

Abstract
The MYC proto-oncogenes encode a family of transcription factors that are among the most commonly activated oncoproteins in human neoplasias. Indeed, MYC aberrations or upregulation of MYC-related pathways by alternate mechanisms occur in the vast majority of cancers. MYC proteins are master regulators of cellular programmes. Thus, cancers with MYC activation elicit many of the hallmarks of cancer required for autonomous neoplastic growth. In preclinical models, MYC inactivation can result in sustained tumour regression, a phenomenon that has been attributed to oncogene addiction. Many therapeutic agents that directly target MYC are under development; however, to date, their clinical efficacy remains to be demonstrated. In the past few years, studies have demonstrated that MYC signalling can enable tumour cells to dysregulate their microenvironment and evade the host immune response. Herein, we discuss how MYC pathways not only dictate cancer cell pathophysiology but also suppress the host immune response against that cancer. We also propose that therapies targeting the MYC pathway will be key to reversing cancerous growth and restoring antitumour immune responses in patients with MYC-driven cancers.
The MYC oncogene (also known as c-MYC) is part of a superfamily of genes with products that are among the most commonly activated in human cancers1–4. MYC is a master regulator of multiple biological programmes and mediates much of its function primarily as a transcription factor that regulates the expression of thousands of genes, either directly or indirectly5,6. In addition, MYC exerts multiple biological effects on cellular programmes that influence both the cell-intrinsic biology as well as the host immunity and tumour microenvironment (TME).
MYC has two paralogues: MYCL and MYCN. The initially held view was that MYC is ubiquitously involved in human cancers (both haematological and solid), whereas L-MYC is associated with small-cell lung cancer and N-MYC with neuroblastoma. However, in the past few years, insights gained from genome-sequencing studies show a broader role for both N-MYC7–10 and L-MYC11–13 in many other cancers3,4 (FIG. 1a). Thus, when considering the entire MYC family, most human cancers harbour the genetic activation of one of its members1–4.
Fig. 1 |. Major genetic alterations involving MYC and its paralogues in human cancers.
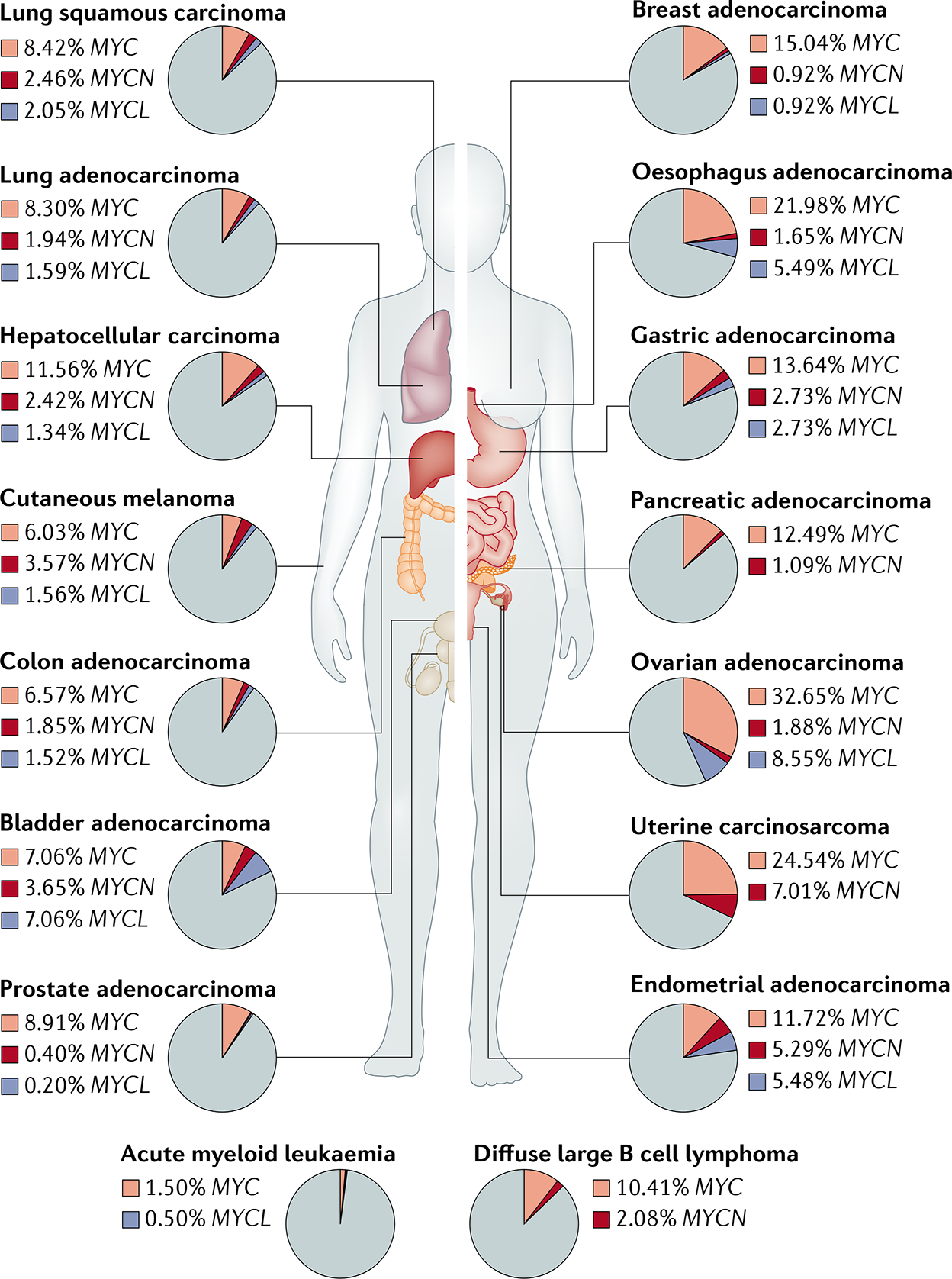
Prevalence of gene amplification of the three MYC paralogues MYC, MYCL and MYCN across 16 major human cancer types in The Cancer Genome Atlas.
A multitude of reports have documented that MYC overexpression can cause tumorigenesis. In a landmark study, Adams et al.14 demonstrated that transgenic overexpression of Myc in mice was sufficient for B cell lymphomagenesis. Subsequently, many other investigators documented that MYC overexpression can causally induce other types of cancer in humans15–17. Moreover, the use of conditional transgenic mouse models has enabled a refined understanding of how MYC activation causes tumorigenesis and how its inactivation can elicit tumour regression. Two general approaches have been used to modulate MYC in preclinical studies: conditional regulation of the expression of a MYC transgene using the Tet system18–23 and conditional expression of a synthetic MYC inhibitor, Omomyc24–26. In these conditional transgenic mouse models, MYC-induced tumours regress rapidly and dramatically upon MYC inactivation18–23. Tumour regression occurs even without influencing physiological endogenous MYC expression or function, in a phenomenon referred to as oncogene addiction27. This phenomenon suggests that targeting MYC could be an effective approach to treat some human cancers. To date, however, no drug has demonstrated efficacious therapeutic targeting of MYC or the MYC pathway, although promising candidates are in development.
Cancer is a complex process that requires a multitude of genetic events and the acquisition of the hallmarks of cancer28. Importantly, MYC activation can contribute to many of these hallmarks, including proliferation, self-renewal, cell survival, genomic instability, metabolism and invasiveness as well as angiogenesis and immune evasion29–31. Thus, MYC seems to have crucial effects in newly transformed cancer cells and also enables an evolving tumour to remodel the TME and evade the host immune response32,33. Details on how MYC biologically modulates cancer cell-intrinsic programmes of cellular proliferation, differentiation, survival and death have been described elsewhere34–38.
In this Review, we focus on the broad clinical importance of MYC and MYC-related oncogenic signalling in the pathogenesis of human cancers. In particular, we provide evidence that MYC is a major driver of human cancer through its effects on tumour cells and also by enabling cancers to evade host immune surveillance. We summarize the studies that have demonstrated rapid and sustained tumour regression in MYC-driven cancers upon MYC inactivation as a consequence of oncogene addiction. Finally, we provide a rationale for and examples of therapeutic strategies targeting the MYC pathway. We conclude that MYC signalling is one of the most important, yet to be successfully targeted, oncogenic pathways in human cancer; thus, mechanistic insights into the tumorigenic roles of this gene family will be key to developing efficacious therapies for cancer.
MYC is often activated in human cancers
Genetic alterations affecting the MYC proto-oncogenes and the MYC-related signalling pathways are among the most common in human cancers1–4 (FIG. 1). MYC can be activated in cancers through multiple genetic, epistatic, epigenetic and post-translational mechanisms (FIG. 2), which vary between cancer types.
Fig. 2 |. Mechanisms leading to MYC activation in human cancers.
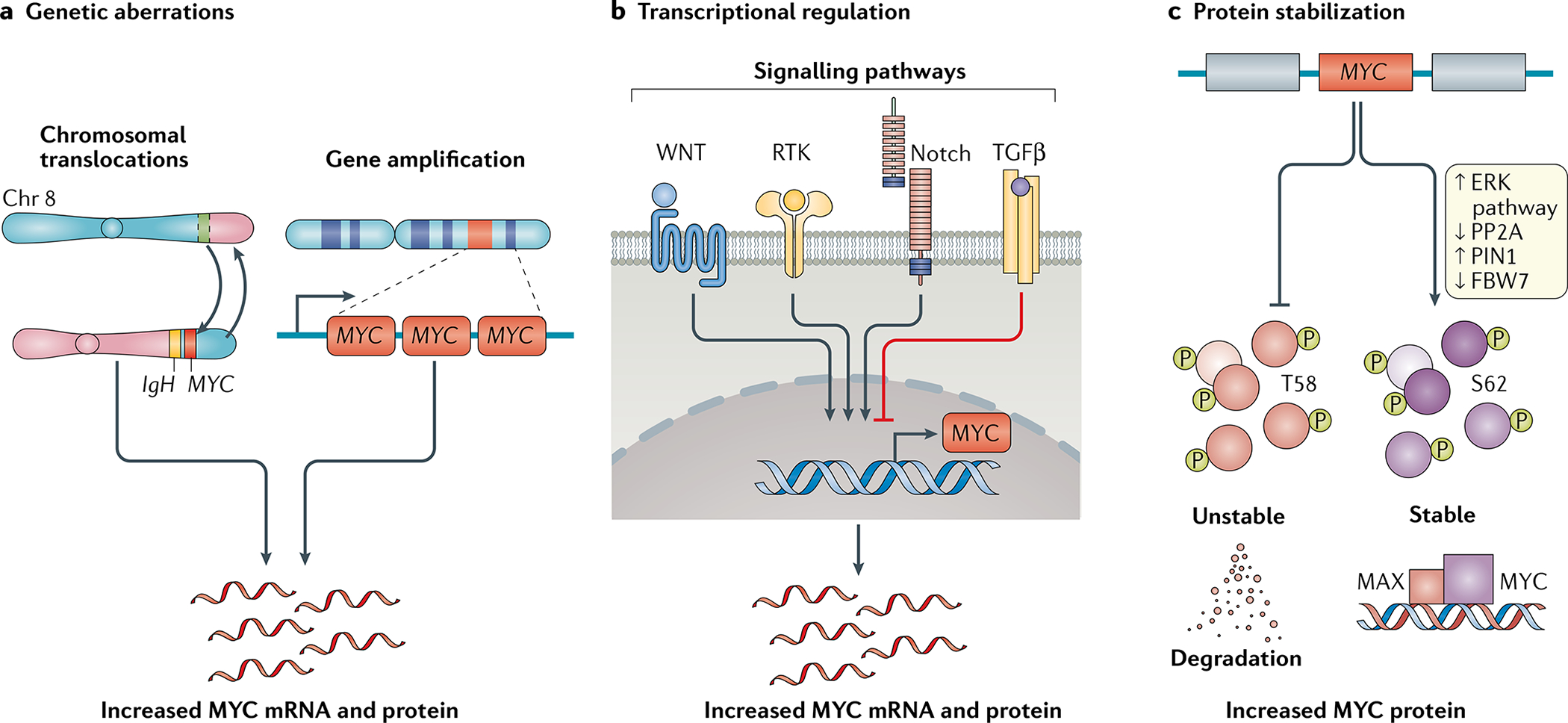
a | Genetic aberrations, such as chromosomal translocations and genomic amplifications, lead to increased MYC mRNA expression. b | Alteration of upstream regulatory pathways can lead to increased or decreased transcription of the MYC oncogene. c | Post-translational modifications of the MYC protein, such as preferential phosphorylation of the serine 62 (S62) residue versus threonine 58 (T58), can block degradation and promote stabilization of MYC, thereby enhancing activation of the MYC pathway.
Genomic alterations, including gene amplification, chromosomal translocations and mutations, can increase MYC expression1,4,39 (FIG. 2a). A pan-cancer assessment of alterations in 33 human cancer types conducted as part of The Cancer Genome Atlas project revealed that MYC or its paralogues MYCL or MYCN are amplified in 28% of tumours3. An analysis encompassing 17 cancer types also showed that MYC is frequently amplified in extrachromosomal DNA during tumour evolution40,41. These amplifications can directly lead to MYC overexpression or, indirectly, to the activation of genes involved in the MYC pathway as demonstrated in 30 of the 33 cancer types analysed in The Cancer Genome Atlas pan-cancer study3. MYC is genetically amplified in many solid tumour types, including breast and liver cancers, and is frequently chromosomally translocated in B cell and T cell leukaemias and lymphomas39,42,43 (FIG. 1). MYC expression can also be increased through the insertion of upstream enhancers by retroviruses44 or epistatically and/or epigenetically through the activation of many other oncogenic pathways, such as those mediated by WNT–β-catenin, SRC, numerous receptor tyrosine kinases and Notch45, or via the loss of tumour suppressors such as APC46 and TGFβ47 (FIG. 2b).
In addition, MYC can be activated posttranslationally through many mechanisms increasing protein stability48,49 (FIG. 2c). MYC has a short half-life that is tightly regulated through phosphorylation and proteasomal degradation. Tumours with stable MYC expression have elevated levels of phospho-serine 62 (P-S62)-MYC and low levels of phospho-threonine 58 (P-T58)-MYC49,50. Mitogenic pathways, such as RAS–MEK–ERK signalling, can increase P-S62 levels and thereby increase MYC stability51. Moreover, MYC mutations affecting the T58 residue can lead to constitutive S62 phosphorylation52. Cancers can also down-regulate PP2A, a serine/threonine phosphatase complex that targets P-S62, leading to MYC accumulation50,53–55. The levels of PIN1, a member of the peptidyl-prolyl cis-trans isomerase (PPIase) family that functionally regulates MYC stability via isomerization, is overexpressed in multiple human cancer types, suggesting that this is another mechanism by which levels of MYC are increased in cancer cells56,57. Finally, FBW7 is a tumour suppressor that controls the proteasomal degradation of many proteins, including MYC58. FBW7 is frequently inactivated in human cancers, such as uterine (18%), colon (16%) or cervical cancer (13%), through deletion, mutation or epigenetic modification59,60, which can promote cancer progression at least in part owing to the resulting increase in MYC levels53.
The MYC superfamily also includes genes encoding multiple other proteins considered to function as transcription factors or co-regulatory proteins and which are very commonly activated in human cancers61. These genes include MAX, which encodes an MYC heterodimer binding partner, and MLX as well as MGA, MXD1, MXD3, MXD4, MXI1, MNT, MLXIP and MLXIPL, which encode proteins that interact with MAX and/or MLX61. Interactions between different members of this superfamily can result in transcriptional activation or repression of specific genes that, in turn, regulate cell-cycle progression and/or cellular transformation. Mutations or deletions in these members of the MYC superfamily have also been reported in human cancers62,63. Therefore, the MYC pathway is dysregulated in the majority of human cancers3,29,64,65. Accordingly, we have suggested that MYC signalling is a molecular hallmark of cancer66.
MYC activation alone is generally not sufficient to induce the neoplastic transformation of non-malignant cells. Indeed, the tumorigenic functions of oncogenic MYC are restrained by many physiological mechanisms that cause cell-cycle arrest, apoptosis and/or cellular senescence. For example, unrestricted MYC activation can induce the expression of TP53 (REF.67) as well as of cell-cycle checkpoint genes (such as CDKN2A)68 and/or regulators of apoptosis (such as BCL2)69. Correspondingly, the inactivation of genes such as TP53 or CDKN2A, the activation of genes such as BCL2, or the deletion of pro-apoptotic genes (for example, that encoding caspase-8 (REF.70)) cooperate with MYC signalling to block cell-cycle arrest, senescence and/or apoptosis, thereby driving cancer initiation. The progressive shortening of the telomeres after every cell cycle eventually halts cell division and is another cellular protective mechanism; however, MYC increases telomerase activity by inducing the expression of a telomerase reverse transcriptase, thus favouring cellular immortality71.
Provocatively, despite activation of MYC signalling being one of the most common genetic events in human tumorigenesis, MYC might not always be essential for tumour initiation. Genetically engineered Myc-haploinsufficient (Myc+/−) mice develop lymphomas at the same rate as Myc-wild-type mice, albeit with reduced cell proliferation and delayed tumour progression72. Regardless, members of the MYC superfamily are commonly activated and thought to be required for tumorigenesis in most human cancers.
MYC oncogene addiction
In 2002, Bernard Weinstein proposed that cancers are ‘oncogene addicted’27 following evidence from both experimental mouse models of MYC-driven cancers and clinical studies of heterogeneous human tumours. A multitude of studies has demonstrated that MYC can act as a cancer driver and that suppression of MYC signalling can result in sustained tumour regression18–23. In transgenic mouse models of MYC-driven cancers (including lymphoma, leukaemia, osteosarcoma, hepatocellular carcinoma (HCC), renal cell carcinoma and lung adenocarcinoma (LAC)), sustained tumour regression can be observed upon MYC inactivation, although the specific consequences depend upon the particular type of cancer18–23. Furthermore, the reversible expression of a dominant-negative MYC mutant and MYC–MAX disruptor, Omomyc, leads to the rapid regression of lung tumours in mouse models24–26. Thus, extensive experimental evidence supports targeting MYC as a potential therapeutic approach against a multitude of human cancers. MYC activation drives cancer progression through two types of mechanisms involving either acquisition of hallmarks of cancer that are intrinsic to tumour cells2,66 or changes in the TME and anticancer immune response32,73.
Effects of MYC activation
Cancer cell-intrinsic mechanisms.
MYC regulates cancer cell-intrinsic processes with several physiological roles such as cell growth, differentiation and metabolism74–77. Such processes are co-opted upon MYC activation in a cancer cell-autonomous manner, thus facilitating cellular transformation (FIG. 3). In non-malignant cells, the expression of MYC is tightly controlled. High levels of MYC expression are, in fact, associated with increased sensitivity to apoptosis78,79. However, MYC activation can overcome these physiological barriers and promote cancer through several interdependent cell-intrinsic mechanisms.
Fig. 3 |. MYC is a grand orchestrator of the hallmarks of cancer.
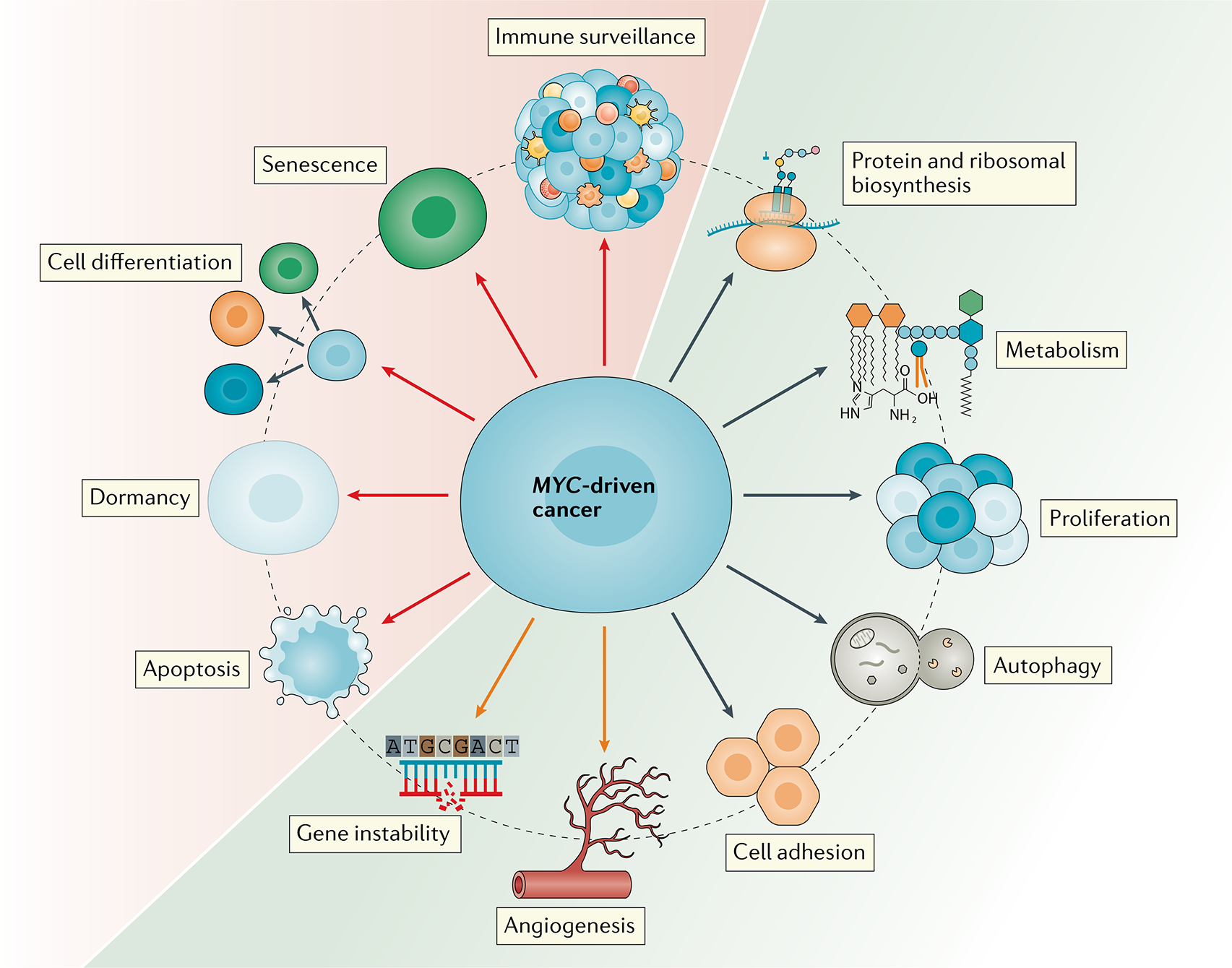
MYC regulates several cancer cell-intrinsic and host-dependent pathways to promote cancer cell growth and survival (green area). Cancer cell-intrinsic processes regulated by MYC include proliferation, metabolism, invasiveness, autophagy, and protein and ribosomal biosynthesis. MYC also simultaneously blocks other cellular protective mechanisms, such as differentiation or senescence, thereby promoting cancer progression (red area). Paradoxically, MYC also induces cellular processes, such as apoptosis and chromosomal instability, that can be detrimental to cancer cell survival (orange arrows). The delicate balance between these events and cellular context ultimately determines cell fate. Furthermore, MYC controls the ability of cancer cells to undergo dormancy and to overcome nutrient-low environments, eventually leading to tumour relapse. To maintain this quiescent state, MYC inhibits several cellular programmes, including cell differentiation and senescence. MYC activation in the cancer cells also drives enhanced angiogenesis, thus promoting tumour progression. Finally, one of the most important functions of MYC is to enable cancer cells to evade and inhibit immune surveillance to safeguard their survival. All of these hallmarks regulated by MYC work in unison to drive cancer progression.
MYC overexpression enforces relentless cellular proliferation by enabling cancer cells to re-enter the cell cycle79 and also accelerates progression through the cell cycle by inhibiting cell-cycle checkpoints37,80,81. In addition, MYC promotes cancer cell growth by inducing a substantial increase in ribosomal and protein biogenesis82–85. MYC alters cancer cell metabolism by globally rewiring multiple metabolic pathways to support rapid growth and proliferation35,86. This function is partially accomplished by facilitating nutrient uptake in cancer cells through transcriptional induction of glucose and glutamine transporters and also of genes involved in glycolysis and glutaminolysis87–89. MYC activates cellular survival programmes through specific effects on DNA replication. In particular, MYC can directly activate the DNA replication machinery90. Of note, Myc is one of the four factors that enable stemness and embryonic programmes of self-renewal in induced pluripotent stem cells91. In addition, MYC can bypass and/or transcriptionally repress cell-cycle checkpoint proteins, such as p15 or p21, in some contexts, thus preventing cellular senescence even in the presence of DNA damage92. MYC activation can also result in genomic instability owing to many types of genomic damage, including DNA breaks, chromosomal translocations, chromosomal gains or losses, aneuploidy, and polyploidy. The mechanisms by which MYC induces genomic damage involve overriding cell-cycle checkpoints (such as transcriptional repression of p21) and blocking DNA double-strand break repair21,93–96 as well as mitochondrial reactive oxygen species generated during MYC-driven cell proliferation97–99. MYC activation also can induce proliferative arrest and endoreduplication as well as genomic instability in vivo100,101.
The ability of oncogenic MYC to elicit cell proliferation seems to depend upon the specific tissue lineage and the developmental context. For example, MYC overexpression in liver cells of embryonic or neonatal mice results in rapid cellular proliferation and rapid formation of hepatoblastomas (a paediatric neoplasm), whereas MYC overexpression in liver cells of adult mice results in cellular hypertrophy, endoreduplication and late-onset HCC100. In summary, MYC activation contributes to both the initiation and maintenance of tumorigenesis through several cancer cell-intrinsic mechanisms.
Effects on the immune response and TME.
Preclinical studies have revealed many mechanisms by which MYC influences the TME, involving effects on host stromal cells, vascular endothelial cells, innate and adaptive immune cells, and, in some cases, specific cytokines and their receptors (FIG. 4).
Fig. 4 |. MYC blocks immune surveillance.
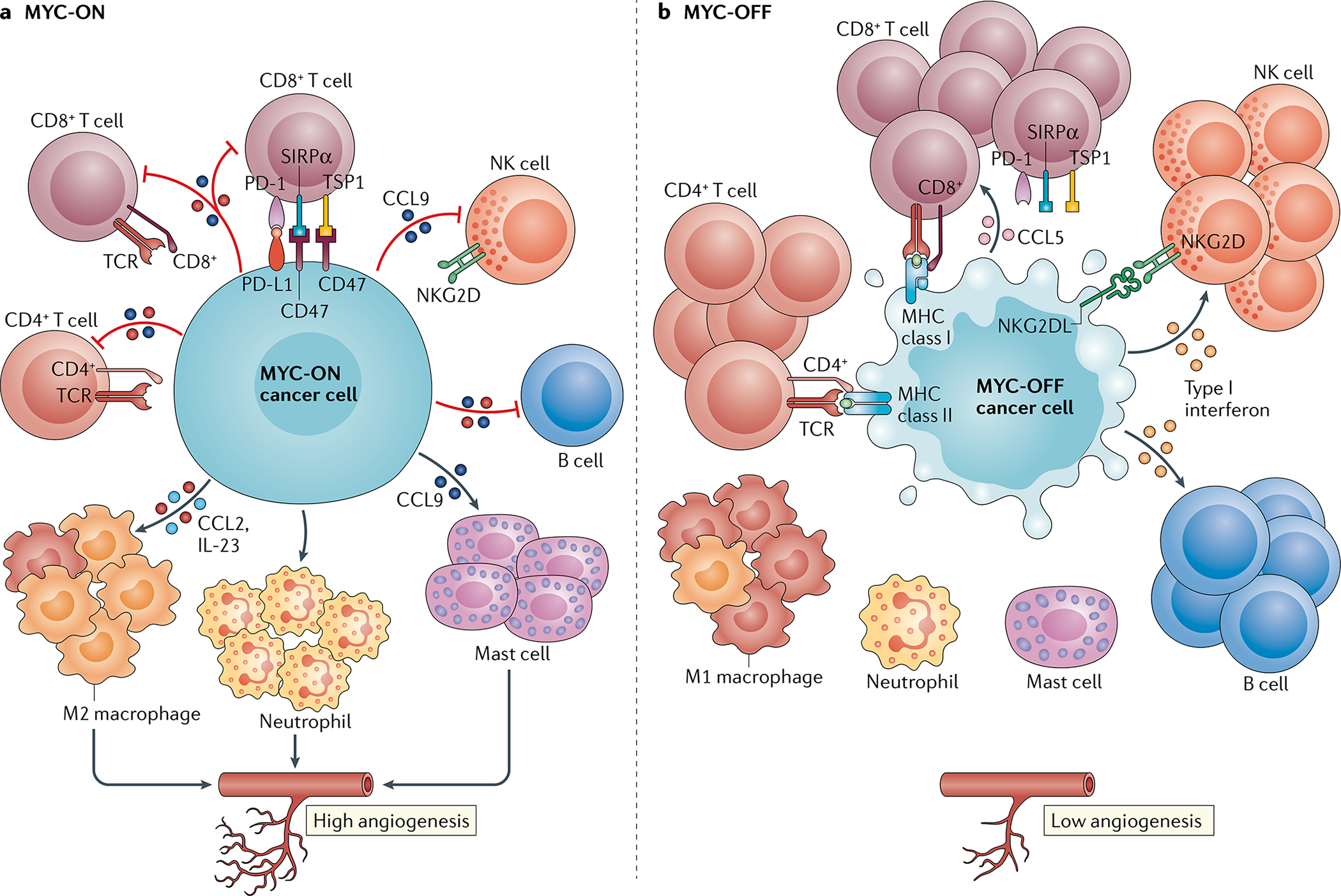
MYC enables cancers to evade the immune system through several distinct mechanisms. a | MYC regulates the expression and production of several immune ligands or receptors and immune effector molecules, such as PD-L1, CD47, MHC classes I and II, and NKG2D. MYC also promotes the expression of several cytokines, such as CCL2, IL-23 and CCL9, which regulate the conversion of antitumour M1 macrophages to pro-tumour M2 macrophages and prevent the activation and recruitment of B cells, natural killer (NK) cells, and CD4+ and CD8+ T cells. CCL9 activates mast cells, which in turn induce angiogenesis. b | Upon inactivation of MYC, the downregulation of PD-L1 and CD47 results in the rapid recruitment and activation of CD8+ T cells and NK cells. The inactivation of MYC also increases the levels of NKG2DL in cancer cells, resulting in NK cell recruitment. The production of most of the cytokines described above decreases upon MYC inactivation. By contrast, the expression of type I interferons and CCL5 increases upon MYC inactivation, resulting in the recruitment and activation of NK cells and B cells and of CD8+ T cells, respectively. Thus, MYC controls the immune status of a tumour by creating an immunosuppressive ‘cold’ tumour microenvironment when activated, which reverts to an immune-sensitive ‘hot’ milieu when inactivated. TSP1, thrombospondin 1.
MYC expression in cancer cells can influence host endothelial cells, resulting in reprogramming of the TME to support cellular proliferation and induction of angiogenesis102–104 (FIG. 3). Moreover, MYC initiates and maintains tumorigenesis through a thrombospondin 1 (TSP1)-regulated angiogenic switch105 (FIG. 3).
MYC-dependent perturbation of the host immune response is causally related to the mechanism of tumorigenesis and is an important mechanism underlying tumour regression upon MYC inactivation106–108. Characterized mechanisms include effects on CD4+ T cells, macrophages, and natural killer (NK) cells and involve cytokines (such as TGFβ, interferons and TSP1) and key immunomodulatory molecules (such as PD-L1, CD47 and MHC class I (MHC I); FIG. 4)106–112. More than 30 years ago, a study described that MYC downregulates the expression of proteins involved in antigen presentation, such as MHC I, thus enabling immune evasion113–115. In addition, MYC induces the secretion of immune inhibitory cytokines from cancer cells, such as TGFβ, which in turn suppresses cytolytic T cell responses116,117. Furthermore, MYC has been found to upregulate the expression of immune-checkpoint proteins, such as PD-L1 and CD47, thus leading to T cell exhaustion107,108,110,118,119. In addition, MYC suppresses NK cell-mediated immune surveillance by transcriptionally repressing STAT1 and STAT2 and the type I interferon pathway107,109,111. Finally, MYC cooperates with TWIST1 to regulate the secretion of CCL2 and IL-13, thereby promoting the recruitment and polarization of immunosuppressive macrophages, which in turn facilitate metastasis120 (FIG. 4).
Effects of MYC inactivation
MYC inactivation can cause tumour regression through effects on tumour cells as well as the host immune system and TME. In preclinical models18–23, MYC inactivation results in a cascade of changes in both tumour cells and the TME that lead to rapid tumour regression and the restoration of regular tissue structures (FIG. 4). The specific kinetics and consequences of MYC inactivation seem to depend upon the tissue of origin of a cancer as well as on the specific genetic context. Thus, in mice with MYC-driven haematological cancers, such as T cell acute lymphoblastic leukaemia, MYC inactivation is associated with rapid proliferative arrest and very rapid tumour regression within a few days, together with robust induction of cellular differentiation, senescence and apoptosis20. By contrast, in mice with MYC-driven mesenchymal-derived tumours, such as osteosarcoma, MYC inactivation results in the robust terminal differentiation of bone cancer into bone18,21. Finally, in mice with MYC-driven epithelial-derived tumours, such as HCC, LAC and renal cell carcinoma, MYC inactivation results in tumour regression but a dormant population of otherwise histologically normal-appearing cells remains19,121,122.
Cancer cell-intrinsic mechanisms.
MYC inactivation leads to the restoration of physiological cell-intrinsic checkpoints on cellular growth and differentiation. In mouse models of MYC-driven cancer, tumour cells undergo proliferative arrest, differentiation, senescence and/or apoptosis upon MYC inactivation18–23,66. These processes seem to occur through the restoration of cell-cycle checkpoints, DNA repair and chromatin remodelling programmes. Thus, the knockdown of cell-cycle checkpoint proteins66, such as p16, or DNA repair regulators, such as p53, impedes tumour regression elicited by MYC inactivation21. MYC inactivation also causes tumour regression through the loss of survival signals with the maintenance of death signals within cancer cells123. The mechanism involves both a TGFβ autocrine mechanism that elicits proliferative arrest and senescence upon MYC inactivation116,123 and the modulation of microRNAs (such as miR17-92, miR-15 and let-7 family members) that epigenetically regulate genes required for cell survival and stemness124–127. Thus, MYC maintains tumorigenesis partially through the coordination of multiple cell-intrinsic biological programmes.
Effects on the immune response and TME.
MYC inactivation can also elicit tumour regression through effects on the host immune response and TME such as the suppression of angiogenesis102,105. Indeed, MYC has a known role in inducing the expression of cytokines that regulate angiogenesis. One such key regulator is the anti-angiogenic factor TSP1, which is transcriptionally repressed by MYC128. Upon MYC inactivation, the upregulation of TSP1 results in the death of endothelial cells within the tumour vasculature and the loss of mean vessel density105.
Within a week of MYC inactivation, host innate immune cells (such as macrophages107,120 and NK cells111,129) and adaptive immune effectors (including CD4+ T cells106,130 and B cells23) are recruited to the tumour bed (FIG. 4). All of these changes in host cellular compartments have been correlated and, in some cases, shown to be causally associated with changes in the levels of specific cytokines (such as CCL2 or IL-1β) and immune checkpoints (for example, PD-L1 or CD47)103,107,108,120. After several weeks of MYC inactivation, complete regression of the tumour occurs together with the restoration of regular tissue architecture and establishment of a durable immune response. Indeed, tumour regression upon MYC inactivation seems to be associated with long-standing immune rejection of the same tumour type106.
Modelling oncogene addiction.
Of note, changes associated with oncogene addiction can be modelled mathematically. For example, one such model was developed to predict how relative changes in survival and death signals affect tumour growth123. In transgenic mouse models of LAC driven by MYC alone, Ras alone or both, oncogene addiction was found to be best described as a consequence of a reduced proliferative response rather than a net increase in apoptosis123. This observation is consistent with the ‘oncogenic shock model’, which proposes that, upon inactivation of oncogenic pathways, pro-survival signals dissipate quickly whereas pro-apoptotic signals persist for longer123,131. Indeed, the genetic context of a tumour and its sensitivity to undergo proliferative arrest and/or apoptosis influences the ability of MYC inactivation to elicit tumour regression. The inactivation of oncogenes other than MYC, such as RAS or ERBB2, can lead to similar phenotypical features of oncogene addiction, suggesting that the mechanisms involved are not necessarily unique to MYC132–134. Lastly, mathematical models have demonstrated mechanisms by which the TME contributes to tumour regression upon oncogene inactivation in transgenic mouse models135.
Mechanisms of host immune evasion
MYC activation in cancer cells can impede the host immune response. Conversely, its inactivation seems to restore antitumour immunity in a sequential manner. In an experimental mouse model of pancreatic cancer, MYC activation led to the rapid induction of the potent pro-inflammatory cytokine IL-1β, driving the increased proliferation of endothelial cells103. CCL2 and CCL5 were also induced, thus triggering the recruitment of mast cells and delaying that of macrophages and neutrophils136. In 2020, Muthalagu et al. reported a novel mechanism whereby MYC and KRAS-G12D cooperatively promote pancreatic cancer progression in mice by repressing the type I interferon pathway111. Targeted suppression of MYC–MIZ1 complex formation restores the activation of this pathway and leads to CXCL13-mediated recruitment of antitumour B cells and NK cells111. Therefore, in multiple mouse models, MYC inactivation is associated with the restoration of an anticancer innate and adaptive immune response.
Some studies have revealed that MYC can regulate the immune response in a manner clearly dependent upon another driver oncogene. For example, in an experimental mouse model of HCC, MYC activation in the context of TWIST1 activation leads to transcriptional changes that confer macrophages from a pro-inflammatory to a prometastatic phenotype mediated by the secretion of CCL2 and IL-13 (REF.120). Importantly, levels of MYC, TWIST1, CCL2 and IL-13 were also directly correlated with a worse prognosis in 33 different human cancers. Moreover, measurement of CCL2 and IL-13 enabled prediction of the invasiveness of HCC in patients with this cancer type. Similarly, MYC and RAS cooperate to elicit an inflammatory phenotype in a mouse model of LAC107 through a process characterized by increased production of CCL9 and IL-23 (REF.107). In this model, PD-L1+ macrophages inhibit T cells and induce angiogenesis. In summary, the specific features of the immune response regulated by MYC seem to be dependent on the specific type of cancer as well as on the genetic events associated with that tumour137.
MYC regulates immune checkpoints
MYC affects the host immune response through the regulation of multiple immune checkpoints, including PD-L1 and CD47. MYC regulates PD-L1 in a complex manner involving multiple mechanisms, including direct transcriptional regulation as well as indirect post-transcriptional regulation108,138,139. In transgenic mouse models of haematological cancers, MYC binds to the promoter region and regulates the transcription of Cd274 (encoding PD-L1)108. The constitutive retrovirally mediated expression of PD-L1 abrogated the ability of MYC inactivation to result in the inhibition of angiogenesis and the induction of cellular senescence and blocked tumour regression108,72,107,140,141. Similarly, in neuroblastoma and ALK-negative anaplastic large cell lymphoma cells, the in vitro inhibition of MYC expression with short-hairpin RNAs or small-molecule inhibitors reduces PD-L1 expression142,143. Thus, the transcriptional regulation of CD274 expression seems to be a conserved mechanism by which MYC promotes immune evasion.
MYC can also regulate PD-L1 indirectly in cooperation with other oncogenes or through other key transcriptional regulators. For example, in a mouse model of triple-negative breast cancer, MYC and p65 cooperate to regulate Cd274 transcription110. In mice with LAC, MYC cooperates with KRAS to overcome translational repression of PD-L1 (REF.107). Increased protein stability of MYC is another mechanism of PD-L1 regulation. Indeed, in a transgenic mouse model of HCC, an engineered increase in MYC protein stability correspondingly increased PD-L1 expression and reduced the tumour infiltration of CD8+ T cells118. In summary, MYC can increase the expression of PD-L1 in cancer cells through multiple mechanisms.
MYC can also regulate other receptors involved in the immune response such as CD47 and MHC I. CD47 is an immune regulatory molecule that provides a ‘do not eat me’ signal to immune cells expressing two types of ligands: TSP1 and tyrosine-protein phosphatase non-receptor type substrate 1 (also known as SIRPα)144,145. In human-derived lymphoma cells, MYC upregulated CD47 and inhibition of MYC signalling with the BET bromodomain inhibitor JQ1 reduced CD47 levels without affecting PD-L1 expression146. Similarly, in a mouse model of cutaneous T cell lymphoma, high levels of MYC induced the transcription of CD47 (REF.147). Interestingly, CD47 has been shown to increase MYC expression in a feedforward loop144,147.
Finally, MYC suppresses the expression of MHC I. More than 30 years ago, MYC was shown to downregulate MHC I in multiple cancers, including neuroblastoma, melanoma and lymphoma113,115,148. A subsequent study in mice showed that this downregulation is associated with a decrease in the recruitment of T cells and NK cells to LACs107. Of note, oncogenic RAS also seems to decrease MHC I expression; this process might occur through its known cooperation with MYC149,150. Together, these studies show that the regulation of immune checkpoints is a crucial mechanism whereby the MYC oncogene remodels the immune TME and facilitates immune evasion.
Immune regulation through metabolism
MYC regulates diverse metabolic pathways in cancer cells151–154. Importantly, immune function is also highly regulated by cellular metabolism155–158. More specifically, MYC influences the host TME directly and indirectly through its effects on metabolism.
MYC expression in cancer cells results in perturbations in metabolic pathways that lead to the release of immunomodulatory molecules. During tumour initiation in mouse models, MYC-driven proliferating cancer cells release metabolites (such as lactate159 and glutamate160,161) that influence the local immune function within the TME. As the tumour grows, these effects could become more general and cause systemic disruption of anticancer immune responses162,163.
MYC-driven cancers have dysregulated glucose, glutamine and lipid metabolism, which could affect the host metabolic homeostasis thereby indirectly influencing the immune system and immunoediting152,164–166. MYC enables a metabolic shift from oxidative phosphorylation to glycolysis (the Warburg effect), upregulates glutaminolysis and increases lipogenesis in cancer cells151–154, which improves cellular fitness and confers a cell survival advantage. However, such an advantage could lead to a competition between cancer and immune cells for key metabolites, thus making immune cells ineffective167. Hence, MYC could have both direct and indirect effects on the host immune system through its effects on cancer metabolism.
The MYC pathway as a therapeutic target
Despite substantial experimental evidence showing that targeting MYC can lead to tumour regression through both direct and indirect effects18–26 and decades of effort towards clinical translation of these findings, MYC has not yet been successfully targeted therapeutically. The difficulties in targeting MYC have been described elsewhere168,169; briefly, some of the reasons include the highly disordered protein structure of MYC and the lack of a binding pocket or specific enzymatic activity. Nevertheless, a variety of strategies to inhibit MYC activity are currently being explored (FIG. 5) in preclinical cancer models or in clinical studies (Supplementary Table 1). Individual approaches have been comprehensively reviewed elsewhere169–175; herein, we provide a broad overview of different strategies for the therapeutic targeting of MYC-driven cancers.
Fig. 5 |. Therapeutic strategies to target MYC-driven tumours.
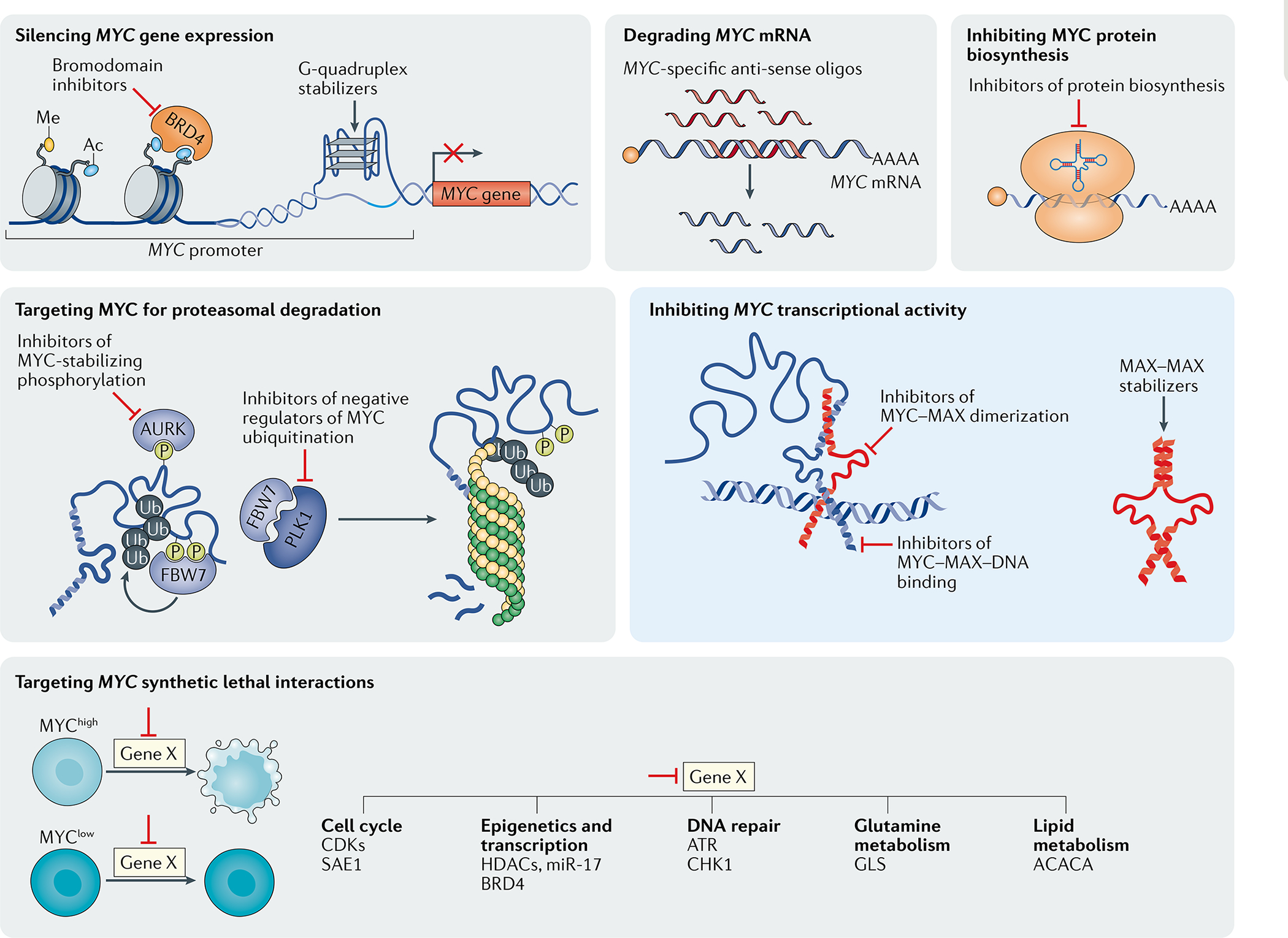
Among the multiple strategies currently explored to target MYC-driven tumours, the majority use indirect approaches (grey boxes) such as those based on inhibiting MYC synthetic lethal genes or interfering with the expression of MYC at the DNA, RNA or protein level. Direct strategies to inhibit MYC (blue box) include approaches using small molecules, peptides or ‘miniproteins’ to inhibit MYC–MAX dimerization, sequester MAX via homodimer stabilization, or interfering with MYC–MAX binding to target DNA sequences. Ac, acetylation; CDKs, cyclin-dependent kinases; HDACs, histone deacetylases; Me, methylation; P, phosphorylation; Ub, ubiquitylation.
Given that MYC functions as a heterodimer with MAX176, small molecules that target the MYC–MAX interface, stabilize MAX–MAX homodimers or disrupt the binding of MYC–MAX to DNA have been used to inhibit MYC signalling. The MYC–MAX disruptors MYCi361 and MYCi975 (REF.177), the MAX–MAX stabilizer KI-MS2-008 (REF.178), disruptors of MYC–MAX binding to its canonical Ebox DNA sequence (such as ME47 (REF.179)), the inhibitor of MYC–DNA binding and MYC transcriptional activity EN4 (REF.180), and Omomyc have all shown activity in preclinical models of Myc-driven cancers24,25. Some of the discovered small molecules and designed miniproteins show efficacy in various preclinical mouse cancer models but clinical evidence is not yet available. To date, Omomyc is the only promising candidate to have entered clinical trials.
Another approach to targeting MYC involves decreasing MYC biosynthesis or altering its stability. For example, inhibitors of the PI3K–AKT–mTOR signalling pathway suppress MYC translation and thus decrease tumoural levels of MYC in a mouse model181. Similarly, silvestrol, a small-molecule inhibitor of the translation initiator eIF4A, reduces MYC translation and inhibits tumour growth in a mouse model of colon cancer182. Moreover, inhibitors of Aurora kinases can induce MYC protein degradation and specifically inhibit MYC protein overexpression in cancer cells without affecting physiological MYC expression in non-malignant cells183,184. PLK1 also regulates MYC protein stability and is another promising therapeutic target185. Finally, PIN1 modulates MYC turnover and transcriptional activity186. Specific PIN1 inhibitors have shown promising therapeutic activity in preclinical models of MYC-driven cancers187.
In the past few years, important developments have occurred in the design of specific protein degraders or proteolysis-targeting chimaeras (PROTACs). These approaches are based on cereblon-mediated protein degradation and provide a new means to directly and specifically target transcription factors, such as MYC and/or its interaction partners, for proteasomal degradation188,189.
Reducing MYC mRNA stability in tumours is another potential strategy to target MYC-driven tumours190. Antisense oligonucleotides have been used in preclinical models to target MYC191–193. For example, a MYC-specific antisense oligonucleotide, MYC-ASO, impairs tumour progression and elicits an antitumour immune response in a primary transgenic mouse model of HCC194.
Another strategy involves targeting upstream regulators to inhibit MYC transcription. For example, BET-motif inhibitors have demonstrated antitumour efficacy that might partially result from the inhibition of Myc transcription195,196. Drugs that stabilize the G-quadruplex structure in the Myc promoter region, such as IZCZ-3, can also inhibit Myc transcription197–201.
Vulnerabilities that might be therapeutically targetable can be identified in screens for MYC-related synthetic lethal interactions — that is, of genes likely to be key mediators of MYC-driven cellular processes required for tumorigenesis. As a transcription factor, MYC drives the expression of a multitude of gene products that are required to initiate and/or maintain cancers2,5,61,202,203, thus diversifying the arsenal of potential drug targets. Synthetic lethal screens204–206 have identified multiple potential targets, such as cyclin-dependent kinases (CDKs)207,208, SAE1/2-mediated sumoylation204 and TNFRSF10B205. In addition, studies have demonstrated the dependency of MYC-driven cancers on a functional DNA damage response mediated by ATR, CHK1 and PRKDC209,210. A novel therapeutic synthetic lethal approach could address the transcription-independent role of MYCN in preventing replication stress-induced DNA damage. Indeed, in complex with Aurora kinase A, MYCN prevents R-loop formation and thus facilitates cell proliferation in a mouse model of neuroblastoma. This process could potentially be exploited therapeutically through the combined inhibition of Aurora kinase A and ATR211. In Myc-driven mouse models of B cell lymphomas or lung cancer, the anti-apoptotic protein MCL1 is a synthetic lethal vulnerability of MYCN. MCL1 inhibitors are currently being tested in clinical trials (Supplementary Table 1)212,213. Of note, these mechanisms are specific to MYCN.
Epigenetic modifiers, such as histone deacetylases (HDACs)214–216 or histone methyltransferases (such as EZH2217), can also be used to target MYC-driven cancers. In non-small-cell lung cancer cell lines, the combination of DNA methyltransferase inhibitors with HDAC inhibitors led to the suppression of MYC signalling and consequent activation of an IFNα/β-based transcriptional programme accompanied by a CCL5-mediated antitumour T cell response140. These investigators were able to demonstrate that epigenetic therapy could deplete MYC, enabling tumours to reverse immune evasion140. Similarly, MYC can promote cancer through miR17-92-mediated regulation of chromatin remodelling104,125,218. The use of anti-miR-17 oligonucleotides delayed tumour progression in a mouse model of liver cancer219. Finally, given that MYC is a key regulator of cellular metabolism19,35,152, targeting MYC synthetic metabolic vulnerabilities, such as SREBP1, is another potential therapeutic strategy153,165. Our laboratory is currently exploiting the concept of synthetic lethality with CRISPR-based functional in vivo genomic screening to identify novel molecular targets in MYC-driven cancers (A.D. and D.W.F., unpublished results).
A final therapeutic approach involves exploiting MYC-induced alterations of immune surveillance. For example, MYC regulates the expression of specific immunomodulatory molecules and cellular immune mechanisms in specific cancers and, thus, molecular and cellular therapies that restore these mechanisms might be particularly effective against MYC-driven cancers. Some evidence supports this strategy, at least experimentally, such that Myc-driven mouse cancers have been shown to be sensitive to the cytokine-mediated restoration of anticancer immune responses220, reinstitution of NK cell-mediated immune surveillance109 or immune-checkpoint inhibition31,108.
Similarly, drugs that target the MYC pathway might sensitize tumours to specific immunotherapies. For example, MYCi361 has demonstrated promising synergy with anti-PD-1 antibodies in a mouse model of Myc-driven prostate cancer177. In addition, in a xenograft model of pancreatic ductal adenocarcinoma, the combination of an anti-PD-L1 antibody and JQ1 had synergistic effects221. Hence, MYC expression might predict response to immunotherapy and targeting the MYC pathway might sensitize cancers to and enhance the efficacy of immunotherapy.
In summary, many strategies can be used to either directly or indirectly target MYC-driven cancers. Ongoing early phase clinical trials are testing direct MYC inhibitors that either silence MYC gene expression, inhibit MYC protein biosynthesis or target MYC for proteasomal degradation (Supplementary Table 1). MYC can also be inhibited indirectly by targeting identified synthetic lethal interactions, with examples including HDACs, CDK4, CDK6, CDK7 and CDK9, DNA repair genes (such as CHK1 and ATR), and anti-apoptotic genes (for example, MCL1; Supplementary Table 1). Furthermore, small-molecule screens have identified compounds that directly target MYC–MAX. Nevertheless, most of these MYC–MAX inhibitors are yet to enter clinical trials. Strategies such as antisense RNA oligonucleotides have been tested in phase I clinical trials but, to our knowledge, have not been further pursued as cancer therapeutics222,223. To date, Omomyc is the only agent directly targeting MYC that has been tested in clinical trials224 (Supplementary Table 1).
We believe that direct targeting of MYC is likely to be more clinically effective than indirect targeting strategies. Nevertheless, several drugs targeting MYC indirectly, either through synthetic lethal interactions or upstream signalling pathways, have reached the clinic (Supplementary Table 1). These agents include CDK4/6 inhibitors and mTOR inhibitors; however, such approaches are likely to be associated with therapeutic resistance. As we have described, MYC controls interconnected cell-intrinsic and cell-extrinsic programmes and, thus, cancer cells will probably be able to bypass the inhibition of any one specific mechanism. Therefore, even the brief or partial depletion of MYC can result in sustained suppression of tumorigenesis in preclinical models18,105,225. We believe that future efforts will make it possible to find agents that directly target MYC and that a therapeutic window exists in which these drugs have clinical activity against cancer while mitigating toxic effects in non-malignant cells.
Future directions
Insights on how MYC regulates both cancer cell-intrinsic processes, such as proliferation or metabolism, and cell-extrinsic phenomena, such as the host immune response and angiogenesis, have important practical implications for the development of therapeutics as well as guiding the selection of therapies. Agents that target MYC could be highly effective for the treatment of some cancer types or as individualized treatments for some patients. To date, no direct MYC inhibitors have been approved for clinical use, although we envision that such agents, once identified, will be effective either alone or in combination with other therapeutic agents to treat MYC-driven cancers. Furthermore, understanding and targeting those MYC functions that are independent of its transcriptional activity might provide alternative therapeutic strategies. Hence, molecular understanding of how MYC causes cancer should enable efforts to identify which patients with MYC-driven cancers are most likely to have a response to specific therapies such as direct MYC inhibitors, MYC synthetic lethal agents, MYC-guided immunotherapies and/or MYC-guided metabolic treatments (FIG. 6). Thus, we envision that patients with tumours could be assigned to MYChigh or MYClow phenotypes defined by genomic events including MYC amplifications and translocations and/or expression of an MYC activation gene signature226,227. Subsequently, deep phenotyping of tumours using different approaches (including DNA and RNA sequencing, mass cytometry, or mass spectrometry) would guide the selection of therapeutic strategies to target MYC (FIG. 6). For example, patients with tumours harbouring MYC amplifications can potentially be treated with direct MYC inhibitors while those with enrichment of synthetic lethal targets of MYC would receive drugs targeting these specific gene products. MYC-directed therapies could be used to sensitize MYChigh immunologically ‘cold’ tumours to immunotherapies, whereas tumours in which MYC predominantly induces metabolic dysregulation can potentially be treated with drugs targeting metabolic pathways (such as lipid metabolism). In summary, discoveries from the past few years elucidating the specific mechanisms by which MYC drives cancer can potentially enable researchers to develop novel therapeutic strategies to target MYC.
Fig. 6 |. Proposed biomarker-stratified therapeutic strategies to target MYC-driven cancers.
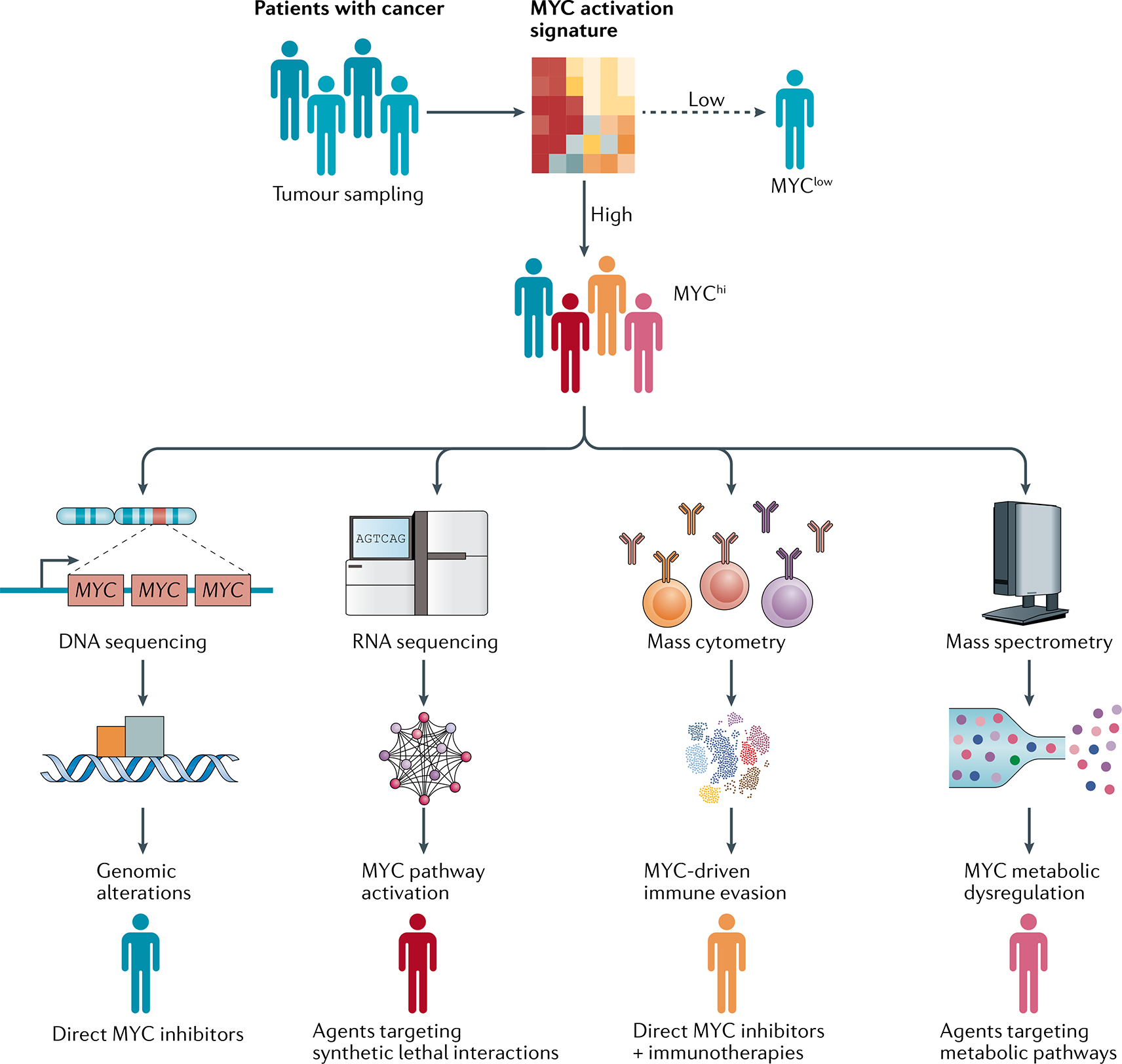
Patients with cancer can be assigned to MYChi and MYClow subgroups defined by enrichment of an MYC activation signature in tumour samples. Patients with MYC-driven tumours can potentially be further classified according to various disease phenotypes on the basis of the dominant mechanism of action of MYC, which would enable stratification to receive different treatments. For example, patients harbouring tumours with MYC amplifications could be treated with direct MYC inhibitors, whereas those with enrichment of synthetic lethal targets of MYC would receive agents targeting these specific gene products. By contrast, MYC-directed therapies could be used to sensitize MYChi immunologically ‘cold’ tumours to immunotherapies, whereas patients with tumours in which MYC prominently induces metabolic dysregulation can be treated with drugs targeting these pathways. In summary, discoveries elucidating the specific mechanisms by which MYC drives cancer are enabling the development of novel and selective therapeutic strategies to target MYC in human cancers.
Conclusions
MYC and other genes in the MYC superfamily are among the most commonly activated signalling mechanisms in human cancer. MYC initiates and maintains cancer through both tumour cell-intrinsic mechanisms and host immune and TME-dependent mechanisms. In experimental models, MYC inactivation can induce rapid tumour regression through effects on tumour cells, which occur as a consequence of oncogene addiction, as well as the restoration of immune responses. To date, however, the MYC pathway remains to be successfully targeted therapeutically. In this Review, we have described multiple approaches for targeting MYC or MYC-regulated pathways that are being evaluated in clinical trials (Supplementary Table 1). Furthermore, we propose that MYC and other members of the MYC pathway could serve as biomarkers to stratify patients for specific therapies (FIG. 6).
MYC is a particularly exciting therapeutic target because it regulates not only tumour cell-intrinsic growth but also the host immune response. Thus, immune surveillance might be restored by targeting the MYC pathway. For more than 30 years, MYC was presumed to contribute to tumorigenesis through its direct effects on cancer cells. Nowadays, the role of MYC as a grand orchestrator of biological processes, not only inherent to tumour cells but also relating to the TME and the host immune system, has become apparent. MYC coordinates intercellular communications that occur between the cancer cells and specific immune cells, thus enabling tumour initiation, progression and metastasis. Experimentally, targeting MYC and the MYC pathway seems to both affect cancer cells and restore antitumour immunity. The evidence of MYC as a grand orchestrator provides further impetus to develop therapeutics to target this otherwise elusive oncogenic pathway.
Supplementary Material
Key points.
The MYC oncogene is activated in the vast majority of cancers by genetic, epigenetic or post-translational mechanisms.
In preclinical models, inactivation of MYC can result in sustained tumour regression owing to oncogene addiction.
MYC activation drives cancer progression through mechanisms involving either the cell-intrinsic acquisition of hallmarks of cancer or dysregulation of the tumour microenvironment and host immune responses.
MYC leads to cancer initiation and maintenance by regulating the host immune system through mechanisms involving immune checkpoints, specific receptors and secreted cytokines.
Currently, no direct inhibitors of MYC are approved; however, many therapeutic agents targeting MYC are under development.
We propose that therapies targeting the MYC pathway will be key to reversing cancerous growth and restoring antitumour immune responses in patients with MYC-driven cancers.
Acknowledgements
R.D. receives grant support from the NIH (grant CA222676). A.D. has received support from Lymphoma Research Foundation. A.S.H. has received support from Lundbreck Foundation. A.M.G. has received support from the NIH (grant CA196585). D.W.F. receives grant support from the NIH (grants CA208735, CA253180, CA188383 and CA184384).
Footnotes
Competing interests
D.W.F. is a consultant for Revolution Medicines, a company developing MYC pathway therapies, co-founder of Bachus and Molecular Decisions, and has had advisory roles for American Gene Technologies, Geron, Moderna and Regulus. The other authors declare no conflicts of interest.
Peer review information
Nature Reviews Clinical Oncology thanks M. Eilers, C.M. Eischen, E.V. Prochownik and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Supplementary information
The online version contains supplementary material available at https://doi.org/10.1038/s41571-021-00549-2.
References
- 1.Meyer N & Penn LZ Reflecting on 25 years with MYC. Nat. Rev. Cancer 8, 976–990 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Dang CV MYC on the path to cancer. Cell 149, 22–35 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaub FX et al. Pan-cancer alterations of the MYC oncogene and its proximal network across The Cancer Genome Atlas. Cell Syst. 6, 282–300.e2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalkat M et al. MYC deregulation in primary human cancers. Genes 8, 151 (2017). [Google Scholar]
- 5.Kress TR, Sabò A & Amati B MYC: connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 15, 593–607 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Conacci-Sorrell M, McFerrin L & Eisenman RN An overview of MYC and its interactome. Cold Spring Harb. Perspect. Med 4, a014357 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williamson D et al. Relationship between MYCN copy number and expression in rhabdomyosarcomas and correlation with adverse prognosis in the alveolar subtype. J. Clin. Oncol 23, 880–888 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Williams RD et al. Molecular profiling reveals frequent gain of MYCN and anaplasia-specific loss of 4q and 14q in Wilms tumor. Genes Chromosomes Cancer 50, 982–995 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Berger A et al. N-Myc–mediated epigenetic reprogramming drives lineage plasticity in advanced prostate cancer. J. Clin. Invest 129, 3924–3940 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rickman DS, Schulte JH & Eilers M The expanding world of N-MYC–driven tumors. Cancer Discov. 8, 150–163 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Cheng J et al. Merkel cell polyomavirus recruits MYCL to the EP400 complex to promote oncogenesis. PLoS Pathog. 13, e1006668 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohshima K et al. Integrated analysis of gene expression and copy number identified potential cancer driver genes with amplification-dependent overexpression in 1,454 solid tumors. Sci. Rep 7, 641 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boutros PC et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat. Genet 47, 736–745 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Adams JM et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 318, 533–538 (1985). [DOI] [PubMed] [Google Scholar]
- 15.Dalla-Favera R et al. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc. Natl Acad. Sci. USA 79, 7824–7827 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gurel B et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod. Pathol 21, 1156–1167 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stock C, Kager L, Fink FM, Gadner H & Ambros PF Chromosomal regions involved in the pathogenesis of osteosarcomas. Genes Chromosomes Cancer 28, 329–336 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Jain M et al. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science 297, 102–104 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Shroff EH et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc. Natl Acad. Sci. USA 112, 6539–6544 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Felsher DW & Bishop JM Reversible tumorigenesis by MYC in hematopoietic lineages. Mol. Cell 4, 199–207 (1999). [DOI] [PubMed] [Google Scholar]
- 21.Wu C-H et al. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc. Natl Acad. Sci. USA 104, 13028–13033 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lourenco C et al. Modelling the MYC-driven normal-to-tumour switch in breast cancer. Dis. Model. Mech 12, dmm038083 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sodir NM et al. MYC instructs and maintains pancreatic adenocarcinoma phenotype. Cancer Discov. 10, 588–607 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Soucek L et al. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 27, 504–513 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jung LA et al. OmoMYC blunts promoter invasion by oncogenic MYC to inhibit gene expression characteristic of MYC-dependent tumors. Oncogene 36, 1911–1924 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Demma MJ et al. Omomyc reveals new mechanisms to inhibit the MYC oncogene.Mol. Cell. Biol 39, e00248–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weinstein IB Cancer: addiction to oncogenes —the Achilles heal of cancer. Science 297, 63–64 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Carroll PA, Freie BW, Mathsyaraja H & Eisenman RN The MYC transcription factor network: balancing metabolism, proliferation and oncogenesis. Front. Med 12, 412–425 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dang CV c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell. Biol 19, 1–11 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Casey SC, Baylot V & Felsher DW The MYC oncogene is a global regulator of the immune response. Blood 131, 2007–2015 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Casey SC, Baylot V & Felsher DW MYC: master regulator of immune privilege. Trends Immunol. 38, 298–305 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Casacuberta-Serra S & Soucek L Myc and Ras, the Bonnie and Clyde of immune evasion. Transl. Cancer Res 7 (Suppl. 4), S457–S459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baluapuri A, Wolf E & Eilers M Target gene-independent functions of MYC oncoproteins. Nat. Rev. Mol. Cell Biol 21, 255–267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stine ZE, Walton ZE, Altman BJ, Hsieh AL & Dang CV MYC, metabolism, and cancer. Cancer Discov. 5, 1024–1039 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bernard S & Eilers M Control of cell proliferation and growth by Myc proteins. Results Probl. Cell. Differ 42, 329–342 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Bretones G, Delgado MD & León J Myc and cell cycle control. Biochim. Biophys. Acta 1849, 506–516 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Levens D ‘You don’t muck with MYC’. Genes Cancer 1, 547–554 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beroukhim R et al. The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turner KM et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543, 122–125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu S et al. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 575, 699–703 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boxer LM & Dang CV Translocations involving c-myc and c-myc function. Oncogene 20, 5595–5610 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Drach J et al. The biology of multiple myeloma. J. Cancer Res. Clin. Oncol 126, 441–447 (2000). [PubMed] [Google Scholar]
- 44.Dudley JP, Mertz JA, Rajan L, Lozano M & Broussard DR What retroviruses teach us about the involvement of c-Myc in leukemias and lymphomas. Leukemia 16, 1086–1098 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Weng AR et al. c-Myc is an important direct target of Notch 1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 20, 2096–2109 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He TC et al. Identification of c-MYC as a target of the APC pathway. Science 281, 1509–1512 (1998). [DOI] [PubMed] [Google Scholar]
- 47.Yagi K et al. c-myc is a downstream target of the Smad pathway. J. Biol. Chem 277, 854–861 (2002). [DOI] [PubMed] [Google Scholar]
- 48.Allen-Petersen BL & Sears RC Mission possible: advances in MYC therapeutic targeting in cancer. BioDrugs 33, 539–553 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sears R et al. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 14, 2501–2514 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arnold HK & Sears RC Protein phosphatase 2A regulatory subunit B56alpha associates with c-myc and negatively regulates c-myc accumulation. Mol. Cell. Biol 26, 2832–2844 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Farrell AS et al. MYC regulates ductalneuroendocrine lineage plasticity in pancreatic ductal adenocarcinoma associated with poor outcome and chemoresistance. Nat. Commun 8, 1728 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang X et al. Phosphorylation regulates c-Myc’s oncogenic activity in the mammary gland. Cancer Res. 71, 925–936 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reavie L et al. Regulation of c-Myc ubiquitination controls chronic myelogenous leukemia initiation and progression. Cancer Cell 23, 362–375 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ruvolo PP The broken ‘Off switch in cancer signaling: PP2A as a regulator of tumorigenesis, drug resistance, and immune surveillance. BBA Clin. 6, 87–99 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kauko O et al. PP2A inhibition is a druggable MEK inhibitor resistance mechanism in KRAS-mutant lung cancer cells. Sci. Transl. Med 10, eaaq1093 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou XZ & Lu KP The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nat. Rev. Cancer 16, 463–478 (2016). [DOI] [PubMed] [Google Scholar]
- 57.Cheng C-W & Tse E PIN 1 in cell cycle control and cancer. Front. Pharmacol 9, 1367 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.King B et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell 153, 1552–1566 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yeh CH, Bellon M & Nicot C FBXW7: a critical tumor suppressor of human cancers. Mol. Cancer 17, 115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cao J, Ge M-H & Ling Z-Q Fbxw7 tumor suppressor: a vital regulator contributes to human tumorigenesis. Medicine 95, e2496 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grandori C, Cowley SM, James LP & Eisenman RN The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol 16, 653–699 (2000). [DOI] [PubMed] [Google Scholar]
- 62.Edelmann J et al. Frequent evolution of copy number alterations in CLL following first-line treatment with FC(R) is enriched with TP53 alterations: results from the CLL8 trial. Leukemia 31, 734–738 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang G & Hurlin PJ MNT and emerging concepts of MNT-MYC antagonism. Genes 8, 83 (2017). [Google Scholar]
- 64.Fredlund E, Ringner M, Maris JM & Påhlman S High Myc pathway activity and low stage of neuronal differentiation associate with poor outcome in neuroblastoma. Proc. Natl Acad. Sci. USA 105, 14094–14099 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Diolaiti D, McFerrin L, Carroll PA & Eisenman RN Functional interactions among members of the MAX and MLX transcriptional network during oncogenesis. Biochim. Biophys. Acta 1849, 484–500 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gabay M, Li Y & Felsher DW MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med 4, a014241 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bailey ST et al. MYC activation cooperates with Vhl and Ink4a/Arf loss to induce clear cell renal cell carcinoma. Nat. Commun 8, 15770 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vaux DL, Cory S & Adams JM Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 335, 440–442 (1988). [DOI] [PubMed] [Google Scholar]
- 69.Teitz T et al. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat. Med 6, 529–535 (2000). [DOI] [PubMed] [Google Scholar]
- 70.Wu KJ et al. Direct activation of TERT transcription by c-MYC. Nat. Genet 21, 220–224 (1999). [DOI] [PubMed] [Google Scholar]
- 71.Hofmann JW et al. Reduced expression of MYC increases longevity and enhances healthspan. Cell 160, 477–488 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Seton-Rogers S Driving immune evasion. Nat. Rev. Cancer 18, 67–67 (2018). [DOI] [PubMed] [Google Scholar]
- 73.Chappell J & Dalton S Roles for MYC in the establishment and maintenance of pluripotency. Cold Spring Harb. Perspect. Med 3, a014381 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pelengaris S & Khan M The many faces of c-MYC. Arch. Biochem. Biophys 416, 129–136 (2003). [DOI] [PubMed] [Google Scholar]
- 75.de la Cova C, Abril M, Bellosta P, Gallant P & Johnston LA Drosophila myc regulates organ size by inducing cell competition. Cell 117, 107–116 (2004). [DOI] [PubMed] [Google Scholar]
- 76.Bettess MD et al. c-Myc is required for the formation of intestinal crypts but dispensable for homeostasis of the adult intestinal epithelium. Mol. Cell. Biol 25, 7868–7878 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Evan GI et al. Induction of apoptosis in fibroblasts by c-myc protein. Cell 69, 119–128 (1992). [DOI] [PubMed] [Google Scholar]
- 78.Shi Y et al. Role for c-myc in activation-induced apoptotic cell death in T cell hybridomas. Science 257, 212–214 (1992). [DOI] [PubMed] [Google Scholar]
- 79.Kaczmarek L, Hyland JK, Watt R, Rosenberg M & Baserga R Microinjected c-myc as a competence factor. Science 228, 1313–1315 (1985). [DOI] [PubMed] [Google Scholar]
- 80.Zindy F et al. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 12, 2424–2433 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Karn J, Watson JV, Lowe AD, Green SM & Vedeckis W Regulation of cell cycle duration by c-myc levels. Oncogene 4, 773–787 (1989). [PubMed] [Google Scholar]
- 82.Rosenwald IB Upregulated expression of the genes encoding translation initiation factors eIF-4E and eIF-2a in transformed cells. Cancer Lett. 102, 113–123 (1996). [DOI] [PubMed] [Google Scholar]
- 83.Boon K et al. N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J. 20, 1383–1393 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schmidt EV The role of c-myc in cellular growth control. Oncogene 18, 2988–2996 (1999). [DOI] [PubMed] [Google Scholar]
- 85.Grewal SS, Li L, Orian A, Eisenman RN & Edgar BA Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat. Cell Biol 7, 295–302 (2005). [DOI] [PubMed] [Google Scholar]
- 86.Dong Y, Tu R, Liu H & Qing G Regulation of cancer cell metabolism: oncogenic MYC in the driver’s seat. Signal Transduct. Target. Ther 5, 124 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim J-W et al. Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol. Cell. Biol 24, 5923–5936 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Osthus RC et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem 275, 21797–21800 (2000). [DOI] [PubMed] [Google Scholar]
- 89.Wise DR et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl Acad. Sci. USA 105, 18782–18787 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dominguez-Sola D et al. Non-transcriptional control of DNA replication by c-Myc. Nature 448, 445–451 (2007). [DOI] [PubMed] [Google Scholar]
- 91.Takahashi K & Yamanaka S Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006). [DOI] [PubMed] [Google Scholar]
- 92.García-Gutiérrez L, Delgado MD & León J MYC oncogene contributions to release of cell cycle brakes. Genes 10, 244 (2019). [Google Scholar]
- 93.Li Z et al. c-Myc suppression of DNA double-strand break repair. Neoplasia 14, 1190–1202 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Felsher DW & Bishop JM Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc. Natl Acad. Sci. USA 96, 3940–3944 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kuzyk A & Mai S c-MYC-induced genomic instability. Cold Spring Harb. Perspect. Med 4, a014373 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Karlsson A et al. Defective double-strand DNA break repair and chromosomal translocations by MYC overexpression. Proc. Natl Acad. Sci. USA 100, 9974–9979 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vafa O et al. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol. Cell 9, 1031–1044 (2002). [DOI] [PubMed] [Google Scholar]
- 98.Dang CV, Li F & Lee LA Could MYC induction of mitochondrial biogenesis be linked to ROS production and genomic instability? Cell Cycle 4, 1465–1466 (2005). [DOI] [PubMed] [Google Scholar]
- 99.Reimann M et al. The Myc-evoked DNA damage response accounts for treatment resistance in primary lymphomas in vivo. Blood 110, 2996–3004 (2007). [DOI] [PubMed] [Google Scholar]
- 100.Beer S et al. Developmental context determines latency of MYC-induced tumorigenesis. PLoS Biol. 2, e332 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ray S et al. MYC can induce DNA breaks in vivo and in vitro independent of reactive oxygen species. Cancer Res. 66, 6598–6605 (2006). [DOI] [PubMed] [Google Scholar]
- 102.Baudino TA et al. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 16, 2530–2543 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shchors K et al. The Myc-dependent angiogenic switch in tumors is mediated by interleukin 1β. Genes Dev. 20, 2527–2538 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dews M et al. The Myc–miR-17-92 axis blunts TGFβ signaling and production of multiple TGFβ-dependent antiangiogenic factors. Cancer Res. 70, 8233–8246 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Giuriato S et al. Sustained regression of tumors upon MYC inactivation requires p53 or thrombospondin-1 to reverse the angiogenic switch. Proc. Natl Acad. Sci. USA 103, 16266–16271 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rakhra K et al. CD4+ T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell 18, 485–498 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kortlever RM et al. Myc cooperates with Ras by programming inflammation and immune suppression. Cell 171, 1301–1315.e14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Casey SC et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352, 227–231 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Swaminathan S et al. MYC functions as a switch for natural killer cell-mediated immune surveillance of lymphoid malignancies. Nat. Commun 11, 2860 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Maeda T et al. MUC1-C induces PD-L1 and immune evasion in triple-negative breast cancer. Cancer Res. 78, 205–215 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Muthalagu N et al. Repression of the type I interferon pathway underlies MYC & KRAS-dependent evasion of NK & B cells in pancreatic ductal adenocarcinoma. Cancer Discov. 10, 872–887 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dhanasekaran R et al. MYC and Twist1 cooperate to drive metastasis by eliciting crosstalk between cancer and innate immunity. eLife 9, e50731 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bernards R, Dessain SK & Weinberg RA N-myc amplification causes down-modulation of MHC class I antigen expression in neuroblastoma. Cell 47, 667–674 (1986). [DOI] [PubMed] [Google Scholar]
- 114.Braun J, Felsher DW & Goodglick LA c-myc, MHCI, and NK resistance in immunodeficiency lymphomas. Ann. N. Y. Acad. Sci 651, 467–469 (1992). [DOI] [PubMed] [Google Scholar]
- 115.Versteeg R, Noordermeer IA, Krüse-Wolters M, Ruiter DJ & Schrier PI c-myc down-regulates class I HLA expression in human melanomas. EMBO J. 7, 1023–1029 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.van Riggelen J et al. The interaction between Myc and Miz1 is required to antagonize TGFbeta-dependent autocrine signaling during lymphoma formation and maintenance. Genes Dev. 24, 1281–1294 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Reimann M et al. Tumor stroma-derived TGF-beta limits myc-driven lymphomagenesis via Suv39h1-dependent senescence. Cancer Cell 17, 262–272 (2010). [DOI] [PubMed] [Google Scholar]
- 118.Xu Y et al. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat. Med 25, 301–311 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Vartuli RL et al. Eya3 promotes breast tumor-associated immune suppression via threonine phosphatase-mediated PD-L1 upregulation. J. Clin. Invest 128, 2535–2550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dhanasekaran R et al. MYC and Twist1 cooperate to drive metastasis by eliciting crosstalk between cancer and innate immunity. eLife 9, e50731 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shachaf CM et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 431, 1112–1117 (2004). [DOI] [PubMed] [Google Scholar]
- 122.Tran PT et al. Combined inactivation of MYC and K-Ras oncogenes reverses tumorigenesis in lung adenocarcinomas and lymphomas. PLoS One 3, e2125 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tran PT et al. Survival and death signals can predict tumor response to therapy after oncogene inactivation. Sci. Transl. Med 3, 103ra99 (2011). [Google Scholar]
- 124.Bui TV & Mendell JT Myc: maestro of microRNAs. Genes Cancer 1, 568–575 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Li Y, Choi PS, Casey SC, Dill DL & Felsher WD MYC through miR-17–92 Suppresses specific target genes to maintain survival, autonomous proliferation, and a neoplastic state. Cancer Cell 26, 262–272 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chang T-C et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat. Genet 40, 43–50 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Adams CM, Hiebert SW & Eischen CM Myc induces miRNA-mediated apoptosis in response to HDAC inhibition in hematologic malignancies. Cancer Res. 76, 736–748 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhou L et al. Silencing of thrombospondin-1 is critical for myc-induced metastatic phenotypes in medulloblastoma. Cancer Res. 70, 8199–8210 (2010). [DOI] [PubMed] [Google Scholar]
- 129.Swaminathan S et al. MYC functions as a master switch for natural killer cell-mediated immune surveillance of lymphoid malignancies. Blood 132, 2619–2619 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Casey SC, Li Y, Fan AC & Felsher DW Oncogene withdrawal engages the immune system to induce sustained cancer regression. J. Immunother. Cancer 2, 24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sharma SV, Fischbach MA, Haber DA & Settleman J ‘Oncogenic shock’: explaining oncogene addiction through differential signal attenuation. Clin. Cancer Res 12, 4392s–4395s (2006). [DOI] [PubMed] [Google Scholar]
- 132.Fisher GH et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 15, 3249–3262 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kohl NE et al. Protein farnesyltransferase inhibitors block the growth of ras-dependent tumors in nude mice. Proc. Natl Acad. Sci. USA 91, 9141–9145 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Moody SE et al. Conditional activation of Neu in the mammary epithelium of transgenic mice results in reversible pulmonary metastasis. Cancer Cell 2, 451–461 (2002). [DOI] [PubMed] [Google Scholar]
- 135.Hori SS et al. A mathematical model of tumor regression and recurrence after therapeutic oncogene inactivation. Sci. Rep 11, 1341 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Soucek L et al. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat. Med 13, 1211–1218 (2007). [DOI] [PubMed] [Google Scholar]
- 137.Mahauad-Fernandez WD & Felsher DW The Myc and Ras partnership in cancer: indistinguishable alliance or contextual relationship? Cancer Res. 80, 3799–3802 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sun L et al. Gastric cancer mesenchymal stem cells derived IL-8 induces PD-L1 expression in gastric cancer cells via STAT3/mTOR-c-Myc signal axis. Cell Death Dis. 9, 928 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kharma B et al. STAT1 drives tumor progression in serous papillary endometrial cancer. Cancer Res. 74, 6519–6530 (2014). [DOI] [PubMed] [Google Scholar]
- 140.Topper MJ et al. Epigenetic therapy Ties MYC depletion to reversing immune evasion and treating lung cancer. Cell 171, 1284–1300.e21 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Coelho MA et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity 47, 1083–1099.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Melaiu O et al. PD-L1 is a therapeutic target of the bromodomain inhibitor JQ1 and, combined with HLA Class I, a promising prognostic biomarker in neuroblastoma. Clin. Cancer Res 23, 4462–4472 (2017). [DOI] [PubMed] [Google Scholar]
- 143.Atsaves V et al. PD-L1 is commonly expressed and transcriptionally regulated by STAT3 and MYC in ALK-negative anaplastic large-cell lymphoma. Leukemia 31, 1633–1637 (2017). [DOI] [PubMed] [Google Scholar]
- 144.Pai S et al. CD47-SIRPα signaling induces epithelial-mesenchymal transition and cancer stemness and links to a poor prognosis in patients with oral squamous cell carcinoma. Cells 8, 1658 (2019). [Google Scholar]
- 145.Kaur S et al. Thrombospondin-1 signaling through CD47 inhibits self-renewal by regulating c-Myc and other stem cell transcription factors. Sci. Rep 3, 1673 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Li W et al. Targeting MYC activity in double-hit lymphoma with MYC and BCL2 and/or BCL6 rearrangements with epigenetic bromodomain inhibitors. J. Hematol. Oncol 12, 73 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kamijo H et al. Thrombospondin-1 promotes tumor progression in cutaneous T-cell lymphoma via CD47. Leukemia 34, 845–856 (2020). [DOI] [PubMed] [Google Scholar]
- 148.Felsher DW, Rhim SH & Braun J A murine model for B-cell lymphomagenesis in immunocompromised. hosts:naturalkillercells arean importantcomponent of host resistance to premalignant B-cell lines. Cancer Res. 50, 7050–7056 (1990). [PubMed] [Google Scholar]
- 149.El-Jawhari JJ et al. Blocking oncogenic RAS enhances tumour cell surface MHC class I expression but does not alter susceptibility to cytotoxic lymphocytes. Mol. Immunol 58, 160–168 (2014). [DOI] [PubMed] [Google Scholar]
- 150.Ji H et al. K-ras activation generates an inflammatory response in lung tumors. Oncogene 25, 2105–2112 (2006). [DOI] [PubMed] [Google Scholar]
- 151.Van Dang C & Kim J-W Convergence of cancer metabolism and immunity: an overview. Biomol. Ther 26, 4–9 (2018). [Google Scholar]
- 152.Gouw AM, Hsieh AL, Stine ZE & Dang CV In: Tumor Cell Metabolism (Mazurek S, Shoshan M eds) 101–122 (Springer, 2015). [Google Scholar]
- 153.Dang CV MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med 3, a014217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gouw AM et al. The MYC oncogene cooperates with sterol-regulated element-binding protein to regulate lipogenesis essential for neoplastic growth. Cell Metab. 30, 556–572.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ricciardi S et al. The translational machinery of human CD4 T cells is poised for activation and controls the switch from quiescence to metabolic remodeling. Cell Metab. 28, 895–906.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Friesen LR, Gu B & Kim CH A ligand-independent fast function of RARa promotes exit from metabolic quiescence upon T cell activation and controls T cell differentiation. Mucosal Immunol. 14, 100–112 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chapman NM, Boothby MR & Chi H Metabolic coordination of T cell quiescence and activation. Nat. Rev. Immunol 20, 55–70 (2020). [DOI] [PubMed] [Google Scholar]
- 158.Kapur S & Pal A Immune cell activation: stimulation, costimulation, and regulation of cellular activation. Immune Response Activation Immunomodul. 10.5772/intechopen.81568 (2019). [DOI] [Google Scholar]
- 159.Shim H et al. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc. Natl Acad. Sci. USA 94, 6658–6663 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Briggs KJ et al. Paracrine Induction of HIF by glutamate in breast cancer: EglN1 senses cysteine. Cell 166, 126–139 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bott AJ et al. Oncogenic Myc induces expression of glutamine synthetase through promoter demethylation. Cell Metab. 22, 1068–1077 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gerriets VA & Rathmell JC Metabolic pathways in T cell fate and function. Trends Immunol. 33, 168–173 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Verbist KC et al. Metabolic maintenance of cell asymmetry following division in activated T lymphocytes. Nature 532, 389–393 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Le A et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 15, 110–121 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Eberlin LS et al. Alteration of the lipid profile in lymphomas induced by MYC overexpression. Proc. Natl Acad. Sci. USA 111, 10450–10455 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Allison KE, Coomber BL & Bridle BW Metabolic reprogramming in the tumour microenvironment: a hallmark shared by cancer cells and T lymphocytes. Immunology 152, 175–184 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kouidhi S, Ben Ayed F & Elgaaied AB Targeting tumor metabolism: a new challenge to improve immunotherapy. Front. Immunol 9, 353 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Massó-Vallés D & Soucek L Blocking myc to treat cancer: reflecting on two decades of omomyc. Cells 9, 883 (2020). [Google Scholar]
- 169.Wolf E & Eilers M Targeting MYC proteins for tumor therapy. Annu. Rev. Cancer Biol 4, 61–75 (2020). [Google Scholar]
- 170.McKeown MR & Bradner JE Therapeutic strategies to inhibit MYC. Cold Spring Harb. Perspect. Med 4, a014266 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Fletcher S & Prochownik EV Small-molecule inhibitors of the Myc oncoprotein. Biochim. Biophys. Acta 1849, 525–543 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Whitfield JR, Beaulieu M-E & Soucek L Strategies to inhibit myc and their clinical applicability. Front. Cell Dev. Biol 5, 10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Carabet LA, Rennie PS & Cherkasov A Therapeutic inhibition of myc in cancer. structural bases and computer-aided drug discovery approaches. Int. J. Mol. Sci 20, 120 (2018). [Google Scholar]
- 174.Madden SK, de Araujo AD, Gerhardt M, Fairlie DP & Mason JM Taking the Myc out of cancer: toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 20, 3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Thng DKH, Toh TB & Chow EK-H Capitalizing on synthetic lethality of MYC to treat cancer in the digital age. Trends Pharmacol. Sci 42, 166–182 (2021). [DOI] [PubMed] [Google Scholar]
- 176.Cascon A & Robledo M MAX and MYC: a heritable breakup. Cancer Res. 72, 3119–3124 (2012). [DOI] [PubMed] [Google Scholar]
- 177.Han H et al. Small-molecule MYC inhibitors suppress tumor growth and enhance immunotherapy. Cancer Cell 36, 483–497.e15 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Struntz NB et al. Stabilization of the max homodimer with a small molecule attenuates Myc-driven transcription. Cell Chem. Biol 26, 711–723.e14 (2019). [DOI] [PubMed] [Google Scholar]
- 179.Lustig LC et al. Inhibiting MYC binding to the E-box DNA motif by ME47 decreases tumour xenograft growth. Oncogene 36, 6830–6837 (2017). [DOI] [PubMed] [Google Scholar]
- 180.Boike L et al. Discovery of a functional covalent ligand targeting an iIntrinsically disordered cysteine within MYC. Cell Chem. Biol 28, 4–13.e17 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Pourdehnad M et al. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc. Natl Acad. Sci. USA 110, 11988–11993 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wiegering A et al. Targeting translation initiation bypasses signaling crosstalk mechanisms that maintain high MYC levels in colorectal cancer. Cancer Discov. 5, 768–781 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Gustafson WC et al. Drugging MYCN through an allosteric transition in Aurora kinase A. Cancer Cell 26, 414–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Yang D et al. Therapeutic potential of a synthetic lethal interaction between the MYC proto-oncogene and inhibition of aurora-B kinase. Proc. Natl Acad. Sci. USA 107, 13836–13841 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Xiao D et al. Polo-like kinase-1 regulates myc stabilization and activates a feedforward circuit promoting tumor cell survival. Mol. Cell 64, 493–506 (2016). [DOI] [PubMed] [Google Scholar]
- 186.Yeh E et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol 6, 308–318 (2004). [DOI] [PubMed] [Google Scholar]
- 187.Farrell AS et al. Pin1 regulates the dynamics of c-Myc DNA binding to facilitate target gene regulation and oncogenesis. Mol. Cell. Biol 33, 2930–2949 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Winter GE et al. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Bondeson DP et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol 11, 611–617 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Koh CM, Sabò A & Guccione E Targeting MYC in cancer therapy: RNA processing offers new opportunities. Bioessays 38, 266–275 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ricker JL, Mata JE, Iversen PL & Gattone VH c-myc antisense oligonucleotide treatment ameliorates murine ARPKD. Kidney Int. 61 (Suppl. 1), S125–S131 (2002). [DOI] [PubMed] [Google Scholar]
- 192.Yu BW, Nguyen D, Anderson S & Allegra CA Phosphorothioated antisense c-myc oligonucleotide inhibits the growth of human colon carcinoma cells. Anticancer. Res 17, 4407–4413 (1997). [PubMed] [Google Scholar]
- 193.Cotter FE Antisense oligonucleotides: considerations for lymphoma therapy. Hematology 1, 53–57 (1996). [DOI] [PubMed] [Google Scholar]
- 194.Dhanasekaran R et al. MYC ASO impedes tumorigenesis and elicits oncogene addiction in autochthonous transgenic mouse models of HCC and RCC. Mol. Ther. Nucleic Acids 21, 850–859 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Filippakopoulos P et al. Selective inhibition of BET bromodomains. Nature 468, 1067–1073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Raina K et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl Acad. Sci. USA 113, 7124–7129 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Felsenstein KM et al. Small molecule microarrays enable the identification of a selective, quadruplex-binding inhibitor of MYC expression. ACS Chem. Biol 11, 139–148 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Brown RV, Danford FL, Gokhale V, Hurley LH & Brooks TA Demonstration that drug-targeted down-regulation of MYC in non-Hodgkins lymphoma is directly mediated through the promoter G-quadruplex. J. Biol. Chem 286, 41018–41027 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Dutta D et al. Cell penetrating thiazole peptides inhibit c-MYC expression via site-specific targeting of c-MYC G-quadruplex. Nucleic Acids Res. 46, 5355–5365 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Hu M-H et al. Discovery of a new four-leaf clover-like ligand as a potent c-MYC transcription inhibitor specifically targeting the promoter G-quadruplex. J. Med. Chem 61, 2447–2459 (2018). [DOI] [PubMed] [Google Scholar]
- 201.Panda D, Saha P, Das T & Dash J Target guided synthesis using DNA nano-templates for selectively assembling a G-quadruplex binding c-MYC inhibitor. Nat. Commun 8, 16103 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Harbor CS, Dang CV & Eisenman RN Myc and the pathway to cancer (Cold Spring Harbor Laboratory Press, 2014). [Google Scholar]
- 203.Harrison C Synthetic lethality enables targeting of MYC. Nat. Rev. Drug Discov 11, 602–602 (2012). [DOI] [PubMed] [Google Scholar]
- 204.Kessler JD et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science 335, 348–353 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Wang Y et al. Synthetic lethal targeting of MYC by activation of the DR5 death receptor pathway. Cancer Cell 5, 501–512 (2004). [DOI] [PubMed] [Google Scholar]
- 206.Chen L et al. CRISPR-Cas9 screen reveals a MYCN-amplified neuroblastoma dependency on EZH2. J. Clin. Invest 128, 446–462 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Chipumuro E et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 159, 1126–1139 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Kwiatkowski N et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 511, 616–620 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Krüger K et al. Multiple DNA damage-dependent and DNA damage-independent stress responses define the outcome of ATR/Chk1 targeting in medulloblastoma cells. Cancer Lett. 430, 34–46 (2018). [DOI] [PubMed] [Google Scholar]
- 210.Murga M et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. Struct. Mol. Biol 18, 1331–1335 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Roeschert I et al. Combined inhibition of Aurora-A and ATR kinases results in regression of MYCN-amplified neuroblastoma. Nat. Cancer 2, 312–326 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Dammert MA et al. MYC paralog-dependent apoptotic priming orchestrates a spectrum of vulnerabilities in small cell lung cancer. Nat. Commun 10, 3485 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Kelly GL et al. Targeting of MCL-1 kills MYC-driven mouse and human lymphomas even when they bear mutations in p53. Genes Dev. 28, 58–70 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Kretzner L et al. Combining histone deacetylase inhibitor vorinostat with aurora kinase inhibitors enhances lymphoma cell killing with repression of c-Myc, hTERT, and microRNA levels. Cancer Res 71, 3912–3920 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Pei Y et al. HDAC and PI3K antagonists cooperate to inhibit growth of MYC-driven medulloblastoma. Cancer Cell 29, 311–323 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Lernoux M et al. Novel HDAC inhibitor MAKV-8 and imatinib synergistically kill chronic myeloid leukemia cells via inhibition of BCR-ABL/MYC-signaling: effect on imatinib resistance and stem cells. Clin. Epigenet 12, 69 (2020). [Google Scholar]
- 217.Zhao X et al. Disruption of the MYC-miRNA-EZH2 loop to suppress aggressive B-cell lymphoma survival and clonogenicity. Leukemia 27, 2341–2350 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.O’Donnell KA, Wentzel EA, Zeller KI, Dang CV & Mendell JT c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435, 839–843 (2005). [DOI] [PubMed] [Google Scholar]
- 219.Dhanasekaran R et al. Anti-miR-17 therapy delays tumorigenesis in MYC-driven hepatocellular carcinoma (HCC). Oncotarget 9, 5517–5528 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Lai I et al. Lipid nanoparticles that deliver IL-12 messenger RNA suppress tumorigenesis in MYC oncogene-driven hepatocellular carcinoma. J. Immunother. Cancer 6, 125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Pan Y et al. Synergistic inhibition of pancreatic cancer with anti-PD-L1 and c-Myc inhibitor JQ1. Oncoimmunology 8, e1581529 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Devi GR et al. In vivo bioavailability and pharmacokinetics of a c-MYC antisense phosphorodiamidate morpholino oligomer, AVI-4126, in solid tumors. Clin. Cancer Res 11, 3930–3938 (2005). [DOI] [PubMed] [Google Scholar]
- 223.Tolcher AW et al. Safety and activity of DCR-MYC, a first-in-class Dicer-substrate small interfering RNA (DsiRNA) targeting MYC, in a phase I study in patients with advanced solid tumors. J. Clin. Orthod 33, 11006–11006 (2015). [Google Scholar]
- 224.Villanueva MT Long path to MYC inhibition approaches clinical trials. Nat. Rev. Drug Discov 10.1038/d41573-019-00055-2 (2019). [DOI]
- 225.Shachaf CM et al. Genomic and proteomic analysis reveals a threshold level of MYC required for tumor maintenance. Cancer Res. 68, 5132–5142 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Jung M et al. A myc activity signature predicts poor clinical outcomes in myc-associated cancers. Cancer Res. 77, 971–981 (2017). [DOI] [PubMed] [Google Scholar]
- 227.Tran D et al. Functional genomics analysis reveals a myc signature associated with a poor clinical prognosis in liposarcomas. Am. J. Pathol 185, 717–728 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.