

Abstract
The continued improvement of combinatorial CRISPR screening platforms necessitates the development of new computational pipelines for scoring combinatorial screening data. Unlike for single-guide RNA (sgRNA) pooled screening platforms, combinatorial scoring for multiplexed systems is confounded by guide design parameters such as the number of gRNAs per construct, the position of gRNAs along constructs, and additional features that may impact gRNA expression, processing or capture. In this protocol we describe Orthrus, an R package for processing, scoring and analyzing combinatorial CRISPR screening data that addresses these challenges. This protocol walks through the application of Orthrus to previously-published combinatorial screening data from the CHyMErA experimental system, a platform we recently developed that pairs Cas9 with Cas12a gRNAs and affords the programmed targeting of multiple genomic sites. We demonstrate Orthrus’ features for screen quality assessment and two distinct scoring modes for dual-guides (dgRNAs) that target the same gene twice or dgRNAs that target two different genes. Running Orthrus requires basic R programming experience and approximately 5–10 minutes of computational time.
EDITORIAL SUMMARY:
This protocol describes the use of Orthrus, an R package for processing, scoring and analyzing combinatorial CRISPR screening data, including data produced by the CHyMErA experimental system.
Introduction
Genetic tools that systematically map genetic interactions are powerful hypothesis-generating technologies for both basic research as well as drug discovery. Genetic interactions (GIs) are defined as the phenomenon by which combinatorial mutations in multiple genes result in phenotypic effects that are greater or less than expected, given the phenotypes of the individual mutants1. Pioneering work in yeast resulted in a nearly-complete map of yeast pairwise GIs, which illuminated the functions of uncharacterized genes, revealed functional connections between pathways2, and enabled the systematic characterization of compound mode-of-action3,4.
The study of human genetic interactions at scale has recently been made possible in human cell culture systems with the development of CRISPR/Cas-enabled genetic screens5–9. These experiments induce mutations across any number of human genes in pooled cell culture and typically work by targeting each gene with a small number of Cas9 guides that are individually expressed in cells. Pooled CRISPR screens can be performed in various genetic backgrounds to identify different effects, such as in cancer cell lines to uncover cancer-specific genetic dependencies10 or in isogenic cell lines to map genetic interactions11. Combinatorial CRISPR screening platforms were also developed to directly identify GIs by knocking multiple genes out through the expression of multiple guides within the same cell. These platforms function by pairing individual Cas9 guide RNAs (gRNAs) with each other, combining orthologous Cas9 gRNAs, or by multiplexing multiple Cas12a gRNAs11–19. We recently developed a novel combinatorial screening platform named Cas Hybrid for Multiplexed Editing and Screening Applications (CHyMErA) that instead targets genes with hybrid guide RNAs (hgRNAs) composed of Cas9 guides fused with one or more additional Cas12a guides, which can subsequently be processed into individual guides by Cas12a’s RNA-processing activity12.
Although accurate scoring of genetic interaction data requires precise quantitative measurements2,11, there is a lack of computational tools that enable quantitative genetic interaction scoring for combinatorial experimental designs. To date, most combinatorial CRISPR screening studies have scored data by taking simple or weighted averages of log fold-change values (LFCs) between start and end read counts for dual guide RNAs (dgRNAs) targeting specific gene pairs with additional corrections based on control guides or the phenotypes of similar dgRNAs13–16. While this approach may be suitable for non-combinatorial screens, different Cas enzymes12, as well as the position of gRNAs on combinatorial guide constructs16, may strongly affect guide efficiency. This necessitates the development of combinatorial scoring methods which take these effects into account.
To address issues arising from the combinatorial nature of data output from CHyMErA and other platforms, we developed a novel scoring method named Orthrus for combinatorial CRISPR screening data. The key feature of Orthrus is that it takes orientation - whether gene A is targeted by a gRNA in position 1 and gene B is targeted by a gRNA in position 2, or vice versa - into account during scoring, which is necessary to consider for Cas9 and Cas12a guides that can cause different fitness effects even when targeting the same gene. This scoring method is bundled in a well-documented R package with a variety of features to simplify combinatorial data processing, quality control and analysis, and is downloadable at https://github.com/csbio/Orthrus. Here, we present the recommended Orthrus workflow, demonstrate its key features on previously-published data from two separate combinatorial screening experiments, discuss important considerations for performing quality-control analyses of screening data, and detail expected results from the application of Orthrus to combinatorial screening data.
Development of the Protocol
The Orthrus package implements the GI scoring schema presented in Gonatopoulos-Pournatzis et al.12, although with several key improvements that include more sensitive scoring, a variety of quality control plots and metrics, and a new user interface. Most broadly, Orthrus presents a consistent user interface for scoring any kind of combinatorial screening data stored in a delimited text file. The code that was used to score data in Gonatopoulos-Pournatzis et al.12 does not generalize to other experiments or combinatorial screening platforms. This new user interface also provides push-button functions that automatically generate a variety of quality control plots and metrics useful for assessing the quality of most types of screening data. In addition, the Orthrus package implements several features absent from the previously published scoring code. These features improve the sensitivity of scoring and user confidence in resulting hits and include guide filtering based on plasmid pool or early timepoint read counts, loess-normalization of residual effects, and moderated t-testing in addition to the original Wilcoxon rank-sum testing. Lastly, given properly-formatted input files, the Orthrus package provides several different scoring interfaces which enable partial or complete automation of the scoring process.
Comparisons with other Methods
We are aware of five existing scoring methods for combinatorial CRISPR screens: ΔLFC, GIMap, π-score, Norm-GI and GEMINI13–15,17,20. All scoring methods compare the null model of multiplicative single-gene effects, typically derived from intragenic guides paired with intergenic controls (exonic-intergenic guides), to the observed effects of exonic-exonic guides. The simplest proposed method is ΔLFC13, which directly performs this comparison while deriving empirical FDRs from permuted data. GIMap, Norm-GI and π-score all perform the same comparison with some additional corrections. GIMap normalizes exonic-intergenic guides to a quadratic fit and residual effects to negative control guide effects17. Norm-GI normalizes residual effects to control guide phenotypes, as well as to the phenotypes of similar guides using a moving average across bins15. The π-score weights exonic-exonic guides, giving higher preference to guides with stronger phenotypes14. GEMINI is a Bayesian approach that explicitly models sample-independent and sample-dependent effects and uses coordinate ascent variational inference to update the posteriors of exonic-intergenic effects20. Of these five scoring methods, only GEMINI exists in a generalized, runnable form as an R package.
Orthrus primarily differs from GEMINI in how it accounts for orientation, whether it computes effects relative to control genes, the types of guides it is designed to score, and the number of auxiliary functions it offers. GEMINI does not account for guide orientation, which is an important consideration for CHyMErA screening data. Furthermore, GEMINI computes p-values and FDRs relative to a specified set of negative control genes, which may be appropriate for whole-genome screens but is not necessarily appropriate for screens performed with specialized libraries. GEMINI similarly computes effect size based on a set of positive control genes, which may or may not be available depending on the library design and interrogated phenotype. Orthrus, on the other hand, does not rely on negative or positive control genes during scoring, and can identify significant hits even for moderate phenotypes due to the use of moderated t-testing. The combination of these choices allows Orthrus to score multiple different types of guides, as it can directly score combinatorial guides against single-targeting controls, as well as chemogenetic screens against non-treated controls or single-targeting guides. Lastly, unlike GEMINI, Orthrus presents the user with a host of data processing, quality control and plotting functions to assess screen quality.
Applications of the Method
The Orthrus package is generally applicable to a variety of combinatorial CRISPR screening settings. While Orthrus’ orientation-based filtering is designed to minimize false positives or discrepancies due to guide orientation, this scoring approach requires double the amount of hypothesis testing per gene pair and may be overly conservative in some settings. For applications such as CHyMErA screens, this conservative approach should be adopted (see Step 15 of Procedure 2). For multiplexing screens with single nucleases (either Cas9 or Cas12a), the user should instead reduce the amount of hypothesis tests performed per gene pair by ignoring orientation-specific effects (see Step 4 of Procedure 3). In addition to flexible scoring functions, Orthrus provides an extensive selection of data processing and quality control functions that are applicable to any combinatorial screening dataset and can be applied irrespective of downstream scoring methods.
Limitations
The Orthrus package currently supports the analysis of data from negative selection CRISPR screens with combinatorial guide libraries targeting A) a single gene of interest twice, B) two different genes, and C) a gene paired with a control region. It has been tested on CHyMErA data for all of these guide types as well as for combinatorial-targeting guides from a different multiplexed Cas12a platform, as described in the Procedures below. Genes of interest could be targeted in either exonic or intronic regions depending on the screens’ experimental design, although Orthrus has only been tested to score the effects of gRNAs targeting exonic regions. While Orthrus is currently the only scoring package that accounts for orientation-specific effects, this feature can be disabled to increase statistical power for screens where guide orientation is less relevant. Thus, Orthrus is flexible enough to score any combinatorial data for all guide types listed above. While Orthrus is not designed to score combinatorial positive selection or drug rescue experiments, a combination of stringent guide filtering and scoring with Wilcoxon rank-sum hypothesis testing may be appropriate for these types of screens. However, further testing on genome-scale combinatorial positive selection screens is necessary to assess Orthrus’ ability to score this type of data.
Expertise Needed to Implement the Protocol
Basic experience with R programming is required. Experience analyzing CRISPR screen data is recommended, but not required.
Input Format
Orthrus requires one mandatory and two optional (but encouraged) tab-separated input files: a mandatory reads file, and the two optional sample and batch files. The reads file contains read count information for all screens, and is required for Orthrus to function. Its construction from raw sequencing data is detailed in Procedure 1. The sample file maps replicate columns to their matching screens, but unlike similar files required by other packages, it also maps screens to other screens they must be normalized against (e.g. reference time point screens, plasmid pools). The batch file maps screens to other screens they must be scored against, such as for drug treatment screens against control screens. While Orthrus provides manual options detailed in Procedure 2 that offer precise input methods for specifying the information contained in sample and batch files, the sample and batch files can drastically reduce the complexity of data processing and scoring - as demonstrated in Procedures 2 and 3 - and are thus highly encouraged. In addition to the detailed descriptions below, please refer to this video tutorial for a conceptual overview of these three input files, which is available to view as Supplementary Video 1 or at https://youtu.be/w8mGWnQ-9Wo.
Reads file.
Like other packages that score CRISPR screening data, Orthrus requires input data formatted as a delimited text file where rows correspond to guides and columns correspond to metadata and raw read counts for all screens. However, unlike alternative scoring methods, Orthrus requires two gene label columns whose position reflects the orientation of each guide. In detail, Orthrus requires the following assumptions about the input file’s format to be met.
All guides in the dataset (every row) must have non-empty labels for the genomic regions they target contained in two separate columns.
These columns must be labeled gene1 and gene2. Although these will typically contain gene symbol annotations, they may contain any identifier desired by the user.
The gene1 column must contain gene labels for the first guide in the dataset and the gene2 column must contain gene labels for the second guide in the dataset. The definition of “first” and “second” is left to the user. For example, the dataset analyzed in Procedure 2 contains genes targeted by Cas9 guide sequences in the gene1 column and Cas12a guide sequences in the gene2 column, whereas the dataset analyzed in Procedure 3 contains genes targeted by Cas12a in both columns.
To enable Orthrus’ default scoring mode, each gene label column must map to a respective guide ID column. For example, the gene1 and gene2 columns for the dataset analyzed in Procedure 2 map to the columns Cas9.Guide and Cpf1.Guide (Cas12a was previously named Cpf1), respectively. Orthrus does not assume a standardized name for guide ID columns, and instead, the user passes in the name of guide ID columns during the processing step. The first guide ID column name passed in maps to gene1 and the second maps to gene2. For libraries where single-targeting guides do not share guide IDs with combinatorial-targeting guides, the less-sensitive Wilcoxon rank-sum scoring approach implemented in Orthrus does not require this information.
For “dual-targeting” guides which target the same gene twice, the gene1 column must contain the name of the targeted gene and the gene2 column must contain the string None.
For single-targeting guides paired with a standardized negative control, such as an intergenic region for the dataset analyzed in Procedure 2 or a non-essential gene for the dataset analyzed in Procedure 3, the control must be named NegControl in the corresponding gene label column.
While these assumptions may require users to manually pre-process their reads file, this format is a concise way to represent both orientation-specific information as well as guide type information for any combinatorial screen. Although alternative scoring methods also require tab-delimited files with identifying gene labels and guide ID columns, they do not take either orientation or guide type into account (to which assumptions 3–6 above relate). An example format for a reads file containing mock data is shown in Table 2. This table contains the gene name columns gene1 and gene2, the guide ID columns Cas9 guide and Cas12a guide, and reads for two technical replicates in the columns T0 reads and T18 reads.
Table 2.
Example of a properly-formatted reads file for mock data of one gene pair. Lines 1 and 2 represent combinatorial-targeting guides knocking out both ARID1A and ARID1B simultaneously, lines 3–6 represent single-targeting guides knocking out either ARID1A (lines 3–4) or ARID1B (lines 5–6) paired with a negative control guide in both orientations, and lines 7–8 represent dual-targeting guides cutting either ARID1A (line 7) or ARID1B (line 8) twice. Guide sequences and read counts are mock data for illustrative purposes.
| gene1 | gene2 | Cas9 guide | Cas12a guide | T0 reads | T18 reads |
|---|---|---|---|---|---|
| ARID1A | ARID1B | AATG | TTGC | 45 | 0 |
| ARID1B | ARID1A | CGAC | TATT | 54 | 1 |
| ARID1A | NegControl | AATG | CGCT | 70 | 60 |
| NegControl | ARID1A | GGTA | TATT | 82 | 75 |
| ARID1B | NegControl | CGAC | CGCT | 61 | 87 |
| NegControl | ARID1B | GGTA | TTGC | 76 | 92 |
| ARID1A | None | AATG | TATT | 91 | 42 |
| ARID1B | None | CGAC | TTGC | 63 | 53 |
Additionally, because the CHyMErA reads file downloadable at https://crispr.ccbr.utoronto.ca/chymera/index.html contains several formatting errors, the script used to reformat the CHyMErA data into the version bundled in the Orthrus package download is uploaded to the Zenodo repository available here and is demonstrated in Procedure 1 Step 6.
Sample file.
The sample file must be a tab-separated file containing exactly three columns named Screen, Replicates, and NormalizeTo. The Screen column contains unique users-pecified labels for each screen. The Replicates column contains a list of read column names for all technical replicates corresponding to that screen in the reads file, separated by semicolons. The NormalizeTo column contains the name of a different screen in the Screen column to normalize the current screen against (e.g. a T0 screen or plasmid pool). The sample file for Procedure 2 is shown in Table 3.
Table 3.
Sample table used in Procedure 2, Step 4B. Each row corresponds to a single screen, with the Replicates column containing the names of its technical replicates separated by semicolons and the NormalizeTo column containing the name of a screen to normalize against (e.g. a T0 screen).
| Screen | Replicates | NormalizeTo |
|---|---|---|
| HAP1_T0 | HAP1.T0 | NA |
| RPE1_T0 | RPE1.T0 | NA |
| HAP1_T12 | HAP1.T12A;HAP1.T12B;HAP1.T12C | HAP1_T0 |
| HAP1_T18 | HAP1.T18A;HAP1.T18B;HAP1.T18C | HAP1_T0 |
| Torin_T12 | HAP1.Torin.T12A;HAP1.Torin.T12B;HAP1.Torin.T12C | HAP1_T0 |
| Torin_T18 | HAP1.Torin.T18A;HAP1.Torin.T18B;HAP1.Torin.T18C | HAP1_T0 |
| RPE1_T18 | RPE1.T18A;RPE1.T18B;RPE1.T18C | RPE1_T0 |
| RPE1_T24 | RPE1.T24A;RPE1.T24B;RPE1.T24C | RPE1_T0 |
Batch file.
The batch file must be a tab-separated file containing exactly two columns named Screen and Control. The Screen column contains labels for screens listed in the Screen column of the sample file which the user wants to score against screens listed in the Control column. To score combinatorial-targeting guides against null models derived from single-targeting guides, instead specify combn in the Control column. The batch file for Procedure 2 is shown in Table 4.
Table 4.
Batch table used in Procedure 2, Step 19. Each row corresponds to a single screen, with the Screen column containing the names of screens defined in the sample table and the Control column containing either the names of screens to score against for the dual-targeted scoring mode or “combn” to score them with the combinatorial-targeting mode.
| Screen | Control |
|---|---|
| Torin_T12 | HAP1_T12 |
| Torin_T18 | HAP1_T18 |
| HAP1_T12 | combn |
| HAP1_T18 | combn |
| RPE1_T18 | combn |
| RPE1_T24 | combn |
| Torin_T12 | combn |
| Torin_T18 | combn |
Experimental Design
Workflow
The recommended workflow for combinatorial CRISPR screen analysis with Orthrus involves the following key steps, as shown in Fig. 1. First, the computational workspace is set up with appropriate variables that describe screen information in a read count matrix. Second, read counts are processed and normalized to sequencing depth as well as to screens from earlier timepoints, if provided. During this step, a variety of quality control (QC) plots are output, which include QC plots for raw read counts as well as for normalized log fold-changes (LFCs). Based on the results of QC plots and metrics, this step may be repeated several times with updated filtering and normalization options. Third, LFCs are scored and analyzed in separate ways for guides that target one gene multiple times (dual-targeted scoring) or guides that target different genes with the same construct (combinatorial scoring).
Figure 1.
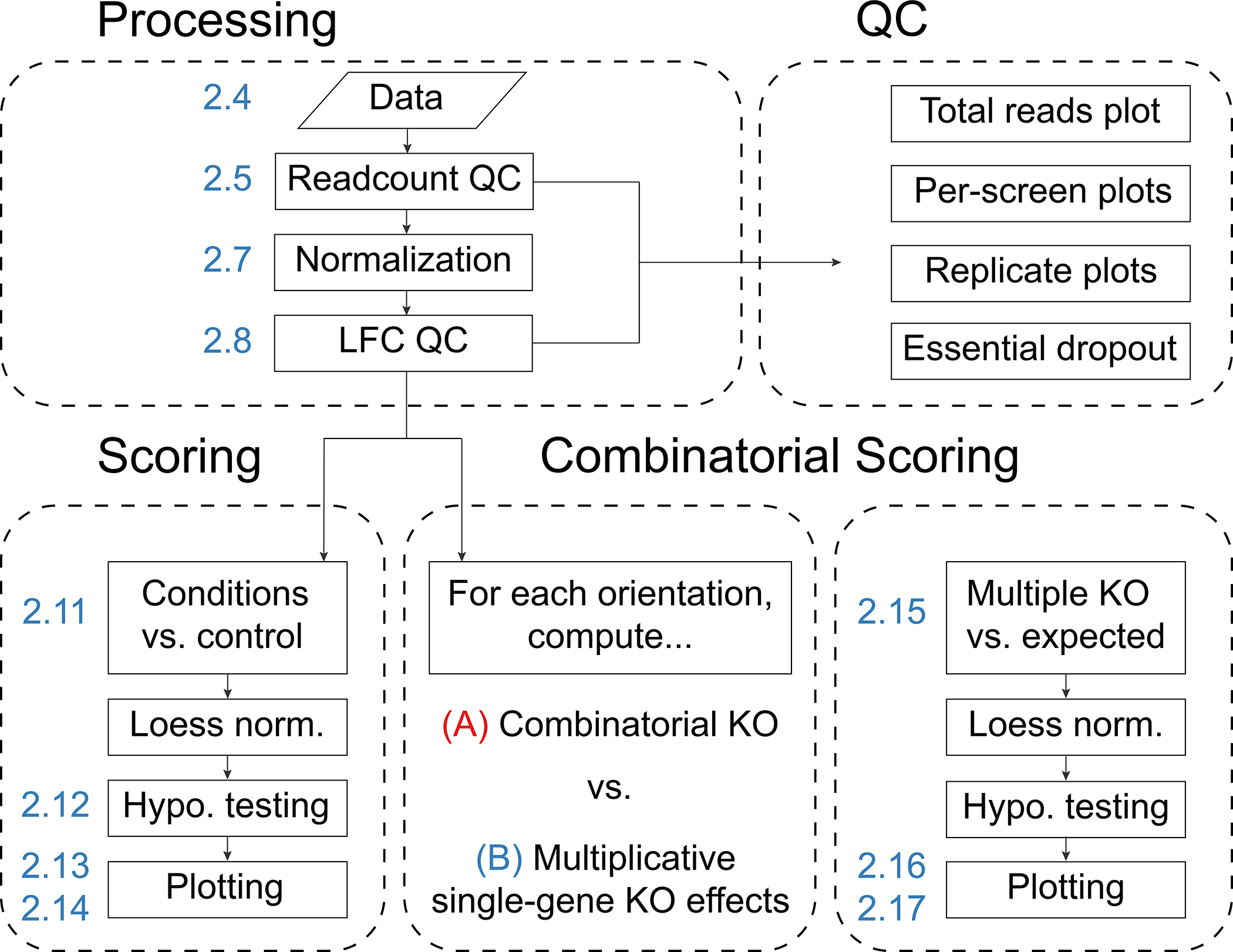
The Orthrus scoring workflow. First, combinatorial screening data is processed and depth-normalized before LFCs between late and early timepoints are computed. During this step, QC plots for raw read count data as well as LFC data are output. Second, data is scored, either for condition screens against control screens (e.g. for drug-treated screens against untreated screens), or for the effects of combinatorial knockouts against expected effects derived from single knockouts. Numeric labels, in blue, indicate the corresponding step of Procedure 2.
Scoring modes
Orthrus offers two primary scoring modes and one additional scoring mode for different types of guides.
The dual-targeting mode scores guides that target one gene multiple times across different conditions. This mode is suitable for drug screening applications. For example, in previous CHyMErA screens12, dual-targeting guides cut each gene in two separate exonic regions, and these guides are scored for differences between drug treated (Torin1) and untreated cells.
As an extension to the dual-targeting scoring mode, the single-targeting mode scores single-targeting guides that cut both a single gene and a control region across different conditions. The gene of interest could be cut in an exonic or intronic region, while the control gRNA typically targets an intergenic region or nonessential gene. This mode uses the same interface as the dual-targeting scoring mode with different parameters.
The combinatorial-targeting mode scores guides that target multiple genes. This mode scores genetic interactions. In previous CHyMErA screens, combinatorial-targeting guides cut two different genes in a single exonic region per gene. The effect of double-knockouts induced by these guides is scored against the estimated effect of double-knockouts derived from single-targeting guides that target each gene separately (while paired with guides targeting control regions - see below). For experiments which investigate intronic function, this mode could instead score the effect of dgRNAs which target intronic regions of multiple genes. This mode can also be configured to ignore orientation-specific effects, which increases statistical power for experiments with few observable orientation-specific signatures.
For a given gene pair’s set of guides, all of Orthrus’ scoring modes compare LFC values for an effect of interest against LFC values for control effects with either moderated t-testing or Wilcoxon rank-sum testing. Users may also loess-normalize their residual effects to account for non-normality in their data. For most purposes, we recommend that users run moderated t-testing with loess-normalization enabled. However, for screens where guide-level residuals cannot be computed because single-targeting controls do not share guide IDs with combinatorial-targeting dgRNAs (see the Input Format subsection below), Wilcoxon rank-sum testing without loess normalization must be applied instead. A description of important parameters and the algorithms applied by Orthrus during the scoring process, as well as their typical use cases, is provided in Table 1.
Table 1.
Select parameters of Orthrus’ normalization and scoring functions with their associated algorithm (when applicable) and a description of their typical use case.
| Parameter | Algorithm | Description | Typical use case |
|---|---|---|---|
| scaling_factor | scaling factor for LFC computation | Scales raw read counts to a default value of 1e6 that forces each screen to the chosen read depth, ensuring comparability across technical replicates. The specific choice of scaling_factor is largely irrelevant | All screens |
| pseudocount | pseudocount for LFC computation | Adds a pseudocount to each raw read count, by default 1, as required to take log2-normalized read counts. Smaller pseudocounts, e.g. between 1 and 5, are advised to avoid de-prioritizing moderate effects | All screens |
| test | “moderated-t” - moderated t-testing | Computes p-values via Empirical Bayes estimate across all residuals fit with separate linear models for each gene pair. Calls limma’s eBayes function on its lmFit function applied to residuals with default parameters for both24 | Most screens |
| test | “rank-sum” - Wilcoxon rank-sum testing | Computes p-values via Wilcoxon rank-sum testing between effect and control LFCs | Combinatorial screens with unpaired controls |
| loess | “TRUE” - Loess normalization with MA transformation | Normalizes by fitting a loess curve with degree 2 and a span of 0.4 to MA-transformed residuals. The MA transformation was originally developed for the analysis of microarray data25. Here, loess fits a trend for the measured residual value ([double mutant - null model] or [condition - control]) vs. the sum of the two values used in computing this residual (eg. [double mutant + null model] or [condition + control]). | Most screens |
| fdr_method | “BY” - Benjamini-Yekutieli FDR correction | Adjusts p-values with Benjamini-Yekutieli FDR correction | Most screens |
| fdr_method | “BH” - Benjamini-Hochberg FDR correction | Adjusts p-values with Benjamini-Hochberg FDR correction | Low-signal screens |
| fdr_method | “bonferroni” - bonferroni FDR correction | Adjusts p-values with Bonferroni multiple hypothesis correction | High-signal screens |
| filter_genes | N/A | Genes to filter out from scoring process | Remove technical controls or flagged genes |
| ignore_orientation | N/A | If TRUE, groups guides from both orientations for each gene pair together to reduce the amount of hypothesis testing by half | Cas12a-Cas12a or low-signal screens |
Scoring interfaces
Orthrus provides three different interfaces to the steps listed in the Workflow subsection: a manual interface that allows fine-grained control over each step, a batch scoring interface for users to process data manually and score it automatically, and a wrapper interface that runs the entire Orthrus pipeline in a single function call. After construction of the reads file from raw sequencing data in Procedure 1, the first interface is demonstrated in Procedure 2, and the latter two interfaces are demonstrated in Procedure 3. All three interfaces to Orthrus are described below, and their corresponding function calls are summarized in Fig. 2.
Figure 2.
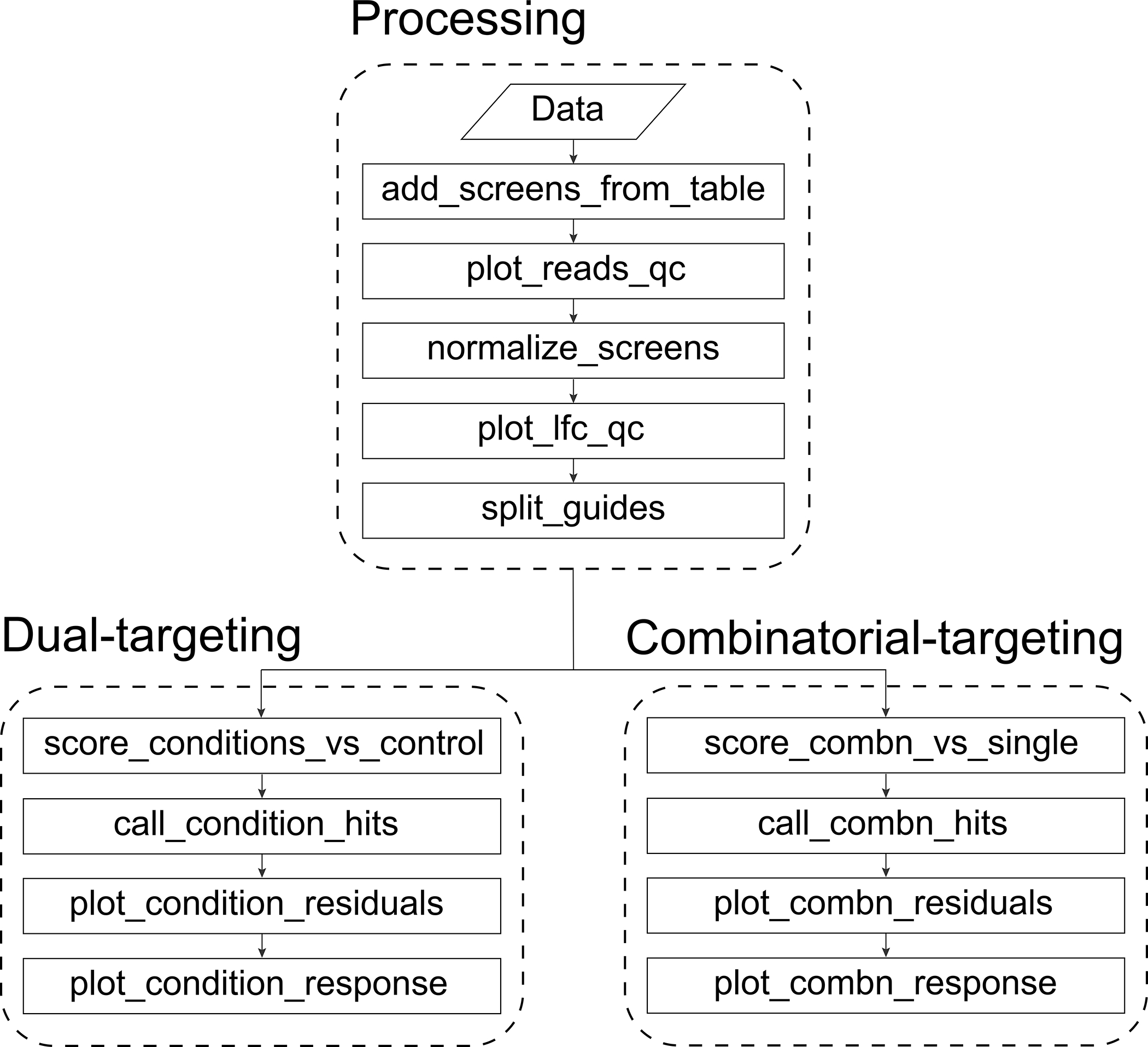
Specific Orthrus functions to call in order for data processing as well as dual-targeting and combinatorial-targeting scoring.
The manual interface is the most verbose and allows users to score specific screens in different ways. For instance, with this interface users may choose to score screens with different FDR thresholds, or may alter the parameters of plots generated for different screens (e.g. to relabel hits in figure legends). This interface requires a minimum of 13 Orthrus function calls for an experiment with both dual-targeting and combinatorial-targeting guides and may require significantly more for complex experimental designs.
The batch scoring interface is more succinct than the manual interface and affords users a similar level of control as the manual interface. Scoring many screens with this interface requires only one line of code, for a minimum (and typically, a maximum) of seven Orthrus function calls across the entire workflow. However, this interface requires that users score all guides of a specific type with the same parameters across all screens. For large-scale experiments where scores should be computed in a standardized way, this behavior is desired.
The wrapper interface allows users to score their data with a single Orthrus function call. While convenient, this interface is only recommended for users with a deep understanding of both their own data as well as how specific parameters applied to different steps of the scoring process affect their results.
For typical experimental designs, the batch scoring interface is recommended. Orthrus separates the processing and scoring steps in order to encourage users to manually examine their data at key breakpoints during an analysis session. Ideally, after running Orthrus’ processing steps, users will refer to the variety of automatically-generated QC plots and metrics that Orthrus outputs to inform their parameter choices. Users will then choose to either proceed to the scoring steps with their current parameter choices or choose to re-process their data with changed parameters (or other additions, such as manual filters for problematic guides). While the manual interface also encourages this behavior, the batch scoring interface is far more succinct and additionally forces the user to choose common parameters for scoring different screens, which facilitates the generation of results that are more comparable across screens.
Preparation stage
During the preparation stage, users first format their reads file, their sample table, and their batch table as described in the Input Format subsection. They load these files into their workspace and proceed to the processing stage.
Processing stage
Users run Orthrus’ processing and QC functions as shown in Steps 5–10 of Procedure 2 or Step 3 of Procedure 3. After running these functions and manually examining QC plots and metrics for their data, users then decide whether or not to change certain parameters (e.g. to filter low-readcount guides) and either re-run this step or proceed to the scoring stage.
Scoring stage
Finally, users call Orthrus’ scoring functions either manually or with the batch scoring interface. The manual interface requires users to dual-targeting, combinatorial-targeting and single-targeting guides separately, but the recommended batch scoring interface allows users to score all three types of guides at the same time.
After scoring their data, users are encouraged to examine their final results and decide to either keep their current parameters or re-run the processing and scoring stages with different parameters. Some of the most consequential parameters include the choice of hypothesis testing (we recommend moderated t-testing as opposed to Wilcoxon rank-sum testing for most screens), whether or not to loess-normalize residual effects, and the choice of FDR and effect size thresholds for hit-calling. Table 1 provides more information on these and other parameters, as well as their recommended use cases.
Chosen data for procedures
The procedures below demonstrate the application of the Orthrus package for analyzing two combinatorial CRISPR screening example datasets. The first dataset analyzed in Procedures 1 and 2 consists of both raw sequencing data as well as a pre-processed reads file from CHyMErA screens described in Gonatopoulos-Pournatzis et al12. The second dataset analyzed in Procedure 3 contains screens from a separate multiplexed Cas12a system described in Dede et al18. All data for Procedures 1 and 3 is downloadable from the Zenodo repository here, whereas the processed reads file and required tables for Procedure 2 are bundled with Orthrus’ download.
Materials
Equipment
Software
-
Procedure 1
Tested with Bowtie 0.12.9, although any Bowtie version 1 is applicablePerl 5Bash
R version 3.6 or greater
-
Procedures 2 and 3
-
R version 3.6 or greater with the packages listed below and in Step 1 of Procedure 2
devtools
Orthrus
ggplot2
ggthemes
pheatmap
PRROC
RColorBrewer
BiocManager
limma
-
Hardware
-
Procedure 1
CPUs: tested on a machine with a single 2.6GHz Intel Core i7 processor
Memory: tested on a machine with 16 GB of RAM, but should run on most machines with 4+ GB of RAM
Operating system: tested on macOS 10.13.6 High Sierra, but should run on most Unix operating systems
An internet connection is required to download the required software, scripts and data
-
Procedures 2 and 3
Memory: at least 4 GB of RAM
Operating system: any operating system capable of running R and installing the packages listed in Step 1 of Procedure 2
An internet connection is required to download the Orthrus package
Data
The CHyMErA dataset analyzed in Procedures 1 and 2 comprises combinatorial CRISPR screens performed with the CHyMErA experimental platform in two different human cell lines, HAP1 and RPE1, across a control and a Torin1-treated condition, with read counts taken at two different timepoints for each cell line (T12 and T18 for HAP1, T18 and T24 for RPE1). While the CHyMErA dataset contains single-targeting, dual-targeting and combinatorial-targeting guides, the multiplexed Cas12a dataset analyzed in Procedure 3 only contains combinatorial-targeting guides that target 400 paralog pairs and control guides that target paralogous genes paired with non-essential genes18. These control guides are treated as single-targeting guides for scoring purposes, as they are analogous to single-targeting guides in Procedure 1 and 2’s CHyMerA dataset that target paralogous genes paired with intergenic regions.
-
Procedure 1
The subset sequencing data processed in Procedure 1 only contains reads from the WT HAP1 T18 screen
Downloadable in the “Procedure1” folder of the Zenodo repository here
-
Procedure 2
The reads file analyzed in this Procedure contains processed reads for all screens and cell lines described above
Each guide in the reads file, represented by a single row, has associated read counts for the knockout of regions specified by a Cas9 and a Cas12a guide sequence
The first set of columns in the dataset contain metadata for guide pairs, and the remaining numeric columns contain raw read counts for the guide pairs across every screen
-
This guide library contains several different types of guides
Dual-targeting guides that target the same gene twice
Combinatorial-targeting guides that target each gene of a paralogous gene pair
Single-targeting guides that target a gene’s exonic region in addition to a relatively distant intergenic region. These are required to score combinatorial-targeting guides for GIs
Bundled with Orthrus’ download
-
Procedure 3
The reads file analyzed in this Procedure contains processed reads for screens performed in A549, HT29 and OVCAR8 cell lines, and is formatted similarly to the reads file in Procedure 2
-
The guide library contains two different types of guides
Combinatorial-targeting guides that target pairs of paralogous genes
Single-targeting guides that target paralogous genes paired with non-essential control genes
Downloadable in the Zenodo repository here
Equipment Setup
Software setup
If R is not already installed, download R version 3.6 or greater from https://cran.rstudio.com/. To use R, we recommend downloading the Rstudio IDE from https://rstudio.com/products/rstudio/download/.
Procedure 1: processing CHyMErA data
Set-up.
Timing: X-Y min.
1. Download required data.
Download the subset sequencing data, library guide sequences, processing scripts and Bowtie. CHyMErA data was processed with Bowtie version 0.12.9, whose download link is below. While other versions of Bowtie 1 are similarly appropriate for processing short sequencing data such as for CHyMErA screens, Bowtie 2 is not recommended.
Download everything in the Zenodo repository located here and unzip the files: https://zenodo.org/record/4527616 Download bowtie version 0.12.9 here: https://sourceforge.net/projects/bowtie-bio/files/bowtie/0.12.9/ Unzip the downloaded bowtie files into a subdirectory of the “Procedure1” folder of the Zenodo repository. Ensure that the “Procedure1” folder is the current working directory and that it contains all files in the “Procedure1” folder of the Zenodo repository. Similarly, ensure that the “Library” folder is a subdirectory of the current working directory, and that it contains all the files in the “Library” folder of the original Zenodo repository linked above.
2. Preprocess sequencing data.
To identify hgRNA barcodes present in a given sample, CHyMErA screening libraries are subjected to paired-end Illumina sequencing to capture both Cas12a and Cas9 guide sequences (Aregger et al. Nature Protocols, under review). To identify guide sequences within sequencing reads for two technical replicates of a single screen (HAP1 T0) using U6 promoter and Cas9 tracrRNA sequences as “anchor” sequences, run the preprocessReadsPE.pl script as follows. This takes two FASTQ sequencing files, representing a single technical replicate of a single screen, as input. It outputs four files, two per FASTQ file. The first, [FILENAME]_preprocessed.fastq, contains reads trimmed down to the guide sequences, and the second, [FILENAME]_failed.fastq, contains full reads where anchor sequences weren’t found. These anchor sequences are hard-coded U6 and tracr sequences in the R1_stem and R2_stem variables, which should be appropriate for all CHyMErA screens, but may be replaced if analyzing data from a different experimental platform. This step takes around 1Mb of memory and five minutes to pre-process 34.4M paired-end reads.
./preprocessReadsPE.pl Moffat_HH-79_S1_R1_001.fastq.gz Moffat_HH-79_S1_R2_001.fastq.gz
3. Align reads with Bowtie.
Next, the reads must be aligned to the screening library, which may be custom or provided by a vendor. The library used for previous CHyMErA screens is contained in the file Human_HybridGuide_Library_v3.txt, and for details on how this library was constructed, please consult Gonatopoulos et al. (2020)12. Perform this alignment with bowtie 0.12.9, using either option A to run on a single core, or option B to run on a cluster if available. The output of this step consists of six files: two contain unmapped reads, two contain mapped reads, one is a .sam file and the last is a log file. The .sam file named “HH-79_aligned.sam” is the only file required by the next step.
(A) Aligning reads on a single core
Change -p [N_CORES] as follows to take advantage of bowtie’s parallel processing to use any number of available CPU cores. On a single core, it takes about 7–8 hours to align 31.5M pre-processed sequence reads.
export BOWTIE_INDEXES=./Library/
bowtie-0.12.9/bowtie -p 1 -v 3 -l 18 --chunkmbs 256 -t
paralog_library_V3 --un HH-79_unmapped.fastq --al
HH-79_mapped.fastq -1
Moffat_HH-79_S1_R1_001_preprocessed.fastq -2
Moffat_HH-79_S1_R2_001_preprocessed.fastq HH-79_aligned.sam
2> HH-79.log
(B) To run this on a cluster, run bowtie with the submitjob command in a Bash for loop. This is useful for processing more than one screen concurrently.
export BOWTIE_INDEXES=./Library/
for f1 in *_R1_001_preprocessed.fastq
do
d=$(echo $f1 | sed -E
‘s/_S[0–9]+_R1_001_preprocessed.fastq//g’)
submitjob -c 6 bowtie-0.12.9/bowtie -p 6 -v 3 -l 18 --chunkmbs
256 -t paralog_library_V3 --un $d”_unmapped.fastq” --al
$d”_mapped.fastq” −1 $f1 −2 ${f1/_R1_/_R2_}
$d”_aligned.sam”
2\> $d”.log”
done
4. Parse alignments to guide counts.
Run parseBowtieOutput.pl as follows to get guide-level read counts for the HAP1 T0 screen. This takes roughly 2 minutes running on a single core and outputs the file “HH-79_counts.txt” required by step 5.
cat HH-79_aligned.sam | perl parseBowtieOutput.pl >
HH-79_counts.txt
5. Merge count files.
Merge all count files in the current working directory into a reads file with mergeAndAnalyzeParalogResults.R as follows. This generates a raw reads file named “rawCounts.txt.”
Rscript mergeChymeraResults.R
6. Re-format counts for Orthrus (optional).
As described in the Input Format section, Orthrus requires reads files to describe orientation in gene symbol columns. Both the initial library file and the file “rawCounts.txt” with appended read counts do not represent orientation in this way. Run the following script to fix this issue for the CHyMErA paralog data. Because the reads output in Step 5 are only for the HAP1 T0 screen, the script “prepChymeraData.R” instead runs on the included file “paralogLibrary_rawCounts.txt.”
Rscript prepChymeraData.R
Procedure 2: scoring CHyMErA data manually
Set-up.
Timing: 1–5 min.
1. Install required R packages.
Before installing Orthrus, install all packages it requires from both the CRAN repository and Bioconductor as follows:
install.packages(“ggplot2”)
install.packages(“ggthemes”)
install.packages(“pheatmap”)
install.packages(“PRROC”)
install.packages(“RColorBrewer”)
if (!requireNamespace(“BiocManager”, quietly = TRUE))
install.packages(“BiocManager”)
BiocManager::install(“limma”)
To install the Orthrus package, install it directly from its Github repository using the install_github command from the devtools package as follows:
install.packages(“devtools”) library(devtools) install_github(“csbio/Orthrus”)
2. Load packages.
After installing the dependencies above, load Orthrus and ggplot into the R environment as follows and rename the example data described above. For a detailed description of the dataset, please consult Gonatopoulos-Pournatzis et al12.
library(orthrus) df <- chymera_paralog
3. Set parameters.
While Orthrus does not require any global parameters, it is helpful to create output folders for various plots, text files and spreadsheets ahead of time, as follows. Many downstream functions will take these as parameters.
output_folder <- file.path(“orthrus_protocol”)
qc_folder <- file.path(output_folder, “qc”)
plot_folder <- file.path(output_folder, “scored”) lfc_folder <- file.path(qc_folder, “lfc_plots”)
if (!dir.exists(output_folder)) { dir.create(output_folder,
recursive = TRUE)}
if (!dir.exists(plot_folder)) { dir.create(plot_folder) }
if (!dir.exists(qc_folder)) { dir.create(qc_folder) }
if (!dir.exists(lfc_folder)) { dir.create(lfc_folder) }
4. Name screens.
Lastly, Orthrus requires the user to associate technical replicate read counts with screen names, so that downstream functions operate on the screen level rather than the replicate level. To do this, build up a list of screen objects either manually with the add_screen function, described in option (A), or automatically from a sample file mapping screen names to replicate columns with the add_screens_from_table function, described in option (B).
(A) Build up a list of screen objects manually
-
i
The add_screen function requires the user to give each screen a name, such as HAP1_T12, and a list of column names corresponding to technical replicates for that screen. To apply this function, list names for T0 screens, which have no technical replicates, and call parameters by their name. For subsequent calls to add_screen, pass previous results in as the first argument to build up the list of screens, as follows:
screens <- add_screen(name = “HAP1_T0”, replicates = “HAP1.T0”) screens <- add_screen(screens, “RPE1_T0”, “RPE1.T0”)
-
ii
To normalize the rest of the screens to their respective T0 screens to get log fold-changes (LFCs), add the name of the screen to normalize against in the final parameter of the add_screen function (the normalize_name parameter) as follows. All screens from later timepoints have three technical replicates, A, B, and C, which are separately normalized to T0s and are automatically averaged farther downstream in the pipeline.
screens <- add_screen(screens, “HAP1_T12”, c(“HAP1.T12A”,
“HAP1.T12B”, “HAP1.T12C”), “HAP1_T0”)
screens <- add_screen(screens, “HAP1_T18”, c(“HAP1.T18A”,
“HAP1.T18B”, “HAP1.T18C”), “HAP1_T0”)
screens <- add_screen(screens, “Torin_T12”, c(“HAP1.Torin.T12A”,
“HAP1.Torin.T12B”, “HAP1.Torin.T12C”), “HAP1_T0”)
screens <- add_screen(screens, “Torin_T18”, c(“HAP1.Torin.T18A”,
“HAP1.Torin.T18B”, “HAP1.Torin.T18C”), “HAP1_T0”)
screens <- add_screen(screens, “RPE1_T18”, c(“RPE1.T18A”,
“RPE1.T18B”, “RPE1.T18C”), “RPE1_T0”)
screens <- add_screen(screens, “RPE1_T24”, c(“RPE1.T24A”,
“RPE1.T24B”, “RPE1.T24C”), “RPE1_T0”)
(B) Build up a list of screen objects manually
Use the add_screens_from_table function as follows. This function requires the user to specify either a dataframe or the path to a tab-separated file mapping screen names to technical replicate names, in addition to another screen to which the given screen should be normalized during LFC computation in Step 7. This sample table is bundled with Orthrus’ download and is described in Table 3, which must include the column names Screen, Replicates and NormalizeTo. To disable LFC computation for specific screens, such as T0 screens, specify NA for those screens in the NormalizeTo column.
sample_table <- chymera_sample_table screens <- add_screens_from_table(sample_table)
Processing.
Timing: 5–30 min.
5. Make read count QC plots.
After associating technical replicate read counts to screen names, make QC plots for pre-normalization read count data. This allows users to investigate potential issues with their screening data, such as low sequencing depth for certain screens or unexpected skew in read count distributions. One function makes all of these QC plots for all screens, which are automatically saved to the qc subfolder created earlier as either png or pdf files, as follows:
plot_reads_qc(df, screens, qc_folder, display_numbers = FALSE, plot_type = “pdf”)
A summary of all plots made by the plot_reads_qc function is contained in Table 5.
Table 5.
Files output by QC functions and their descriptions in Procedure 2, Steps 5 and 8 and Procedure 3, Step 4.
| QC function | Output file names | Description |
|---|---|---|
| plot_reads_qc | [SCREEN]_raw_reads_histogram | Histograms of log-scaled read counts |
| plot_reads_qc | total_reads | Total read counts for all screens with hypothetical coverage appended |
| plot_reads_qc | reads_heatmap | Pearson correlation between log-scaled readcounts |
| plot_lfc_qc | [REPLICATE1]_vs_[REPLICATE2]_replicate_comparison | Scatterplot of LFCs between all replicates |
| plot_lfc_qc | replicate_pcc | Tab-delimited file of correlations for all replicates |
? Troubleshooting
6. Examine read count QC plots.
Closely examine of all output QC plots to reveal screening issues that need to be manually addressed by the user. For example, certain screens may be heavily skewed towards guides with unexpectedly high or low read counts. Additionally, for typical experiments the user should expect T0 read counts to cluster separately from later timepoint replicates in the log-normalized read count heatmap of Pearson correlations between screens. T0 replicates that do not cluster separately could implicate overarching screen quality issues.
PAUSEPOINT:
A manual examination of all read count-based QC plots and metrics should be performed before proceeding.
7. Normalize read counts and compute LFCs.
Normalize read counts based on sequencing depth and compute guide LFCs with the normalize_screens function, as follows. All screens passed into the function will be log2-scaled and depth-normalized, including T0 screens. LFC values will additionally be computed for all screens with associated normalized_name parameters. In this procedure, LFCs for the RPE1_T18 and RPE_T24 screens will be computed relative to the RPE_T0 screen, and similarly for HAP1_T12, HAP1_T18, Torin_T12 and Torin_T18 to the HAP1_T0 screen.
While the normalize_screens function normalizes against early-timepoint screens with multiple replicates by computing the mean log2-scaled reads across early-timepoint replicates before LFC computation, because neither dataset analyzed in this protocol includes replicates for early-timepoint screens, this behavior is not demonstrated. Additionally, while it is recommended to normalize chemogenetic screens against early-timepoint screens to compute LFCs before comparing drug treatment screens to control screens, Orthrus’ downstream scoring functions are applicable to log2-normalized reads as well as LFCs.
In addition to guide normalization and LFC computation, the normalize_screens function also automatically removes guides that are over or under-expressed at earlier timepoints. For this procedure, to remove guides with less than 30 read counts in any T0 screen, pass a list of both T0 screens into the filter_names parameter and set the min_reads parameter to 30. Note that this value or more conservative values such as 40–60 are recommended for most screens, regardless of library size. Both coverage and the standard deviation of gRNA abundance for early-timepoint or plasmid pool data, however, can be taken into account when setting this parameter. Because this relationship can be complex, please refer to Imkeller et al. (2020)21. No guides in this library are over-expressed (defined by the default value of 10,000 reads for the max_reads parameter), so although over-expressed guides are also automatically filtered out this will not affect the data.
df <- normalize_screens(df, screens, filter_names = c(“HAP1_T0”, “RPE1_T0”), min_reads = 30, max_reads = 10000)
? Troubleshooting
8. Make LFC QC plots.
While read count QC plots allow for birds-eye views of screening data, LFC-based QC reveals specific quality information on individual guides and also outputs important quantitative QC metrics. To generate LFC-based QC plots and metrics for all screens, call the plot_lfc_qc function, as follows:
plot_lfc_qc(df, screens, qc_folder, display_numbers = FALSE, plot_type = “png”, negative_controls = c(“NT”))
A summary of all plots made by the plot_lfc_qc function is contained in Table 5.
9. Examine LFC QC plots.
Like for read count QC plots, closely examine all output QC plots to reveal important screen quality issues. Replicate comparison plots can reveal specific guides that are unexpectedly over or under-represented, or which appear to be outliers for a specific technical replicate but not others. Points are colored by whether or not both gRNAs for each dgRNA target either a non-essential gene as defined by Hart et al. (2014) or a gene in the list specified by the user in the negative_controls parameter22. More generally, they also show technical replicates that do not appear to correlate with each other or are skewed in problematic ways. Quantitative information on replicate Pearson and Spearman correlations is also contained in the output file “replicate_cor.tsv.”
Because most libraries are designed with sets of positive and negative control essential genes, Orthrus reports the area under ROC curves for essential-gene dropout for all technical replicates. Specifically, essential genes are defined by the CEG2 core essential gene set and nonessential genes are defined by Hart et al. 201422. Gene effects for essential genes are computed based on the dropout of guides that target essential genes twice, two different essential genes, or an essential gene and an intergenic region, and similarly for non-essential genes. The area under ROC curves for essential genes compared to both nonessential genes as well as all other genes is reported in the output file “essential_PR_QC.tsv.” While for most negative selection whole-genome screens one would expect AUC values > 0.9, for specialized guide libraries in which one expects many other strong negative fitness effects outside of essential gene pairs this AUC value may be substantially lower. Moreover, because AUC values for comparisons against reference nonessential genes are typically higher than AUC values for comparisons against all genes not in the essential set, the latter AUC value tends to be more predictive of screen quality issues.
PAUSEPOINT:
A manual examination of all read LFC-based QC plots and metrics should be performed before proceeding.
? Troubleshooting
10. Parse gene pairs by type.
To support Orthrus’ two different scoring modes, split the guide dataframe into different types of guides based on orientations defined by gene symbol columns named gene1 and gene2 with the split_guides function, as follows. The relationship between gene symbols and orientation, as well as their required formatting, is explained in the “Input Format” section of the Introduction. If guides are mapped to unique identifiers such as guide sequences, additionally pass in column names for those identifiers such that the column name passed in first corresponds to the gene1 column and the column name passed in second corresponds to the gene2 column. This enables loess-correction with moderated t-testing, the default scoring modes supported by Orthrus, in Steps 11 and 15. If unspecified, the user must default to Wilcoxon rank-sum testing, which is not recommended for most experimental designs.
guides <- split_guides(df, screens, “Cas9.Guide”, “Cpf1.Guide”) dual <- guides[[“dual”]] single <- guides[[“single”]] paralogs <- guides[[“combn”]]
The output of this process is three separate lists, where each element contains all guides targeting a single gene pair. Dual-targeting guides are contained in the dual list, single-targeting guides are contained in the single list, and combinatorial-targeting guides are contained in the paralogs list.
Dual-targeted scoring.
Timing: 5–10 min.
11. Score guides targeting the same gene twice.
Currently, the Orthrus package supports scoring dual-targeting guides by comparing one or more condition screens against a single control screen. This is performed via moderated t-testing for each gene pair, condition, and orientation against corresponding guides in the control screen. As in Aregger et al.11, guide-level residuals are corrected with loess-normalization before performing hypothesis testing to account for skewed and non-normal distributions. Scoring guides in this way results in both an effect size measure, based on the mean of loess-normalized residuals between condition and control LFCs, as well as a measure of statistical significance (a p-value from moderated t-testing on these residuals).
One function, score_conditions_vs_control, performs comparisons for all gene pairs using the dual list of guides defined above. Score the provided dataset for Torin1-specific effects in HAP1 cells by comparing Torin1 effects at each timepoint against untreated HAP1 effects using the following code, specifying moderated t-testing (instead of Wilcoxon rank-sum testing, which is supported but not recommended due to its reduced statistical power) and loess normalization. Additionally, pass in “NT” to the filter_genes parameter to ignore non-targeting control genes during scoring. This parameter may contain any number of genes stored in a character vector, but for the CHyMErA library only “NT” needs to be specified.
temp <- score_conditions_vs_control(dual, screens, “HAP1_T12”,
“Torin_T12”, test = “moderated-t”, loess = TRUE,
min_guides = 3, filter_genes = c(“NT”))
dual_scores1 <- temp[[“scored_data”]]
residuals1 <- temp[[“residuals”]]
temp <- score_conditions_vs_control(dual, screens, “HAP1_T18”,
“Torin_T18”, test = “moderated-t”, loess = TRUE,
min_guides = 3, filter_genes = c(“NT”))
dual_scores2 <- temp[[“scored_data”]]
residuals2 <- temp[[“residuals”]]
The above code returns a list of two dataframes. The first dataframe in the list, named scored_data, contains effect size and FDR values for all gene pairs comparing Torin effects to WT Hap1 effects at either T12 or T18, as well as many additional columns described in Table 6. It is important to note that NA values in this dataframe represent genes that have too few guides remaining post-filtering based on T0 read counts and specified filter genes, for a default threshold of 3 guides per gene pair. The second dataframe in the list, residuals, contains guide-level residual values for detailed examination of a given gene pair’s results, and is used for residual plotting functions in Step 13.
Table 6.
Columns contained in scored data output from the dual-targeting scoring mode in Procedure 2, Steps 11 and 12 and Procedure 3, Step 4. SCREEN placeholders represent the names of all scored condition and control screens, whereas CONDITION and CONTROL placeholders represent the names of condition and control screens, respectively.
| Scored data column | Description |
|---|---|
| gene1 | Gene symbol targeted by the first guide |
| gene2 | Gene symbol targeted by the second guide |
| n_[SCREEN] | Number of guides post-filtering for the screen |
| mean_[SCREEN] | Mean LFC across all guides |
| variance_[SCREEN] | Variance for all guides |
| differential_[CONDITION]_vs_[CONTROL] | Loess-adjusted (if specified) differential between mean condition and control LFCs |
| pval_[CONDITION]_vs_[CONTROL] | P-value between loess-adjusted residuals for condition and control |
| fdr_[CONDITION]_vs_[CONTROL] | FDR-adjusted p-value |
| significant_[CONDITION]_vs_[CONTROL] | Significance calls returned from call_significant_response |
| effect_type_[CONDITION]_vs_[CONTROL] | Effect type calls returned from call_significant_response |
? Troubleshooting
12. Call significant effects for dual-targeting guides.
After scoring data, for a typical experiment the user would like to reduce a large list of significant hits down to a ranked list of high-priority hits. Prioritize hits using the function call_condition_hits. Call this function with a given FDR threshold and differential effect threshold to call significant positive or negative hits as follows.
Hits are called based on two criteria. Gene pairs with a) an FDR less than the given FDR threshold and b) an absolute value of their loess-normalized residuals that is greater than the given differential effect threshold will be called as significant positive or negative hits. When calling this function, the user may also choose to rename positive and negative hits.
dual_scores1 <- call_condition_hits(dual_scores1, “HAP1_T12”,
“Torin_T12”, neg_type = “Sensitizer”, pos_type =
“Suppressor”, fdr_threshold = 0.1, differential_threshold =
0.5)
dual_scores2 <- call_condition_hits(dual_scores2, “HAP1_T18”,
“Torin_T18”, neg_type = “Sensitizer”, pos_type =
“Suppressor”, fdr_threshold = 0.1, differential_threshold =
0.5)
write.table(dual_scores1, file.path(output_folder,
“dual_targeting_gene_calls_t12.tsv”), sep = “\t”,
row.names = FALSE, col.names = TRUE, quote = FALSE)
write.table(dual_scores2, file.path(output_folder,
“dual_targeting_gene_calls_t18.tsv”), sep = “\t”,
row.names = FALSE, col.names = TRUE, quote = FALSE)
With scoring complete, write the data to file as shown above.
13. Plot residual effects (optional).
After scoring data, users might like to see all guide-level LFC values for significant hits to visually confirm that certain genes possess consistent effects across most guides. Use the function plot_condition_residuals as follows to automatically generate these plots for all hits called with call_condition_hits, and output them in a sorted order to the given folder. Make LFC plots for Torin-specific significant hits for T12 and T18.
plot_condition_residuals(dual_scores1, residuals1, “HAP1_T12”, “Torin_T12”, file.path(lfc_folder, “dual_lfc_t12”),
neg_type
= “Sensitizer”, pos_type = “Suppressor”, plot_type = “png”) plot_condition_residuals(dual_scores2, residuals2, “HAP1_T18”, “Torin_T18”, file.path(lfc_folder, “dual_lfc_t18”),
neg_type
= “Sensitizer”, pos_type = “Suppressor”, plot_type = “png”)
For this data, the plot “neg_1_HECTD1_None.png” refers to the top negative hit in terms of differential effect (marked by “neg_1” in the filename) for Torin T18 against WT Hap1. Similarly, the file “pos_2_EED_None.png” refers to the second-highest ranked positive hit in terms of differential effect.
14. Plot condition response.
Finally, generate plots for each condition against the control screen with plot_condition_response, as follows. This outputs two plots, a scatterplot and a volcano plot, to a given folder. The volcano plot displays either -log10(p-value) or -log2(FDR) on the y-axis based on whether “pval” or “FDR” is passed to the parameter volcano_type, respectively. These volcano plots may be helpful to determine effect size and fdr thresholds for specific datasets. As for generating residual plots, ensure that the chosen names for negative and positive effects are passed into the function.
plot_condition_response(dual_scores1, “HAP1_T12”, “Torin_T12”,
plot_folder, neg_type = “Sensitizer”, pos_type =
“Suppressor”, volcano_type = “FDR”, plot_type = “pdf”)
plot_condition_response(dual_scores2, “HAP1_T18”,
“Torin_T18”, plot_folder, neg_type =
“Sensitizer”, pos_type =
“Suppressor”, volcano_type = “FDR”, plot_type = “pdf”)
PAUSEPOINT:
A manual examination of all dual-targeting scoring output should be performed before proceeding.
Combinatorial scoring.
Timing: 5–10 min.
15. Score guides targeting multiple genes.
Orthrus also supports scoring guides that target multiple genes, such as paralog pairs, by comparing the effect of double-knockouts against an expected model derived from single-knockout effects. In detail, all guides that target gene A with Cas9 and gene B with Cas12a comprise the observed model for one orientation. All multiplicative combinations (additive in log-space) of single-targeting guides that target gene A with Cas9, target gene B with Cas12a, and match guide sequences with the observed model comprise the expected model for the same orientation (Fig. 3). Like for dual-targeted scoring, the residuals between the observed and expected models are computed for each orientation and loess-normalized before performing moderated t-testing for each gene pair and condition.
Figure 3.

Schematic demonstrating how Orthrus accounts for guide orientation during combinatorial scoring by scoring guides from different orientations separately. Orientation is represented as guides that occupy different positions along a guide construct that targets both gene A and gene B. For both orientations, combinatorial knockouts are compared to expected effects derived from the sum of matching single knockout LFCs. After hypothesis testing, filters for absolute value of effect size, FDR and whether or not both orientations’ effects have the same effect sign are applied to call significant hits.
One function, score_combn_vs_single, scores combinatorial-targeting guides for any number of conditions by passing in the “dual” list of guides defined above. Call this function and score the provided dataset for combinatorial-targeting genetic interactions for the HAP1 T12 and the RPE1 T24 screens as follows. As for dual-targeted scoring, additionally pass in “NT” to the filter_genes parameter to ignore non-targeting control genes during scoring. Write scores to file afterwards.
screens_to_score <- c(“HAP1_T12”, “HAP1_T18”, “RPE1_T18”,
“RPE1_T24”, “Torin_T12”, “Torin_T18”)
temp <- score_combn_vs_single(paralogs, single, screens,
screens_to_score, test = “moderated-t”,
return_residuals = TRUE, filter_genes = c(“NT”))
paralog_scores <- temp[[“scored_data”]]
paralog_residuals <- temp[[“residuals”]]
paralog_scores <- call_combn_hits(paralog_scores,
screens_to_score, neg_type = “Negative GI”, pos_type =
“Positive GI”, fdr_threshold = 0.2, differential_threshold
=
0.5)
write.table(paralog_scores, file.path(output_folder,
“paralog_gene_calls.tsv”), sep = “\t”, row.names = FALSE,
col.names = TRUE, quote = FALSE)
Like for score_conditions_vs_control, the above code returns a list of two dataframes. The first dataframe in the list, named scored_data, contains FDR values for all gene pairs comparing observed double-knockout effects to single-knockout effects as described in Procedure 2 Steps 11–12 for all six screens. It also contains many additional columns for each screen, listed in Table 7, and like for score_conditions_vs_control NA values in this dataframe represent genes that have too few guides remaining post-filtering based on T0 read counts. This guide threshold is also controlled by the min_guides parameter. The second dataframe in the list, residuals, contains guide-level residual values to enable the detailed examination of a given gene pair’s results. For analyzing combinatorial-targeting CHyMErA data, we recommend raising the FDR threshold from the default of 0.1 to 0.2 with the fdr_threshold parameter as shown above. The specific choice of threshold is flexible, however, and is dependent on the expected signal-to-noise ratio in the dataset.
Table 7.
Columns contained in scored data output from the combinatorial-targeting scoring mode obtained in Procedure 2, Steps 15 and 19, and Procedure 3, Step 4. SCREEN placeholders represent the names of all scored screens.
| Scored data column | Description |
|---|---|
| gene1 | Gene symbol targeted by the first guide |
| gene2 | Gene symbol targeted by the second guide |
| n_combn_[SCREEN] | Number of combinatorial-targeting guides post-filtering for the screen |
| n_single_[SCREEN] | Number of single-targeting guides post-filtering for the screen |
| mean_combn_[SCREEN] | Mean LFC across all combinatorial-targeting guides and both orientations |
| mean_single_[SCREEN] | Mean LFC across all combinations of single-targeting LFC sums that match combinatorial-targeting guide IDs, for both orientations |
| var_combn_[SCREEN] | Variance for all combinatorial-targeting guides across both orientations |
| var_single_[SCREEN] | Variance for all combinations of single-targeting LFC sums across both orientations |
| orientation_agree_[SCREEN] | True if both orientations’ differential effects have the same sign, false otherwise. |
| differential_combn_vs_single_[SCREEN] | Loess-adjusted (if specified) differential between mean combn and single LFCs |
| pval1_combn_vs_single_[SCREEN] | P-value between loess-adjusted residuals for the first orientation of combn and single LFCs |
| pval2_combn_vs_single_[SCREEN] | P-value between loess-adjusted residuals for the second orientation of combn and single LFCs |
| fdr1_combn_vs_single_[SCREEN] | FDR-adjusted p-value for the first orientation |
| fdr2_combn_vs_single_[SCREEN] | FDR-adjusted p-value for the second orientation |
| significant_[SCREEN] | Significance calls returned from call_significant_response_combn |
| effect_type_[SCREEN] | Effect type calls returned from call_significant_response_combn |
? Troubleshooting
16. Plot combinatorial residual effects (optional).
After scoring data, some users would like to see guide-level residual LFC values for significant combinatorial hits. Like for dual-targeted scoring, the function plot_combn_residuals automatically generates these plots for all hits called with call_combn_hits, and outputs them in sorted order to the given folder as described in Step 13. Make LFC plots for HAP1_T12 and RPE1_T24 significant hits as follows:
residual_folder <- file.path(lfc_folder, “HAP1_T12_combn”) plot_combn_residuals(paralog_scores, paralog_residuals,
“HAP1_T12”, residual_folder, neg_type = “Negative GI”,
pos_type = “Positive GI”)
residual_folder <- file.path(lfc_folder, “RPE1_T24_combn”)
plot_combn_residuals(paralog_scores, paralog_residuals,
“RPE1_T24”, residual_folder, neg_type = “Negative GI”,
pos_type = “Positive GI”)
17. Plot condition response.
FInally, generate plots for each condition against the control screen as well as volcano plots for each screen with plot_combn_response, as follows. Ensure that the chosen names for negative and positive effects are passed into the function. For plotting effects from the Torin screen, additionally set the color of hits also significant in the WT HAP1 screens to gray by specifying the name of the respective control screen in the filter_name parameter.
plot_combn_response(paralog_scores, “HAP1_T12”, loess = TRUE,
plot_folder, neg_type = “Negative GI”, pos_type =
“Positive GI”)
plot_combn_response(paralog_scores, “HAP1_T18”, loess = TRUE,
plot_folder, neg_type = “Negative GI”, pos_type =
“Positive GI”)
plot_combn_response(paralog_scores, “RPE1_T18”, loess = TRUE,
plot_folder, neg_type = “Negative GI”, pos_type =
“Positive GI”)
plot_combn_response(paralog_scores, “RPE1_T24”, loess = TRUE,
plot_folder, neg_type = “Negative GI”, pos_type =
“Positive GI”)
plot_combn_response(paralog_scores, “Torin_T12”, loess = TRUE,
plot_folder, neg_type = “Negative GI”, pos_type =
“Positive GI”, filter_name = “HAP1_T12”)
plot_combn_response(paralog_scores, “Torin_T18”, loess = TRUE,
plot_folder, neg_type = “Negative GI”, pos_type =
“Positive GI”, filter_name = “HAP1_T18”)
PAUSEPOINT:
A manual examination of all combinatorial-targeting scoring output should be performed before proceeding.
Single-targeting scoring.
Timing: 1–5 min.
18. Score guides targeting the same gene twice.
Orthrus allows users to score single-targeting guides, the same guides contained in the “single” list used to construct the expected guide set for combinatorial scoring, by treating these as a single orientation for the dual-targeting scoring mode. To score single-targeting guides in this way, call score_conditions_vs_control with the separate_orientation argument set to TRUE, as follows.
This returns a list of two dataframes of scored data, one for each orientation. For this library, the first dataframe in the list contains scores for guides where Cas9 targets an exonic region, and the second dataframe contains scores where Cas12a targets an exonic region. Due to the CHyMErA library design that aimed to target each gene with three Cas9 guides and five Cas12a guides, as well as due to guides filtered from their low representation in T0 screens, the scored Cas9 single-targeting data contains too few significant hits. Accordingly, analyze only the single-targeting Cas12a data.
After scoring single-targeting guides, remove scored genes with too few guides remaining after T0 read count filters, and call significant hits with desired FDR and read count thresholds as follows:
single_scores <- score_conditions_vs_control(single, screens, “HAP1_T18”, “Torin_T18”, separate_orientation = TRUE) single_scores <- single_scores[[2]] [[“scored_data”]] to_keep <- !is.na(single_scores$n_HAP1_T18) cat(paste(“Removing”, nrow(single_scores) - sum(to_keep), “sparse single-targeting genes\n”)) single_scores <- single_scores[to_keep,] single_scores <- call_condition_hits(single_scores, “HAP1_T18”, “Torin_T18”, neg_type = “Sensitizer”, pos_type = “Suppressor”, fdr_threshold = 0.2, differential_threshold = 0.5) write.table(single_scores, file.path(output_folder, “single_targeting_gene_calls_t18.tsv”), sep = “\t”, row.names = FALSE, col.names = TRUE, quote = FALSE)
PAUSEPOINT:
A manual examination of all single-targeting scoring output should be performed before proceeding.
? Troubleshooting
Batch scoring.
19. Score dual and combinatorial-targeting guides in batch (optional).
Instead of scoring data manually, Orthrus provides the option to score different types of guides at the same time using its batch scoring mode. Use the following two function calls. This requires the creation of a batch file formatted as described in the Input Format section of the Introduction that maps screens to other screens they should be scored against, or to “combn” to perform combinatorial scoring. This batch table is bundled with Orthrus’ download, and is also shown in Table 4. The expected output of this process is largely equivalent to the output of Steps 5–17 above, with key differences including the use of standardized FDR and effect size thresholds across both dual-targeted and combinatorial-targeted scoring and that single-targeting scores are not computed automatically.
batch_table <- chymera_batch_table
batch_output_folder <- file.path(“orthrus_protocol_batch”)
if (!dir.exists(batch_output_folder))
{ dir.create(batch_output_folder) }
score_conditions_batch(dual, screens, batch_table, output_folder, test = “moderated-t”, loess = TRUE, filter_genes = c(“NT”), neg_type = “Sensitizer”, pos_type = “Suppressor”, fdr_threshold = 0.1, differential_threshold = 0.5)
score_combn_batch(paralogs, single, screens, batch_table, output_folder, test = “moderated-t”, loess = TRUE, filter_genes = c(“NT”),
neg_type = “Sensitizer”, pos_type = “Suppressor”, fdr_threshold = 0.2, differential_threshold = 0.5)
Procedure 3: analyzing dual Cas12a gRNA data with the batch scoring interface
Set-up.
Timing: 5–10 min.
1. Install and load required R packages.
As in Procedure 1, before installing Orthrus, install all packages it requires from both the CRAN repository and Bioconductor. Then load devtools to install Orthrus from the development Github repository, and finally load both Orthrus and ggplot2 into the R environment, as follows:
install.packages(“devtools”)
install.packages(“ggplot2”)
install.packages(“ggthemes”)
install.packages(“pheatmap”)
install.packages(“PRROC”)
install.packages(“RColorBrewer”)
install.packages(“stringr”)
if (!requireNamespace(“BiocManager”, quietly = TRUE))
install.packages(“BiocManager”)
BiocManager::install(“limma”)
library(devtools)
install_github(“csbio/Orthrus”)
library(orthrus)
library(stringr)
2. Download and load datasets.
Download and unzip the required datasets for this procedure from the Zenodo repository. Ensure that the working directory in R contains a subdirectory named “dede_input” with input files that mirror the contents of the Zenodo repository. Load the reads file containing three combinatorial screens with two technical replicates each performed with a dual-Cas12a system18, in addition to sets of reference essential and nonessential gene standards used to process the data and to calculate QC metrics.
Download and unzip the zenodo directory from the following link as follows and ensure that the folder “dede_input” is a subdirectory of the current working directory: https://zenodo.org/record/4527616 input_folder <- file.path(“dede_input”) prepped_file <- file.path(input_folder, “prepped_dede_paralog.tsv”) df <- read.csv(file.path(input_folder, “original_dede_paralog.txt”), sep = “\t”, header = TRUE, stringsAsFactors = FALSE) essentials <- read.csv(file.path(input_folder, “control_essentials.csv”), header = TRUE, stringsAsFactors = FALSE) nonessentials <- read.csv(file.path(input_folder, “control_nonessentials.csv”), header = TRUE, stringsAsFactors = FALSE)
3. Prep readcount dataset.
Instead of using intergenic-targeting guides like the CHyMErA library, this library targets nonessential genes as negative controls. To allow Orthrus to recognize this experimental design during scoring, after splitting the dataset’s single gene symbol and guide ID columns into two, rename all nonessential-targeting guides as “NegControl” in both gene symbol columns, as follows. Because gene symbols in this dataset accurately reflect guide orientations, further alterations to the gene symbol columns are unnecessary.
# Preps dataset essentials <- unlist(essentials) nonessentials <- unlist(nonessentials) split <- str_split_fixed(df$GENE, “:”, 2) df$gene1 <- gsub(“\\..*”, ““, split[,1]) df$gene2 <- gsub(“\\..*”, ““, split[,2]) df$gene1[df$gene1 %in% nonessentials] <- “NegControl” df$gene2[df$gene2 %in% nonessentials] <- “NegControl” # Adds guide columns split <- str_split_fixed(df$GENE_CLONE, “_”, 4) df$Guide1 <- split[,2] df$Guide2 <- split[,4] # Writes to file write.table(df, prepped_file, sep = “\t”, row.names = FALSE, col.names = TRUE, quote = FALSE)
4. Score combinatorial-targeting guides.
Orthrus provides two ways to automatically score data: a batch scoring mode that first requires the user to process their data with Orthrus manually, and a wrapper function that runs the entire Orthrus pipeline in a single function call. Use option A to apply the batch scoring mode to the Dede et al. dataset, and option B to apply the wrapper function to the Dede et al. dataset. Both outputs are equivalent except for the choice of whether or not to loess-normalize residuals: option A demonstrates the output of loess-normalization, and option B demonstrates the linear fit computed without loess-normalization. For this dataset, loess normalization is not recommended as a visual examination of loess-correction on scored data displays overfitting for points with a wide spread in the bottom two quadrants of each scatterplot, implicating the presence of both false positives and false negatives (Fig. 4).
Figure 4.
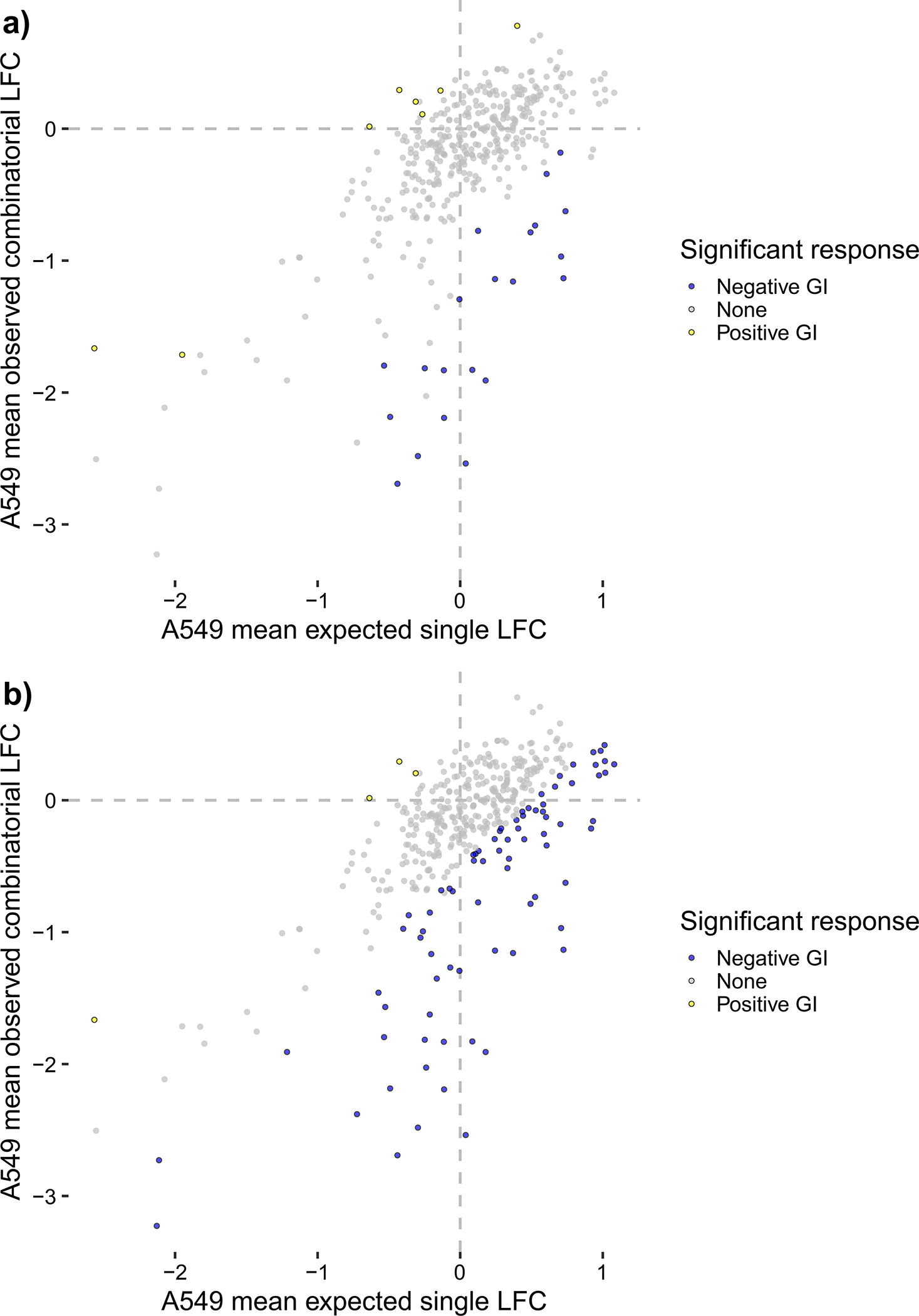
Summary plots of mean LFC, colored by significant effects, for all n = 403 gene pairs with combinatorial- and single-targeting guides for the Dede et al. A549 screen analyzed in Procedure 3. Gene-level GIs are shown as colored points that significantly deviate from the computed null model. Blue points are negative GIs with mean residual effects < −0.5 and Benjamini and Yekutieli FDRs < 0.2, while yellow points are positive GIs with mean residual effects > 0.5 and Benjamini and Yekutieli FDRs < 0.2. (a) shows scores with loess-normalization enabled for 8 positive GIs and 21 negative GIs, and (b) shows scores with loess-normalization disabled for 4 positive GIs and 78 negative GIs. For this dataset, we conclude that loess-normalization is not recommended due to potential false positives and negatives introduced in the bottom two quadrants of (a).
(A) Batch scoring mode
i) To run batch scoring, process the Dede et al. dataset as described below, which corresponds to Steps 4–10 of Procedure 2 (with a few changes as explained further below). Afterwards, call the score_combn_batch function to automatically score combinatorial-targeting guides in the dataset. This function requires that the user first specify either a dataframe or a batch .tsv file that maps screen names to their respective controls (for dual-targeted scoring) or to a derived null model from single-targeting effects (for combinatorial scoring). This batch table is located in the “dede_input” folder of the Zenodo repository here. An additional function, score_conditions_batch, scores dual-targeting guides automatically and takes the same batch file as input. However, because the Dede et al. dataset contains no dual-targeting guides, call score_conditions_batch to demonstrate its use but expect no output from it.
Three changes to parameters compared to values shown in Procedure 2 are suggested to accurately score combinatorial-targeting guides in this dataset. First, change the list of negative controls to include “NegControl.” Second, because the early-timepoint readcounts to normalize against for this dataset consist of plasmid pool readcounts with relatively high sequencing depth and low dropout, the min_reads parameter described in Step 7 of Procedure 1 can be safely tightened to 40 reads instead of the default 30 reads (this threshold could be further tightened if the user desires). Third, set the ignore_orientation parameter to TRUE to enable combinatorial-scoring that aggregates guides across both orientations before running moderated t-testing. This reduces the amount of t-testing from two tests per gene pair to one test per gene pair, and is advised for this dataset because the dual-Cas12a combinatorial system is less likely to be influenced by guide orientation than the CHyMErA experimental system. By default, hits from this scoring mode are still filtered based on whether or not the signs of their orientation-specific effects agree. The results of this step for the A549 screen are shown in Fig 10A.
Figure 10.
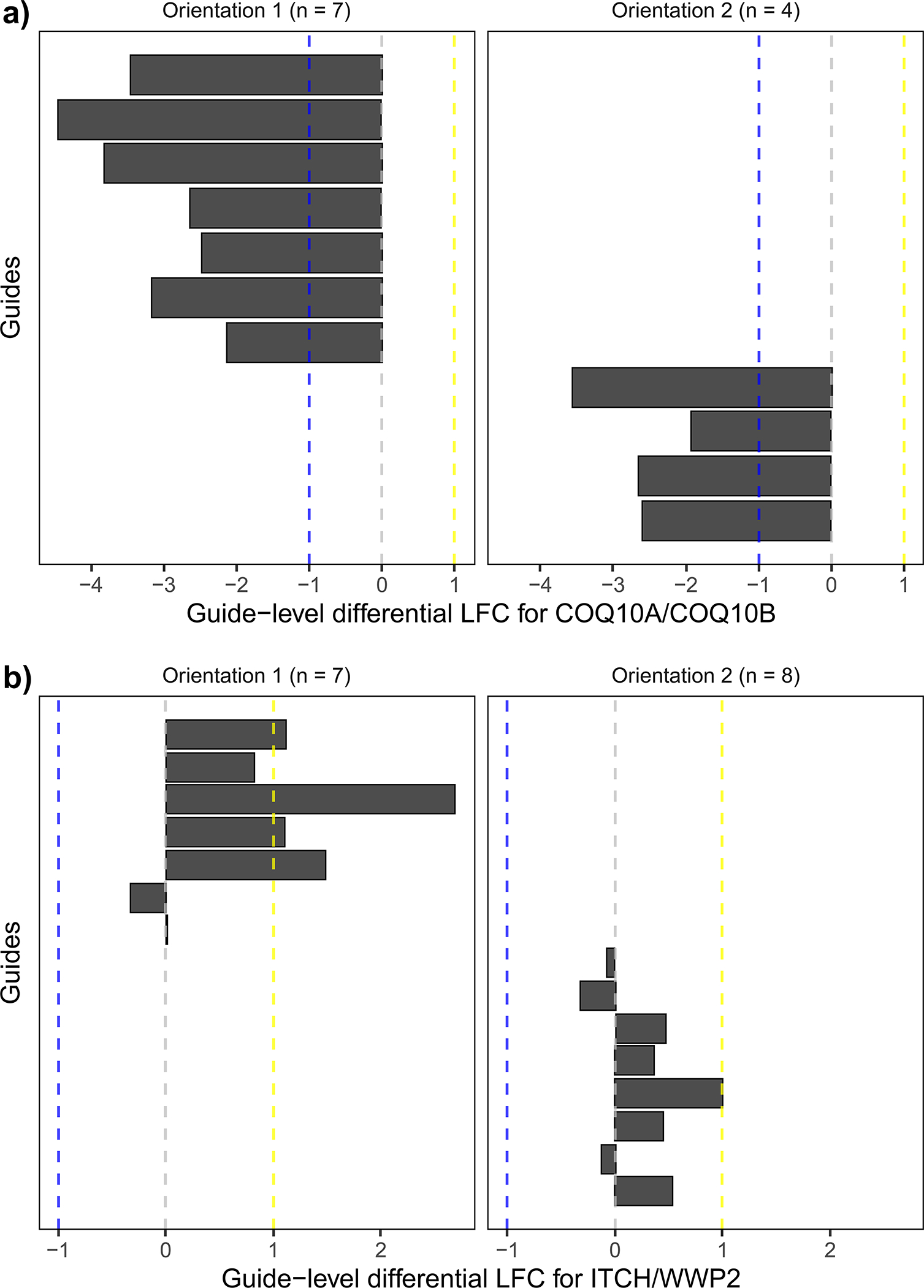
Differential LFC for WT HAP1 guides comprising two significant hits of the scored combinatorial-targeting guides at T12 from the ChyMErA dataset analyzed in Procedure 2, Steps 15–17. Genes were scored based on deviations from a loess-corrected null model, so the agreement of individual guides of interesting hits should be examined to qualitatively confirm hit quality. (a) is the top-ranked negative hit, COQ10A and COQ10B, whose guides in both orientations strongly agree. (b) is the top-ranked positive hit, ITCH and WWP2, with agreement for five out of seven guides for orientation 1 and agreement for five out of eight guides for orientation 2 The remaining guides show weak phenotypes, indicating poor performance for those guides and good agreement for all other guides.
# Sets important paths
sample_file <- file.path(input_folder, “dede_sample_table.tsv”)
batch_file <- file.path(input_folder, “dede_batch_table.tsv”)
output_folder <- file.path(“dede_output_batch”)
qc_folder <- file.path(output_folder, “qc”)
if (!dir.exists(output_folder)) { dir.create(output_folder) }
if (!dir.exists(qc_folder)) { dir.create(qc_folder) }
# Processes data
screens <- add_screens_from_table(sample_file)
plot_reads_qc(df, screens, qc_folder, display_numbers = TRUE, plot_type = “png”)
df <- normalize_screens(df, screens, filter_names = “T0”, min_reads = 40)
plot_lfc_qc(df, screens, qc_folder, display_numbers = TRUE, plot_type = “png”, negative_controls = c(“NegControl”))
guides <- split_guides(df, screens, “Guide1”, “Guide2”) dual <- guides[[“dual”]]
single <- guides[[“single”]]
combn <- guides[[“combn”]]
# Scores data with batch scoring functions
score_conditions_batch(dual, screens, batch_file, output_folder, test = “moderated-t”, loess = FALSE)
score_combn_batch(combn, single, screens, batch_file, output_folder, test = “moderated-t”, loess = FALSE, filter_genes = c(“NegControl”), neg_type = “Sensitizer”, pos_type = “Suppressor”, fdr_threshold = 0.2, differential_threshold = 0.5)
(B) Using the wrapper function
i) To automatically run the entire Orthrus package with the wrapper function, call orthrus_wrapper as follows and pass in paths to the properly-formatted reads file, the sample file, and the batch file downloaded in Step 2. This automatically outputs all plots, metrics, and scored data discussed for the data processing and combinatorial-scoring steps of Procedure 2 to a specified output folder. In addition, for this step set the loess flag to FALSE to disable the loess-correction of residuals. The results for this step are shown in Fig. 10B.
output_folder <- file.path(“dede_output”)
sample_file <- file.path(input_folder, “dede_sample_table.tsv”)
batch_file <- file.path(input_folder, “dede_batch_table.tsv”)
orthrus_wrapper(prepped_file, sample_file, batch_file,
output_folder, id_col1 = “Guide1”, id_col2 = “Guide2”,
filter_names = “T0”, min_reads = 40,
display_numbers = TRUE, negative_controls = nonessentials,
test = “moderated-t”, loess = FALSE, fdr_method = “BY”,
fdr_threshold = 0.2, differential_threshold = 0.5,
plot_type = “png”, ignore_orientation = TRUE)
? Troubleshooting
Troubleshooting
See Table 8.
Table 8. Troubleshooting Table.
Common screen quality issues reflected by Orthrus’ output as well as common issues encountered by users.
| Procedure - Step | Problem | Possible reasons | Solutions |
|---|---|---|---|
| 2–7, 3–4 | Many guides with too few reads in early-timepoint screens are filtered out | Various experimental issues early on in the screen, such as with T0 samples |
Orthrus’ scoring accounts for this, but users may consider applying the “ignore_orientation” flag during scoring |
| 2–9, 3–4 | Technical replicates correlate poorly with each other | Mislabelling in the sample file or any number of experimental issues | Fix technical replicate labels in the sample file, consider filtering early-timepoint guides more stringently, or redo the problematic screen |
| 2–9, 3–4 | Guides appear to drop out stochastically between replicates in QC scatterplots | Dosage for a drug screen was too high | Remove guides that completely drop out in any late-timepoint replicate, or redo the problematic screen |
| 2–5, 3–4 | The function plot_reads_qc returns the error “Error in check_screen_para ms(df, screens) : replicate [REPLICATE] not in df, remove screen [REPLICATE] with remove_screens” | Replicate name set in add_screen function does not exist in input dataframe | Remove screen containing the offending replicate from the screen list with the function remove_screen and readd with correct replicate names |
| 2–11, 2–15, 2–18, 3–4 | Many rows in scored data contain NA values | Guide filtering based on early-timepoint readcounts removed too many guides to score gene pairs with NA values (gene pairs with fewer guides than the min_guides parameter are not scored) | Consider relaxing guide filtering threshold, lowering the min_guides parameter, or investigating issues in early-timepoint screens |
| 2–11, 2–15, 2–18, 3–4 | Scoring takes much longer than expected given the number of screens | Control gene pairs with many guides are not filtered out |
Add controls such as “NT” guides to the filter_genes scoring parameter in a vector |
Timing
Procedure 1 demonstrates Orthrus’ ability to process data for 92,746 guides across six screens. On a Windows machine running R with a single core and 16 GB of memory, the procedure run as a script took slightly under 5 minutes of runtime. Because Orthrus can be run on different screens sequentially, memory usage is not anticipated to be a bottleneck.
To test Orthrus’ runtime for different numbers of screens, we ran Orthrus on a single screen duplicated 5, 10, 20, 50 and 100 times and timed the processing (Procedure 2, Steps 1–10), dual-targeting guide scoring (Procedure 2, Steps 11–14), and combinatorial scoring (Procedure 2, Steps 15–17) stages separately. For the processing stage, all guides were processed in the same function calls. However, because guide scoring doesn’t take into account information between screens, both guide scoring stages were run as a loop to score each screen separately. Timing results are summarized in Table 9.
Table 9.
Time taken during different steps of the pipeline for processing increasing numbers of screens. Values obtained using the script test_at_scale.R, which implements Procedure 2 in a loop and is available in the Zenodo repository here.
| Screens | Processing time (min) | Dual-targeted scoring time (min) | Combinatorial scoring time (min) |
|---|---|---|---|
| 1 | 1.66 | 1.14 | 0.40 |
| 5 | 3.86 | 6.57 | 2.10 |
| 10 | 5.60 | 10.28 | 3.28 |
| 20 | 8.99 | 19.43 | 5.71 |
| 50 | 20.11 | 48.42 | 14.76 |
Anticipated Results
All anticipated results for the protocol, as well as an R script containing the provided code, are available at https://zenodo.org/record/4527616.
Procedure 1
The files output by Procedure 1 are as follows. Step 2 outputs four files, two per FASTQ file, which contain reads trimmed down to the guide sequences and full reads where anchor sequences weren’t found. These are named based on the input .fastq.gz files with the format [FILENAME]_preprocessed.fastq and [FILENAME]_failed.fastq, respectively. Step 3 outputs six files, where two contain unmapped reads, two contain mapped reads, one is a.sam file and the last is a log file. If the bowtie command run in this step fails for any reason, the log file will note the error. Otherwise, it will log how long bowtie took to run at different stages and the numbers of reads processed, reads with alignments, and reads that failed to align. Out of 31.5M reads processed, about 18M reads should align and 13M reads should fail to align. The unmapped and mapped reads files are intermediate files not required by subsequent steps. Step 4 outputs the file “HH-79_counts.txt” which is processed by Step 5 into the file “rawCounts.txt.” Lastly, Step 6 is optional because the output file is included in the Orthrus R package, but is named “procedure1_reads.tsv.”
Procedure 2
Key results for the processing phase are QC plots, which are listed in Table 5. These include, among many other plots, a plot of total reads and a heatmap of LFCs across all replicates. The total reads for each screen should correspond to expected reads based on sequencing depth for each screen, which are plotted as dashed horizontal lines and can be set earlier in the pipeline when adding screens (Fig. 5). To interpret the heatmap, for typical experimental designs the most important features to examine are whether or not T0 screens cluster separately from other screens and whether or not technical replicates cluster within their respective screens (Fig. 6). The output AUC values for recovering essential genes should hover close to one for whole-genome screens as aforementioned, although for this highly specialized library with few essential genes and many strong expected effects we see values between 0.6 and 0.7 (Table 10).
Figure 5.
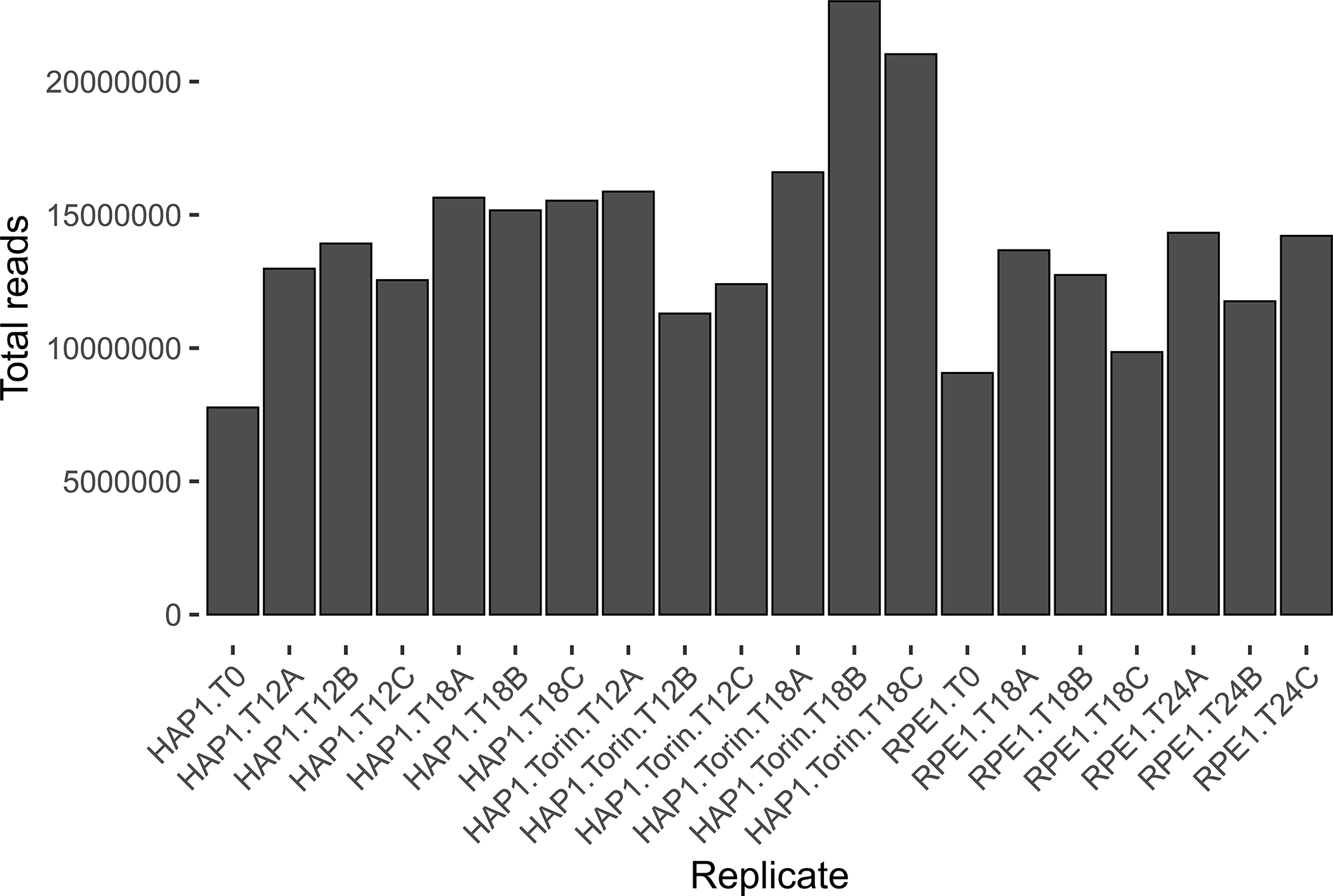
Total read counts for all technical replicates in the example CHyMErA dataset(n = 20), output in Procedure 2 step 5.
Figure 6.
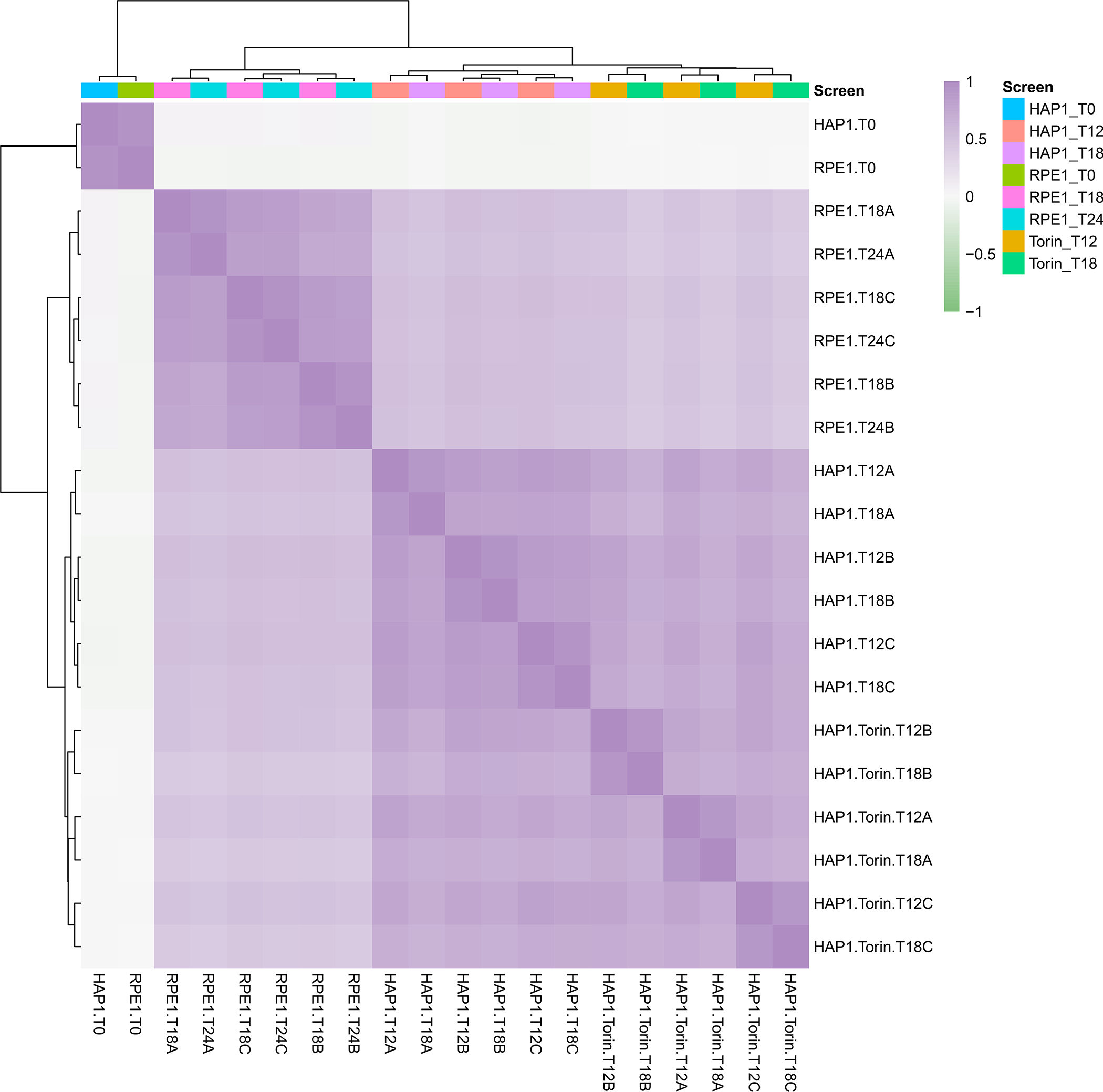
Heatmap of Pearson correlations between LFCs for all technical replicates in the example CHyMErA dataset (n = 20), output in Procedure 2 Step 8. Well-correlated screens cluster together depending on the main sources of variation in the dataset. Here, RPE1 screens cluster separately from the HAP1 screens, which are further clustered into WT HAP1 screens and HAP1 + Torin1 screens.
Table 10.
Evaluation of recovery of essential gene LFC values for all technical replicates compared to all other genes (reported as AUC values, area under the receiver operating characteristic curve values) from the ChyMErA dataset analyzed in Procedure 2, Step 8.
| Technical replicate | Essential gene recovery AUC |
|---|---|
| HAP1 T0 | 0.51 |
| RPE1 T0 | 0.51 |
| HAP1 T12A | 0.70 |
| HAP1 T12B | 0.70 |
| HAP1 T12C | 0.71 |
| HAP1 T18A | 0.69 |
| HAP1 T18B | 0.70 |
| HAP1 T18C | 0.70 |
| HAP1 + Torin1 T12A | 0.68 |
| HAP1 + Torin1 T12B | 0.68 |
| HAP1 + Torin1 T12C | 0.68 |
| HAP1 + Torin1 T18A | 0.67 |
| HAP1 + Torin1 T18B | 0.67 |
| HAP1 + Torin1 T18C | 0.67 |
| RPE1 T18A | 0.60 |
| RPE1 T18B | 0.60 |
| RPE1 T18C | 0.61 |
| RPE1 T24A | 0.61 |
| RPE1 T24B | 0.61 |
| RPE1 T24C | 0.61 |
Dual-targeted scoring should output scored Torin-specific GIs in HAP1 cells for two timepoints, T12 and T18. These results are plotted in the files “torin_vs_hap1_t12.png” and “torin_vs_hap1_t18.png,” and the scored data is contained in the files “dual_targeted_gene_calls_t12.tsv” and “dual_targeted_gene_calls_t18.tsv.” These output plots should resemble a slightly-skewed fit between data that’s mostly correlated with many outliers representing genetic interactions. The null model, indirectly shown by gray, non-interacting genes that correlate well across both screens, reflects the loess-normalization of residual values performed to account for skewed data or nonlinear trends (Fig. 7).
Figure 7.
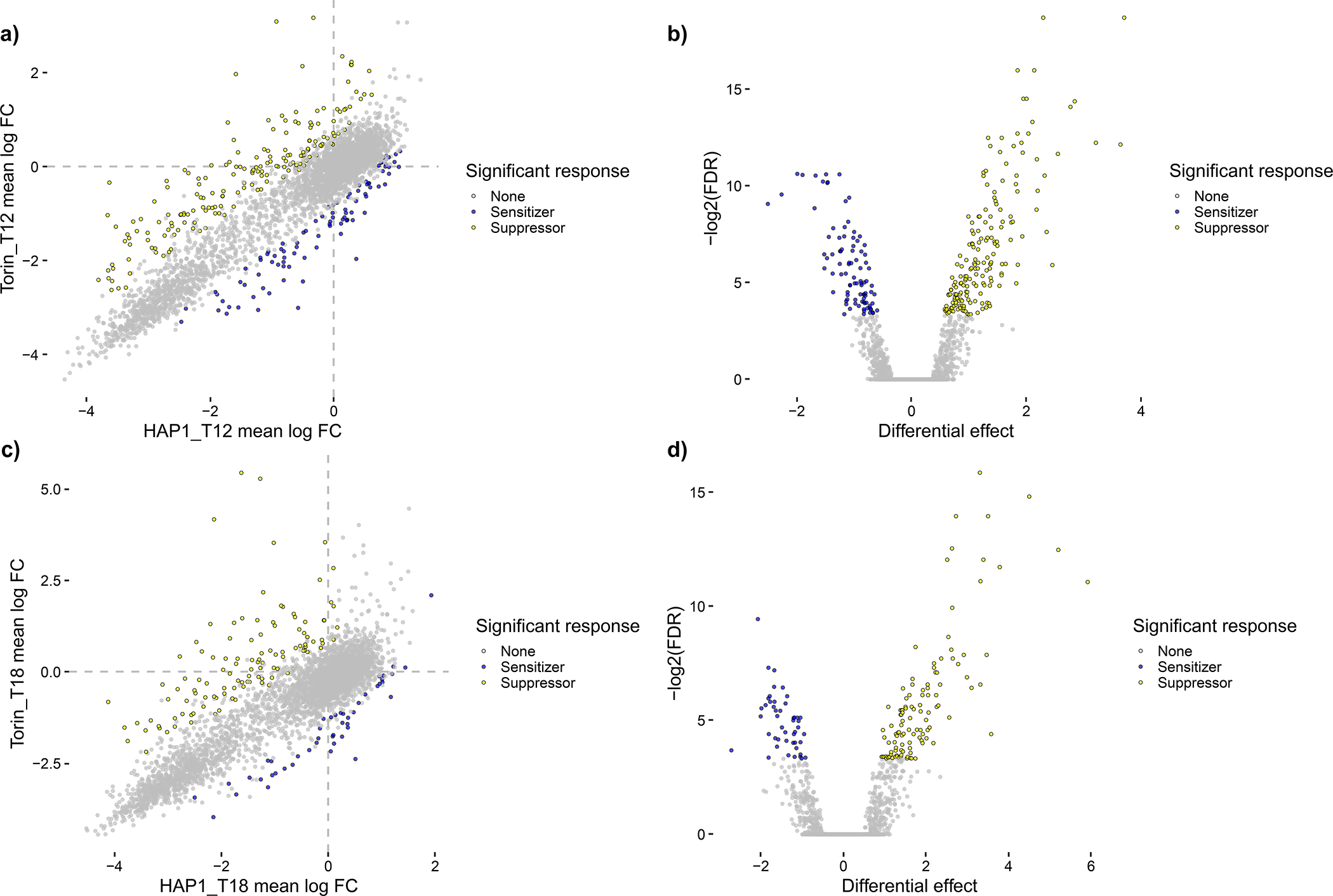
Summary plots of mean LFC, colored by significant effects, for WT HAP1 and HAP1 + Torin1 screening data for all n = 3,870 gene pairs with dual-targeting guides analyzed in Procedure 2, Steps 11–14. Gene-level drug-gene interactions, where sensitizer interactions indicate that the gene’s knockout confers increased sensitivity to Torin-1 and suppressor interactions indicate that the knockout bypasses potentially deleterious effects of Torin-1 on cell fitness, are shown as colored points that significantly deviate from the computed null model. Blue points are sensitizing interactions with mean residual effects < −0.5 and Benjamini and Yekutieli FDRs < 0.1, while yellow points are suppressor interactions with mean residual effects > 0.5 and Benjamini and Yekutieli FDRs < 0.1. (a) and (b) show scores for T12 data in a scatter plot and a volcano plot, respectively, for 182 suppressor interactions and 93 sensitizer interactions. (c) and (d) show scores for T18 data in a scatter plot and a volcano plot, respectively, for 114 suppressor interactions and 47 sensitizer interactions. These plots allow users to contextualize effect size, effect strength, and statistical significance for both WT HAP1 data at T12 and WT RPE1 data at T24.
Residual LFC values across all guides for all significant hits are also output from dual-targeted scoring (automatically with batch and wrapper scoring), and they should reflect consistent positive and negative effects for well-performing guides (Fig. 7). While context affects the definition of positive and negative effects, for chemogenetic screens negative effects typically represent sensitizers and positive effects represent suppressors, and for genetic interaction screens negative effects typically represent synthetic sick or lethal interactions while positive effects represent buffering interactions23. The top-ranked negative hit in WT HAP1 screening data at T18, HECTD1 (Fig. 8A), shows three such well-performing guides with consistent differential effects. On the other hand, the fourth-ranked negative hit, INPPL1 (Fig. 8B), shows mostly positive differential effects despite being called a negative hit. This is a red flag for the quality of this hit, and closer examination reveals that it is called as a result of how the loess-normalized null model poorly fits the handful of points with the strongest negative and positive expected effects. This hit should be ignored, and the user can consider tightening the effect size threshold to account for this during scoring. As another example, the eighth-ranked negative hit TAF5L (Fig. 8C) shows another case where 3 of its 4 guides display strong negative phenotypes, but the fourth showed little phenotype. This is not a red flag for hit quality, but could be a red flag for the guide with the smallest differential effect.
Figure 8.
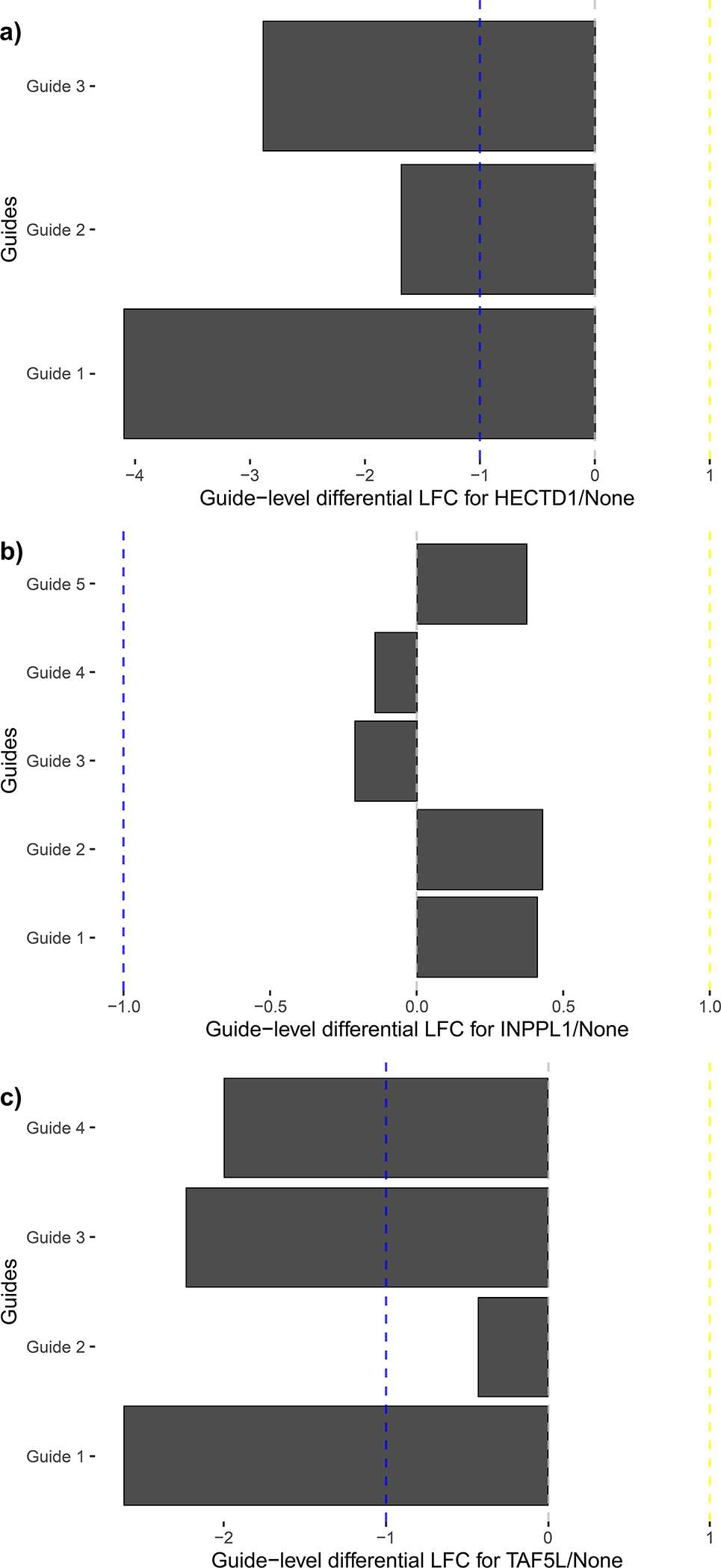
Differential LFC for WT HAP1 guides from the ChyMErA dataset analyzed in Procedure 2 comprising three significant hits of the scored dual-targeting guides at T18. Genes were scored based on deviations from a loess-corrected null model, so the agreement of individual guides of interesting hits should be examined to qualitatively confirm hit quality. (a) is the top-ranked negative hit, HECTD1, with strong agreement for all three guides. (b) is the fourth-ranked negative hit, INPPL1, whose guides indicate a lower hit quality due to three out of five guides possessing positive residual LFCs. (c) is the eighth-ranked negative hit TAF5L, with strong agreement for three guides and no phenotype for the fourth.
Combinatorial scoring should output scored interactions for roughly ~700 paralog gene pairs for all six screens. Similarly, six plots summarizing scored paralog gene pairs that resemble dual-targeted scoring plots should be output, one for each screen, with the scored data available in the file “paralog_gene_calls.tsv.” Two of these plots, for WT HAP1 T12 and WT RPE1 T24 data are shown in Fig. 9. Plots of residual effect values across guides for all significant hits for the HAP1 with Torin T18 and untreated HAP1 screens should also be output in the subfolders “HAP1_T18_combn” and “Torin_T18_combn” of the qc folder, respectively. The top negative and positive hits for WT HAP1 T12 data are shown in Fig. 10.
Figure 9.
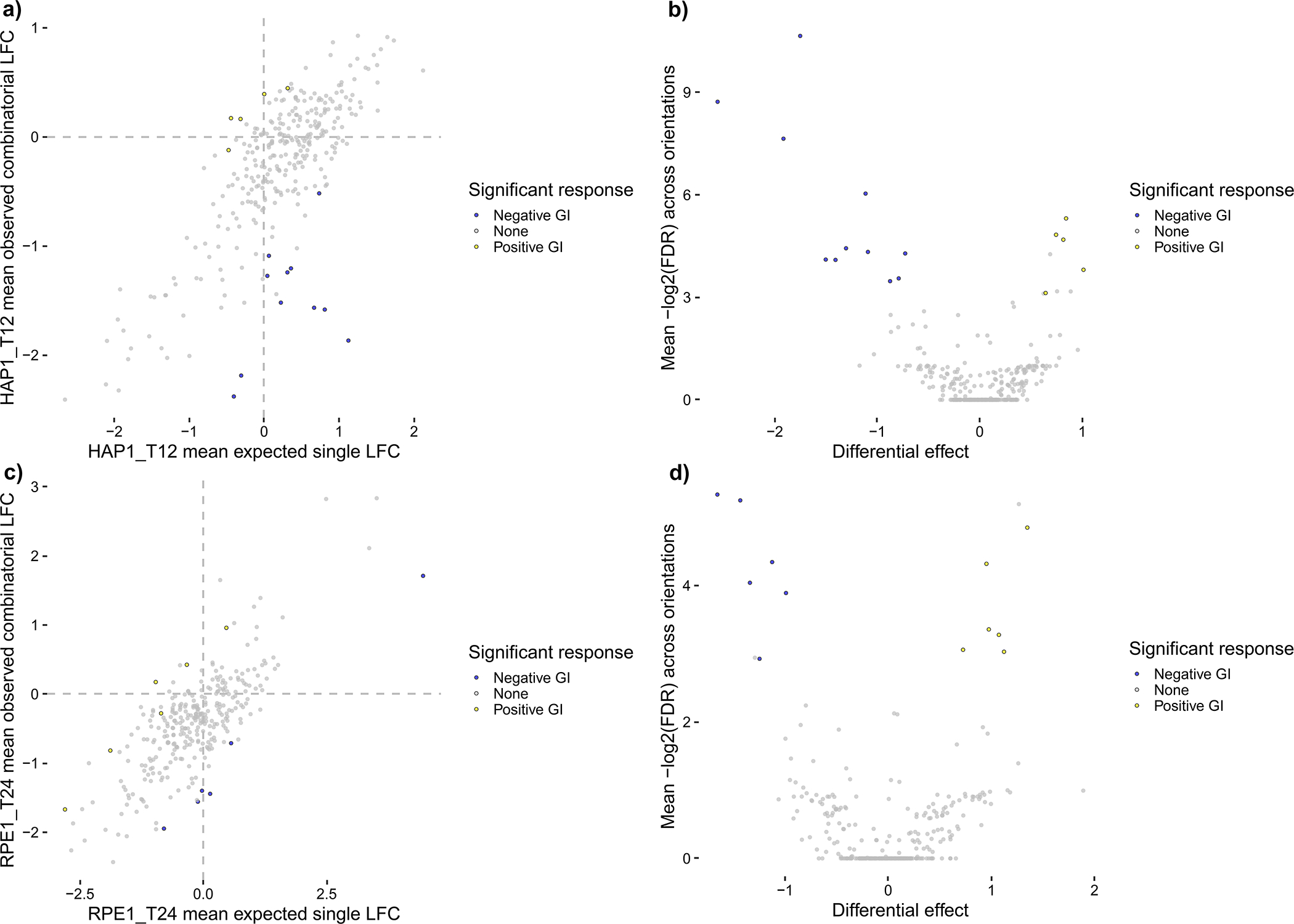
Summary plots of mean LFC, colored by significant effects, for all n = 313 CHyMErA gene pairs with combinatorial- and single-targeting guides analyzed in Procedure 2, Steps 15–17. Gene-level GIs are shown as colored points that significantly deviate from the computed null model. Blue points are negative GIs with mean residual effects < −0.5 and Benjamini and Yekutieli FDRs < 0.2, while yellow points are positive GIs with mean residual effects > 0.5 and Benjamini and Yekutieli FDRs < 0.2. (a) and (b) show scores for WT HAP1 data at T12 in a scatter plot and a volcano plot, respectively, for 5 positive GIs and 11 negative GIs. (c) and (d) show scores for WT RPE1 data at T24 in a scatter plot and a volcano plot, respectively, for 6 positive GIs and 6 negative GIs. These plots allow users to contextualize effect size, effect strength, and statistical significance for both WT HAP1 data at T12 and WT RPE1 data at T24.
To examine specific hits for data scored with Orthrus, we recommend the following process. First, establish clear statistical significance and effect size thresholds when calling significant hits, which may be different for different guide libraries and experiments. Second, to look at either negative (e.g. synthetic lethal) or positive (e.g. buffering) genetic interactions, subset the scored data to those that are labelled as negative or positive hits in the “effect_type_[CONDITION]” column. Third, sort the data by the differential effect column, which for dual-targeting scored data is “differential_[CONDITION]_vs_[CONTROL]” and for combinatorial-targeting scored data is “differential_combn_vs_single_[CONDITION].”
The output for batch scoring performed in Step 19 mirrors the output for the manual scoring performed in Steps 11–17, but contains additional residual LFC plot folders, as Steps 13 and 16 only generated residual LFC plots for four out of eight screens.
Procedure 3
The anticipated results for Procedure 3 mirror the anticipated results for Procedure 2, although dual-targeting plots and scores are not output because the analyzed guide library only contains combinatorial-targeting and single-targeting guides. Like for Procedure 2, all anticipated results are provided at https://zenodo.org/record/4527616.
A handful of differences between the anticipated output of the two Procedures exist. First, the scored data for Procedure 3 is only contained in the file “combn_gene_calls.tsv.” Accordingly, the “plots” folder only contains three corresponding scatterplots and three volcano plots, one for each screen. Second, because the library contains fewer than 10 guides that dual-target one non-essential gene or combinatorially-target two non-essential genes in the Hart et al. 2014 non-essential standard22, the file “essential_PR_QC.tsv” reports NA values for the AUC of recovering essential genes compared to non-essential genes under ROC curves. Third, we chose not to apply loess-normalization to score the dataset because of the wide spread of points at the left tail at the plot with both strong phenotypes. Loess typically overfits points such as these, and for this dataset a visual examination of the A549 screen shows that the linear fit to residuals compared to loess-correction avoids both likely false positives and false negatives (Fig. 10). The scored data shown in the plots folder thus represents plots scored against a linear fit with Step 4B as opposed to loess-corrected residuals calculated in Step 4A.
Supplementary Material
Supplementary Video 1. Tutorial on how to format the reads file, sample and batch tables for running Orthrus.
Acknowledgements
We are grateful to K. Lin for his help testing the Orthrus package. H.N.W, M.B. and C.L.M. were partially supported by grants from the National Science Foundation (1818293) and the National Institutes of Health (R01HG005084, R01HG005853). T.G.-P. was supported by the NIH Earl Stadtman Investigator Program and the NIH Distinguished Scholars Program. M.A. was supported by a Swiss National Science Foundation fellowship (P300PA_164667). M.B. was supported by the German Research Foundation DFG (Bi 2086/1–1). B.J.B was supported by a Canadian Institutes for Health Research Foundation grant (FDN-148434) and by an Ontario Institute of Regenerative Medicine grant. J.M. was supported by grants from the Canadian Institutes for Health Research (MOP-142375) and by Genome Canada (OGI-157).
Footnotes
Competing Financial Interests
A patent application (no. GB 1907733.8) describing the development and applications of CHyMErA, to the University of Toronto and T.G.-P., M.A., K.R.B., J.M. and B.J.B., is pending. J.M. previously performed sponsored research for Repare Therapeutics.
Code Availability
The Orthrus package and example dataset is available at https://github.com/csbio/Orthrus. All code presented from all procedures is available in separate scripts along with their expected output, and are provided under a CC-BY 4.0 license at https://zenodo.org/record/4527616. Code contained in this repository generated each panel of Figures 4–10.
Related links
Gonatopoulos-Pournatzis, et al. Genetic interaction mapping and exon-resolution functional genomics with a hybrid Cas9–Cas12a platform. Nat. Biotechnol. 38, 638–648 (2020). https://doi.org/10.1038/s41587-020-0437-z
TWEET: Orthrus; an R package for processing, scoring and analyzing combinatorial CRISPR screening data.
Data Availability
The example dataset is downloadable with the Orthrus package at https://github.com/csbio/Orthrus. The expected output from all Procedures is provided under a CC-BY 4.0 license at https://zenodo.org/record/4527616.
References
- 1.VanderSluis B et al. Integrating genetic and protein–protein interaction networks maps a functional wiring diagram of a cell. Curr. Opin. Microbiol. 45, 170–179 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Costanzo M et al. A global genetic interaction network maps a wiring diagram of cellular function. Science 353, aaf1420–aaf1420 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simpkins SW et al. Predicting bioprocess targets of chemical compounds through integration of chemical-genetic and genetic interactions. PLOS Comput. Biol. 14, e1006532–e1006532 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piotrowski JS et al. Functional annotation of chemical libraries across diverse biological processes. Nat. Chem. Biol. 13, 982–993 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shalem O et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang T, Wei JJ, Sabatini DM & Lander ES Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hart T et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 163, 1515–1526 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Ruiz S et al. A Genome-wide CRISPR Screen Identifies CDC25A as a Determinant of Sensitivity to ATR Inhibitors. Mol. Cell 62, 307–313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wong ASL et al. Multiplexed barcoded CRISPR-Cas9 screening enabled by CombiGEM. Proc. Natl. Acad. Sci. 113, 2544–2549 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsherniak A et al. Defining a Cancer Dependency Map. Cell 170, 564–576.e16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aregger M et al. Systematic mapping of genetic interactions for de novo fatty acid synthesis identifies C12orf49 as a regulator of lipid metabolism. Nat. Metab. 2, 499–513 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonatopoulos-Pournatzis T et al. Genetic interaction mapping and exon-resolution functional genomics with a hybrid Cas9–Cas12a platform. Nat. Biotechnol. 38, 638–648 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Najm FJ et al. Orthologous CRISPR-Cas9 enzymes for combinatorial genetic screens. Nat. Biotechnol. 36, 179–189 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen JP et al. Combinatorial CRISPR-Cas9 screens for de novo mapping of genetic interactions. Nat. Methods 14, 573–576 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han K et al. Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat. Biotechnol. 35, 463–474 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gier RA et al. High-performance CRISPR-Cas12a genome editing for combinatorial genetic screening. Nat. Commun. 11, 3455 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horlbeck MA et al. Mapping the Genetic Landscape of Human Cells. Cell 174, 953–967.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dede M, McLaughlin M, Kim E & Hart T Multiplex enCas12a screens detect functional buffering among paralogs otherwise masked in monogenic Cas9 knockout screens. Genome Biol. 21, 262 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Replogle JM et al. Combinatorial single-cell CRISPR screens by direct guide RNA capture and targeted sequencing. Nat. Biotechnol. 38, 954–961 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zamanighomi M et al. GEMINI: A variational Bayesian approach to identify genetic interactions from combinatorial CRISPR screens. Genome Biol. 20, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Imkeller K, Ambrosi G, Boutros M & Huber W gscreend: modelling asymmetric count ratios in CRISPR screens to decrease experiment size and improve phenotype detection. Genome Biol. 21, 53 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hart T, Brown KR, Sircoulomb F, Rottapel R & Moffat J Measuring error rates in genomic perturbation screens: gold standards for human functional genomics. Mol. Syst. Biol. 10, 733 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boucher B & Jenna S Genetic interaction networks: better understand to better predict. Front. Genet 4, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ritchie ME et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang YH et al. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 30, e15 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Video 1. Tutorial on how to format the reads file, sample and batch tables for running Orthrus.
Data Availability Statement
The example dataset is downloadable with the Orthrus package at https://github.com/csbio/Orthrus. The expected output from all Procedures is provided under a CC-BY 4.0 license at https://zenodo.org/record/4527616.