

Abstract
Chemoradiotherapy with cisplatin and etoposide is a curative management regimen for both small and non-small cell lung cancers. While the treatment regimen is effective, it also has a high toxicity profile. One potential strategy to improve the therapeutic ratio of chemoradiation is to utilize nanotherapeutics. Nanoparticle formulation of cisplatin and etoposide, however, is challenging due to the significant mismatch in chemical properties of cisplatin and etoposide. Herein we report the formulation of a polymeric nanoparticle formulation of cisplatin and etoposide using a prodrug approach. We synthesized a hydrophobic platinum prodrug, which was then co-delivered with etoposide using a nanoparticle. Using mouse models of lung cancer, we demonstrated that dual-drug loaded nanoparticles are significantly more effective than small molecule chemotherapy in chemoradiotherapy. These results support further investigation of nanoparticle-based drug formulations of combination chemotherapies and the use of nanotherapeutics in chemoradiotherapy.
Statement of Significance
The treatment of lung cancer often involves a combination of chemotherapy and radiation. While it can be effective, it also has a high toxicity profile. Preferential delivery of chemotherapeutics to the tumor while avoiding normal tissue would improve efficacy and lower toxicity. While this is challenging with conventional drug delivery technologies, nanotechnology offers a unique opportunity. In this study, we have engineered nanoparticles that are loaded with combination chemotherapeutics and showed such nanotherapeutics are more effective and less toxic than free chemotherapeutics in chemoradiotherapy. Our work highlights the importance and potential of nanoformulations of combination chemotherapy in chemoradiotherapy and cancer treatment. This approach can be translated clinically and it can have a significant impact on cancer treatment.
Keywords: Nanomedicine, Nanoparticle, Lung cancer, Chemoradiotherapy, Combination drug delivery
1. Introduction
Lung cancer is the second most common type of cancer for both men and women, behind only prostate and breast cancer, respectively. But with an estimated 229,000 new cases and over 135,000 deaths in the US in 2020, lung cancer is the leading cause of cancer deaths in the US, attributing to more cancer-related deaths than breast and prostate cancer combined [1]. Two main subtypes of lung cancer include small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). One of the curative treatments for both SCLC and NSCLC is chemoradiotherapy (CRT) [2–4]. In these CRT regimens, cisplatin (CP) with etoposide (ET) drug combinations are commonly utilized with radiotherapy [5–13]. While effective, this treatment is also highly toxic [13–17]. Thus, there has been strong interest in further improving the CRT regimen.
One strategy to improve CRT is to utilize nanotherapeutics. Nanoparticles (NPs) have been shown to act as effective drug delivery systems (DDSs), improving toxicity profiles and treatment efficacy [18]. Dual-drug loaded NPs (DNPs) can offer synergistic drug delivery with improved efficacy over free drugs [19–22]. By engineering NPs to contain a payload with a controlled drug ratio, any additive or synergistic effects of the drugs can be maximized. These drug loaded NPs also result in decreased toxicity profiles and fewer side effects since a larger portion of the drug is delivered to the tumor. This occurs through preferential accumulation of NPs at the tumor due to the enhanced permeability and retention (EPR) effect [18]. Our research investigated NP formulations of CP and ET to improve the efficacy of this combined regimen while limiting off target effects when using this drug combination in conjunction with radiotherapy (XRT) against NSCLC.
The large differences in CP and ET’s aqueous/organic phase solubility and drug loading ratios make their co-delivery challenging. To overcome this solubility difference, we employed a proven method for the modification of CP with a fatty acid to make a more hydrophobic cisplatin prodrug (CPP) to allow co-loading with ET [23–27]. We chose a biocompatible poly(lactic-co-glycolic acid)–polyethylene glycol (PLGA–PEG) polymer NP as the DDS. Using multiple NSCLC cell lines, we assessed the benefit that NPs can provide CRT using a dual-drug CP plus ET combination through in vitro cytotoxicity and in vivo tumor inhibition (Fig. 1). We found that DNPs co-loaded with a precise ratio of CP plus ET improved CRT over both free drug combinations and mixed single drug loaded NPs (MNPs).
Fig. 1.
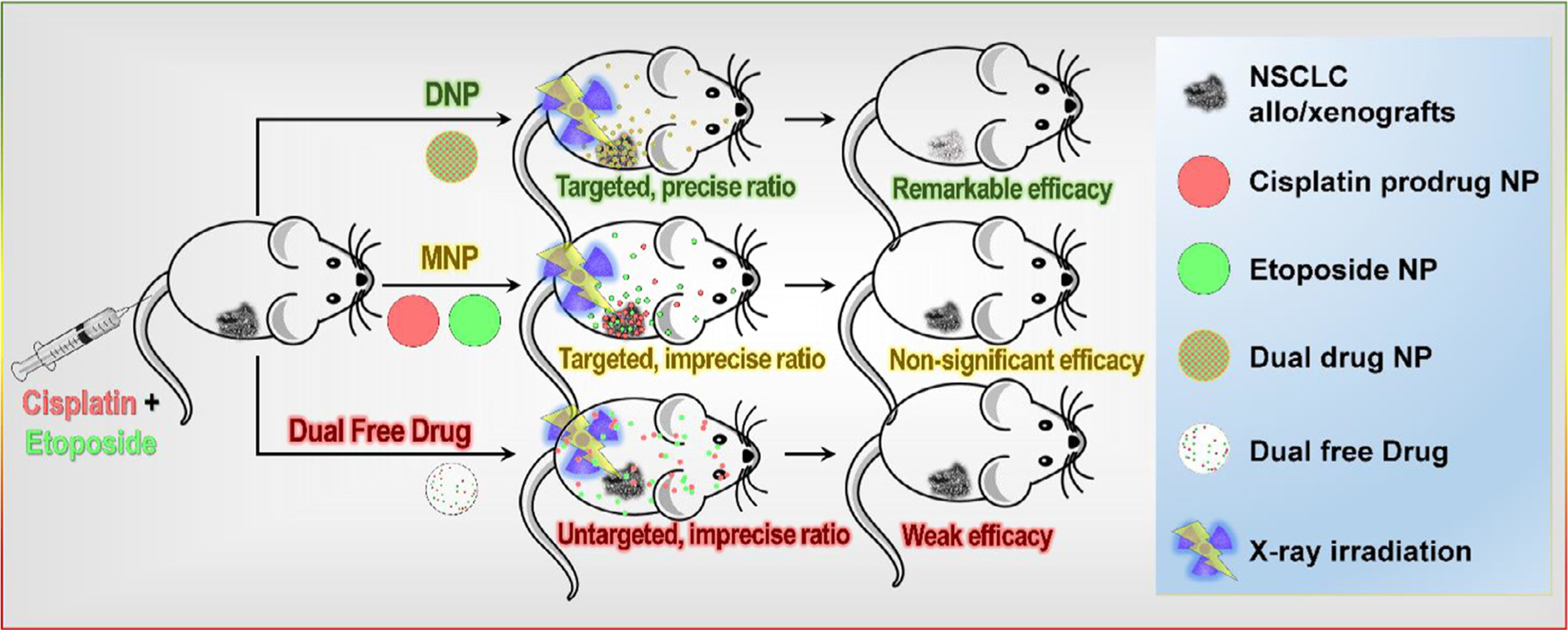
Co-delivery of CPP and ET using PLGA–PEG DNPs improves chemoradiotherapy. DNPs both preferentially accumulate at the tumor, and also deliver a precise drug ratio to tumor cells simultaneously through all NPs being loaded with the desired dual-drug ratio.
2. Materials and methods
2.1. Materials
ET was obtained from APExBIO. PEG-PLGA (3600–30,000 MW, 50:50 LA:GA) was acquired from PolySciTech. Poly-(vinyl alcohol) (6,000 MW, 80% hydrolyzed) was acquired from Polysciences, Inc. Cell culture reagents were purchased from Gibco by Life Technologies. CellTiter 96® AQueous MTS Reagent Powder was purchased from Promega. Anti-Cisplatin modified DNA Antibody was purchased from Abcam. Cleaved Caspase-3 (Asp175) Antibody and anti-rat IgG Alexafluor®555 Antibodies were purchased from Cell Signaling. Hydrogen peroxide was purchased from Fisher Chemical. TritonX100 was purchased from PerkinElmer Life and Analytical Sciences. Crystal Violet was purchased from Thermo Fisher Scientific. All other chemicals were acquired from Sigma Aldrich and used without further purification.
2.2. Cell culture
The non-small cell lung cancer cell lines A549 (ATCC® CCL-185TM) and H460 (ATCC® HTB-177TM) were obtained from American Type Culture Collection (ATCC) supplied by the Tissue Culture Facility at the UNC Lineberger Comprehensive Cancer Center. The 344SQ cell line was kindly provided by Professor Chad Pecot’s lab. Cells were cultured in a complete growth medium made of DMEM (A549) or RPMI-1640 (H460 & 344SQ) medium supplemented with 10% (v v−1) fetal bovine serum and 1% (v v−1) penicillin/streptomycin.
2.3. CPP synthesis
CPP was synthesized as previously described elsewhere [23–27]. Verification of CPP synthesis was determined by 1H NMR (Fig. S1) and mass spectrometry using positively charged electrospray with a measured m/z of 608.1587 and a 1.3 ppm error with a [M+Na]+ adduct ionization [28].
2.4. Animal maintenance
6–8 week old male nude mice weighing 27–32 g were supplied by the University of North Carolina animal facility and maintained under pathogen-free conditions in the Center for Experimental Animals (an AAALAC accredited experimental animal facility). The animal use protocol (15–301.0) was approved by the University of North Carolina Institutional Animal Care and Use Committee and conformed to the Guide for the Care and Use of Laboratory Animals [29].
2.5. Synthesis of NPs
NPs were synthesized via nanoprecipitation. Drug and polymer stock solutions were prepared in advance. ET was dissolved in acetonitrile (ACN) at a concentration of 2.5 mg mL−1. CPP was dissolved in acetone at a concentration of 2 mg mL−1. mPEG-PLGA (M.W. mPEG 3,600 & PLGA 30,000) was dissolved in ACN at a concentration of 20 mg mL−1. Drug stock solutions were combined at varying ratios as needed, with the drug solution (volume varies) then mixed with 100 μL of polymer stock solution at a 1:1 ratio. ACN was added to make a final drug-polymer solution of 500 μL. The solution was then added drop-wise into 0.5% poly-(vinyl alcohol) deionized water (2 mL) to form a NP suspension. To remove organic solvents, the suspension was evaporated for 3 h at room temperature under an air extraction arm while stirring at 600 rpm. Next, the suspension was concentrated by ultra-centrifugation using an Amicon Ultra-4 filter (MWCO-100 kDa) at 1000 g for 15 min (EMD Millipore, Billerica, MA, USA). The concentrated NP suspension was then washed with deionized water, concentrated twice, and finally suspended in PBS.
2.6. Characterization of NPs
Size and zeta potential were determined by dynamic light scattering (DLS) (Malvern Instruments Ltd, Worcestershire, UK) with a concentrated NP suspension (10 μL) added to a cuvette with deionized water (990 μL). All tests were performed in triplicate. Transmission electron microscopy (TEM) images were obtained via a JEOL JEM 1230 TEM. Prior to imaging, concentrated NP suspensions were diluted 100x with deionized water then loaded on a 400-mesh carbon film copper grid for 1 min to adsorb. The grid was negatively stained with 2% potassium phosphotungstate (pH 7.0) for 1 min. Excess stain was removed by capillary action by touching the edge of the grid with filter paper. The grid was air dried before loading into the TEM. The stability of DNP size was evaluated under varying biological conditions including deionized water, pH 7.4 PBS, pH 6.8 PBS, DMEM medium, and DMEM medium with 10% v/v normal human serum. All tests were performed in triplicate. DNP size was first measured to establish a baseline, then 2 mg of DNPs were added to 10 mL of each solution and measured immediately after mixing, then incubated at 37 °C for 24 h with aliquots removed and measured at 1.5, 6, and 24 h. While our other NP characterization studies herein relied on reported average NP size, the addition of serum included significant measurements of particles below 40 nm in scans performed without NPs but serum only. For this reason, we instead evaluated stability based on the NP distribution peak size.
2.7. Determination of drug loading in NPs
To determine drug loading, NP samples (40 μL) were mixed with ACN (200 μL), vortexed, and sonicated for 10 min to completely break down the NP and release all drug. The treated samples were loaded and measured via a Shimadzu SPD-M20A high performance liquid chromatography (HPLC) equipped with a diode array detector and a Chromolith Fast Gradient RP-18e 50 × 2mm column (EMD. Millipore, Billerica, MA, USA). Samples (10 μL) were injected into the HPLC and eluted using a binary solvent system (phase A and B: A for Water, B for ACN) at a flow rate of 0.25 mL min−1. The linear gradient program was set as follows: 0 to 20 min, 0% to 100% B; 20 to 25 min, 100% B; 25 to 30 min, 50% B; 30 to 35 min, 0% B. The column and sample temperature were maintained at 30 °C and 4 °C respectively during determination. ET (retention time 7.5 min) and CPP (retention time 12.4 min) were monitored at 220 nm.
2.8. In vitro drug release
In vitro drug-release profiles of drug loaded NPs were acquired under sink condition. NP suspensions (composed of 0.5 mL concentrated NP suspension with 1 mL deionized water) were split into aliquots (50 μL) and added into Slide-A-Lyzer MINI dialysis microtubes (MWCO 20 kDa, Pierce, Rockford, IL, USA). These microtubes were then dialyzed in PBS (4 L) under gentle stirring at 37 °C. NP samples (40 μL) were collected at the indicated times and measured by HPLC as previously described. Percentage of drug released was calculated by (1 − Ct/C0) × 100%, where C0 and Ct represent the remaining drug concentration at time 0 and t respectively.
2.9. In vitro cytotoxicity
Cells were plated in 96-well plates (3600 cells well−1) and allowed to recover overnight. Cells were then washed with PBS and treated with varying concentrations and ratios of free drugs or drug encapsulated NPs at 37 °C for 2 h. Cells were then washed with PBS 3 times and allowed to grow in complete cell culture medium for 72 h. After incubation, cell viability was analyzed by an MTS assay per vendors instructions (Promega). Absorbance was recorded at 492 nm via a microtiter plate reader (Molecular Devices Corporation, California, USA). The IC50 values of different treatments were calculated by fitting the dose-dependent cell viabilities to a four-parameter logistic model using the MasterPlex 2010 software pack (MiraiBio Group, Hitachi Solutions America, Ltd.).
2.10. Clonogenic survival assay
Cells (200, 300, 400, 800, 2000, and 5000 cells well−1) were seeded in 6-well plates and incubated in complete cell culture medium for 2 h for attachment. Cells were then washed with PBS and treated with either PBS, free drug, MNPs, or DNPs for 2 h (drugs were added at their IC20 dose, NPs at a 1:1.8 ET:CP/CPP ratio). Cells were washed with PBS three times and complete cell culture medium was added. Cells in 6-well plates were irradiated (X-RAD 320, Precision X-ray Inc., North Branford, CT, USA) according to the number of cells seeded (1, 2, 4, 6, and 8 Gy for 300, 400, 800, 2000, and 5000 cells well−1 respectively), with the 200 cells well−1 plate receiving no irradiation. The irradiated cells were allowed to grow for 14 days to generate colonies. 6-well plates with colonies were washed with PBS three times, fixed and stained with a mixture of 10% formalin and 0.5% crystal violet for 1 h, gently rinsed with tap water, and then dried in normal air at room temperature. All colonies with over 50 cells were counted. Plating efficiency was calculated via the number of colonies formed divided by the number of cells seeded. Surviving fraction (SF) of different irradiation doses was calculated by plating efficiency of irradiation treatment divided by that of non-irradiation treatment. D0 and N were acquired by fitting the calculated SF to the single-hit multitarget model [SF = 1 − (1 − e−D/D0)N, where D represents the irradiation dose, D0 the average lethal dose, and N the extrapolation number] using GraphPad Prism version 6.01 for Windows (GraphPad Software, La Jolla California USA). Sensitivity enhancement ratio (SER) was calculated using D0 of untreated divided by that of drug treated cells.
2.11. In vivo anticancer efficacy
Murine tumor models were formed by injecting 800,000 cells (0.8 × 106 cells suspended in 0.2 mL of 50% RPMI-1640 medium and 50% Matrigel® v v−1) into the right flank. Tumors were allowed to grow to 80–150 mm3 before initiating treatment. Mice were divided into six groups with six mice per group. All mice received 200 μL tail vein injections on day 0 with either (1) PBS, (2) mixture of free ET (1.25 mg kg−1) and CP (1.16 mg kg−1), (3) PBS, (4) mixture of free ET (1.25 mg kg−1) and CP (1.16 mg kg−1), (5) MNPs (1.25 mg kg−1 ET SNP and 2.25 mg kg−1 CPP SNP), or (6) DNPs (ET 1.25 mg kg−1 and CPP 2.25 mg kg−1). NP drug doses listed denote drug dose, not including weight of polymer in NP. CP dosing is based on low-dose CP [30,31], CPP dosing is the molecular weight ratio of CPP/CP, and ET dosing is at the optimally identified ET:CPP synergistic molar ratio of 1:1.8 as identified in previous experiments. (Mice in groups 3, 4, 5, and 6 received X-ray irradiation (5 Gy) on day 0 (3 h after injection), day 1, and day 2. Tumor length (L) and width (W) were measured, and tumor volume was calculated using: (L*W2)/2, with L ≥ W. Body weight and tumor volume were measured and recorded on day 0, day 1, and every other day after day 1. Mice were humanely euthanized using CO2 inhalation once tumor dimensions were larger than 2 cm in any direction, if body weight loss exceeded 20%, or if the University of North Carolina core veterinarians determined significant deterioration in general health.
2.12. In vivo hematological toxicity, hepatotoxicity and nephrotoxicity assay
In vivo toxicity was determined by sacrificing two randomly selected mice from each group on day 3. Circulating blood (0.5 mL) was collected via cardiac puncture. For hematological toxicity, whole-blood (0.5 mL) was stored in a heparin pre-treated stopper covered tube at 4 °C and analyzed for white and red blood cell counts. For hepatotoxicity and nephrotoxicity, whole-blood (1 mL) was transferred to an eppendorf tube (2 mL) and centrifuged at 900 g for 10 min to separate the plasma. The whole-blood and the isolated plasma was analyzed by the Department of Pathology & Laboratory Medicine, Animal Histopathology & Laboratory Medicine Core, University of North Carolina, for blood cell counts, aspartate aminotransferase (AST), blood urea nitrogen (BUN), and creatinine (Crea) levels.
2.13. Immunofluorescent imaging
Tumors were excised from one euthanized mouse in each arm on day 3 after initial treatment. The tissue was washed with PBS, fixed in formalin (10% v v−1), dehydrated, and then embedded in paraffin. Blank paraffin slides were hydrated and then microwaved twice for 10 min in sodium citrate solution (0.01 μm, pH 6.0). The slides were cooled to room temperature and washed with PBS. The slides were then treated with a PBS solution containing hydrogen peroxide (3% v v−1) and Triton X-100 (0.5% w v−1) and subsequently washed three times with PBS.
Tumor sections were mounted and then blocked with bovine serum albumin (BSA, 10% w v−1) for 1 h. The slides were treated with 1:400 PBS diluted anti-cisplatin modified DNA antibody (Abcam, ab103261) or cleaved caspase-3 (Asp175) antibody (Cell Signaling, 9661S) overnight at 4 °C. Secondary antibody was then added with Anti-rat IgG (H+L) (Alexa Fluor® 555 Conjugate, Cell Signaling, 4417S) for anti-cisplatin modified DNA antibody or F(ab’)2-goat anti-rabbit IgG (H+L) cross-adsorbed secondary antibody (Alexa Fluor 594, Thermo Fisher Scientific, A11072) for anti-cleaved caspase-3 (Asp175) antibody. After 1 h, slides were washed with PBS (0.2% v v−1, Tween 20) and sections were treated with DAPI (1 μg mL−1, Thermo Fisher Scientific, D1306) for 20 min. Slides were washed with PBS and then sealed with fluoromount™ aqueous mounting medium (Sigma-Aldrich, F4680–25ML) and a cover glass. Immunofluorescence images were obtained via a laser scanning confocal microscope (ZEISS, LSM 700).
2.14. Statistical methods
All experiments were performed at least three times and expressed as mean ± SD for in vitro (n = 3) or mean ± SEM for in vivo (n = 6) studies. Statistical significance was determined using two-tailed Student’s t-test except in the clonogenic study where the SERs were compared using a one-way analysis of variance (ANOVA) followed by a Tukey test and the in vivo efficacy study in which a single-tailed Student’s t-test was employed. Differences were considered significant when P < .05. All statistical analyses were completed in GraphPad Prism version 6.01 for Windows.
3. Results
3.1. Drug loading and release kinetics in vitro
Nanoprecipitation was used to formulate PEG-PLGA NPs loaded with ET and/or CPP. ET and CPP are encapsulated inside the hydrophobic NP core during nanoprecipitation. CP will not readily load into PLGA NPs so CPP was used instead to improve encapsulation. CPP can be reduced back to CP by tumor overexpressed intracellular mercaptans such as glutathione or ascorbate [32].
We examined varying drug feed ratios (wt% of drug vs polymer) (CPP 2.5–12.5%, ET 7.5–50%) to determine the optimal NP drug loading and loading ratio between ET and CPP. Encapsulation efficiency (wt% of drug encapsulated in NP vs initial feed amount) and drug loading wt% (wt% of drug vs total NP) were characterized for each formulation, first in SNPs, then in DNPs (Fig. 2, Table S1, and Table S2). In SNPs, CPP loading increased with feed ratio, reaching a maximum drug loading of 7.1 ± 0.2% at a feed ratio of 12.5%. ET SNP drug loading followed a similar trend, increasing with feed ratio and reaching a maximum drug loading of 6.8 ± 0.2% at a feed ratio of 50%. ET feed ratio was held at 25% for all further DNP studies to obtain significant ET drug loading. CPP and ET were then co-loaded to form DNPs with the CPP feed ratio ranging from 2.5–12.5%. The resulting DNP loadings illustrated in Fig. 2c and d allowed us to synthesize DNPs with ET:CPP molar ratios from 2.5:1 to 1:5. These formulations were used for in vitro efficacy studies to identify the most effective formulation.
Fig. 2.
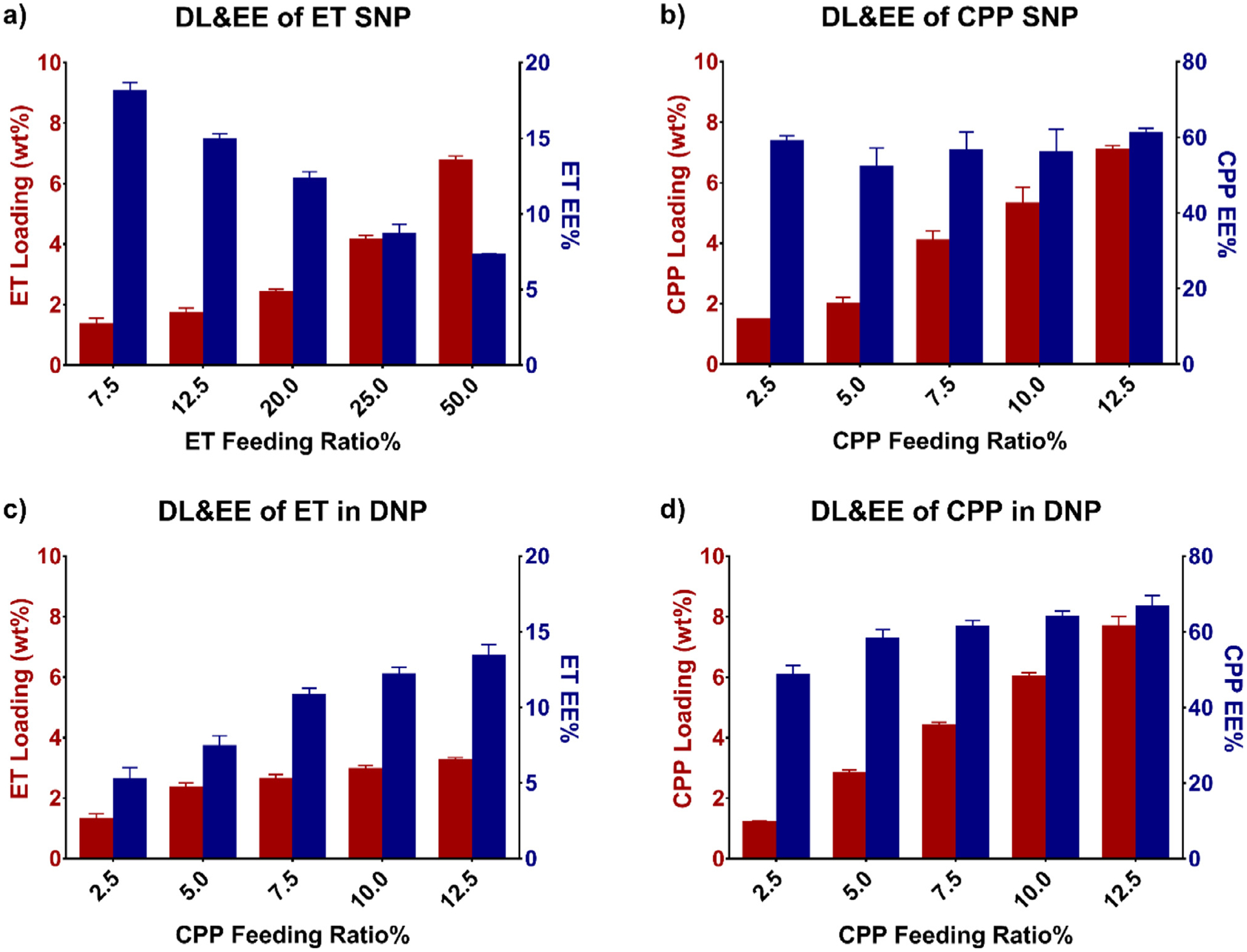
Encapsulation efficiency (EE) and drug loading wt% (DL) of a) ET SNP, b) CPP SNP, c) ET in DNPs, and d) CPP in DNPs. a) ET encapsulation efficiency decreased, but total drug loading increased as the feed ratio was increased. b) While CPP drug loading increased with feed ratio, encapsulation efficiency remained consistent throughout. In c) and d) the ET feed ratio was kept constant at 25% in DNPs and the CPP feed ratio was varied. A CPP feed ratio of 7.5% produced the loaded 1:1.8 ET:CPP molar ratios used in all further studies in vivo.
NP size, polydispersity index (PDI), and zeta potential were also measured at each feed ratio by TEM and DLS (Fig. S2, Fig. S3, Table S3, and Table S4). ET SNPs were smaller (~70 nm) than those of CPP SNPs (~100 nm) and DNPs (~100 nm) at all feed ratios. All ET SNPs maintained a PDI below 0.1, while the PDI of CPP SNPs and DNPs increased with CPP feed ratios of 7.5% or greater. Final formulations kept the CPP feed ratio below 7.5% to keep particle size uniform for all NPs. DLS sizes were verified with TEM analysis of particle morphology, which revealed approximately spherical particles ranging from 60 to 150 nm (Fig. S3). Zeta potentials of all three NP formulations were measured, ranging from −15 to −35 mV.
Stability of DNPs was measured in varying biological media through sequential NP size measurements (Fig. S4). DNP size varied minimally after synthesis (~70nm) through 24 h of incubation with deionized water, normal PBS at pH 7.4, acidic PBS at pH 6.8, DMEM with no serum added, or DMEM with 10% v/v serum added. These studies showed that DNPs’ size was stable for 24 h in these conditions. This data is consistent with previous reports [33].
Drug release kinetics of SNPs and DNPs were evaluated under physiologic sink conditions (Fig. S5). ET released more quickly than CPP in all NP formulations, and in DNPs both drugs released more slowly than their SNP counterparts (CPP SNP t1/2 = 7.2 h vs DNP t1/2 = 11.1 h, ET SNP t1/2 = 2.4 h vs DNP t1/2 = 3.1 h).
3.2. In vitro cytotoxicity
Three NSCLC cell lines: A549, H460, and 344SQ, were used for in vitro studies. IC50 values of cell lines treated with single or dual free drugs, SNPs, MNPs, or DNPs are presented in Fig. 3a and Table S5. In all cell lines, CPP and ET loaded NPs were more potent than their free drug counterparts (Fig. 3a). The IC50 drug concentration of each cell line for CPP SNPs were 0.86, 0.33, and 0.31 μm respectively, and the ET SNPs were 78, 5.5, and 6.0 μm respectively. Due to the 13-fold higher ET SNP IC50 concentration for A549, this cell line was excluded from all subsequent in vitro and in vivo work as this concentration level is not feasible for in vivo studies.
Fig. 3.
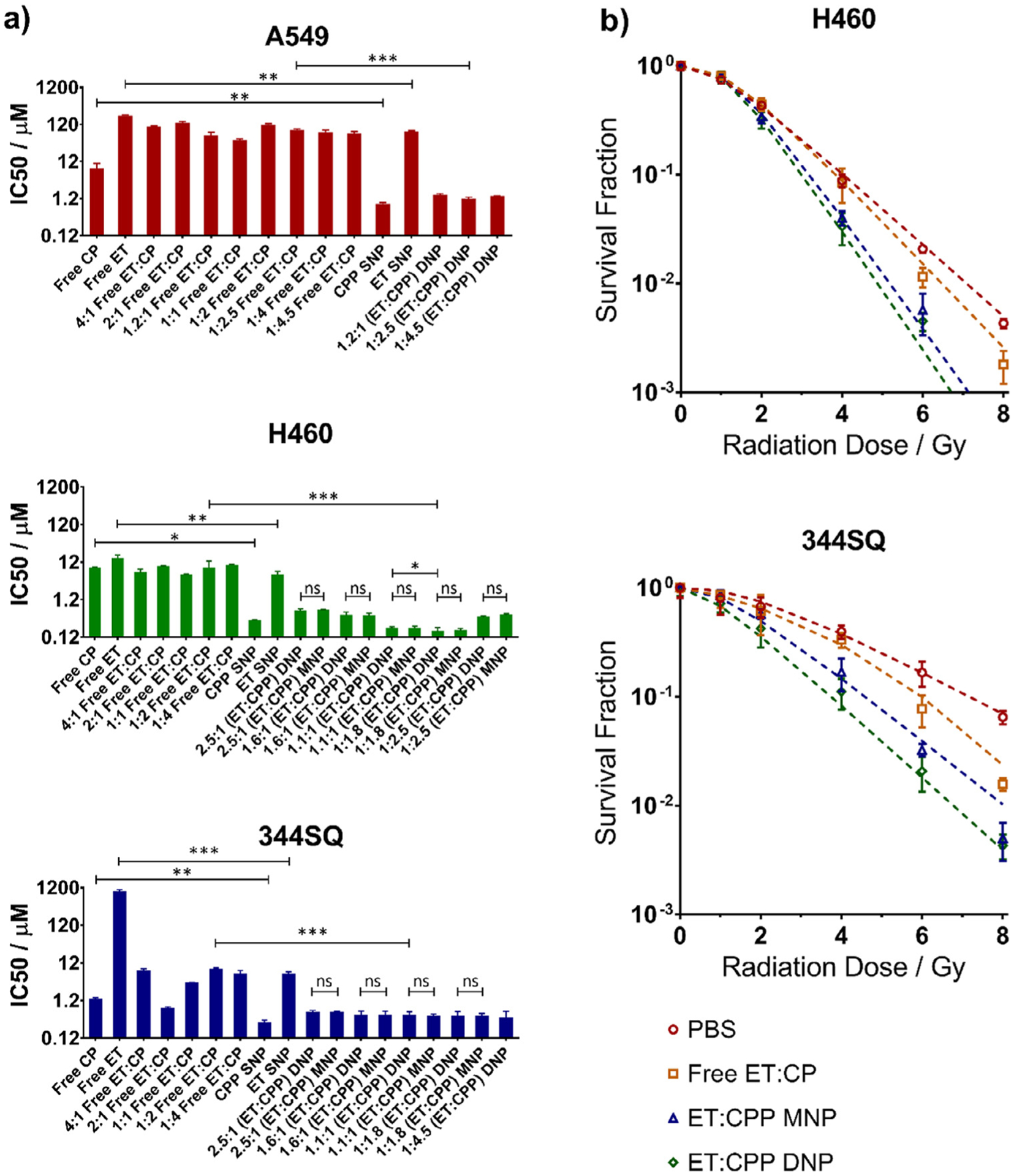
a) In vitro cytotoxicity of varying ET:CPP formulations including individual free drugs, dual free drugs, MNPs, and DNPs showing the most significant improvement for NPs at an ET:CPP ratio of 1:1.8 in H460 cells. b) Clonogenic assay of PBS only treatment, dual free drugs, MNPs, and DNPs resulting in modestly improved SERs for MNPs and DNPs versus dual free drugs in the 344SQ cell lines, and significantly improved SERs in the H460 cell line. No significant difference in SER was determined between MNPs and DNPs. Cells were treated with the IC20 dose of their respective drug combination 2 h prior to radiation treatment with all dual-drug treatments at the ET:CPP ratio of 1:1.8. (*P < 0.05, **P < 0.01, ***P < 0.001, ns = not significant).
For each cell line, we tested multiple mixed drug ratios in addition to the individual free drugs. Free drug ET:CP ratios were varied from 4:1 to 1:4 in each cell line, and NP formulations were tested both as MNPs and DNPs with ET:CPP ratios ranging from 2.5:1 to 1:4.5. IC50 values of all DNP drug ratios in the A549 and 344SQ cell lines were slightly larger than CPP SNPs (1.4- to 1.7-fold for A549, 1.3- to 1.9-fold for 344SQ), but significantly lower than ET SNPs (51- to 65-fold for A549, 10- to 15-fold for 344SQ). Differences in IC50 values between DNPs and MNPs at each drug ratio in the H460 and 344SQ cell lines were not significant. While there was no significant difference between the 1:1.8 ET:CPP loaded DNP and any other DNP group in the 344SQ cell line, the IC50 level minimally decreased as CPP loading increased, suggesting additive effects in this cell line. The lowest IC50 from any group was the DNP with an ET:CPP ratio of 1:1.8 in the H460 cell line. This was significantly lower than the IC50 of CPP SNPs and ET SNPs (2- and 32-fold IC50 improvements, with P < .01 and P < .001 respectively) but was not significantly lower than its 1:1.8 MNP counterpart. Additionally, the combination indexes of these DNPs in H460 and 344SQ cell lines were found to be 0.44 and 1.06 at ED50 respectively, suggesting synergistic or at least additive effects of DNPs at this ratio (Table S6) [34]. Due to these synergistic effects and the significantly enhanced cytotoxicity as evidenced by IC50 values at the 1:1.8 ET:CPP ratio, we chose to use this DNP drug ratio for all further experiments.
Typical first line therapy for locally advanced NSCLC consists of regular doses of 1.4 mg kg−1 of both CP and ET in an average 70 kg, 1.9 m2 person [9–11,35,36]. Our treatments were purposefully kept below this level in order to better monitor the benefits of one treatment arm over another. We used a single treatment of 1.25 mg kg−1 ET and 1.16 mg kg−1 CP as this was the optimal ratio we found in our in vitro work.
To examine the effects of these NPs with radiotherapy, they were studied using clonogenic survival assays (Fig. 3b). The sensitivity enhancement ratio (SER), a measure of radiosensitizer potency, was calculated for all treatment arms in the H460 and 344SQ cell lines (Fig. S9). These cell lines were each treated with dual free drugs or NP formulations and then exposed to increasing doses of radiation. IC20 drug levels were used in favor of higher IC levels to prevent complete cell death when coupled with radiotherapy, allowing increased detection of discernible differences between treatment arms. In the 344SQ cell line, the free drug SER was similar to PBS. The SER of both MNPs and DNPs was much better than PBS (1.6 ± 0.1 and 1.6 ± 0.2 vs 1.0 ± 0.3) (P < .01), but only modestly better than free drugs (1.1 ± 0.5) (P = .047 and P = .055) In the H460 cell line the free drug SER compared to PBS was significantly improved (1.3 ± 0.1 versus 1.0 ± 0.1) (P < .05), as was the SER of MNPs and DNPs (1.57 ± 0.07 and 1.65 ± 0.07) compared to both PBS and free drugs (P < .05 MNP vs free drug and P < .01 DNP vs free drug). While neither cell line showed a significant difference in SER between MNPs and DNPs, these values indicate radiosensitization from free drugs in the H460 cell line and NP formulations in both cell lines.
3.3. In vivo efficacy
We then evaluated the efficacy of our NP DDS in combination with XRT in murine models using both the H460 xenograft and 344SQ allograft. Mice were either in a PBS control group or one of five treatment arms including dual free drugs, PBS+XRT, dual free drugs+XRT, MNPs+XRT, or DNPs+XRT. Drug dose, drug ratio (ET:CP/CPP = 1:1.8), and fractionated irradiation doses (5 Gy × 3d) were kept the same in applicable treatments. As shown in the tumor growth curves (Fig. 4), all drug loaded NPs significantly reduced tumor growth in both cell lines when compared to PBS only. The MNPs also significantly reduced tumor growth compared to free drugs with XRT in the 344SQ model (P < .05), but not in the H460 model. DNPs provided even greater reductions in tumor growth, significantly so when compared to free drugs in both the H460 and 344SQ models (P < .05 and P < .01). When compared to MNPs, the DNPs again provided significant tumor growth reductions in both cell lines (P < .05 in both).
Fig. 4.
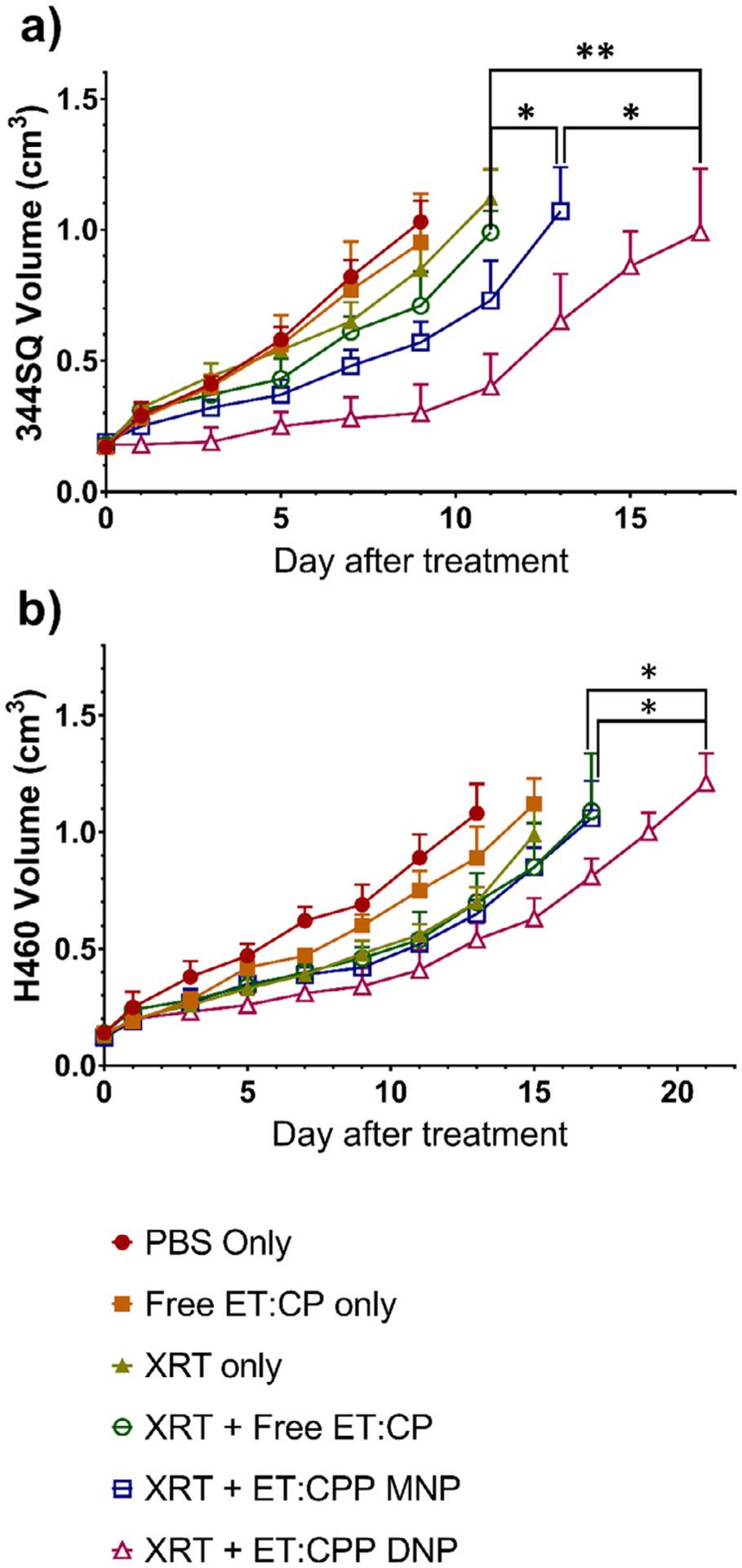
In vivo efficacy of NPs in a) 344SQ and b) H460 models showing significant tumor growth inhibition from MNPs versus dual free drugs in the 344SQ model, and from DNPs versus both MNPs and dual free drugs in both models. Mice were treated once tumor volume reached 80–150 mm3. Data are represented as tumor volume (cm3) after treatment (*P < 0.05, **P < 0.01).
Tumor histology was analyzed for both cleaved caspase 3, a marker for early-stage apoptosis, and CP-DNA complexes, indicative of increased CP delivery (Fig. S8a, c, e, and g). Tissue sections from NP treatment arms (MNP and DNP) depicted a significant increase in CP-DNA adduct formation versus dual free drugs (Fig. S8b and f). This indicated increased delivery of drug to tumor tissue by NPs resulting in less systemic accumulation, and improved localization of CP to the nucleus when using NP delivery vehicles. Increases in the intensity of the apoptosis marker cleaved caspase 3 reflect the enhanced killing of tumor cells by XRT combined with our NP regimen (Fig. S8d and h).
While NPs can increase delivery to tumors by way of the EPR effect, there will also be significant distribution to clearance organs such as the liver and kidneys [37–41]. We examined if our nanoformulations caused significant toxicity to those off-target organs. Despite the potent efficacy in vivo of our DNP+XRT regimen, nephrotoxicity and hepatotoxicity in our PLGA-PEG formulated drug combinations were found to be low based on liver and kidney function indicators including aspartate aminotransferase (AST), blood urea nitrogen (BUN), and creatinine (Crea) (Table S7 and S8). Additionally, the WBC of NP groups remained within the range of other IV treatment groups. While there was some loss of body weight following XRT, animals recovered to normal levels within approximately one week (Fig. S6 and S7).
4. Discussion
Combination CRT with CP plus ET is one of the primary treatment methods for SCLC and unresectable locally advanced NSCLC due to its superior efficacy over alternative regimens such as carboplatin with paclitaxel, despite CP plus ET’s higher toxicity. As this combination has been previously shown to be synergistic [42], we developed a biocompatible PLGA-PEG delivery vehicle which could be loaded at varying drug ratios to improve the efficacy of CRT with CP plus ET. All of our NP formulations showed improved dose response curves and IC50 values in vitro over free drugs with no significant difference in IC50 values between MNPs and DNPs implying consistent cellular uptake of all NPs. DNPs, however, showed significantly greater efficacy in vivo than MNPs and free drugs, indicating that only DNPs were able to continue to deliver both drugs at the precise synergistic ratio. This results from varying systemic distribution of each free drug or individually loaded NP in the MNP combination, causing a matching variation in drug ratio in tissues and tumor due to differences in characteristics such as size and surface properties (Fig. S2, Table S3). By maintaining a precise ratio of drugs that are delivered simultaneously through co-loading, our DNPs allow both drugs to act synergistically with no change in drug mechanism. While this type of synergy has been demonstrated in other studies with alternate drug combinations [19–22], our study demonstrated in vivo efficacy with a single treatment administration when others have required up to three sequential administrations. Moreover, this regimen also allowed lower CPP dosing when coupled with ET. Alternative future approaches for dual drug delivery could include incorporation of drugs loaded into gold microplates as these have been shown to offer significant drug loading [43].
In addition to improving efficacy through controlled loading ratios, low systemic toxicity was observed, similar to other studies where the observed tissue toxicity of PLGA based NPs in brain, heart, spleen, thymus, liver, kidney, and lung tissue has been shown to be no greater than typical incidental findings and that PLGA NPs are biodegradable non-toxic particles [44,45]. NPs have also been shown to reduce hepatotoxicity of NP delivered drugs due to macrophage uptake of NPs [46].
There have been significant recent advances in nanomedicine approaches including organic/inorganic hybrid NPs, artificial intelligence/machine learning to model nanomedicine, and dual drug delivery through NPs [43,47,48]. The success of precisely delivered ratios of dual drugs through NPs has also been seen in the clinic through the liposomal formulation of daunorubicin and cytarabine in VYXEOS, where a fixed 1:5 molar ratio provided synergistic treatment benefits/improved survival in acute myeloid leukemia [49]. While there have been a number of nanotherapeutics approved by the FDA for clinical use, this liposomal formulation is the only dual-drug loaded nanotherapeutic to date and the only nanotherapeutic that has shown improved efficacy/survival over its small molecule counterpart in the clinical setting [50,51]. Additionally, as acute myeloid leukemia is a cancer of the blood and bone marrow, VYXEOS does not benefit from the EPR effect. Clinical data from VYXEOS suggest that dual drug delivery can be a key strategy to improve therapeutic efficacy of nanotherapeutics. It is important to note that many other groups, including ourselves, have reported dual-drug NPs and have shown these agents are highly effective in the preclinical setting. However, there has been a lack of studies examining the use of dual-drug NPs in the chemoradiotherapy setting. Our report on NP ET/CPP in chemoradiation fills this gap and further enhances the clinical translation potential of dual-drug nanotherapeutics. As seen in our results, specific drug ratio at the tumor cells is critical to obtain synergy [21,22].
5. Conclusion
Chemoradiotherapy is a key treatment for lung cancer. Our study has demonstrated a novel strategy to further improve the therapeutic ratio of CRT for lung cancer. By engineering a nanotherapeutic that contains both CPP and ET, we increased the therapeutic efficacy of CRT in two murine lung cancer models. Further-more, we showed the treatment did not induce additional toxicity when compared to a standard CRT regimen with small molecule drugs. Our DNP highlights the potential of dual drug delivery in nanomedicine.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health/National Cancer Institute (R01CA178748-01, and U54CA198999 for Carolina Center of Cancer Nanotechnology Excellence (CCNE)-Nano Approaches to Modulate Host Cell Response for Cancer Therapy). Zachary Rodgers was supported by the Carolina Cancer Nanotechnology T32 Training Program (C-CNTP, NIH-1T32CA196589), C. Tilden Hagan IV was supported by the National Institute of Health Medical Scientist Training Program (T32 GM008719), and Maofan Zhang was supported by the CMU promising young teacher supportive plan (QGZ2018085).
Abbreviations:
- ACN
acetonitrile
- AST
aspartate aminotransferase
- BUN
blood urea nitrogen
- CP
cisplatin
- CPP
cisplatin prodrug
- Crea
creatinine
- CRT
chemoradiotherapy
- DDS
drug delivery system
- DL
drug loading
- DLS
dynamic light scattering
- DNP
dual-drug loaded nanoparticle
- EE
encapsulation efficiency
- EPR
enhanced permeability and retention
- ET
etoposide
- FR
feed ratio
- MNP
mixed single drug loaded nanoparticle
- NP
nanoparticle
- NSCLC
non-small cell lung cancer
- PDI
polydispersity index
- PLGA-PEG
poly(lactic-co-glycolic acid)-polyethylene glycol
- SCLC
small cell lung cancer
- SER
sensitivity enhancement ratio
- SNP
single drug loaded nanoparticle
- TEM
transmission electron microscopy
- XRT
radiotherapy
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
AZW is cofounder of Capio Biosciences and Archimmune Therapeutics. Neither is relevant to this work.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.actbio.2021.02.001.
References
- [1].Siegel RL, Miller KD, Jemal A, Cancer statistics, 2020, CA Cancer J. Clin 70 (2020) 7–30, doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- [2].Urvay SE, Yucel B, Erdis E, Turan N, Prognostic factors in stage III non-small-cell lung cancer patients, Asian Pac. J. Cancer Prev 17 (2016) 4693, doi: 10.22034/APJCP.2016.17.10.4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Movsas B, Scott C, Sause W, Byhardt R, Komaki R, Cox J, Johnson D, Lawton C, Dar AR, Wasserman T, Roach M, Lee JS, Andras E, The benefit of treatment intensification is age and histology-dependent in patients with locally advanced non-small cell lung cancer (NSCLC): a quality-adjusted survival analysis of radiation therapy oncology group (RTOG) chemoradiation studies, Int. J. Radiat. Oncol. Biol. Phys 45 (1999) 1143–1149, doi: 10.1016/S0360-3016(99)00325-9. [DOI] [PubMed] [Google Scholar]
- [4].Pignon JP, Tribodet H, v Scagliotti G, Douillard JY, Shepherd FA, Stephens RJ, Dunant A, Torri V, Rosell R, Seymour L, Spiro SG, Rolland E, Fossati R, Aubert D, Ding K, Waller D, le Chevalier T, Group LC, Lung adjuvant cisplatin evaluation: a pooled analysis by the LACE collaborative group, J. Clin. Oncol 26 (2008) 3552–3559, doi: 10.1200/JCO.2007.13.9030. [DOI] [PubMed] [Google Scholar]
- [5].Arriagada R, Kramar A, le Chevalier T, de Cremoux H, Competing Events determining relapse-free survival in limited small-cell lung carcinoma, J. Clin. Oncol 10 (1992) 447–451, doi: 10.1200/JCO.1992.10.3.447. [DOI] [PubMed] [Google Scholar]
- [6].Chan BA, Coward JIG, Chemotherapy advances in small-cell lung cancer, J. Thoracic Dis 5 (2013) S565, doi: 10.3978/j.issn.2072-1439.2013.07.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Turrisi AT, Kim K, Blum R, Sause WT, Livingston RB, Komaki R, Wagner H, Aisner S, Johnson DH, Twice-daily compared with once-daily thoracic radiotherapy in limited small-cell lung cancer treated concurrently with cisplatin and etoposide, N. Engl. J. Med 340 (1999) 265–271, doi: 10.1056/NEJM199901283400403. [DOI] [PubMed] [Google Scholar]
- [8].Takada M, Fukuoka M, Kawahara M, Sugiura T, Yokoyama A, Yokota S, Nishiwaki Y, Watanabe K, Noda K, Tamura T, Fukuda H, Saijo N, Phase III study of concurrent versus sequential thoracic radiotherapy in combination with cisplatin and etoposide for limited-stage small-cell lung cancer: results of the Japan Clinical Oncology Group Study 9104, J. Clin. Oncol 20 (2002) 3054–3060, doi: 10.1200/JCO.2002.12.071. [DOI] [PubMed] [Google Scholar]
- [9].Rusch VW, Giroux DJ, Kraut MJ, Crowley J, Hazuka M, Winton T, Johnson DH, Shulman L, Shepherd F, Deschamps C, Livingston RB, Gandara D, Induction chemoradiation and surgical resection for superior sulcus non-small-cell lung carcinomas: long-term results of Southwest Oncology Group trial 9416 (Intergroup trial 0160), J. Clin. Oncol 25 (2007) 313–318, doi: 10.1200/JCO.2006.08.2826. [DOI] [PubMed] [Google Scholar]
- [10].Hanna N, Neubauer M, Yiannoutsos C, McGarry R, Arseneau J, Ansari R, Reynolds C, Govindan R, Melnyk A, Fisher W, Richards D, Bruetman D, Anderson T, Chowhan N, Nattam S, Mantravadi P, Johnson C, Breen T, White A, Einhorn L, Phase III study of cisplatin, etoposide, and concurrent chest radiation with or without consolidation docetaxel in patients with inoperable stage III non-small-cell lung cancer: the Hoosier Oncology Group and U.S. Oncology, J. Clin. Oncol 26 (2008) 5755–5760, doi: 10.1200/JCO.2008.17.7840. [DOI] [PubMed] [Google Scholar]
- [11].Albain KS, Crowley JJ, Turrisi AT, Gandara DR, Farrar WB, Clark JI, Beasley KR, Livingston RB, Concurrent cisplatin, etoposide, and chest radiotherapy in pathologic stage IIIB non-small-cell lung cancer: a Southwest oncology group phase II study, SWOG 9019, J. Clin. Oncol 20 (2002) 3454–3460, doi: 10.1200/JCO.2002.03.055. [DOI] [PubMed] [Google Scholar]
- [12].Wakelee H, Kelly K, Edelman MJ, 50 Years of progress in the systemic therapy of non-small cell lung cancer, Am. Soc. Clin. Oncol. Educ. Book (2014) 177–189, doi: 10.14694/EdBook_AM.2014.34.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tam K, Daly M, Kelly K, Treatment of locally advanced non-small cell lung cancer, Hematol. Oncol. Clin. North Am 31 (2017) 45–57, doi: 10.1016/j.hoc.2016.08.009. [DOI] [PubMed] [Google Scholar]
- [14].Muscato JJ, Cirrincione C, Clamon G, Perry MC, Omura G, Berkowitz I, Reid T, Herndon JE, Green MR, Etoposide (VP-16) and cisplatin at maximum tolerated dose in non-small cell lung carcinoma: a Cancer and Leukemia Group B study, Lung Cancer 13 (1995) 285–294, doi: 10.1016/0169-5002(95)00501-3. [DOI] [PubMed] [Google Scholar]
- [15].Rossi A, di Maio M, Chiodini P, Rudd RM, Okamoto H, Skarlos DV, Früh M, Qian W, Tamura T, Samantas E, Shibata T, Perrone F, Gallo C, Gridelli C, Martelli O, Lee SM, Carboplatinor cisplatin-based chemotherapy in first-line treatment of small-cell lung cancer: the COCIS meta-analysis of individual patient data, J. Clin. Oncol 30 (2012) 1692–1698, doi: 10.1200/JCO.2011.40.4905. [DOI] [PubMed] [Google Scholar]
- [16].Crona DJ, Faso A, Nishijima TF, McGraw KA, Galsky MD, Milowsky MI, A systematic review of strategies to prevent cisplatin-induced nephrotoxicity, Oncologist 22 (2017) 609–619, doi: 10.1634/theoncologist.2016-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhu R, Wang Q, Zhu Y, Wang Z, Zhang H, Wu B, Wu X, Wang S, pH sensitive nano layered double hydroxides reduce the hematotoxicity and enhance the anticancer efficacy of etoposide on non-small cell lung cancer, Acta Biomater 29 (2016) 320–332, doi: 10.1016/j.actbio.2015.10.029. [DOI] [PubMed] [Google Scholar]
- [18].Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R, Nanocarriers as an emerging platform for cancer therapy, Nat. Nanotechnol 2 (2007) 751–760, doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- [19].Kemp JA, Shim MS, Heo CY, Kwon YJ, Combo” nanomedicine: Co-delivery of multi-modal therapeutics for efficient, targeted, and safe cancer therapy, Adv. Drug Deliv. Rev 98 (2016) 3–18, doi: 10.1016/j.addr.2015.10.019. [DOI] [PubMed] [Google Scholar]
- [20].Ma L, Kohli M, Smith A, Nanoparticles for combination drug therapy, ACS Nano 7 (2013) 9518–9525, doi: 10.1021/nn405674m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tian J, Min Y, Rodgers Z, Au KM, Hagan CT, Zhang M, Roche K, Yang F, Wagner K, Wang AZ, Wang AZ, Co-delivery of paclitaxel and cisplatin with biocompatible PLGA-PEG nanoparticles enhances chemoradiotherapy in non-small cell lung cancer models, J. Mater. Chem. B 5 (2017) 6049–6057, doi: 10.1039/C7TB01370A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang M, Hagan CT, Min Y, Foley H, Tian X, Yang F, Mi Y, Au KM, Medik Y, Roche K, Wagner K, Rodgers Z, Wang AZ, Nanoparticle co-delivery of wortmannin and cisplatin synergistically enhances chemoradiotherapy and reverses platinum resistance in ovarian cancer models, Biomaterials 169 (2018) 1–10, doi: 10.1016/j.biomaterials.2018.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Xu X, Xie K, Zhang XQ, Pridgen EM, Park GY, Cui DS, Shi J, Wu J, Kantoff PW, Lippard SJ, Langer R, Walker GC, Farokhzad OC, Enhancing tumor cell response to chemotherapy through nanoparticle-mediated codelivery of siRNA and cisplatin prodrug, Proc. Natl. Acad. Sci. USA 110 (2013) 18638–18643, doi: 10.1073/pnas.1303958110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Johnstone TC, Lippard SJ, The effect of ligand lipophilicity on the nanoparticle encapsulation of Pt(IV) prodrugs, Inorg. Chem 52 (2013) 9915–9920, doi: 10.1021/ic4010642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Xiao H, Song H, Zhang Y, Qi R, Wang R, Xie Z, Huang Y, Li Y, Wu Y, Jing X, The use of polymeric platinum(IV) prodrugs to deliver multinuclear platinum(II) drugs with reduced systemic toxicity and enhanced antitumor efficacy, Biomaterials 33 (2012) 8657–8669, doi: 10.1016/j.biomaterials.2012.08.015. [DOI] [PubMed] [Google Scholar]
- [26].Xiao H, Song H, Yang Q, Cai H, Qi R, Yan L, Liu S, Zheng Y, Huang Y, Liu T, Jing X, A prodrug strategy to deliver cisplatin(IV) and paclitaxel in nanomicelles to improve efficacy and tolerance, Biomaterials 33 (2012) 6507–6519, doi: 10.1016/j.biomaterials.2012.05.049. [DOI] [PubMed] [Google Scholar]
- [27].Xiao H, Qi R, Liu S, Hu X, Duan T, Zheng Y, Huang Y, Jing X, Biodegradable polymer - cisplatin(IV) conjugate as a pro-drug of cisplatin(II), Biomaterials 32 (2011) 7732–7739, doi: 10.1016/j.biomaterials.2011.06.072. [DOI] [PubMed] [Google Scholar]
- [28].Wan X, Min Y, Bludau H, Keith A, Sheiko SS, Jordan R, Wang AZ, Sokolsky-Papkov M, Kabanov A.v., Drug combination synergy in worm-like polymeric micelles improves treatment outcome for small cell and non-small cell lung cancer, ACS Nano 12 (2018) 2426–2439, doi: 10.1021/acsnano.7b07878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals, 8th ed., National Academies Press (US), Washington (DC), 2011. Available from: https://www.ncbi.nlm.nih.gov/books/NBK54050/, doi: 10.17226/12910. [DOI] [Google Scholar]
- [30].Ou W, Ye S, Yang W, Wang Y, Ma Q, Yu C, Shi H, Yuan Z, Zhong G, Ren J, Zhu W, Wei Y, Enhanced antitumor effect of cisplatin in human NSCLC cells by tumor suppressor LKB1, Cancer Gene Ther. 19 (2012) 489–498, doi: 10.1038/cgt.2012.18. [DOI] [PubMed] [Google Scholar]
- [31].Dieleman EMT, Uitterhoeve ALJ, van Hoek MW, van Os RM, Wiersma J, Koolen MGJ, Kolff MW, Koning CCE, Adam JA, Verberne HJ, Annema JT, Rasch CRN, Concurrent daily cisplatin and high-dose radiation therapy in patients with stage III non-small cell lung cancer, Int. J. Radiat. Oncol. Biol. Phys 102 (2018) 543–551, doi: 10.1016/j.ijrobp.2018.07.188. [DOI] [PubMed] [Google Scholar]
- [32].Wexselblatt E, Gibson D, What do we know about the reduction of Pt(IV) prodrugs? J. Inorg. Biochem (2012) 220–229, doi: 10.1016/j.jinorgbio.2012.06.013. [DOI] [PubMed] [Google Scholar]
- [33].Fang C, Bhattarai N, Sun C, Zhang M, Functionalized nanoparticles with long-term stability in biological media, Small (Weinheim an Der Bergstrasse, Germany) 5 (2009) 1637, doi: 10.1002/SMLL.200801647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chou TC, Talalay P, Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors, Adv. Enzyme. Regul 22 (1984) 27–55, doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- [35].Henderson LW, The problem of peritoneal membrane area and permeability, Kidney Int. 3 (1973) 409–410, doi: 10.1038/ki.1973.63. [DOI] [PubMed] [Google Scholar]
- [36].Gandara DR, Chansky K, Albain KS, Leigh BR, Gaspar LE, Lara PN, Burris H, Gumerlock P, Kuebler JP, Bearden JD, Crowley J, Livingston R, Consolidation docetaxel after concurrent chemoradiotherapy in stage IIIb non-small-cell lung cancer: phase II southwest oncology group study S9504, J. Clin. Oncol 21 (2003) 2004–2010, doi: 10.1200/JCO.2003.04.197. [DOI] [PubMed] [Google Scholar]
- [37].Rafiei P, Haddadi A, Docetaxel-loaded PLGA and PLGA-PEG nanoparticles for intravenous application: pharmacokinetics and biodistribution profile, Int. J. Nanomed 12 (2017) 935–947, doi: 10.2147/ijn.s121881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Semete B, Booysen L, Kalombo L, Ramalapa B, Hayeshi R, Swai HS, Effects of protein binding on the biodistribution of PEGylated PLGA nanoparticles post oral administration, Int. J. Pharm 424 (2012) 115–120, doi: 10.1016/j.ijpharm.2011.12.043. [DOI] [PubMed] [Google Scholar]
- [39].Au KM, Min Y, Tian X, Zhang L, Perello V, Caster JM, Wang AZ, Improving cancer chemoradiotherapy treatment by dual controlled release of Wortmannin and Docetaxel in polymeric nanoparticles, ACS Nano 9 (2015) 8976–8996, doi: 10.1021/acsnano.5b02913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Caster JM, Yu SK, Patel AN, Newman NJ, Lee ZJ, Warner SB, Wagner KT, Roche KC, Tian X, Min Y, Wang AZ, Effect of particle size on the biodistribution, toxicity, and efficacy of drug-loaded polymeric nanoparticles in chemoradiotherapy, Nanomedicine 13 (2017) 1673–1683, doi: 10.1016/j.nano.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Li M, Panagi Z, Avgoustakis K, Reineke J, Physiologically based pharmacokinetic modeling of PLGA nanoparticles with varied mPEG content, Int. J. Nanomed 7 (2012) 1345–1356, doi: 10.2147/ijn.s23758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Reboul FL, Radiotherapy and chemotherapy in locally advanced non-small cell lung cancer: preclinical and early clinical data, Hematol. Oncol. Clin. North Am 18 (2004) 41–53 https://www.ncbi.nlm.nih.gov/pubmed/15005280. [DOI] [PubMed] [Google Scholar]
- [43].Singh AV, Batuwangala M, Mundra R, Mehta K, Patke S, Falletta E, Patil R, Gade WN, Biomineralized anisotropic gold microplate-macrophage interactions reveal frustrated phagocytosis-like phenomenon: a novel paclitaxel drug delivery vehicle, ACS Appl. Mater. Interfaces 6 (2014) 14679–14689, doi: 10.1021/am504051b. [DOI] [PubMed] [Google Scholar]
- [44].Khan I, Joshi G, Nakhate KT, Kumar Ajazuddin R, Gupta U, Nano-co-delivery of berberine and anticancer drug using PLGA nanoparticles: exploration of better anticancer activity and in vivo kinetics, Pharm. Res 36 (2019), doi: 10.1007/s11095-019-2677-5. [DOI] [PubMed] [Google Scholar]
- [45].Fonseca-Gomes J, Loureiro JA, Tanqueiro SR, Mouro FM, Ruivo P, Carvalho T, Sebastião AM, Diógenes MJ, Pereira MC, In vivo bio-distribution and toxicity evaluation of polymeric and lipid-based nanoparticles: a potential approach for chronic diseases treatment, Int. J. Nanomed 15 (2020) 8609–8621, doi: 10.2147/IJN.S267007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Yang F, Medik Y, Li L, Tian X, Fu D, Brouwer KLR, Wagner K, Sun B, Sendi H, Mi Y, Wang AZ, Nanoparticle drug delivery can reduce the hepatotoxicity of therapeutic Cargo, Small 16 (2020) 1906360, doi: 10.1002/smll.201906360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Singh AV, Ansari MHD, Rosenkranz D, Maharjan RS, Kriegel FL, Gandhi K, Kanase A, Singh R, Laux P, Luch A, Artificial intelligence and machine learning in computational nanotoxicology: unlocking and empowering nanomedicine, Adv. Healthc. Mater 9 (2020) 1901862, doi: 10.1002/adhm.201901862. [DOI] [PubMed] [Google Scholar]
- [48].Zhang C, Chen W, Zhang T, Jiang X, Hu Y, Hybrid nanoparticle composites applied to photodynamic therapy: strategies and applications, J. Mater. Chem. B 8 (2020) 4726–4737, doi: 10.1039/d0tb00093k. [DOI] [PubMed] [Google Scholar]
- [49].Blair HA, Adis drug evaluation daunorubicin/cytarabine liposome: a review in acute myeloid leukaemia, Drugs 78 (2018) 1903–1910, doi: 10.1007/s40265-018-1022-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ventola CL, Progress in nanomedicine: approved and investigational nanodrugs, P and T. 42 (2017) 742–755. /pmc/articles/PMC5720487/?report=abstract (accessed September 19, 2020). [PMC free article] [PubMed] [Google Scholar]
- [51].Anselmo AC, Mitragotri S, Nanoparticles in the clinic: An update, Bioeng. Transl. Med 4 (2019), doi: 10.1002/btm2.10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.