediated autoimmune pain and immunotherapies. J Neurol Neurosurg Psychiatry 2020; 91: 177–188. [DOI] [PubMed] [Google Scholar]
- 61. Gao YJ, Zhang L, Ji RR. Spinal injection of TNF‐α‐activated astrocytes produces persistent pain symptom mechanical allodynia by releasing monocyte chemoattractant protein‐1. Glia 2010; 58: 1871–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Blake KJ, Jiang XR, Chiu IM. Neuronal regulation of immunity in the skin and lungs. Trends Neurosci 2019; 42: 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Solinski HJ, Kriegbaum MC, Tseng P‐Y et al. Nppb neurons are sensors of mast cell‐induced itch. Cell Rep 2019; 26: 3561–3573.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang F, Trier AM, Li F et al. A basophil‐neuronal axis promotes itch. Cell 2021; 184: 422–440.e417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Xie AX, Madayag A, Minton SK, McCarthy KD, Malykhina AP. Sensory satellite glial Gq‐GPCR activation alleviates inflammatory pain via peripheral adenosine 1 receptor activation. Sci Rep 2020; 10: 14181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Feld M, Garcia R, Buddenkotte J et al. The pruritus‐ and TH2‐associated cytokine IL‐31 promotes growth of sensory nerves. J Allergy Clin Immunol 2016; 138: 500–508.e524. [DOI] [PubMed] [Google Scholar]
- 67. Pogatzki‐Zahn EM, Pereira MP, Cremer A et al. Peripheral sensitization and loss of descending inhibition is a hallmark of chronic pruritus. J Invest Dermatol 2020; 140: 203–211.e204. [DOI] [PubMed] [Google Scholar]
- 68. Steinhoff M, Vergnolle N, Young SH et al. Agonists of proteinase‐activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med 2000; 6: 151–158. [DOI] [PubMed] [Google Scholar]
- 69. Steinhoff M, Neisius U, Ikoma A et al. Proteinase‐activated receptor‐2 mediates itch: a novel pathway for pruritus in human skin. J Neurosci 2003; 23: 6176–6180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Buhl T, Ikoma A, Kempkes C et al. Protease‐activated receptor‐2 regulates neuro‐epidermal communication in atopic dermatitis. Front Immunol 2020; 11: 1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Poole DP, Amadesi S, Veldhuis NA et al. Protease‐activated receptor 2 (PAR2) protein and transient receptor potential vanilloid 4 (TRPV4) protein coupling is required for sustained inflammatory signaling. J Biol Chem 2013; 288: 5790–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Buddenkotte J, Stroh C, Engels IH et al. Agonists of proteinase‐activated receptor‐2 stimulate upregulation of intercellular cell adhesion molecule‐1 in primary human keratinocytes via activation of NF‐kappa B. J Invest Dermatol 2005; 124: 38–45. [DOI] [PubMed] [Google Scholar]
- 73. Lim KM, Park YH. Development of PAC‐14028, a novel transient receptor potential vanilloid type 1 (TRPV1) channel antagonist as a new drug for refractory skin diseases. Arch Pharm Res 2012; 35: 393–396. [DOI] [PubMed] [Google Scholar]
- 74. Chiu IM, Heesters BA, Ghasemlou N et al. Bacteria activate sensory neurons that modulate pain and inflammation. Nature 2013; 501: 52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Deftu AF, Filippi A, Shibsaki K, Gheorghe RO, Chiritoiu M, Ristoiu V. Chemokine (C‐X‐C motif) ligand 1 (CXCL1) and chemokine (C‐X‐C motif) ligand 2 (CXCL2) modulate the activity of TRPV1+/IB4+ cultured rat dorsal root ganglia neurons upon short‐term and acute application. J Physiol Pharmacol 2017; 68: 385–395. [PubMed] [Google Scholar]
- 76. Qu Y, Wang G, Sun X, Wang K. Inhibition of the warm temperature‐activated Ca2+‐permeable transient receptor potential vanilloid TRPV3 channel attenuates atopic dermatitis. Mol Pharmacol 2019; 96: 393–400. [DOI] [PubMed] [Google Scholar]
- 77. Lin S‐H, Steinhoff M, Ikoma A et al. Involvement of TRPV1 and TDAG8 in pruriception associated with noxious acidosis. J Invest Dermatol 2017; 137: 170–178. [DOI] [PubMed] [Google Scholar]
- 78. Pollema‐Mays SL, Centeno MV, Ashford CJ, Apkarian AV, Martina M. Expression of background potassium channels in rat DRG is cell‐specific and down‐regulated in a neuropathic pain model. Mol Cell Neurosci 2013; 57: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kühn H, Kappes L, Wolf K et al. Complementary roles of murine NaV1.7, Na V 1.8 and Na V 1.9 in acute itch signalling. Sci Rep 2020; 10: 2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ma H, Gao T, Jakobsson JET et al. The neuropeptide Y Y2 receptor is coexpressed with Nppb in primary afferent neurons and Y2 activation reduces histaminergic and IL‐31‐induced itch. J Pharmacol Exp Ther 2020; 372: 73–82. [DOI] [PubMed] [Google Scholar]
- 81. Choi JE, Di Nardo A. Skin neurogenic inflammation. Semin Immunopathol 2018; 40: 249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Solinski HJ, Dranchak P, Oliphant E et al. Inhibition of natriuretic peptide receptor 1 reduces itch in mice. Sci Transl Med 2019; 11: eaav5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Guseva D, Rüdrich U, Kotnik N et al. Neuronal branching of sensory neurons is associated with BDNF‐positive eosinophils in atopic dermatitis. Clin Exp Allergy 2020; 50: 577–584. [DOI] [PubMed] [Google Scholar]
- 84. Bigliardi‐Qi M, Lipp B, Sumanovski LT, Buechner SA, Bigliardi PL. Changes of epidermal mu‐opiate receptor expression and nerve endings in chronic atopic dermatitis. Dermatology 2005; 210: 91–99. [DOI] [PubMed] [Google Scholar]
- 85. Liu J, Li A‐R, Wang Y et al. Discovery of AMG 853, a CRTH2 and DP dual antagonist. ACS Med Chem Lett 2011; 2: 326–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Retamal JS, Ramírez‐García PD, Shenoy PA, Poole DP, Veldhuis NA. Internalized GPCRs as potential therapeutic targets for the management of pain. Front Mol Neurosci 2019; 12: 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yuan W, Chen Y, Zhou Y et al. Formononetin attenuates atopic dermatitis by upregulating A20 expression via activation of G protein–coupled estrogen receptor. J Ethnopharmacol 2021; 266: 113397. [DOI] [PubMed] [Google Scholar]
- 88. Schmelz M. Itch processing in the skin. Front Med (Lausanne) 2019; 6: 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Liu T, Ji RR. New insights into the mechanisms of itch: are pain and itch controlled by distinct mechanisms? Pflugers Arch 2013; 465: 1671–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Yosipovitch G, Berger T, Fassett MS. Neuroimmune interactions in chronic itch of atopic dermatitis. J Eur Acad Dermatol Venereol 2020; 34: 239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Roosterman D, Goerge T, Schneider SW, Bunnett NW, Steinhoff M. Neuronal control of skin function: the skin as a neuroimmunoendocrine organ. Physiol Rev 2006; 86: 1309–1379. [DOI] [PubMed] [Google Scholar]
- 92. Usoskin D, Furlan A, Islam S et al. Unbiased classification of sensory neuron types by large‐scale single‐cell RNA sequencing. Nat Neurosci 2015; 18: 145–153. [DOI] [PubMed] [Google Scholar]
- 93. Nguyen MQ, von Buchholtz LJ, Reker AN, Ryba NJ, Davidson S. Single‐nucleus transcriptomic analysis of human dorsal root ganglion neurons. Elife 2021; 10: e71752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Klein A, Solinski HJ, Malewicz NM et al. Pruriception and neuronal coding in nociceptor subtypes in human and nonhuman primates. Elife 2021; 10: e64506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Tavares‐Ferreira D, Shiers S, Ray PR et al. Spatial transcriptomics of dorsal root ganglia identifies molecular signatures of human nociceptors. Sci Transl Med 2022; 14: eabj8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Mishra SK, Hoon MA. The cells and circuitry for itch responses in mice. Science 2013; 340: 968–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Handwerker HO. Frontiers in neuroscience itch hypotheses: from pattern to specificity and to population coding. In: Carstens E, Akiyama T (eds). Itch: Mechanisms and Treatment. Boca Raton, FL: CRC Press/Taylor & Francis Group LLC, 2014. [PubMed] [Google Scholar]
- 98. Silverberg JI, Gelfand JM, Margolis DJ et al. Pain is a common and burdensome symptom of atopic dermatitis in United States adults. J Allergy Clin Immunol Pract 2019; 7: 2699–2706.e2697. [DOI] [PubMed] [Google Scholar]
- 99. Wagner KM, Gomes A, McReynolds CB, Hammock BD. Soluble epoxide hydrolase regulation of lipid mediators limits pain. Neurotherapeutics 2020; 17: 900–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ji RR, Xu ZZ, Wang X, Lo EH. Matrix metalloprotease regulation of neuropathic pain. Trends Pharmacol Sci 2009; 30: 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Jin Y, Wei S, Liu TT, Qiu CY, Hu WP. Acute P38‐mediated enhancement of P2X3 receptor currents by TNF‐α in rat dorsal root ganglion neurons. J Inflamm Res 2021; 14: 2841–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Steinhoff M, Oaklander AL, Szabó IL, Ständer S, Schmelz M. Neuropathic itch. Pain 2019; 160(Suppl 1): S11–S16. [DOI] [PubMed] [Google Scholar]
- 103. Barr TP, Hrnjic A, Khodorova A, Sprague JM, Strichartz GR. Sensitization of cutaneous neuronal purinergic receptors contributes to endothelin‐1–induced mechanical hypersensitivity. Pain 2014; 155: 1091–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kreß L, Hofmann L, Klein T et al. Differential impact of keratinocytes and fibroblasts on nociceptor degeneration and sensitization in small fiber neuropathy. Pain 2021; 162: 1262–1272. [DOI] [PubMed] [Google Scholar]
- 105. Sharma JN, Al‐Sherif GJ. Pharmacologic targets and prototype therapeutics in the kallikrein‐kinin system: bradykinin receptor agonists or antagonists. ScientificWorldJournal 2006; 6: 1247–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Liu T, Ji RR. Frontiers in neuroscience Toll‐like receptors and itch. In: Carstens E, Akiyama T (eds). Itch: Mechanisms and Treatment. Boca Raton, FL: CRC Press/Taylor & Francis Group LLC, 2014. [PubMed] [Google Scholar]
- 107. Braz J, Solorzano C, Wang X, Basbaum AI. Transmitting pain and itch messages: a contemporary view of the spinal cord circuits that generate gate control. Neuron 2014; 82: 522–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Andersen HH, Akiyama T, Nattkemper LA et al. Alloknesis and hyperknesis‐mechanisms, assessment methodology, and clinical implications of itch sensitization. Pain 2018; 159: 1185–1197. [DOI] [PubMed] [Google Scholar]
- 109. Ferretti E, Corcione A, Pistoia V. The IL‐31/IL‐31 receptor axis: general features and role in tumor microenvironment. J Leukoc Biol 2017; 102: 711–717. [DOI] [PubMed] [Google Scholar]
- 110. Lejeune D, Dumoutier L, Constantinescu S, Kruijer W, Schuringa JJ, Renauld JC. Interleukin‐22 (IL‐22) activates the JAK/STAT, ERK, JNK, and p38 MAP kinase pathways in a rat hepatoma cell line. Pathways that are shared with and distinct from IL‐10. J Biol Chem 2002; 277: 33676–33682. [DOI] [PubMed] [Google Scholar]
- 111. O'Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK‐STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med 2015; 66: 311–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wohlmann A, Sebastian K, Borowski A, Krause S, Friedrich K. Signal transduction by the atopy‐associated human thymic stromal lymphopoietin (TSLP) receptor depends on Janus kinase function. Biol Chem 2010; 391: 181–186. [DOI] [PubMed] [Google Scholar]
- 113. Amano W, Nakajima S, Kunugi H et al. The Janus kinase inhibitor JTE‐052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J Allergy Clin Immunol 2015; 136: 667–677.e667. [DOI] [PubMed] [Google Scholar]
- 114. Krishnamurthy P, Kaplan MH. STAT6 and PARP family members in the development of T cell‐dependent allergic inflammation. Immune Netw 2016; 16: 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Clarysse K, Pfaff CM, Marquardt Y et al. JAK1/3 inhibition preserves epidermal morphology in full‐thickness 3D skin models of atopic dermatitis and psoriasis. J Eur Acad Dermatol Venereol 2019; 33: 367–375. [DOI] [PubMed] [Google Scholar]
- 116. Yeo H, Ahn SS, Lee YH, Shin SY. Regulation of pro‐opiomelanocortin (POMC) gene transcription by interleukin‐31 via early growth response 1 (EGR‐1) in HaCaT keratinocytes. Mol Biol Rep 2020; 47: 5953–5962. [DOI] [PubMed] [Google Scholar]
- 117. Gross AR, Theoharides TC. Chondroitin sulfate inhibits secretion of TNF and CXCL8 from human mast cells stimulated by IL‐33. BioFactors 2019; 45: 49–61. [DOI] [PubMed] [Google Scholar]
- 118. Petra AI, Tsilioni I, Taracanova A, Katsarou‐Katsari A, Theoharides TC. Interleukin 33 and interleukin 4 regulate interleukin 31 gene expression and secretion from human laboratory of allergic diseases 2 mast cells stimulated by substance P and/or immunoglobulin E. Allergy Asthma Proc 2018; 39: 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Harrington R, Al Nokhatha SA, Conway R. JAK inhibitors in rheumatoid arthritis: an evidence‐based review on the emerging clinical data. J Inflamm Res 2020; 13: 519–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Wong ITY, Tsuyuki RT, Cresswell‐Melville A, Doiron P, Drucker AM. Guidelines for the management of atopic dermatitis (eczema) for pharmacists. Can Pharm J (Ott) 2017; 150: 285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Chong M, Fonacier L. Treatment of eczema: corticosteroids and beyond. Clin Rev Allergy Immunol 2016; 51: 249–262. [DOI] [PubMed] [Google Scholar]
- 122. Hong J, Buddenkotte J, Berger TG, Steinhoff M. Management of itch in atopic dermatitis. Semin Cutan Med Surg 2011; 30: 71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Kobyletzki L, Thomas K, Schmitt J et al. What factors are important to patients when assessing treatment response: an international cross‐sectional survey. Acta Derm Venereol 2017; 97: 86–90. [DOI] [PubMed] [Google Scholar]
- 124. Simpson EL, Guttman‐Yassky E, Margolis DJ et al. Association of inadequately controlled disease and disease severity with patient‐reported disease burden in adults with atopic dermatitis. JAMA Dermatol 2018; 154: 903–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Nakagawa H, Nemoto O, Igarashi A, Saeki H, Kaino H, Nagata T. Delgocitinib ointment, a topical Janus kinase inhibitor, in adult patients with moderate to severe atopic dermatitis: a phase 3, randomized, double‐blind, vehicle‐controlled study and an open‐label, long‐term extension study. J Am Acad Dermatol 2020; 82: 823–831. [DOI] [PubMed] [Google Scholar]
- 126. Product label (US): OPZELURA™ (ruxolitinib) cream, for topical use. https://www.opzelura.com/prescribing‐information.pdf [Accessed April 27 2022].
- 127. Vu YH, Hashimoto‐Hachiya A, Takemura M et al. IL‐24 Negatively regulates keratinocyte differentiation induced by tapinarof, an aryl hydrocarbon receptor modulator: implication in the treatment of atopic dermatitis. Int J Mol Sci 2020; 21: 9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Hamilton JD, Suárez‐Fariñas M, Dhingra N et al. Dupilumab improves the molecular signature in skin of patients with moderate‐to‐severe atopic dermatitis. J Allergy Clin Immunol 2014; 134: 1293–1300. [DOI] [PubMed] [Google Scholar]
- 129. Prescribing information (US): ADBRY™ (tralokinumab‐ldrm) injection, for subcutaneous use. https://www.leo‐pharma.us/Files/Billeder/US%20Website%20Product%20PIs/AdbryPI.pdf
- 130. Product label (EU): Adtralza 150 mg solution for injection in pre‐filled syringe. https://www.ema.europa.eu/en/documents/product‐information/adtralza‐epar‐product‐information_en.pdf
- 131. Simpson EL, Forman S, Silverberg JI et al. Baricitinib in patients with moderate‐to‐severe atopic dermatitis: results from a randomized monotherapy phase 3 trial in the United States and Canada (BREEZE‐AD5). J Am Acad Dermatol 2021; 85: 62–70. [DOI] [PubMed] [Google Scholar]
- 132. Product label (US): CIBINQO™ (abrocitinib) tablets, for oral use. https://cdn.pfizer.com/pfizercom/USPI_Med_Guide_CIBINQO_Abrocitinib_tablet.pdf
- 133. Product label (EU): Cibinqo film‐coated tablets. https://www.ema.europa.eu/en/documents/product‐information/cibinqo‐epar‐product‐information_en.pdf
- 134. Product label (UK): Cibinqo 100 mg film‐coated tablets. https://www.medicines.org.uk/emc/product/12873/smpc
- 135. Guttman‐Yassky E, Teixeira HD, Simpson EL et al. Once‐daily upadacitinib versus placebo in adolescents and adults with moderate‐to‐severe atopic dermatitis (Measure Up 1 and Measure Up 2): results from two replicate double‐blind, randomised controlled phase 3 trials. Lancet 2021; 397: 2151–2168. [DOI] [PubMed] [Google Scholar]
- 136. Reich K, Teixeira HD, de Bruin‐Weller M et al. Safety and efficacy of upadacitinib in combination with topical corticosteroids in adolescents and adults with moderate‐to‐severe atopic dermatitis (AD Up): results from a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet 2021; 397: 2169–2181. [DOI] [PubMed] [Google Scholar]
- 137. Simpson EL, Sinclair R, Forman S et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate‐to‐severe atopic dermatitis (JADE MONO‐1): a multicentre, double‐blind, randomised, placebo‐controlled, phase 3 trial. Lancet 2020; 396: 255–266. [DOI] [PubMed] [Google Scholar]
- 138. Silverberg JI, Simpson EL, Thyssen JP et al. Efficacy and safety of abrocitinib in patients with moderate‐to‐severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol 2020; 156: 863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Bieber T, Simpson EL, Silverberg JI et al. Abrocitinib versus placebo or dupilumab for atopic dermatitis. N Engl J Med 2021; 384: 1101–1112. [DOI] [PubMed] [Google Scholar]
- 140. Crowley EL, Nezamololama N, Papp K, Gooderham MJ. Abrocitinib for the treatment of atopic dermatitis. Expert Rev Clin Immunol 2020; 16: 955–962. [DOI] [PubMed] [Google Scholar]
- 141. Yang X, Kambe N, Takimoto‐Ito R, Kabashima K. Advances in the pathophysiology of atopic dermatitis revealed by novel therapeutics and clinical trials. Pharmacol Ther 2021; 224: 107830. [DOI] [PubMed] [Google Scholar]
- 142. Han H, Roan F, Ziegler SF. The atopic march: current insights into skin barrier dysfunction and epithelial cell‐derived cytokines. Immunol Rev 2017; 278: 116–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Bissonnette R, Papp KA, Poulin Y et al. Topical tofacitinib for atopic dermatitis: a phase IIa randomized trial. Br J Dermatol 2016; 175: 902–911. [DOI] [PubMed] [Google Scholar]
- 144. Papp K, Szepietowski JC, Kircik L et al. Efficacy and safety of ruxolitinib cream for the treatment of atopic dermatitis: results from two phase 3, randomized, double‐blind studies. J Am Acad Dermatol 2021; 85: 863–872. [DOI] [PubMed] [Google Scholar]
- 145. Li H, Zuo J, Tang W. Phosphodiesterase‐4 inhibitors for the treatment of inflammatory diseases. Front Pharmacol 2018; 9: 1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Paller AS, Stein Gold L, Soung J, Tallman AM, Rubenstein DS, Gooderham M. Efficacy and patient‐reported outcomes from a phase 2b, randomized clinical trial of tapinarof cream for the treatment of adolescents and adults with atopic dermatitis. J Am Acad Dermatol 2021; 84: 632–638. [DOI] [PubMed] [Google Scholar]
- 147. Wollenberg A, Blauvelt A, Guttman‐Yassky E et al. Tralokinumab for moderate‐to‐severe atopic dermatitis: results from two 52‐week, randomized, double‐blind, multicentre, placebo‐controlled phase III trials (ECZTRA 1 and ECZTRA 2). Br J Dermatol 2021; 184: 437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Vazquez ML, Kaila N, Strohbach JW et al. Identification of N‐{cis‐3‐[Methyl(7H‐pyrrolo[2,3‐d]pyrimidin‐4‐yl)amino]cyclobutyl}propane‐1‐sulfonamide (PF‐04965842): a selective JAK1 clinical candidate for the treatment of autoimmune diseases. J Med Chem 2018; 61: 1130–1152. [DOI] [PubMed] [Google Scholar]