

PURPOSE
Activating mutations in PIK3CA are observed across multiple tumor types. The NCI-MATCH (EAY131) is a tumor-agnostic platform trial that enrolls patients to targeted therapies on the basis of matching genomic alterations. Arm Z1F evaluated copanlisib, an α and δ isoform–specific phosphoinositide 3-kinase (PI3K) inhibitor, in patients with PIK3CA mutations (with or without PTEN loss).
PATIENTS AND METHODS
Patients received copanlisib (60 mg intravenous) once weekly on days 1, 8, and 15 in 28-day cycles until progression or toxicity. Patients with KRAS mutations, human epidermal growth factor receptor 2–positive breast cancers, and lymphomas were excluded. The primary end point was centrally assessed objective response rate (ORR); secondary end points included progression-free survival, 6-month progression-free survival, and overall survival.
RESULTS
Thirty-five patients were enrolled, and 25 patients were included in the primary efficacy analysis as prespecified in the Protocol. Multiple histologies were enrolled, with gynecologic (n = 6) and gastrointestinal (n = 6) being the most common. Sixty-eight percent of patients had ≥ 3 lines of prior therapy. The ORR was 16% (4 of 25, 90% CI, 6 to 33) with P = .0341 against a null rate of 5%. The most common reason for protocol discontinuation was disease progression (n = 17, 68%). Grade 3/4 toxicities observed were consistent with reported toxicities for PI3K pathway inhibition. Sixteen patients (53%) had grade 3 toxicities, and one patient (3%) had grade 4 toxicity (CTCAE v5.0). Most common toxicities include hyperglycemia (n = 19), fatigue (n = 12), diarrhea (n = 11), hypertension (n = 10), and nausea (n = 10).
CONCLUSION
The study met its primary end point with an ORR of 16% (P = .0341) with copanlisib showing clinical activity in select tumors with PIK3CA mutation in the refractory setting.
INTRODUCTION
Phosphoinositide 3-kinase (PI3K) is involved in tumor proliferation and survival. Activation of the PI3K/AKT/mTOR pathway through PIK3CA mutations or PTEN loss is observed in many human cancers.1,2 Class I PI3K is a heterodimer with a regulatory and a catalytic subunit. The catalytic subunit, p110, has four isoforms: α, encoded by PIK3CA; β, encoded by PIK3CB; γ, encoded by PIK3CG, and δ, encoded by PIK3CD. Among solid tumors, mutations involving the kinase and helical domains of the p110α isoform are the most common, leading to aberrant pathway activation.
CONTEXT
Key Objective
PIK3CA is the most commonly mutated oncogene with alterations observed across multiple tumor types including breast, ovarian, and colon. Consequently, targeting PIK3CA has been of major clinical interest. However, initial efforts targeting PIK3CA mutations have yielded mixed results because of lack of isoform specificity of inhibitors and dose-limiting toxicities. The NCI-MATCH is a tumor-agnostic platform trial that enrolls patients on the basis of matching genomic alterations. Current study evaluated copanlisib, an α and δ isoform–specific phosphoinositide 3-kinase inhibitor, in patients with PIK3CA mutations.
Knowledge Generated
The study met its primary end point with an objective response rate of 16% (P = .0341), with copanlisib showing promising clinical activity in select tumors with PIK3CA mutation in the late-line, refractory setting.
Relevance
The current study suggests that copanlisib could be a viable therapeutic option for select tumors with PIK3CA mutations.
Clinical trials have suggested that activating PIK3CA mutations predict for increased sensitivity to therapies targeting the PI3K/AKT/mTOR signaling pathway.3-5 Patients with PIK3CA mutations and/or PTEN aberrations treated with PI3K/Akt/mTOR inhibitors had a higher clinical response rate (18%) compared with wild-type (WT) PIK3CA patients (6%). However, initial clinical efforts targeting PIK3CA mutations yielded mixed results because of lack of isoform specificity of inhibitors and poor therapeutic index. Recently, alpelisib, an alpha-selective PI3K inhibitor, was US Food and Drug Administration–approved for PIK3CA-mutated, hormone receptor–positive, metastatic breast cancers in combination with fulvestrant.4
Copanlisib is a class I inhibitor of PI3K with inhibitory activity predominantly against PI3Kα (half maximal inhibitory concentrations [IC50] = 0.5 nM) and PI3Kδ (IC50 = 0.7 nM) compared with PI3Kβ (IC50 = 3.7 nM) and PI3Kγ (IC50 = 6.4 nM) isoforms.6 Copanlisib had mean IC50 values of 19 nM against cell lines with PIK3CA-activating mutations, while the activity in WT PIK3CA was about 40-fold less (average IC50 = 774 nM).6 Copanlisib (Aliqopa) was approved by the US Food and Drug Administration for the treatment of adult patients with relapsed follicular lymphoma who have received at least two prior systemic therapies.7
Launched in 2015, the National Cancer Institute—Molecular Analysis for Therapy Choice (NCI-MATCH) is a signal-finding, molecularly matched study for patients with refractory cancers. The primary aim was to establish whether patients with advanced cancer would derive clinical benefit from treatments selected on the basis of their tumor molecular profile regardless of histology. Patients with actionable molecular alterations of interest are treated with matching therapies in independent subprotocols. Here, we report the results of the NCI-MATCH Subprotocol Z1F study, evaluating copanlisib in tumors with PIK3CA mutations.
PATIENTS AND METHODS
Study Design
NCI-MATCH was designed and coadministered by the Division of Cancer Treatment and Diagnosis, NCI, and the ECOG-ACRIN Cancer Research Group (ECOG-ACRIN) with the participation of the NCI National Clinical Trials Network and NCI Community Oncology Research Program.8 Patients with histologically documented solid tumors, lymphomas, or multiple myelomas who had progressed after at least one line of standard systemic therapy or for whom no standard curative therapy exists were eligible. Molecular alterations were considered actionable if they met definitions as outlined previously.8 Patients were required to have either a core biopsy or available archived tumor tissue obtained within the past 6 months for assessment. All tumors had central review of pathology classification performed at the ECOG-ACRIN Central Biorepository and Pathology Facility at MD Anderson Cancer Center and were coded with International Classification of Diseases for Oncology 3.1. Tumor profiling was accomplished as described previously.9 Actionable mutations were assessed using a next generation sequencing panel (Oncomine AmpliSeq panel, Thermo Fisher Scientific, Waltham, MA) of 143 genes, including single-nucleotide variants (SNVs), insertion and deletions (indel), amplifications, and selected fusions. Immunohistochemistry assays for PTEN, MLH1, and MSH2 were also performed. After the end of central profiling of 5,540 tumor samples in May 2017, patients were accepted if they had eligible molecular alterations identified by one of the 25 Clinical Laboratory Improvement Amendments–accredited laboratories approved to identify eligible patients. Molecular abnormalities identified by outside assays were subsequently confirmed by NCI central assay. Patients were assigned using a prospectively defined NCI-designed informatics rules algorithm (MATCHBOX), as previously described.10 The study was performed in accordance with provisions of the Declaration of Helsinki and Good Clinical Practice guidelines.
Patient Eligibility and Selection
Adult patients (age ≥ 18 years) with any solid tumor or myeloma who had progressed on standard treatment or without prior therapy if no curative treatment existed were eligible. Patients were required to have an activating mutation in PIK3CA in their tumor (Appendix Table A1, online only). Patients with coexisting PTEN loss on next generation sequencing were allowed. Patients were required to have measurable disease and have an Eastern Cooperative Oncology Group performance status of 0-1 with acceptable organ function.11-13 Patients with KRAS mutations, human epidermal growth factor receptor 2–positive breast cancers, and lymphomas were excluded. Patients with uncontrolled type I or II diabetes mellitus (HbA1c > 8.5%) and those who received prior copanlisib or other PI3K, Akt, or mTOR inhibitors were excluded. There was no limitation on the number of prior treatments received. The study (ClinicalTrials.gov identifier: NCT02465060) was approved by the NCI Central Institutional Review Board, the institutional review board of record for all participating institutions. All patients signed a written informed consent before participation.
Study Therapy and Assessments
Copanlisib was administered intravenously at a dose of 60 mg over an hour once weekly on days 1, 8, and 15 of a 28-day treatment cycle until progression or toxicity. All patients were required to have a normal ECG within 8 weeks before treatment assignment. Tumor response was assessed using revised RECIST criteria version 1.1.11 Tumor assessments were carried out every two cycles. Adverse events (AEs) were reported according to the NCI Common Terminology Criteria for Adverse Events (CTCAE) version 5.0.
Statistical Considerations
The primary objective was to evaluate the objective response rate (ORR). Allowing for a 10% ineligibility rate, 35 patients were to be accrued, to reach 31 eligible and treated patients. With this design, the power is 91.8% to conclude an agent is promising if the true response rate is 0.25 with a type 1 error rate of 1.8% (one-sided) under the null response rate of 5%. If the observed ORR were ≥ 5 of 31 (16%), it would then be concluded that the agent has promising clinical activity and is worthy of further investigation. Patients who were enrolled on the basis of the NCI-MATCH assay or on the basis of outside assays with molecular abnormalities confirmed by the NCI-MATCH assay were included in the primary analyses. As specified in the MATCH protocol, if fewer than 31 patients were in the primary analysis population (n = 25), primary efficacy was assessed using a 5% one-sided exact binomial test of the null hypothesis that the response rate was ≤ 5%. Secondary objectives were to report the proportion of patients who were progression-free at 6 months (PFS6), the progression-free survival (PFS), toxicity assessment, and evaluation of predictive biomarkers (comutations or other factors that potentially predict for response). PFS was defined as time from treatment start to disease progression or death from any cause; overall survival (OS) was defined as time from treatment start to death from any cause. PFS and OS were estimated using the Kaplan-Meier method. Secondary analysis results by combining all eligible and treated patients (n = 28) are included in the Appendix (online only).
RESULTS
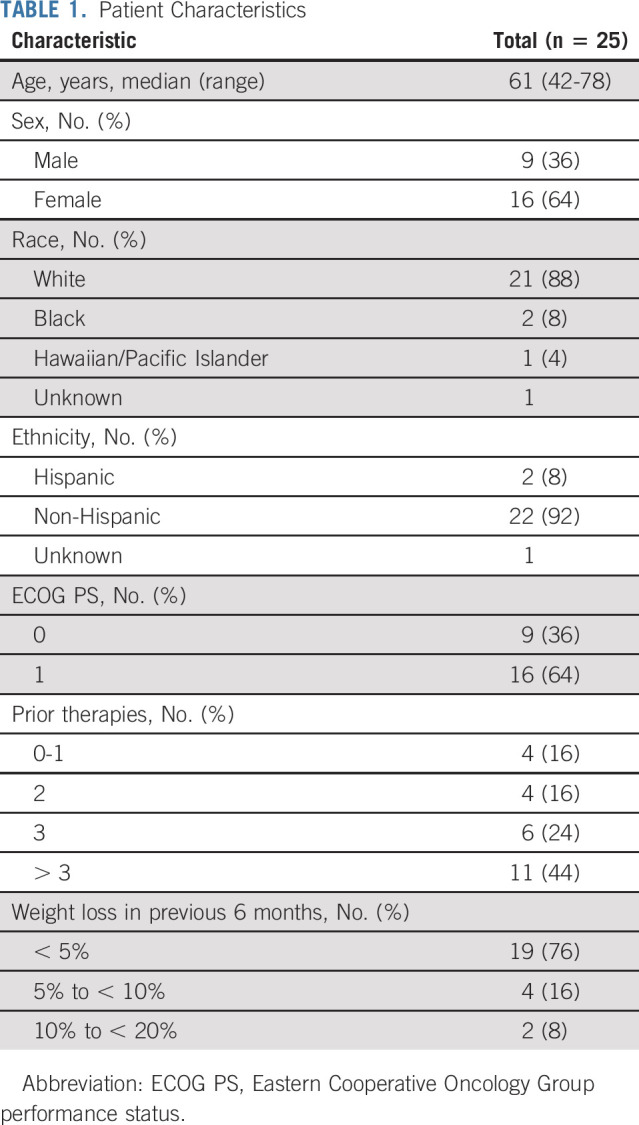
Patient Characteristics and Demographics
Forty-eight patients were assigned to the Z1F arm, of which 35 were enrolled in the study (Fig 1). Five patients did not start therapy and two patients were considered ineligible (one had pretreatment creatinine clearance outside of eligibility range and the other had baseline AE greater than threshold for eligibility). Out of the 28 patients eligible for treatment, three patients were not included in the primary analysis, as mutational status was not confirmed at NCI central laboratory (did not have enough tissue or biopsy was not feasible). Per protocol, 25 patients were included in the primary analysis (who were eligible and treated, and outside assay results confirmed by the central NCI-MATCH assay). Data on the 28-patient cohort (eligible and treated population) are presented in Appendix Figures A2-A4 and Table A4 (online only). Patient characteristics for 25 evaluable patients are shown in Table 1. Median age was 61 years. Sixty-four percent of patients had Eastern Cooperative Oncology Group performance status of 1. The study enrolled patients who were heavily pretreated with 68% of them having received ≥ 3 prior lines of therapy. Appendix Table A2 (online only) shows the histopathologic classification of tumors for the 25 evaluable patients. The study enrolled patients with more than 20 different histologies, with gynecologic (n = 6), gastrointestinal (n = 6), and genitourinary (n = 4) being the most common tumors.
FIG 1.

Flowchart. Out of 35 enrolled patients, five patients did not start therapy. Among 30 that began treatment, 28 were considered evaluable for response (one patient had pretreatment creatinine clearance outside of eligibility range and the other had baseline AE greater than threshold for eligibility). Twenty-five patients were included in the primary efficacy analysis (three patients were excluded as mutational status was unable to be confirmed by the central assay). AE, adverse event; CrCl, creatinine clearance; NCI, National Cancer Institute.
TABLE 1.
Patient Characteristics
Appendix Table A3 (online only) summarizes the distribution of PIK3CA alterations for the 25 evaluable patients. Twenty-four patients had PIK3CA SNVs, whereas one patient had both an SNV and PIK3CA gene amplification. Helical domain PIK3CA mutations were predominant with variants at E545 and H1047 being the most common (Fig 2). The landscape of co-occurring mutations is shown in Appendix Figure A1 (online only). The most frequent co-occurring mutations were TP53 (N = 11, 44%), PTEN (N = 3, 12%), and ARID1A (N = 3, 12%). The median number of genomic alterations was 3 (range 1-9). Nine patients had ≥ 4 genomic alterations in their tumors.
FIG 2.
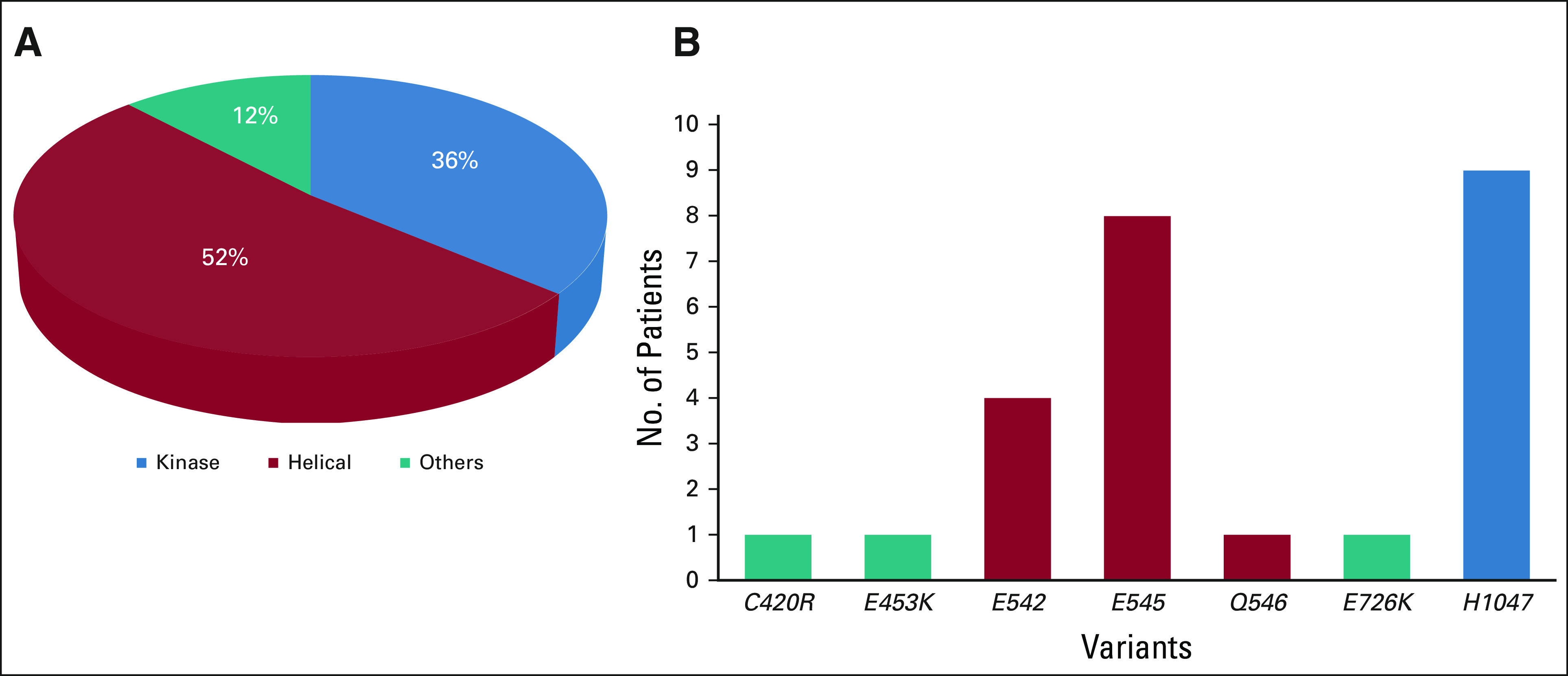
(A) Distributions of PIK3CA mutations across helical and kinase domains. (B) Distribution of specific variants in PIK3CA.
Twenty-five patients were included in the primary efficacy analysis (Table 2). One patient withdrew from study during cycle 1, with no further follow-up data available. The ORR was 16% (90% CI: 6%-33%), the one-sided exact binomial test P value was .0341 under the null hypothesis of ORR of 5% (ie, Pr(k ≥ 4 | ORR = 5%) was 3.41%), with four patients having a partial response (PR). For the cohort of 28 patients (eligible and treated), ORR was 14% (90% CI, 5 to 30; P = .0491; Appendix Table A4). The waterfall plot depicting the best tumor change from baseline in target lesion size for the 22 patients (out of 25 patients included in the primary efficacy analysis) who had subsequent tumor measurement is shown in Figure 3. Clinical benefit rate, defined as complete response, PR, or stable disease (SD) for ≥ 6 months was 36% (9 of 25, 90% CI, 20 to 54). Two patients with PR are still on study treatment and have received 28 and 29 cycles at the time of data cut off (January 6, 2021). Figure 4 shows the duration of treatment on the study for 25 analyzable patients. The median time on treatment was 1.7 months (range: 0.2-26.0 months). Appendix Table A5 (online only) shows details on patients who had confirmed PR or SD for > 6 months. PRs were observed in endometrial adenocarcinoma, clear cell carcinoma of anterior abdominal wall, ameloblastoma of the mandible, and low-grade myxoid liposarcoma (MLS). The patient with MLS and another with ameloblastoma of the mandible are still on treatment. Clinical responses were observed in patients with helical (E545K and E542K) and kinase (H1047R) domain PIK3CA mutations. Among nine patients with H1047 variants, two PR and two SD were observed; whereas among eight patients with E545 variants, one PR and four SD were noted. One PR and two SD were observed among four patients with E542 variants (Fig 2, Appendix Table A5). For patients with PR (n = 4), the median number of prior therapies was 1 (range 1-4). For patients with SD for at least 6 months (n = 5), the median number of prior therapies was 3 (range 2-4). For patients without clinical benefit (n = 16), the median number prior therapies was 3.5 (range 1-6). Estimated 6-month PFS rate was 38% (90% CI, 22 to 53), with a median PFS of 3.4 months (90% CI, 1.8 to 6.6). The estimated 6-month OS rate was 50% (90% CI, 32 to 65), and the median OS was 5.9 (90% CI, 4.9 to 13.7) months (Fig 5).
TABLE 2.
Tumor Response Assessment (n = 25)
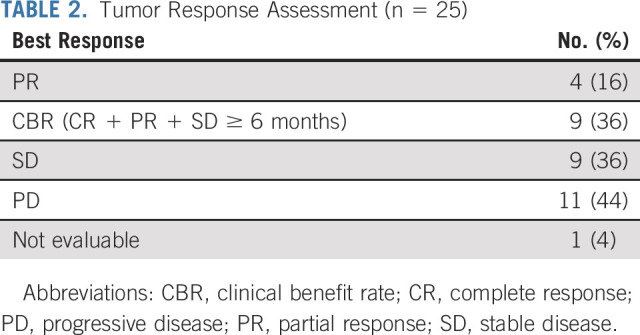
FIG 3.

Best percentage change from baseline. Of the 25 patients included in the primary efficacy analysis, 22 patients had lesions measurable after baseline and are included. Four patients had PR. GU, genitourinary; Gyn, gynecologic; PD, progressive disease; PR, partial response; SD, stable disease.
FIG 4.
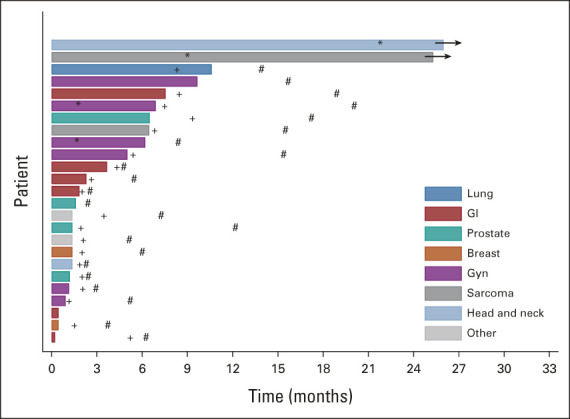
Duration of treatment. Twenty-five patients included in the primary efficacy analysis are shown here. Occurrence of response (*); disease progression (+); and death (#). Two patients are still on study treatment. GU, genitourinary; Gyn, gynecologic.
FIG 5.
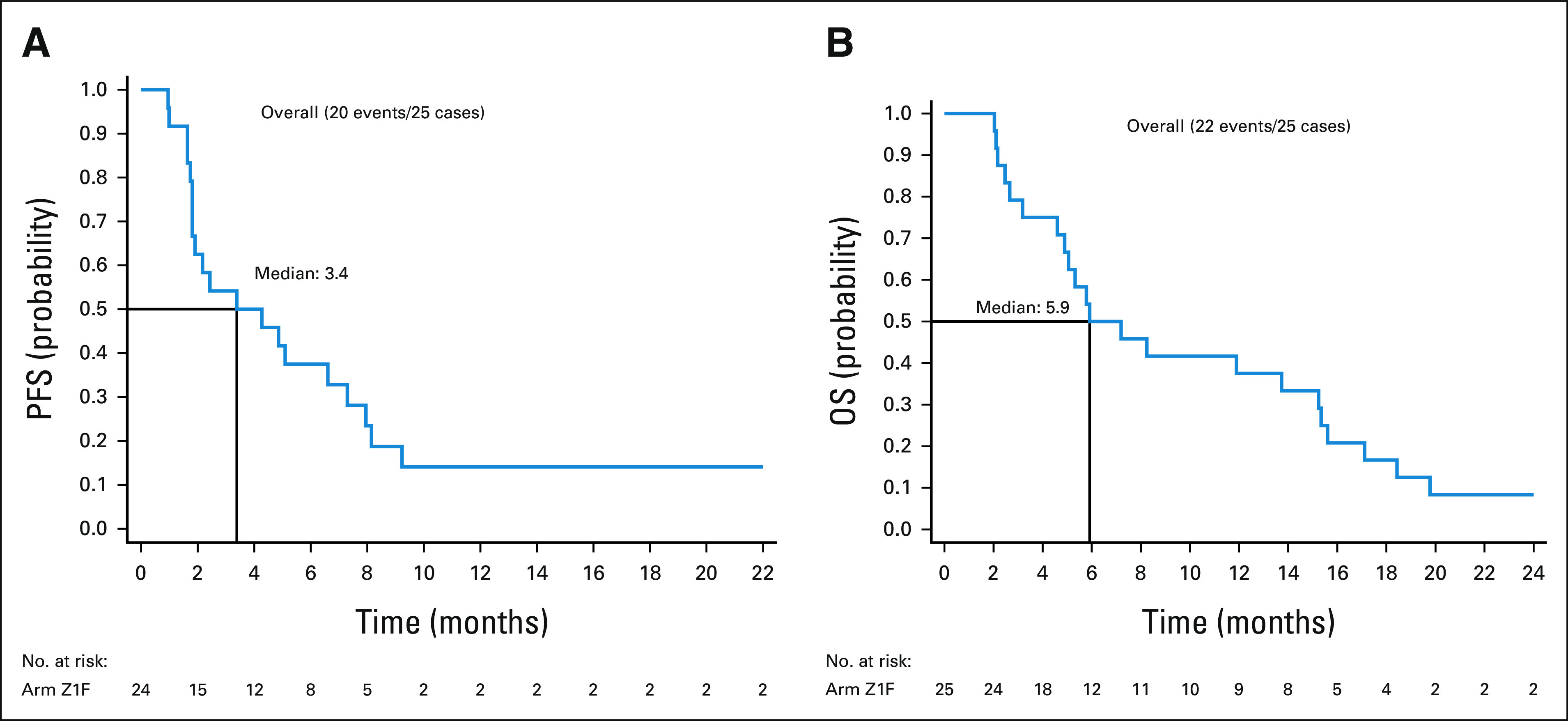
(A) PFS and (B) OS. OS, overall survival; PFS, progression-free survival.
Of the 35 patients enrolled, 30 were included in the toxicity analysis. Treatment-related toxicities seen in ≥ 10% and/or grade 3 severity are listed (Appendix Table A6, online only). The most common toxicities included hyperglycemia (n = 19), fatigue (n = 12), diarrhea (n = 11), hypertension (n = 10), nausea (n = 10), and maculopapular rash (n = 9). Grade 1/2 hyperglycemia was seen in 11 patients, and grade 3 hyperglycemia was observed in seven patients. Nine patients had grade 2 hypertension. Three patients discontinued study drug because of toxicity. Of the 23 evaluable patients who had discontinued protocol therapy, the median number of cycles was 2 (range: 1-10). The most common reason for treatment discontinuation was disease progression (n = 17, 68%).
DISCUSSION
PIK3CA mutations are frequently observed across multiple tumors including breast, endometrium, ovarian, and colon cancer. However, targeting PIK3CA has been hampered because of lack of selectivity of inhibitors and dose-limiting toxicities. Also, PIK3CA mutations are very heterogeneous, and their oncogenicity depends on the location of mutation in the gene.14,15 Moreover, PIK3CA mutations are often observed to coexist with other pathway alterations (eg, KRAS), which can affect clinical response.5 Patients with PIK3CA mutations and/ or PTEN aberrations with concurrent KRAS mutations had shorter median PFS (1.8 months v 2.9 months) as well as inferior response rates (4% v 24%), compared with patients without KRAS mutations.5 Consequently, patients with PIK3CA mutations with concurrent KRAS mutations were excluded.
A previous NCI-MATCH study (arm I) evaluated taselisib in tumors with PIK3CA mutations, with no objective responses observed.16 In a phase I basket study of taselisib in solid tumors with PIK3CA mutations, an ORR of 9% was reported.17 Although all known activating PIK3CA mutations annotated in MATCHBOX as actionable were considered eligible, it should be noted that PRs and SD ≥ 6 months were observed only in helical and kinase domain mutations. It has been reported that patients with double PIK3CA mutations in cis may respond better to PI3Ka inhibitors compared with single-hotspot mutations.18,19 However, in a triplet study of palbociclib, taselisib, and fulvestrant in PIK3CA-mutant breast cancers, patients with double mutations had shorter PFS.20 In our study, no double mutations in PIK3CA (one patient had both SNV and amplification) were observed.
A major limitation of PI3K inhibitors is the role of PI3Ka in normal tissue homeostasis. In general, toxicity depends on the isoform targeted, with hyperglycemia, diarrhea, rash, and fatigue being the common adverse effects. The adverse effects observed in the study were consistent with those previously described with copanlisib.21 In contrast to other PI3K inhibitors, copanlisib was largely well tolerated. Approximately 10% of patients discontinued copanlisib because of adverse effects, whereas the discontinuation rates with other PI3K inhibitors have ranged from 20% to 25%.4,22
MLS are aggressive tumors with a propensity for high rates of local recurrence and distant metastases.23 These tumors account for approximately 5%-10% of all soft tissue sarcomas and are typically characterized by t(12; 16) translocation resulting in expression of the chimeric fusion protein, FUS-DDIT3. Systemic therapy options are limited and include conventional anthracycline-based chemotherapy, eribulin, and recently trabectedin.24 Activation of PI3K/Akt/mTOR pathway through mutations in PIK3CA or PTEN loss has been shown to be essential in MLS.25,26 Approximately one third of MLS display alterations in PIK3CA, AKT1, and PTEN, with PIK3CA mutations observed in nearly 15% of all MLS.25 Patients whose tumors harbor mutations in PIK3CA have shorter disease-specific survival than those with WT PIK3CA.23 Preclinical studies have shown that use of PI3K inhibitors was associated with significant tumor growth inhibition.25 Among the MLS enrolled in the study, one had PR and the other had SD > 6 months. The patient who achieved PR is still on treatment, suggesting dependence of PI3K pathway in these rare tumors. Considering the observed clinical activity, PI3K inhibitors could be a potential targeted therapy option for patients with MLS.
Ameloblastomas are rare, locally destructive tumors of the jaw. Although mutations in SMO are common in ameloblastomas of the maxilla (40%), mutations in BRAF (approximately 50%) are typically observed in tumors of the mandible.27,28 Mutations in PIK3CA (< 3%) and FGFR2 (6%) have also been described to co-occur in the background of SMO or BRAF mutations.29,30 In our patient with mandibular ameloblastoma, in addition to PIK3CA, BRAF V600E and MED12 mutations were also observed. This patient was not previously treated with a BRAF inhibitor. Incidentally, loss of MED12 (component of the transcriptional mediator complex) has been shown to mediate resistance to BRAF inhibitors through activation of transforming growth factor-β by compensatory ERK1/2 signaling.31 The patient achieved PR and is still on treatment. Considering the prolonged clinical activity of copanlisib, PI3K inhibition in these rare tumors could potentially be clinically meaningful, notwithstanding the presence of BRAF driver mutation.
Interestingly, among three patients with coexisting ARID1A loss, one PR and two SD > 6 months were observed. ARID1A (the adenine and thymine–rich interacting domain-containing protein 1A) is the most frequently mutated SWItch/Sucrose Non-Fermentable complex gene across cancer types.32 ARID1A mutations have been reported in ovarian clear cell carcinomas (up to 50%) and ovarian endometrioid carcinomas (30%) and less commonly in breast, lung, and pancreatic cancers.33,34 Co-occurrence of PIK3CA and ARID1A mutations has been observed in ovarian and endometrial cancers as well as in gastric and colon cancers.35 In a phase I study of copanlisib in patients with solid tumors, the only patient who had PR had a concurrent mutation in PIK3CA and ARID1A.21 The mechanisms that characterize the interaction between these mutations are not clear, although changes in cell differentiation and EMT have been implicated.36 PIK3IP1 (phosphoinositide 3-kinase-interacting protein 1), a negative regulator of the PI3K pathway, has recently been recognized as a target of ARID1A.37 ARID1A loss has been shown to decrease its association with the PIK3IP1 promoter and suppress the expression of PIK3IP1.38 Thus, we speculate ARID1A loss could lead to activation of PI3K pathway and render tumors sensitive to PI3K pathway inhibitors. The clinical activity of copanlisib in patients with coexistent PIK3CA mutation and ARID1A loss has led to a proposed trial of PI3K inhibitor in patients with coexisting PIK3CA and ARID1A mutations.
Among three patients with concomitant PTEN loss, SD > 6 months was observed with copanlisib. Loss of PTEN has been associated with decreased clinical benefit in patients with PIK3CA mutations and treated with PI3Kα-specific inhibitors. Acquisition of PTEN alterations has been described as a mechanism of acquired resistance to alpelisib with activation of downstream signaling through the PI3Kβ isoform.39 Similarly, PTEN loss-of-function mutations at baseline were associated with rapid disease progression on alpelisib and aromatase inhibitors in hormone receptor–positive metastatic breast cancer patients.40 Prolonged tumor response observed in our study in patients with coexistent PTEN loss suggests that the clinical activity of copanlisib is not limited by PTEN loss, in contrast to PI3Kα-specific inhibitors.
The Z1F study met its primary end point with copanlisib, showing promising clinical activity in select tumors with PIK3CA mutation in the late-line, refractory setting. Further study of copanlisib in rational combinations is warranted.
ACKNOWLEDGMENT
We would like to thank all the patients who participated and their families, as well as coinvestigators, nurses, study coordinators, and operations staff at each of the clinical sites. We also thank Dr Austin Doyle, Cancer Therapy Evaluation Program, National Cancer Institute, Bethesda, MD.
APPENDIX
FIG A1.

Distributions of comutations in patients with PIK3CA mutations.
FIG A2.

Best percentage change from baseline in patient cohort (n = 28). Of the 28 evaluable patients, 24 had lesions measurable after baseline and are included (one patient without target lesion at baseline, and three patients without target lesion at baseline and without any follow-up imaging assessments were not included). GU, genitourinary; Gyn, gynecologic; PD, progressive disease; PR, partial response; SD, stable disease.
FIG A3.
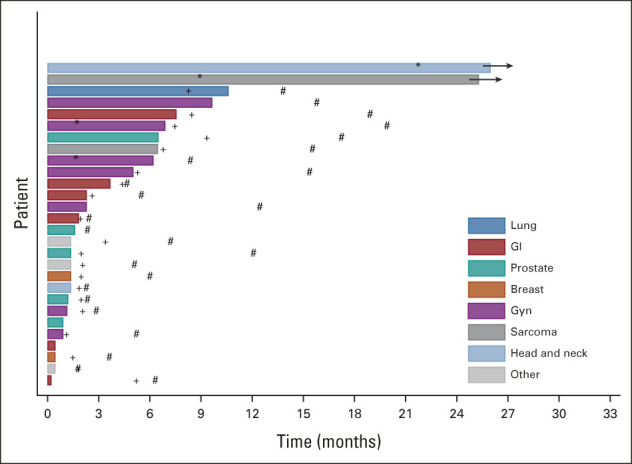
Duration of treatment in patient cohort (n = 28). Occurrence of response (*); disease progression (+); and death (#). Two patients are still on study treatment. GU, genitourinary; Gyn, gynecologic.
FIG A4.
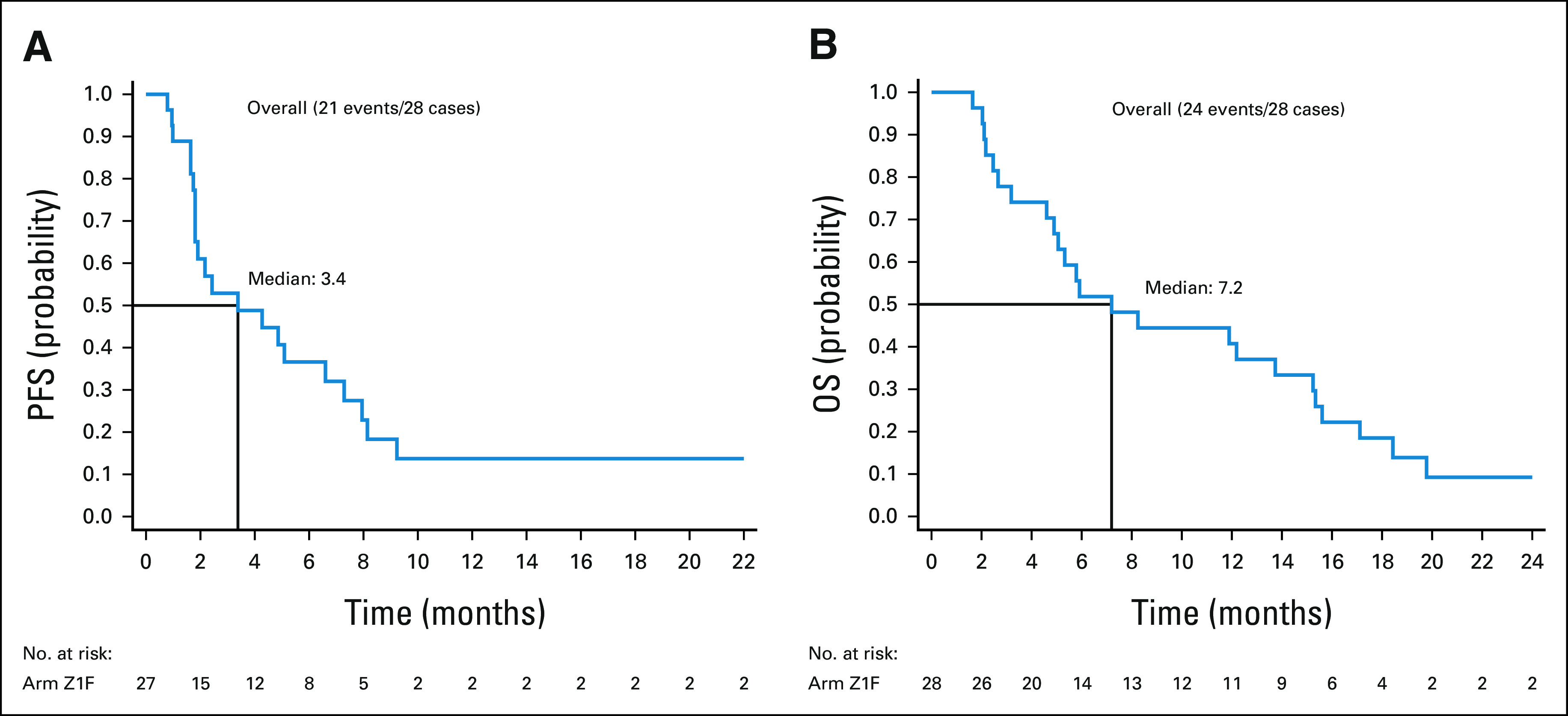
(A) PFS and (B) OS for 28 patients (eligible and treated). OS, overall survival; PFS, progression-free survival.
TABLE A1.
List of Actionable PIK3CA Mutations for Z1F Subprotocol

TABLE A2.
Histopathologic Classification

TABLE A3.
Distribution of PIK3CA Mutations in Analyzable Patients

TABLE A4.
Tumor Response Assessment in Patient Cohort (n = 28)

TABLE A5.
Patients With Confirmed PR or SD > 6 Months

TABLE A6.
Treatment-Related Toxicities

Senthil Damodaran
Research Funding: EMD Serono (Inst), Guardant Health (Inst), Taiho Pharmaceutical (Inst), Novartis (Inst), Sermonix Pharmaceuticals (Inst)
Fengmin Zhao
This author is a member of the Journal of Clinical Oncology Editorial Board. Journal policy recused the author from having any role in the peer review of this manuscript.
Dustin A. Deming
Consulting or Advisory Role: Bayer, Promega, Array BioPharma, Lilly, Pfizer
Research Funding: Merck (Inst), Bristol Myers Squibb (Inst), Genentech (Inst), Revolution Medicines (Inst), Millennium (Inst), Bayer
Edith P. Mitchell
Leadership: Corvus Pharmaceuticals
Honoraria: Sanofi, Exelixis
Consulting or Advisory Role: Genentech, Novartis, Merck, Bristol Myers Squib
Speakers' Bureau: Ipsen
Research Funding: Genentech (Inst), sanofi (Inst)
Robert J. Gray
Research Funding: Agios, Amgen, AstraZeneca, Bristol Myers Squibb, Boehringer Ingelheim, Celgene, Genentech/Roche, Genomic Health, Genzyme, GlaxoSmithKline, Janssen-Ortho, Onyx, Pfizer, Sequenta, Syndax, Novartis, Takeda, AbbVie, Sanofi, Merck Sharp & Dohme
Lisa M. McShane
This author is a member of the Journal of Clinical Oncology Editorial Board. Journal policy recused the author from having any role in the peer review of this manuscript.
Larry V. Rubinstein
This author is a member of the Journal of Clinical Oncology Editorial Board. Journal policy recused the author from having any role in the peer review of this manuscript.
P. Mickey Williams
Research Funding: Illumina (Inst)
Patents, Royalties, Other Intellectual Property: I was a coinventor of the DLBCL cell of origin patent recently filed by the NIH
Stanley R. Hamilton
Research Funding: Minerva Biotechnologies, Intima
Carlos L. Arteaga
Stock and Other Ownership Interests: Provista Diagnostics
Consulting or Advisory Role: Novartis, Lilly, Sanofi, Radius Health, Taiho Pharmaceutical, Puma Biotechnology, Merck, Origimed, Immunomedics, Daiichi Sankyo, Athenex, Astrazeneca, Arvinas
Research Funding: Pfizer, Lilly, Takeda
Other Relationship: Susan G. Komen for the Cure
Lyndsay N. Harris
Patents, Royalties, Other Intellectual Property: Philips Healthcare
Peter J. O'Dwyer
Consulting or Advisory Role: Genentech
Research Funding: Bristol Myers Squibb (Inst), Pfizer (Inst), Novartis (Inst), Genentech (Inst), Mirati Therapeutics (Inst), Celgene (Inst), GlaxoSmithKline (Inst), BBI Healthcare (Inst), Pharmacyclics (Inst), Five Prime Therapeutics (Inst), Forty Seven (Inst), Amgen (Inst), H3 Biomedicine (Inst), Taiho Pharmaceutical (Inst), Array BioPharma (Inst), Lilly/ImClone (Inst), Syndax (Inst), Minneamrita Therapeutics (Inst)
Expert Testimony: Lilly, Dai-ichi Sankyo
Alice P. Chen
Uncompensated Relationships: Frontiers in Medicine
Keith T. Flaherty
Stock and Other Ownership Interests: Clovis Oncology, Loxo, X4 Pharma, Strata Oncology, PIC Therapeutics, Shattuck Labs, Apricity Health, Oncoceutics, FOGPharma, Tvardi Therapeutics, Checkmate Pharmaceuticals, Kinnate Biopharma, Scorpion Therapeutics, ALX Oncology, xCures, Monopteros Therapeutics, Vibliome Therapeutics, Transcode Therapeutics, Soley Therapeutics
Consulting or Advisory Role: Novartis, Lilly, Oncoceutics, Tvardi Therapeutics, Takeda, Boston Biomedical, Debiopharm Group, FOGPharma
No other potential conflicts of interest were reported.
DISCLAIMER
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
PRIOR PRESENTATION
Presented (oral) at ASCO Virtual Meeting, May 29, 2020.
SUPPORT
This study was coordinated by the ECOG-ACRIN Cancer Research Group (Peter J. O'Dwyer, MD and Mitchell D. Schnall, MD, PhD, Group Co-Chairs) and supported by the National Cancer Institute of the National Institutes of Health under the following award numbers: U10CA180820, U10CA180794, UG1CA233329, U10CA233302, UG1CA233277, UG1CA233180, UG1CA233341, UG1CA189821, and U10CA180868.
CLINICAL TRIAL INFORMATION
DATA SHARING STATEMENT
Data used in this publication will be shared via the appropriate NCI-controlled access data sharing platform(s).
AUTHOR CONTRIBUTIONS
Conception and design: Senthil Damodaran, Dustin A. Deming, Edith P. Mitchell, John J. Wright, Robert J. Gray, Lisa M. McShane, Larry V. Rubinstein, David R. Patton, P. Mickey Williams, Stanley R. Hamilton, Barbara A. Conley, Peter J. O'Dwyer, Alice P. Chen, Keith T. Flaherty
Financial support: David R. Patton
Administrative support: David R. Patton, Stanley R. Hamilton, Lyndsay N. Harris
Provision of study materials or patients: David R. Patton, Stanley R. Hamilton
Collection and assembly of data: Senthil Damodaran, Dustin A. Deming, Robert J. Gray, Victoria Wang, David R. Patton, P. Mickey Williams, Stanley R. Hamilton, Jennifer M. Suga
Data analysis and interpretation: Senthil Damodaran, Fengmin Zhao, Dustin A. Deming, Victoria Wang, Lisa M. McShane, David R. Patton, P. Mickey Williams, Stanley R. Hamilton, Carlos L. Arteaga, Lyndsay N. Harris, Peter J. O'Dwyer, Keith T. Flaherty
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Phase II Study of Copanlisib in Patients With Tumors With PIK3CA Mutations: Results From the NCI-MATCH ECOG-ACRIN Trial (EAY131) Subprotocol Z1F
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Senthil Damodaran
Research Funding: EMD Serono (Inst), Guardant Health (Inst), Taiho Pharmaceutical (Inst), Novartis (Inst), Sermonix Pharmaceuticals (Inst)
Fengmin Zhao
This author is a member of the Journal of Clinical Oncology Editorial Board. Journal policy recused the author from having any role in the peer review of this manuscript.
Dustin A. Deming
Consulting or Advisory Role: Bayer, Promega, Array BioPharma, Lilly, Pfizer
Research Funding: Merck (Inst), Bristol Myers Squibb (Inst), Genentech (Inst), Revolution Medicines (Inst), Millennium (Inst), Bayer
Edith P. Mitchell
Leadership: Corvus Pharmaceuticals
Honoraria: Sanofi, Exelixis
Consulting or Advisory Role: Genentech, Novartis, Merck, Bristol Myers Squib
Speakers' Bureau: Ipsen
Research Funding: Genentech (Inst), sanofi (Inst)
Robert J. Gray
Research Funding: Agios, Amgen, AstraZeneca, Bristol Myers Squibb, Boehringer Ingelheim, Celgene, Genentech/Roche, Genomic Health, Genzyme, GlaxoSmithKline, Janssen-Ortho, Onyx, Pfizer, Sequenta, Syndax, Novartis, Takeda, AbbVie, Sanofi, Merck Sharp & Dohme
Lisa M. McShane
This author is a member of the Journal of Clinical Oncology Editorial Board. Journal policy recused the author from having any role in the peer review of this manuscript.
Larry V. Rubinstein
This author is a member of the Journal of Clinical Oncology Editorial Board. Journal policy recused the author from having any role in the peer review of this manuscript.
P. Mickey Williams
Research Funding: Illumina (Inst)
Patents, Royalties, Other Intellectual Property: I was a coinventor of the DLBCL cell of origin patent recently filed by the NIH
Stanley R. Hamilton
Research Funding: Minerva Biotechnologies, Intima
Carlos L. Arteaga
Stock and Other Ownership Interests: Provista Diagnostics
Consulting or Advisory Role: Novartis, Lilly, Sanofi, Radius Health, Taiho Pharmaceutical, Puma Biotechnology, Merck, Origimed, Immunomedics, Daiichi Sankyo, Athenex, Astrazeneca, Arvinas
Research Funding: Pfizer, Lilly, Takeda
Other Relationship: Susan G. Komen for the Cure
Lyndsay N. Harris
Patents, Royalties, Other Intellectual Property: Philips Healthcare
Peter J. O'Dwyer
Consulting or Advisory Role: Genentech
Research Funding: Bristol Myers Squibb (Inst), Pfizer (Inst), Novartis (Inst), Genentech (Inst), Mirati Therapeutics (Inst), Celgene (Inst), GlaxoSmithKline (Inst), BBI Healthcare (Inst), Pharmacyclics (Inst), Five Prime Therapeutics (Inst), Forty Seven (Inst), Amgen (Inst), H3 Biomedicine (Inst), Taiho Pharmaceutical (Inst), Array BioPharma (Inst), Lilly/ImClone (Inst), Syndax (Inst), Minneamrita Therapeutics (Inst)
Expert Testimony: Lilly, Dai-ichi Sankyo
Alice P. Chen
Uncompensated Relationships: Frontiers in Medicine
Keith T. Flaherty
Stock and Other Ownership Interests: Clovis Oncology, Loxo, X4 Pharma, Strata Oncology, PIC Therapeutics, Shattuck Labs, Apricity Health, Oncoceutics, FOGPharma, Tvardi Therapeutics, Checkmate Pharmaceuticals, Kinnate Biopharma, Scorpion Therapeutics, ALX Oncology, xCures, Monopteros Therapeutics, Vibliome Therapeutics, Transcode Therapeutics, Soley Therapeutics
Consulting or Advisory Role: Novartis, Lilly, Oncoceutics, Tvardi Therapeutics, Takeda, Boston Biomedical, Debiopharm Group, FOGPharma
No other potential conflicts of interest were reported.
REFERENCES
- 1.Engelman JA, Chen L, Tan X, et al. : Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 14:1351-1356, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Samuels Y, Wang Z, Bardelli A, et al. : High frequency of mutations of the PIK3CA gene in human cancers. Science 304:554, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Janku F, Tsimberidou AM, Garrido-Laguna I, et al. : PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Mol Cancer Ther 10:558-565, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andre F, Ciruelos E, Rubovszky G, et al. : Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N Engl J Med 380:1929-1940, 2019 [DOI] [PubMed] [Google Scholar]
- 5.Janku F, Hong DS, Fu S, et al. : Assessing PIK3CA and PTEN in early-phase trials with PI3K/AKT/mTOR inhibitors. Cell Rep 6:377-387, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu N, Rowley BR, Bull CO, et al. : BAY 80-6946 is a highly selective intravenous PI3K inhibitor with potent p110alpha and p110delta activities in tumor cell lines and xenograft models. Mol Cancer Ther 12:2319-2330, 2013 [DOI] [PubMed] [Google Scholar]
- 7.Dreyling M, Santoro A, Mollica L, et al. : Phosphatidylinositol 3-kinase inhibition by copanlisib in relapsed or refractory indolent lymphoma. J Clin Oncol 35:3898-3905, 2017 [DOI] [PubMed] [Google Scholar]
- 8.Flaherty KT, Gray RJ, Chen AP, et al. : Molecular landscape and actionable alterations in a genomically guided cancer clinical trial: National Cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH). J Clin Oncol 38:3883-3894, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lih CJ, Harrington RD, Sims DJ, et al. : Analytical validation of the next-generation sequencing assay for a nationwide signal-finding clinical trial: Molecular analysis for therapy choice clinical trial. J Mol Diagn 19:313-327, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flaherty KT, Gray R, Chen A, et al. : The molecular analysis for therapy choice (NCI-MATCH) trial: Lessons for genomic trial design. J Natl Cancer Inst 112:1021-1029, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eisenhauer EA, Therasse P, Bogaerts J, et al. : New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer 45:228-247, 2009 [DOI] [PubMed] [Google Scholar]
- 12.Wen PY, Macdonald DR, Reardon DA, et al. : Updated response assessment criteria for high-grade gliomas: Response assessment in neuro-oncology working group. J Clin Oncol 28:1963-1972, 2010 [DOI] [PubMed] [Google Scholar]
- 13.Kumar S, Paiva B, Anderson KC, et al. : International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol 17:e328-e346, 2016 [DOI] [PubMed] [Google Scholar]
- 14.Chaussade C, Cho K, Mawson C, et al. : Functional differences between two classes of oncogenic mutation in the PIK3CA gene. Biochem Biophys Res Commun 381:577-581, 2009 [DOI] [PubMed] [Google Scholar]
- 15.Gymnopoulos M, Elsliger MA, Vogt PK: Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci U S A 104:5569-5574, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krop IE, Jegede O, Grilley-Olson JE, et al. : Results from molecular analysis for therapy choice (MATCH) arm I: Taselisib for PIK3CA-mutated tumors. J Clin Oncol 36, 2018. (suppl 15; abstr 101) [Google Scholar]
- 17.Jhaveri K, Chang MT, Juric D, et al. : Phase I basket study of taselisib, an isoform-selective PI3K inhibitor, in patients with PIK3CA-mutant cancers. Clin Cancer Res 27:447-459, 2021 [DOI] [PubMed] [Google Scholar]
- 18.Zhang M, Jang H, Nussinov R: PI3K driver mutations: A biophysical membrane-centric perspective. Cancer Res 81:237-247, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vasan N, Razavi P, Johnson JL, et al. : Double PIK3CA mutations in cis increase oncogenicity and sensitivity to PI3Kalpha inhibitors. Science 366:714-723, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pascual J, Lim JSJ, Macpherson IR, et al. : Triplet therapy with palbociclib, taselisib, and fulvestrant in PIK3CA-mutant breast cancer and doublet palbociclib and taselisib in pathway-mutant solid cancers. Cancer Discov 11:92-107, 2021 [DOI] [PubMed] [Google Scholar]
- 21.Morschhauser F, Machiels JP, Salles G, et al. : On-target pharmacodynamic activity of the PI3K inhibitor copanlisib in paired biopsies from patients with malignant lymphoma and advanced solid tumors. Mol Cancer Ther 19:468-478, 2020 [DOI] [PubMed] [Google Scholar]
- 22.Baselga J, Faye Dent S, Cortes J, et al. : Phase III study of taselisib (GDC-0032) + fulvestrant (FULV) v FULV in patients (pts) with estrogen receptor (ER)-positive, PIK3CA-mutant (MUT), locally advanced or metastatic breast cancer (MBC): Primary analysis from SANDPIPER. J Clin Oncol 36, 2018. (suppl 18; abstr LBA1006) [Google Scholar]
- 23.Manji GA, Schwartz GK: Managing liposarcomas: Cutting through the fat. JCO Oncol Pract 12:221-227, 2016 [DOI] [PubMed] [Google Scholar]
- 24.Demetri GD, von Mehren M, Jones RL, et al. : Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: Results of a phase III randomized multicenter clinical trial. J Clin Oncol 34:786-793, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trautmann M, Cyra M, Isfort I, et al. : Phosphatidylinositol-3-kinase (PI3K)/Akt signaling is functionally essential in myxoid liposarcoma. Mol Cancer Ther 18:834-844, 2019 [DOI] [PubMed] [Google Scholar]
- 26.Demicco EG, Torres KE, Ghadimi MP, et al. : Involvement of the PI3K/Akt pathway in myxoid/round cell liposarcoma. Mod Pathol 25:212-221, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sweeney RT, McClary AC, Myers BR, et al. : Identification of recurrent SMO and BRAF mutations in ameloblastomas. Nat Genet 46:722-725, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kurppa KJ, Caton J, Morgan PR, et al. : High frequency of BRAF V600E mutations in ameloblastoma. J Pathol 232:492-498, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gultekin SE, Aziz R, Heydt C, et al. : The landscape of genetic alterations in ameloblastomas relates to clinical features. Virchows Arch 472:807-814, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown NA, Rolland D, McHugh JB, et al. : Activating FGFR2-RAS-BRAF mutations in ameloblastoma. Clin Cancer Res 20:5517-5526, 2014 [DOI] [PubMed] [Google Scholar]
- 31.Rosell R: Mediating resistance in oncogene-driven cancers. N Engl J Med 368:1551-1552, 2013 [DOI] [PubMed] [Google Scholar]
- 32.Mittal P, Roberts CWM: The SWI/SNF complex in cancer - biology, biomarkers and therapy. Nat Rev Clin Oncol 17:435-448, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu JN, Roberts CW: ARID1A mutations in cancer: Another epigenetic tumor suppressor? Cancer Discov 3:35-43, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wiegand KC, Shah SP, Al-Agha OM, et al. : ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med 363:1532-1543, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Su YF, Tsai EM, Chen CC, et al. : Targeted sequencing of a specific gene panel detects a high frequency of ARID1A and PIK3CA mutations in ovarian clear cell carcinoma. Clin Chim Acta 494:1-7, 2019 [DOI] [PubMed] [Google Scholar]
- 36.Chandler RL, Damrauer JS, Raab JR, et al. : Coexistent ARID1A-PIK3CA mutations promote ovarian clear-cell tumorigenesis through pro-tumorigenic inflammatory cytokine signalling. Nat Commun 6:6118, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He X, Zhu Z, Johnson C, et al. : PIK3IP1, a negative regulator of PI3K, suppresses the development of hepatocellular carcinoma. Cancer Res 68:5591-5598, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bitler BG, Aird KM, Garipov A, et al. : Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med 21:231-238, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Juric D, Castel P, Griffith M, et al. : Convergent loss of PTEN leads to clinical resistance to a PI(3)Kalpha inhibitor. Nature 518:240-244, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Razavi P, Dickler MN, Shah PD, et al. : Alterations in PTEN and ESR1 promote clinical resistance to alpelisib plus aromatase inhibitors. Nat Cancer 1:382-393, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data used in this publication will be shared via the appropriate NCI-controlled access data sharing platform(s).