

Amyotrophic lateral sclerosis (ALS) is associated with proteostasis collapse: ALS is an unrelenting neurodegenerative disease that is characterized by the loss of motor neurons in the brain and spinal cord, resulting in the progressive atrophy, and eventual paralysis, of skeletal muscles. Death due to respiratory failure usually occurs within 2–5 years from symptom onset. Approximately 90% of ALS cases are of unknown etiology and are termed sporadic ALS (sALS). The remaining 10% of ALS cases present with a family history (familial ALS; fALS) and are associated with genetic mutations in a range of over 20 functionally heterogeneous genes. Regardless of disease origin, the pathological hallmark of ALS is the accumulation of ubiquitylated protein inclusions in motor neurons and surrounding glial cells. The presence of these inclusions, compromised largely of misfolded and aggregated proteins, implies a collapse in proteostasis. Proteostasis refers to the maintenance of the proteome in a state of balance or equilibrium so that a cell can perform its proper function. Central to the maintenance of proteostasis are the predominant protein degradation pathways, the ubiquitin-proteasome system (UPS) and the autophagy system. In recent years, the identification of numerous ALS associated genes involved in protein degradation systems, including VCP, SQSTM1, UBQLN2, OPTN, TBK1, CCNF, DNAJC7 and CYLD have strengthened the association between proteostasis failure, in particular protein degradation, and ALS. The accumulating evidence implicating protein degradation pathways as a cause of ALS raises two critical questions; are mutations in genes within functional pathways distinct from protein degradation such as C9orf72, TARDBP, FUS and SOD1 in any way related to the UPS or autophagy dysfunction that causes ALS and why are motor neurons selectively affected? Understanding the unique vulnerability of motor neurons may provide additional insight into the pathogenesis of this currently incurable disease.
Ubiquitin is critical to cellular function: Ubiquitin is a small 76 amino acid polypeptide abundantly expressed in all cell types. Through a highly ordered three-step process, ubiquitin is covalently bound to lysine residues on a target substrate. As ubiquitin itself contains seven lysine residues and an N-terminal methionine, these can become further sites of ubiquitylation resulting in a variety of polyubiquitin chains which act to specify the localization and cellular function of the protein. The most abundant ubiquitin linkages are K48 chains, the canonical signal directing proteins to the 26S proteasome, in line with the predominant protein degradation role of ubiquitin. Other linkages are found on proteins involved in a range of cellular functions including DNA repair (K6 and K63), protein trafficking (K33), endocytosis (monoubiquitination) and innate immunity (K63 and K27). To add further complexity to the system of ubiquitylation, chains can also contain heterogeneous linkages where different linkages are present within the one chain. Further, ubiquitin itself can undergo post-translational modification. This broad diversity of ubiquitin signaling or the “ubiquitin code” is essential for the selectivity and specificity of targeting proteins.
Neurons, in particular, have a unique dependence on ubiquitylation as it is essential for synapse formation, function and pruning, excitability and neurotransmission (reviewed in Hallengren et al., 2013). It also regulates inflammatory pathways, an area of particular importance given the association of neuroinflammation with neurodegenerative disorders such as ALS. Inside cells, ubiquitin exists in a dynamic equilibrium between “free” or unconjugated monomeric ubiquitin and ubiquitin conjugated to proteins in monomeric or in chain form. These pools are tightly regulated through a combination of gene expression, polyubiquitin chain formation and removal of ubiquitin from substrate proteins by a group of over 100 deubiquitinating enzymes. Maintaining an adequate supply of free ubiquitin is critical for normal cellular function, as well as a response to stress or altered conditions. Any stress which results in transiently or persistently elevated levels of misfolded protein creates an environment of proteotoxic stress which cells need to manage. We and others have found that motor neurons in particular are uniquely susceptible to proteotoxic stress.
Motor neurons have a metastable proteome: Ubiquitin-positive inclusions within motor neurons and adjacent cells are a characteristic hallmark of ALS, regardless of disease origin. Inclusions not only contain a primary misfolded protein, such as TDP-43, FUS or SOD1, but also a range of other proteins, including ubiquitin; many of which are not known native interactors of the initial inclusion-forming protein. Furthermore, they share few characteristics other than co-aggregation within inclusions. Previous work in other neurodegenerative diseases, including Alzheimer’s, Huntington’s and Parkinson’s diseases, have identified that co-aggregating proteins with plaques, neurofibrillary tangles and Lewy bodies primarily contain supersaturated proteins (Ciryam et al., 2013). Supersaturated proteins are defined as those that are expressed at higher levels than their predicted solubility based on either their unfolded (σu) or folded (σf) state. Furthermore, these proteins are metastable and therefore are at increased risk of aggregation, particularly under conditions of stress or ageing. We recently found that whilst co-aggregating ALS inclusion associated proteins are not supersaturated when their expression is calculated based on averaged scores from a range of tissues, they are, however, deemed supersaturated when calculations use their expression from spinal cord motor neurons (Ciryam et al., 2017). The proteins found in this metastable sub-proteome are overrepresented in pathways known to be disrupted in ALS; proteasome function, mitochondrial function, protein transport and mRNA processing. Furthermore, supersaturation scores calculated from the proteome of vulnerable spinal cord motor neurons and resistant oculomotor neurons identified that motor neurons had a subtle but overall higher median supersaturation score than oculomotor neurons (Yerbury et al., 2019). In conjunction with an increased supersaturated proteome, transcriptomic and gene ontology analysis found genes involved in ubiquitin-mediated proteolysis were downregulated in spinal motor neurons compared to oculomotor neurons (Brockington et al., 2013). Collectively, this work suggests the possibility that an initial misfolded and aggregating protein in spinal cord motor neurons might precipitate a series of uncontrolled aggregation events of otherwise uninvolved proteins, but which are critically needed in essential cellular pathways. The mechanism driving this apparently accumulating aggregation process lies in the metastability of supersaturated proteins, which by definition requires energy, in this case in the form of the protein quality control system, to keep them in a soluble state. However, as spinal cord motor neurons have a reduced UPS capacity compared to other neuronal cell types, it is likely that the maintenance of such a supersaturated proteome is balanced on a razor’s edge and that this may provide an explanation for their vulnerability in ALS.
Evidence ALS associated misfolded proteins disrupt ubiquitin homeostasis: ALS-associated proteins TDP-43, FUS and SOD1 are supersaturated within motor neurons, highlighting their susceptibility to aggregation and potential to disrupt ubiquitin homeostasis. Our previous work has demonstrated that although these proteins aggregate via distinct pathways in motor neuron-like (NSC-34) cells, ubiquitin is consistently incorporated into TDP-43, SOD1 and FUS inclusions (Farrawell et al., 2018, 2020). The presence of ubiquitin within inclusions has been known for decades. However, more recently, dual labelling of sALS postmortem spinal cord tissue showed ubiquitin is diffusely present in motor neurons in the absence of TDP-43 inclusions, whilst increased ubiquitin is colocalised with TDP-43 aggregates in cells containing inclusions, suggesting that the aggregation of ALS-associated proteins, such as TDP-43, may perturb ubiquitin homeostasis (Farrawell et al., 2020). In line with this idea, we observed increased UPS dysfunction in NSC-34 cells expressing mutant TDP-43M337V, FUSR495X and SOD1A4V compared to their wild-type forms using a fluorescent ubiquitin reporter, containing the CL1 degron peptide. Furthermore, fluorescence recovery after nuclear photobleaching (FRANP), a technique which exploits the nuclear pore as a filter for free monomeric ubiquitin, showed TDP-43M337V, FUSR495X and SOD1A4V expressing cells containing inclusions have reduced levels of free monomeric ubiquitin compared to cells expressing soluble TDP-43, FUS and SOD1 (Farrawell et al., 2018, 2020). Moreover, using the tUI-HA probe, which explicitly binds to free ubiquitin, we were able to confirm cells expressing TDP-43M337V and FUSR495X exhibited lower levels of free ubiquitin compared to cells without inclusions or cells expressing their wild-type forms (Farrawell et al., 2020). Collectively, this data is consistent with the notion that the aggregation of ALS-associated proteins depletes the free monomeric ubiquitin pool.
To further investigate the extent of how ALS-associated aggregates alter ubiquitin homeostasis, we employed a proteomic approach to investigate ubiquitylated proteins in NSC-34 cells expressing mutant SOD1A4V. We observed a 5-fold increase in the number of exclusively ubiquitylated proteins compared to SOD1WT. Pathway enrichment analysis identified these proteins as involved in metabolic pathways, the UPS, in addition to ribosome and mRNA processing. Furthermore, calculations of the supersaturation score of ubiquitylated proteins identified in SOD1A4V cells revealed these proteins possessed a supersaturation score 10x (σu) and 25x (σf) higher than the whole proteome (Farrawell et al., 2018). This suggests that in the presence of aggregated proteins, ubiquitin pools are impaired and re-distributed, either to ubiquitin targeting vulnerable supersaturated and aggregation-prone proteins or potentially redistributed to cellular pathways associated with ALS. As growing evidence implicates disturbed ubiquitin homeostasis in ALS, our attention must begin to turn to how to target the UPS as a therapeutic avenue.
Restoring ubiquitin homeostasis as a therapeutic strategy to treat ALS: While it is yet to be confirmed if the protein aggregates present in ALS affected motor neurons are the culprit of proteasomal impairment or a symptom, the presence of ubiquitin in these aggregates and the role of this phenomenon in motor neuron dysfunction has not been fully appreciated. This sequestration of ubiquitin, combined with alterations in the ubiquitylated proteome observed when aggregates are present, suggests restoring ubiquitin homeostasis holds promise as a therapeutic target. Strategies that reduce the accumulation of misfolded protein would address the question of causation and if misfolded proteins are reduced by enhancing the efficiency of the UPS this would concomitantly replenish ubiquitin monomers to the cellular pool. There are several attractive targets for this strategy. Delivery of disease-specific E3 ligases would increase ubiquitination of target proteins, directing them to the proteasome for degradation. This has been successfully trialled in a Parkinson’s disease model with AAV delivery of the E3 ligase, Parkin (Yasuda et al., 2007). Silencing or inhibiting the proteasomal DUB, USP14 has promoted degradation of ataxin-3, TDP-43 and tau, while increasing expression of proteasomal subunits or identifying small molecule activators of the UPS could also upregulate this rate-limiting step [reviewed in (Kors et al., 2019)]. Inspired by progress in the cancer field, PROTACs or proteolysis targeting chimeras are gaining traction in neurodegenerative research. PROTACs are bifunctional molecules that simultaneously bind a target protein and an E3 ligase, accelerating the protein’s degradation. PROTACs for TDP-43, tau, α-synuclein and Huntingtin protein have shown promise in various cellular and mouse models [reviewed in Schmidt et al. (2021)].
Given accumulating evidence that the UPS is compromised in ALS, attempting to use this system to reduce protein misfolding and replenish ubiquitin may meet obstacles. An alternative strategy that does not depend on UPS function is to prevent the translation of targeted proteins via antisense oligonucleotides or siRNA molecules (reviewed in Bennett, 2019). An antisense oligonucleotide for SOD1 is currently in phase II clinical trials and has been shown to effectively reduce cerebrospinal fluid SOD1 concentrations (Miller et al., 2020). One deficiency of this method is the inability of these molecules to differentiate between wild-type and mutant versions of the protein and it is yet to be established if removal of the multitude of wild-type ALS associated proteins will not result in detrimental consequences. Antibodies capable of recognizing only the misfolded form of target proteins would overcome this obstacle and with advances in technology improving access across the blood brain barrier, this field is showing promise
Despite decades of research, ALS remains a devastatingly cruel disease with no effective treatment. Considering the integral and diverse role ubiquitin signaling plays in neuronal cell function, it is probable that any disruption in the availability of this protein will have detrimental downstream consequences across a range of cellular pathways. We have shown that ubiquitin homeostasis is disrupted in cells expressing ALS associated proteins. The reduction of free monomeric ubiquitin and the modification of the ubiquitylated proteome indicate a cell in ubiquitin flux (Figure 1). Whether this disruption is the cause of proteostasis collapse in motor neurons remains to be determined but the clearance of ubiquitylated protein aggregates and strategies to enhance ubiquitin driven protein degradation remain promising therapeutic targets to reduce neuronal degeneration.
Figure 1.
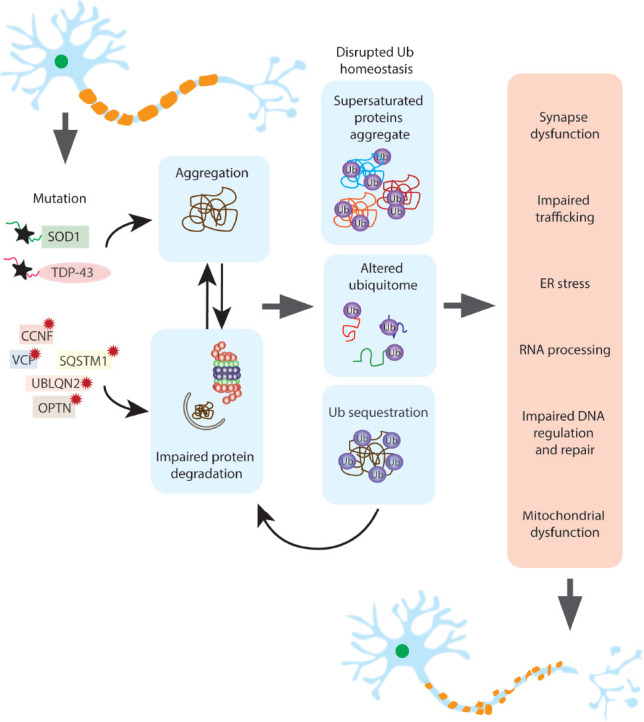
Disruptions to ubiquitin homeostasis may be responsible for motor neuron degeneration.
Mutations in ALS associated proteins including SOD1, TDP-43, FUS and C9ORF72 result in protein aggregation and proteasome and autophagy dysfunction. In addition, mutations in genes directly involved in protein degradation pathways such as OPTN, VCP and UBLQN2 also cause impaired proteostasis and protein aggregation. The consequence of aggregation and impaired proteostasis is the disruption of ubiquitin homeostasis through several mechanisms; sequestration of ubiquitin in the aggregates themselves, the precipitation of supersaturated proteins out of the soluble cellular matrix and their subsequent ubiquitinylation and the change in ubiquitylated proteins as a result of the presence of aggregates. This generates a feedforward loop where ubiquitin homeostasis disruptions cause further impairment of the proteostasis systems. In the final stage of this fatal cascade, the aggregation of integral proteins and the changes in ubiquitylation resulting in disruptions to protein supply in other cellular pathways leads to the degeneration and eventual death of the motor neuron. CCNF: Cyclin F; ER: endoplasmic reticulum; OPTN: optineurin; SQSTM1: sequestosome 1; SOD1: superoxide dismutase 1; TDP-43: TAR DNA-binding protein-43; UBLQN2: ubiquilin 2; VCP: valosin-containing protein.
This work was supported by an NHMRC Investigator Grant (No. 1194872, to JJY), a NHMRC Dementia Teams Grant (No. 1095215 both to JJY and NEF), a UOW-Yerbury Family Scholarship (to CGC), and a Fight MND Drug Development Grant (to JJY).
Additional files: Open peer review report 1 (80.8KB, pdf) .
Footnotes
Availability of data and materials: All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open peer reviewer: Fuminori Tokunaga, Osaka City University, Japan; Nico Dantuma, Karolinska Institutet, Sweden.
P-Reviewers: Dantuma N, Tokunaga F; C-Editors: Zhao M, Liu WJ, Wang L; T-Editor: Jia Y
References
- 1.Bennett CF. Therapeutic antisense oligonucleotides are coming of age. Annu Rev Med. 2019;70:307–321. doi: 10.1146/annurev-med-041217-010829. [DOI] [PubMed] [Google Scholar]
- 2.Brockington A, Ning K, Heath PR, Wood E, Kirby J, Fusi N, Lawrence N, Wharton SB, Ince PG, Shaw PJ. Unravelling the enigma of selective vulnerability in neurodegeneration:motor neurons resistant to degeneration in ALS show distinct gene expression characteristics and decreased susceptibility to excitotoxicity. Acta Neuropathol. 2013;125:95–109. doi: 10.1007/s00401-012-1058-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ciryam P, Tartaglia Gian G, Morimoto Richard I, Dobson Christopher M, Vendruscolo M. Widespread aggregation and neurodegenerative diseases are associated with supersaturated proteins. Cell Rep. 2013;5:781–790. doi: 10.1016/j.celrep.2013.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ciryam P, Lambert-Smith IA, Bean DM, Freer R, Cid F, Tartaglia GG, Saunders DN, Wilson MR, Oliver SG, Morimoto RI, Dobson CM, Vendruscolo M, Favrin G, Yerbury JJ. Spinal motor neuron protein supersaturation patterns are associated with inclusion body formation in ALS. Proc Natl Acad Sci U S A. 2017;114:E3935–3943. doi: 10.1073/pnas.1613854114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farrawell NE, Lambert-Smith I, Mitchell K, McKenna J, McAlary L, Ciryam P, Vine KL, Saunders DN, Yerbury JJ. SOD1A4V aggregation alters ubiquitin homeostasis in a cell model of ALS. J Cell Sci. 2018;131:jcs209122. doi: 10.1242/jcs.209122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farrawell NE, McAlary L, Lum JS, Chisholm CG, Warraich ST, Blair IP, Vine KL, Saunders DN, Yerbury JJ. Ubiquitin homeostasis is disrupted in TDP-43 and FUS cell models of ALS. iScience. 2020;23:101700. doi: 10.1016/j.isci.2020.101700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hallengren J, Chen PC, Wilson SM. Neuronal ubiquitin homeostasis. Cell Biochem Biophys. 2013;67:67–73. doi: 10.1007/s12013-013-9634-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kors S, Geijtenbeek K, Reits E, Schipper-Krom S. Regulation of proteasome activity by (post-) transcriptional mechanisms. Front Mol Biosci. 2019;6:48. doi: 10.3389/fmolb.2019.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller T, Cudkowicz M, Shaw PJ, Andersen PM, Atassi N, Bucelli RC, Genge A, Glass J, Ladha S, Ludolph AL, Maragakis NJ, McDermott CJ, Pestronk A, Ravits J, Salachas F, Trudell R, Van Damme P, Zinman L, Bennett CF, Lane R, et al. Phase 1-2 trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med. 2020;383:109–119. doi: 10.1056/NEJMoa2003715. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt MF, Gan ZY, Komander D, Dewson G. Ubiquitin signalling in neurodegeneration:mechanisms and therapeutic opportunities. Cell Death Differ. 2021;28:570–590. doi: 10.1038/s41418-020-00706-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yasuda T, Miyachi S, Kitagawa R, Wada K, Nihira T, Ren YR, Hirai Y, Ageyama N, Terao K, Shimada T. Neuronal specificity of α-synuclein toxicity and effect of Parkin co-expression in primates. Neuroscience. 2007;144:743–753. doi: 10.1016/j.neuroscience.2006.09.052. [DOI] [PubMed] [Google Scholar]
- 12.Yerbury JJ, Ooi L, Blair IP, Ciryam P, Dobson CM, Vendruscolo M. The metastability of the proteome of spinal motor neurons underlies their selective vulnerability in ALS. Neurosci Lett. 2019;704:89–94. doi: 10.1016/j.neulet.2019.04.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.