

Abstract
Recombination-mediated genetic engineering, also known as recombineering, is the genomic incorporation of homologous single-stranded or double-stranded DNA into bacterial genomes. Recombineering and its derivative methods have radically improved genome engineering capabilities, perhaps none more so than multiplex automated genome engineering (MAGE). MAGE is representative of a set of highly multiplexed single-stranded DNA-mediated technologies. First described in Escherichia coli, both MAGE and recombineering are being rapidly translated into diverse prokaryotes and even into eukaryotic cells. Together, this modern set of tools offers the promise of radically improving the scope and throughput of experimental biology by providing powerful new methods to ease the genetic manipulation of model and non-model organisms. In this Primer, we describe recombineering and MAGE, their optimal use, their diverse applications and methods for pairing them with other genetic editing tools. We then look forward to the future of genetic engineering.
Many cellular properties are multifactorial and emerge from systems-level interactions encoded across multiple genomic loci. Disentangling the specific biological factors that encode observed behaviours is a defining challenge of biology. To do this, scientists must be able to perturb the cell and measure the effects of these perturbations on its physiology. The advent of high-throughput omics technologies — genomics, transcriptomics, proteomics and metabolomics — has provided vast data sets that have led to a more comprehensive understanding of complex biological networks and the biomolecules that govern their collective behaviour. For instance, high-throughput DNA sequencing of a growing number of organisms has revealed their genomic blueprint and has helped to establish a fundamental understanding of genetic variation associated with phenotypic diversity. However, elucidating a causal understanding between genotype and phenotype requires the development of genome modification technologies that are able to introduce targeted genetic changes at the gene, network and whole-genome scale.
Chemical and transposon mutagenesis techniques have been widely used to mutagenize target genomes and are usually coupled to a screen or selection to enrich for an altered phenotype. These techniques are commonly used to tease apart the workings of cellular processes that are encoded by multiple genetic components1. The principal drawbacks of these methods are that they are untargeted and introduce additional genetic modifications unrelated to the selected phenotype, which can confound efforts to link targeted modifications to their associated phenotypes. By contrast, homologous recombination-based methods are used to introduce targeted insertions or deletions of specific genetic elements, but are driven by inefficient native recombination machinery and require customized DNA constructs that contain large regions of homology (often 500–2,000 bp) to drive genetic modifications2.
Homologous recombination.
A type of genetic recombination by which nucleotide sequences are exchanged between molecules that share similar or identical sequences.
Recent advances in gene editing technologies are ushering in a new era in which precise genomic manipulation is becoming feasible across diverse biological organisms. The most widespread gene editing methods currently are nuclease-based and rely on the introduction of DNA double-strand breaks (DSBs)3,4. As the introduction of DSBs is toxic to prokaryotes, these approaches have primarily been used in eukaryotes where a DSB can be rescued by either non-homologous end-joining (NHEJ) or homologous recombination if a homologous DNA template is naturally available or exogenously delivered. Zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) both use engineered protein domains fused to the FokI endonuclease catalytic domain to introduce targeted DNA DSBs5–7. Each zinc-finger module recognizes a 3-bp sequence, whereas TALENs have single-base resolution6. Distinct from ZFNs and TALENs, CRISPR–Cas9 and related proteins are repurposed components of bacterial innate immune systems that are directed through the pairing of the nuclease Cas9 with short guide RNAs (gRNAs) to recognize precise DNA elements via Watson–Crick base pairing8–10. CRISPR–Cas9 approaches have been demonstrated in many organisms, allowing the creation of genetic knockouts by NHEJ and precise gene editing via homology-directed repair (HDR)4.
DNA double-strand breaks.
(DSBs). Simultaneous breaks in both strands of a DNA helix.
Non-homologous end-joining.
(NHEJ). The repair of double-strand DNA breaks by direct ligation of cut DNA ends without a homologous template.
Short guide RNAs.
(gRNAs). Molecules that bind to and then guide Cas9 or a similar protein to a targeted genomic locus by nucleotide base pairing.
Homology-directed repair.
(HDR). The repair of double-strand DNA breaks using a homologous template.
Despite their rapid development and broad utility in eukaryotes, gene editing technologies that rely on the DSB mechanism are limiting for a few key reasons. First, DSBs are cytotoxic in both prokaryotes and eukaryotes, despite quite extensive eukaryotic DSB repair mechanisms, and this lethality is magnified when DSBs are introduced at multiple sites11. Second, many changes introduced by DSB repair are subject to additional unwanted insertions or deletions (indels) during HDR12–14. For many genetic elements (for example, genes, promoters and non-coding RNAs), DNA sequence motifs are sensitive to small sequence changes, meaning that indels and imprecise editing will interfere with their proper functioning. Lastly, DSB repair mechanisms in most eukaryotic cells exhibit a low frequency of HDR relative to NHEJ, necessitating screening, which severely limits throughput. This imprecision prevents more complex applications, in which numerous modifications can be made to a single cell or across a population of cells to produce combinatorial genetic diversity for the exploration of broad genetic landscapes15.
Combinatorial genetic diversity.
A population of cells that has been diversified through genetic engineering to include individual cells that each contain multiple modifications to their genome. These modifications are randomly introduced from a pool of potential modifications, creating combinatorial diversity in the population.
The recent development of base editing16,17 and prime editing18 methods has addressed some of the above shortfalls as they avoid the introduction of DSBs. These methods use either a partially inactivated Cas9 nickase17 or a fully inactivated19 nuclease-deficient Cas9 variant fused to an accessory protein. In the case of base editing, a partially or fully inactivated Cas9 can be used and the accessory protein is a tRNA deaminase domain that chemically modifies a DNA base, permitting A to G and C to T transition mutations. In the case of prime editing, a Cas9 nickase is required, the accessory protein is a reverse transcriptase and the gRNA must be expanded to include two distinct domains: one that anneals to the targeted genomic site and a second that serves as a template for the synthesis of cDNA that contains the desired genomic edit. Limitations remain for both methods: base editing displays significant off-target activity, including on untargeted mRNA, and is limited in its spectrum of possible mutations, whereas prime editing displays inconsistent editing outcomes and necessitates the design and delivery of multiple molecular parts. Furthermore, delivery of multiple gRNAs to a single cell for combinatorial editing is limited, although improvements in gRNA array technology have been made20. Base editing and prime editing have both been used to revert disease-relevant single-nucleotide polymorphisms in human cell lines, setting the stage for clinical applications21,22. Although base editing and prime editing represent key advances in gene editing technology, neither provides a pathway towards genome-scale reverse genetics or the generation of combinatorial genetic diversity at base-pair resolution, which are defining challenges for the field.
Base editing.
A method that fuses a Cas9 nickase to a deaminase domain. The Cas9 is directed by a guide RNA to a target site on the genome, whereupon the deaminase will edit within a window of DNA bases.
Prime editing.
A method whereby a Cas9 nickase is fused to a reverse transcriptase and a guide RNA is fused to a repair template. The Cas9 nickase nicks the target DNA strand, is then resected by host proteins and the reverse-transcribed DNA is used as a repair template, conveying the specified modification.
Cas9 nickase.
A Cas9 variant that has been partially deactivated so that it cuts one strand of a double-stranded helix, creating a ‘nick’ instead of a double-strand break.
Reverse transcriptase.
An enzyme that transcribes RNA into cDNA.
Single-nucleotide polymorphisms.
Any number of substitutions of single nucleotides at specific genomic locations.
Reverse genetics.
Classical genetics is the prediction of allelic determinants of phenotypic variation by genetic analysis. Reverse genetics is the creation of genetic variation and subsequent phenotypic characterization of these known allelic variants.
Recombineering harnesses phage-derived proteins to create universally targetable and scarless modifications to chromosomal DNA, integrating either single-stranded DNA (ssDNA)23 or double-stranded DNA (dsDNA)24 (through an ssDNA intermediate25) into a replicating chromosome (FIG. 1). Classically, recombineering of dsDNA was carried out in Escherichia coli by expressing either the Red operon from coliphage λ26 or RecET from the Rac prophage27. The Red operon comprises three genes: λ exo, which encodes Exo, a 5′ to 3′ dsDNA exonuclease that loads Redβ onto resected ssDNA28,29; λ bet, which encodes Redβ, a single-stranded DNA-annealing protein (SSAP) that anneals ssDNA to genomic DNA at the replication fork30; and λ gam, which encodes Gam, a nuclease inhibitor that protects linear dsDNA from degradation31. Whereas all three proteins are needed for efficient dsDNA recombineering, Redβ alone, the SSAP, was found to be sufficient for ssDNA recombineering23. Customized dsDNA cassettes or synthetic ssDNA oligodeoxynucleotides (oligos), which are the carriers of new genetic information in recombineering, are introduced into a cell population by electroporation and are designed to have homology to a complementary target sequence in the genome at both their 5′-terminal and 3′-terminal arms. Once inside the cell, SSAPs anneal ssDNA to complementary genomic DNA as the chromosome separates into leading and lagging strands at the replication fork. ssDNA that has been annealed at the replication fork acts as a primer for genomic replication, and is thereby incorporated into the nascent copy of the genome. If the genomic modification is not corrected, it is then stably inherited after another round of replication.
Fig. 1 |. Single-stranded and double-stranded recombineering and MAGE.
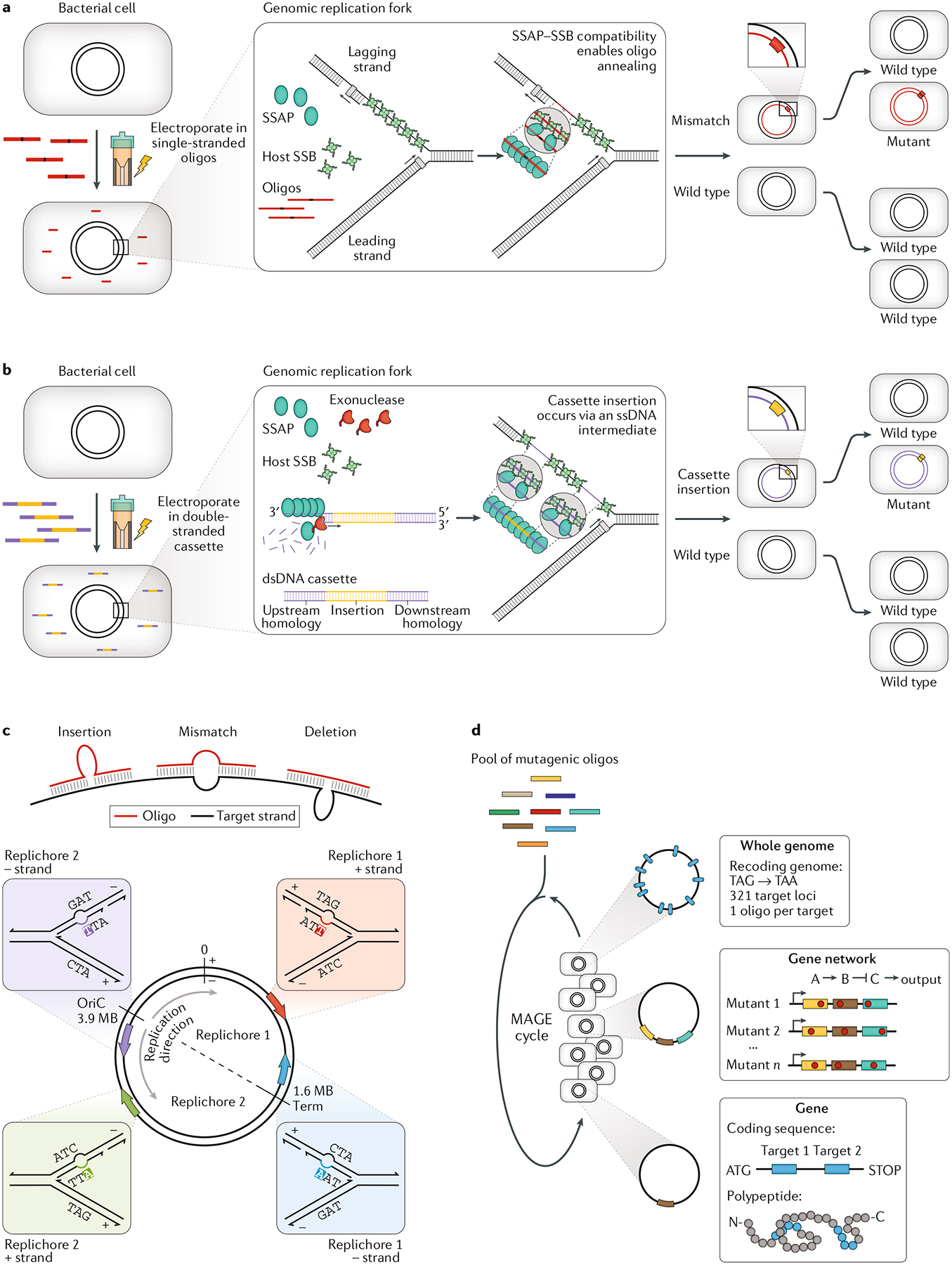
a | In single-stranded recombineering, single-stranded DNA (ssDNA; typically synthetic oligodeoxynucleotides (oligos)) is the carrier of new genetic information. First, ssDNA that is targeted to the lagging strand of the replication fork is electroporated into a cell. Once inside the cell, ssDNA is thought to be bound by an exogenously expressed phage single-stranded DNA-annealing protein (SSAP) and annealed at the replication fork through a specific interaction between the SSAP and the host bacterial single-stranded DNA-binding protein (SSB). The annealed ssDNA then primes synthesis of the nascent genome, incorporating user-defined modifications in the process. After the first round of replication there is one wild-type copy of the genome and one chimeric copy. A second round of replication would then afford one mutant genome and three wild-type copies. This supports the theory that recombineering efficiency should top out at 25%, although higher efficiencies have recently been demonstrated, with editing presumably occurring over multiple rounds of genomic replication. b | Recombineering using double-stranded DNA (dsDNA) as the template works much the same, except that an additional phage protein is required. An exogenously expressed phage exonuclease degrades one strand of a dsDNA cassette, loading the SSAP onto the exposed strand. This ssDNA strand is then annealed at the lagging strand of the replication fork and recombineering proceeds as in part a. Often, dsDNA will be designed to contain a selectable marker, as integration of a long strand is much less efficient than small modifications. c | Oligos can precisely create insertions, mismatches or deletions in genomic DNA. These can have various uses when targeted at different genetic elements. d | Because efficiency is around an order of magnitude higher when targeting the lagging strand of the replication fork, it is important to understand which replichore a target modification is being directed to. This will determine whether to target the positive (+) strand or the negative (−) strand. e | Multiplex automated genome engineering (MAGE) offers two conceptual advances: a pool of diverse oligos is used, and many cycles of editing are conducted to saturate mutations within a population. Applications of MAGE range from whole-genome recoding and allele tracking to mutagenesis of gene clusters and saturating mutagenesis of a single gene. OriC, genomic origin of replication.
Single-stranded DNA-annealing protein.
(SSAP). A protein that speeds the specific annealing of two strands of single-stranded DNA (ssDNA), sometimes also interacting with proteins coating ssDNA to allow annealing to proceed.
Multiplex automated genome engineering (MAGE) builds upon recombineering by targeting the precise modifications introduced by ssDNA recombineering at many genomic loci in a single cell or throughout a population of cells, generating combinatorial genetic diversity32,33. MAGE oligos can be designed to skip, mispair or add bases with respect to the target region, causing a deletion, mismatch or insertion, respectively, and can target loci across the genome with single-base resolution. As this process is easily repeated and single oligo incorporation events are independent of one other, MAGE is automatable and uniquely able to generate large populations of cells containing combinatorial genomic diversity that can sample vast genotypic and phenotypic landscapes. MAGE has been demonstrated as a capable tool for introducing large numbers of precise edits into a single cell, which it does over sequential rounds of editing34. The efficiency of MAGE has been improved since its initial development35, and its utility is being demonstrated in a growing number of prokaryotes36 and eukaryotes32. With its powerful genome engineering capabilities, MAGE has been used for the molecular evolution of single genes37, pathway diversification to alter cellular metabolism32,33 and whole-genome recoding34,38,39. Furthermore, in addition to genomic targets, recombineering and MAGE have been used to target bacterial artificial chromosomes, plasmids and viral genomes40–43.
Multiplex automated genome engineering.
(MAGE). An umbrella term referring to techniques that involve single-stranded DNA-mediated recombineering at multiple sites.
ssDNA recombineering.
Recombineering using single-stranded DNA (ssDNA) as the carrier of genetic information.
Whole-genome recoding.
The replacement of a codon with one or multiple alternative codons systematically throughout a genome.
Bacterial artificial chromosome.
A large circular DNA element distinct from the bacterial chromosome that replicates from a plasmid origin.
This Primer aims to give the reader a thorough understanding of recombineering, MAGE and their many powerful derivative techniques that together form a growing, recombineering-based genome editing toolkit. We begin by discussing the development of MAGE in both prokaryotes and eukaryotes, then discuss experimental design and considerations for host engineering and SSAP identification, move into a thorough analysis of applications given varying levels of allelic recombination frequency (ARF) and finish with a view of where the future lies for improved genome writing and genome editing technologies. We do not extensively cover the mechanism of recombineering; this subject is well covered in a classic Review44. We also do not discuss nuclease-based genome editing techniques, except as a point of reference to help the reader draw an easy comparison between competing technologies; we refer readers to a recent, thorough review of CRISPR–Cas9-based genome editing technologies45.
Allelic recombination frequency.
(ARF). The fraction of a cell population that successfully inherits a specified modification after a genetic editing technique such as multiplex automated genome engineering is carried out.
Experimentation
Recombineering of marked dsDNA was first described in 1998 (REFS26,27), recombineering of markerless ssDNA followed in 2001 (REF.23) and MAGE was developed 8 years later in 2009 (REF.33). All of these milestones were achieved in E. coli. We detail here the technical advances in recombineering that enabled the development of MAGE, improvements to the technology since that time and efforts to broaden the range of species in which recombineering and MAGE are available. In the past few years, efforts to broaden the host range of these techniques have accelerated, giving rise to the first description of MAGE in a eukaryotic host32, which is given its own subsection below.
Recombineering and MAGE in prokaryotes
MAGE is essentially multiplex oligo-mediated recombineering, and enables rapid, automated and high-throughput genome editing in E. coli33; however, in its standard form, MAGE has several limitations. It is optimized for laboratory E. coli strains, demands prior genetic modification of the host bacterium and is prone to the accumulation of off-target mutations34. In this section, we summarize the methodological advances that have been made to address these issues and discuss experimental design considerations for recombineering and MAGE in prokaryotes.
MAGE differs conceptually from simple ssDNA recombineering because, instead of a single oligo, it uses pools of oligos or other carriers of genetic information, and multiple cycles of editing are performed to increase the penetration of mutations into a population of edited cells. When first described in E. coli, MAGE depended primarily on three technical advances: oligo-mediated recombineering with Redβ, the SSAP from the Red operon of coliphage λ23; chemical modification of oligos to include two phosphorothioate bonds at the 5′ end of the oligo to resist nuclease degradation and improve the lifetime of the oligo in the cell33; and avoidance of host methyl-directed mismatch repair (MMR) by disruption of mutS, which recognizes mispaired bases46 (FIG. 2). Two of the limitations of MAGE mentioned above — the need to modify a targeted bacterium and the accumulation of off-target mutations — are direct consequences of this last advance, which seeks to avoid MMR by prior modification of the target bacterium into a hypermutator strain.
Fig. 2 |. Optimizing the ARF in bacteria.

There are several factors that help improve the allelic recombination frequency (ARF), which are applicable across most bacterial species. a | Oligodeoxynucleotides (oligos) are designed containing phosphorothioate (PT) bonds, shown as asterisks. b | A population of bacteria are transformed with oligos through electroporation. The percentage of cells successfully transformed presents an upper bound for the ARF. Once inside the host cell, all free single-stranded DNA (ssDNA) is bound by bacterial single-stranded DNA-binding protein (SSB). c | Host nucleases degrade ssDNA in most bacteria, so protection with PT bonds is important. Two PT bonds at the 5′ end of the oligo usually provide adequate protection. d | Optimization of protein production can improve the ARF significantly. Some factors to consider are codon optimization, promoter strength and ribosome binding site strength. e | Orders of magnitude improvement can be gained by expressing a host-optimized single-stranded DNA-annealing protein (SSAP). A specific interaction with a host SSB determines SSAP compatibility. f | After a modification has been made to the genomic DNA, a mismatched base pair will be present. There are several possible strategies to prevent error correction by endogenous mismatch repair (MMR) machinery. Transient expression of a dominant-negative MutL (MutL-DN) causes chain termination and failure to recruit MutH. This optimal route is depicted here, but alternatives are available including MutH or MutS disruption, and the modification of multiple (4+) consecutive bases, which would then not be efficiently recognized by MutS. g | Serial enrichment for efficient recombineering (SEER) is a method to identify a host-optimized SSAP. A diverse library of SSAP variants is encoded on a plasmid, transformed into the target bacterial host, enriched through a series of antibiotic selections and deep sequenced to identify the most successful SSAP variants. Oligos used in the enrichment steps each contain an allelic modification to a host gene that confers resistance to a common antibiotic. Ab R, antibiotic resistance gene; NGS, next-generation sequencing; Ori, origin of replication.
MMR functions to reduce the ARF by recognizing mispairing between a modified nascent strand and the parental strand. The unmethylated nascent strand is then nicked, degraded and re-synthesized, efficiently eliminating recombinant alleles and restoring the original sequence47,48. MMR thereby reduces the frequency of successful genomic modification events, typically by more than 100-fold46. Early MMR avoidance strategies sought to knock out critical components of host MMR machinery, for example by generating ΔmutS strains. These strains display a hypermutator phenotype, with an approximately 50-fold to 1,000-fold increase in their genomic mutation rate leading to the rapid accumulation of off-target mutations49,50. Such a strain was used to facilitate the replacement of all TAG stop codons in E. coli. Following 25–30 cycles of MAGE in a ΔmutS E. coli strain, cells were found to carry more than 350 off-target mutations34,38. Such undesired mutations have the potential to muddle the analysis of the phenotypic effects of on-target genetic modifications. To avoid MMR during MAGE cycles without permanently disabling the cellular machinery, several strategies have been adopted — including modification of oligo design, diversion of MMR and transient suppression of MMR.
The simplest strategy for MMR avoidance is to chemically modify or alternatively design oligos. The MMR system in E. coli recognizes mismatches containing common transition mutations. Mismatches that rarely occur during DNA replication — such as A·G and C·C mismatches or four-nucleotide strings of unmatched bases — remain undetected49,51,52. These recognition biases can be exploited to design specific oligos that avoid correction in MMR-enabled cells, by targeting uncommon transversion mutations or extending mismatch length53–56. Introduction of a C·C mismatch 6 bases away from a desired mutation increased the efficiency of recombineering 30-fold in E. coli55. However, this strategy has severe limitations in the scope and precision of possible genomic modifications. Specific transitions are often desired and multiple consecutive base changes may be undesirable because of the decreased ARF or for numerous biological reasons including altered mRNA stability, codon usage or gene expression levels. Alternatively, oligos can be designed to include chemically modified nucleotides with unnatural DNA back-bone distortions that are not recognized by MMR51,57. This method increases allelic replacement efficiencies by up to 20-fold in E. coli and is expected to be broadly applicable, but it adds extra cost to oligo synthesis and the effects of these modified bases on bacterial cells have not been fully characterized51.
Two additional MMR avoidance techniques endeavour to disrupt MMR only transiently, with the reasoning that reducing MMR activity only during allelic replacement would allow MMR to recommence its genome maintenance activities during the growth phases of MAGE. The first technique relies on diversion of MMR by providing an overabundance of substrate to occupy MMR machinery. Nucleotide analogues have been employed in this manner, such as 2-aminopurine, which mispairs with cytosine during DNA replication, leading to the depletion of a central MMR protein (MutL)46,58. However, 2-aminopurine is itself mutagenic and this method reduces the ARF59,60. The second and more successful method for transient MMR disruption is to impair the pathway genetically. Several strategies have been pursued, including modifying MMR proteins to contain temperature-sensitive defects61,62, overexpression of Dam methylase63,64 and expression of a dominant-negative MutL variant known as MutL-DN, which in E. coli corresponds to an E32K mutation36,65. This last strategy has been the most effective, with expression of E. coli MutL-DN essentially mimicking the effects of a mutS deletion in E. coli36. Importantly, transient inhibition of MMR by MutL-DN exhibits a low off-target mutational burden. In an experiment in E. coli in which 24 sequential cycles of MAGE were run with MutL-DN expressed either transiently from a plasmid or in a ΔmutS background, 84 off-target mutations were observed in the ΔmutS strain whereas none were observed in the strain with MutL-DN transient expression36.
As new techniques helped to circumvent MMR, a final limitation to widespread adoption of MAGE was its narrow host range. MAGE depends on high-ARF recombineering being available in a host organism. Redβ has long been the standard SSAP for recombineering in E. coli, but studies that have tried to produce high ARFs in other bacteria quickly ran into trouble as Redβ seems to be limited in its effectiveness to a narrow range of species closely related to E. coli66–68. A recent study found that this narrow host tropism is due to the specific recognition of the carboxy-terminal tail of bacterial single-stranded DNA-binding protein (SSB) by the phage-derived SSAP68 (FIG. 2a). Searches for SSAP homologues in new bacterial species were met with mixed success for many years, but the serial enrichment for efficient recombineering (SEER) method has been recently developed to quickly screen hundreds of diverse SSAP homologues67 (FIG. 2b). Briefly, plasmid libraries containing phylogenetically diverse SSAP variants are constructed and used to transform a target bacterial strain. Successive recombineering cycles are then run, in which the bacteria are transformed with an oligo that encodes an antibiotic-resistant phenotype and selected for on the relevant antibiotic. The surviving population is thereby enriched for SSAP variants that most efficiently incorporate an allelic modification into the host bacterium. Variants from the RecT or ERF protein families were found to be the most promising source of bacterial SSAPs67. Using SEER, an SSAP variant (CspRecT) with approximately double the efficacy of Redβ in E. coli was isolated, and another (PapRecT) was identified that improved recombineering frequency in Pseudomonas aeruginosa. This method has the potential to easily identify efficient SSAPs for many new bacterial hosts67,68.
Single-stranded DNA-binding protein.
(SSB). An essential protein that binds to single-stranded DNA, protecting it and coordinating chromosome replication, and that is preserved throughout all domains of life.
Serial enrichment for efficient recombineering.
(SEER). A method for screening a large library of single-stranded DNA-annealing proteins to identify variants that perform efficiently in a given host.
In moving high-ARF recombineering into new bacterial species, there were several follow-on concerns after identification of an optimal SSAP variant. First, it is not a given that MMR avoidance by expression of E. coli MutL-DN will function in unrelated species. The glutamic acid at position 32 in E. coli MutL is, however, well conserved36, and analogous mutations of mutL in other bacterial species achieved similar MMR avoidance — specifically, an E36K mutation in both P. aeruginosa and Pseudomonas putida, an E32K mutation in Vibrio cholerae and an E33K mutation in Lactococcus lactis67–69. Furthermore, it is important to work in a bacterial strain that is efficiently electroporated if oligos are the carrier of new genetic information, and the ability to express an SSAP from a plasmid from a strong inducible promoter is helpful (FIG. 2a). For a list of MAGE-capable strains and plasmids available for use in multiple organisms, see TABLE 1.
Table 1 |.
Plasmid and strain resources for recombineering
| Name | Genotype | AddgeneID | Organisms tested in | Comments |
|---|---|---|---|---|
| Escherichia coli EcNR2 | MG1655 Δ(ybhB-bioAB)::[λcI857 N(cro-ea59)::tetR-bla] ΔmutS::cat | #26931 | E. coli MG1655-derived strain | E. coli strain with integrated recombineering functions, suitable for dsDNA recombineering, high off-target mutagenesis, restricted to <32 °C |
| Broad SSAP Library | p15a: 232-member SSAP library under pBAD induction | #164653 | E. coli in current plasmid | Has been cloned into different backbones and tested in Agrobacterium tumefaciens, Lactococcus lactis, Mycobacterium smegmatis and Staphylococcus aureus |
| pORTMAGE2, pORTMAGE3 or pORTMAGE4 | pBBR1: λcI857-pL λ gam, λ bet, λ exo, (Ec)mutL-DN | #72677, #72678, #72679 | E. coli, Citrobacter freundii, Klebsiella pneumoniae, Salmonella enterica | High ARF, suitable for dsDNA recombineering, broad host range origin, low off-target mutagenesis, restricted to <32 °C |
| pORTMAGE311B | RSF1010: xylS-Pm λbet, (Ec) mutL-DN | #120418 | E. coli, C. freundii, K. pneumoniae, S. enterica | High ARF, low off-target mutagenesis, broad host range origin |
| pORTMAGE-Pa1 | pBBR1: xylS-Pm (Pap)recT, (Pa) mutL-DN | #138475 | Pseudomonas aeruginosa | High ARF, broad host range origin |
| pSEVA2314-rec2-MutLE36K (HEMSE) | pBBR1: λcI857-pL (Ppp)erf, (Pp) mutL(E36K) | seva.cnb.csic.es183 | Pseudomonas putida | Low ARF, broad host range origin, restricted to <32 °C |
| pORTMAGE-Ec1 | RSF1010: xylS-Pm (Csp)recT, (Ec)MutL-DN | #138474 | E. coli, C. freundii, K. pneumoniae, S. enterica | Ultra-high ARF, broad host range origin, slightly elevated off-target mutagenesis |
| pJP042 | SH71rep: SppKR-Pspp (Lrp)recT | NA | Lactobacillus reuteri | High ARF, restricted to lactic acid bacteria |
| pJP005 | repA, repC: PnisA (Lrp)recT | NA | L. lactis | High ARF, restricted to lactic acid bacteria |
| pCN-EF2132tet | pT181cop-634ts: Ptet (Efp)recT | #107191 | S. aureus | Ultra-low ARF, restricted to S. aureus and <32 °C |
| pKM444 | oriE, oriM: Ptet (Mp)recT-bxb1 | #108319 | M. smegmatis, Mycobacterium tuberculosis | Ultra-low ARF, restricted to mycobacteria |
ARF, allelic recombination frequency; dsDNA, double-stranded DNA; NA, not available; SSAP, single-stranded DNA-annealing protein.
MAGE in eukaryotes
While the potential of MAGE was being demonstrated in E. coli, efforts were undertaken to develop an analogous technology in eukaryotes. Although eukaryotes have different chromosomal replication processes and machinery, efforts in the model yeast Saccharomyces cerevisiae have gained some significant ground. Early attempts to perform oligo-mediated recombineering in S. cerevisiae resulted in an ARF of 10−6–10−5, limiting their application to modifications that could be selectively enriched70. However, the same mutational signatures observed in oligo recombineering experiments in E. coli24,27,46 are also observed in S. cerevisiae: recombination is more efficient with oligos targeting the lagging strand of the replication fork and is highly dependent on MMR activity. These similarities implicated a similar strand annealing mechanism through which mutagenic oligos could integrate. The development of yeast oligo-mediated recombineering improved the ARF to 2%; this was achieved by transformation via electroporation of oligos of optimal length and concentration for recombineering, and by overexpressing Rad51-dependent homologous recombination factors Rad51 and Rad54 in an MMR-deficient background71. Surprisingly, neither Rad51 nor Rad54 are directly involved in single-stranded DNA annealing, the presumed mechanism by which oligos integrate.
Single-stranded DNA annealing.
The annealing of two strands of single-stranded DNA by base pairing.
Building on yeast oligo-mediated recombineering, and borrowing the technique of co-selection MAgE as first demonstrated in E. coli72, eukaryotic MAGE (eMAGE) shows an oligo-mediated ARF of up to 40% at specific loci in S. cerevisiae32. As in E. coli, eMAGE relies on SSAPs to anneal exogenously introduced oligos on the lagging strand of a replicon and is consistent with mutational signatures and recombination outcomes observed in prior work70,71. In early iterations of its development, eMAGE ARFs were improved by the overexpression of either Rad51-dependent homologous recombination or Rad51-independent proteins.
Co-selection MAGE.
A multiplex genome engineering technique in which a target modification that does not confer a selective phenotype is made in close proximity to one that does, allowing enrichment of both modifications in comparison with an unselected population.
Without selection, editing efficiencies under optimized conditions are too low for recombineering regardless of whether genetic backgrounds biasing Rad51-dependent homologous recombination or Rad51-independent single-stranded DNA annealing are used. A co-selection strategy was implemented to overcome low editing efficiencies and improve ARF in a new genetic background by decoupling the incorporation of a desired mutation from its selection32. In this scheme, a locus to be targeted for editing by oligos is cloned near an endogenous origin of replication and is flanked by a dual selectable marker (FIG. 3). Placement near an origin of replication facilitates the prediction of the direction of genome replication and, therefore, also helps to define leading versus lagging strands. This in turn informs strand-specific oligo design. Improvement in the ARF when targeting lagging versus leading strands is less pronounced in eukaryotes (2-fold to 5-fold)32 than in bacteria (8-fold to 50-fold)46. That lagging strand stringency is less pronounced in model eukaryotes than in bacteria suggests more flexibility in oligo design for organisms with difficult to predict or relaxed replication origin consensus sequences, such as humans. The selectable marker and the proximal locus can then be targeted by a pair of oligos, and selection for a mutation in the marker results in efficient co-selection for a mutation in the adjacent locus. Deletion of Rad51-independent HDR factors Rad52 and Rad59 — but not Rad51 — greatly impairs the co-selection ARF. These observations contrast with the single-locus ARF, where the effect of Rad51 on the ARF is stimulatory rather than inhibitory32,71. Because Rad52 and Rad59 participate in Okazaki fragment processing and post-replication repair, these findings support a model in which oligo incorporation is replication-dependent and Rad51-independent, as in bacterial recombineering73,74 (FIG. 3).
Fig. 3 |. Eukaryotic MAGE.
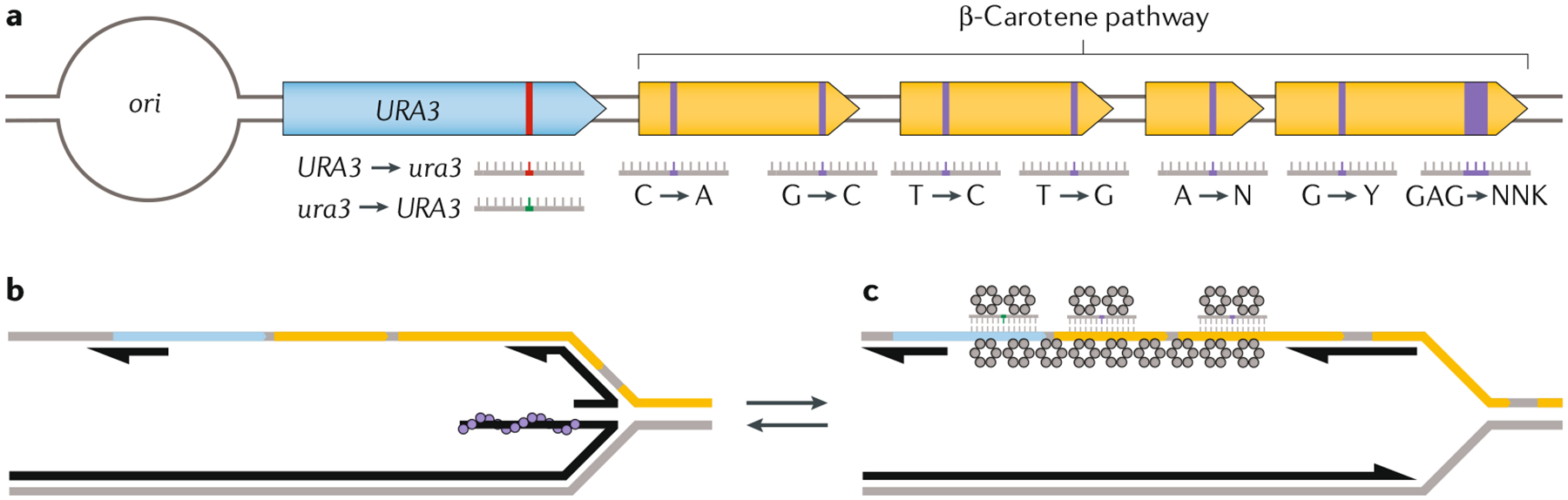
a | Schematic of a eukaryotic multiplex automated genome engineering (eMAGE) locus. An origin of replication (ori) is cloned immediately upstream of a locus of interest and a selectable marker so that the leading and lagging strands of either are predictable. The selectable marker used in this illustration is URA3, with an in-frame stop codon indicated in red. Correction of this stop codon allows cells to survive in the absence of supplemented uracil or uridine. The correction of this in-frame stop codon with an oligodeoxynucleotide (oligo) can be selected for, which enriches populations that have successfully made this modification for incorporation of oligos that confer targeted modifications (purple) into the adjacent locus of interest. b,c | Recombination pathways compete for oligo incorporation. Replication fork stalling or collapse can activate one of several DNA damage tolerance pathways or require fork restart. Rad51 (grey circles) is recruited to a stalled or collapsed replisome, mediating strand invasion and fork restart (b). Rad52 (grey hexamers) is involved in a recombination salvage mechanism, whereby annealing can occur at stalled replication forks (c). This Rad52-directed mechanism is the presumed recombination pathway responsible for oligo incorporation in eMAGE. We present a picture of either mechanism, as mechanistic details of eukaryotic recombineering remain to be worked out.
Origin of replication.
The site at which proteins involved in genome replication begin the synthesis of a new genomic copy.
As with the development of bacterial recombineering, multiplex editing achieved with eMAGE depended on prior inactivation of MMR, requiring the use of a S. cerevisiae mutator strain. To improve the eMAGE ARF while minimizing unintended secondary mutations, several genetic and experimental improvements were recently implemented: a dominant negative MMR mutant was inducibly expressed to transiently suppress MMR during editing75; a second dual selectable marker was introduced adjacent to the opposite flank of the eMAGE locus; and oligo concentrations and ratios were optimized. Synthesis of these considerations into a single, optimized eMAGE platform enables an ARF of up to 90%, reduces the spontaneous mutation rate 17-fold and permits multisite incorporation of oligos across a locus up to 20 kb in length75.
eMAGE was developed to apply iterative and combinatorial genome editing to S. cerevisiae for applications ranging from protein engineering to metabolic pathway optimization. Regardless of the application, a typical eMAGE experiment requires the integration of target DNA adjacent to both an origin of replication and a dual selectable marker in an MMR-deficient background. The importance of a dual selectable marker (for example, URA3) is highlighted by the cyclical nature of an eMAGE experiment: because each cycle requires the selection of a recombination event at the same locus, a dual selectable marker can be targeted both with loss-of-function and gain-of-function mutations during sequential rounds of co-selection.
Depending on the intended application and scale of editing, an eMAGE experiment can proceed in one of three directions, in order of increasing genetic diversity generated: diversifying a single target site with a degenerate oligo; editing many target sites within the co-selectable locus using combinations of oligos; and diversifying many target sites within the co-selectable locus using combinations of degenerate oligos (FIG. 3). In each case, S. cerevisiae is grown and transformed in 2-day or 3-day cycles, during which cells are electroporated with one oligo targeting the dual selectable markers and one or more oligos targeting the locus of interest. Each round of eMAGE can introduce substitutions, insertions and deletions and can generate populations of S. cerevisiae with more than 105 unique genotypes32. Iterative rounds of mutagenesis have employed combinations of unique oligos to generate combinatorial genomic diversity across a population of cells, the utility of which has been demonstrated by rapidly evolving a heterologous multigene biosynthetic pathway for altered carotenoid production32.
Results
Significant progress has been made in expanding the number of species in which MAGE is possible, and in streamlining laboratory techniques. Here, we describe the successes in enabling MAGE in diverse species, methods for isolating modified cells and techniques for working with large populations of edited cells.
Editing frequency across species
As advanced recombineering-based techniques have been developed for E. coli, progress is also being made towards adapting recombineering to new bacterial hosts. Here, we describe the results achieved to date and the bacterial species that have been targeted by researchers.
Redβ from coliphage λ has long been the most widely used protein for E. coli recombineering23, and along with RecT from the Rac prophage of E. coli27, these proteins are best categorized as part of a family of viral SSAPs called the RecT family76. Redβ and RecT, however, do not function well in bacterial species outside the family Enterobacteriaceae, because of a specific interaction of SSAPs with the C-terminal tail of the host SSB29,68. By screening RecT homologues from phages known to infect a species of interest, researchers have found SSAPs that allow recombineering in new bacterial families, with the most successful examples occurring in Lactobacillaceae54, Mycobacteriaceae77, Pseudomonadaceae78 and Vibrionaceae79. Due to challenges in the predictability of viral SSAPs’ tropism, screening a library of phylogenetically diverse SSAPs is the best method for maximizing the ARF for a given host67. TABLE 2 presents organisms in which recombineering has been demonstrated, the highest frequency reported at a single site and the optimal SSAP. When planning an experiment in bacterial or eukaryotic species in which recombineering has not yet been demonstrated, the first step is to run SEER67 against a library of SSAP homologues that can be procured from Addgene (TABLE 1) to identify a high-efficiency SSAP variant for the species of interest.
Table 2 |.
Organisms in which recombineering has been reported, along with the SSAP homologue used
| Species | SSAP (reference name) | UniParc ID | Highest reported oligo ARF | Ref. |
|---|---|---|---|---|
| Citrobacter freundii | CspRecT | UPI0001837D7F | 3.6 × 10−1 | 67 |
| Escherichia coli | CspRecT | UPI0001837D7F | 5.1 × 10−1 | 67 |
| Klebsiella pneumoniae | CspRecT | UPI0001837D7F | 1.1 × 10−1 | 67 |
| Lactobacillus reuteri | LrpRecT (RecT1) | UPI00006BE92C | 1.1 × 10−1 | 184 |
| Lactococcus lactis | LrpRecT (RecT1) | UPI00006BE92C | 1.2 × 10−1 | 184 |
| Pseudomonas aeruginosa | PapRecT | UPI0001E9E6CB | 1.5 × 10−1 | 67 |
| Corynebacterium glutamicum | CapRecT (SSAP-5) | UPI00019D7E17 | 2.1 × 10−2 | 185 |
| Mycoplasma pneumoniae | BpRecT (gp35) | UPI000009B1BC | 2.7 × 10−2 | 126 |
| Pseudomonas putida | PppERF (Rec2) | UPI0002A3D976 | 5.8 × 10−2 | 186 |
| Salmonella enterica | Redβ | UPI0000030D3E | 3.2 × 10−2 | 36 |
| Shewanella oneidensis | SpRecT (w3 beta) | UPI00005FCAB4 | 5.0 × 10−2 | 187 |
| Caulobacter crescentus | Redβ + PaSSB | UPI0000030D3E | 4.4 × 10−5 | 68 |
| Legionella pneumophila | LppRecT (ORF C) | UPI00000BCD4C | 5 × 10−4 | 188 |
| Lactobacillus rhamnosus | LrpRecT (RecT1) | UPI00006BE92C | 7.7 × 10−4 | 68 |
| Pseudomonas syringae | PspRecT (Psyr_2820) | UPI000050D92E | 2.4 × 10−4 | 189 |
| Saccharomyces cerevisiae | Redβ | UPI0000030D3E | 1.0 × 10−3 | 32 |
| Staphylococcus aureus | EfpRecT (EF2132) | UPI000005C42D | 1.4 × 10−4 | 56 |
| Acinetobacter baumannii | AbpRecT (NIS123_2461) | UPI000277BE9F | ND | 190 |
| Bacillus subtilis | BpRecT (gp35) | UPI000009B1BC | ND | 191 |
| Burkholderia sp. | BpRecT (Redβ7029) | UPI000655597E | ND | 192 |
| Clostridium acetobutylicum | CppRecT (CPF0939) | UPI0000DB59B0 | ND | 193 |
| Lactobacillus brevis | LbpRecT (LVISKB_1732) | UPI0002C4A04B | ND | 194 |
| Lactobacillus casei | LcpRecT (LCABL_13050) | UPI000176A57E | ND | 195 |
| Lactobacillus plantarum | LppRecT_2 (lp_0641) | UPI00000109A3 | ND | 196 |
| Mycobacterium smegmatis | MpRecT (gp61) | UPI000017D497 | ND | 130 |
| Mycobacterium tuberculosis | MpRecT (gp61) | UPI000017D497 | ND | 130 |
| Photorhabdus luminescens | PlpRecT (plu2935) | UPI00001D37D8 | ND | 197 |
| Vibrio natriegens | VcmRecT (SXT-Beta) | UPI00000B0129 | ND | 79 |
| Zymomonas mobilis | EcpRecT | UPI0000030D3A | ND | 198 |
ARF is reported for oligo-mediated recombineering only, without any form of selection. ARF, allelic recombination frequency; ND, not determined; SSAP, single-stranded DNA-annealing protein; oligo, oligodeoxynucleotide.
Isolating modified cells
Absent a genetic change that is directly selectable, successfully modified cells will need to be identified by other means. Depending on the chosen recombineering strategy, the frequency of genomic editing and whether or not a co-selective marker is used, different methods may be optimal for isolating modified cells from a mixed population that contain a desired genomic edit. The most straightforward method is to use allele-specific PCR80 amplification of the targeted genomic locus. This method requires the design of three primers: one universal primer that binds upstream or downstream of the targeted locus, and two allele-specific primers that are designed so that their 3′ nucleotide binds to either a wild-type or a modified genomic nucleotide (FIG. 4a). The 3′ nucleotide of the allele-specific primer allows discrimination between modified and wild-type alleles at the proper melting temperature (identified by temperature gradient PCR). By designing amplicon lengths so that each amplicon runs at an easily distinguishable band size on a DNA gel, this technique can be used to diagnose up to ten simultaneous genetic modifications from the PCR amplification of a bacterial colony, in a technique sometimes called multiple allele-specific colony PCR34. Importantly, proofreading polymerases should not be used for allele-specific PCR, as they contain a 3′ to 5′ exonuclease domain that will excise the discriminating 3′ nucleotide of an unmatched allele-specific primer81. Furthermore, newer variations on allele-specific PCR have been developed that improve on its ability to discern allelic variants82,83. Another available technique for discriminating by PCR amplification between modified or wild-type cells is high-resolution melting84. High-resolution melting — which, like allele-specific PCR, can be run from a bacterial colony — amplifies a short locus centred around the targeted mutation and then screens for small differences in the melt curves of the PCR amplicons using a DNA intercalating dye. Differences in the melt curves will usually be apparent between any two differing alleles.
Fig. 4 |. Reading out MAGE results.
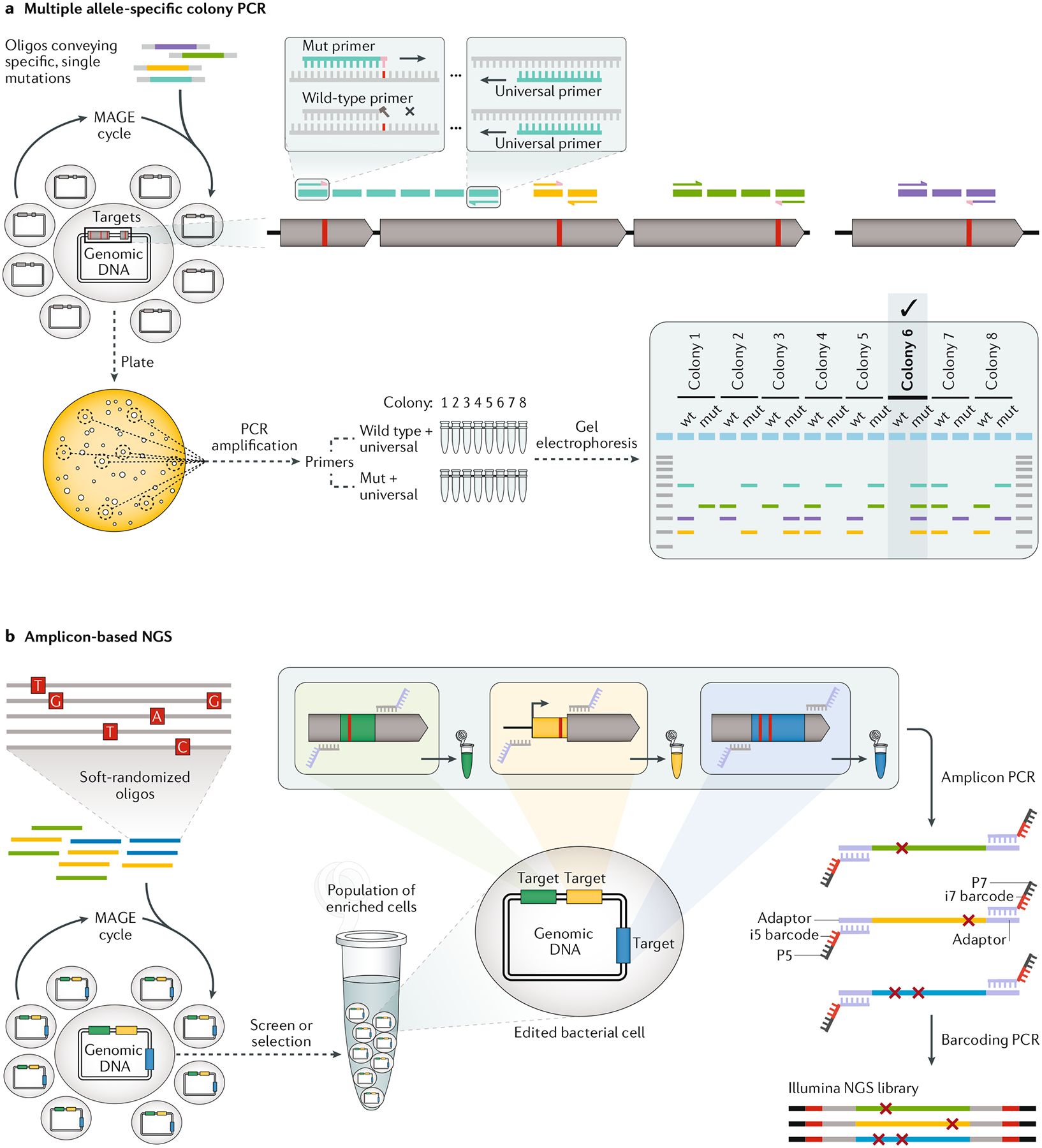
a | Multiple allele-specific colony PCR is a method for quickly identifying edited clonal populations. First, three primers are designed for each targeted modification. Forward primers bind to either the wild-type (wild-type primer) or edited (Mut primer) DNA, whereas a third reverse primer (universal primer) will be paired with both forward primers. Disambiguation is strongest when the 3′-terminal base of the forward primers is designed to anneal to the targeted base modification. Here, the Mut primer is depicted to have a pink terminal base that pairs successfully with the mutated red base, whereas the wild-type primer has a grey terminal base that does not pair, blocking elongation of the primer by DNA polymerase in the PCR reaction. After numerous multiplex automated genome engineering (MAGE) cycles, the edited population is plated out for single colonies, and two separate PCR reactions are run for each colony (wild type + universal and Mut + universal). On an electrophoresis gel, a DNA band should appear only for the allele that is present in the clonal population. Multiple alleles can be combined into a single PCR reaction if the amplicons are designed to have different lengths so that they are easily differentiated by gel electrophoresis. Colony 6, with four mut bands, has successfully incorporated all of the targeted allelic modifications, whereas every other colony shows at least one wild-type band (wt). b | Amplicon-based next-generation sequencing (NGS) for screening and selecting targeted mutations introduced by MAGE-based strategies such as MAGE sequencing or directed evolution with random genomic mutations (DIvERGE; pictured here). Illumina NGS libraries can be easily created in two PCR steps from a population of edited or edited and then enriched cells. First, amplicon PCR mixes a population of cells separately with primers to amplify each targeted locus (green, yellow or blue) and at the same time affix an adapter sequence. Amplicon PCR reactions are run separately for each targeted locus, but the population of cells is pooled in the reaction. Second, barcoding PCR is run on each amplified locus to add primers that bind to the adapter region and affix a unique barcode and sequences for binding to a flow cell. Oligo, oligodeoxynucleotide.
To increase the frequency of modified cells within a population, common strategies employ selection or counter-selection to enrich for edited cells. First, antibiotic selection is typically used for recombineering with dsDNA cassettes (FIG. 1b), meaning that successful integration of the dsDNA can be directly selected for. In the case of recombineering with oligos, oligonucleotide-mediated recombineering followed by Bxb1 integrase targeting (ORBIT) integrates a selective marker into a genomic locus via an oligo-encoded recombinase site, and so can be used for quickly isolating deletions of genes or operons, which are usually lower-frequency events than single base-pair edits85. Alternatively, antibiotic co-selection can enrich for successfully modified cells by co-transforming two oligos, one that makes a desired edit and a second that makes a small modification close to the desired edit. With the idea that successful incorporation of the selective edit indicates a cell that is actively replicating that region of the chromosome, selecting for the resistant phenotype will increase the frequency of the desired edit in the resistant population, a technique known as co-selection MAGE72. Finally, if recombineering is combined with CRISPR counter-selection, in which a gRNA is designed to make a DSB in only unmodified cells, then efficiencies can be close to 100%, meaning that only a few colonies would need to be screened to confirm a correctly modified cell.
Single and multilocus library analysis
Next-generation sequencing (NGS) is often used to analyse allelic frequencies in a population of cells that has been diversified by MAGE. Genetic variation targeted to a single locus or to a few loci can be read easily by NGS of PCR amplicons. Primers are commonly designed to bind to a genomic locus and include a 5′ universal Illumina adaptor sequence. After primary amplification of the locus, a second round of PCR is run with limited cycles to add NGS barcodes and a standard motif to bind to a flow cell (FIG. 4b). Genetic loci longer than 550 bp (or the maximum available short-read sequencing length, allowing for some overlap between forward and reverse reads) should either be split into segments and individually sequenced by Illumina or, if epistatic effects are of interest between mutations distal from each other in the primary sequence space, long-read sequencing techniques can be used, such as those offered by Pacific Biosciences or Oxford Nanopore43,86.
It is often of interest to target variation broadly throughout the genome, rather than at just one or a few genomic loci. For applications that range from whole-organism recoding to metabolic engineering, whole-genome sequencing is often the best read-out. Several cycles of MAGE (usually between 3 and 20 depending on the ARF and the number of desired mutations) can be run using a diverse pool of oligos that specify variation broadly throughout the genome. This diversified population can be subject to selection if applicable and plated for single colonies. These colonies are then submitted to whole-genome sequencing and clonal variants that incorporate many desired mutations can be identified.
Applications
ARF influence on available applications
The suitable applications for a genome engineering technology are dependent on specific metrics that include the editing frequency, multiplexability, off-target effects, the types of genomic scar introduced and sequence constraints that limit where modifications can be made. Recombineering differentiates itself from competing technologies by offering few sequence constraints and scarless editing, and MAGE builds on this with unmatched multiplexability. Methodological transformations over the past decade have made efficiency gains that improved multiplex editing53,67,87, reduced off-target effects36,61 and expanded its availability to new species32,36,67. Still, MAGE requires at least transient disabling of MMR pathways, and so remains constrained by low level, off-target effects, in addition to being dependent on the availability of efficient recombineering in each organism and on our ability to synthesize and deliver target-specific oligos.
The ARF — the fraction of cells that receive a targeted edit after one round of single-site recombineering — is the most salient factor for selecting applications of recombineering. The ARF varies tremendously by organism and by construct, so we group applications here by ARF value to help users plan experiments accordingly. ARFs in various organisms and with varying recombineering methods span a range of efficiencies, from far below 1% to approximately 50% (TABLES 1,2). To provide application guidelines, we define ARF ranges as ultra-low frequency (<1%), low frequency (1–10%), high frequency (10–25%) and ultra-high frequency (>25%). We hope that the guidance in this section will enable new users to understand and apply MAGE and related recombineering-based techniques in a wide variety of organisms.
Ultra-low frequency (ARF <1%).
Most reports of adapting recombineering to new species of bacteria either do not report population-level efficiencies or report efficiency well below 1% (TABLE 2). We categorize these cases as exhibiting ultra-low efficiency, making isolation of cells containing a desired edit the primary concern. For cases in which a trait cannot be selected for, the scarcity of desired mutants generated by ultra-low ARF exacerbates the trade-off between the time required to generate variants and the time required to isolate them. Although desired clones can be isolated using techniques such as multiple allele-specific colony PCR34 (FIG. 4a) or high-resolution melting86, a frequency of less than 1% implies that more than 100 colonies must be screened to identify a single edited clone. To mitigate the need for extensive screening, one can perform multiple MAGE cycles, shifting the burden of time from screening to editing. For cases in which the desired mutation decreases fitness, however, cycles of outgrowth and editing will serve as a counter-selection, making this strategy unworkable. When mutations are not expected to decrease strain fitness, based on cycling efficiency equations88 we recommend five cycles of MAGE per desired mutation to integrate one to three multiplexed mutations.
Despite the poor ARF of some species, there remain many worthwhile and feasible applications of ultra-low-frequency recombineering, including translational knockouts33, codon substitutions89,90 and user-defined allelic modifications91,92. Even in systems that offer other gene editing tools, such as CRISPR–Cas-based techniques or more established homologous recombination-based methods, ultra-low-efficiency recombineering may be the preferred option for mutagenesis as it is capable of precision genomic editing and can generate pools of cells in which the edited subpopulation will contain varied genotypes. Furthermore, recombineering lacks the random mutation-generating mechanisms of nuclease-based technologies93 and the sequence limitations of CRISPR94,95.
To improve the apparent ARF of ultra-low-efficiency recombineering, it can be paired with techniques such as CRISPR–Cas9 counter-selection56,96,97 or recombinase-based positive selection83. Library generation is even possible as long as the generation of desired mutants is coupled with appropriate selections, such as antibiotic resistance36,72, fluorescence-based sorting43,98 or CRISPR–Cas9-based counter-selection99–101. By eliminating nearly all wild-type cells, these techniques can isolate rare variants from vast populations. With a mutation-generating frequency of 10−3, for instance, a 1 million-member library can be readily generated in a single cycle of MAGE on a population of 109 bacterial cells. Antibiotic resistance was used as a selective mechanism to study a library of this size composed of spectinomycin binding-site mutants in L. lactis68. This study found a previously unknown RpsE variant that would not have been readily accessible by directed evolution methods for two reasons: it requires a minimum of three nucleotide changes, and each of the two amino acid mutations provides no improved resistance individually, making the effect epistatic to either mutation. This variant is as fit as wild-type L. lactis in laboratory culture and is able to grow rapidly in 100 μg/ml spectinomycin68. Additionally, CRISPR–Cas9-based counter-selection is routinely used with libraries of this size, in the form of CRISPR-optimized MAGE recombineering (CRMAGE)99 or CRISPR-assisted MAGE (CRAM)39, quickly becoming a fundamental tool for microbial functional genomics. Owing to constraints encountered when simultaneously expressing multiple gRNAs20,102, CRISPR–Cas9 counter-selection remains most commonly employed at only a few loci at a time.
Ultimately, with the appropriate selection or counter-selection tools, ultra-low-efficiency MAGE is uniquely suited for targeted genome editing and library generation in non-model microorganisms. Furthermore, users should be able to readily improve the ARF above 1% in most targeted organisms to access applications that are highlighted in the next sections.
Low frequency (ARF 1–10%) and high frequency (ARF 10–25%).
ARFs above 1% have been reported in 11 bacterial species to date, and as scientists screen for host-active SSAPs and develop more efficient methodologies in non-model bacteria, this number should continue to grow. Increased ARF decreases the cycling and colony screening requirements for isolating modified cells and dramatically increases attainable library sizes. The applications are similar for both low-frequency and high-frequency MAGE, but experimental time frames will differ, to account for cycling and screening, as will the library sizes that can be readily attained. With low-frequency MAGE, selection and counter-selection strategies play a larger role in experimental design and additional cycling will often be desired, whereas for high-frequency MAGE, although selection strategies remain important, wild-type cells will represent only a small fraction of total cells with proper cycling. In this section we describe techniques that are not useful at ultra-low frequency but have powerful applications when the ARF rises to low frequency and perform even more robustly at high frequency. These elevated ARFs have only been available in E. coli and a select few other bacteria until recently67.
The first report of MAGE in 2009 described a robust single-locus ARF of between 15 and 25% with most single mismatch oligo designs (high frequency)33, and first envisioned the genome as a massively editable template. MAGE was initially applied to metabolic engineering and is well suited for this task because the optimization of metabolic pathways often requires 10–30 modifications to native genetic elements in a microbial chassis. Wang et al.72, with the goal of optimizing lycopene biosynthesis, used a pool of oligos to alter the ribosome binding sites of 20 genes and to introduce in-frame stop codons to 4 other genes. They succeeded in quickly isolating a strain with five mutations that quadrupled lycopene biosynthesis. Computational workflows can be used in tandem with low-efficiency or high-efficiency MAGE to predict and validate mutations that improve metabolic pathways103.
In addition to optimizing bioproduction, MAGE at these frequencies can be used to create specialized chassis organisms for various synthetic biology applications. The E. coli reduced aromatic aldehyde reduction (RARE) strain was designed to eliminate endogenous aldehyde reductases that rapidly convert aromatic aldehydes into their corresponding alcohols104. The strain was first constructed using P1 phage transduction to knock out seven genes, a process requiring several months. Recently, this task was repeated with newly developed tools; using only 15 MAGE cycles, researchers reproduced this months-long effort in a single week (unpublished data). In another recent example of strain engineering, MAGE with CRISPR–Cas9 counter-selection was used to eliminate all mobile genetic elements from the chemically competent E. coli strain BL21(DE3)101. Compared with a previous effort that required years of experimental effort105, this method allowed the inactivation of 30 genomic insertion sequence elements in just weeks. Some examples of host-engineering efforts that MAGE could rapidly accelerate include knockout of proteases and nucleases to improve protein expression106–108, engineering cellular redox potential to enable cytoplasmic disulfide bond formation109–111 and opening up orthogonal protein glycosylation pathways112.
Another powerful application of MAGE at these frequencies is in vivo protein engineering and directed evolution experiments. In contrast with many currently available in vivo diversification technologies, MAGE allows bias-free exploration of protein sequence space with full user control. By targeting the active site of an aminoacyl-tRNA synthetase, MAGE was used to isolate highly active variants together with corresponding tRNAs, improving non-standard amino acid incorporation37. Similarly, MAGE can be used to perform mutational scanning at genomic targets (FIG. 5). The impact of codon usage on mRNA structure was investigated by single-codon scanning mutagenesis (FIG. 5a) of infA with the fitness landscape read out by deep sequencing the locus after continuous exponential growth113. A complementary technique, directed evolution with random genomic mutations (commonly known as DIvERGE) employs a set of partially overlapping, soft-randomized oligos to tile a target locus (FIG. 5b). These oligos incorporate a low level of degeneracy at each nucleotide position, and so can introduce random mutations throughout a gene with a small number of oligos (FIG. 5). DIvERGE was used to discover a pair of gyrA and parC mutations that confer resistance to gepotidacin, a new antibiotic in phase II clinical trials for treatment of Neisseria gonorrhoeae infections86,114. These techniques offer precise targeting of mutational load, enable multiple rounds of diversification and selection, and are capable of producing libraries that far outpace the size available from traditional plasmid-based cloning. Because library size is limited only by the ARF in a given host, with high-efficiency MAGE it is easy in most academic laboratories to generate libraries with degeneracy in the order of 109–1010 protein variants, levels that are not easily attainable by other means115,116.
Fig. 5 |. Library-scale genome diversification using MAGE-seq and DIvERGE.
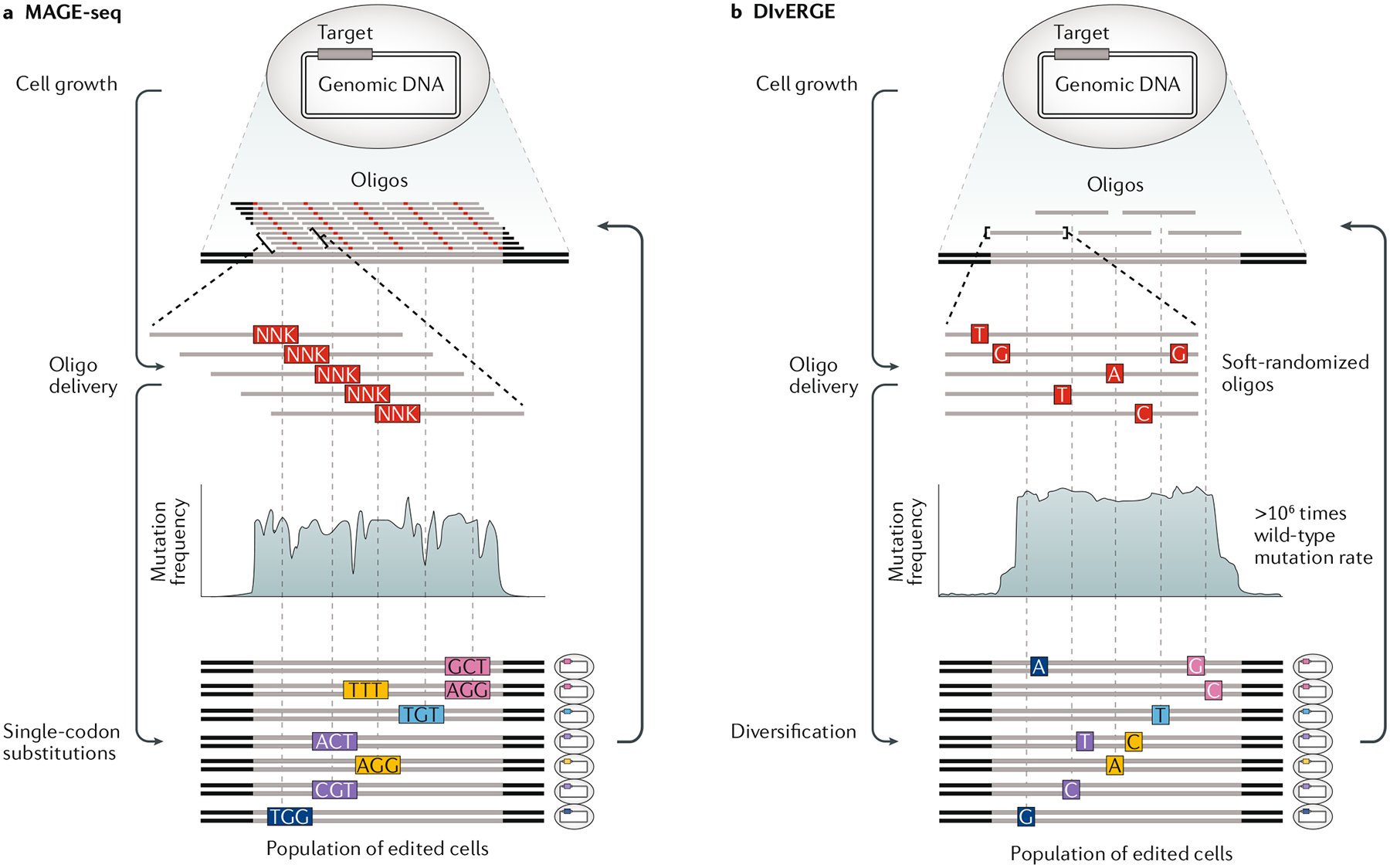
a | Multiplex automated genome engineering with amplicon deep sequencing (MAGE-seq) is based on scanning codon mutagenesis, wherein an oligodeoxynucleotide (oligo) with a degenerate NNK codon is designed for each codon within a targeted gene region. This batch of oligos is pooled and delivered as a library, subjected to multiple MAGE cycles and then a desired phenotype is screened or selected for. The population of enriched cells is then genotyped by amplicon deep sequencing. b | In directed evolution with random genomic mutations (DIvERGE), soft-randomized oligos are designed to tile a targeted genomic locus. As the soft-randomized oligos are incorporated, they will introduce mutations at random, and, depending on the allelic recombination frequency (ARF), this will enable a highly elevated mutation rate to be targeted with precision throughout the genome.
The intended modification plays a significant role in determining ARF. Point mutations have higher integration frequencies than insertions or deletions67,88, and insertion length is capped by oligo synthesis constraints and the need for approximately 30 bp of upstream and downstream homology. This leaves short regulatory modifications or protein tags as such as polyhistidine tags (His-tags), degradation tags or short promoters as the primary insertion targets72. Previously, His-tags have been appended to 38 genes over 110 MAGE cycles across 9 strains to reconstitute translation machinery in a cell-free environment117. A related application that could be facilitated by MAGE is the appending of degradation tags to protein targets to allow for dynamic metabolic control and to optimize the pathway yields for products such as myo-inositol118 and poly(3-hydroxybutyrate)119.
Ultra-high frequency (ARF >25%).
Given a classically understood mechanism of recombineering23, the upper bound for the single-locus ARF was thought to be 25% (REF.120) as four strands of chromosomal DNA are present in a dividing cell and only one is targeted by oligo-mediated recombineering. A method for improved screening of SSAPs recently reported single-locus ARFs that meaningfully surpassed this limit, which is hypothesized to be because editing can occur over multiple cell generations or at multiple replication forks within a single cell. Layering a highly active SSAP on top of many other methodological improvements enabled ARFs as high as 36% in Citrobacter freundii and 51% in E. coli67 — a feat enabled by SEER and available to all through the plasmid construct pORTMAGE-Ec1, which encodes the SSAP variant CspRecT (Accession no. 138474 on Addgene).
Ultra-high ARFs have only recently been reported67, so applications are still exploratory. It is safe to predict that ARFs >25% will further improve the many existing applications of MAGE, including library generation capacity, chassis development efficiency, continuous protein engineering and more. We expect that ultra-high ARFs should reasonably enable, for instance, introduction of more than 30 mutations over 10 cycles in E. coli in 1 week based on cycling equations88. Similarly, multiple proteins or operons could realistically be targeted with DIvERGE86. Indeed, improvements in multiplex editing seem to scale more dramatically than single-locus ARF improvements. For example, DIvERGE using CspRecT expressed by pORTMAGE-Ec1 demonstrated an approximate tenfold increase in efficiency over DIvERGE using the legacy SSAP Redβ; however, CspRecT showed only a twofold improvement when performance was measured at a single locus67. The library size thus attainable with DIvERGE powered by CspRecT vastly exceeds the potential of other targeted genome diversification tools because it can target precise windows of DNA, unlike EvolvR116, and there are no multiplexing limitations as with other nuclease-based editors121,122. Additionally, high-efficiency MAGE will expedite future efforts that aim to create novel genomically recoded organisms34,123,124. With ultra-high ARFs, the generation of sufficient genomic diversity is no longer limiting, and instead experiments will be limited by a screening mechanism or by population size itself.
Advanced recombineering techniques
Early technologies employing recombineering have given rise to more sophisticated and scalable methods such as MAGE or DIvERGE33,86, and below we highlight how the genome editing toolkit can be further extended by pairing these techniques with complementary molecular tools.
CRISPR–MAGE.
The widely used CRISPR–Cas9 system has been paired with recombineering as a counter-selection tool to increase the apparent ARF9,56,97,125,126. A programmable gRNA in the ribonucleoprotein complex directs the Cas9 endonuclease to generate a site-specific DSB, which can be lethal to bacterial cells that lack efficient HDR127. In this way, the lethality of Cas9-generated DSBs is used as a counter-selection strategy to selectively kill unedited cells. CRISPR–Cas9 cleavage was first leveraged to introduce different types of mutations (insertions, deletions and point mutations) into E. coli by co-introduction of a Cas9 targeting construct and an editing template. Successfully modified cells would both incorporate a desired mutation and eliminate the Cas9/gRNA cleavage target96. Similar CRISPR-recombineering strategies for large-scale deletions up to 19.4 kb and insertions up to 8 kb have been demonstrated128,129.
To couple iterative CRISPR–Cas9 counter-selection with MAGE, a two-plasmid system was developed that expresses Cas9, Redβ, Dam (an MMR suppressor) and RecX (a RecA inhibitor, which increases the lethality of Cas9 counter-selection) on one vector and two gRNA arrays on another99 (FIG. 6a). Self-targeting gRNAs facilitated rapid curing of the gRNA array (92–96% of cells lost the plasmid in 2–3 h), which in turn allowed transformation of new gRNA arrays and oligos for cyclic editing. With CRMAGE, ARFs of up to 98% can be obtained in a single round of recombineering. We expect that a future improvement will be the use of highly multiplexed, non-repetitive single gRNA arrays20.
Fig. 6 |. Advanced techniques pair MAGE with other tools.

a | CRISPR multiplex automated genome engineering (CRMAGE) operates via a two-plasmid system. Cas9 and the recombineering proteins Dam, a single-stranded DNA-annealing protein (SSAP) and RecX are expressed on an episomally maintained vector. A second vector contains trans-activating crispr RNA (tracrRNA) and a CRISPR array with genomic-targeting (green) and self-targeting (grey and brown) CRISPR RNAs (crRNAs). First, oligodeoxynucleotide (oligo) editing templates are transformed into cells, these templates are incorporated into the host genome by recombineering and the successfully edited cells are selected for with induction of the CRISPR targeting system. Cas9/short guide RNA (gRNA) fails to recognize edited target sequence but creates double-strand breaks in unedited targets, resulting in cell death and selection for edited cells. b | Oligonucleotide-mediated recombineering followed by Bxb1 integrase targeting (ORBIT) can create genetic knockouts or fusions depending on the integrating plasmid selected and the oligo design. To fuse green fluorescent protein (GFP) to a gene of interest, an oligo encoding an attP (red) site and a plasmid containing GFP (green), a hygromycin resistance marker (blue) and an attB site (yellow) are co-transformed into a cell. After successful incorporation of the oligo by recombineering, Bxb1 integrase (Int) integrates the GFP plasmid at the attP site, creating a carboxy-terminal gene fusion of GFP to the target gene (grey). A similar strategy can be used to perform targeted gene deletion. c | Replicon excision for enhanced genome engineering through programmed recombination (REXER) efficiently integrates long synthetic DNA into Escherichia coli genomes. A bacterial artificial chromosomes (BAC) (grey) containing an edited template (red) is transformed into E. coli. CRISPR–Cas9 is then expressed and excises the edited template along with the −2 (sacB; orange) and +2 (CamR; green) selection markers from the transformed BAC. In step 1, homology arms facilitate replacement of genomic DNA (black) and the −1 (rpsL; yellow) and +1 (KanR; blue) selection markers with both negative and positive selection pressures. Step 2 uses a new BAC with a new editing template and the −1, +1 markers to replace the previously incorporated −2, +2 markers. This process can be repeated for de novo synthesis of a synthetic E. coli genome (genome stepwise interchange synthesis (GENESIS)). abR, antibiotic resistance marker; Ori, origin of replication.
ORBIT.
Site-specific recombinases are powerful genetic tools, although their application is limited to specific recognition motifs. These motifs are short enough, however, to be encoded onto an oligo, which allows the action of these recombinases to be accurately targeted across the genome with oligo-mediated recombineering. This was first demonstrated in mycobacteria, which had been difficult to genetically manipulate by oligo recombineering, in part due to the inefficiency of available SSAPs and high levels of illegitimate recombination130. The electroporation of linear dsDNA frequently results in unwanted non-homologous, ectopic integration events131,132. By merging two recombination systems, ORBIT overcomes these hurdles83. ORBIT relies on an initial recombineering step that site-specifically incorporates the Bxb1 attP sequence into the genome, followed by a subsequent Bxb1-mediated plasmid integration into the attP site that allows the low-frequency oligo recombineering step to be positively selected (FIG. 6b). First, an oligo containing the Bxb1 attP sequence and site-specific homology arms is integrated into the genome by the Che9 SSAP, which is from the RecT protein family. Next, a non-replicating plasmid containing the desired cargo, an antibiotic resistance marker and the Bxb1 attB sequence is integrated at the attP site by the Bxb1 phage integrase (Int). By co-transforming the oligo and cargo plasmid into cells expressing Che9 SSAP and Int, antibiotic-resistant clones can be produced in a single step. As cargo plasmids only require the attB site for integration, once a particular functionality has been created in a non-replicating plasmid, it can be reused to systematically perform operations such as promoter replacement, gene knockouts and protein tagging at different target genes. This pairing of site-specific recombinases with recombineering has so far only been demonstrated in mycobacteria, but has the potential to be widely applicable to various biological systems.
REXER.
By pairing dsDNA recombineering with CRISPR–Cas9, recombineering can be leveraged to perform large-scale genomic rearrangements. These operations require the host-active SSAP to be paired with an appropriate exonuclease, usually found within the same bacteriophage operon. Exo and Redβ (an SSAP) from coliphage λ have been shown to interact, and Exo is presumed to load Redβ onto newly exposed ssDNA as it degrades the opposite strand of dsDNA29 (FIG. 1b). Traditional recombineering methods show markedly low incorporation rates for dsDNA longer than 5 kb (REFS129,133). Replicon excision for enhanced genome engineering through programmed recombination (REXER) uses a large plasmid or bacterial artificial chromosome and CRISPR–Cas9-mediated recombineering to overcome this barrier and deliver long dsDNA cargo133. In its most commonly applied form, REXER2, a dual (positive and negative) selection strategy allows for sequential, large-scale genomic DNA replacements133. By selecting first for the insertion of the introduced template and then against the presence of the unedited genomic sequence, gene replacements of >100 kb were achieved with nearly 100% efficiency (FIG. 6c). This process can be performed iteratively with a scheme called genome stepwise interchange synthesis (GENESIS) for whole-genome replacement. REXER has been used to systematically recode essential operons in the E. coli genome to determine allowed and disallowed synonymous coding reassignments, which in turn provided valuable insights for reprogramming the E. coli genetic code123,134.
Retron recombineering.
Until recently, MAGE and other recombineering methods required the exogenous delivery of editing templates, most typically oligos delivered through electroporation. This ultimately limits continuous operation and prevents the fully autonomous execution of directed evolution experiments. This limitation has been overcome with retrons, a class of bacterial retro-elements that produce ssDNA in vivo135–137 and have been linked to phage defence138. Retrons typically encode the specialized RNA primer-template msr-msd — where msr remains RNA, while msd is reverse-transcribed into DNA — and a retron-specific reverse transcriptase. The msr-msd RNA adopts a unique secondary structure that is recognized by the reverse transcriptase. The msd RNA then serves as a template for reverse transcription, with RNase H degrading the RNA, and ultimately yields a covalently linked RNA–DNA hybrid molecule called multicopy satellite DNA. Retrons can be reprogrammed to produce custom ssDNA in living cells by replacing a region of msd with a desired sequence. Synthetic cellular recorder integrating biological events (SCRIBE) was the first platform to use the in vivo production of ssDNA templates for recombineering139 (FIG. 7a). By placing the Ec86 (Eco1) retron under an IPTG-inducible promoter, gene editing is effectively coupled to a chemical signal, repurposing the host cell’s genome as a recorder of biological events or signals. Increasing the expression of retron elements and the removal of exoX and mutS from the host led to a 78-fold improvement in the efficiency of retron-mediated recombineering140–142. This was coupled with the placement of the msr-msd element under the control of a mutagenic T7 RNA polymerase-driven promoter to allow error-prone multicopy satellite DNA generation, and thereby the constant diversification of a genomic region, with a 190-fold higher mutation rate than the genomic background140 (FIG. 7b). Most recently, further improvements to the ARF of retron-mediated recombineering enabled the exploration of phenotype to genotype relationships from a multi-million-member retron library142. Because retrons are maintained within a lineage of cells, and can be made to edit that lineage of cells very efficiently, they serve as an identifier or ‘barcode’ that can be used to identify mutant lineages within pools. This techinque thus enables genome-scale reverse genetics experiments, wherein many retrons specify many mutations within a pool, and these barcodes can be tracked by amplicon sequencing of the retron plasmid. In one example, random genomic fragments were cloned in a massively parallel experiment into retron cassettes. These retrons served as the carriers of new genetic information upon their delivery into wild-type E. coli cells, and this allowed every base in the genome to be queried for changes leading to phenotypic effects, in this case their contribution to trimethoprim resistance. This technique for genome-scale reverse genetics in bacteria is called retron library recombineering (RLR)142 (FIG. 7c).
Fig. 7 |. Retrons allow recombineering without exogenously delivered DNA.

a | Synthetic cellular recorder integrating biological events (SCRIBE) uses retrons to produce multicopy satellite DNA (msDNA) in vivo, which is used as a substrate for recombineering. The presence of an induction molecule triggers expression of a reverse transcriptase (RT; orange) and a retron transcript (msr/msd). The msd portion of the retron transcript is reverse transcribed by the RT to create msDNA. This msd region can be modified to include a customized loop that carries a user-defined mutant allele. A single-stranded DNA-annealing protein (SSAP) then mediates incorporation of the msDNA into the nascent copy of the genome at the lagging strand of the replication fork. b | Error-prone T7 RNA polymerase (RNAP) can be used to produce mutagenized msr-msd transcripts. These transcripts are reverse transcribed to produce a library of msDNA editing templates that contain random mutations for continuous evolution of a target sequence. c | Retron library recombineering is a technique for genome-scale reverse genetics. Synthetic or natural DNA variation is encoded into the retron msd element on a plasmid library. This plasmid library is transferred to a population of bacteria, the retron directs a mutation into the genome and the population is taken through a screen or selection. The prevalence of each allele within a population can be tracked by sequencing of a plasmid amplicon containing the retron msd. NSG, next-generation sequencing; ssDNA, single-stranded DNA.
Reproducibility and data deposition
As MAGE can greatly accelerate experimental throughput and facilitate bacterial genomic diversification, it is important to consider the practical ramifications for the laboratory. A greater emphasis on stock nomenclature, organization and genotyping is necessary, as well as clear and easily communicable pipelines for computational workflows.
Strain storage
The primary outputs of MAGE experiments are modified strains, and the phenotypic measurements and sequencing data from those strains. It is quite easy to rapidly build up a large collection of strains, and so a careful cataloguing system is essential for any laboratory or individual researcher who does extensive experimental work in this area. First, we recommend whole-genome shotgun sequencing and keeping a cryogenic stock of any strain that will be the focus of future genomic manipulation. Next, it is good practice once a MAGE cycle has completed to cryogenically stock strains while genetic alterations are confirmed by Sanger or NGS. In this way, strains will not build up any additional mutations through unnecessary passaging or storage at stationary phase. Finally, proper sample storage is of particular importance when dealing with populations of cells that contain anywhere from thousands to billions of distinct genotypes that are output from MAGE, DIvERGE, RLR and related library-building techniques. The initial, diverse population of cells should be stored cryogenically before any form of selection is applied to it, ideally after 8–16 h of outgrowth or longer depending on the generation time. It is important to store a sufficient amount of cells to be able to reconstitute the naive population, so if a library size is on the order of 109 cells, at least 1 ml of bacterial culture should be frozen. Cells should also be cryogenically stored after a selection is applied, allowing for experiments to be performed later on identical bacterial populations and for sequencing to be repeated if necessary, adding robustness and reproducibility to an experimental pipeline. As with any publicly funded work, important strains should be deposited with Addgene or another similar repository.
Data deposition and code availability
Standards and practices from institutions, journals or funding sources that detail expectations for electronic data availability should be followed rigorously. This is common best practice for omics studies that involve large sets of raw sequencing data and a code base that is used to perform analyses on these data. We recommend depositing all raw NGS data along with experimental details and strain information in an open-access data-base, such as the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) or the European Nucleotide Archive (ENA), and thoroughly commenting on and cleaning up a code base before making it available on GitHub.
Limitations and optimizations
Despite the decades-long development of oligo-mediated recombineering for microbial DNA editing143,144, there remain some limitations and areas for further improvement. First and foremost, SSAPs display host-tropic activity, limiting recombineering to organisms for which a functional SSAP homologue has been identified. Of equal importance to the selection of a proper SSAP, maximization of the ARF often involves the optimization of some more mundane parameters.
When optimizing the ARF in a target organism, two factors that seem to have great influence are the electroporation efficiency and SSAP expression (FIG. 2). To optimize the electroporation efficiency, an array of variables should be screened that include the following: media compositions, such as osmotic protectants or glycine to weaken the cell wall145,146; growth conditions, such as the growth temperature and optical density at harvest147,148; harvesting variables, such as the temperature of the competent cell preparation, wash buffer composition and the duration and speed of centrifugation149,150; and electroporation settings, including the voltage, resistance and cuvette gap width147,151. To optimize SSAP expression levels, genetic elements should be optimized for high expression, making plasmid-based expression preferable to genomic integration of the SSAP cassette. A plasmid should be constructed that includes an inducible promoter, an optimized ribosome binding site152 and a codon-optimized SSAP. It is preferable to test a few ribosome binding site designs and codon-optimization schemes as these elements are difficult to predict a priori; for instance, in a recent study a codon-optimized version of Redβ was found to be far inferior to the native sequence67. This plasmid should then be tested with various induction regimens, by varying the concentration of the induction molecule as well as the duration of protein expression and the optical density of the bacterial culture at induction. For a list of available plasmid resources, see TABLE 1.
Electroporation has been successful in delivering oligos into many organisms, but efficient electroporation procedures have not yet been established in many important hosts and environmental isolates. Methods such as RLR142 and REXER133 should be adaptable to species that uptake DNA most readily by conjugation or natural competence. The continued development of electroporation protocols in new species and exploration of alternative modes of DNA delivery such as biolistics153 and sonoporation154 may help expedite the broad utility of recombineering methods.
Once the ARF is optimized experimentally in a target system, oligo design and manufacture can present challenges. The primary considerations in oligo design are ensuring that the oligo targets the lagging strand, contains the desired mutation and avoids potential design flaws such as repetitive sequences or long mononucleotide strings. To target oligos to the lagging strand of an open replication fork, which is usually at least an order of magnitude more efficient than targeting the leading strand46, the direction of replication at the targeted genomic locus must be known in advance. However, replication dynamics can be challenging to predict, especially in eukaryotic systems. Users can identify replichores based on software tools155,156, manual annotation36,157 or experimentation43. Once directionality is determined, some additional challenges can be posed by target sequence composition. Short regions of homology are sufficient for recombination33,53, manipulating repetitive sequence-containing loci can result in unwanted recombination or decreased efficiency34 and targets with high GC content or internal repeats can render editing oligos prone to secondary structures that exhibit large, negative Gibbs free-energy changes with self-folding, which consequently decreases editing efficiency33. Online oligo design tools such as MERLIN or MODEST can help a user to avoid some of these problems157,158.
In addition to oligo design, the manufacture of mutation-carrying oligos can pose limitations on the speed, cost-effectiveness and fidelity of recombineering. MAGE requires the manufacture of oligos of 70–90 nucleotides in relatively large quantities (for example, >100 pmol), but the chemical production of such oligos remains time-consuming and error-prone. Studies have shed light on DNA synthesis as the primary source of error in oligo recombineering34,42. Despite continual improvements to industrial DNA manufacturing, single-base deletions remain frequent during chemical oligo synthesis86,159,160. The genomic incorporation of these erroneous sequences results in unwanted deletions and, in a worst-case scenario, frameshift mutations at the target genomic locus. Newer oligo synthesis techniques are mitigating this problem to some degree. Products such as large, 2,000-plus member Oligo Pools from Twist Bioscience can provide a low error rate (1:2,000 nucleotides), offer oligos up to 300 bp and give full control over library diversity. Vendor-provided quantities are low for now (~0.2 fmol per oligo), but various amplification strategies have been demonstrated already for chip-synthesized oligos87,161. Alternatively, oPools from Integrated DNA Technologies offers up to 50 pmol per oligo; however, this platform has a higher synthesis error rate and offers less sequence diversity for a given synthesis price.
As electroporation and protein expression are improved and oligo manufacturing is optimized, there remains the possible limitation of an upper bound on the ARF. During exponential growth, bacterial cells can contain as many as eight active replication forks within a single cell, as the generation time is often shorter than the amount of time it takes to copy a bacterial genome162. Each active fork can undergo oligo incorporation, which after segregating during cell division can lead to unedited, homozygous and heterozygous cells53,163,164. In the case where a mutation confers a dominant phenotype, the apparent ARF declines during recovery until complete segregation of mutated and wild-type genomes occurs, whereas the opposite is true of a recessive phenotype163. As each oligo is only incorporated into one of the four strands present at each replication fork, it has been theorized that the ARF should top out at 25% (REF.120). Promisingly, recent studies have surmounted this theo retical limit, which implies that editing can occur at multiple forks over successive rounds of genome replication67. Future improvements that increase the half-life of oligos33,53,165 or that allow continuous in vivo ssDNA production and incorporation141,142 will continue to push the upper bounds of the accessible ARFs of ultra-high-frequency MAGE.
Finally, MAGE can be a time-intensive endeavour, and despite the simplicity of many of the experimental procedures involved and the possibility of automation33, the robotized execution of such experiments remains poorly developed. Automated devices for electrotransformation of bacteria have been developed33,166,167, but these methods are either restricted to non-continuous operation or can only manipulate small population sizes, limiting the efficient exploration of genomic mutational space. Additionally, whereas most laboratory strains are well suited for such iterative planktonic growth, biofilm formation, precipitation or cellular changes in the case of certain environmental or clinical isolates can prevent repeated growth and electroporation. Consequently, most recombineering experiments are still performed manually.
Outlook
Simplifying genetic engineering
Recombineering was developed two decades ago to ease genetic modifications in E. coli. It allows gene knockouts to be made with dsDNA cassettes24,168 and point mutations with oligos23 (FIG. 1). Recombineering is more streamlined and efficient than older homologous recombination methods, which are still in use today in many bacteria that require donor plasmids with long homology arms and rely on the host’s DNA repair and recombination machinery169. Newer methods that are built on recombineering, such as MAGE, recombineering with CRISPR counter-selection, ORBIT and REXER, have continued to ease precise genomic modifications from single-nucleotide polymorphisms to megabase-scale rearrangements. In eukaryotes, recombineering-based tools that target kilobase-scale modifications are still in active development.
Tremendous progress has been made in the development of tools that perform singleplex genetic manipulations such as DNA insertions and deletions, protein fusions and tagging, and point mutations. In most eukaryotic cells, gene ablations with CRISPR-based methods are relatively straightforward, as NHEJ can be leveraged to disrupt a targeted locus. DNA insertions and point mutations in eukaryotic cells, however, are lower-frequency events that rely on homologous recombination and template-directed repair. Newer methods such as prime editing mitigate this to some degree, and by pairing CRISPR with recombineering tools it may be possible to further improve these methods. CRISPR-based editing is less promising as a stand-alone method in prokaryotes, however, as DSBs are usually lethal, making CRISPR more useful as a complementary tool to other gene editing methods. CRISPR is most effective in prokaryotes, in fact, when paired with recombineering, enabling otherwise low-frequency, scarless editing events to be selected for. ORBIT, an alternative method not based on CRISPR, pairs recombineering with site-specific recombinases to allow positive selection of large, kilobase-scale edits using only oligos83. Methods based on recombineering technology offer unmatched power and flexibility when properly optimized for a given host, however, as many of these methods have only been demonstrated in single host species, important work lies ahead in applying these tools to new human commensals, pathogens and environmental bacteria to further our understanding of these species.
For large-scale genomic manipulations, pairing CRISPR with dsDNA recombineering has produced results that are not possible by either method alone. REXER, the best example of the power of this pairing, was used to efficiently replace multiple 100-kb segments of the E. coli genome, contributing to the assembly of a completely synthetic, recoded genome123,124. In the next decade, similar methods could be used to recode species relevant to industrial biotechnology, where monocultures are susceptible to phage infection; environmental engineering, where biocontainment of genetically engineered organisms is a large concern; and human health, where biocontained human commensals could have therapeutic potential. In building future synthetic genomes, MAGE will be useful as a quick diagnostic, error-correction and trouble-shooting tool, helping to decide between recoding schemes, gauging phage resistance and deciding which biocontainment strategies work best38,89,90. Importantly, recombineering-based methods such as REXER and RLR require only plasmid-based elements, and so all of the necessary components can be conjugated into hosts of interest. This raises the exciting possibility that phages or conjugative bacteria could be used to deliver the components required for gene editing to microorganisms within a complex community, similar to gene therapy for eukaryotic cells.
Multiplex recombineering
The development of high ARFs in E. coli was a direct catalyst for the development of MAGE and other multiplex recombineering-based technologies. Although there are now solutions to achieve library-scale screening of genomic variants by coupling low-frequency (ARF 1–10%) or ultra-low-frequency (ARF <1%) MAGE with an appropriate selection strategy, the project of unlocking high ARF in species important to the furthering of science and human well-being will be of continued importance. We have discussed many relevant uses that have been demonstrated for MAGE-driven complex phenotype optimization in fields from metabolic engineering33,72,170 to antibiotic resistance68,86,142,171. At the same time, new screening and automation technologies are being developed that will complement our ability to build genetic diversity into populations of cells. Protein-based biosensors designed to recognize a diverse array of metabolites172,173 will provide flexibility in manipulating general metabolism by allowing detection of intermediate products and pathways, whereas laboratory automation166,167 and continuous culture systems174,175 will allow our capacity to screen traits that can be tied to strain fitness to be readily scaled. As companion screening and selection technologies improve and new tools and techniques continue to increase the ARFs of a diverse set of species, the applications of multiplex recombineering technologies will grow ever more impactful.
In E. coli and C. freundii, recombineering has now been optimized to operate with exceptional efficiency. A newly discovered SSAP variant, CspRecT, raises the ARF to as high as 50% in some cases, allowing for a new category of ultra-high-frequency (ARF >25%) MAGE67. This will serve to supercharge a host of techniques that are already among the most powerful options for targeted genome engineering. For instance, retron recombineering neared 100% ARF using CspRecT in E. coli and RLR, which is built on this foundation, enables the phenotypic tracking of millions of mutant alleles, and has the potential to identify genetic drivers of complex phenotypes142. Additionally, MAGE has enormous potential to improve protein engineering37,86,113. Due to the exponential nature of combinatorial diversity in protein sequence space, it is challenging to comprehensively sample protein libraries that target degeneracy to as few as five positions. Ultra-high-frequency MAGE promises to navigate deep mutational landscapes, accessing a precise slice of an impossibly large combinatorial space that can be directed by bioinformatics and computational learning68,176,177. This contrasts with methods that perform random walks through this same combinatorial space, such as error-prone mutagenesis178,179 and phage-assisted continuous evolution180,181. As fitness landscapes are often epistatic, multiple rounds of mutagenesis should be more effective at navigating them than single libraries182. By integrating computer-guided oligo design with multiple cycles of editing and phenotypic read-outs, MAGE permits a user to computationally generate models of a fitness landscape, test these models and rapidly iterate to explore rich areas of protein landscapes as the design algorithm improves or learns.
MAGE is unique in its ability to edit genomic DNA. With MAGE, users can target mutation to any genomic region, precisely define mutations and genetic diversity, and create populations of genetic variants that are constrained in size and diversity by only the population size itself. Although recombineering has been improved over the course of two decades, there remain many avenues for continued research and development of related technologies. Pushing ARF higher in most prokaryotes is likely to require, among other factors, SSAP variant screening and improved efficiency of DNA delivery (BOX 1), whereas in eukaryotes our understanding of the dynamics of host replication and repair machinery and the access of oligos to the replication fork remains incomplete. The continued development of simple and high-throughput genome editing technologies holds the promise to substantially improve our understanding of complex microbial systems and to engineer novel proteins and strains for biotechnology and for improving human health.
Box 1 |. Outstanding challenges in recombineering.
Despite recent discoveries, our understanding of both the determinants of the allelic recombination frequency (ARF) and its ceiling remains incomplete. Looking forward, future studies that focus on the exploration of chromosomal dynamics and replication, specific molecular interactions at the replication fork, high-efficiency single-stranded DNA-annealing proteins (SSAPs), dual-strand targeting and oligo delivery, degradation and manufacturing can be expected to further improve recombineering systems. A focus on non-model microorganisms and eukaryotic systems will be particularly relevant to broadening the reach of multiplex automated genome engineering (MAGE) and its powerful derivative technologies.
In the coming years, advanced DNA manufacturing will supply users with rapid production of high-fidelity, made-to-order oligonucleotides. Improvements to manufactured oligodeoxynucleotide (oligo) length, fidelity, turnaround time and cost promise to radically improve the power, speed and scope of the techniques discussed here. Furthermore, library-generation methods such as MAGE and directed evolution with random genomic mutations (DIvERGE) will be made more powerful by the cost-effective synthesis of large, diverse oligo pools and soft-randomized oligos.
Acknowledgements
The authors thank J. Aach for helpful insights. Funding for this research was provided by the US Department of Energy (DOE) under grant DE-FG02-02ER63445 (G.M.C). The authors acknowledge support from the National Institute of General Medical Sciences of the National Institutes of Health (NIH) under a Chemistry–Biology Interface Training Grant that supported M.A.J. (Award Number T32GM133395). The study was supported by the following research grants: European Research Council (ERC) H2020-ERC-2014-CoG 648364 — Resistance Evolution (C.P.); ‘Célzott Lendület’ Programme of the Hungarian Academy of Sciences LP-2017–10/2017 (C.P.); ‘Élvonal’ KKP 126506 (C.P.); and GINOP-2.3.2–15–2016–00014 (EVOMER, to C.P.). P.N.C. was supported by Physical and Engineering Biology training grant 5T32EB019941-05. A.N. was supported by an EMBO LTF 160-2019 Long-Term fellowship. A.D.E. and K.J. acknowledge funding from the Air Force Office of Scientific Research (FA9550-14-1-0089) and the Welch Foundation (F-1654).
RELATED LINKS
European Nucleotide Archive (ENA): https://www.ebi.ac.uk/ena/browser
GitHub: https://github.com
National Center for Biotechnology Information (NCBI)
Sequence Read Archive (SRA): https://www.ncbi.nlm.nih.gov/sra
Footnotes
Competing interests
T.M.W, G.T.F. and G.M.C. are inventors on a patent application related to serial enrichment for efficient recombineering (SEER) and new single-stranded DNA-annealing protein (SSAP) discovery. A.N. and C.P. are inventors on a patent related to directed evolution with random genomic mutations (DIvERGE) (US10669537B2: Mutagenizing Intracellular Nucleic Acids). F.J.I. and G.M.C. are inventors on a MAGE patent, which has been licensed. F.J.I. is an inventor on a patent application related to eukaryotic MAGE. The remaining authors declare no competing interests.
References
- 1.Simon R, Priefer U & Pühler A A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Nat. Biotechnol 1, 784–791 (1983). [Google Scholar]
- 2.Ye B et al. Unmarked genetic manipulation in Bacillus subtilis by natural co-transformation. J. Biotechnol 284, 57–62 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Chandrasegaran S & Carroll D Origins of programmable nucleases for genome engineering. J. Mol. Biol 428, 963–989 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doudna JA & Charpentier E Genome editing. The new frontier of genome engineering with CRISPR–Cas9. Science 346, 1258096 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Gersbach CA Genome engineering: the next genomic revolution. Nat. Methods 11, 1009–1011 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Gaj T, Gersbach CA & Barbas CF ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31, 397–405 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim H & Kim J-S A guide to genome engineering with programmable nucleases. Nat. Rev. Genet 15, 321–334 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Cong L et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jinek M et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mali P et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jakočiūnas T et al. Multiplex metabolic pathway engineering using CRISPR/Cas9 in Saccharomyces cerevisiae. Metab. Eng 28, 213–222 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Fu Y et al. High-frequency off-target mutagenesis induced by CRISPR–Cas nucleases in human cells. Nat. Biotechnol 31, 822–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inui M et al. Rapid generation of mouse models with defined point mutations by the CRISPR/Cas9 system. Sci. Rep 4, 5396 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paquet D et al. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 533, 125–129 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Maruyama T et al. Increasing the efficiency of precise genome editing with CRISPR–Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol 33, 538–542 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gaudelli NM et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anzalone AV et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith CJ et al. Enabling large-scale genome editing at repetitive elements by reducing DNA nicking. Nucleic Acids Res 48, 5183–5195 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reis AC et al. Simultaneous repression of multiple bacterial genes using nonrepetitive extra-long sgRNA arrays. Nat. Biotechnol 37, 1294–1301 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Zeng Y et al. Correction of the Marfan syndrome pathogenic FBN1 mutation by base editing in human cells and heterozygous embryos. Mol. Ther 26, 2631–2637 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zeng J et al. Therapeutic base editing of human hematopoietic stem cells. Nat. Med 26, 535–541 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ellis HM, Yu D, DiTizio T & Court DL High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl Acad. Sci. USA 98, 6742–6746 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article was the first to thoroughly examine the possibility of recombineering with ssDNA as a template.
- 24.Yu D et al. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl Acad. Sci. USA 97, 5978–5983 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mosberg JA, Lajoie MJ & Church GM λ red recombineering in Escherichia coli occurs through a fully single-stranded intermediate. Genetics 186, 791–799 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murphy KC Use of bacteriophage λ recombination functions to promote gene replacement in Escherichia coli. J. Bacteriol 180, 2063–2071 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Buchholz F, Muyrers JP & Stewart AF A new logic for DNA engineering using recombination in Escherichia coli. Nat. Genet 20, 123–128 (1998). [DOI] [PubMed] [Google Scholar]
- 28.Little JW An exonuclease induced by bacteriophage λ. II. Nature of the enzymatic reaction. J. Biol. Chem 242, 679–686 (1967). [PubMed] [Google Scholar]
- 29.Caldwell BJ et al. Crystal structure of the Redβ C-terminal domain in complex with λ exonuclease reveals an unexpected homology with λ Orf and an interaction with Escherichia coli single stranded DNA binding protein. Nucleic Acids Res 47, 1950–1963 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Z, Karakousis G, Chiu SK, Reddy G & Radding CM The β protein of phage λ promotes strand exchange. J. Mol. Biol 276, 733–744 (1998). [DOI] [PubMed] [Google Scholar]
- 31.Murphy KC λ Gam protein inhibits the helicase and chi-stimulated recombination activities of Escherichia coli RecBCD enzyme. J. Bacteriol 173, 5808–5821 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barbieri EM, Muir P, Akhuetie-Oni BO, Yellman CM & Isaacs FJ Precise editing at DNA replication forks enables multiplex genome engineering in eukaryotes. Cell 171, 1453–1467.e13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article describes eMAGE, the first instance of MAGE in a eukaryotic cell, leveraging co-selection to improve the ARF.
- 33.Wang HH et al. Programming cells by multiplex genome engineering and accelerated evolution. Nature 460, 894–898 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the original article describing MAGE as a method for multiplex genome editing.
- 34.Isaacs FJ et al. Precise manipulation of chromosomes in vivo enables genome-wide codon replacement. Science 333, 348–353 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carr PA et al. Enhanced multiplex genome engineering through co-operative oligonucleotide co-selection. Nucleic Acids Res 40, e132 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nyerges Á et al. A highly precise and portable genome engineering method allows comparison of mutational effects across bacterial species. Proc. Natl Acad. Sci. USA 113, 2502–2507 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work first describes the transient suppression of MMR by expression of a dominant negative MutL.
- 37.Amiram M et al. Evolution of translation machinery in recoded bacteria enables multi-site incorporation of nonstandard amino acids. Nat. Biotechnol 33, 1272–1279 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lajoie MJ et al. Genomically recoded organisms expand biological functions. Science 342, 357–360 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This landmark article is the first to report a fully recoded organism, in this case an E. coli strain with 321 TAG stop codon reassignments, produced with MAGE.
- 39.Napolitano MG et al. Emergent rules for codon choice elucidated by editing rare arginine codons in Escherichia coli. Proc. Natl Acad. Sci. USA 113, E5588–5597 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swaminathan S et al. Rapid engineering of bacterial artificial chromosomes using oligonucleotides. Genesis 29, 14–21 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Thomason LC, Costantino N, Shaw DV & Court DL Multicopy plasmid modification with phage λ Red recombineering. Plasmid 58, 148–158 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oppenheim AB, Rattray AJ, Bubunenko M, Thomason LC & Court DL In vivo recombineering of bacteriophage λ by PCR fragments and single-strand oligonucleotides. Virology 319, 185–189 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Hueso-Gil A, Nyerges Á, Pál C, Calles B & de Lorenzo V Multiple-site diversification of regulatory sequences enables interspecies operability of genetic devices. ACS Synth. Biol 9, 104–114 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Court DL, Sawitzke JA & Thomason LC Genetic engineering using homologous recombination. Annu. Rev. Genet 36, 361–388 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Anzalone AV, Koblan LW & Liu DR Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol 38, 824–844 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Costantino N & Court DL Enhanced levels of λ Red-mediated recombinants in mismatch repair mutants. Proc. Natl Acad. Sci. USA 100, 15748–15753 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the first article to describe evasion of mismatch repair as an effective strategy to improve the ARF.
- 47.Au KG, Welsh K & Modrich P Initiation of methyl-directed mismatch repair. J. Biol. Chem 267, 12142–12148 (1992). [PubMed] [Google Scholar]
- 48.Burdett V, Baitinger C, Viswanathan M, Lovett ST & Modrich P In vivo requirement for RecJ, ExoVII, ExoI, and ExoX in methyl-directed mismatch repair. Proc. Natl Acad. Sci. USA 98, 6765–6770 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schaaper RM & Dunn RL Spectra of spontaneous mutations in Escherichia coli strains defective in mismatch correction: the nature of in vivo DNA replication errors. Proc. Natl Acad. Sci. USA 84, 6220–6224 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iyer RR, Pluciennik A, Burdett V & Modrich PL DNA mismatch repair: functions and mechanisms. Chem. Rev 106, 302–323 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Wang HH, Xu G, Vonner AJ & Church G Modified bases enable high-efficiency oligonucleotide-mediated allelic replacement via mismatch repair evasion. Nucleic Acids Res 39, 7336–7347 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Modrich P Mechanisms and biological effects of mismatch repair. Annu. Rev. Genet 25, 229–253 (1991). [DOI] [PubMed] [Google Scholar]
- 53.Sawitzke JA et al. Probing cellular processes with oligo-mediated recombination and using the knowledge gained to optimize recombineering. J. Mol. Biol 407, 45–59 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Pijkeren J-P & Britton RA High efficiency recombineering in lactic acid bacteria. Nucleic Acids Res 40, e76 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work is one of the first and best instances of the screening of a small group of SSAPs to permit high-frequency MAGE in a non-E. coli bacterium, here L. lactis and Lactobacillus reuteri.
- 55.Binder S, Siedler S, Marienhagen J, Bott M & Eggeling L Recombineering in Corynebacterium glutamicum combined with optical nanosensors: a general strategy for fast producer strain generation. Nucleic Acids Res 41, 6360–6369 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Penewit K et al. Efficient and scalable precision genome editing in Staphylococcus aureus through conditional recombineering and CRISPR/Cas9-mediated counterselection. mBio 9, e00067 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Ravesteyn TW et al. LNA modification of single-stranded DNA oligonucleotides allows subtle gene modification in mismatch-repair-proficient cells. Proc. Natl Acad. Sci. USA 113, 4122–4127 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matic I, Babic A & Radman M 2-Aminopurine allows interspecies recombination by a reversible inactivation of the Escherichia coli mismatch repair system. J. Bacteriol 185, 1459–1461 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pitsikas P, Patapas JM & Cupples CG Mechanism of 2-aminopurine-stimulated mutagenesis in Escherichia coli. Mutat. Res 550, 25–32 (2004). [DOI] [PubMed] [Google Scholar]
- 60.Ang J et al. Mutagen synergy: hypermutability generated by specific pairs of base analogs. J. Bacteriol 198, 2776–2783 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nyerges Á et al. Conditional DNA repair mutants enable highly precise genome engineering. Nucleic Acids Res 42, e62 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hong ES, Yeung A, Funchain P, Slupska MM & Miller JH Mutants with temperature-sensitive defects in the Escherichia coli mismatch repair system: sensitivity to mispairs generated in vivo. J. Bacteriol 187, 840–846 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lennen RM et al. Transient overexpression of DNA adenine methylase enables efficient and mobile genome engineering with reduced off-target effects. Nucleic Acids Res 44, e36 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang H, Wolff E, Kim M, Diep A & Miller JH Identification of mutator genes and mutational pathways in Escherichia coli using a multicopy cloning approach. Mol. Microbiol 53, 283–295 (2004). [DOI] [PubMed] [Google Scholar]
- 65.Aronshtam A & Marinus MG Dominant negative mutator mutations in the mutL gene of Escherichia coli. Nucleic Acids Res 24, 2498–2504 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ricaurte DE et al. A standardized workflow for surveying recombinases expands bacterial genome-editing capabilities. Microb. Biotechnol 11, 176–188 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wannier TM et al. Improved bacterial recombineering by parallelized protein discovery. Proc. Natl Acad. Sci. USA 117, 13689–13698 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work describes SEER, a method for adapting MAGE to new bacterial species, and the improvement of ARF to ultra-high frequency in E. coli and C. freundii.
- 68.Filsinger G et al. Characterizing the portability of RecT-mediated oligonucleotide recombination. Nat. Chem. Biol 10.1038/s41589-020-00710-5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work describes a molecular basis for the host tropism displayed by SSAPs, namely their interaction with the host SSB.
- 69.Aparicio T, Nyerges A, Martínez-García E & de Lorenzo V High-efficiency multi-site genomic editing of Pseudomonas putida through thermo-inducible ssDNA recombineering. iScience 23, 100946 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Storici F, Lewis LK & Resnick MA In vivo site-directed mutagenesis using oligonucleotides. Nat. Biotechnol 19, 773–776 (2001). [DOI] [PubMed] [Google Scholar]
- 71.DiCarlo JE et al. Yeast oligo-mediated genome engineering (YOGE). ACS Synth. Biol 2, 741–749 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article is the first to explore recombineering in eukaryotes, focusing on Rad51 expression and MMR avoidance.
- 72.Wang HH et al. Genome-scale promoter engineering by co-selection MAGE. Nat. Methods 9, 591–593 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee M et al. Rad52/Rad59-dependent recombination as a means to rectify faulty Okazaki fragment processing. J. Biol. Chem 289, 15064–15079 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Arbel M, Bronstein A, Sau S, Liefshitz B & Kupiec M Access to PCNA by Srs2 and Elg1 controls the choice between alternative repair pathways in Saccharomyces cerevisiae. mBio 11, e00705–e00720 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liang Z, Metzner E & Isaacs FJ Advanced eMAGE for highly efficient combinatorial editing of a stable genome. Preprint at bioRxiv 10.1101/2020.08.30.256743 (2020). [DOI] [Google Scholar]
- 76.Iyer LM, Koonin EV & Aravind L Classification and evolutionary history of the single-strand annealing proteins, RecT, Redβ, ERF and RAD52. BMC Genom 3, 8 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.van Kessel JC & Hatfull GF Recombineering in Mycobacterium tuberculosis. Nat. Methods 4, 147–152 (2007). [DOI] [PubMed] [Google Scholar]
- 78.Aparicio T, Jensen SI, Nielsen AT, de Lorenzo V & Martínez-García E The Ssr protein (T1E_1405) from Pseudomonas putida DOT-T1E enables oligonucleotide-based recombineering in platform strain P. putida EM42. Biotechnol. J 11, 1309–1319 (2016). [DOI] [PubMed] [Google Scholar]
- 79.Lee HH, Ostrov N, Gold MA & Church GM Recombineering in Vibrio natriegens. Preprint at bioRxiv 10.1101/130088 (2017). [DOI] [Google Scholar]
- 80.Wu DY, Ugozzoli L, Pal BK & Wallace RB Allele-specific enzymatic amplification of β-globin genomic DNA for diagnosis of sickle cell anemia. Proc. Natl Acad. Sci. USA 86, 2757–2760 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Johnson KA The kinetic and chemical mechanism of high-fidelity DNA polymerases. Biochim. Biophys. Acta 1804, 1041–1048 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lefever S et al. Cost-effective and robust genotyping using double-mismatch allele-specific quantitative PCR. Sci. Rep 9, 2150 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Imyanitov EN et al. Improved reliability of allele-specific PCR. BioTechniques 33, 484–490 (2002). [DOI] [PubMed] [Google Scholar]
- 84.Słomka M, Sobalska-Kwapis M, Wachulec M, Bartosz G & Strapagiel D High resolution melting (HRM) for high-throughput genotyping-limitations and caveats in practical case studies. Int. J. Mol. Sci 18, 2316 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Murphy KC et al. ORBIT: a new paradigm for genetic engineering of mycobacterial chromosomes. mBio 9, e01467–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This inventive article describes the pairing of recombineering with site-specific recombinases to ease genomic deletions and fusions in mycobacteria.
- 86.Nyerges Á et al. Directed evolution of multiple genomic loci allows the prediction of antibiotic resistance. Proc. Natl Acad. Sci. USA 115, E5726–E5735 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article describes DIvERGE, an important technique for diversification of targeted genomic loci.
- 87.Bonde MT et al. Direct mutagenesis of thousands of genomic targets using microarray-derived oligonucleotides. ACS Synth. Biol 4, 17–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang HH & Church GM Multiplexed genome engineering and genotyping methods applications for synthetic biology and metabolic engineering. Meth. Enzymol 498, 409–426 (2011). [DOI] [PubMed] [Google Scholar]
- 89.Mandell DJ et al. Biocontainment of genetically modified organisms by synthetic protein design. Nature 518, 55–60 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rovner AJ et al. Recoded organisms engineered to depend on synthetic amino acids. Nature 518, 89–93 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sandberg TE et al. Evolution of Escherichia coli to 42 °C and subsequent genetic engineering reveals adaptive mechanisms and novel mutations. Mol. Biol. Evol 31, 2647–2662 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wannier TM et al. Adaptive evolution of genomically recoded Escherichia coli. Proc. Natl Acad. Sci. USA 115, 3090–3095 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pattanayak V, Guilinger JP & Liu DR Determining the specificities of TALENs, Cas9, and other genome-editing enzymes. Meth. Enzymol 546, 47–78 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rees HA & Liu DR Publisher correction: base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet 19, 801 (2018). [DOI] [PubMed] [Google Scholar]
- 95.Mougiakos I, Bosma EF, de Vos WM, van Kranenburg R & van der Oost J Next generation prokaryotic engineering: the CRISPR–Cas toolkit. Trends Biotechnol 34, 575–587 (2016). [DOI] [PubMed] [Google Scholar]
- 96.Jiang W, Bikard D, Cox D, Zhang F & Marraffini LA RNA-guided editing of bacterial genomes using CRISPR–Cas systems. Nat. Biotechnol 31, 233–239 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Oh J-H & van Pijkeren J-P CRISPR–Cas9-assisted recombineering in Lactobacillus reuteri. Nucleic Acids Res 42, e131 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Higgins SA, Ounkap S & Savage DF Rapid and programmable protein mutagenesis using plasmid recombineering. ACS Synth. Biol 10.1021/acssynbio.7b00112 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ronda C, Pedersen LE, Sommer MOA & Nielsen AT CRMAGE: CRISPR optimized MAGE recombineering. Sci. Rep 6, 19452 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Oesterle S, Gerngross D, Schmitt S, Roberts TM & Panke S Efficient engineering of chromosomal ribosome binding site libraries in mismatch repair proficient Escherichia coli. Sci. Rep 7, 12327 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Umenhoffer K et al. Genome-wide abolishment of mobile genetic elements using genome shuffling and CRISPR/Cas-assisted MAGE allows the efficient stabilization of a bacterial chassis. ACS Synth. Biol 6, 1471–1483 (2017). [DOI] [PubMed] [Google Scholar]
- 102.Ding T et al. Reversed paired-gRNA plasmid cloning strategy for efficient genome editing in Escherichia coli. Microb. Cell Fact 19, 63 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Farasat I et al. Efficient search, mapping, and optimization of multi-protein genetic systems in diverse bacteria. Mol. Syst. Biol 10, 731 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kunjapur AM, Tarasova Y & Prather KLJ Synthesis and accumulation of aromatic aldehydes in an engineered strain of Escherichia coli. J. Am. Chem. Soc 136, 11644–11654 (2014). [DOI] [PubMed] [Google Scholar]
- 105.Pósfai G et al. Emergent properties of reduced-genome Escherichia coli. Science 312, 1044–1046 (2006). [DOI] [PubMed] [Google Scholar]
- 106.Grodberg J & Dunn JJ ompT encodes the Escherichia coli outer membrane protease that cleaves T7 RNA polymerase during purification. J. Bacteriol 170, 1245–1253 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Studier FW, Daegelen P, Lenski RE, Maslov S & Kim JF Understanding the differences between genome sequences of Escherichia coli B strains REL606 and BL21(DE3) and comparison of the E. coli B and K-12 genomes. J. Mol. Biol 394, 653–680 (2009). [DOI] [PubMed] [Google Scholar]
- 108.Borja GM et al. Engineering Escherichia coli to increase plasmid DNA production in high cell-density cultivations in batch mode. Microb. Cell Fact 11, 132 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Derman AI, Prinz WA, Belin D & Beckwith J Mutations that allow disulfide bond formation in the cytoplasm of Escherichia coli. Science 262, 1744–1747 (1993). [DOI] [PubMed] [Google Scholar]
- 110.Bessette PH, Aslund F, Beckwith J & Georgiou G Efficient folding of proteins with multiple disulfide bonds in the Escherichia coli cytoplasm. Proc. Natl Acad. Sci. USA 96, 13703–13708 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lobstein J et al. SHuffle, a novel Escherichia coli protein expression strain capable of correctly folding disulfide bonded proteins in its cytoplasm. Microb. Cell Fact 11, 56 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yates LE et al. Glyco-recoded Escherichia coli: recombineering-based genome editing of native polysaccharide biosynthesis gene clusters. Metab. Eng 53, 59–68 (2019). [DOI] [PubMed] [Google Scholar]
- 113.Kelsic ED et al. RNA structural determinants of optimal codons revealed by MAGE-Seq. Cell Syst 3, 563–571.e6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Scangarella-Oman NE et al. In vitro activity and microbiological efficacy of gepotidacin from a phase 2, randomized, multicenter, dose-ranging study in patients with acute bacterial skin and skin structure infections. Antimicrob. Agents Chemother 64, e01302–e01319 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Garst AD et al. Genome-wide mapping of mutations at single-nucleotide resolution for protein, metabolic and genome engineering. Nat. Biotechnol 35, 48–55 (2017). [DOI] [PubMed] [Google Scholar]
- 116.Halperin SO et al. CRISPR-guided DNA polymerases enable diversification of all nucleotides in a tunable window. Nature 560, 248–252 (2018). [DOI] [PubMed] [Google Scholar]
- 117.Wang HH et al. Multiplexed in vivo His-tagging of enzyme pathways for in vitro single-pot multienzyme catalysis. ACS Synth. Biol 1, 43–52 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Brockman IM & Prather KLJ Dynamic knockdown of E. coli central metabolism for redirecting fluxes of primary metabolites. Metab. Eng 28, 104–113 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Durante-Rodríguez G, de Lorenzo V & Nikel PI A post-translational metabolic switch enables complete decoupling of bacterial growth from biopolymer production in engineered Escherichia coli. ACS Synth. Biol 7, 2686–2697 (2018). [DOI] [PubMed] [Google Scholar]
- 120.Pines G, Freed EF, Winkler JD & Gill RT Bacterial recombineering: genome engineering via phage-based homologous recombination. ACS Synth. Biol 4, 1176–1185 (2015). [DOI] [PubMed] [Google Scholar]
- 121.Choudhury A et al. CRISPR/Cas9 recombineering-mediated deep mutational scanning of essential genes in Escherichia coli. Mol. Syst. Biol 16, e9265 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bao Z, Cobb RE & Zhao H Accelerated genome engineering through multiplexing. Wiley Interdiscip. Rev. Syst. Biol. Med 8, 5–21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fredens J et al. Total synthesis of Escherichia coli with a recoded genome. Nature 569, 514–518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ostrov N et al. Design, synthesis, and testing toward a 57-codon genome. Science 353, 819–822 (2016). [DOI] [PubMed] [Google Scholar]
- 125.Jiang Y et al. Multigene editing in the Escherichia coli genome via the CRISPR–Cas9 system. Appl. Environ. Microbiol 81, 2506–2514 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Piñero Lambea C et al. Mycoplasma pneumoniae genome editing based on oligo recombineering and Cas9-mediated counterselection. ACS Synth. Biol 10.1021/acssynbio.0c00022 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cui L & Bikard D Consequences of Cas9 cleavage in the chromosome of Escherichia coli. Nucleic Acids Res 44, 4243–4251 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pyne ME, Moo-Young M, Chung DA & Chou CP Coupling the CRISPR/Cas9 system with λ Red recombineering enables simplified chromosomal gene replacement in Escherichia coli. Appl. Environ. Microbiol 81, 5103–5114 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Li Y et al. Metabolic engineering of Escherichia coli using CRISPR–Cas9 meditated genome editing. Metab. Eng 31, 13–21 (2015). [DOI] [PubMed] [Google Scholar]
- 130.van Kessel JC & Hatfull GF Efficient point mutagenesis in mycobacteria using single-stranded DNA recombineering: characterization of antimyco-bacterial drug targets. Mol. Microbiol 67, 1094–1107 (2008). [DOI] [PubMed] [Google Scholar]
- 131.Aldovini A, Husson RN & Young RA The uraA locus and homologous recombination in Mycobacterium bovis BCG. J. Bacteriol 175, 7282–7289 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kalpana GV, Bloom BR & Jacobs WR Insertional mutagenesis and illegitimate recombination in mycobacteria. Proc. Natl Acad. Sci. USA 88, 5433–5437 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang K et al. Defining synonymous codon compression schemes by genome recoding. Nature 539, 59–64 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chin JW Reprogramming the genetic code. Science 336, 428–429 (2012). [DOI] [PubMed] [Google Scholar]
- 135.Lampson BC, Inouye M & Inouye S Retrons, msDNA, and the bacterial genome. Cytogenet. Genome Res 110, 491–499 (2005). [DOI] [PubMed] [Google Scholar]
- 136.Simon AJ, Ellington AD & Finkelstein IJ Retrons and their applications in genome engineering. Nucleic Acids Res 47, 11007–11019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yee T, Furuichi T, Inouye S & Inouye M Multicopy single-stranded DNA isolated from a gram-negative bacterium, Myxococcus xanthus. Cell 38, 203–209 (1984). [DOI] [PubMed] [Google Scholar]
- 138.Millman A et al. Bacterial retrons function in anti-phage defense. Cell 10.1016/j.cell.2020.09.065 (2020). [DOI] [PubMed] [Google Scholar]
- 139.Farzadfard F & Lu TK Genomically encoded analog memory with precise in vivo DNA writing in living cell populations. Science 346, 1256272 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Simon AJ, Morrow BR & Ellington AD Retroelement-based genome editing and evolution. ACS Synth. Biol 7, 2600–2611 (2018). [DOI] [PubMed] [Google Scholar]
- 141.Farzadfard F, Gharaei N, Citorik RJ & Lu TK Efficient retroelement-mediated DNA writing in bacteria. Preprint at bioRxiv 10.1101/2020.02.21.958983 (2020). [DOI] [PubMed] [Google Scholar]
- 142.Schubert MG et al. High throughput functional variant screens via in-vivo production of single-stranded DNA. Preprint at bioRxiv 10.1101/2020.03.05.975441 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gallagher RR, Li Z, Lewis AO & Isaacs FJ Rapid editing and evolution of bacterial genomes using libraries of synthetic DNA. Nat. Protoc 9, 2301–2316 (2014). [DOI] [PubMed] [Google Scholar]
- 144.Sharan SK, Thomason LC, Kuznetsov SG & Court DL Recombineering: a homologous recombination-based method of genetic engineering. Nat. Protoc 4, 206–223 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Holo H & Nes IF High-frequency transformation, by electroporation, of Lactococcus lactis subsp. cremoris grown with glycine in osmotically stabilized media. Appl. Environ. Microbiol 55, 3119–3123 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Shepard BD & Gilmore MS Electroporation and efficient transformation of Enterococcus faecalis grown in high concentrations of glycine. Methods Mol. Biol 47, 217–226 (1995). [DOI] [PubMed] [Google Scholar]
- 147.Dower WJ, Miller JF & Ragsdale CW High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res 16, 6127–6145 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Okamoto A, Kosugi A, Koizumi Y, Yanagida F & Udaka S High efficiency transformation of Bacillus brevis by electroporation. Biosci. Biotechnol. Biochem 61, 202–203 (1997). [DOI] [PubMed] [Google Scholar]
- 149.Wards BJ & Collins DM Electroporation at elevated temperatures substantially improves transformation efficiency of slow-growing mycobacteria. FEMS Microbiol. Lett 145, 101–105 (1996). [DOI] [PubMed] [Google Scholar]
- 150.Tu Q et al. Room temperature electrocompetent bacterial cells improve DNA transformation and recombineering efficiency. Sci. Rep 6, 24648 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.McIntyre DA & Harlander SK Genetic transformation of intact Lactococcus lactis subsp. lactis by high-voltage electroporation. Appl. Environ. Microbiol 55, 604–610 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Salis HM The ribosome binding site calculator. Meth. Enzymol 498, 19–42 (2011). [DOI] [PubMed] [Google Scholar]
- 153.Liu J et al. Genome-scale sequence disruption following biolistic transformation in rice and maize. Plant Cell 31, 368–383 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Miller DL, Pislaru SV & Greenleaf JE Sonoporation: mechanical DNA delivery by ultrasonic cavitation. Somat. Cell Mol. Genet 27, 115–134 (2002). [DOI] [PubMed] [Google Scholar]
- 155.Gao F & Zhang C-T Ori-Finder: a web-based system for finding oriCs in unannotated bacterial genomes. BMC Bioinform 9, 79 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sernova NV & Gelfand MS Identification of replication origins in prokaryotic genomes. Brief. Bioinform 9, 376–391 (2008). [DOI] [PubMed] [Google Scholar]
- 157.Bonde MT et al. MODEST: a web-based design tool for oligonucleotide-mediated genome engineering and recombineering. Nucleic Acids Res 42, W408–W415 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Quintin M et al. Merlin: computer-aided oligonucleotide design for large scale genome engineering with MAGE. ACS Synth. Biol 5, 452–458 (2016). [DOI] [PubMed] [Google Scholar]
- 159.Hecker KH & Rill RL Error analysis of chemically synthesized polynucleotides. Biotechniques 24, 256–260 (1998). [DOI] [PubMed] [Google Scholar]
- 160.Temsamani J, Kubert M & Agrawal S Sequence identity of the n-1 product of a synthetic oligonucleotide. Nucleic Acids Res 23, 1841–1844 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Schmidt TL et al. Scalable amplification of strand subsets from chip-synthesized oligonucleotide libraries. Nat. Commun 6, 8634 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Nordström K & Dasgupta S Copy-number control of the Escherichia coli chromosome: a plasmidologist’s view. EMBO Rep 7, 484–489 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Reynolds TS & Gill RT Quantifying impact of chromosome copy number on recombination in Escherichia coli. ACS Synth. Biol 4, 776–780 (2015). [DOI] [PubMed] [Google Scholar]
- 164.Boyle NR, Reynolds TS, Evans R, Lynch M & Gill RT Recombineering to homogeneity: extension of multiplex recombineering to large-scale genome editing. Biotechnol. J 8, 515–522 (2013). [DOI] [PubMed] [Google Scholar]
- 165.Parekh-Olmedo H, Drury M & Kmiec EB Targeted nucleotide exchange in Saccharomyces cerevisiae directed by short oligonucleotides containing locked nucleic acids. Chem. Biol 9, 1073–1084 (2002). [DOI] [PubMed] [Google Scholar]
- 166.Moore JA et al. Automated electrotransformation of Escherichia coli on a digital microfluidic platform using bioactivated magnetic beads. Biomicrofluidics 11, 014110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Madison AC et al. Scalable device for automated microbial electroporation in a digital microfluidic platform. ACS Synth. Biol 6, 1701–1709 (2017). [DOI] [PubMed] [Google Scholar]
- 168.Datsenko KA & Wanner BL One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl Acad. Sci. USA 97, 6640–6645 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jasin M & Schimmel P Deletion of an essential gene in Escherichia coli by site-specific recombination with linear DNA fragments. J. Bacteriol 159, 783–786 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Liang L et al. CRISPR EnAbled Trackable genome Engineering for isopropanol production in Escherichia coli. Metab. Eng 41, 1–10 (2017). [DOI] [PubMed] [Google Scholar]
- 171.Szili P et al. Rapid evolution of reduced susceptibility against a balanced dual-targeting antibiotic through stepping-stone mutations. Antimicrob. Agents Chemother 63, e00207–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Zhang J, Jensen MK & Keasling JD Development of biosensors and their application in metabolic engineering. Curr. Opin. Chem. Biol 28, 1–8 (2015). [DOI] [PubMed] [Google Scholar]
- 173.Glasgow AA et al. Computational design of a modular protein sense-response system. Science 366, 1024–1028 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hoffmann SA, Wohltat C, Müller KM & Arndt KM A user-friendly, low-cost turbidostat with versatile growth rate estimation based on an extended Kalman filter. PLoS ONE 12, e0181923 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wong BG, Mancuso CP, Kiriakov S, Bashor CJ & Khalil AS Precise, automated control of conditions for high-throughput growth of yeast and bacteria with eVOLVER. Nat. Biotechnol 36, 614–623 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Yang KK, Wu Z & Arnold FH Machine-learning-guided directed evolution for protein engineering. Nat. Methods 16, 687–694 (2019). [DOI] [PubMed] [Google Scholar]
- 177.Biswas S, Khimulya G, Alley EC, Esvelt KM & Church GM Low-N protein engineering with data-efficient deep learning. Preprint at bioRxiv 10.1101/2020.01.23.917682 (2020). [DOI] [PubMed] [Google Scholar]
- 178.Beckman RA, Mildvan AS & Loeb LA On the fidelity of DNA replication: manganese mutagenesis in vitro. Biochemistry 24, 5810–5817 (1985). [DOI] [PubMed] [Google Scholar]
- 179.Skandalis A, Encell LP & Loeb LA Creating novel enzymes by applied molecular evolution. Chem. Biol 4, 889–898 (1997). [DOI] [PubMed] [Google Scholar]
- 180.Badran AH et al. Continuous evolution of Bacillus thuringiensis toxins overcomes insect resistance. Nature 533, 58–63 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hu JH et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Reetz MT, Prasad S, Carballeira JD, Gumulya Y & Bocola M Iterative saturation mutagenesis accelerates laboratory evolution of enzyme stereoselectivity: rigorous comparison with traditional methods. J. Am. Chem. Soc 132, 9144–9152 (2010). [DOI] [PubMed] [Google Scholar]
- 183.Martínez-García E, Aparicio T, Goñi-Moreno A, Fraile S & de Lorenzo V SEVA 2.0: an update of the Standard European Vector Architecture for de-/reconstruction of bacterial functionalities. Nucleic Acids Res 43, D1183–D1189 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.van Pijkeren J-P, Neoh KM, Sirias D, Findley AS & Britton RA Exploring optimization parameters to increase ssDNA recombineering in Lactococcus lactis and Lactobacillus reuteri. Bioengineered 3, 209–217 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chang Y, Wang Q, Su T & Qi Q The efficiency for recombineering is dependent on the source of the phage recombinase function unit. Preprint at bioRxiv 10.1101/745448 (2019). [DOI] [Google Scholar]
- 186.Aparicio T et al. Mismatch repair hierarchy of Pseudomonas putida revealed by mutagenic ssDNA recombineering of the pyrF gene. Environ. Microbiol 22, 45–58 (2020). [DOI] [PubMed] [Google Scholar]
- 187.Corts AD, Thomason LC, Gill RT & Gralnick JA A new recombineering system for precise genome-editing in Shewanella oneidensis strain MR-1 using single-stranded oligonucleotides. Sci. Rep 9, 1–10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bryan A & Swanson MS Oligonucleotides stimulate genomic alterations of Legionella pneumophila. Mol. Microbiol 80, 231–247 (2011). [DOI] [PubMed] [Google Scholar]
- 189.Swingle B, Bao Z, Markel E, Chambers A & Cartinhour S Recombineering using RecTE from Pseudomonas syringae. Appl. Environ. Microbiol 76, 4960–4968 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Tucker AT et al. Defining gene–phenotype relationships in Acinetobacter baumannii through one-step chromosomal gene inactivation. mBio 5, e01313–01314 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sun Z et al. A high-efficiency recombineering system with PCR-based ssDNA in Bacillus subtilis mediated by the native phage recombinase GP35. Appl. Microbiol. Biotechnol 99, 5151–5162 (2015). [DOI] [PubMed] [Google Scholar]
- 192.Wang X et al. Discovery of recombinases enables genome mining of cryptic biosynthetic gene clusters in Burkholderiales species. Proc. Natl Acad. Sci. USA 115, E4255–E4263 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Dong H, Tao W, Gong F, Li Y & Zhang Y A functional recT gene for recombineering of Clostridium. J. Biotechnol 173, 65–67 (2014). [DOI] [PubMed] [Google Scholar]
- 194.Huang H, Song X & Yang S Development of a RecE/T-assisted CRISPR–Cas9 toolbox for Lactobacillus. Biotechnol. J 14, e1800690 (2019). [DOI] [PubMed] [Google Scholar]
- 195.Xin Y, Guo T, Mu Y & Kong J Identification and functional analysis of potential prophage-derived recombinases for genome editing in Lactobacillus casei. FEMS Microbiol. Lett 364, fnx243 (2017). [DOI] [PubMed] [Google Scholar]
- 196.Yang P, Wang J & Qi Q Prophage recombinases-mediated genome engineering in Lactobacillus plantarum. Microb. Cell Fact 14, 154 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Yin J et al. A new recombineering system for Photorhabdus and Xenorhabdus. Nucleic Acids Res 43, e36 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Wu Y et al. RecET recombination system driving chromosomal target gene replacement in Zymomonas mobilis. Electron. J. Biotechnol 30, 118–124 (2017). [Google Scholar]