

Abstract
The construction of fully bio-based epoxy resins (EP) has been of particular interest in both academia and industrial circles for years; among these, epoxidized soybean oil (ESO) derived thermosets have received the most attention, but they usually exhibit poor performance due to their flexible fatty chains. Herein, tannic acid (TA), with its great degree of functionality and massive aromatic structures, was chosen as the multi-phenol curing agent for ESO to prepare fully bio-based EP thermosets with a high relaxation temperature and satisfactory mechanical properties. As a natural 2-substituted imidazole-containing substance, histidine (H) was used as the curing accelerator under moderate curing conditions (120–180 °C). This EP system showed high curing activity and a good curing degree while operating. The cured thermosets were found to be thermally stable (T5% > 270 °C) and displayed a high relaxation temperature (77 °C) with a tensile strength of 23 MPa. Preliminary adhesion tests showed that the cured product exhibited a high lap-shear strength of about 19 MPa in adhesion failure mode. Taking these advantages into account, this kind of fully bio-based EP could introduce more chances for versatile applications, such as being used in structural materials and construction adhesives.
Fully bio-based EP thermosets from epoxidized soybean oil and tannic acid were prepared for versatile applications under moderate curing conditions.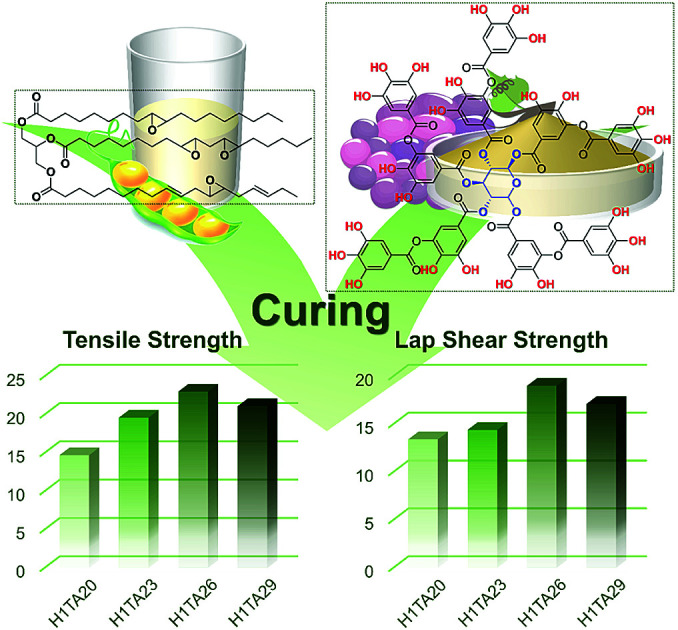
Introduction
Cured from different epoxy resins (EP) with multi-epoxide groups in the presence of apposite curing agents (or hardeners), epoxy thermosets represent an important class of thermosetting polymers, which have found versatile applications, including in adhesives and coatings, and for electrical and electronic purposes, as well as being used in high-performance composites, owing to their excellent mechanical strength, thermal and chemical resistance, strong adhesion, and low shrinkage after curing.1–4 Globally, EP accounts for approximately 70% of the market of thermosetting polymers;5 the gross output of EP has been ever-increasing and was evaluated to have reached 3 million tons by 2017.6 Unfortunately, it is noteworthy that about 90% of EP is derived from the petroleum-based and non-renewable diglycidyl ether of bisphenol A (DEGBA), thus the rapid growth of EP causes the depletion of fossil resources. Moreover, the raw materials for DGEBA are bisphenol A (BPA) and epichlorohydrin (ECH), which can also cause some negative effects on living organisms and ecosystems that cannot be ignored.7,8 As a result, awareness about environmental deterioration and dependency on a fossil feedstock that is depleting forces both academia and industrial circles to find a solution in order to design a more sustainable future. In the EP field in particular, exploring sustainable epoxy thermosets, including both EP and curing agents from renewable resources, has been an area of ever-increasing concern in recent years.9–12 Different biomass resources, through years of research, such as plant oils,13–16 isosorbide,17,18 itaconic acid,19,20 lignin derivatives,21 vanillin,22 and sugar,23–25 have been used to design and prepare bio-based epoxy thermosets.
Among the different kinds of renewable raw materials, epoxidized soybean oil (ESO) has attracted great attention in both scientific and industrial areas for fabricating lubricants, coatings, paints and bioplastics in recent years, making it an ideal renewable alternative to fossil based epoxies.26–32 Nevertheless, the long aliphatic chains in ESO endow resulting materials with specific characteristics such as elasticity and flexibility, but also a relatively low glass transition temperature and poor mechanical strength, considerably limiting their practical application.33–36
Generally, the final properties of epoxy thermosets depend strongly on their chemical structures, which are determined by the types of both EP and curing agents used. In order to construct high-performance ESO-based thermosets, it is necessary to find a naturally rigid structure to compensate for the deficiencies of ESO. Among the different renewable substances with aromatic structures, lignin and tannic acid (TA) are the two most important and widely available ones; these bio-based polyphenols continue to generate ever-increasing interest for the development of aromatic polymer architectures or additives for material formulations. Meanwhile, these bio-based polyphenols can also be utilized to prepare EP as a substitute to BPA. Many chemical modifications of lignin and TA have been described for these purposes.37–39 As a type of bio-based polyphenol, TA has abundant reactive terminal phenolic hydroxyl groups with a hyperbranched aromatic/alicyclic polyester core, which can make up for the flexible chain defects of ESO and form highly crosslinked networks. The chemical formula for commercial TA is often given as C76H52O46, which corresponds with decagalloyl glucose,40 as shown in Scheme 1. Architecturally, it can be supposed that, as either a starting material for EP or a type of polyphenol curing agent, the multi-functional groups and rigid ring structure of TA will impart high density and strong behavior on resulting materials.
Scheme 1. Typical chemical structure of TA (C76H52O46).

For instance, Aouf et al.41,42 prepared a novel bio-based epoxy thermoset from natural polyphenols (TA and gallic acid, which is found both free and as part of hydrolyzable TA), where a two-step synthesis involving the allylation of OH groups followed by the epoxidation of the resulting double bonds was reported. Preliminary results showed that this new EP based on polyphenols displayed interesting physical and mechanical properties close to those of commercial DGEBA. Shibata et al.43,44 explored TA as a curing agent with glycerol polyglycidyl ether (GPE), sorbitol polyglycidyl ether (SPE), and ESO to form biobased EP thermosets. The most balanced thermal and mechanical properties were obtained at a 1 : 1 epoxy (with either GPE or SPE) to hydroxyl (TA) ratio; whereas, a slightly higher ratio was necessary for ESO (1 : 1.4), due to additional steric hindrance from the epoxy groups along the fatty chains of ESO. As a result, the Tg value, tensile strength, and modulus of the ESO thermoset were much lower than those of the GPE and SPE systems. Even worse, the relatively low reactivity of the polyphenol groups in TA let to a high curing temperature of 210 °C for the ESO/TA thermoset, which is higher than the boiling point of ESO (150 °C at 0.53 kPa),45 and thermal/thermo-oxidative decomposition at such high temperatures for these aliphatic chains of ESO is also worthy of consideration. In this case, curing accelerators are extremely necessary for the aforementioned system.
To the best of our knowledge, examples are lacking of EP thermosets prepared from TA and ESO through moderate curing addition with appropriate curing accelerators. In this work, fully bio-based EP thermosets were prepared through mixing TA/ESO in different mass ratios in the presence of histidine (H) as a curing accelerator at relatively moderate temperatures. The performance of the cured resins was controlled by adjusting the ratio of TA to ESO. The curing behavior and comprehensive properties of such fully bio-based EP thermosets were investigated. Consequently, the obtained thermosets displayed high thermal stability and satisfactory tensile strength and lap shear strength, indicating their potential for versatile applications.
Experimental section
Reagents and materials
Tannic acid (98%, AR), ethanol and acetone (AR) were provided by Kelong Chemical Reagent Co., Ltd. (Chengdu, China). Epoxidized soybean oil with an average number of epoxy groups per molecule of 4 was provided by Aladdin Chemical Reagent Co., Ltd. (Shanghai, China). Histidine (99%, AR) was provided by the Energy Chemical Co., Ltd. (Shanghai, China). All chemicals were used without further purification.
Curing
TA was formulated with ESO at different mass ratios of TA/ESO (20%, 23%, 26% and 29%; and the relevant molar ratios of –OH/epoxide were 0.8, 1.0, 1.2, and 1.4, respectively) via a solvent blending method to achieve homogeneous mixtures. Briefly, stoichiometric TA and ESO or E51 were put into a 100 mL glass scintillation vial followed by the addition of ethanol to dissolve and mix the monomers. Ethanol was then removed via rotary evaporation at 50 °C, and the resulting EP was dried under high vacuum as a viscous liquid for later use. Bubbles were removed from the viscous liquid EP in a vacuum oven and 1% H was added; the component was then poured into a Teflon mold. Then, the system was pre-cured at 120 °C for 2 h and post-cured at 180 °C for 5 h for further testing. Samples were coded as Hx/TAy, where x = 0 or 1 and y = 20, 23, 26 or 29 with regard to the mass ratios of H and TA, respectively.
Preparation for lap shear adhesion tests
Single lap shear strips were clipped from a stainless steel slice with dimensions of 150 mm (length) × 25 mm (width) × 2.0 mm (thickness). Prior to thermal curing, single-lap-joint specimens were prepared through loading appropriate amounts of viscous Hx/TAy samples between the stainless steel strips with a bonding area of about 13 mm × 25 mm. To ensure a uniform bonding thickness, slides with a thickness of 0.2 mm were placed in the resin bonding area. Test specimens were put into an oven to cure the resin for 2 h at 120 °C; the samples were compressed using a vulcanizing machine and then subjected to a post-curing step for 5 h at 180 °C.
Differential scanning calorimetry (DSC) characterization
DSC thermograms were recorded using a TA Q200 DSC (TA, USA) under a N2 atmosphere. About 4 mg of sample was placed in an aluminum DSC pan at 40 °C for 1 min and this was then heated up to 300 °C at a constant rate; after keeping the sample at this temperature for 1 min, it was finally cooled to 40 °C at the same rate. The final heating curves were recorded for thermal transition analysis, including curing condition and non-isothermal curing kinetics investigations. The isothermal TA-ESO curing kinetics were also investigated by monitoring the heat flow evolution during curing with a TA Q200 DSC apparatus. The isothermal runs were performed at temperatures ranging from 140 to 200 °C for TA-ESO without H and from 80 to 140 °C for H-accelerated TA-ESO curing.
Fourier transform infrared (FT-IR) characterization
Samples were prepared as KBr pellets and spectra were recorded using a spectrophotometer (Nicolet 6700, USA) over a wavenumber range from 4000 to 400 cm−1. The resolution and number of scanning cycles were 4 cm−1 and 32, respectively.
Gel fraction measurements
Powdered cured samples wrapped in dried filter paper were precisely weighed (M1), then extracted with acetone for 48 h under reflux using a Soxhlet extractor, and finally dried at 80 °C under vacuum for 6 h and weighed (M3): M2 is the quality of the dry filter paper. Each group of the cured samples was tested in triplicate. The gel content is calculated using eqn (1).
 |
1 |
Dynamic mechanical analysis (DMA)
The dynamic mechanical behaviors of tested samples, including storage moduli and tan δ values, were measured using a TA Instruments DMA Q800 analyzer via a three-point bending model with an oscillation frequency of 1 Hz. The testing temperature ranged from 0 to 160 °C at a heating rate of 5 °C min−1. Cured samples with dimensions of 20 mm (length) × 10 mm (width) × 4 mm (thickness) were used for testing.
Thermogravimetric analysis (TGA)
The thermal stability of the cured samples was evaluated using TG 209 F1 apparatus (NETZSCH, Germany). Samples (about 5 mg) were heated from 40 to 700 °C at a heating rate of 10 °C min−1 under N2 at a flow rate of 50 mL min−1.
Tensile tests
Tensile testing was conducted using an Instron Universal Testing Machine (Model 3366, Instron Engineering Corp., Canton, MA, USA) with a 1 KN sensor and a speed of 1 mm min−1 at room temperature. Reactive mixtures containing ESO, TA and H were cured on a Teflon plate. After being cured, samples were cut into dumbbell shapes with a width and thickness of 4.0 and 2.0 mm, according to GB/T 1040.2-2006 (plastics – determination of tensile properties). The initial distance between the grips was 20 mm, and at least five measurements were performed for each sample.
Characterization of adhesion ability
Adhesive lap shear testing was conducted in tension mode on an Instron Universal Testing Machine with a load cell (10 kN) at a free crosshead speed of 5 mm min−1, according to ASTM D1002. Each group of cured samples was tested in quadruplicate. The lap shear strength was calculated using eqn (2).
 |
2 |
Results and discussion
Curing
Fig. 1a shows DSC heating scans of H0/TA23 (the molar ratio of –OH/epoxide is 1) and H1/TA23 at a heating rate of 9 °C min−1. The exothermic peak temperature of H0/TA23 was 210 °C with an exothermic area ranging from 130 to 300 °C. Polyphenol curing agents can polymerize with epoxides via ring-opening to form polyethers upon heating (Scheme 2a). Unfortunately, as illustrated both in the DSC scans and the existing literature,42 the relatively low reactivity of the polyphenol groups in TA, and the additional steric hindrance of the epoxy groups along the fatty chains of ESO resulted in extremely low curing efficiency. In contrast, for H1/TA23, the exothermic peak temperature significantly decreased to 150 °C and the curing process finished in the temperature range of 120 to 180 °C, which was much narrower than the curing exothermic peak of H0/TA23, indicating that the presence of H could significantly improve the curing reactivity and efficiency. The curing reaction of EP with 2,4-substituted imidazole has been well established before,46–48 as shown in Scheme 2b. At the very beginning, pyridine-type nitrogen opened the oxirane ring via nucleophilic addition to form the 1 : 1 adduct. Next, intramolecular H+ transfer changed pyrrole-type nitrogen into pyridine-type nitrogen; then the newly generated pyridine-type nitrogen reacted with an oxirane ring to form the 1 : 2 adduct containing a reactive alkoxide anion (–O–) and hydroxyl (–OH). Finally, polymerization and further crosslinking were induced by alkoxides and hydroxyl groups via etherification reactions. Besides being the main curing agent, imidazole could also act as a curing accelerator for different curing agents, such as phenols.49–52 In this case, as a result of the high reactivity of nucleophilic addition between pyridine-type nitrogen in the imidazole ring and epoxy, a ring-opening reaction was firstly initiated to form alkoxide anions, as illustrated in Scheme 2b. Afterward, in the presence of phenolic hydroxyl, anionic homo-polymerization (etherification propagation) was suppressed, since the alkaline alkoxide anions were much stronger than the phenoxide anion, suggesting that the reaction rate of the acid–base reaction that formed the imidazole/phenol complex was much higher than that of etherification (Scheme 2c). Comparatively, for ring-opening polymerization and further crosslinking, the generated phenoxide anions in the imidazole/phenol complexes exhibited much higher reactivities than the phenols themselves. As a consequence, the curing reaction could be effectively accelerated by histidine (Scheme 2d). As indicated from the DSC heating scans, temperatures of 120 and 180 °C were selected for the pre-curing and post-curing of Hx/TAy.
Fig. 1. (a) DSC curves of H0/TA23 and H1/TA23 (at a heating rate of 9 °C min−1). (b) FT-IR spectra of ESO and ternary H1/TA23 after pre-curing and post-curing. (c) The gel fractions of cured thermosets with different TA content: the error bars represent standard deviation from three specimens. (d) FT-IR spectra of cured thermosets with different TA content.

Scheme 2. The proposed curing process of ESO. (a) The reaction between phenol and epoxy via anion addition; (b) the reaction between 2,4-substituted imidazole and epoxy via two-step nucleophilic addition; (c) initiation by an MZ/phenol complex and a 1 : 1 adduct/phenol complex generated from ionic interactions between TA and the pyridine-type nitrogen in histidine (H) and a 1 : 1 adduct of H and epoxy, respectively, and (d) a schematic diagram of the curing networks of ESO and TA at the presence of H.

FT-IR was used to assess the degree of the curing reactions. FT-IR spectra of the H1/TA23 ternary mixture after pre-curing and post-curing were recorded and are shown in Fig. 1b. For comparison, the FT-IR spectrum of pristine ESO was also recorded. As can be observed, the peak ascribed to the oxirane rings at 840 cm−1 gradually disappeared after thermal curing, and the peak from the –CH2– vibration at 2920 cm−1 remained nearly unchanged after thermal curing. Compared with pristine ESO, the absorption peak intensity of the epoxy groups showed no obvious reduction after the pre-curing process (120 °C for 2 h), which meant that a considerable amount of epoxy groups remained. After being heated at 180 °C for 5 h, the epoxy groups almost disappeared after the post-curing process. From calculations, the ratio of the absorbance of the epoxy groups to the –CH2– groups was 0.16 in ESO, and 0.080 and 0.030 for H1/TA23 samples after pre-curing and post-curing, respectively. These results indicate that residual epoxy groups were continuously reduced, as the epoxy groups underwent complicated curing reactions, as shown in Scheme 2, where two types of crosslinking networks were established: the first was generated from the reaction between epoxy and alkoxide anions, initiated by imidazole, while the second one relied on abundant reactive terminal phenolic hydroxyls with hyperbranched aromatic/alicyclic polyester cores, which exhibited potential positive effects on the further properties of the thermosets.
Gel content, as a key indicator relating to the formation of a desired crosslinked network, presents a direct relationship with the final properties of cured samples. Herein, the gel content values of thermosets with different TA content were examined and they are shown in Fig. 1c. It is clear that all the samples showed a high gel content ranging between 83.1 to 88.2%, which indicated that ESO was well cured by TA in the presence of H. The gel content increased slightly with increasing TA content, and further decreased when the TA content was 29 wt%, this could be extracted with acetone during Soxhlet extraction, resulting in the observed decreased gel content for H1/TA29. Further evidence could be seen from Fig. 1d that the H1/TA20 and H1/TA23 cured products maintain a small number of epoxy groups, while the epoxy groups in the H1/TA26 cured product completely disappeared. According to former research from a kinetics study of imidazole-cured epoxy-phenol resins,51,52 as the phenol concentration is increased and that of imidazole is decreased in turn, the polarity of the curing system is raised and consequently the reaction constant for chain propagation through phenoxide anion becomes larger, suggesting a positive effect on curing (Scheme 2c). However, as the TA content is further increased, the initiation of epoxy-alkoxide anions is delayed by the formation of imidazole/phenol complexes. Furthermore, the steric hindrance from phenol hydroxyls around the aromatic core in TA becomes more obvious as the TA content reaches its maximum. As a result, H1/TA29 cured products maintained a small number of epoxy groups, indicating that curing was incomplete due to an excessive amount of TA in the H1/TA29 samples.
To further investigate the curing kinetics, the non-isothermal curing behavior of H0/TA23 and H1/TA23 was investigated via DSC at different heating rates from 6 to 15 °C min−1. Fig. 2a and b show non-isothermal curing scans of H0/TA23 and H1/TA23 systems as typical examples for comparison. It's obvious that the exothermic peak temperature (Tp) increased with an increase in the heating rate. The activation energies of H0/TA23 and H1/TA23 for the curing reaction were determined from the peak temperatures (Tp) at different heating rates from the non-isothermal curing scans. According to Kissinger's method, the activation energy can be calculated from the peak temperatures at different heating rates following the Kissinger equation53 (eqn (3))
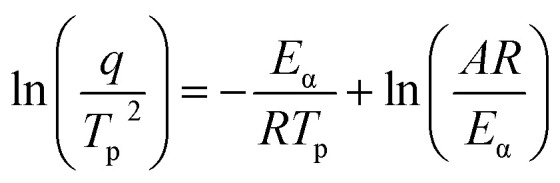 |
3 |
where q represents the heating rate of the non-isothermal curing scan, Eα denotes the activation energy, A indicates a pre-exponential factor and R stands for the gas constant (8.314 J mol−1 K−1). As shown in Fig. 2c, Eα was calculated from the slope of a linear fitting plot of ln(q/Tp2) vs. 1/Tp. Similarly, Ozawa's theory54 can also be used to calculate the activation energy, as presented in eqn (4)
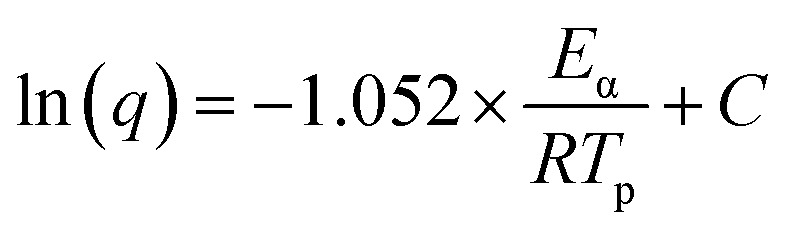 |
4 |
where C is a constant. Accordingly, Eα could be calculated from the slope of a linear fitting plot of ln(q) vs. 1/Tp, as shown in Fig. 2d. For H0/TA23, Eα values were calculated as 62.7 kJ mol−1 (Kissinger method) and 67.6 kJ mol−1 (Ozawa method), much higher than the calculated Eα values for a sample of H1/TA23 (46.4 kJ mol−1 and 50.4 kJ mol−1). These results suggest that the 2-substituted imidazole group in H could considerably accelerate the curing reaction, which is in good agreement with the aforementioned results (Fig. 1a). Based on this, moderate curing conditions for this kind of fully bio-based epoxy thermoset were established, which greatly reduced the curing temperature for traditional phenol/ESO thermoset systems.
Fig. 2. A non-isothermal curing kinetics study. (a) DSC curves of H0/TA23 at different heating rates (6, 9, 12, and 15 °C min−1); (b) DSC curves of H1/TA23 at different heating rates (6, 9, 12, and 15 °C min−1); (c) a linear plot of ln(q/Tp2) versus 1/Tp according to Kissinger's method; and (d) a linear plot of ln(q) versus 1/Tp according to Ozawa's method.

Isothermal curing kinetics were also investigated by monitoring the heat flow evolution during curing.55,56 The isothermal runs were performed at temperatures ranging from 140 to 200 °C for TA-ESO without H and from 80 to 140 °C for H-accelerated TA-ESO curing. The results of heat flow analysis, including the activation energies for H0/TA and H1/TA composition are shown in the ESI.† These results suggested that the 2-substituted imidazole group in H considerably improved the reactivity of the curing reaction, which was in good agreement with the aforementioned non-isothermal results, as summarized in Fig. 2.
Thermal properties of the cured epoxy thermosets
DMA was used to determine the relaxation temperature (Tα), modulus values, and to calculate the crosslinking densities of the cured samples. Herein, curves of tan δ and the storage modulus values of Hx/TAy samples as a function of temperature are illustrated in Fig. 3, in which the peak values of tan δ were used to determine the Tα values of the cured samples. The modulus values at a temperature of Tα + 60 °C were used to calculate the crosslinking densities of the cured samples according to eqn (5)57,58
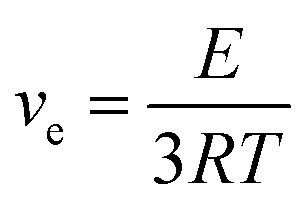 |
5 |
where E stands for the storage modulus of a cured sample in the rubbery plateau region, T denotes the absolute temperature, and R is the gas constant. As presented in Table 2, generally, the relaxation temperature (Tα) and storage modulus values at 25 °C of the cured thermosets firstly increased with increasing TA content: the H1/TA26 sample possessed the highest modulus of 1103 MPa at room temperature (25 °C), corresponding to its highest Tα value of 77 °C. However, the modulus and Tα values of the H1/TA29 cured product were slightly lower than those of H1/TA26 samples. The crosslinking densities of the cured thermosets were calculated from the modulus of the rubber platform, which decreased as the TA content further increased from 26% to 29%. This suggested that the Tα and modulus values of the crosslinked thermosets not only related to the rigidity of the chain segments but were also related to the crosslinking density, in accordance with the results from gel fraction and FT-IR studies, as illustrated in Fig. 1c and d.
Fig. 3. DMA curves of the cured thermosets.

Thermogravimetric data for the cured thermosets.
| Sample | T 5% a (°C) | T max b (°C) | R c (%/min) | W 700 d (%) |
|---|---|---|---|---|
| H1/TA20 | 306.7 | 387.5 | −11.8 | 6.4 |
| H1/TA23 | 291.6 | 381.5 | −11.5 | 6.5 |
| H1/TA26 | 287.1 | 381.9 | −10.1 | 8.3 |
| H1/TA29 | 270.7 | 380.4 | −9.5 | 8.7 |
T 5% denotes the temperature of 5 wt% sample weight loss.
T max defines the temperature of maximum weight loss.
R refers to the weight loss rate at Tmax.
W 700 stands for the residual weight at 700 °C.
The glass transition (Tg) values of the cured samples were investigated via DSC, as listed in Table 1. Very similar to the tendency of Tα, Tg values of thermosets cured with different TA content amounts firstly increased to a maximum (63 °C for H1/TA26), then decreased due to a lower crosslinking density. It should also be noted that the Tg (or Tα) values were much higher than for other ESO-derived thermosets (Table 4).
Thermal properties data for the cured thermosets.
| Sample | T α a (°C) | T g b (°C) | Modulus at 25 °C (MPa) | Modulus at Tα + 60 °C (MPa) | ν e (mol m−3) |
|---|---|---|---|---|---|
| H1/TA20 | 66 | 54 | 630 | 6.23 | 626 |
| H1/TA23 | 72 | 62 | 937 | 5.75 | 569 |
| H1/TA26 | 77 | 63 | 1103 | 4.54 | 444 |
| H1/TA29 | 74 | 57 | 1081 | 1.76 | 203 |
T α is defined as the peak temperature of tan δ, recorded via DMA.
T g denotes the glass transition temperature, tested via DSC.
A comparison between results from this work and previous work: only the best values from each system are summarized.
| Formulation | Curing temperature (°C) | Thermal and mechanical properties | Reference | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Epoxy resin | Curing agent | Accelerator | T α a (°C) | T g (°C) | T 5% (°C) | Tensile strength (MPa) | Lap shear strength (MPa) | ||
| ESO | Dicarboxyl-terminated polyamide | n/a | 210 | 23 | 3.5 | 381 | 25.8 | n/a | 36 |
| ESO | Tannic acid | n/a | 210 | 57 | n/a | n/a | 12.7 | n/a | 44 |
| Henkel LOCTITE® EA 3982 | 125 | n/a | 89b | n/a | 57.0 | 13.8 | 57 | ||
| Henkel LOCTITE® EA 3984 | 125 | n/a | 74b | n/a | 38.0 | 13.8 | 58 | ||
| ESO | Tannic acid | Histidine | 120; 180 | 77 | 63 | 287 | 23.0 | 19.0 | This work |
T α represents the tan δ peak temperature from DMA studies.
The glass transition temperature is tested according to ASTM E228.
The thermal stabilities of the cured samples under nitrogen were investigated via thermogravimetric analysis (TGA). Thermogravimetric (TG) and derivative thermogravimetric (DTG) curves are presented in Fig. 4, and the corresponding data are listed in Table 2. The degradation temperatures at 5 wt% weight loss (T5%, defined as the initial decomposition temperature) of the cured materials were around 300 °C. T5% of the thermosets generally decreased with increasing TA content. This might be due to lower cross-linking density in network formed at higher –OH/epoxide ratios (unreacted phenol hydroxyls could remain in the networks). However, the excess of epoxide at lower –OH/epoxide ratios could potentially still participate in polymerization via primary and secondary hydroxyl groups from the ring-opening of ESO. The weight loss rate at the temperature of maximum weight loss decreased and the residual weight at 700 °C was higher with increasing TA content. That is, the thermal stability at high temperatures was enhanced by introducing the TA component. Although the TA content affected the thermal stability of the thermosets, it is worth noting that all samples showed good thermal stability since they all had an onset temperature of about 300 °C.
Fig. 4. TG (a) and DTG (b) curves of the cured thermosets under a N2 atmosphere.

Tensile properties of the cured epoxy thermosets
In general, the chemical structures of the curing agent and EP exhibit significant effects on the crosslinking density of the polymer network and hence determine the mechanical properties of the resulting thermosets. As shown in Fig. 5 and Table 3, the tensile properties of cured thermosets increase and then decrease as the mass ratio of TA : ESO increases from 20% to 29%, and the highest tensile strength of 23 MPa was obtained by the curing product H1/TA26. As illustrated in the aforementioned results, due to the delaying effect of the formation of imidazole/phenol complexes on the initiation of epoxy-alkoxide anions, and the steric hindrance of the phenol hydroxyls around the aromatic-aliphatic polyester core in TA, the H1/TA29 sample exhibited a relatively lower crosslinking density, therefore the tensile strength slightly decreased with a large sacrifice of elongation at break, indicating that high mechanical strength could be simultaneously attributed either to the rigid structure or to the highly crosslinking density. The tensile modulus of the resulting polymers increased as the TA content increased, which was in accordance with the increase of the rigid component. The mechanical performance of this fully bio-based epoxy thermoset was lower than those of commercial products from petroleum-derived feedstocks, which was likely due to the presence of an aliphatic epoxy structure. Combining their mechanical performance and thermal properties, it was worth noting that the comprehensive properties of TA-cured ESO thermosets were better than those of other ESO-based thermosets (Table 4).
Fig. 5. Representative stress–strain curves from the cured thermosets at different mass ratios.

Tensile data from the cured thermosets.
| Sample | Tensile strength (MPa) | Tensile modulus (MPa) | Elongation at break (%) |
|---|---|---|---|
| H1/TA20 | 14.7 ± 1.4 | 181 ± 23 | 35 ± 3 |
| H1/TA23 | 19.6 ± 2.1 | 376 ± 15 | 33 ± 4 |
| H1/TA26 | 23.0 ± 1.8 | 434 ± 20 | 28 ± 2 |
| H1/TA29 | 21.1 ± 1.6 | 450 ± 26 | 6 ± 2 |
Adhesion strength of the cured epoxy thermosets
The lap shear adhesion strength of the cured thermosets was measured to evaluate their potential application as structural adhesives, as shown in Fig. 6a. The cured epoxy thermosets were placed between two stainless steel adherends and pulled to failure in two typical modes: the first was adhesion failure, where failure occurred between the adhesive and the surface of the adherends (i), suggesting the adhesion forces holding the adhesive and substrates together, including hydrogen bonding, electrostatic interactions, dispersion, or mechanical interlocking became the failure points; and the second was cohesive failure (ii), indicating that the adhesive itself was broken under external stress, which relies on intra-/intermolecular forces and the viscoelastic properties of the polymers. In this case, where the tensile strength of the H1/TA26 sample was 23 MPa, the bonding area after pulling the two adherends apart was examined, as shown in Fig. 6b, and residual adhesive remained on one adherend surface, indicating typical adhesive failure; the adhesive sample itself could resist the shear force in other words. Representative force–extension curves from adhesive single-lap-joints made from Hx/TAy are shown in Fig. 6c. For a more detailed comparison, the adhesion strengths were calculated from the force–extension curves and are illustrated in Fig. 6d. Similar to the tendency of tensile strength, the lap shear strength increased and then decreased as the mass ratio of TA/ESO gradually increased from 20% to 29%, and the adhesive H1/TA26 had the highest lap shear adhesion strength value of 19 MPa. This value was higher than the manufacturer-claimed lap shear strength of the LOCTITE® epoxy adhesives 3892 and 3894 (13.8 MPa)59,60 used for multiple surface bonding (Table 4), suggesting potential application as a structural adhesive. Generally, a higher loss modulus (viscous modulus) in an adhesive usually results in low cohesion; while higher storage modulus (elastic modulus) values usually result in adhesion failure. The observed high lap shear adhesion strength was to some extent attributed to rigid moieties in the curing networks. Also the high polar group content, including residual phenol hydroxyls, ether-linkages and aromatic-aliphatic esters, created high adhesion forces on the surface of stainless steel.
Fig. 6. Lap shear bonding test data. (a) Polymer glue is placed between two adherends and pulled to failure: (i) adhesion failure is failure between the adhesive and the surface of the adherends; while (ii) cohesion failure is failure within the adhesive. (b) A photograph of a lap shear test specimen (H1/TA26) after pulling apart. (c) Representative force–extension curves for an adhesive single-lap-joint made from Hx/TAy. Maximum adhesion is indicated by the peak of the extension (in millimeters) versus force (in newtons) plot. (d) The adhesion lap shear strength values of Hx/TAy cured at different ratios. Error bars represent standard deviation from four specimens.

Conclusions
For decades, bio-based EP derived from sustainable resources such as polyphenols has offered tremendous potential for creating new polymeric materials. In this work, we reported a facile way of fabricating fully bio-based and high-performance epoxy thermosets through the curing of ESO with TA. To accelerate the curing reaction, a natural imidazole-containing substance, histidine (H), was introduced herein. The process for the preparation of EP was environmentally friendly, without the use of any toxic chemicals, showing great potential for manufacturing sustainable materials. As a highly efficient curing accelerator, H significantly reduced the activation energy and increased the reaction rate, and DSC experiments revealed that the samples (Hx/TAy) could be thermally cured at ∼120–180 °C, which were much lower temperatures than for examples reported previously. The thermosets were thermally stable with an initial decomposition temperature higher than 270 °C. Both tensile testing and thermal mechanical analysis suggested that the thermosets displayed satisfactory mechanical and thermal properties (tensile strength of 23 MPa and relaxation temperature of 77 °C), which were attributed to a high crosslinking density and rigid segments. At the same time, thermosets of H1/TA26 exhibited the highest adhesion lap shear strength of 19 MPa, which was comparable to commercial adhesives. With excellent comprehensive properties, this kind of fully bio-based EP can have more chance of achieving versatile applications, such as use in structural materials and construction adhesives, and this is also a helpful and fruitful attempt for the future design and fabrication of sustainable materials.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
Financial support from the National Natural Science Foundation of China (grant no. 51761135132 and 51773137) and the Sichuan Province Youth Science and Technology Innovation Team (grant no. 2017TD0006) is sincerely acknowledged.
Electronic supplementary information (ESI) available: Isothermal curing kinetics. See DOI: 10.1039/c8ra03874k
Notes and references
- Raquez J. M. Deléglise M. Lacrampe M. F. Krawczak P. Prog. Polym. Sci. 2010;35:487–509. doi: 10.1016/j.progpolymsci.2010.01.001. [DOI] [Google Scholar]
- Montarnal D. Capelot M. Tournilhac F. Leibler L. Science. 2011;334:965–968. doi: 10.1126/science.1212648. [DOI] [PubMed] [Google Scholar]
- Wan J. Zhao J. Gan B. Li C. Molinaaldareguia J. Zhao Y. Pan Y. T. Wang D. Y. ACS Sustainable Chem. Eng. 2016;4:2869–2880. doi: 10.1021/acssuschemeng.6b00479. [DOI] [Google Scholar]
- Pan Y. T. Wan J. Zhao X. Li C. Wang D. Y. Chem. Eng. J. 2017;33:1222–1231. doi: 10.1016/j.cej.2017.08.059. [DOI] [Google Scholar]
- Auvergne R. Caillol S. David G. Boutevin B. Pascault J. P. Chem. Rev. 2014;114:1082–1115. doi: 10.1021/cr3001274. [DOI] [PubMed] [Google Scholar]
- Ma S. Li T. Liu X. Zhu J. Polym. Int. 2016;65:164–173. doi: 10.1002/pi.5027. [DOI] [Google Scholar]
- Ranjit N. Siefert K. Padmanabhan V. J. Perinatol. 2010;30:2–9. doi: 10.1038/jp.2009.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada H. Tokunaga T. Liu X. Takayanagi S. Matsushima A. Shimohigashi Y. Environ. Health Perspect. 2008;116:32–38. doi: 10.1289/ehp.10587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baroncini E. A. Kumar Yadav S. Palmese G. R. Stanzione J. F. J. Appl. Polym. Sci. 2016;133:44103–44122. doi: 10.1002/app.44103. [DOI] [Google Scholar]
- Ding C. Matharu A. S. ACS Sustainable Chem. Eng. 2014;2:2217–2236. doi: 10.1021/sc500478f. [DOI] [Google Scholar]
- Gandini A. Green Chem. 2011;13:1061–1083. doi: 10.1039/C0GC00789G. [DOI] [Google Scholar]
- Thakur M. K. Thakur V. K. Gupta R. K. Pappu A. ACS Sustainable Chem. Eng. 2016;4:1–17. doi: 10.1021/acssuschemeng.5b01327. [DOI] [Google Scholar]
- Xia Y. Larock R. C. Green Chem. 2010;12:1893–1909. doi: 10.1039/C0GC00264J. [DOI] [Google Scholar]
- Dan R. Mustata F. Tudorachi N. Musteata V. E. Rosu L. Varganici C. D. RSC Adv. 2015;5:45679–45687. doi: 10.1039/C5RA17550J. [DOI] [Google Scholar]
- Llevot A. J. Am. Oil Chem. Soc. 2016;94:1–18. [Google Scholar]
- Li C. Li Y. Cai X. Wang H. Bossmann S. H. Sung J. Sun X. S. ACS Sustainable Chem. Eng. 2016;4:794–800. doi: 10.1021/acssuschemeng.5b00806. [DOI] [Google Scholar]
- Chrysanthos M. Galy J. Pascault J. P. Polymer. 2011;52:3611–3620. doi: 10.1016/j.polymer.2011.06.001. [DOI] [Google Scholar]
- Ma S. Webster D. C. Jabeen F. Macromolecules. 2016;49:3780–3788. doi: 10.1021/acs.macromol.6b00594. [DOI] [Google Scholar]
- Ma S. Liu X. Jiang Y. Tang Z. Zhang C. Zhu J. Green Chem. 2013;15:245–254. doi: 10.1039/C2GC36715G. [DOI] [Google Scholar]
- Li P. Ma S. Dai J. Liu X. Jiang Y. Wang S. Wei J. Chen J. Zhu J. ACS Sustainable Chem. Eng. 2016;5:1228–1236. doi: 10.1021/acssuschemeng.6b02654. [DOI] [Google Scholar]
- Zhao S. Abu-Omar M. M. Biomacromolecules. 2015;16:2025–2031. doi: 10.1021/acs.biomac.5b00670. [DOI] [PubMed] [Google Scholar]
- Wang S. Ma S. Xu C. Liu Y. Dai J. Wang Z. Liu X. Chen J. Shen X. Wei J. Zhu J. Macromolecules. 2017;50:1892–1901. doi: 10.1021/acs.macromol.7b00097. [DOI] [Google Scholar]
- Ma S. Webster D. C. Macromolecules. 2015;48:7127–7137. doi: 10.1021/acs.macromol.5b01923. [DOI] [Google Scholar]
- Zhang Q. Molenda M. Reineke T. M. Macromolecules. 2016;49:8397–8406. doi: 10.1021/acs.macromol.6b01861. [DOI] [Google Scholar]
- Pan X. Sengupta P. Webster D. C. Biomacromolecules. 2011;12:2416–2422. doi: 10.1021/bm200549c. [DOI] [PubMed] [Google Scholar]
- Li A. Li K. ACS Sustainable Chem. Eng. 2014;2:2090–2096. doi: 10.1021/sc5003853. [DOI] [Google Scholar]
- Wu Y. Li A. Li K. J. Am. Oil Chem. Soc. 2015;92:111–120. doi: 10.1007/s11746-014-2563-6. [DOI] [Google Scholar]
- Li A. Li K. RSC Adv. 2015;5:85264–85271. doi: 10.1039/C5RA15144A. [DOI] [Google Scholar]
- Zhao T.-H. Wu Y. Li Y.-D. Wang M. Zeng J.-B. ACS Sustainable Chem. Eng. 2017;5:1938–1947. doi: 10.1021/acssuschemeng.6b02684. [DOI] [Google Scholar]
- Li A. Li K. RSC Adv. 2014;4:21521–21530. doi: 10.1039/C4RA03557G. [DOI] [Google Scholar]
- Nasiri M. Reineke T. M. Polym. Chem. 2016;7:5233–5240. doi: 10.1039/C6PY00700G. [DOI] [Google Scholar]
- Clark A. J. Seng S. H. Polym. Chem. 2014;5:3238–3244. doi: 10.1039/C3PY01527K. [DOI] [Google Scholar]
- Wang X. L. Chen L. Wu J. N. Fu T. Wang Y. Z. ACS Sustainable Chem. Eng. 2017;5:3353–3361. doi: 10.1021/acssuschemeng.6b03201. [DOI] [Google Scholar]
- Teng G. Soucek M. D. J. Am. Oil Chem. Soc. 2000;77:381–387. doi: 10.1007/s11746-000-0062-0. [DOI] [Google Scholar]
- Yang D. Peng X. Zhong L. Cao X. Chen W. Zhang X. Liu S. Sun R. Carbohydr. Polym. 2014;103:198–206. doi: 10.1016/j.carbpol.2013.12.043. [DOI] [PubMed] [Google Scholar]
- Jian X. Y. An X. P. Li Y. D. Chen J. H. Wang M. Zeng J. B. Macromolecules. 2017;50:5729–5738. doi: 10.1021/acs.macromol.7b01068. [DOI] [Google Scholar]
- Duval A. Averous L. ACS Sustainable Chem. Eng. 2017;5:330–340. [Google Scholar]
- Zhao S. Abu-Omar M. M. ACS Sustainable Chem. Eng. 2017;5:5059–5066. doi: 10.1021/acssuschemeng.7b00440. [DOI] [Google Scholar]
- Duval A. Averous L. Chemsuschem. 2017;10:984–992. doi: 10.1002/cssc.201700066. [DOI] [PubMed] [Google Scholar]
- Ejima H. Richardson J. J. Liang K. Best J. P. van Koeverden M. P. Such G. K. Cui J. Caruso F. Science. 2013;341:154–157. doi: 10.1126/science.1237265. [DOI] [PubMed] [Google Scholar]
- Aouf C. Lecomte J. Villeneuve P. Dubreucq E. Fulcrand H. Green Chem. 2012;14:2328–2336. doi: 10.1039/C2GC35558B. [DOI] [Google Scholar]
- Cao L. Liu X. Na H. Wu Y. Zheng W. Zhu J. J. Mater. Chem. A. 2013;1:5081–5088. doi: 10.1039/C3TA01700A. [DOI] [Google Scholar]
- Shibata M. Teramoto N. Makino K. J. Appl. Polym. Sci. 2011;120:273–278. doi: 10.1002/app.33082. [DOI] [Google Scholar]
- Nouailhas H. Aouf C. Guerneve C. L. Caillol S. Boutevin B. Fulcrand H. J. Polym. Sci., Part A: Polym. Chem. 2011;49:2261–2270. doi: 10.1002/pola.24659. [DOI] [Google Scholar]
- Jin H. Zhang Y. Wang C. Sun Y. Yuan Z. Pan Y. Xie H. Cheng R. J. Therm. Anal. Calorim. 2014;117:773–781. doi: 10.1007/s10973-014-3849-5. [DOI] [Google Scholar]
- Vidil T. Tournilhac F. Musso S. Robisson A. Leibler L. Prog. Polym. Sci. 2016;62:126–179. doi: 10.1016/j.progpolymsci.2016.06.003. [DOI] [Google Scholar]
- Ham Y. R. Kim S. H. Shin Y. J. Lee D. H. Yang M. Min J. H. Shin J. S. J. Ind. Eng. Chem. 2010;16:556–559. doi: 10.1016/j.jiec.2010.03.022. [DOI] [Google Scholar]
- Xu Y. J. Wang J. Tan Y. Qi M. Chen L. Wang Y. Z. Chem. Eng. J. 2018;337:30–39. doi: 10.1016/j.cej.2017.12.086. [DOI] [Google Scholar]
- Chen Y. C. Chiu W. Y. Lin K. F. J. Polym. Sci., Part A: Polym. Chem. 2015;37:3233–3242. doi: 10.1002/(SICI)1099-0518(19990815)37:16<3233::AID-POLA21>3.0.CO;2-A. [DOI] [Google Scholar]
- And Y. C. C. Chiu W. Y. Macromolecules. 2000;33:6672–6684. doi: 10.1021/ma9909758. [DOI] [Google Scholar]
- Chen Y. C. Chiu W. Y. Polymer. 2001;42:5439–5448. doi: 10.1016/S0032-3861(01)00024-6. [DOI] [Google Scholar]
- Wong F. F. Lin C. M. Chen K. L. Shen Y. H. Huang J. J. Macromol. Res. 2010;18:324–330. doi: 10.1007/s13233-010-0416-1. [DOI] [Google Scholar]
- Kissinger H. E. J. Res. Natl. Bur. Stand. 1956;57:217–221. doi: 10.6028/jres.057.026. [DOI] [Google Scholar]
- Ozawa T. J. Therm. Anal. 1976;9:369–373. doi: 10.1007/BF01909401. [DOI] [Google Scholar]
- Reisen R., Thermosets, in Application Handbook Thermal Analysis, Mettler-Toledo GmbH, 2006 [Google Scholar]
- Lin K. Heath W. H. Torkelson J. M. Polymer. 2015;81:70–78. doi: 10.1016/j.polymer.2015.10.068. [DOI] [Google Scholar]
- Scanlan J. J. Polym. Sci. 1960;43:23–33. doi: 10.1002/pol.1960.1204314219. [DOI] [Google Scholar]
- Hill L. W. Prog. Org. Coat. 1997;31:235–243. doi: 10.1016/S0300-9440(97)00081-7. [DOI] [Google Scholar]
- Available online, https://tds.henkel.com/tds5/Studio/ShowPDF/EA%203982-EN?pid=EA%203982&format=MTR&subformat=REAC&language=EN&plant=WERCS, (accessed on April, 2018)
- Available online, https://tds.henkel.com/tds5/Studio/ShowPDF/EA%203984-EN?pid=EA%203984&format=MTR&subformat=REAC&language=EN&plant=WERCS, (accessed on April, 2018)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.