

Abstract
White matter (WM) is injured in most strokes, which contributes to functional deficits during recovery. Casein kinase 2 (CK2) is a protein kinase that is expressed in brain, including WM. To assess the impact of CK2 inhibition on axon recovery following oxygen glucose deprivation (OGD), mouse optic nerves (MONs), which are pure WM tracts, were subjected to OGD with or without the selective CK2 inhibitor CX-4945. CX-4945 application preserved axon function during OGD and promoted axon function recovery when applied before or after OGD. This protective effect of CK2 inhibition correlated with preservation of oligodendrocytes and conservation of axon structure and axonal mitochondria. To investigate the pertinent downstream signaling pathways, siRNA targeting the CK2α subunit identified CDK5 and AKT as downstream molecules. Consequently, MK-2206 and roscovitine, which are selective AKT and CDK5 inhibitors, respectively, protected young and aging WM function only when applied before OGD. However, a novel pan-AKT allosteric inhibitor, ARQ-092, which targets both the inactive and active conformations of AKT, conferred protection to young and aging axons when applied before or after OGD. These results suggest that AKT and CDK5 signaling contribute to the WM functional protection conferred by CK2 inhibition during ischemia, while inhibition of activated AKT signaling plays the primary role in post-ischemic protection conferred by CK2 inhibition in WM independent of age. CK2 inhibitors are currently being used in clinical trials for cancer patients; therefore, our results will provide rationale for repurposing these drugs as therapeutic options for stroke patients by adding novel targets.
Keywords: Stroke, white matter, myelin, oligodendrocyte, aging
Introduction
Casein kinase 2 (CK2) is a tetrameric protein kinase composed of two catalytic α-subunits (α and α’) and two regulatory β-subunits1. CK2 regulates many cellular functions that are important for brain development2 and cellular homeostasis3,4. Despite the initial lack of substrate following its discovery, CK2 was subsequently shown to phosphorylate numerous substrates, including other protein kinases, thus acting as a “master regulator” among protein kinases5,6 using both ATP and GTP7. Upregulation of CK2 activity is associated with many diseases, such as cancers8, cardiac hypertrophy9,10, and ischemic injury11,12. CK2 is expressed in brain and is associated with cellular injury12, and indeed CK2 has been reported to be a neuroprotectant by directly modulating NADPH oxidase activity during cerebral ischemia13. CK2 is also expressed in glial cells such as oligodendrocytes14 and a brief and moderate AMPA receptor activation in rat oligodendrocyte cell cultures triggers CK2 activity to mediate injury15,16. Consequently, CK2 inhibitors applied before AMPA receptor activation alleviated AMPA-mediated excitotoxic oligodendrocyte death16.
Stroke typically affects both the neurons in brain gray matter as well as glial cells and axons in brain white matter (WM). WM constitutes half of human brain volume and injury to axons largely translates into functional loss observed after a stroke. Because axons are natural extensions of neuronal cell bodies, it is often misconceived that similar mechanisms mediate injury to both portions of the cellular structure. This perception has been fortified by the fact that the rodent brain contains only ~10% WM and because commonly-used stroke models in rodents spare the corpus callosum, which is the main WM tract. Therefore, ischemic brain injury in rodent models overwhelmingly reflects the outcome of neuronal (gray matter) injury. WM is composed of axons, oligodendrocytes, microglia, and astrocytes17,18. Thus, WM is composed of a complex cellular environment in which glial cell-cell interactions intricately maintain axon function. As a result, mechanisms of WM injury significantly differ from gray matter. Consequently, protective interventions in neurons may become ineffective at promoting recovery of, or even become injurious to, WM. Therefore, an ideal stroke therapeutic must be directed towards both neuronal and axon-glia protection. The role of kinases remains unexplored in WM function and injury mechanisms; hence an intriguing question remains as to whether or not CK2, which has been previously shown to act as a master regulator and a neuroprotectant11,12, can protect WM against ischemia. The use of optic nerve as a pure WM tract provides a unique platform to test WM-specific injury mechanisms in isolation19–25. To our knowledge, this is the first demonstration that inhibition of a protein kinase before or after injury is protective of WM structure and function. Using specific small molecule inhibitors that cross the blood-brain barrier (BBB)26–29, we show that CK2 inhibition recruits both the AKT and CDK5 signaling pathways to confer protection to both young and aging WM structure and function when applied before injury, while AKT signaling specifically contributes to post-ischemic protection. We therefore suggest that CK2 inhibitors, which are currently in phase I-II clinical trials for cancer therapy, could be repurposed to provide a novel therapeutic target for ischemic stroke patients. The role of CK2 signaling in WM injury will also be applicable to other neurodegenerative conditions that are characterized by axonal injury, such as traumatic brain injury, Alzheimer’s disease, spinal cord injury, and multiple sclerosis.
Materials and Methods
Animals and chemicals
All experimental procedures were approved by The Institutional Animal Care and Use Committee (IACUC) of the Cleveland Clinic. The sources of the chemicals for electrophysiology experiments have been described in detail previously19. Selective kinase inhibitors against CK2 (CX-4945), cyclin-dependent kinase 5 (CDK5, roscovitine), the inactive conformation of AKT (MK-2206), and both active and inactive conformation of AKT (ARQ-092) were purchased from Selleckchem.com (Houston, TX). C57BL/6J adult male mice (young: 2–3 months of age; aging: 12 months of age and 20 months of age) were purchased from Jackson Laboratory (USA) and housed in the Cleveland Clinic animal facility under a 12 hour-light/dark cycle with food and water available ad libitum. Mice expressing neuronal mitochondrial-targeted CFP on a C57BL/6 background (Thy-1 mito-CFP30) were originally received by the senior author at the University of Washington and were later brought to and bred in the Cleveland Clinic animal facility.
Electrophysiology and oxygen glucose deprivation
Electrophysiology experiments in mouse optic nerves (MONs) were performed as previously described19. Briefly, MONs were obtained from adult male C57BL/6J mice (The Jackson Laboratory, USA) or Thy-1 mito-CFP mice at 2–3 months of age. Following CO2 asphyxiation, MONs were freed from their dural sheaths, placed in a Haas top perfusion chamber (Harvard Apparatus, Holliston, MA), and superfused with artificial cerebrospinal fluid (ACSF) with the following composition (in mM): 124 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 23 NaHCO3, and 10 d-glucose. All experiments were performed at 37°C. Glass suction electrodes filled with ACSF were used for stimulating (Isostim520; WPI, Sarasota, FL) and for recording compound action potentials (CAPs). The recording electrode was connected to an Axoclamp 900A amplifier (Molecular Devices, LLC, Sunnyvale, CA) and signals were amplified 20 or 50 times, filtered at 3 kHz, and acquired at 20–30 kHz (SR560, Stanford Research Systems, Sunnyvale, CA). Stimulus pulse strength (30 μs duration) was adjusted to evoke the maximum CAP possible, and then increased another 25% for supra-maximal stimulation. During experiments, the supramaximal CAP was elicited every 30s. MONs were allowed to equilibrate for at least 15 min in the recording chamber in normal ACSF. Oxygen glucose deprivation (OGD) was induced by switching to a glucose-free ACSF (replaced with equimolar sucrose to maintain osmolarity) and to a gas mixture containing N2 (95%) / CO2 (5%). Following 60 min of OGD, ACSF and O2 (95%) were restored and CAPs were recorded for up to 5 h.
Optic nerve function was monitored quantitatively as the area under the supramaximal CAP using Clampfit (version 10.2, Molecular Devices). The CAP area, which quantifies CAP amplitude and duration, is proportional to the total number of excited axons and represents a convenient and reliable means of monitoring optic nerve axon function 19,31,32. Another important factor in defining the CAP area is axon diameter, as larger axons are expected to generate larger CAP areas due to influx of Na variation in membrane potentials, and concentrations and densities of Na channels subtypes 33. Therefore, the CAP area is the best indicator of complex spatiotemporal summation of action potentials from individual axons 19. For comparing the CAP area between control and experimental groups, an average of 10 CAPs at baseline was measured. CAP areas from each experiment were normalized to baseline control levels for studying recovery after OGD. Experiments in the same group were pooled together, and the percentage of recovery was calculated in the normalized group’s time course. The recovery was monitored throughout and a 5 h recovery time point was used for comparison between groups.
Immunohistochemistry
Immunohistochemistry (IHC) experiments were conducted by standard methods used routinely in our laboratory22,23,25,34 with MONs collected after 5 h following OGD. Briefly, MONs were perfusion-fixed (4% paraformaldehyde and 0.025% glutaraldehyde) and cryosectioned. Sections of 16 μm thickness were blocked in 5% normal goat/donkey (50% by volume) serum (Sigma-Aldrich, St-Louis, MO) and 0.3% Triton X-100 (Sigma-Aldrich, St-Louis, MO) for 60 min at room temperature. Then, sections were incubated in primary antibody overnight at 4°C (see Table 1 for antibodies and dilutions used). Secondary antibodies used were Donkey anti-rabbit Cy5, anti-mouse Cy5, anti-mouse Cy3, and anti-rabbit Cy3 (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) prepared in 2% normal goat serum for 2 h.
Table 1.
Primary antibody list
| Antigen | Host | Catalog # | Dilution | Source |
|---|---|---|---|---|
|
| ||||
| APC | mouse | OP80 | 1:100 | Calbiochem |
| CK2-α | mouse | AB70774 | 1:500 | Abcam |
| GFAP | rabbit | 22522 | 1:15 | Immunostar |
| NF-200 | rabbit | N4142 | 1:100 | Sigma-Aldrich |
| Olig-2 | rabbit | AB9610 | 1:500 | EMD Millipore |
| PLP | rabbit | AB28486 | 1:250 | Abcam |
| SMI-31 | mouse | SMI31R | 1:100 | Biolegend |
| Sytox Green | 57020 | 1:25000 | EMD Millipore | |
Imaging and analysis
A Leica DMI6000 inverted confocal laser-scanning microscope (Buffalo Grove, IL) was used for digital image acquisition except for CFP imaging, where a Leica TCS SP511 upright multiphoton laser scanning microscope was used (Buffalo Grove, IL). Argon and He–Ne lasers were used to excite Sytox Green/488nm, Cy3/543 nm, and Cy5/647nm. Two to three adjacent sections from each MON were imaged for a total of five areas of interest (AOI). A total of 10–16 optical sections of 1μm thickness at 1024 X 1024 pixel size were collected in the z-axis from a single microscopic field using a 40X (HCX APO, water immersion; numerical aperture, 0.80; Leica, Buffalo Grove, IL) objective lens under fixed gain, laser power, pinhole, and photomultiplier tube (PMT) settings. CFP (+) mitochondrial pixel intensity was quantified in MONs among different conditions as reported previously25,34. CFP multiphoton laser wavelength was set at 900 nm and resolution was set at 512 X 512 pixels. The objective lens used was 25X (HCX IR APO, water immersion; numerical aperture, 0.95). Leica imaging software (LAS-AF ver. 2.7; Buffalo Grove, IL) was used to process images, following which ImageJ software35 was used to confirm co-localization and also to create projection images from image stacks. The Z-stacks obtained were used to create orthogonal views using ImageJ software in order to confirm co-localization between different channels.
Preparation of mouse optic nerve lysates and Western blotting
MON samples (two pairs) were collected at the end of experiments and homogenized (W-220F sonicator, Qsonica, LCC, Newtown, CT) on ice in 75 μL of Cell Lysis Buffer (Cell Signaling Technology, Danvers, MA) with protease inhibitor and phosphatase inhibitor cocktails (Sigma-Aldrich, St-Louis, MO). Samples were then centrifuged at 14,000 rpm for 10 min, the supernatant was collected, and the protein concentration was estimated using a bicinchoninic acid assay (BCA; Thermo Fisher Scientific, Grand Island, NY). Protein lysates were prepared in 4X laemmli sample buffer (Bio-Rad, Hercules, CA) and 2-mercaptoethanol (Sigma-Aldrich, St-Louis, MO) at a 10:1 ratio and incubated at 70°C for 10 minutes. Equal amounts (20 μg) of protein were loaded into each well of stain-free 4–20% Mini-Protean TGX gels (Bio-Rad, Hercules, CA). After gel electrophoresis and transfer, blots were blocked in 5% milk powder in TBS-Tween (0.1%) and incubated with rabbit GSK3β (1:1000; Cell Signaling Technology, Danvers, MA; 12456S), rabbit pGSK3β Ser9 (1:1000; Cell Signaling Technology, Danvers, MA; 5558S) and mouse β-actin (1:20,000; Sigma-Aldrich, St-Louis, MO; A5316) antibodies overnight at 4ºC. Blots were incubated with the secondary antibodies goat anti-mouse HRP (1:10,000; BioRad Hercules, CA; 1721011EDU) and goat anti-rabbit HRP (1:10,000; Thermo Fisher Scientific, Grand Island, NY; 31466) for 2 hours at RT. Chemiluminescence was detected using Clarity Western ECL substrate (Bio-Rad, Hercules, CA), imaged using a Bio-Rad Chemidoc MP, and analyzed using Image Lab software (ver. 6.0).
Cell cultures and Western blotting
HEK293T cells were cultured in DMEM with high glucose medium (with Sodium pyruvate; Sigma; D5671) supplemented with 10% fetal bovine serum (FBS; Gibco, ThermoFisher Scientific, Grand Island, NY; 10082147) plus 1X antibiotic-antimycotic solution (Gibco, ThermoFisher Scientific, Grand Island, NY; 15240062). MO3.13 cells were cultured in DMEM medium with high glucose lacking sodium pyruvate supplement (Sigma-Aldrich, St. Louis, MO; D5796) added with 10% FBS and 1X antibiotic-antimycotic solution. Transfections of HEK293T and MO3.13 cells were performed by Lipofectamine® RNAiMAX Transfection Reagent (ThermoFisher Scientific, Grand Island, NY; 13778100) following manufacturer’s instructions using siRNAs to Lipofectamine ratio of 1:2. Before transfection, cells were plated in 60mm culture dishes overnight. The next morning, cells at 60–70% confluency were transfected with 75 pmol siRNAs smart pool targeting human casein kinase 2 alpha gene (CSNK2A1, Dharmacon, Lafayette, CO; M-003475–03-0005). For the experiment, non-targeting siRNA pool#1 (Dharmacon, Lafayette, CO; D-001206–13-05) was used as negative control. After 4 days post-treatment, total protein was extracted from transfected HEK293T and MO3.13 cells in RIPA lysis buffer (ThermoFisher Scientific, Grand Island, NY; 89900) and supplemented with 1X protease and phosphatase inhibitor cocktails (ThermoFisher Scientific, Grand Island, NY; 1861281). 40μg of total protein was resolved on 4–12% SDS-PAGE gels and transferred to PVDF membranes (Invitrogen, ThermoFisher Scientific, Grand Island, NY; IB24002). Membranes was blocked in 5% nonfat dry milk in TBS-Tween and incubated overnight at 4°C with following primary antibodies: mouse anti-CK2α (1:1000; Abcam, Cambridge, MA; ab70774); mouse anti-CDK5 (1:1000, MilliporeSigma, Burlington, MA; 05–364), mouse anti-AKT1 (1:500; Abcam, Cambridge, MA; ab89402), rabbit pAKT1 (1:500; Abcam, Cambridge, MA; ab133458), rabbit GSK3β (1:1000; Cell Signaling Technology, Danvers, MA, 12456S), rabbit pGSK3β (1:1000; Cell Signaling Technology, Danvers, MA; 5558S), mouse anti-GAPDH (1:5000, Millipore Sigma, Burlington, MA; MAB374), and mouse actin (1:10000, Millipore Sigma; MAB1501 ). After washing in TBST, blots were incubated with peroxidase-conjugated anti-mouse (1:2500; GE Healthcare, Little Chalfont, England; NA931V) and anti-rabbit IgG (1:2500; GE Healthcare Little Chalfont, England; NA934V) for 1 hour at RT. Chemiluminescence was detected using Clarity Western ECL substrate (Bio-Rad Laboratories, Hercules, CA; 170–5061) and imaged using a Bio-Rad Chemidoc MP, and analyzed using Image Lab software (ver. 6.0).
Statistical analysis
All summarized data are presented as mean ± SEM. Graphpad Prism version 5.0 (Graphpad Software, Inc., La Jolla, CA) was used for statistical analysis. Statistical comparisons of the means were performed using unpaired two-tailed Student’s t-tests (Figures 5B, 5C, 5D, 5F, 5H, 6B, 6D, 7B, 7D, 10C and 10D) or one-way ANOVAs with Newman-Keuls post-hoc tests (Figure 1C, 1D, 2B, 5J, 8B, 8D, 9B, 10B, 10F and 10H) as appropriate. Differences were considered to be significant at p < 0.05. The n values indicate the number of optic nerves. The p values and significance are indicated individually for each figure in the text.
Figure 5. CK2 signals via CDK5 and AKT1S129/GSK3βS9.

siRNA against CK2α1 subunits in HEK293T cells (A) and MO3.13 cells (E) effectively suppressed CK2α1 subunit levels. Loss of CK2α1 is associated with significant reductions in CDK5 (HEK293T, A and B; MO3.13, E and F) as well as AKT1S129 phosphorylation (HEK293T, A; MO3.13, E) compared to total AKT1 (HEK293T, B; MO3.13, F). Furthermore, a decrease in CK2α1 subunit levels led to a decrease in GSK3βS9 phosphorylation (A and G) compared to total GSK3β (D and H). Interestingly, an increase in GSK3βS9 was observed in MONs following OGD and reperfusion; however, this was attenuated with CX-4945 (5 μM) pre-treatment (I and J). * p < 0.05, ** p < 0.01, and *** p < 0.001, unpaired Student’s two-tailed t-test or one-way ANOVA with Newman-Keuls post hoc test.
Figure 6. Roscovitine, a CDK5 inhibitor, and MK-2206, a pan-AKT inhibitor, promote axon function recovery following OGD.

Pre-treatment with Roscovitine (5 μM) (Roscovitine, Blue; A and B) applied before OGD promoted a consistent and sustained CAP area recovery. Pre-treatment with MK-2206 (5 μM) (MK-2206, Maroon C and D) applied before OGD promoted a consistent and sustained CAP area recovery. ** p < 0.01, unpaired Student’s two-tailed t-test.
Figure 7. Roscovitine or MK-2206 applied after OGD did not promote axon function recovery.

Roscovitine (5 μM) applied after OGD did not promote CAP area recovery (Roscovitine, Green; A and B). MK-2206 (5 μM) applied after OGD did not promote CAP area recovery (MK-2206, Salmon; C and D). NS, p > 0.05, unpaired Student’s two-tailed t-test.
Figure 10. CK2 inhibition promotes recovery in aging axons by differentially regulating the CDK5 and AKT pathways.

Pre-treatment with CX-4945 (5 μM) (CK2, red; A and B) applied before OGD promoted consistent and sustained CAP area recovery in aging axons. ARQ-092, which is an inhibitor of activated AKT, promoted axon function recovery when applied as a pre-treatment and as a post-treatment in both 12-month-old (12 m) and 20-month-old (20 m) animals. Pre-treatment with Roscovitine (5 μM) (Roscovitine, Blue; Graph C and inset) and MK-2206 (5 μM) (MK-2206, Maroon; Graph D and inset) before OGD promoted a consistent and sustained CAP area recovery. Pre-treatment with ARQ-092 (500 nM, dark blue, E, F, G and H) applied before OGD promoted a consistent and sustained CAP area recovery. ARQ-092 (500 nM, Brown, E, F, G and H) applied after OGD promoted consistent and sustained CAP area recovery. * p <0.05, ** p < 0.01 and *** p < 0.001, unpaired Student’s two-tailed t-test or one-way ANOVA with Newman-Keuls post hoc test.
Figure 1. The CK2 small molecule inhibitor, CX-4945, promotes function recovery in young axons following OGD.

Pre-treatment with CX-4945 (5 μM) (CK2, red; A and B) applied before OGD promoted a consistent and sustained CAP area recovery. A) Examples of CAP traces taken at baseline (a), OGD (b), and recovery (c) for control and CX-4945 (5 μM). Arrow shows sustained CAP at the end of OGD following CX-4945. B) Time course shows minimal recovery following OGD (black); pre-treatment with CX-4945 (5 μM, red) preserved CAP area during OGD and improved axon function following recovery. C) CX-4945 improved CAP area recovery at 1 and 5 μM, but conferred no protection at 10 μM, when compared to control CAP area recovery. D) CX-4945 had no effect on optic nerve baseline excitability. Scale bar = 1 ms. ** p < 0.01, one-way ANOVA with Newman-Keuls post hoc test.
Figure 2. CX-4945 applied after OGD promotes function recovery in young axons.

A) CX-4945 (5 μM, Magenta) applied after OGD promoted a consistent and sustained CAP area recovery. Insets shows examples of CAP traces taken at baseline (a), OGD (b), and recovery (c). B) CX-4945 improved CAP area recovery at 1 and 5 μM, but conferred no protection at 10 μM, when compared to control CAP area recovery. Scale bar = 1 ms. ** p < 0.001, one-way ANOVA with Newman-Keuls post hoc test.
Figure 8. ARQ-092, a novel pan-AKT inhibitor, promotes axonal recovery when applied pre- and post-OGD.

ARQ-092, which is an inhibitor of activated AKT, promoted axon function recovery when applied as a pre-treatment and as a post-treatment. A) Pre-treatment with ARQ-092 (500 nM, dark blue) applied before OGD promoted consistent and sustained CAP area recovery. B) ARQ-092 improved CAP area recovery at 250 nM and 500 nM when compared to control CAP area recovery. C) ARQ-092 (500 nM, brown) applied after OGD promoted consistent and sustained CAP area recovery. D) ARQ-092 improved CAP area recovery at 250 and 500 nM compared to control CAP area recovery. * p <0.05 and ** p < 0.01, one-way ANOVA with Newman-Keuls post hoc test.
Figure 9. CK2 inhibition preserves Thy-1 mito-CFP (+) mitochondria.

A) Two-photon confocal images of axonal CFP (+) mitochondria in MONs from Thy-1 mito-CFP mice after control, OGD, and OGD + CX-4945. OGD attenuated CFP fluorescence; in addition, mitochondria became smaller and less numerous. CX-4945 pre-treatment preserved CFP (+) mitochondria fluorescence and morphology. B) Histogram summarizing the data. Scale bar = 10 μm. *** p < 0.001, one-way ANOVA with Newman-Keuls post hoc test.
Results
CK2 inhibition preserves young WM function during OGD
To determine the impact of CK2 inhibition on axonal electrical activity following ischemia, MONs were initially superfused with control ACSF (30 min), then subjected to OGD (60 min), and then oxygen and glucose were reintroduced in the reperfusion period (~5 h). OGD gradually depressed axon function and caused complete loss of CAP area in 30.5 min, followed by 18 ± 3% recovery (Fig. 1A and B, black traces and time course). The selective CK2 inhibitor CX-4945 (5 μM) was included in the ACSF 30 min before OGD, during OGD, and maintained for 30 min following OGD during the reperfusion period. CX-4945 (5 μM) prevented the complete loss of CAP area during OGD (12 ± 5%, p <0.05, n = 5, Fig. 1A, arrow) and improved CAP area recovery to 62 ± 12% (n = 5, p <0.01; Fig. 1A and 1B). We observed that CX-4945 at 5 μM afforded the most protection, as less protection was observed with 1 μM or 10 μM CX-4945 (Fig. 1C). Because CK2 has been reported to regulate the expression and organization of Na+ and K+ channels at the nodes of Ranvier36,37, we confirmed that the axon protection mediated by CX-4945 was not due to alterations in axonal excitability. To this end, no changes in CAP area or shape were observed following 60 min of CX-4945 applied at different concentrations (Fig. 1D) during maximal stimulation, suggesting that CK2 inhibition did not alter axonal excitability.
To determine the clinical relevance and translational value of CK2 inhibition, we applied CX-4945 for 45 min after the end of OGD (Fig. 2A). Expectedly, OGD suppressed CAP area completely; however, CAP area recovered to 44 ± 4% (p <0.001, n=7). Similar to CX-4945 pre-application, post-OGD application of CX-4945 at 5 μM conferred the greatest amount of protection to CAP area recovery. CAP area recovery also improved when CX-4945 was applied after OGD at 1 μM or 10 μM, albeit to a lesser extent (Fig. 2B). These results show that CK2 inhibition provides a clinically-relevant window of opportunity to attenuate ischemic WM injury.
The CK2α subunit is expressed in glial cell compartments
We evaluated the expression and localization of CK2 in MONs using immunohistochemistry and glial cell-specific antibodies in conjunction with confocal imaging to support a biological basis for CK2 inhibitor action in our optic nerve preparation. The expression of CK2α subunit co-localized with GFAP (+) astrocyte nuclei and some processes (Fig. 3, top row) and also with NF-200 (+) axons (Fig. 3, bottom row). Almost all Olig2 (+) oligodendrocytes were strongly immunoreactive for CK2α (Fig. 3, second row). Immunolabeling was also evident on PLP (+) myelin (Fig. 3, third row). This extensive expression of CK2α in glial cells in addition to axons implicate them as cellular targets of CX-4945.
Figure 3. CK2α subunits are expressed in optic nerve astrocytes, myelin sheath, and oligodendrocytes.
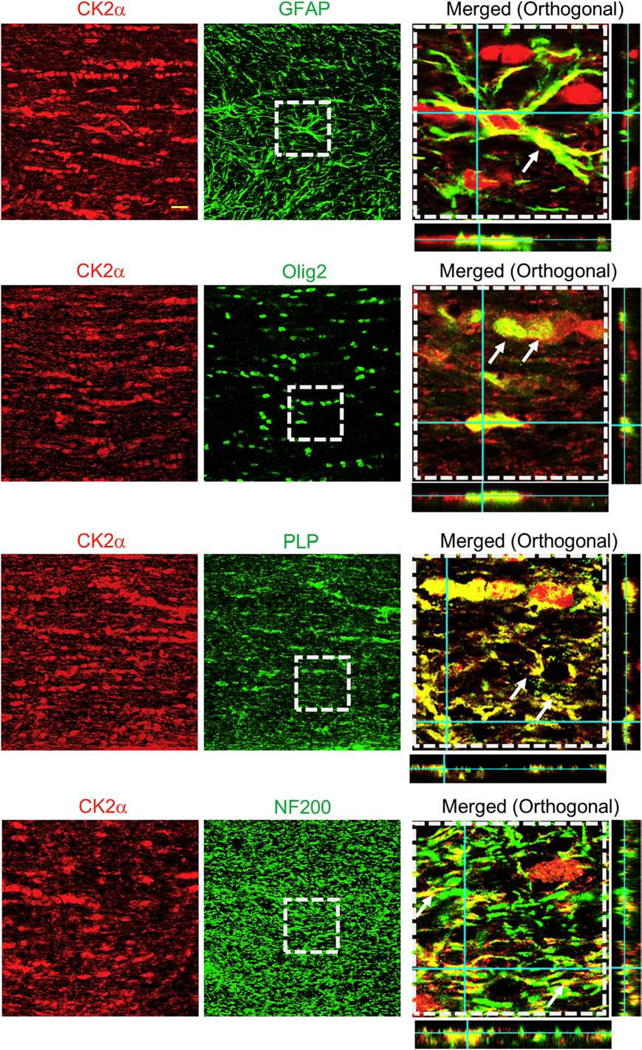
To identify the cellular expression of CK2α subunits in mouse optic nerve, CK2α was co-immunolabeled with glial fibrillary acidic protein (GFAP, astrocytes, top row), oligodendrocyte lineage transcription factor 2 (Olig2, oligodendrocytes, second row), myelin proteolipid protein (PLP, myelin, third row), and neurofilament protein (NF200, axons, bottom row). Note that the merged images (xy, xz and yz orthogonal view) in the right panels are enlarged areas (50 μm x 50 μm) indicated by the squares with dashed lines in the middle panels. Scale bar = 20 μm.
CK2 inhibition preserves WM structural integrity
We further verified the protective effect of CK2 inhibition that was revealed by electrophysiological analysis in a series of imaging studies (Fig. 4). We assessed the impact of CK2 inhibition on nuclear morphology, oligodendrocytes, and the axon cytoskeleton, which are the critical elements that show widespread injury after ischemia20,22,24,38. Under control conditions, oligodendrocyte nuclei labeled with Sytox were observed as blue and containing three to five nucleoli (Fig. 4, Control, white arrows). OGD caused extensive loss of APC (+) oligodendrocytes (magenta) with increased pyknotic nuclei characterized by small dense bright Sytox (+) nuclei in MONs imaged 5 h after 60 min of OGD (Fig. 4, OGD) as reported previously39. Similarly, the intensity and characteristic linear structure of SMI-31 (+) axonal labeling under control conditions (Fig. 4, Control) were completely disrupted after OGD (Fig. 4, OGD)22. Treatment of MONs with CX-4945 (5 μM) preserved oligodendrocytes and attenuated axonal injury and nuclei to an extent that was similar to control conditions (Fig. 4, OGD + CX-4945). These results suggest that inhibition of CK2α in astrocytes, oligodendrocytes, and/or the myelin sheath accounts for the improved oligodendrocyte survival following CK2 inhibition, thus improving axonal structural and functional recovery following OGD.
Figure 4. CK2 inhibition prevents OGD-induced axon injury and oligodendrocyte death.
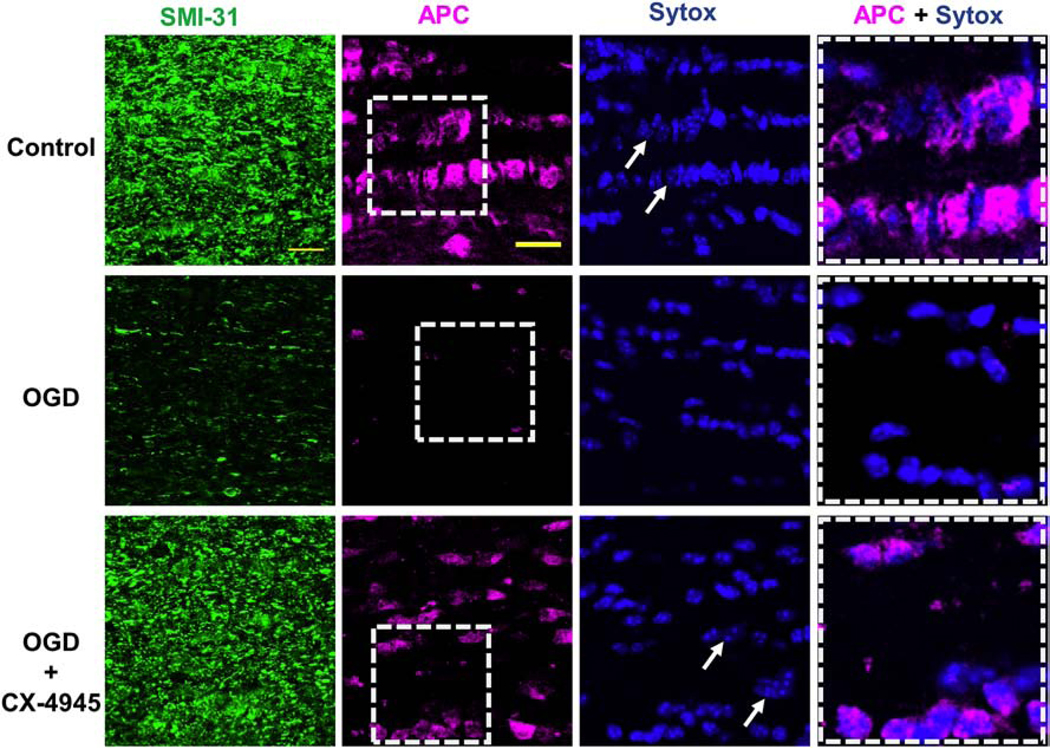
OGD (1 h) caused widespread loss of SMI-31 (+) axons (green) and adenomatous polyposis coli (APC (+), oligodendrocytes, magenta), while CX-4945 (5 μM) prevented axonal injury and oligodendrocyte loss. Glial nuclei were labeled with Sytox (blue, arrows). Note that the merged images in the right panels are enlarged areas indicated by the squares with dashed lines in the middle panels. Scale bar = 20 μm.
CK2 signals via the CDK5 and AKT/GSK3β pathways
CK2 signaling is an important and novel target for improving axonal recovery and it has effects when modified before or after ischemic injury, providing a wide therapeutic window and a high level of translational value. To identify the downstream molecules that mediate protection of WM against ischemia following CK2 inhibition, we performed experiments using siRNA directed against the CK2α subunit in lysates prepared from HEK293T cells and MO3.13 oligodendrocyte cells. Treatment with siRNA led to a profound reduction in CK2α subunit expression compared to scrambled (control) siRNA treatment (Fig. 5A, 5E and 5G). To assess the downstream signaling components altered by this loss of CK2 expression, protein levels of CDK5, AKT1, pAKT1S129, GSK3β, and pGSK3βS9 were determined. Results showed that suppressing CK2α signaling resulted in reduction of CDK5 levels (Fig 5C and 5F). Reduction of CK2α subunits also resulted in a concomitant significant reduction in CK2-specific AKT1S129 phosphorylation (pAKT1)40 (Fig. 5B and 5F). Consistent with these findings, decreased levels of CK2α subunits in both HEK293T and MO3.13 cell lines led to a significant decrease in GSK3β phosphorylation at S9 (GSK3βS9), which is a downstream effector of AKT41 (Fig. 5D and 5H). An increase in GSK3β phosphorylation at S9 has been shown to have an inhibitory effect on its downstream signaling42,43. Our results, therefore, provide proof-of-principle that loss of CK2 signaling leads to downregulation of CDK5 and regulation of downstream signaling through the AKT/GSK3β pathways.
To determine whether the protective effect of CK2 inhibition on axon function after ischemia was due to regulation of GSK3β, lysates obtained from MONs were probed with antibodies against GSK3β and pGSK3βS9 (Fig. 5I and 5J). Phosphorylation of the S9 residue of GSK3β has been reported to be differentially regulated during ischemia and reperfusion, where GSK3β is dephosphorylated and activated during ischemia, but phosphorylated and inhibited during reperfusion44. MONs showed a two-fold increase in GSK3βS9 phosphorylation after ischemia (1.9 ± 0.1%; p < 0.001, n=8) compared to controls (1.0 ± 0.02%; n=8); which was attenuated when MONs were treated with CX-4945 (Fig. 5I and 5J; 1.2 ± 0.06%; p < 0.001, n=8). These results suggest that the protective effect of CK2 inhibition during WM ischemia is mediated by regulating downstream GSK3β activity.
CK2 inhibition exerts post-ischemic protection via AKT signaling
CK2 inhibition promoted axon function recovery when applied before or after ischemia (Figs. 1 and 2) and experiments that downregulated CK2α levels using siRNA showed that these protective effects of CK2 inhibition can be mediated via CDK5 or AKT signaling. To further validate the signaling pathway(s) that mediated the protective effects of CK2 inhibition, specifically the signaling mediators that exerted post-ischemic protection to WM, we used specific small molecule inhibitors while monitoring the functional integrity of optic nerve axons. CDK5 is a CK2 effector that is expressed at nodes of Ranvier57,56 and in oligodendrocytes58. Roscovitine, which is a selective CDK5 small molecule inhibitor, was applied before OGD, and it prevented complete loss of CAP area during OGD (8 ± 4%; p < 0.05, n=6) and it improved CAP area recovery to 53 ± 7% after OGD (n=6, p < 0.01; Fig. 6A and 6B). Similarly, MK-2206, which is a selective AKT small molecule inhibitor, was applied before OGD and it prevented complete loss of CAP area during OGD (12 ± 5%; p < 0.05, n=5) and promoted axon functional recovery to 48 ± 4% after OGD (n=6, p < 0.01; Fig. 6C and 6D). Because both CDK5 and AKT inhibition prevented complete loss of CAP area during OGD and also improved CAP recovery following OGD, these results suggest that CDK5 and AKT are both molecular targets that contribute to WM ischemic injury.
To determine whether CDK5 and AKT inhibition provide post-injury protection, roscovitine or MK-2206 was applied for 45 min after the end of OGD (60 min). To our surprise, neither roscovitine (Fig. 7A and 7B) nor MK-2206 (Fig. 7C and 7D) afforded post-ischemic protection to axon function when applied after OGD. Because MK-2206 targets the inactive conformation of AKT, where the PH domain engages the kinase domain, thus preventing phosphorylation and activation45,46, we further tested the possibility that AKT inhibition after OGD required an inhibitor that targets the active conformation of AKT to confer post-OGD protection. For these experiments, we examined the effectiveness of a new selective allosteric pan-AKT inhibitor, ARQ-092, which targets the inactive and active conformations of AKT47,48 with equal potency. When applied before OGD, ARQ-092 (500 nM) improved CAP area recovery to 54 ± 1% (p < 0.01, Fig. 8A and 8B), which is similar to the effect of MK-2206. However, in contrast to MK-2206, ARQ-092 promoted axon function recovery to 43 ± 4% when applied after the end of OGD (p < 0.01, Fig. 8C and 8D). This evidence suggests that CDK5 and AKT inhibition are sufficient to protect axon function against ischemia when applied before injury, while inhibition of the active conformation of AKT is necessary to exert post-ischemic protection.
CK2 inhibition preserves mitochondrial integrity in WM
We previously reported that in WM, post-injury protection correlated with conservation of mitochondrial integrity during ischemic injury22,24,49. Therefore, we postulated that the protective effects of CK2 inhibition in promoting axon function recovery after ischemia would correlate with axonal mitochondrial preservation. Using MONs obtained from Thy-1 mito-CFP mice30, we monitored mitochondrial fluorescence during control conditions and during OGD with or without CX-4945 treatment. Mitochondria in control MONs displayed short tubular morphology (Fig. 9A, left panel). After OGD, there was a dramatic loss of mitochondrial CFP (+) fluorescence (Fig. 9A, middle panel). The remaining mitochondria exhibited a small punctate morphology that is typical of ischemia-induced mitochondrial fission39,50,51. Pretreatment of MONs with CX-4945 effectively attenuated the loss of mitochondrial fluorescence and preserved mitochondrial morphology (Fig. 9A, right panel, and 9B). These findings confirm our previous reports that approaches conferring post-ischemic protection to WM function correlate with preserved mitochondrial integrity.
CK2 inhibition preserves aging WM function during OGD
WM becomes increasingly susceptible to ischemic injury with age52,53. We have previously reported structural and functional changes to aging axons, myelin, and mitochondria that underlie the increased vulnerability of aging WM to ischemia21,24,54. Therefore, the effects of blocking CK2 and its downstream targets on axon function after OGD were determined in MONs obtained from 12-month-old (Fig. 10A–F) and 20-month-old male C57BL/6J mice (Fig. 10G–H). In these aging MONs, CAP area showed minimal recovery (8 ± 2%, n = 7) following OGD (60 min; Fig. 10A, black time course and Fig. 10B, black histogram). CX-4945 (5 μM) prevented complete loss of CAP area during OGD (12 ± 4%, p <0.01, n = 5, Fig. 10A, arrow) and improved CAP area recovery to 32 ± 3% (n = 5, p <0.001; Fig. 10A and 10B). Interestingly, post-OGD application of CX-4945 at 5 μM promoted CAP area recovery to 18 ± 3% (n = 3, p <0.01; Fig. 10A and 10B). Similar to young WM, we examined whether CDK5 and AKT inhibition promoted axon function recovery by using roscovitine or MK-2206 application before, during, and after OGD (60 min) in MONs obtained from 12-month-old mice. In contrast to young WM, roscovitine application before OGD in aging WM did not prevent the complete loss of CAP area during OGD; however, it improved CAP area recovery to 31 ± 6% after OGD (n = 4, p < 0.01; Fig. 10C). MK-2206, on the other hand, preserved CAP area during OGD in aging WM (11 ± 3%; p < 0.001, n = 4; Fig. 10D) and further promoted axon functional recovery to 39 ± 5% after OGD (n = 4, p < 0.001; Fig. 10D). Thus, CDK5 and AKT are both molecular targets contributing to ischemic injury in aging WM. In young WM, post-OGD protection was prominent when ARQ-092, which targets the inactive and active conformations of AKT47,48 with equal potency, was applied (Fig. 8C and 8D), but not with MK-2206 (Fig. 7C and 7D). Hence, we examined the effect of active AKT inhibition in aging WM. When applied before OGD, ARQ-092 (500 nM) improved CAP area recovery to 40 ± 4% (n = 4, p < 0.001, Fig. 10E and 10F) in 12-month-old WM and 33 ± 4% (n = 4, p < 0.001, Fig. 10E and 10F) in 20-month-old WM. Additionally, ARQ-092 promoted axon function recovery when applied after the end of OGD in both 12-month-old (28 ± 3%, n = 4, p < 0.001, Fig. 10G and 10H) and 20-month-old (20 ± 4%, n = 6, p < 0.01, Fig. 10G and 10H) WM. These findings suggest that in both young and aging WM, CK2 inhibition is a common target to improve axon function recovery and further that CDK5 and AKT are the effector molecules that mediate the protective effects. More importantly, the active conformation of AKT provides a universal target to improve functional recovery across these age groups.
Discussion
In this study, we show that CK2, which is a master kinase, is robustly expressed in glial cells and axons of WM and that CK2 inhibition before or after an ischemic episode promotes functional recovery of young and aging WM axons and preserves WM structure. Functional recovery mediated by CK2 inhibition correlated with preservation of oligodendrocytes, axon structure, and maintenance of axonal mitochondria. The main downstream mediators of the protective effects of CK2 inhibition were the CDK5 and AKT/GSK3ß signaling pathways. More importantly, while inhibition of CDK5 promoted axon function recovery when applied as a pretreatment, inhibition of the active conformation of AKT was necessary to promote functional recovery when applied after ischemia, suggesting that a clinically-relevant widow of opportunity exists for the use of CK2 inhibitors in ameliorating ischemic injury in WM.
One novel finding in our study is that CK2 inhibition preserved axon function and structure in WM against ischemia. Consistent with these findings, oligodendrocytes, astrocytes, myelin, and axons were found to express CK2α. The robust expression pattern of CK2α in glial cells and components is intriguing and suggests an extensive kinase regulation of WM structure and function37,55–57 and a significant role during ischemia13,58. These results are consistent with previous reports that CK2 levels and activity are increased in cerebral ischemia11,12,59, as well as in kidneys exposed to ischemia12. The evidence that CK2α is expressed in immature oligodendrocytes60 combined with our results suggest that CK2α is an important molecular target in WM for development and acute injury in experimental models43. Furthermore, because CK2α was shown to be expressed in primary human astrocytes57, CK2 inhibition may have important clinical implications. Future experiments will investigate whether glial cell-specific CK2 inhibition can confer protection to WM integrity.
However, in contrast, CK2 inhibition was not protective of neurons in an in vivo model of transient focal ischemia13. There may be several reasons to explain the lack of protection to neurons against ischemia. First, we used CX-4945, which is a specific small molecule inhibitor that crosses the BBB to block CK2α29, as opposed to inhibitor TBCA13, which was used in that study and could inhibit other protein kinases61,62. Second, some CK2 inhibitors, even those closely related chemically, have been reported to generate reactive oxygen species63. Third, strains of mice may differ in their sensitivity to ischemia or drugs (C57BL/6J versus CD1 mice13,64). Finally, the role of CK2 in neurons and in myelinated axons and glia may be different during an ischemic episode. This provides further evidence that mechanisms of ischemic injury are not the same in gray and WM portions of the brain, and therefore that assessing therapeutic interventions in both neuronal cell bodies and in axons is essential in order to achieve global protection and restoration of function in the brain after stroke.
A provocative aspect of our study was the demonstrated efficacy of CK2 inhibition by CX-4945 when administered after an ischemic episode. Ischemic injury in young WM follows a sequential order initiated by loss of ionic homeostasis leading to excitotoxicity and then merging into oxidative injury (Fig. 11); ionic deregulation directly impairs axon excitability, function, and structure due to toxic accumulation of Na+ and Ca2+ 65–68, whereas excessive glutamate accumulation overactivates AMPA/Kainate receptors, causing oligodendrocyte death and myelin disruption. Finally, the oxidative pathway attacks WM constituents via formation of reactive oxygen species69–71. Because functional protection was evident when CX-4945 was applied before or after glutamate accumulation, this finding may suggest that CX-4945 simultaneously targets the excitotoxic and oxidative pathways and may have multiple distinct sites of action: one related to glutamate accumulation and the other involving a post-excitotoxic mechanism, as we have previously shown22. This suggestion is supported by the observation that axon function was preserved during OGD and recovery was 50% higher with pre-injury CK2 inhibitor application compared to post-injury application. Axon function solely relies on local ATP production to maintain excitability via regulation of Na+-K+ ATPase activity. OGD causes a prominent reduction in ATP levels and loss of CFP (+) mitochondria, while CK2 inhibition resulted in sustained CFP (+) mitochondria22,23,25,72. Because CK2 is abundantly expressed by astrocytes, it is plausible that Na+-dependent glutamate release is modified, secondary to preservation of ATP levels and Na+ levels54, thus leading to reduced excitotoxic injury to oligodendrocytes. Alternatively, a reduction in AMPA/Kainate receptor signaling on oligodendrocytes may lead to reduced Ca2+ and Na+ entry, thus ameliorating injury18,73. Furthermore, CK2 phosphorylation of AMPA receptor GluA1 subunit regulates its membrane expression74 and regulation of oligodendrocyte NMDA receptor subunits may alter oligodendrocyte sensitivity to glutamate73,75. Regulation of oligodendrocyte NMDA receptors modulates metabolic support and interactions between oligodendrocytes and axons to impact functional recovery76. Furthermore, CK2 was shown to phosphorylate the NMDA receptor subunit GluN2B, leading to its internalization and replacement with GluN2A-containing NMDA receptors in postsynaptic densities to regulate synaptic activity in neurons77,78. Further experiments are currently underway to assess these possibilities. CK2 inhibition promoted axon function recovery, prevented oligodendrocyte death and axonal damage, and preserved axonal mitochondrial integrity against ischemia. Although the effectiveness of CDK5 and AKT inhibition remains to be quantified based on axonal recovery, we propose WM integrity is equally protected as CK2 inhibition since these are the downstream signaling pathways activated via CK2.
Figure 11. CK2 inhibition confers white matter functional protection against ischemia by differentially regulating the CDK5 and AKT signaling pathways.

Ischemic injury in young WM follows a sequential order initiated by loss of ionic homeostasis leading to excitotoxicity, and then merging into oxidative injury. Our findings suggest that ischemia in WM results in CK2 activation. Functional protection was evident when CX-4945, a CK2 inhibitor, was applied before or after glutamate accumulation, which may suggest that CX-4945 could simultaneously target the excitotoxic and oxidative pathways. CK2 recruits CDK5 and AKT/GSK3β signaling to mediate WM ischemic injury in a spatiotemporal manner such that CDK5 signaling becomes important during ischemia, while AKT signaling emerges as the main pathway during the reperfusion period following ischemia.
Another significant finding in our study was the demonstration that the axon-protective action associated with CK2 inhibition correlated with preservation of mitochondrial structure and function in axons (Fig. 11). Because of the consistent protection conferred by CK2 inhibition during OGD, improved axon function after injury may be a result of attenuated oxidative injury during the spatiotemporal progression of WM injury. The main evidence supporting this concept is that preservation of axonal mitochondrial integrity correlated with axon function recovery. We previously reported that loss of CFP (+) fluorescence in Thy-1 mito-CFP mice is an indicator of decreased ATP production22,25 and subsequently interventions that conserved mitochondrial integrity restored ATP production. CDK5 is suggested to have a direct impact on mitochondrial dynamics and function by increasing the generation of reactive oxygen species and phosphorylation of the mitochondrial fission protein Drp-1, resulting in mitochondrial defects79–84 and acting as a downstream signaling pathway upon CK2 activation. On the other hand, CDK5 inhibition following the ischemic period failed to exert protection to axon function, suggesting that CDK5 signaling during ischemia, but not during the recovery phase, is important to alleviate oxidative injury. The evidence that inhibition of active AKT confers post-ischemic protection to axon function suggests a novel effect of PTEN/AKT pathway activation in mediating mitochondrial injury via regulation of GSK3β in ischemic WM. AKT has been reported to upregulate GLT-1 in astrocytes85–87, which would contribute to increased glutamate release during OGD and ATP depletion, as well as enhanced excitotoxicity21. Consistent with this, AKT inhibition with MK-2206 or ARQ-092 promoted axon function recovery when applied before injury. Note that post-ischemic injury requires blockade by AQ-092, which is a specific blocker for the active form of AKT47,48, suggesting that AKT mediates ischemic WM injury once activated. Therefore, it is necessary to target this active form of AKT to promote recovery. Moreover, a member of the AKT/GSK3ß signaling pathway, the GSK3β isoform, is proposed as an important therapeutic target for cerebral ischemia88,89 and an intriguing relationship between GSK3β and mitochondria is emerging such that GSK3β inhibition reduces the generation of mitochondrial reactive oxygen species and neuronal damage90. GSK3β has been reported to play a dual role in myocardial ischemia, where the kinase is activated during ischemia and inhibited during reperfusion, and inhibition of GSK3β provided protection against ischemic injury44. We observed a similar increase in GSK3βS9 phosphorylation following reperfusion in WM indicative of inactivation of the kinase. CK2 inhibition during ischemia attenuated the inactivation of GSK3β during reperfusion and improved axon function recovery. Our results suggest that CK2 inhibition protects axonal structural integrity and function by either directly maintaining axonal metabolism and/or indirectly by preserving oligodendrocyte health. In addition, future studies will be important to verify whether GSK3β is a universal target that protects both gray and WM against stroke.
It is intriguing that inhibition of the CDK5 and AKT pathways provides similar functional protection to aging axons. The impact of aging on WM is of general interest because the global effects of aging on myelinated nerve fibers are more intricate and extensive than those in cortical gray matter. Aging axons are larger, have thicker myelin, and are characterized by longer and thicker mitochondria that correlate with lower ATP levels and increased generation of nitric oxide, protein nitration, and lipid peroxidation24. Moreover, disruption in Ca2+ homeostasis and defective unfolded protein responses in aging axons are common24. Consequently, excitotoxic and oxidative injury dominate ischemic injury mechanisms in aging WM and interventions that protect young WM become ineffective or impede recovery in aging WM. Because ARQ-092 provides post-ischemic protection to aging axon function, even to MONs obtained from old mice (20-months-old), we propose that AKT is a common molecular target to protect WM function independent of age.
Our findings concerning the effectiveness of CK2 inhibitors have implications for treating various forms of brain injury. The WM is injured in most strokes, contributing to the disability associated with clinical deficits. Because CK2 inhibition provides post-ischemic protection and protects young and aging WM, CK2 inhibitors may provide a suitable option to protect WM in the clinical manifestations of stroke to facilitate successful translation of experimental stroke research to clinical trials. It is also of interest whether CK2 inhibition confers preconditioning of WM to subsequent ischemia to be considered as a preventive therapeutic. These studies are currently being conducted.
Conclusions
CK2 inhibition protects young and aging WM function against an ischemic episode by preserving oligodendrocytes and axonal structure by maintaining mitochondrial integrity. CK2 recruits CDK5 and AKT/GSK3ß signaling to mediate WM ischemic injury in a differential spatiotemporal manner such that CDK5 signaling becomes important during ischemia, while AKT signaling emerges as the main pathway during the reperfusion period following ischemia (Fig. 11). Subsequently, interventions selectively targeting the activated form of AKT confer post-ischemic functional recovery in young and aging WM. These findings warrant evaluation of the roles of CDK5 and AKT signaling in other WM-related diseases such as multiple sclerosis, traumatic brain injury, and spinal cord injury, as well as in neurodegenerative diseases such as Alzheimer’s disease and in cerebral WM injury.
Highlights.
CK2α subunit is expressed in glial cells and axons in WM.
CK2 inhibition preserves young and aging WM function during ischemia.
CK2 signaling differentially regulates the CDK5 and AKT/GSK3β signaling pathways to protect WM.
CDK5 and AKT inhibition protect axon function against ischemia when applied to both young and aging WM before injury.
AKT inhibition selectively confers post-ischemic protection to both young and aging WM, providing a common therapeutic target independent of age.
Acknowledgments
This study was supported by the NIH NINDS (1R21NS094881) to SB and SB; NIH NIA (AG033720) to SB and NIH NS096148 (RD).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: None
References
- 1.Pinna LA & Allende JE Protein kinase CK2 in health and disease: Protein kinase CK2: an ugly duckling in the kinome pond. Cell Mol Life Sci 66, 1795–1799 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lou DY et al. The alpha catalytic subunit of protein kinase CK2 is required for mouse embryonic development. Mol Cell Biol 28, 131–139 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blanquet PR Casein kinase 2 as a potentially important enzyme in the nervous system. Prog Neurobiol 60, 211–246 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Brunet S, Emrick MA, Sadilek M, Scheuer T. & Catterall WA Phosphorylation sites in the Hook domain of CaVβ subunits differentially modulate CaV1.2 channel function. J. Mol. Cell. Cardiol 87, 248–56 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meggio F. & Pinna L. a. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 17, 349–68 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Trembley JH et al. Emergence of Protein Kinase CK2 as a Key Target in Cancer Therapy. Biofectors. 36, 187–95 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Litchfield DW Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem. J 369, 1–15 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sarno S. et al. Toward the rational design of protein kinase casein kinase-2 inhibitors. Pharmacol Ther 93, 159–168 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Hauck L. et al. Protein kinase CK2 links extracellular growth factor signaling with the control of p27(Kip1) stability in the heart. Nat Med 14, 315–324 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Eom GH et al. Casein kinase-2alpha1 induces hypertrophic response by phosphorylation of histone deacetylase 2 S394 and its activation in the heart. Circulation 123, 2392–2403 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Hu BR & Wieloch T. Casein kinase II activity in the postischemic rat brain increases in brain regions resistant to ischemia and decreases in vulnerable areas. J Neurochem 60, 1722–1728 (1993). [DOI] [PubMed] [Google Scholar]
- 12.Ka SO et al. The protein kinase 2 inhibitor tetrabromobenzotriazole protects against renal ischemia reperfusion injury. Sci Rep 5, 14816 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim GS, Jung JE, Niizuma K. & Chan PH CK2 is a novel negative regulator of NADPH oxidase and a neuroprotectant in mice after cerebral ischemia. J. Neurosci 29, 14779–89 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huillard E. et al. Disruption of CK2beta in embryonic neural stem cells compromises proliferation and oligodendrogenesis in the mouse telencephalon. Mol Cell Biol 30, 2737–2749 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Canedo-Antelo M, Matute C. & Sanchez-Gomez MV Protein kinase CK2 and JNK modulate pro-apoptotic effector activation in AMPA-induced excitotoxicity in oligodendrocytes. Neurogune 2nd Basque Neuroscience meeting 1748 (2014). [Google Scholar]
- 16.Canedo-Antelo M, Llavero F, Zugaza J, Matute C. & Sanchez-Gomez M. Inhibition of Casein Kinase 2 reduces AMPA-induced oligodendrocyte death through JNK signaling and ER stress regulation. XII European Meeting on Glial Cells in Health and Disease T05–05b (2015). [Google Scholar]
- 17.Fields RD White Matter. Sci. Am 298, 54–61 (2008). [PubMed] [Google Scholar]
- 18.Baltan S. Ischemic injury to white matter: an age-dependent process. Neuroscientist 15, 126–133 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Stys PK, Ransom BR & Waxman SG Compound action potential of nerve recorded by suction electrode: a theoretical and experimental analysis. Brain Res 546, 18–32 (1991). [DOI] [PubMed] [Google Scholar]
- 20.Tekkok SB, Ye Z. & Ransom BR Excitotoxic mechanisms of ischemic injury in myelinated white matter. J Cereb Blood Flow Metab 27, 1540–1552 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Baltan S. et al. White matter vulnerability to ischemic injury increases with age because of enhanced excitotoxicity. J Neurosci 28, 1479–1489 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baltan S, Murphy SP, Danilov CA, Bachleda A. & Morrison RS Histone deacetylase inhibitors preserve white matter structure and function during ischemia by conserving ATP and reducing excitotoxicity. J Neurosci 31, 3990–3999 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baltan S. Histone deacetylase inhibitors preserve function in aging axons. J Neurochem 123 Suppl, 108–115 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stahon KE et al. Age-related changes in axonal and mitochondrial ultrastructure and function in white matter. J. Neurosci 36, 9990–10001 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murphy SP et al. MS-275, a class I histone deacetylase inhibitor, protects the p53-deficient mouse against ischemic injury. J Neurochem 129, 509–515 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vita M. et al. Tissue distribution, pharmacokinetics and identification of roscovitine metabolites in rat. Eur J Pharm Sci 25, 91–103 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Sallam H. et al. Age-dependent pharmacokinetics and effect of roscovitine on Cdk5 and Erk1/2 in the rat brain. Pharmacol Res 58, 32–37 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Cheng Y. et al. MK-2206, a novel allosteric inhibitor of Akt, synergizes with gefitinib against malignant glioma via modulating both autophagy and apoptosis. Mol Cancer Ther 11, 154–164 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng Y. et al. Targeting protein kinase CK2 suppresses prosurvival signaling pathways and growth of glioblastoma. Clin Cancer Res 19, 6484–6494 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Misgeld T, Kerschensteiner M, Bareyre FM, Burgess RW & Lichtman JW Imaging axonal transport of mitochondria in vivo. Nat Methods 4, 559–561 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Cummins KL, Perkel DH & Dorfman LJ Nerve fiber conduction-velocity distributions. I. Estimation based on the single-fiber and compound action potentials. Electroencephalogr. Clin. Neurophysiol 46, 634–46 (1979). [DOI] [PubMed] [Google Scholar]
- 32.Cummins KL, Dorfman LJ & Perkel DH Nerve fiber conduction-velocity distributions. II. Estimation based on two compound action potentials. Electroencephalogr. Clin. Neurophysiol 46, 647–58 (1979). [DOI] [PubMed] [Google Scholar]
- 33.Hodgkin AL & Katz B. The effect of sodium ions on the electrical activity of the giant axon of the squid. J. Physiol 108, 37–77 (1949). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baltan S, Bachleda A, Morrison RS & Murphy SP Expression of histone deacetylases in cellular compartments of the mouse brain and the effects of ischemia. Transl. Stroke Res 2, 411–23 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schneider CA, Rasband WS & Eliceiri KW NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cerda O. & Trimmer JS Activity-dependent phosphorylation of neuronal Kv2.1 potassium channels by CDK5. J Biol Chem 286, 28738–28748 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brechet A. et al. Protein kinase CK2 contributes to the organization of sodium channels in axonal membranes by regulating their interactions with ankyrin G. J Cell Biol 183, 1101–1114 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tekkök SB & Goldberg MP Ampa/kainate receptor activation mediates hypoxic oligodendrocyte death and axonal injury in cerebral white matter. J. Neurosci 21, 4237–48 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baltan S, Murphy SP, Danilov CA, Bachleda A. & Morrison RS Histone deacetylase inhibitors preserve white matter structure and function during ischemia by conserving ATP and reducing excitotoxicity. J. Neurosci 31, 3990–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Di Maira G. et al. Protein kinase CK2 phosphorylates and upregulates Akt/PKB. Cell Death Differ 12, 668–677 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Manning BD & Toker A. AKT/PKB Signaling: Navigating the Network. Cell 169, 381–405 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.He J-X, Gendron JM, Yang Y, Li J. & Wang Z-Y The GSK3-like kinase BIN2 phosphorylates and destabilizes BZR1, a positive regulator of the brassinosteroid signaling pathway in Arabidopsis. Proc. Natl. Acad. Sci. U. S. A 99, 10185–90 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang G. et al. HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3β/PTEN/Akt axis. Proc. Natl. Acad. Sci. U. S. A 112, 2853–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhai P, Sciarretta S, Galeotti J, Volpe M. & Sadoshima J. Differential roles of GSK-3β during myocardial ischemia and ischemia/reperfusion. Circ. Res 109, 502–11 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barnett SF et al. Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem. J 385, 399–408 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hirai H. et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 9, 1956–1967 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Lapierre JM et al. Discovery of 3-(3-(4-(1-Aminocyclobutyl)phenyl)-5-phenyl-3H-imidazo[4,5-b]pyridin-2-yl)pyridin-2-amine (ARQ 092): An Orally Bioavailable, Selective, and Potent Allosteric AKT Inhibitor. J. Med. Chem 59, 6455–6469 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Yu Y. et al. Targeting AKT1-E17K and the PI3K/AKT pathway with an allosteric AKT inhibitor, ARQ 092. PLoS One 10, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baltan S. Age-dependent mechanisms of white matter injury after stroke. in White matter injury in stroke and CNS disease (eds. Baltan S, Carmichael ST, Matute C, Xi G. & Zhang JH) 373–403 (Springer US, 2014). [Google Scholar]
- 50.Bastian C, Politano S, Day J, Mccray A. & Brunet S. Mitochondrial dynamics and preconditioning in white matter. Cond. Med 1, (2018). [PMC free article] [PubMed] [Google Scholar]
- 51.Bastian C. et al. NOS3 inhibition preserves young and aging white matter structure and function during ischemia by conserving ATP and mitochondrial dynamics. J. Neurosci In press, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baltan S. et al. White matter vulnerability to ischemic injury increases with age because of enhanced excitotoxicity. J. Neurosci 28, 1479–89 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baltan S. Histone deacetylase inhibitors preserve function in aging axons. J. Neurochem 123, 108–115 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baltan S. Excitotoxicity and mitochondrial dysfunction underlie age-dependent ischemic white matter injury. Adv. Neurobiol 11, 151–70 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moreno FJ, Diaz-Nido J, Jimenez JS & Avila J. Distribution of CK2, its substrate MAP1B and phosphatases in neuronal cells. Mol Cell Biochem 191, 201–205 (1999). [PubMed] [Google Scholar]
- 56.Yoshimura T. & Rasband MN Axon initial segments: Diverse and dynamic neuronal compartments. Current Opinion in Neurobiology 27, 96–102 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rosenberger AFN et al. Increased occurrence of protein kinase CK2 in astrocytes in Alzheimer’s disease pathology. J. Neuroinflammation 13, 4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shichinohe H. et al. Neuroprotective effects of cilostazol are mediated by multiple mechanisms in a mouse model of permanent focal ischemia. Brain Res. 1602, 53–61 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Kim SO et al. Ischemia induced activation of heat shock protein 27 kinases and casein kinase 2 in the preconditioned rabbit heart. Biochem Cell Biol 77, 559–567 (1999). [PubMed] [Google Scholar]
- 60.Huillard E. et al. Disruption of CK2 β in Embryonic Neural Stem Cells Compromises Proliferation and Oligodendrogenesis in the Mouse Telencephalon. Mol. Cell. Biol 30, 2737–2749 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pagano MA et al. Optimization of protein kinase CK2 inhibitors derived from 4,5,6,7-tetrabromobenzimidazole. J. Med. Chem 47, 6239–47 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Pagano MA et al. Tetrabromocinnamic acid (TBCA) and related compounds represent a new class of specific protein kinase CK2 inhibitors. Chembiochem 8, 129–39 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Schneider CC, Hessenauer A, Gotz C. & Montenarh M. DMAT, an inhibitor of protein kinase CK2 induces reactive oxygen species and DNA double strand breaks. Oncol Rep 21, 1593–1597 (2009). [DOI] [PubMed] [Google Scholar]
- 64.Sheldon RA, Sedik C. & Ferriero DM Strain-related brain injury in neonatal mice subjected to hypoxia-ischemia. Brain Res. 810, 114–22 (1998). [DOI] [PubMed] [Google Scholar]
- 65.Stys PK, Ransom BR & Waxman SG Effects of polyvalent cations and dihydropyridine calcium channel blockers on recovery of CNS white matter from anoxia. Neurosci Lett 115, 293–299 (1990). [DOI] [PubMed] [Google Scholar]
- 66.Fern R, Ransom BR & Waxman SG Voltage-gated calcium channels in CNS white matter: role in anoxic injury. J. Neurophysiol 74, 369–77 (1995). [DOI] [PubMed] [Google Scholar]
- 67.Wolf JA, Stys PK, Lusardi T, Meaney D. & Smith DH Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J. Neurosci 21, 1923–30 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Underhill SM & Goldberg MP Hypoxic injury of isolated axons is independent of ionotropic glutamate receptors. Neurobiol Dis 25, 284–290 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oka A, Belliveau MJ, Rosenberg PA & Volpe JJ Vulnerability of oligodendroglia to glutamate: pharmacology, mechanisms, and prevention. J. Neurosci 13, 1441–53 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Back SA et al. Selective vulnerability of preterm white matter to oxidative damage defined by F2-isoprostanes. Ann. Neurol 58, 108–20 (2005). [DOI] [PubMed] [Google Scholar]
- 71.Juurlink BH Response of glial cells to ischemia: roles of reactive oxygen species and glutathione. Neurosci. Biobehav. Rev 21, 151–66 (1997). [DOI] [PubMed] [Google Scholar]
- 72.Baltan S, Morrison RS & Murphy SP Novel protective effects of histone deacetylase inhibition on stroke and white matter ischemic injury. Neurotherapeutics 10, 798–807 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baltan S. Age-specific localization of NMDA receptors on oligodendrocytes dictates axon function recovery after ischemia. Neuropharmacology (2015). doi:S0028-3908(15)30107-6 [pii] 10.1016/j.neuropharm.2015.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lussier MP, Gu X, Lu W. & Roche KW Casein kinase 2 phosphorylates GluA1 and regulates its surface expression. Eur J Neurosci 39, 1148–1158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Spitzer S, Volbracht K, Lundgaard I. & Káradóttir RT Glutamate signalling: A multifaceted modulator of oligodendrocyte lineage cells in health and disease. Neuropharmacology 110, 574–585 (2016). [DOI] [PubMed] [Google Scholar]
- 76.Saab AS et al. Oligodendroglial NMDA Receptors Regulate Glucose Import and Axonal Energy Metabolism. Neuron 91, 119–32 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sanz-Clemente A, Gray JA, Ogilvie KA, Nicoll RA & Roche KW Activated CaMKII couples GluN2B and casein kinase 2 to control synaptic NMDA receptors. Cell Rep 3, 607–614 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sanz-Clemente A, Matta JA, Isaac JT & Roche KW Casein kinase 2 regulates the NR2 subunit composition of synaptic NMDA receptors. Neuron 67, 984–996 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Klinman E. & Holzbaur EL Stress-Induced CDK5 Activation Disrupts Axonal Transport via Lis1/Ndel1/Dynein. Cell Rep 12, 462–473 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cherubini M, Puigdellivol M, Alberch J. & Gines S. Cdk5-mediated mitochondrial fission: A key player in dopaminergic toxicity in Huntington’s disease. Biochim Biophys Acta 1852, 2145–2160 (2015). [DOI] [PubMed] [Google Scholar]
- 81.Jahani-Asl A. et al. CDK5 phosphorylates DRP1 and drives mitochondrial defects in NMDA-induced neuronal death. Hum Mol Genet 24, 4573–4583 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Park J. et al. Loss of mitofusin 2 links beta-amyloid-mediated mitochondrial fragmentation and Cdk5-induced oxidative stress in neuron cells. J Neurochem 132, 687–702 (2015). [DOI] [PubMed] [Google Scholar]
- 83.Morel M, Authelet M, Dedecker R. & Brion JP Glycogen synthase kinase-3beta and the p25 activator of cyclin dependent kinase 5 increase pausing of mitochondria in neurons. Neuroscience 167, 1044–1056 (2010). [DOI] [PubMed] [Google Scholar]
- 84.Sun KH, de Pablo Y, Vincent F. & Shah K. Deregulated Cdk5 promotes oxidative stress and mitochondrial dysfunction. J Neurochem 107, 265–278 (2008). [DOI] [PubMed] [Google Scholar]
- 85.Zhang X. et al. Ginsenoside Rd promotes glutamate clearance by up-regulating glial glutamate transporter GLT-1 via PI3K/AKT and ERK1/2 pathways. Front Pharmacol 4, 152 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ji YF et al. Upregulation of glutamate transporter GLT-1 by mTOR-Akt-NF-small ka, CyrillicB cascade in astrocytic oxygen-glucose deprivation. Glia 61, 1959–1975 (2013). [DOI] [PubMed] [Google Scholar]
- 87.Li LB et al. Regulation of astrocytic glutamate transporter expression by Akt: evidence for a selective transcriptional effect on the GLT-1/EAAT2 subtype. J Neurochem 97, 759–771 (2006). [DOI] [PubMed] [Google Scholar]
- 88.Koh S-H, Yoo AR, Chang D-I, Hwang SJ & Kim SH Inhibition of GSK-3 reduces infarct volume and improves neurobehavioral functions. Biochem. Biophys. Res. Commun 371, 894–9 (2008). [DOI] [PubMed] [Google Scholar]
- 89.Cowper-Smith CD, Anger GJA, Magal E, Norman MH & Robertson GS Delayed administration of a potent cyclin dependent kinase and glycogen synthase kinase 3 beta inhibitor produces long-term neuroprotection in a hypoxia-ischemia model of brain injury. Neuroscience 155, 864–75 (2008). [DOI] [PubMed] [Google Scholar]
- 90.Valerio A. et al. Glycogen synthase kinase-3 inhibition reduces ischemic cerebral damage, restores impaired mitochondrial biogenesis and prevents ROS production. J. Neurochem 116, 1148–59 (2011). [DOI] [PubMed] [Google Scholar]