

Abstract
Purpose
In vitro maturation (IVM) of human oocytes offers an invaluable opportunity for infertility treatment. However, in vitro matured oocytes often show lower developmental abilities than their in vivo counterparts, and molecular mechanisms underlying successful maturation remain unclear. In this study, we investigated gene expression profiles of in vitro matured oocytes at the single‐cell level to gain mechanistic insight into IVM of human oocytes.
Methods
Human oocytes were retrieved by follicular puncture and in vitro matured. In total, 19 oocytes from 11 patients were collected and subjected to single‐cell RNA‐seq analyses.
Results
Global gene expression profiles were similar among oocytes at the same maturation stage, while a small number of oocytes showed distinct transcriptomes from those at the corresponding maturation stage. Differential gene expression analysis identified hundreds of transcripts that dynamically altered their expression during IVM, and we revealed molecular pathways and upstream regulators that may govern oocyte maturation. Furthermore, oocytes that were delayed in their maturation showed distinct transcriptomes. Finally, we identified genes whose transcripts were enriched in each stage of oocyte maturation.
Conclusions
Our work uncovers transcriptomic changes during human oocyte IVM and the differential gene expression profile of each oocyte.
Keywords: delay in oocyte maturation, human oocyte, oocyte maturation, single‐cell RNA‐sequencing, transcriptome
In vitro maturation of human oocytes is used for infertility treatment, but molecular mechanisms underlying successful maturation remain unclear. In this study, we reveal transcriptomes of human oocytes during in vitro maturation by singlecell RNA‐seq analyses and identify maturation stage‐specific transcripts.

1. INTRODUCTION
The first successful pregnancy of an in vitro matured human oocyte after in vitro fertilization (IVF) was reported in 1991. 1 Although the clinical application of in vitro maturation (IVM) has increased since the first successful birth resulting from in vitro matured oocytes, 1 , 2 IVM technology is not widely used in infertility treatment due to its poor outcome. Importantly, it is generally regarded that the quality of embryos generated from IVM oocytes is worse than that of embryos from in vivo matured oocytes in multiple aspects, such as developmental competence and subsequent pregnancy rates. 3 , 4 Therefore, oocytes other than the metaphase II (MII) stage at the time of oocyte retrieval are discarded in many fertility clinics. However, IVM of oocytes offers a unique opportunity. For example, for women diagnosed with polycystic ovary syndrome, the injection of human chorionic gonadotropin for ovulation can increase the risk of ovarian hyper stimulation syndrome. This risk can be prevented by performing IVM after collecting non‐MII oocytes. 5 In addition, ovarian tissue is cryopreserved prior to cancer treatment for ensuring fertility of patients who might lose gonadal functions due to the treatment. The introduction of these fertility preservation techniques has provided the possibility of obtaining immature oocytes from very small antral follicles and cryopreserving them after IVM. 6 There are reports of pregnancy using embryos obtained from IVM of these oocytes. 7 , 8 , 9 Thus, IVM of immature oocytes plays a key role in warranting fertility for some patients.
Despite the fact that IVM of human oocytes is an invaluable choice for human reproductive technologies, the pregnancy rate of IVM‐derived oocytes is still low. 3 , 10 Moreover, the developmental ability of IVM oocytes is variable. 3 It is extremely important to learn about the nature of this variability of oocyte quality. The quality of oocytes is profoundly affected by gene expression during oocyte growth. 11 , 12 As a result, the gene expression pattern of MII oocytes with a high developmental capacity seems different from that with a low developmental ability. 13 , 14 However, it remains unclear when the gene expression pattern of human MII oocytes is established during maturation and how variable transcriptomes are found.
Ttranscriptome of oocytes at different stages has first been studied by microarrays. 15 , 16 , 17 One report has revealed that in vitro matured MII oocytes show an over‐abundance of detected transcripts, as compared with in vivo‐derived MII oocytes, 16 suggesting a different transcriptome pattern between in vitro‐ and in vivo‐matured oocytes. The same paper has also suggested that germinal vesicle (GV) and metaphase I (MI) oocytes show similar gene expression profiles. 16 Recently, single‐cell RNA‐seq has been applied to reveal transcriptomes of human oocytes. 18 , 19 , 20 , 21 Zhang et al. 18 reported transcriptomic profiles of human oocytes during folliculogenesis. Differential gene expression related to energy metabolism has been identified between in vitro‐ and in vivo‐matured oocytes. 19 Yu et al. 20 have shown that fewer transcripts are expressed in matured oocytes than immature GV oocytes. The reduced transcripts in MI and MII oocytes are linked to the degradation of maternal RNA for subsequent embryonic development, and the global reduction of mRNA during oocyte maturation is detected as oocytes at these stages are transcriptionally inactive and de novo transcription is not observed. It is also known that polyadenylation of maternal mRNA during oocyte maturation controls protein translation for successful oocyte maturation and subsequent development. 22 , 23 Therefore, the dynamics of polyadenylated maternal RNA need to be determined during human oocyte maturation.
In this study, transcriptomes for human oocytes at different maturation stages were examined using single‐cell RNA‐seq, which captures polyadenylated RNA, and differentially expressed genes were identified. Surplus oocytes, which are not used for infertility treatment, were subjected to this study. Immature oocytes at the time of oocyte retrieval were used for IVM, and oocytes at the GV, MI, and MII stages were subjected to RNA‐seq. Moreover, oocytes that reached each stage with a significant delay were also collected for the comparison. By comparing the transcriptomes of GV, MI, and MII oocytes, we found genes and pathways whose expression was dynamically altered during maturation. Our study deepens the molecular understanding of human oocyte maturation and provides an important platform to search for marker genes that reflect oocytes at different maturation states.
2. MATERIALS AND METHODS
2.1. Human oocyte
Informed consent for this prospective study was obtained from all participants. Surplus oocytes remaining after infertility treatment by IVF or intracytoplasmic sperm injection (ICSI) were used for this study. The oocyte samples for this study were collected from 11 patients in 2019. After aspirating follicles with a fine needle under the ovarian echo, the cumulus oocyte complexes (COCs) were picked up under a stereomicroscope. COCs from infertile patients were scheduled for ICSI, and surrounding cumulus cells were removed with multipurpose handling medium (MHM; Scientific's, 90163) containing 20 IU/ml Cumulus Remover (Recombinant hyaluronidase; Kitazato, 94114). After denudation, oocyte morphology was examined and an oocyte at the MII stage was used for infertility treatment. Oocytes other than the MII stage were subjected to IVM. The collected and denuded oocytes were incubated in the Sydney IVF fertilization medium (Cook medical, K‐sifm‐100) at 37°C under 5% CO2 and 5% O2 in air for IVM, and oocytes that had matured to the MII stage by 16:00 on the same day of oocyte retrieval were used for the treatment of IVF or ICSI. 24 , 25 The remaining oocytes that had not reached the MII stage by 16:00, which are usually treated as surplus oocytes, were further cultured for this study. A few hours after the incubation, some oocytes were matured to MII oocytes, and such matured oocytes were subjected to sampling for RNA‐seq analyses. At that time, GV and MI oocytes were also sampled. For MII oocytes, zona pellucida piercing was performed on the denuded oocytes with LYKOS Clinical IVF laser system and Clinical Laser Software Legacy 5.12 (Hamilton Thorne, 40025) and the first polar body was biopsied with micropipette (CooperSurgical, MPBFP30). Zona was lysed with acidic‐tyrode (Merck, T1788) for 20–30 s, and washed in Phosphate‐buffered saline (PBS; Nacalai Tesque, 07269‐84) containing 0.1% w/v bovine serum albumin (BSA; Merck, NM‐126609‐5GMCN). The zona‐ and polar body‐free oocytes suspended in 1 μl of 0.1% w/v BSA/PBS were moved into 9.5 µl of 1× Reaction buffer from SMART‐seq v4 Ultra Low Input RNA Kit (Takara Bio, 24888N), in which 10× lysis buffer (Takara Bio, 635013) and 2 unit ribonuclease inhibitor (Thermo Fisher Scientific, 10777‐019) were mixed. GV and MI oocytes were similarly sampled for RNA‐seq analyses, except that polar body removal was not carried out. Some oocytes that remained immature at the time of the above‐mentioned sampling for RNA‐seq analyses were left in the maturation medium overnight, and we examined the maturation state on the next day of the oocyte retrieval (9:00 am). Such oocytes were sampled as those which were delayed in their maturation by following the same sampling method as described above. The classification criteria for GV, MI, and MII oocytes are explained in the corresponding result section. This study was performed within the guidelines established by the Clinical Research Ethics Review Committee of Mie University Hospital (H2018‐066) and was registered in the University Hospital Medical Information Network Clinical Trials Registry in Japan (UMIN000034811). In addition, this study was registered for the research dealing with human sperm, ova, and fertilized ova with the Japan Society of Obstetrics and Gynecology. This study was conducted in accordance with the Declaration of Helsinki.
2.2. Nuclear staining
Nuclear staining was performed using the surplus oocytes at each stage as follows. Oocytes were incubated in the fixation medium, 4% w/v paraformaldehyde (Nacalai tesque, Japan, 26126‐25), for 30 min at room temperature, followed by three washes with 0.2% w/v BSA/PBS. After permeabilization with 0.2% v/v Triton X‐100/PBS for 30 min at room temperature, they were washed with 0.2% w/v BSA/PBS. Then, nuclei were stained with 1 μg/ml DAPI (Nacalai tesque, 11034‐56) in 0.2% w/v BSA/PBS solution for 10 min, followed by washes with 0.2% w/v BSA/PBS. The fluorescence signals were observed using a confocal microscope (Carl Zeiss, LSM800), equipped with a laser module (405/488/561/640 nm) and GaAsP detector. Z‐slice thickness was determined using the optimal interval function in the ZEN software (Carl Zeiss).
2.3. Library preparation for RNA‐seq
A single oocyte at each stage was subjected to cDNA synthesis and amplification using SMART‐Seq® v4 Ultra® Low Input RNA Kit (Takara Bio, Z4889N) according to the vendor's instruction with some modifications. Briefly, collected surplus oocytes were subjected to micromanipulation to remove their sibling first polar bodies, and then, zona pellucida was chemically removed as described above. After removing the polar body and the zona pellucida, oocytes were washed with 0.1% w/v BSA/PBS, and then transferred to a 0.2‐ml tube containing 9.5 μl of reaction buffer to make the final volume of 10.5 μl. After cell lysis in the buffer, 1 μl of the solution was removed and, instead, 1 μl of the diluted ERCC RNA Spike‐In Mix was added (diluted for 350 pg of total RNA according to the vender's instruction; Thermo Fisher Scientific, 4456740). The lysed RNA solution with the spike‐in RNA was subjected to reverse transcription. The produced cDNA was amplified by PCR with 16 cycles. The amplified cDNA was purified using AMPure XP beads (Beckman Coulter, A63882). The purified cDNA was measured on the Bioanalyzer (Agilent) using High Sensitivity DNA Kit (Agilent, 5067‐4626), and the normalized volume of cDNA was subjected to library preparation using Nextera XT DNA Library Preparation Kit (Illumina, FC‐131‐1024) by following the vendor's instruction. The obtained library was quality‐checked by Bioanalyzer and quantified using Qubit (Thermo Fisher Scientific). The experiments have been approved by the Clinical Research Ethics Review Committee of Kindai University at the Department of Biology‐Oriented Science and Technology (H30‐1‐006) and we followed the university's guidelines to store the sequencing data.
2.4. Sequencing data filtering
Paired end sequencing (50 bp + 25 bp) was carried out using the Illumina NextSeq system. Fastq files from Illumina sequencing were filtered for low‐quality reads by sliding window trimming (window size 10, <QV20), and low‐quality bases were trimmed from the ends of the reads (<QV20) using Trimmomatic. 26 Reads of less than 20 bases and unpaired reads were removed. Furthermore, adaptor, polyA, polyT, and polyG sequences were removed using trim_galore and cutadapt. 27 The sequencing reads were then mapped to the human genome (hg19) using STAR. 28 Reads on annotated genes were counted using featureCounts by taking multiple mapped reads into account. 29
2.5. RNA‐seq analysis
RNA‐seq reads were visualized using Integrative Genome Viewer. 30 Fragments per kilobase of exon per million mapped reads (FPKM) values were calculated from mapped reads by normalizing to total counts and transcript. Differentially expressed genes (DEGs; p adj < 0.05, 4‐fold difference in FRKM values or p < 0.05, 4‐fold difference in FRKM values) were then identified using DESeq2 31 by applying ERCC‐based normalization. An unsupervised hierarchical clustering of the read count values was performed using hclust in TCC (unweighted pair group method with arithmetic mean: UPGMA). Gene ontology (GO) terms showing over‐representation of genes that were up‐ or downregulated were detected using DAVID tools (p < 0.05). 32 , 33 Furthermore, the gene list of DEGs was compared with the categorized gene lists found in a paper. 18 Each gene list was further subjected to an Ingenuity Pathway Analysis (IPA; Qiagen, https://www.qiagenbioinformatics.com/products/ingenuity‐pathway‐analysis). Using IPA, enriched canonical pathways, upstream transcriptional regulators, gene regulatory networks, and diseases and biological functions in the identified DEGs were investigated. Expression levels of DEGs were displayed in heatmaps with z‐scores obtained from FPKM values of genes in the DEG lists by using heatmapper. 34 The DEG lists consist of genes that satisfy the condition p value < 0.05 and log2FC > 2 or log2FC < −2.
2.6. Statistical analyses
Significant GO terms were identified using the modified Fisher Exact p‐values according to the DAVID functional annotation tool. In IPA, p‐values were obtained according to the statistical methods applied to each analysis explained in the vendor's instruction. A value of p < 0.05 was considered significant. Number of replicates is indicated in the figure legends.
2.7. Data availability
The dataset generated during this study are available in the Supplementary Tables and in the Gene Expression Omnibus (GEO) public repository under accession GSE166533. All the other data are available from the corresponding author upon reasonable request.
3. RESULTS AND DISCUSSION
3.1. RNA‐seq analyses of human oocytes during in vitro maturation
To reveal transcriptomes of human oocytes during IVM, single‐cell RNA‐seq analyses were performed using oocytes at different stages. In total, 19 oocyte samples, collected from 11 patients aged between 30 and 39, were subjected to RNA‐seq analyses (Table S1). Oocytes were classified into three different stages; the immature GV stage, maturing oocytes from the GV to MII stage, and the matured MII stage (Figure 1A). Maturing oocytes from the GV to MII stage were described as the MI, and many of them showed condensed chromosomes after DNA staining (Figure 1A,B). As shown in Figure 1A, human oocytes were first collected for infertility treatment and oocytes at the MII stage or that reached the MII stage until 0–7 h after the oocyte retrieval were used for IVF or ICSI. Oocytes that do not reach the MII stage by then are not used for infertility treatment in Mie University Hospital and these surplus oocytes were analyzed. On the same day of oocyte retrieval, oocytes at the GV, MI, and MII stages were morphologically judged at 7.5–9 h after oocyte retrieval (GV×4, MI×3, and MII×3, respectively; in total, 10 oocytes are in this category) and sampling was carried out (Figure 1A). Some remaining oocytes were further cultured for 15–16.5 h, and oocytes that reached each developmental stage were collected. These oocytes were called as delayed GV, MI, and MII oocytes (dGV×3, dMI×3, and dMII×3, respectively; in total, 9 oocytes were in this category). To note, it has been shown that human MI oocytes cultured in vitro for 2–26 h developed to good‐quality embryos after ICSI and successful implantation was observed using these in vitro matured oocytes. 25 Therefore, it is expected that the oocytes collected under this experimental scheme retain a degree of developmental competence to develop to good‐quality embryos, although in vitro matured oocytes show the lower developmental potential than in vivo counterparts. 25 Successful collection of oocytes at different stages was confirmed with DNA staining (Figure 1B). The collected oocytes were subjected to single‐cell RNA‐seq by taking advantage of the SMART‐seq2 procedure. 35 After sequencing, 13 799 742 ± 4 127 048 reads were obtained and 85.6 ± 2.0% of reads were uniquely mapped to the human hg19 genome (Table S2, mean ±SD). We detected an average of 12 915 ± 1112 genes (FPKM > 1) per oocyte (Table S2). The number of expressed genes (FPKM > 1) decreased as oocyte maturation proceeded (Figure 1C and Table S2, p < 0.05). However, in vitro matured MII oocytes did not show a sharp decrease in the number of expressed genes as compared with that of MI oocytes (Figure 1C and Table S2). It has been reported that, in in vitro matured oocytes, maternal transcripts are not efficiently degraded as compared with their in vivo counterparts. 36 Therefore, the relatively high number of transcripts in the in vitro matured MII oocytes (Figure 1C) might be explained by the incomplete degradation of maternally stored RNA. On the other hand, MI oocytes collected on the day of oocyte collection might include those matured in vivo and remained at the MI stage, as opposed to MII oocytes that are all matured in vitro. Therefore, the low number of transcripts in MI oocytes sampled on the day of oocyte collection might be attributed to the marked degradation of maternal RNA during in vivo maturation.
FIGURE 1.
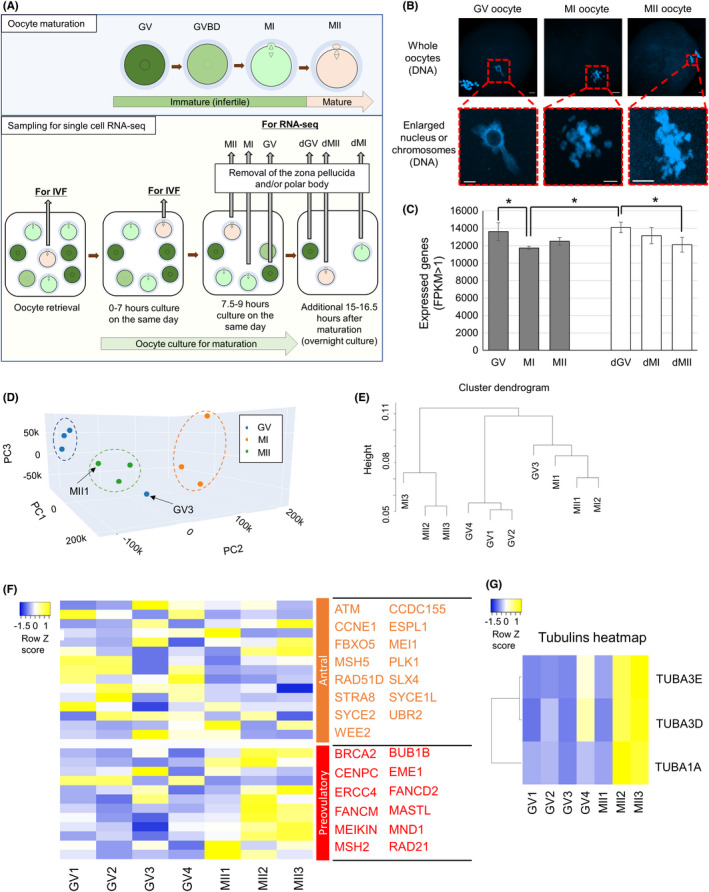
Transcriptomic changes of human oocytes during in vitro maturation. (A) A schematic diagram of human oocyte retrieval for single‐cell RNA‐seq analyses. Nineteen oocytes were collected from 11 patients. (B) Representative images of human oocytes at different stages. The bottom panels show higher magnification images of the regions indicated in the top panels by dashed boxes. DNA was stained by DAPI. Scale bars represent 20 μm for whole oocyte images and 10 μm for enlarged images. (C) The number of expressed genes in oocytes at different maturation stages (FPKM: Fragments per kilobase of exon per million mapped reads >1). Mean ± SD are shown. *p < 0.05 (Fisher's LSD test). (D) Principal component analysis (PCA) of the gene expression profiles (3–4 biological replicates per each maturation stage). Blue dots: GV samples, Orange dots: MI samples, and Green dots: MII samples. (E) Hierarchical clustering dendrogram generated using the gene expression profiles of human oocytes at different maturation stages. Spearman correlation was used. (F) A heatmap depicting the gene expression levels of meiosis‐related genes that have been reported to be abundantly detected in antral oocytes or preovulatory oocytes. 18 Color key with z‐score is shown. (G) A heatmap depicting the gene expression levels of Tubulin‐related genes. dGV, oocytes that were at the germinal vesicle stage the day after oocyte retrieval; dMI, oocytes that were at the metaphase I stage the day after oocyte retrieval; dMII, oocytes that were at the metaphase II stage the day after oocyte retrieval; GV, oocytes at the germinal vesicle stage; GVBD, germinal vesicle breakdown; IVF, in vitro fertilization; MI, maturating oocytes at the metaphase I stage; MII, oocytes at the metaphase II stage
We then compared transcriptomes during oocyte maturation. Generally, oocytes derived from the same stage resemble each other in their transcriptomes (Figure S1A). This notion is also supported by principal component analysis (PCA) (Figure 1D). Oocytes at different stages were also separated by a hierarchical clustering analysis (Figure 1E). Of note, a small number of oocytes were not well classified into the corresponding oocyte stages; GV3 rather exhibited a higher correlation with MI oocytes (Figure 1D,E and Figure S1A) and MII1 showed a closer transcriptome to that of the GV or MI state (Figure 1E). These results suggest that oocytes classified according to the morphological criteria are not always similar in their transcriptomes when gene expression is judged at the single‐cell level.
We next sought to identify differentially expressed genes (DEGs) between GV and MII oocytes as this comparison should represent transcriptomic changes that happen during in vitro maturation. When a stringent criterion is used (p adj < 0.05, 4‐fold difference), only 1 DEG was identified. This is probably due to the variable transcriptome of some individual oocytes such as MII1 and GV3 (Figure 1D,E and Figure S1A). Then, we used another condition to find DEGs between oocytes at different stages (p < 0.05, 4‐fold difference), and 324 DEGs were found; 142 genes were upregulated in MII oocytes, while 182 genes were downregulated at the MII stage (Table S3). Genes that were previously shown to be involved in oocyte meiosis and subsequent embryonic development such as ASF1B, ASNA1, CDC45, and MOS were indeed upregulated in MII oocytes (Table S3). 37 , 38 , 39 , 40 GO analyses were performed using these gene sets, and several significantly enriched terms were identified (Figure S1B). GO terms such as microtubule‐based process and cytoskeleton organization were enriched in MII‐upregulated genes (Figure S1B). We compared these GO terms with a previously published dataset investigating transcriptomic changes of oocytes during human folliculogenesis. 18 GO terms such as microtubule‐based process and actin cytoskeleton organization are enriched in DEGs of human in vivo oocytes obtained from antral follicles when compared with in vivo oocytes of other follicular stages. 18 It is expected that the DEGs of our in vitro matured MII oocytes should resemble those of in vivo oocytes collected from preovulatory follicles, rather than antral follicles, if in vitro maturation is successful. We therefore hypothesized that this GO outcome might be attributed to the incomplete maturation of our in vitro matured oocytes as compared with in vivo matured oocytes, and examined previously reported meiosis‐related genes whose expression increases toward the preovulatory stage. 18 Many meiotic marker genes that should be higher in in vivo preovulatory oocytes than antral oocytes were upregulated in MII oocytes, while some were not (Figure 1F). Furthermore, transcripts of meiosis‐related genes whose expression should be higher in oocytes from antral follicles 18 were not fully downregulated in our in vitro matured MII oocytes (Figure 1F), reminiscent of incomplete degradation of oocyte RNA during in vitro maturation. These results suggest that dynamic changes in expression of meiosis‐related genes were not entirely, but partially complete in in vitro matured oocytes. It is also noteworthy that, among the DEG list, tubulins (TUBA1A, TUBA3E, TUBA3D) showed upregulation during in vitro maturation (Figure 1G), in accordance with the GO outcome (Figure S1B). Translational regulation of these genes might play a role in in vitro oocyte maturation.
To further characterize transcriptomic changes during in vitro maturation of human oocytes, DEGs identified by comparing GV and MII oocytes were subjected to Ingenuity Pathway Analysis (IPA). IPA of DEGs revealed the significantly changed canonical pathways and predicted upstream regulators. As upstream regulators, factors that have been shown to play a role in oocyte maturation such as insulin, NOBOX, PRKAA1, progesterone, and quercetin were identified (Table S4). 41 , 42 , 43 , 44 Interestingly, hypoxia‐inducible factor 1a (HIF1A) target genes were inhibited in MII oocytes presumably due to the effect of HIF1A proteins stored in oocytes (Figure 2A). HIF1A is involved in the hypoxia‐induced dormant state of oocytes 45 and is upregulated from the primary oocytes stage. 18 Injection of an inhibitor that suppresses HIF1A activity into mice reduces the quality of oocytes by impairing the meiotic apparatus, suggesting a defect in spindle assembly and actin dynamics. 46 In good agreement with the previous study, 46 we have also identified genes related to actin dynamics such as VASP as downstream targets for HIF1A (Figure 2A). Therefore, repression of HIF1A target genes might be a part of successful oocyte maturation. HIF is also known as a key player for metabolic reprogramming in concert with Sirtuin. 47 Sirt1‐Nrf2‐Cyclin B1/CDK1 signaling pathway is important for oocyte meiosis by regulating spindle/chromosome organization. 48 Importantly, canonical pathway analyses showed that Sirtuin signaling genes were enriched in DEGs (Figure 2B; green arrow). Taken together, HIF1A and Sirtuin signaling pathway might be an important player for in vitro oocyte maturation in human.
FIGURE 2.
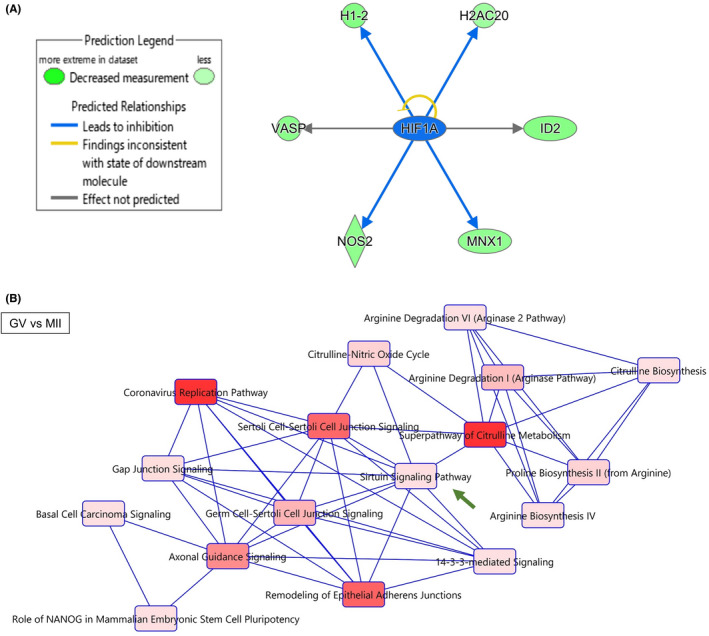
Gene regulatory networks that are associated with dynamic transcriptome changes during human oocyte maturation. (A) Downstream targets of HIF1A were found in the DEG list by Ingenuity Pathway Analysis (IPA). Blue arrows mean “Leads to inhibition,” and black arrows mean “Effect not predicted.” Molecules colored green are downregulated in MII samples. (B) Canonical pathways predicted by IPA using the DEG list of GV versus MII. Significant terms are indicated in red colors: the darker the color, the more significant the term is (p < 0.05, Fisher's exact test). Closely related terms are connected to each other. A green arrow indicates Sirtuin signaling pathway as explained in the relevant text
3.2. Differentially expressed genes between normal and delayed oocytes
We next examined transcriptomic profiles of oocytes that were significantly delayed to reach MI and MII stages (Figure 1A; dMI and dMII, respectively) or GV oocytes that remained immature (Figure 1A; dGV). These types of oocytes are not used for human infertility treatment, but their developmental ability and similarity to the early matured oocytes have not been elucidated well. PCAs showed that transcriptomes of dGV were similar to those of GV oocytes (Figure 3A; +dGV). In contrast, dMI samples were well separated from MI (Figure 3A; +dMI). Regarding transcriptomes of dMII, two of them were close to MII, but one of dMII samples (dMII3) showed a distinct transcriptomic pattern (Figure 3A; +dMII). A hierarchical clustering analysis also supported these notions (Figure S2A).
FIGURE 3.
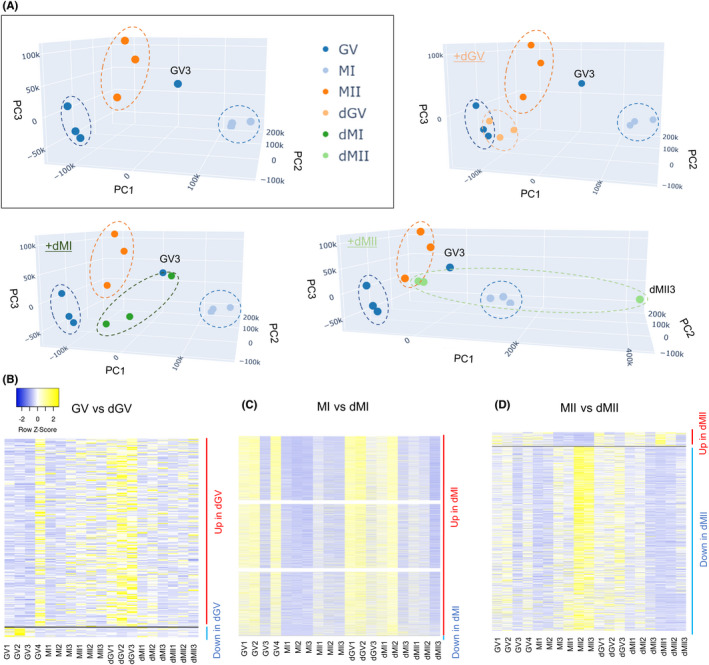
Human oocytes that are delayed in their maturation show different transcriptomes from those matured in an earlier timing. (A) PCA of the gene expression profiles (3–4 biological replicates per each category). The top left image in the rectangle includes samples collected on the day for oocyte retrieval (Figure 1A) (GV, MI, and MII). Nine delayed oocytes were collected from seven patients. Blue dots: GV samples, light blue dots: MI samples, dark orange dots: MII samples, light orange dots: dGV samples, dark green dots: dMI samples, and light green dots: dMII samples. (B–D) Heatmaps depicting the gene expression levels of DEGs identified in the indicated comparisons. (B) shows DEGs in dGV (GV vs. dGV), (C) shows those in dMI (MI vs. dMI), and (D) shows those in dMII (MII vs dMII) samples. Color key with z‐score is shown
We then identified DEGs specifically up‐ or downregulated in delayed oocytes. When a stringent condition is used (p adj < 0.05, 4‐fold difference), 0, 3610, and 32 DEGs were identified by comparing GV vs dGV, MI vs dMI, and MII vs dMII stage oocytes, respectively. Then, we used another condition to find DEGs between oocytes at different stages (p < 0.05, 4‐fold difference), and 244, 4937, and 805 DEGs were identified by comparing GV vs. dGV, MI vs. dMI, and MII vs. dMII stage oocytes, respectively. To be consistent with the analyses performed for comparing GV and MII oocytes in Figures 1 and 2, the later condition for finding DEGs (p < 0.05, 4‐fold difference) was used for further investigation. DEGs were grouped into upregulated and downregulated genes in each comparison (Figure 3B–D, and Table S5); 232 (GV vs. dGV, up in dGV), 4936 (MI vs. dMI, up in dMI), and 57 (MII vs. dMII, up in dMII) upregulated genes were found, while 12 (GV vs. dGV, down in dGV), 1 (MI vs. dMI, down in dMI), and 748 (MII vs. dMII, down in dMII) genes were downregulated.
The comparison between GV and dGV did not result in the identification of many specific gene sets that were disturbed in dGV samples (Table S5), suggesting that transcripts are relatively stable during the overnight incubation of GV oocytes. We then focused on the comparison between MI and dMI oocytes. As discussed above, MI oocytes here might contain those matured in vivo, while dMI should be in vitro cultured and/or matured overnight as only immature oocytes are left at the end of the day for the oocyte collection. GO analyses were performed using these gene sets (Figure S2B). Many specific terms were identified in upregulated genes of dMI samples, and especially, genes involved in mitochondrial regulation were misregulated (Figure S2B). IPA predicted gene networks that were enriched in DEGs of dMI oocytes, and molecular pathways related to cell death were found (Figure S3, dotted square). Together, dMI oocytes might have activated apoptotic pathways.
Next, we performed GO and IPA analyses using DEGs identified between MII and dMII samples. The comparison between MII and dMII by GO analysis shows that the term “ubiquitin protein ligase binding” was identified as most significant in the molecular function using the upregulated gene list in MII oocytes (Figure S2C). Previous studies indicate that genes involved in ubiquitin‐mediated proteolysis are upregulated in MII oocytes. 18 , 20 Together, delayed MII oocytes might fail to upregulate genes related to the ubiquitin proteosome pathway, which is required for subsequent embryonic development. Furthermore, IPA of DEGs revealed canonical pathways and predicted upstream regulators, which were significantly affected by delayed maturation. In canonical pathway analysis, altered pathways between MII and dMII (Figure 4A) were well overlapped with the molecular pathways identified by comparing GV and MII (Figure 2B). This result suggests that genes and related pathways, which are activated during in vitro maturation, tend to be misregulated in delayed MII oocytes. Furthermore, genes related to Sirtuin Signaling Pathway were identified by comparing MII vs dMII stage oocytes (Figure 4A, green arrow); Of note, CYC1, SDHD, and tubulin genes (TUBA1A, TUBA1B, TUBA3C/TUBA3D, TUBA3E, TUBA4A), all involved in Sirtuin Signaling Pathway, were downregulated in delayed MII oocytes (Table S5). As the upstream regulators in the comparison between MII and dMII (Table S6), SMARCA4 was detected and target genes of SMARCA4 were downregulated in delayed MII oocytes (Figure 4B). In good line with our observation, SMARCA4 was shown to be accumulated in aged preovulatory oocytes, 49 which might induce repression of its target genes. Furthermore, an appropriate amount of SMARCA4 transcripts is important for gene expression regulation and embryonic development. 50 Oocytes of delayed maturation might accumulate SMARCA4, which in turn results in misregulated gene expression.
FIGURE 4.
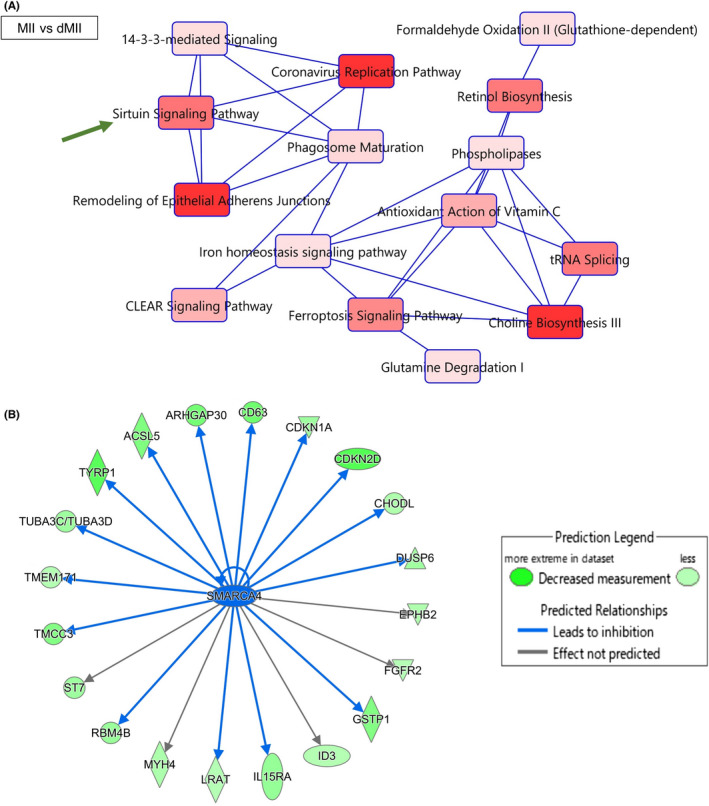
Gene regulatory networks are altered in the human oocytes that are delayed in their maturation. (A) Canonical pathways predicted by IPA using the differentially expressed gene (DEG) list of MII versus dMII. Significant terms are indicated in red colors: the darker the color, the more significant the term is (p < 0.05, Fisher's exact test). Closely related terms are connected to each other. A green arrow indicates Sirtuin signaling pathway as explained in the relevant text. (B) Downstream targets of SMARCA4 were found in the DEG list by IPA. Gene regulatory networks of SMARCA4 in human oocytes were predicted. SMARCA4 affects molecules indicated at the end of arrows, blue arrows mean ‘Leads to inhibition’, black arrows mean “Effect not predicted.” Molecules colored green are downregulated in dMII samples
3.3. Potential marker genes for oocytes at different maturation stages
We next sought to find marker genes that were specifically expressed in each stage of oocytes during maturation. First, we tried to find transcripts whose expression was altered upon the initiation of in vitro maturation. DEGs were identified by comparing the gene expression levels of oocytes at the GV stage with those at the MI and MII stages. This analysis led to the identification of 1889 upregulated transcripts in GV oocytes (p < 0.05, 4‐fold difference; Figure 5A and Table S7). In the meantime, the downregulated genes in GV oocytes as compared with MI and MII oocytes often contained genes that were highly expressed in MII oocytes (Figure 5A). This analysis also implies that transcriptome of each oocyte is somewhat variable; for example, GV3 and MII1 exhibited different patterns of expression from other oocytes at the corresponding stages (Figure 5A). Moreover, delayed MII oocytes did not fully express genes that were abundant in MII2 and MII3 (Figure 5A). Taken together, we identify transcripts whose expression is altered upon the initiation of in vitro maturation of human oocytes and the expression of these transcripts can vary in each oocyte. We next performed unsupervised clustering of genes based on their expression levels and 20 clusters were generated (Figure 5B). For identifying GV‐enriched transcripts, we focused on the cluster 5, which showed strong expression in GV samples, especially in GV1 and GV2 (Figure 5B). These genes were compared with our upregulated DEG list in GV samples (GV vs MI and MII), and 144 genes were identified (Figure 5C and Table S8).
FIGURE 5.
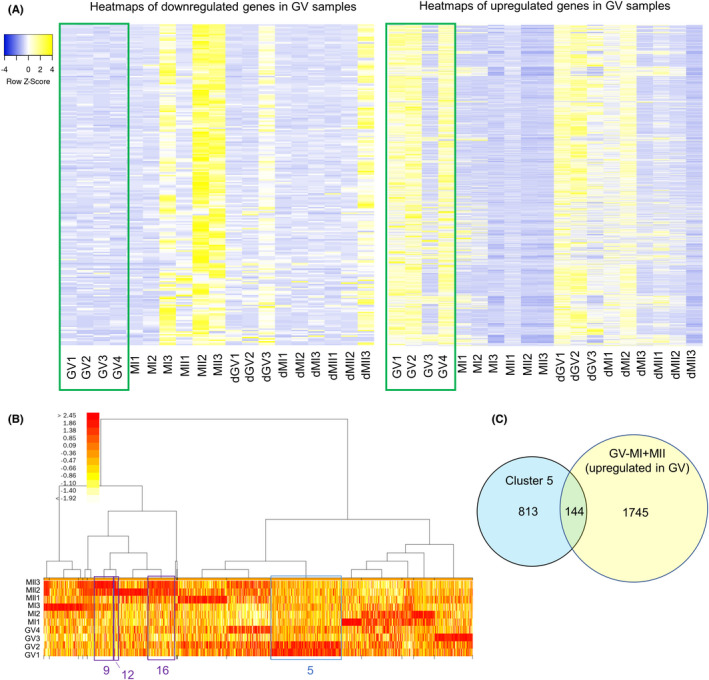
Identification of transcript whose expression is specifically expressed in human GV oocytes. (A) Heatmaps depicting the gene expression levels of DEGs identified by comparing immature GV oocytes (green box) and other oocytes (MI and MII oocytes). The left shows downregulated genes, while the right represents upregulated genes in GV oocytes. (B) A heatmap of gene expression levels after clustering based on expression patterns. Twenty clusters were generated and clusters 5, 9, 12, and 16 are indicated. Color key indicates log2FC(FoldChange). (C) A venn diagram showing the numbers of total and overlapping genes between genes that belong to cluster 5 and upregulated genes in the GV versus MI and MII comparison
We then searched for transcripts uniquely expressed in matured MII oocytes. Clusters 9, 12, and 16 showed strong expression in MII oocytes, especially in MII2 and MII3 (Figure 5B). When genes belonging to these clusters were compared with our upregulated DEG list in MII samples (GV vs MII), 52 genes were identified (Figure 6A, 28 + 13 + 11 genes). These genes (Table S9) may serve as MII‐enriched markers during oocyte maturation. We then examined expression of these marker genes in delayed MII samples, which are supposed to show a lower developmental ability than oocytes that reached the MII stage on the day of oocyte retrieval. Interestingly, almost all of these marker genes were downregulated in delayed MII oocytes (Figure 6B), suggesting that these might be good candidates as a marker to represent MII oocytes with high quality. Especially, ASF1B and BMP15 were highly expressed only in MII2 and MII3 samples (Figure 6B). 51 , 52 Finally, we performed unsupervised clustering of all oocyte samples (Figure S4), and clusters 6 and 12 were identified as highly abundant in MII2 and MII3 samples (Table S10). Among them, APOB and TNFRSF10C were identified as MII‐enriched markers, and therefore might serve as a biomarker for selecting MII oocytes with high‐quality. APOB knockout mice have been reported to show embryonic lethality in homozygous. 53 In addition, it has been reported that a positive correlation is observed between the APOB concentration in follicular fluid and the embryo quality such as fertilization rates and embryonic developmental potentials. 54 Furthermore, TNFRSF10C (TRAILR3) protein is decreased from a medium containing human blastocysts, suggesting TNFRSF10C is utilized for embryonic development. 55
FIGURE 6.
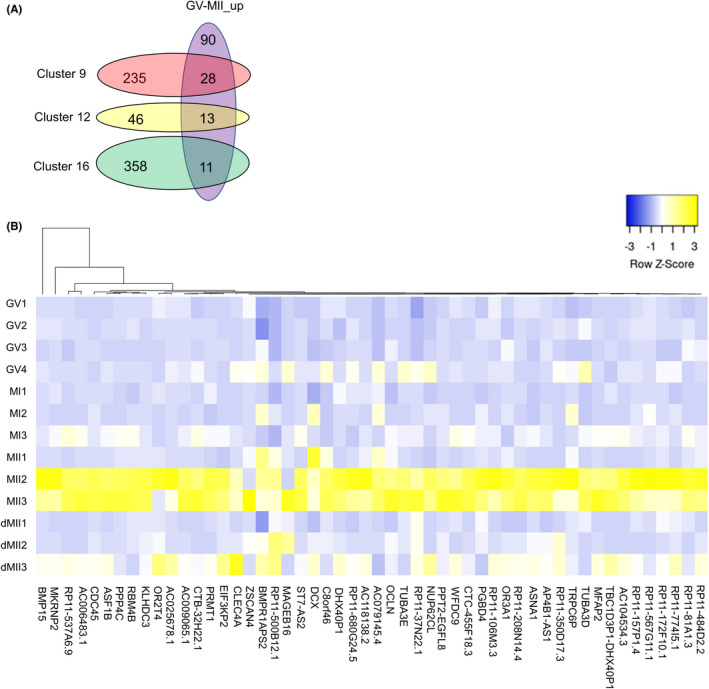
Marker genes for in vitro matured human MII oocytes and potential biomarkers for MII oocytes with high quality. (A) Venn diagrams showing the numbers of total and overlapping genes among genes that belong to clusters 9, 12, and 16, and upregulated genes in the GV versus MII comparison. (B) A heatmap depicting the gene expression levels of candidate genes for human MII oocytes with high quality. Gene symbols are listed. Color key with z‐score is shown
In this study, we identified poly(A)+ transcripts expressed in human oocytes after in vitro culture for maturation, and revealed maternal transcripts whose expression fluctuated during IVM. Furthermore, we showed potential marker genes enriched in in vitro matured MII oocytes of high quality. In the meantime, there are some important limitations when interpreting the outcome of our transcriptomic data. First, we did not perform a large‐scale study due to the limited availability of human oocytes. Second, oocytes matured to the MII stage after 7.5–9 h of IVM were regarded as a model oocyte with normal in vitro maturation as this is the earliest possible timing of oocyte collection for this research. These oocytes collected after 7.5–9 h of IVM are a few hours delayed in their maturation as compared with oocytes used for infertility treatment. We cannot exclude the possibility that this extra incubation might have diminished the quality of oocytes, although this is unlikely as human oocytes derived from 7 to 11 h of in vitro maturation show the similar developmental potential to those from 2 to 7 h of in vitro maturation. 25 Nevertheless, the transcriptomic data shown here provide gene expression information related to IVM of human oocytes, thus contributing to the understanding of human oocyte maturation at the molecular level. Future studies will further reveal the function of our identified marker transcripts for successful maturation and their feasibility as oocyte maturation stage‐specific markers in humans.
CONFLICT OF INTEREST
Hiroki Takeuchi, Mari Yamamoto, Megumi Fukui, Akihiro Inoue, Tadashi Maezawa, Mikiko Nishioka, Eiji Kondo, Tomoaki Ikeda, Kazuya Matsumoto, and Kei Miyamoto declare that they have no conflict of interest.
ETHICAL APPROVAL
This study was conducted after approval by the Clinical Research Ethics Review Committee of Mie University Hospital (H2018‐066), conducted within the guidelines established by the Ethics Committee of the Japan Society of Obstetrics and Gynecology, and registered to the University Hospital Medical Information Network Clinical Trials Registry in Japan. Written informed consent was obtained from all patients for being included in the study. Clinical trial registry: UMIN000045607.
HUMAN RIGHTS STATEMENTS AND INFORMED CONSENT
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964 and its later amendments.
Supporting information
Fig S1‐S4
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Table S7
Table S8
Table S9
Table S10
ACKNOWLEDGMENTS
We thank K.K. DNAFORM (Yokohama, Japan) for RNA‐seq analyses. We thank Ms N Backes Kamimura and Mr James Horvat for proof reading. This research was supported by JSPS KAKENHI Grant Numbers JP17H05045, JP19H05271, JP19H05751, JP20K21376 to K. Mi., by The Naito Foundation to K. Mi., by Takeda Science Foundation to K. Mi, by a Kindai University Research Grant (19‐II‐1) to K. Mi., and by Young Researcher Support Project of Mie University to H.T.
Takeuchi H, Yamamoto M, Fukui M, et al. Single‐cell profiling of transcriptomic changes during in vitro maturation of human oocytes. Reprod Med Biol. 2022;21:e12464. doi: 10.1002/rmb2.12464
Hiroki Takeuchi and Mari Yamamoto are contributed equally to this work.
Gene Expression Omnibus (GEO) public repository under accession GSE166533.
REFERENCES
- 1. Cha KY, Koo JJ, Ko JJ, Choi DH, Han SY, Yoon TK. Pregnancy after in vitro fertilization of human follicular oocytes collected from nonstimulated cycles, their culture in vitro and their transfer in a donor oocyte program. Fertil Steril. 1991;55(1):109‐113. [DOI] [PubMed] [Google Scholar]
- 2. Basatemur E, Sutcliffe A. Health of IVM children. J Assist Reprod Genet. 2011;28(6):489‐493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nogueira D, Sadeu JC, Montagut J. In vitro oocyte maturation: current status. Semin Reprod Med. 2012;30(3):199‐213. [DOI] [PubMed] [Google Scholar]
- 4. Walls ML, Hunter T, Ryan JP, Keelan JA, Nathan E, Hart RJ. In vitro maturation as an alternative to standard in vitro fertilization for patients diagnosed with polycystic ovaries: a comparative analysis of fresh, frozen and cumulative cycle outcomes. Hum Reprod. 2015;30(1):88‐96. [DOI] [PubMed] [Google Scholar]
- 5. Son W‐Y, Henderson S, Cohen Y, Dahan M, Buckett W. Immature oocyte for fertility preservation. Front Endocrinol (Lausanne). 2019;10:464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fasano G, Moffa F, Dechène J, Englert Y, Demeestere I. Vitrification of in vitro matured oocytes collected from antral follicles at the time of ovarian tissue cryopreservation. Reprod Biol Endocrinol. 2011;9:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Prasath EB, Chan MLH, Wong WHW, et al. First pregnancy and live birth resulting from cryopreserved embryos obtained from in vitro matured oocytes after oophorectomy in an ovarian cancer patient. Hum Reprod. 2014;29(2):276‐278. [DOI] [PubMed] [Google Scholar]
- 8. Segers I, Mateizel I, Van Moer E, et al. In vitro maturation (IVM) of oocytes recovered from ovariectomy specimens in the laboratory: a promising “ex vivo” method of oocyte cryopreservation resulting in the first report of an ongoing pregnancy in Europe. J Assist Reprod Genet. 2015;32(8):1221‐1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Uzelac PS, Delaney AA, Christensen GL, Bohler HCL, Nakajima ST. Live birth following in vitro maturation of oocytes retrieved from extracorporeal ovarian tissue aspiration and embryo cryopreservation for 5 years. Fertil Steril. 2015;104(5):1258‐1260. [DOI] [PubMed] [Google Scholar]
- 10. Chen SU, Chen HF, Lien YR, Ho HN, Chang HC, Yang YS. Schedule to inject in vitro matured oocytes may increase pregnancy after intracytoplasmic sperm injection. Arch Androl. 2000;44(3):197‐205. [DOI] [PubMed] [Google Scholar]
- 11. Evsikov AV, Marín de Evsikova C. Gene expression during the oocyte‐to‐embryo transition in mammals. Mol Reprod Dev. 2009;76(9):805‐818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fan Y, Zhao H‐C, Liu J, et al. Aberrant expression of maternal Plk1 and Dctn3 results in the developmental failure of human in‐vivo‐ and in‐vitro‐matured oocytes. Sci Rep. 2015;5:8192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fan X, Zhang X, Wu X, et al. Single‐cell RNA‐seq transcriptome analysis of linear and circular RNAs in mouse preimplantation embryos. Genome Biol. 2015;16:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yanez LZ, Han J, Behr BB, Pera RAR, Camarillo DB. Human oocyte developmental potential is predicted by mechanical properties within hours after fertilization. Nat Commun. 2016;7:10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Assou S, Anahory T, Pantesco V, et al. The human cumulus–oocyte complex gene‐expression profile. Hum Reprod. 2006;21(7):1705‐1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jones GM, Cram DS, Song BI, et al. Gene expression profiling of human oocytes following in vivo or in vitro maturation. Hum Reprod. 2008;23(5):1138‐1144. [DOI] [PubMed] [Google Scholar]
- 17. Kocabas AM, Crosby J, Ross PJ, et al. The transcriptome of human oocytes. Proc Natl Acad Sci USA. 2006;103(38):14027‐14032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Y, Yan Z, Qin Q, et al. Transcriptome landscape of human folliculogenesis reveals oocyte and granulosa cell interactions. Mol Cell. 2018;72(6):1021‐1034.e4. [DOI] [PubMed] [Google Scholar]
- 19. Zhao H, Li T, Zhao Y, et al. Single‐cell transcriptomics of human oocytes: environment‐driven metabolic competition and compensatory mechanisms during oocyte maturation. Antioxid Redox Signal. 2019;30(4):542‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yu B, Doni Jayavelu N, Battle SL, et al. Single‐cell analysis of transcriptome and DNA methylome in human oocyte maturation. PLoS One. 2020;15(11):e0241698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Llonch S, Barragán M, Nieto P, et al. Single human oocyte transcriptome analysis reveals distinct maturation stage‐dependent pathways impacted by age. Aging Cell. 2021;20(5):e13360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mendez R, Murthy KGK, Ryan K, Manley JL, Richter JD. Phosphorylation of CPEB by Eg2 mediates the recruitment of CPSF into an active cytoplasmic polyadenylation complex. Mol Cell. 2000;6(5):1253‐1259. [DOI] [PubMed] [Google Scholar]
- 23. Yang Q, Allard P, Huang M, Zhang W, Clarke HJ. Proteasomal activity is required to initiate and to sustain translational activation of messenger RNA encoding the stem‐loop‐binding protein during meiotic maturation in mice. Biol Reprod. 2010;82(1):123‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. De Vos A, Van de Velde H, Joris H, Van Steirteghem A. In‐vitro matured metaphase‐I oocytes have a lower fertilization rate but similar embryo quality as mature metaphase‐II oocytes after intracytoplasmic sperm injection. Hum Reprod. 1999;14(7):1859‐1863. [DOI] [PubMed] [Google Scholar]
- 25. Vanhoutte L, De Sutter P, Van der Elst J, Dhont M. Clinical benefit of metaphase I oocytes. Reprod Biol Endocrinol. 2005;3:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114‐2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Martin M. Cutadapt removes adapter sequences from high‐throughput sequencing reads. Embnet J. 2011;17(1):10. [Google Scholar]
- 28. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics. 2013;29(1):15‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liao Y, Smyth GK, Shi W. eatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923‐930. [DOI] [PubMed] [Google Scholar]
- 30. Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44‐57. [DOI] [PubMed] [Google Scholar]
- 33. Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Babicki S, Arndt D, Marcu A, et al. Heatmapper: web‐enabled heat mapping for all. Nucleic Acids Res. 2016;44(W1):W147‐W153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Okuno T, Li WY, Hatano Y, et al. Zygotic nuclear F‐actin safeguards embryonic development. Cell Rep. 2020;31(13):107824. [DOI] [PubMed] [Google Scholar]
- 36. Conti M, Franciosi F. Acquisition of oocyte competence to develop as an embryo: integrated nuclear and cytoplasmic events. Hum Reprod Update. 2018;24(3):245‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Messiaen S, Guiard J, Aigueperse C, et al. Loss of the histone chaperone ASF1B reduces female reproductive capacity in mice. Reproduction. 2016;151(5):477‐489. [DOI] [PubMed] [Google Scholar]
- 38. Tachibana K, Mori M, Matsuhira T, et al. Initiation of DNA replication after fertilization is regulated by p90Rsk at pre‐RC/pre‐IC transition in starfish eggs. Proc Natl Acad Sci. 2010;107(11):5006‐5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang Y‐L, Zheng W, Ren P, et al. Biallelic mutations in MOS cause female infertility characterized by human early embryonic arrest and fragmentation. EMBO Mol Med. 2021;13(12):e14887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mukhopadhyay R, Ho Y‐S, Swiatek PJ, Rosen BP, Bhattacharjee H. Targeted disruption of the mouse Asna1 gene results in embryonic lethality. FEBS Lett. 2006;580(16):3889‐3894. [DOI] [PubMed] [Google Scholar]
- 41. Kurowska P, Mlyczyńska E, Estienne A, et al. Expression and impact of vaspin on in vitro oocyte maturation through MAP3/1 and PRKAA1 signalling pathways. Int J Mol Sci. 2020;21(24):9342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cao Y, Zhao H, Wang Z, et al. Quercetin promotes in vitro maturation of oocytes from humans and aged mice. Cell Death Dis. 2020;11(11):965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang H, Jo Y‐J, Oh JS, Kim N‐H. Quercetin delays postovulatory aging of mouse oocytes by regulating SIRT expression and MPF activity. Oncotarget. 2017;8(24):38631‐38641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tripurani SK, Lee K‐B, Wang L, et al. A novel functional role for the oocyte‐specific transcription factor newborn ovary homeobox (NOBOX) during early embryonic development in cattle. Endocrinology. 2011;152(3):1013‐1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shimamoto S, Nishimura Y, Nagamatsu G, et al. Hypoxia induces the dormant state in oocytes through expression of Foxo3. Proc Natl Acad Sci USA. 2019;116(25):12321‐12326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li C, Liu Z, Li W, et al. The FSH‐HIF‐1α‐VEGF pathway is critical for ovulation and oocyte health but not necessary for follicular growth in mice. Endocrinology. 2020;161(4):bqaa038. [DOI] [PubMed] [Google Scholar]
- 47. Zwaans BMM, Lombard DB. Interplay between sirtuins, MYC and hypoxia‐inducible factor in cancer‐associated metabolic reprogramming. Dis Model Mech. 2014;7(9):1023‐1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ma R, Liang W, Sun Q, et al. Sirt1/Nrf2 pathway is involved in oocyte aging by regulating Cyclin B1. Aging. 2018;10(10):2991‐3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Demond H, Trapphoff T, Dankert D, et al. Preovulatory aging in vivo and in vitro affects maturation rates, abundance of selected proteins, histone methylation pattern and spindle integrity in murine oocytes. PLoS One. 2016;11(9):e0162722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Magnani L, Cabot RA. Manipulation of SMARCA2 and SMARCA4 transcript levels in porcine embryos differentially alters development and expression of SMARCA1, SOX2, NANOG, and EIF1. Reproduction. 2009;137(1):23‐33. [DOI] [PubMed] [Google Scholar]
- 51. Su Y‐Q, Sugiura K, Wigglesworth K, et al. Oocyte regulation of metabolic cooperativity between mouse cumulus cells and oocytes: BMP15 and GDF9 control cholesterol biosynthesis in cumulus cells. Development. 2007;135(1):111‐121. [DOI] [PubMed] [Google Scholar]
- 52. Awe JP, Byrne JA. Identifying candidate oocyte reprogramming factors using cross‐species global transcriptional analysis. Cell Reprogram. 2013;15(2):126‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Farese RV, Ruland SL, Flynn LM, Stokowski RP, Young SG. Knockout of the mouse apolipoprotein B gene results in embryonic lethality in homozygotes and protection against diet‐induced hypercholesterolemia in heterozygotes. Proc Natl Acad Sci. 1995;92(5):1774‐1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Scalici E, Bechoua S, Astruc K, et al. Apolipoprotein B is regulated by gonadotropins and constitutes a predictive biomarker of IVF outcomes. Reprod Biol Endocrinol. 2016;14(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dominguez F, Gadea B, Esteban FJ, Horcajadas JA, Pellicer A, Simon C. Comparative protein‐profile analysis of implanted versus non‐implanted human blastocysts. Hum Reprod. 2008;23(9):1993‐2000. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S4
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Table S7
Table S8
Table S9
Table S10
Data Availability Statement
The dataset generated during this study are available in the Supplementary Tables and in the Gene Expression Omnibus (GEO) public repository under accession GSE166533. All the other data are available from the corresponding author upon reasonable request.