

Spinal muscular atrophy (SMA) is a neuromuscular disease caused by the homozygous mutation or deletion of the survival motor neuron 1 (SMN1) gene. A second copy, SMN2, is similar to SMN1, but produces only ~10% SMN protein because of a single-point mutation (C > T) in coding exon 7 causing a splicing defect which leads to the exclusion of exon 7, resulting in a majority (~90%) of transcripts lacking exon 7 that translate into mutant SMN (SMNΔ7) protein. SMA is caused by chronic low levels of SMN and is characterized by the degeneration of the spinal cord motor neurons leading to symmetrical skeletal muscle atrophy, respiratory failure, and death (Ahmad et al., 2012). Chronic low levels of SMN cause the accumulation of pathogenic R-loops and double-stranded breaks (DSBs) in DNA, leading to genomic instability and neurodegeneration in SMA (Kannan et al., 2018). The severity of SMA disease correlates inversely with SMN levels. The SMN2 gene is a promising target to produce higher levels of SMN by enhancing its expression. Cellular and molecular factors that may regulate the expression of SMN genes are slowly emerging, but precise molecular mechanisms which directly influence SMN expression are unclear. This perspective is focused on the potential role of the zinc finger protein ZPR1 as a molecular factor that may regulate the levels of mammalian SMN genes expression in vivo under the normal and disease conditions through a novel mechanism involving the resolution of R-loops that are formed during transcription (Kannan et al., 2020). The potential of ZPR1 as a therapeutic target for developing a new treatment is discussed in the context of currently available treatments for SMA.
In humans, ZPR1 gene is located on Chr11q23.2. The ZPR1 gene is evolutionarily conserved in eukaryotes ranging from yeast to mammals, and encodes a protein with two Cystine 4 (C4)-type Zn2+ fingers, each with unique helix-loop-helix motifs that may mediate its interaction with protein(s) and nucleic acids (Mishra et al., 2007). A genetic study has shown that mutation of the ZPR1 gene causes embryonic lethality in mice (Gangwani et al., 2005). Biochemical studies have provided insight into the function of ZPR1 in cell and neuron survival. ZPR1 interacts with SMN protein and its interaction with SMN is disrupted in the cells derived from severe SMA type I patients (Gangwani et al., 2001). ZPR1 mediates receptor tyrosine kinase signaling by binding to the cytoplasmic domain of epidermal growth factor receptor and platelet-derived growth factor receptor that are highly conserved among the family of receptor tyrosine kinases. In quiescent mammalian cells, ZPR1 binds to inactive epidermal growth factor receptor and platelet-derived growth factor receptor, upon mitogen or serum treatment, ZPR1 translocates from the cytoplasm to the nucleus and triggers mitosis suggesting that ZPR1 may play an important role in nuclear processes involving the expression of genes required for the cell growth and proliferation (Gangwani, 2006). ZPR1 deficiency causes defects in splicing and transcription (Gangwani et al., 2001; Gangwani, 2006). Reduced Zpr1 gene dosage in mice results in the downregulation of ZPR1 levels with aging, leading to the loss of motor neurons in the lumbar region of the spinal cord, axonal degeneration, peripheral (femoral and phrenic) nerve pathology, and mild SMA-like phenotype (Doran et al., 2006). Notably, reduced Zpr1 gene dosage in SMA mice results in downregulation of SMN protein levels and causes an increase in the loss of motor neurons, which increases respiratory distress and disease severity, and reduces the lifespan of SMA mice (Ahmad et al., 2012). Further, in vitro experiments show that ZPR1 overexpression rescues axonal growth defects and stimulates neurite growth of motor neurons derived from SMA mice (Ahmad et al., 2012). The selective inactivation of Zpr1 in cholinergic motor neurons results in developmental defects and perinatal lethality due to respiratory failure in mice (Genabai et al., 2017). In SMA patients, death is caused by respiratory failure; however, factors that contribute to respiratory failure in SMA are unclear. Notably, ZPR1-deficiency in motor neurons causes downregulation of developmen-related Hox genes, including HoxA5, which is required for the maintenance and function of phrenic nerve motor neurons critical for regulating respiration in mammals, suggesting that ZPR1 is critical for respiration and may contribute to SMA pathogenesis (Genabai et al., 2017). ZPR1 interacts in vivo with SMN and is required for the accumulation of SMN in sub-nuclear bodies, including gems and Cajal bodies (Gangwani et al., 2001, 2005). Recent data show that ZPR1 interacts in vivo and in vitro with RNAPII and is a part of transcription complexes (Kannan et al., 2020). A recent study demonstrated that SMN interacts with RNAPII and is a part of transcription complexes, and the disruption of RNAPII-SMN complexes by a mutation in RNAPII causes defects in transcription termination (Zhao et al., 2016). The direct interaction of ZPR1 with RNAPII suggests that ZPR1 and SMN interaction may be mediated in vivo by RNAPII. ZPR1 and SMN may play distinct roles in mRNA biogenesis including snRNP assembly and pre-mRNA splicing (Gangwani et al., 2001). ZPR1 may play a role in the regulation of pre-mRNA transcription by contributing to the resolution of co-transcriptional R-loops. Accumulation of R-loops is a major cause of DNA damage and genomic instability leading to human diseases, including neurodegenerative disorders (Kannan et al., 2020).
The binding of ZPR1 with RNAPII and its in vivo association with genomic SMN (5q13) locus suggest that ZPR1 may be a part of core transcription complexes (Kannan et al., 2020). However, whether ZPR1 is a specific trans-acting factor or transcriptional enhancer that has a specific DNA binding site, or has broad specificity and interacts with nucleic acids as a part of core transcription complexes and regulates transcription itself is unclear and requires further studies. Currently available data demonstrate that ZPR1 may play a role in the resolution of R-loops that are formed during transcription and regulate transcription; however, the correlation between the key molecular steps involved in R-loop resolution and the modulation of ZPR1-dependent transcriptional regulation remains to be examined. Nonetheless, knockdown of ZPR1 in HeLa cells results in accumulation of R-loops and DNA damage, which is consistent with data on the accumulation of R-loops in SMA patient cells that have low levels of ZPR1 suggesting that ZPR1 deficiency causes R-loop accumulation and contributes to genomic instability leading to neurodegeneration in SMA (Kannan et al., 2018). Because ZPR1 interacts with RNAPII and is a part of core transcription complexes, it is possible that ZPR1 may be required for the resolution of co-transcriptional R-loops. The graphical illustration of the molecular mechanism of SMN upregulation by ZPR1 and the rescue of SMA phenotype is presented in Figure 1.
Figure 1.
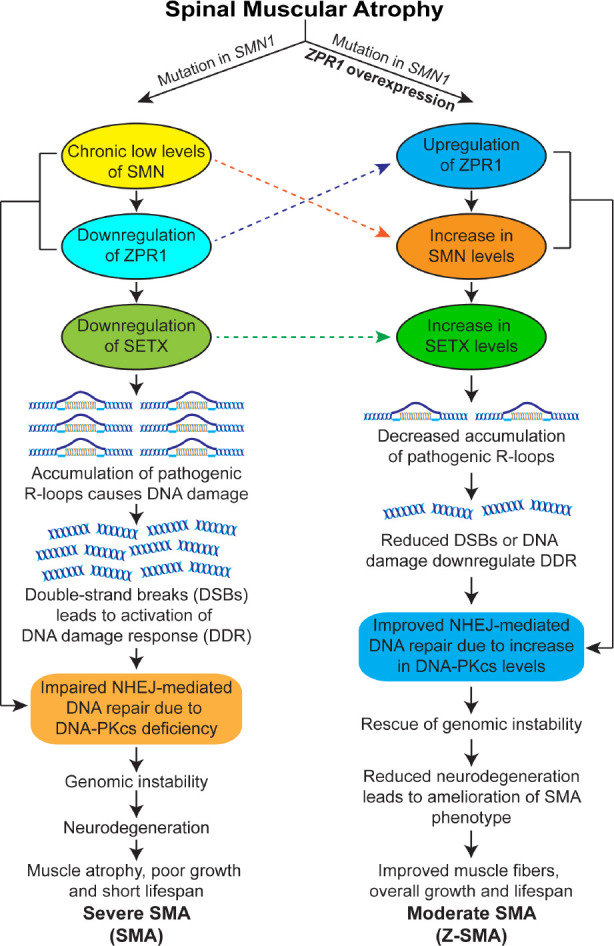
Mechanism of ZPR1-dependent rescue of spinal muscular atrophy disease phenotype.
Mutation in the survival motor neuron 1 (SMN1) gene leads to chronic low levels of the SMN protein. SMN deficiency causes downregulation of ZPR1 and senataxin (SETX), key proteins involved in R-loop resolution. Deficiency of these critical factors results in the accumulation of pathogenic R-loops, causing DSBs and leading to the activation of DNA damage response pathways, genomic instability and neurodegeneration in SMA. Furthermore, the chronic low levels of SMN and ZPR1 result in the deficiency of DNA-dependent protein kinase catalytic subunit (DNA-PKcs), which is critical for non-homologous end joining (NHEJ)-mediated DNA repair. Impairment of NHEJ leads to genomic instability, particularly in neurons (non-proliferating), which rely primarily on NHEJ-mediated DSB repair. R-loop-mediated genomic instability causes degeneration of the spinal cord motor neurons, leading to severe SMA disease with clinical manifestations, including muscle atrophy, reduced growth, and a short life expectancy. ZPR1 overexpression increases the levels of SMN and SETX, leading to decreased accumulation of pathogenic R-loops and rescue of DNA damage in SMA. ZPR1-dependent decrease in R-loop accumulation rescues genomic instability, prevents neurodegeneration, and ameliorates the severity of disease from severe to moderate SMA phenotype. DDR: DNA damage response; DNA-PKcs: DNA-dependent protein kinase catalytic subunit; DSBs: double-stranded breaks; NHEJ: non-homologous end joining; R-loops: RNA-DNA hybrids formed during transcription; SETX: senataxin; SMA: spinal muscular atrophy; SMN: survival motor neuron; Z-SMA: SMA mice with ZPR1 overexpression; ZPR1: zinc finger protein 1.
Modulation of ZPR1 levels in vitro by knockdown or overexpression in cultured cells causes downregulation or upregulation of recombinant SMN1-Luc and SMN2-Luc expression levels, respectively, suggesting a direct correlation between ZPR1 and SMN levels. This is supported by in vivo data of ZPR1 overexpression in SMA mice (Z-SMA, SMA mice with ZPR1 overexpression) on upregulation of SMN2 transcription and increase in SMN levels in Z-SMA mice. Z-SMA mice show ~3-fold increase in ZPR1 expression results in ~3-fold increase in SMN levels, which is further supported by in vitro data on upregulation of endogenous SMN2 transcription and SMN levels by ZPR1 in SMA patient fibroblast that show ~4–5-fold increase in ZPR1 expression results in an equivalent increase in SMN transcript and protein levels. The higher levels of SMN transcripts and protein in SMA patient cells complemented with recombinant ZPR1 compared to Z-SMA mice could be because of the three copies of SMN2 present in GM03813 and GM09677 cell lines (Kannan et al., 2020). These findings demonstrate that ZPR1 is a positive transcriptional regulator of the SMN2 gene. The data from recent studies, including (i) the effect of in vitro ZPR1 overexpression on the increase in SMN levels in SMA patient cells and the rescue of axonal growth defects in SMN-deficient motor neurons derived from SMA mice, and (ii) the effect of reduced Zpr1 gene dosage in Zpr1–/+ mice on neurodegeneration and development of mild SMA-like disease, (iii) reduced Zpr1 gene dosage in SMA mice results in increased disease severity with reduced lifespan, and (iv) the effect of in vivo ZPR1 overexpression in SMA mice results in amelioration of disease phenotype and increase in the lifespan SMA mice demonstrate that the modulation of ZPR1 levels regulates SMN levels in vivo under disease conditions and suggest that ZPR1 may be a true modifier of SMA (Doran et al., 2006; Ahmad et al., 2012; Kannan et al., 2020). Overall, findings of genetic studies support the idea that ZPR1 could be exploited for its therapeutic potential to treat SMA using the pharmacological method to upregulate ZPR1 expression, which in turn elevates SMN levels and opens the door of screening small molecule libraries to identify potent compounds that may upregulate ZPR1 levels.
Currently, there are three methods approved by the Food and Drug Administration in the United States for the treatment of SMA. The first method developed and approved in 2016 was based on the correction of SMN2 mRNA splicing by including exon 7 using an antisense oligonucleotide that targets the intronic splicing silencer N1 and this drug is commercially known as Spinraza or Nusinersen. The second method developed and approved in 2019 to restore SMN protein levels using non-replicating self-complementary adeno-associated viral vector carrying SMN1 gene (scAAV9-SMN1)-based gene therapy is commercially known as AVXS-101 or Zolegensma (omasemnogene abeparvovec). Recently (August, 2020), a third method approved was an orally deliverable small organic molecule that corrects the splicing of the SMN2 gene by increasing the inclusion of exon 7 in the mRNA, which is commercially known as Evrysdi or Risdiplam (Singh et al., 2020). The development of three methods of treatments for SMA represents excellent efforts and impressive progress by the scientific community to reduce the burden of illness on society. However, there are some challenges and gaps in the knowledge about important aspects of current SMA treatments, including post-treatment outcomes and side effects specifically associated with aging of groups of patients either treated at pre- or post-symptomatic stages, responders and non-responders, multi-organ rescue, and restoration of optimal amounts of SMN in a genetically diverse population with a broad spectrum of SMA disease severity are reviewed in detail in (Wirth, 2021). Recently developed SMA treatments have shown improvement in health, life style,and in the survival of SMA patients. However, these treatments are far from achieving the full rescue of SMA disease. Nevertheless, current SMA treatments have provided a solid foundation for developing new strategies to achieve the full rescue of SMA disease. Studies with animal models and clinical trials of patients indicate that the full rescue of SMA disease may require the development of additional SMN-dependent, SMN-independent and combinatorial methods. In addition to ZPR1, two other SMN-dependent modifiers were identified, namely Stasimon and Z+-Agrin. Several SMN-independent modifiers, including SETX (Kannan et al., 2018), plastin 3 (PLS3), and neurocalcin delta (NCALD) have been identified, which are discussed in detail in a recent review (Wirth, 2021). Clinical therapies with SMN-independent methods such as Reldesemtiv, a troponin activator of fast skeletal muscles, and a monoclonal antibody (SRK015) inhibiting myostatin, which facilitates muscle growth and differentiation have not yet gained enough momentum (Wirth, 2021). Notably, an increase in ZPR1 levels (~2–3-fold) is required to elevate SMN levels in different cells/tissues and rescue cellular/organism SMA phenotype (Kannan et al., 2020). Interestingly, in vivo ZPR1 overexpression > 3-fold did not cause toxicity or any adverse effects under normal and disease conditions in mouse models (Kannan et al., 2020). Thus, the recent identification and characterization of ZPR1 as an SMN-dependent modifier of SMA severity is of great potential to develop new combinatorial and standalone therapies by elevating SMN levels in a controlled fashion using the ZPR1-dependent method specifically in patients with mild forms of SMA Type II, III and IV to avoid unanticipated toxic effects of uncontrolled SMN overexpression. Two independent treatment methods could be developed to elevate SMN protein levels in a ZPR1-dependent manner using (a) small cell permeable organic molecule-based approach to increase ZPR1 mRNA expression and protein levels and (b) scAAV9-ZPR1-based gene therapy.
This work was supported by National Institutes of Health grant (R01 NS115834) awarded to LG. JC is a Research Mentee supported by NIH Diversity Supplement (3R01NS115834-01A1S1).
Footnotes
C-Editors: Zhao M, Zhao LJ, Qiu Y; T-Editor: Jia Y
References
- 1.Ahmad S, Wang Y, Shaik GM, Burghes AH, Gangwani L. The zinc finger protein ZPR1 is a potential modifier of spinal muscular atrophy. Hum Mol Genet. 2012;21:2745–2758. doi: 10.1093/hmg/dds102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doran B, Gherbesi N, Hendricks G, Flavell RA, Davis RJ, Gangwani L. Deficiency of the zinc finger protein ZPR1 causes neurodegeneration. Proc Natl Acad Sci U S A. 2006;103:7471–7475. doi: 10.1073/pnas.0602057103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gangwani L. Deficiency of the zinc finger protein ZPR1 causes defects in transcription and cell cycle progression. J Biol Chem. 2006;281:40330–40340. doi: 10.1074/jbc.M608165200. [DOI] [PubMed] [Google Scholar]
- 4.Gangwani L, Flavell RA, Davis RJ. ZPR1 is essential for survival and is required for localization of the survival motor neurons (SMN) protein to Cajal bodies. Mol Cell Biol. 2005;25:2744–2756. doi: 10.1128/MCB.25.7.2744-2756.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gangwani L, Mikrut M, Theroux S, Sharma M, Davis RJ. Spinal muscular atrophy disrupts the interaction of ZPR1 with the SMN protein. Nat Cell Biol. 2001;3:376–383. doi: 10.1038/35070059. [DOI] [PubMed] [Google Scholar]
- 6.Genabai NK, Kannan A, Ahmad S, Jiang X, Bhatia K, Gangwani L. Deregulation of ZPR1 causes respiratory failure in spinal muscular atrophy. Sci Rep. 2017;7:8295. doi: 10.1038/s41598-017-07603-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kannan A, Bhatia K, Branzei D, Gangwani L. Combined deficiency of Senataxin and DNA- PKcs causes DNA damage accumulation and neurodegeneration in spinal muscular atrophy. Nucleic Acids Res. 2018;46:8326–8346. doi: 10.1093/nar/gky641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kannan A, Jiang X, He L, Ahmad S, Gangwani L. ZPR1 prevents R-loop accumulation, upregulates SMN2 expression and rescues spinal muscular atrophy. Brain. 2020;143:69–93. doi: 10.1093/brain/awz373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mishra AK, Gangwani L, Davis RJ, Lambright DG. Structural insights into the interaction of the evolutionarily conserved ZPR1 domain tandem with eukaryotic EF1A, receptors, and SMN complexes. Proc Natl Acad Sci U S A. 2007;104:13930–13935. doi: 10.1073/pnas.0704915104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh RN, Ottesen EW, Singh NN. The first orally deliverable small molecule for the treatment of spinal muscular atrophy. Neurosci Insights. 2020;15:2633105520973985. doi: 10.1177/2633105520973985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wirth B. Spinal muscular atrophy:in the challenge lies a solution. Trends Neurosci. 2021;44:306–322. doi: 10.1016/j.tins.2020.11.009. [DOI] [PubMed] [Google Scholar]
- 12.Zhao DY, Gish G, Braunschweig U, Li Y, Ni Z, Schmitges FW, Zhong G, Liu K, Li W, Moffat J, Vedadi M, Min J, Pawson TJ, Blencowe BJ, Greenblatt JF. SMN and symmetric arginine dimethylation of RNA polymerase II C-terminal domain control termination. Nature. 2016;529:48–53. doi: 10.1038/nature16469. [DOI] [PubMed] [Google Scholar]