

Abstract
Previously, we demonstrated that upstream stimulatory factor 2 (USF2) mediates high glucose-induced thrombospondin1 (TSP1) gene expression and TGF-β activity in glomerular mesangial cells and plays a role in diabetic renal complications. In the present studies, we further determined the molecular mechanisms by which high glucose levels regulate USF2 gene expression. In primary rat mesangial cells, we found that glucose treatment time and dose-dependently upregulated USF2 expression (mRNA and protein). By using cycloheximide to block the de novo protein synthesis, similar rate of USF2 degradation was found under either normal glucose or high glucose conditions. USF2 mRNA stability was not altered by high glucose treatment. Furthermore, high glucose treatment stimulated USF2 gene promoter activity. By using the luciferase-promoter deletion assay, site-directed mutagenesis, and transactivation assay, we identified a glucose-responsive element in the USF2 gene promoter (−1,740 to −1,620, relative to the transcription start site) and demonstrated that glucose-induced USF2 expression is mediated through a cAMP-response element-binding protein (CREB)-dependent transactivation of the USF2 promoter. Furthermore, siRNA-mediated CREB knock down abolished glucose-induced USF2 expression. Taken together, these data indicate that high glucose levels upregulate USF2 gene transcription in mesangial cells through CREB-dependent transactivation of the USF2 promoter.
Keywords: glucose, USF2, mesangial cells, gene transcription
Diabetic renal complications (diabetic nephropathy) is the single most common cause of renal failure in the United States [Susztak et al., 2003; Bloomgarden, 2005], which is characterized by both glomerulosclerosis with thickening of the glomerular basement membrane and mesangial matrix expansion, and tubulointerstitial fibrosis. Diabetic end stage renal disease remains a significant cause of mortality and morbidity for both type 1 and type 2 diabetic patients [Susztak et al., 2003]. A relationship between hyperglycemia and diabetic nephropathy has been established in both animal and clinical studies [Ishimura et al., 1992; Skyler, 1996]. Hyperglycemia is known to play a critical role in the pathogenesis of diabetic nephropathy by stimulation of the pro-fibrotic growth factor-transforming growth factor-β (TGF-β) expression, leading to excessive extracellular matrix deposition in the diabetic kidney. Studies have demonstrated that high glucose conditions (30 mM glucose) stimulated increases in TGF-β bioactivity in mesangial cells is mediated by the matricellular protein thrombospondin1 (TSP1), which converts the latent TGF-β to its active form [Poczatek et al., 2000]. We also demonstrated that the transcription factor, Upstream Stimulatory Factor 2 (USF2), binds to an 18 bp sequence in the TSP1 gene promoter and regulates high glucose-induced TSP1 expression in mesangial cells. Moreover, overexpression of USF2 stimulates TSP1 expression, TGF-β activity and extracellular matrix protein expression, suggesting that USF2 is an important regulator of glucose-induced TSP1-dependent TGF-β activity and may contribute to the development of diabetic nephropathy.
USF2 was initially characterized as a transcription factor implicated in the regulation of the adenovirus major late promoter. In mammals, USF2 is ubiquitously expressed with a molecular weight of 44 kDa. It belongs to the Myc family of transcription factors characterized by a basic/helix loop helix/leucine zipper domain responsible for dimerization and DNA binding. It can form homo- and heterodimers and recognize in vitro a CACGTG core sequence termed E box. Through binding to E boxes of target genes, USF2 has been demonstrated to regulate expression of many genes [Rippe et al., 1997; Vallet et al., 1997; Qian et al., 1999; Kingsley-Kallesen et al., 2001; Nicolas et al., 2001; Bidder et al., 2002; Zhu et al., 2005; Chen et al., 2006]. In our previous studies, we demonstrated that USF2 mediated high glucose conditions-induced TSP1 gene expression in mesangial cells. In addition, glucose exposure stimulated nuclear accumulation of USF2 protein. However, the molecular mechanisms governing USF2 expression in response to glucose is not clear and remains to be investigated.
In this study, we analyzed the transcriptional regulation of USF2 expression in response to high glucose stimulation in primary rat mesangial cells. We identified a glucose-responsive element in the USF2 promoter and demonstrated that glucose-induced USF2 expression is mediated through a cAMP-response element-binding protein (CREB)-dependent transactivation of the USF2 promoter.
MATERIALS AND METHODS
Cell Cultures
Rat mesangial cells (RMCs) were the generous gift from Dr. Anne Woods, University of Alabama at Birmingham [Wang et al., 2002]. Cells were cultured in RPMI 1640 medium supplemented with 20% heat-activated fetal bovine serum, 5 mM d-glucose, 2 mM l-glutamine, 1% (v/v) nonessential amino acids, 2 mM sodium pyruvate, 10 μg/ml transferrin, 5 ng/ml sodium selenite, and 0.6 international U/ml insulin. RMCs were passaged at 80% confluence. Experiments in this study were performed on cells between the 3rd and 10th passages.
Western Blot Analysis
Rat mesangial cells were cultured and treated with normal or high glucose media for indicated time points. After treatment, cells were harvested and nuclear protein was prepared as described previously [Wang et al., 2004]. Equal amounts of protein were subjected to SDS–PAGE gel (12%) under reducing conditions. After electrophoretic transfer to nitrocellulose membranes and blocking, the membranes were incubated with rabbit anti-mouse USF2 antibody (1:500) (Santa Cruz) for 1 h at room temperature. After intensive washing, secondary antibody (1:10,000 dilution) was used for the detection of immunoreactive bands with the enhanced chemiluminescence detection system (Pierce). Equal loading and transfer of protein samples were assayed by staining the blots with Ponceau S.
Northern Blot Analysis and Real-Time PCR
Total RNA from cultured mesangial cells was extracted using RNeasy mini kit (Qiagen). For northern blot analysis, equal amounts of total RNA (10 μg) were denatured, electrophoresed, and transferred to nylon membranes. RNA was fixed by UV-cross-linking. The USF2 probe (mouse USF2 cDNA, generously provided by Dr. Sawadogo at the University of Texas MD Anderson Cancer Center) was radiolabeled by Random Prime DNA-labeling kit (Roche Molecular Biochemicals) with [α−32P]dCTP. The membrane was prehybridized in ExpressHyb hybridization solution (CLONTECH, Palo Alto, CA) for 30 min at 68°C, and hybridization was carried out at 68°C for 1 h in fresh hybridization solution with denatured probe. The membrane was washed three times with 2× SSC (1× SSC = 0.15 M NaCl and 0.015 M sodium citrate), 0.05% SDS for 15 min at room temperature, and then twice with 0.2× SSC, 0.1% SDS at 50°C for 20 min. Blots were−developed, and films were scanned. The absorbance units were corrected for the β-actin levels.
Real-time quantitative RT-PCR analyses were performed using SYBR Green PCR Master Mix kit with a MyiQ real-time PCR thermal cycler (Bio-Rad). Reaction conditions were 50°C for 2 min, 94°C for 10 min, followed by 35 cycles of 94°C for 30 s, and 55°C for 30 s. All reactions were performed in triplicate in a final volume of 25 μl. Standard curves were generated using 18S RNA primers. Dissociation curves were run to detect nonspecific amplification, and we confirmed that single products were amplified in each reaction. The quantities of each test gene and internal control 18S RNA were then determined from the standard curve using the MyiQ system software, and mRNA expression levels of test genes were normalized to 18S levels. The primers used for PCR ofUSF2 (295 bp) were 5’ ACAGACCAGAGCCTACAGGC −3’ and 5’-AAGCCTTGGACAGGATGCCC-3’.
USF2 Promoter-Reporter Constructs
A 2.4 kb region of the mouse USF2 gene promoter was amplified by PCR using mouse genomic DNA as a template with the primers of USF2-P1: 5’-cgggtaccAGCTCAGGTGAGGCCTTGGGGAATCC-3’(containing Kpn I site) and USF2-P2: 5’-gctaagcttGGGGGGGGCGGGGGGGAGGGGAG −3’(containing Hind III site). The PCR product was digested, inserted between the Kpn I and Hind III sites of the luciferase pGL3-Basic vector (Promega), and sequenced.
USF2 (−2,400) was used as a template.Five deletion mutants of USF2 (−1,742), USF2 (−1,620), USF2 (−1,209), USF2 (−837), and USF2 (−430) were prepared using forward primers derived from different 5’ positions (the numbers in parentheses contain Kpn I site) of the USF2 promoter and the same reverse primer containing Hind III site. The PCR products were digested with Kpn I and Hind III and subcloned into pGL3-basic vector, and their sequences were confirmed by sequencing.
Plasmids, Transient Transfection, and Luciferase Assay
Primary rat mesangial cells were seeded into six well plates at a density of 3 × 104 cells/ml for 1 day and made quiescent in serum-free RPMI 1640 media for 2 days. Then cells were transiently transfected using Effectene transfection reagent (Qiagen) with 1 μg of USF2 promoter luciferase reporter plasmid, 100 ng of dominant-negative CREB construct (A-CREB) provided by Dr. Dennis Bruemmer (University of Kentucky) [Watson et al., 2002] or the HSF, c-myb or CRE-mutated USF2 promoter constructs (generated by using a QuikChange™ site-directed mutagenesis kit (Stratagene, La Jolla, CA)). A-CREB was constructed by fusing a designed acidic amphipathic extension onto the N terminus of the CREB leucine zipper domain. The acidic extension of A-CREB interacts with the basic region of CREB forming a coiled-coil extension of the leucine zipper and thus prevents the basic region of wild-type CREB from binding to DNA [Ahn et al., 1998]. pRL-SV40 (0.02 μg) (Promega) was used as an internal control for transfection. Transfected cells were treated with normal (5 mM) or high (30 mM) glucose for 1 day, and the luciferase activities were assayed using the dual-luciferase assay kit (Promega) according to the manufacturer’s directions.
Chromatin Immunoprecipitation Assay
The chromatin immunoprecipitation assay was conducted as described by Upstate Biotechnology with some modifications. In brief, primary mesangial cells were cultured in a 10-cm culture dish. After being made quiescent in serum-free RPMI 1640 media for 2 days, cells were treated with normal glucose (5 mM) or high glucose (30 mM) for 24 h. After treatment, protein-DNA complexes were fixed by 1% formaldehyde in phosphate-buffered saline. The fixed cells were washed and lysed in SDS lysis buffer with protease inhibitors and sonicated on ice. After centrifugation at 500g for 1 min, one portion of the precleared supernatant was used as DNA input control, and the remaining supernatant was subdivided into aliquots and then incubated overnight at 4°C with non-immune rabbit immunoglobulin G (IgG; Santa Cruz) or anti-CREB antibody (Cell Signaling). The immunoprecipitated complexes of antibody-protein-DNA were collected using a protein A slurry, washed successively with low salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl (pH 8.1), 150 mM NaCl), high salt buffer (same as the low-salt buffer but with 500 mM NaCl), LiCl buffer(0.25 M LiCl, 1% Nonidet P-40, 1% deoxycho-late, 1 mM EDTA, 10 mM Tris-HCl (pH 8.1)), and then eluted with elution buffer (1% SDS, 100 mM NaHCO3). The cross-linking of protein-DNA complexes was reversed by incubation with 5 M NaCl at 65°C for 4 h, and DNA was digested with 10 mg of proteinase K (Sigma)/ml for 1 h at 45°C. The DNA was then extracted with phenol-chloroform, and the purified DNA pellet was resuspended in H2O and subjected to PCR amplification with the forward primer, 5’-AACATGGGAGCAGGAA-3’, and the reverse primer, 5’-GAAGGACTAAGTCACCT-3’, which were specifically designed from the USF2 promoter. The 124-bp PCR products were resolved by 2% agarose-ethidium bromide gel electrophoresis, visualized by UV.
Knockdown of CREB Expression by siRNA
Rat mesangial cells were plated on six well plates at a density of 3 × 104 and transiently transfected with 100 nM of CREB siRNA or control siRNA using Effectene transfection reagent (Qiagen). Following transfection, cells were treated with serum-free media for 24 h and then treated with 5 or 30 mM glucose for 24 h. Nuclear protein was prepared as described previously [Wang et al., 2004] and analyzed for CREB and USF2 expression by Western blotting. Anti-CREB antibody was purchased from Cell Signaling. Pre-designed siRNA targeting to rat CREB (catalog # MU-091740–00) and control siRNA (catalog # D-001210-02) were purchased from Dharmacon RNA Technologies.
mRNA Stability Assay
Rat mesangial cells were made quiescent in serum free media for 48 h and then treated with 5 or 30 mM glucose for 24 h. After 24 h, media were removed and actinomycin D (5 μg/ml) was added (designated as t = 0). After different periods of incubation, cells were harvested. Northern analysis and hybridization for USF2 and β-actin were performed as described previously. Measurement of the ratio of USF2/actin at t = 0 (from actinomycin D treatment) was assigned a relative value of 100%.
Protein Stability Assay
Quiescent rat mesangial cells were treated with 5 or 30 mM glucose media for 24 h. After 24 h, media were removed and cycloheximide (10 μg/ml) was added. After different periods of incubation, cells were harvested. Cell lysates were prepared and Western blotting with the USF2 antibody was performed. The intensities of Western blot signals were quantitated by densitometry using NIH Image software. Measurement of the ratio of USF2/actin at t = 0 (from cycloheximide treatment) was assigned a relative value of 100%.
Statistical Analysis
Data are expressed as the mean ± SE. Statistical evaluation of the data was performed using the unpaired t-test, considering the P value of <0.05 as significant.
RESULTS
High Glucose Exposure Dose and Time-Dependently Upregulated USF2 Protein Levels in Primary Rat Mesangial Cells
To determine how high glucose levels regulate USF2 expression, primary rat mesangial cells (RMCs) were cultured and exposed to different concentrations of glucose for 24 h. The protein levels of USF2 in nuclear preparation were determined by Western blotting. As shown in Figure 1, glucose treatment upregulated USF2 expression in a dose-dependent manner. The maximum upregulation of USF2 was achieved by 30 mM glucose treatment. Mannitol (25 mM +5 Mm d-glucose) treatment had no effect on USF2 expression (data not shown), suggesting that glucose-induced USF2 expression in mesangial cells is not due to osmolarity change. In addition, we also observed the effect of high glucose (30 mM) on USF2 expression under different exposure time. As shown in Figure 2, high glucose exposure time-dependently upregulated USF2 protein levels in mesangial cells with maximal induction after 24 h high glucose stimulation.
Fig. 1.

High glucose exposure dose-dependently upregulated USF2 protein levels in rat mesangial cells. Primary rat mesangial cells (p4) were cultured and made quiescent. Cells were then treated with different concentrations of d-glucose media for 24 h. USF2 protein levels in nuclear extracts were determined by Western blotting. Relative USF2 levels were determined by scanning densitometry of immunoblots and normalized to β-actin levels. Results are the mean ±SE (n = 3). *P < 0.05 versus mM.
Fig. 2.
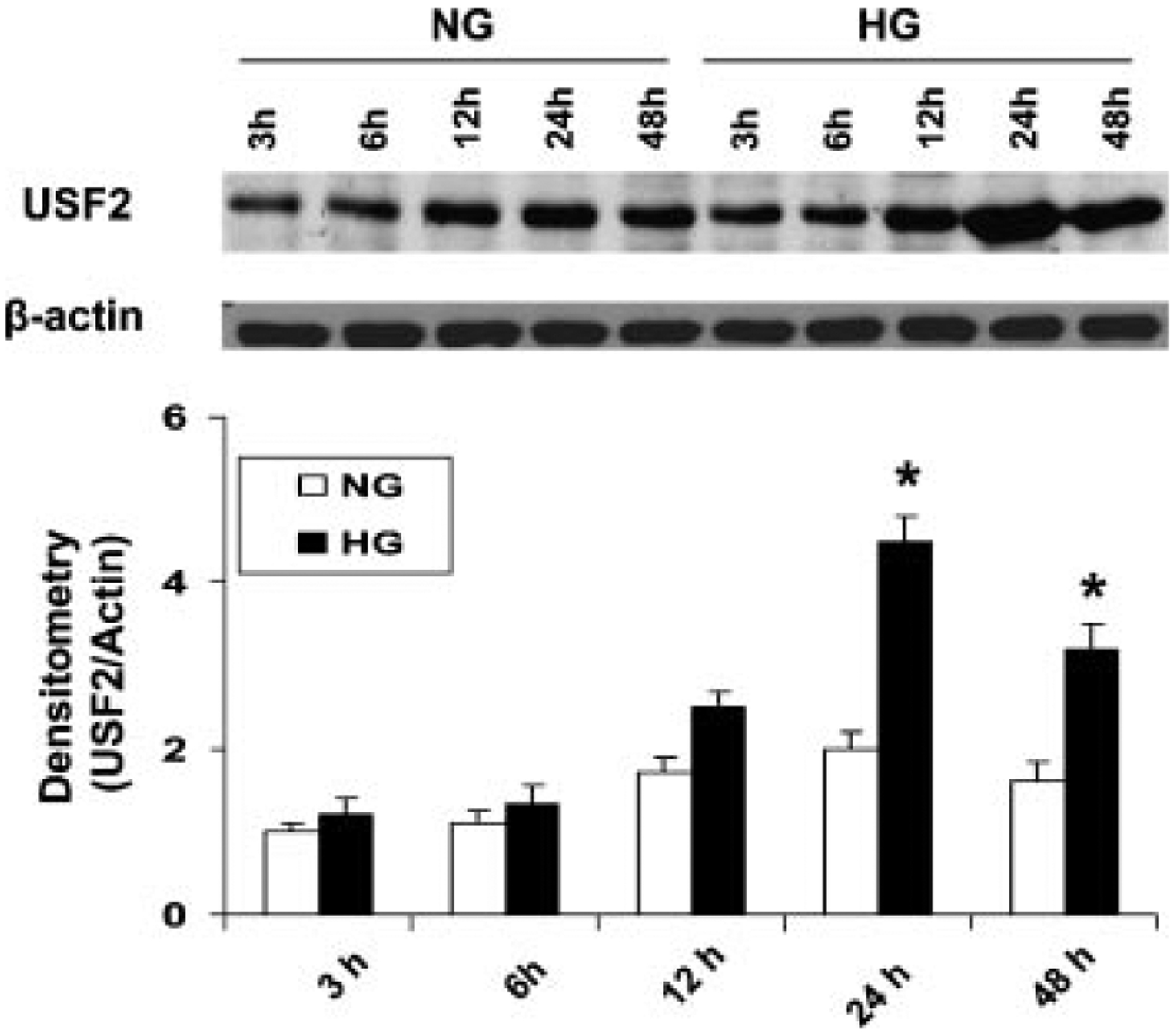
High glucose exposure time-dependently upregulated USF2 protein levels in rat mesangial cells. Quiescent mesangial cells were treated with 5 mM (NG) or 30 mM (HG) glucose media for indicated time period. Then cells were harvested. USF2 protein levels in nuclear extracts were determined by Western blotting. Relative USF2 levels were determined by scanning densitometry of immunoblots and normalized to β-actin levels. Results are the mean ±SE (n = 3). *P < 0.05 versus NG.
High Glucose Exposure did not Alter USF2 Protein Stability
Next, we observed whether high glucose exposure modifies the USF2 protein stability, contributing to the increased USF2 protein levels in mesangial cells. To determine USF2 protein turnover, rat mesangial cells were treated with normal (5 mM glucose) or high glucose (30 mM glucose) media for 24 h, and cycloheximide (10 μg/ml) was added to block the de novo protein synthesis. After indicated time points, cells were harvested and nuclear extracts were prepared. The USF2 protein levels in nuclear preparation were determined by Western blotting. As shown in Figure 3, there was no difference of the rate of USF2 degradation in cells treated with either 5 mM glucose or with 30 mM glucose. The half-life of USF2 is approximately 8 h in mesangial cells. These data indicate that high glucose stimulates USF2 expression is not through altering USF2 protein stability, suggesting that post-translational mechanisms may not involve in glucose-induced USF2 expression.
Fig. 3.
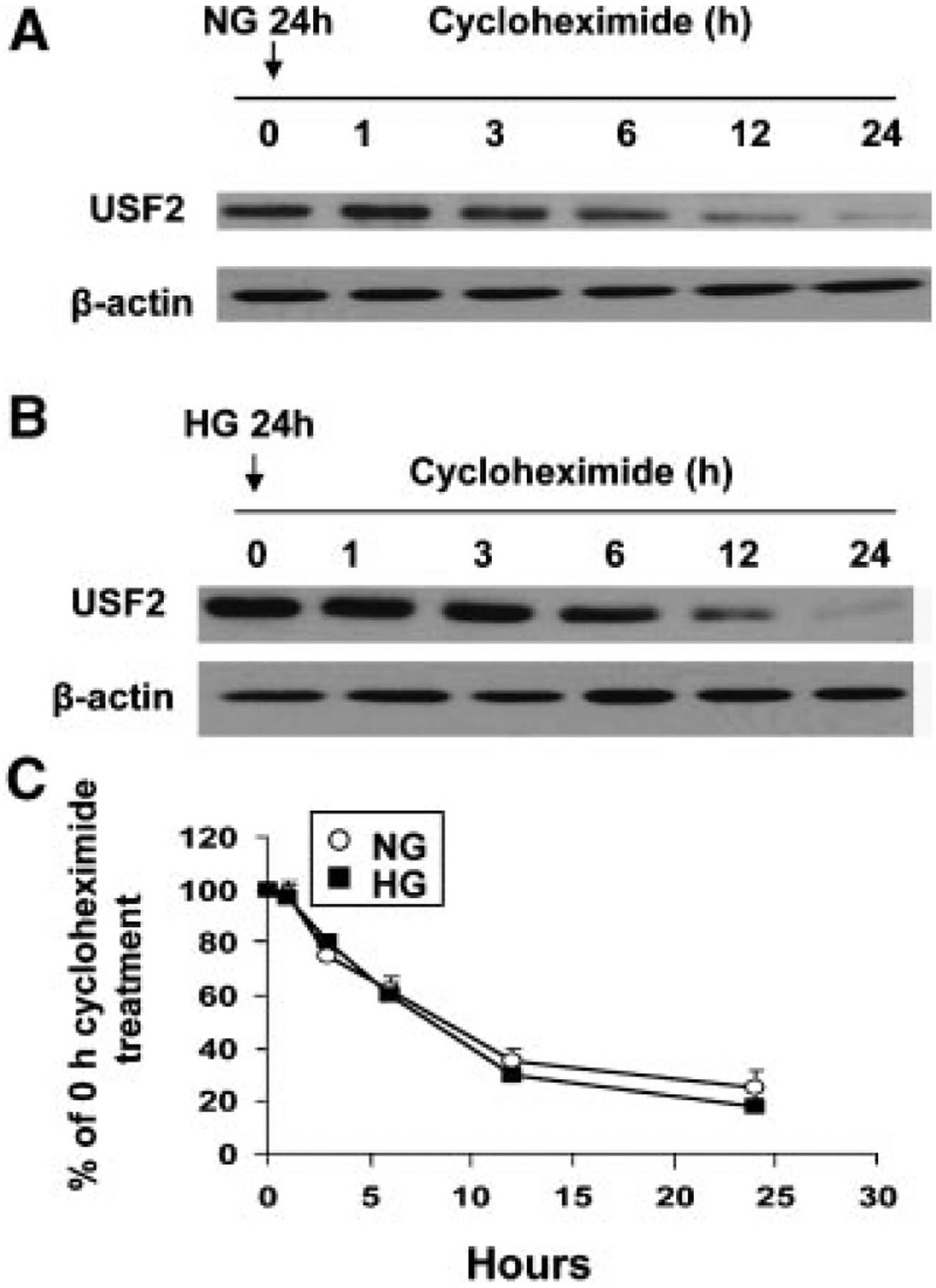
High glucose exposure had no effect on USF2 protein stability. Quiescent rat mesangial cells were treated with 5 mM (NG) (A) or 30 mM (HG) (B) glucose media for 24 h. Cells were then treated with cycloheximide (10 μg/ml) for indicated time period. After treatment, nuclear extracts were prepared and USF2 protein levels were determined by Western blotting. (C) Relative USF2 levels were determined by scanning densitometry of immunoblots and normalized to β-actin levels. Measurement of the ratio of USF2/actin at time = 0 was assigned a relative value of 100%. The results are expressed as mean ±SE of three separate experiments.
High Glucose Exposure Stimulated USF2 Gene Expression in Mesangial Cells at the Transcriptional Level
The above results suggest that post-translational regulation may not involve in glucose-induced USF2 expression. To determine the transcriptional regulation of USF2 expression by high glucose exposure, first, we determined the USF2 mRNA levels in mesangial cells after high glucose exposure by real-time PCR. As shown in Figure 4, high glucose exposure increased USF2 mRNA levels in mesangial cells in a time-dependent manner. We also observed the effect of high glucose exposure on USF2 mRNA stability by adding actinomycin D to inhibit de novo RNA synthesis. As shown in Figure 5, USF2 mRNA stability was unaffected by high glucose exposure. Next, we determined the effect of high glucose exposure on the transcriptional activity of the USF2 promoter. With the transfection of a mouse full length USF2 promoter-luciferase reporter construct(2.4 kb) to rat mesangial cells, we found that 24 h high glucose exposure significantly stimulated the transcriptional activity of the USF2 promoter (Fig. 6A). Together, these data suggest that high glucose exposure stimulated USF2 gene expression in mesangial cells at the transcriptional level.
Fig. 4.
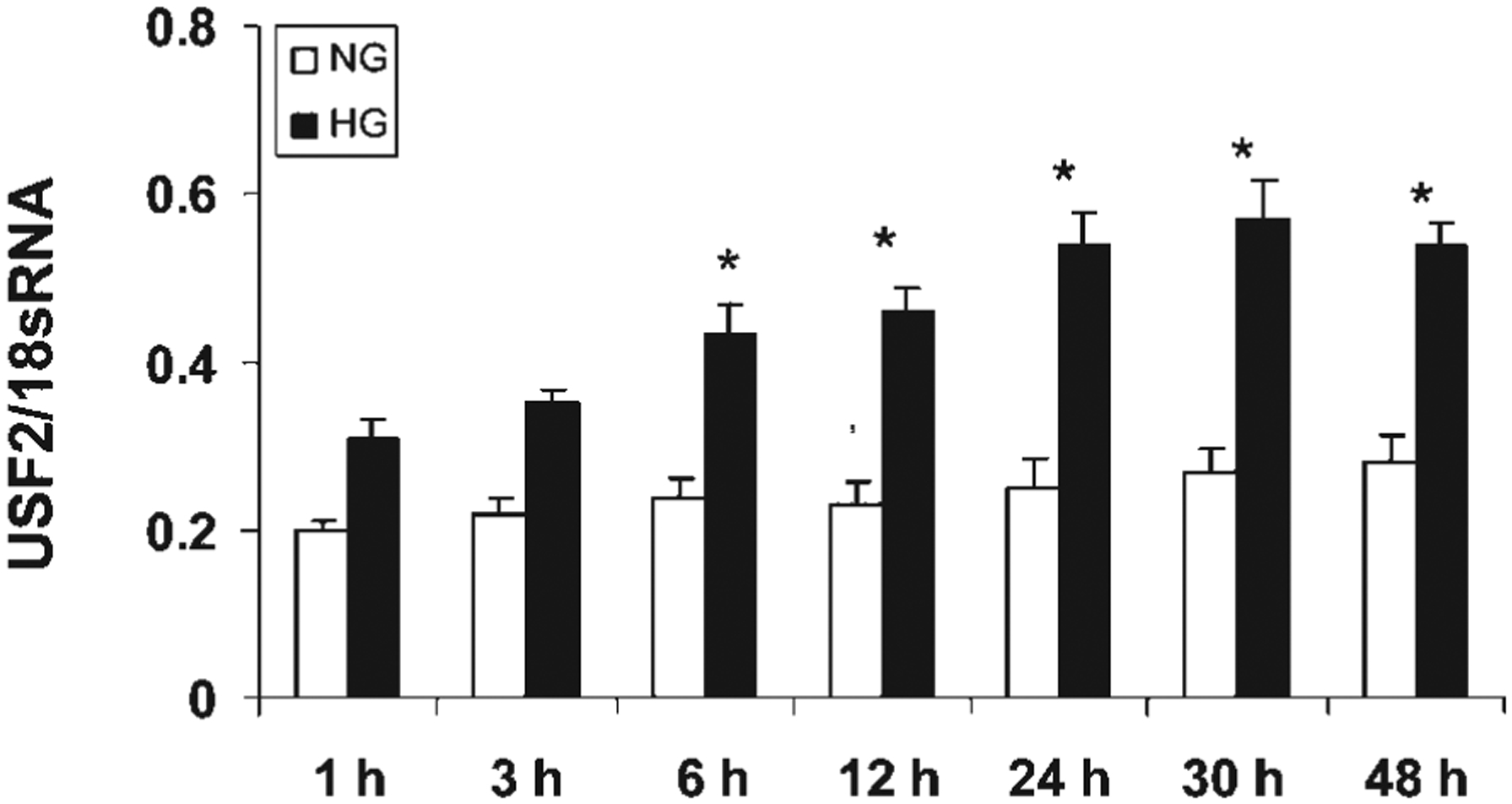
High glucose exposure time-dependently stimulated USF2 mRNA levels in rat mesangial cells. Quiescent rat mesangial cells were treated normal (5 mM) or high glucose (30 mM) media for indicated time points. After treatment, cells were harvested and total RNA was extracted. USF2 mRNA levels were determined by real-time PCR as described in Materials and Methods. The results are expressed as mean ±SE of three separate experiments. *P < 0.05 versus NG.
Fig. 5.

High glucose exposure had no effect on USF2 mRNA stability. Quiescent rat mesangial cells were treated with actinomycin D (5 μg/ml) in the presence of normal (5 mM) or high glucose (30 mM) media for indicated period. After treatment, cells were harvested and total RNA was extracted. Northern analysis and hybridization for USF2 and β-actin were performed as described in Materials and Methods to measure the rate of decay of USF2 mRNA and the half-life. Measurement of the ratio of USF2/actin at time = 0 was assigned a relative value of 100%. The results are expressed as mean ±SE of three separate experiments.
Fig. 6.
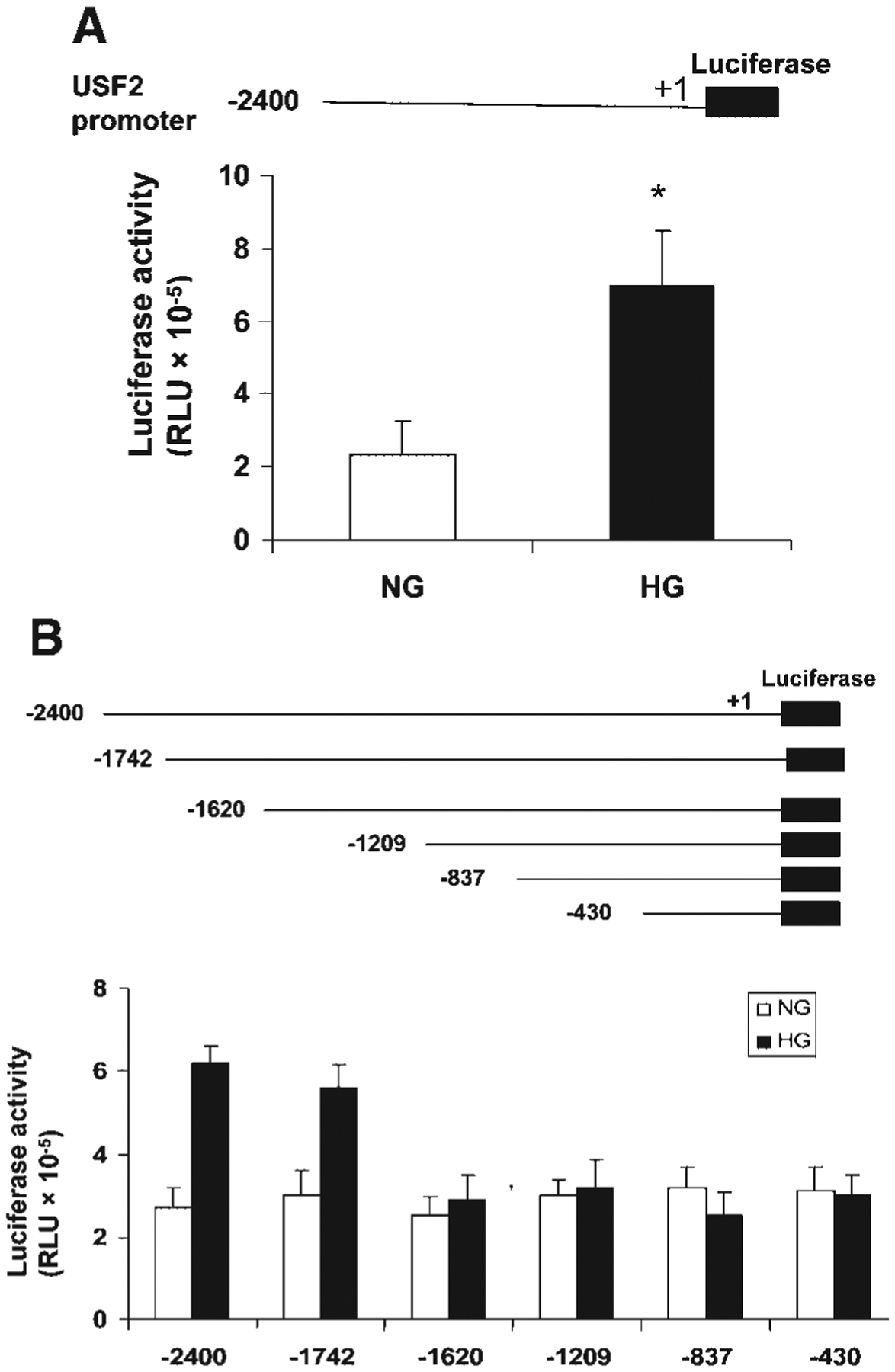
High glucose exposure stimulated the USF2 gene promoter activity and the region between −1,742 and −1,620 was involved. Quiescent rat mesangial cells were transiently transfected with mouse USF2 promoter luciferase construct (USF2 (−2,400)) (A) and a series deletion of USF2 promoter constructs (B) and then treated with normal or high glucose media for 24 h. After 24 h, cells were harvested and lysed. The luciferase activity was measured and normalized to Renilla luciferase levels. Data are shown with the mean ±SE of triplicates of three separate experiments. *P < 0.05 for HG versus NG.
Glucose-Mediated Increases in USF2 Promoter Activity in Mesangial Cells was Supported by the USF2 Promoter Region −1,742 to −1,620
The above result showed that high glucose exposure stimulated USF2 promoter (−2,400 bp) activity. To identify the promoter elements that regulate USF2 gene transcription in mesangial cells in response to high glucose concentrations, a series of USF2 promoter-luciferase reporter constructs was generated and transiently transfected into mesangial cells. The promoter activity was measured by assaying the luciferase activity as described under “Materials and Methods” and normalized to Renilla luciferase activity. As shown in Figure 6B, the longest construct, USF2 (−2,400), gave rise to a 2.4-fold increase in USF2 promoter activity in response to high glucose levels. Deletion of a 122 bp region (−1,742 to −1,620) totally abolished high glucose responsiveness, suggesting that this 122 bp region is important for high glucose-induced USF2 transcription.
Glucose-Mediated Increases in USF2 Promoter Activity in Mesangial Cells Depended on CRE Site Located Between −1,742 and −1,620
To determine the transcription factors that mediate glucose-induced USF2 gene transcription in mesangial cells, we performed a computer analysis using the GCG program, which revealed that this 122 bp (−1,742 to −1,620 site) contains putative binding sequences for several transcription factors, including HSF, c-Myb, and CREB (cAMP-response element-binding protein). To identify which transcription factors might be important for glucose-mediated regulation of USF2 transcription, we introduced point mutation to HSF, c-Myb or CRE binding sites in the USF2 (−1,742) promoter-luciferase construct. As shown in Figure 7, mutation of two HSF sites or c-Myb site had no effect on glucose-induced USF2 promoter activity. However, mutation of CREB binding site abrogated high glucose-induced USF2 promoter activity, suggesting that CREB is involved in high glucose-mediated increase in USF2 transcription.
Fig. 7.
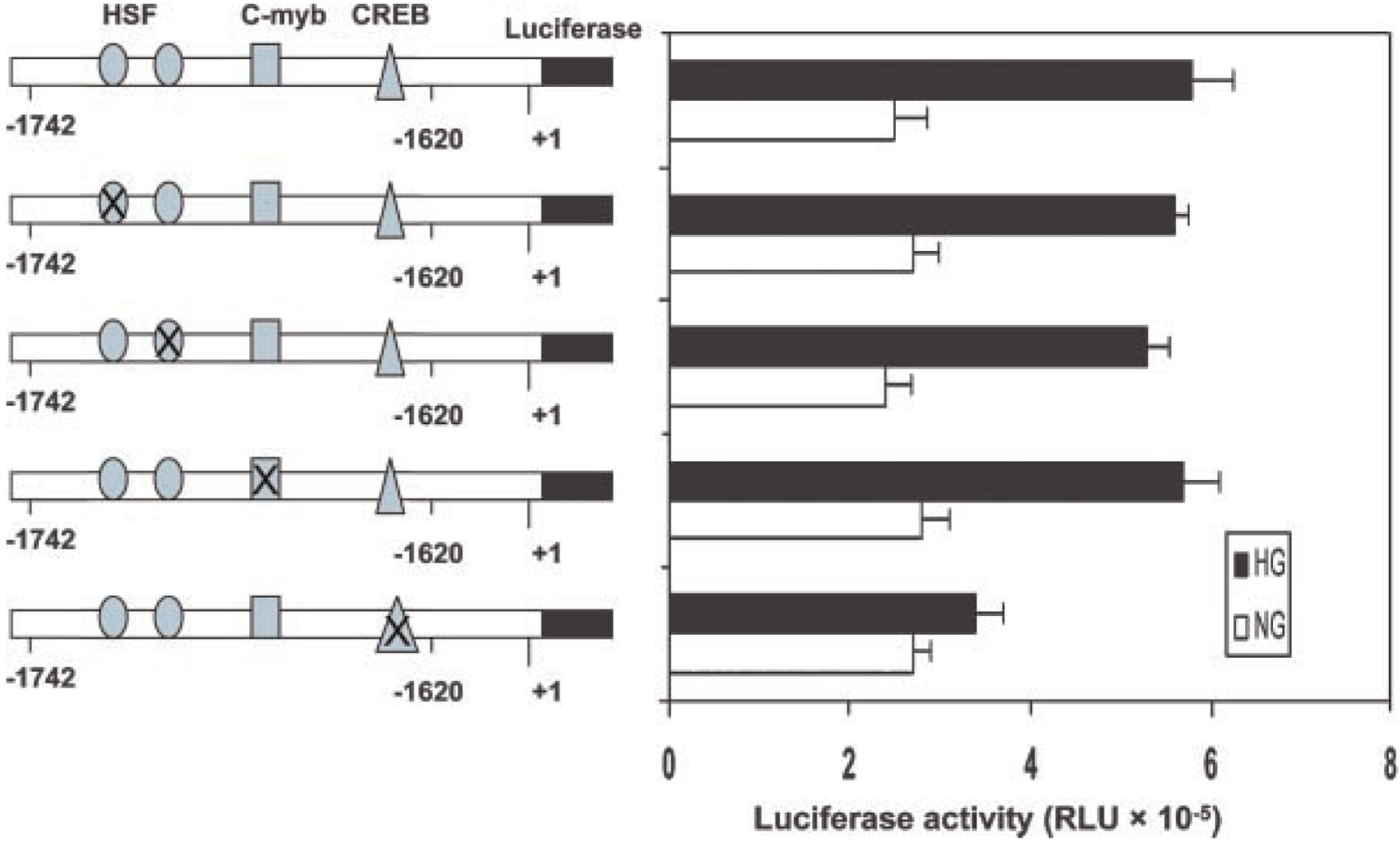
Glucose-stimulated increases in the USF2 promoter activity was mediated by a CRE site located between −1,742 and −1,620 of the USF2 promoter. Quiescent rat mesangial cells were transiently transfected with −1,742 bp USF2 promoter constructs bearing mutations of the HSF, c-Myb, and CRE sites in the presence of normal or high glucose media for 24 h. The plasmid pRL-SV40 was used as internal control. After 24 h, cells were harvested and lysed. The luciferase activity was measured and normalized to Renilla luciferase level. Data are shown with the mean ±SE of triplicates of three separate experiments.
CREB Bound to the USF2 Promoter and Mediated High Glucose-Induced USF2 Expression
We determined whether CREB binds to the CRE site located between −1,742 and −1,620 of the endogenous USF2 promoter. Chromatin immunoprecipitation with an antibody for total CREB and subsequent PCR amplification, using primer pairs that cover the CRE site located between −1,742 and −1,620, demonstrated that CREB bound to the USF2 promoter. This binding was enhanced by high glucose treatment (Fig. 8).
Fig. 8.

Chromatin immunoprecipitation analysis demonstrated the interaction of CREB with the USF2 promoter. A: Schematic representation showing CREB binding site. Two arrows indicate primers used for amplifying the region from −1,742 to −1,620 spanning the CRE site. B: Formaldehyde-cross-linked chromatin isolated from primary mesangial cells was immunoprecipitated (IP) with anti-CREB antibody and subjected to PCR as described under “Materials and Methods.” PCR products were electrophoresed on 2% agarose gel. The experiments were repeated for three times, and the representative result is shown.
To further determine the role of CREB for the transcriptional induction of USF2, we performed promoter activity assay using USF2 (−1,742) construct cotransfected with a eukaryotic expression vector overexpressing a dominant-negative CREB mutant. The effect of high glucose-mediated increase in USF2 promoter activity was abolished by over-expressing dominant-negative CREB (A-CREB) in mesangial cells (Fig. 9A). Next, we used siRNA to knock down CREB to further confirm the role of CREB for high glucose-mediated increase in USF2 expression. As shown in Figure 9B, transfection of mesangial cells with siRNA-CREB resulted in a significant inhibition of CREB expression, which was associated with a substantial decrease in glucose-induced USF2 expression. Taken together, these experiments suggest that CREB binds to the USF2 promoter and mediates glucose-induced USF2 expression in mesangial cells.
Fig. 9.
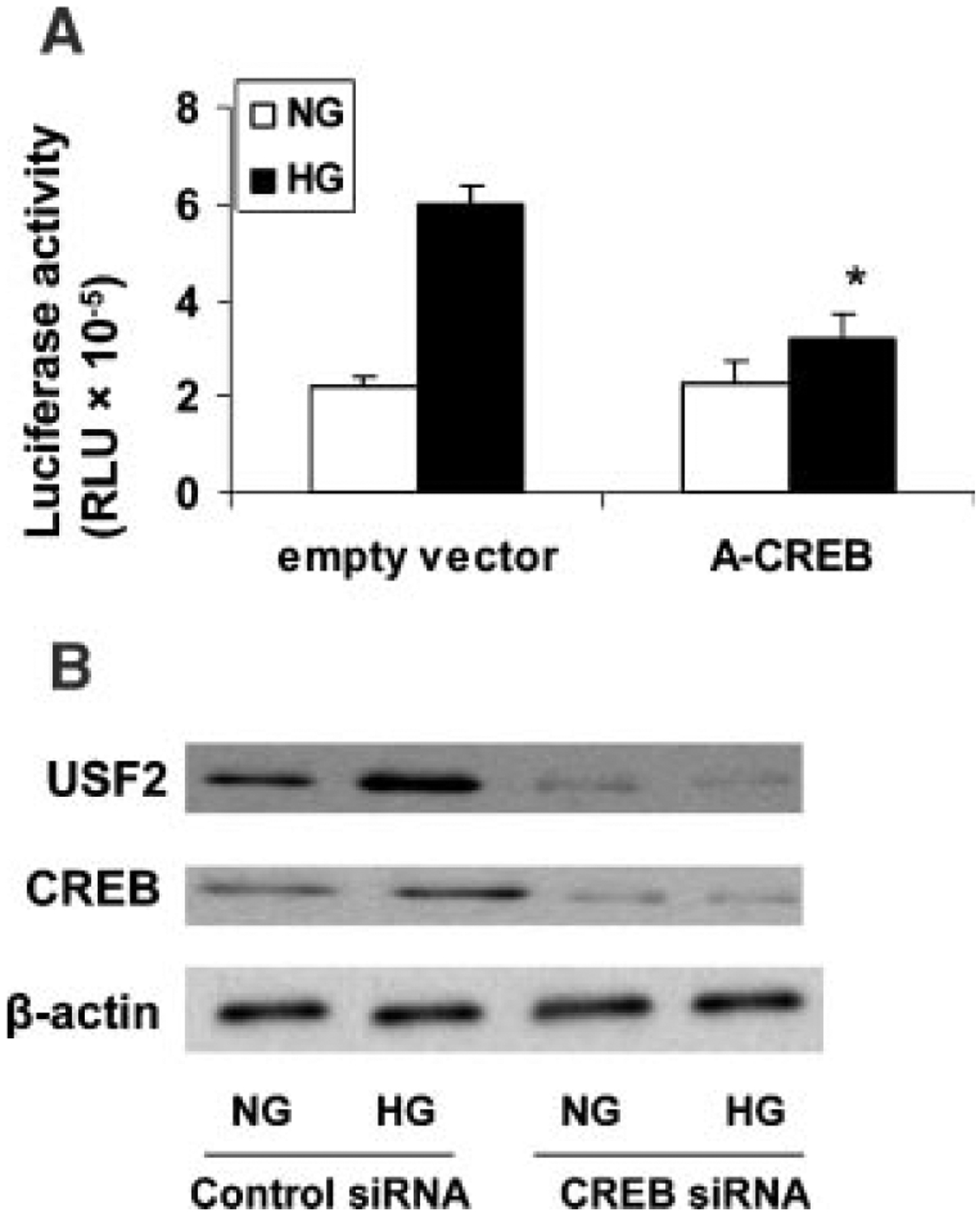
CREB mediated glucose-induced USF2 expression in mesangial cells. A: Quiescent rat mesangial cells were transiently cotransfected with −1,742 bp USF2 promoter construct and an empty control vector or an expression vector overexpressing a dominant-negative CREB mutant (A-CREB) in the presence of normal or high glucose media for 24 h. After 24 h, cells were harvested and lysed. The luciferase activity was measured and normalized to Renilla luciferase level. Data are shown with the mean ±SE of triplicates of three separate experiments. *P < 0.05 versus HG. B: Mesangial cells were transfected with control siRNA or CREB-siRNA in the presence of normal or high glucose for 24 h. Nuclear proteins were analyzed for USF2 and total CREB protein expression by Western blotting. β-actin was used as internal control. The representative blots were shown from three separate experiments.
DISCUSSION
USF2 belongs to the Myc family of transcription factors characterized by a basic/helix loop helix/leucine zipper domain responsible for dimerization and DNA binding [Sawadogo and Roeder, 1985]. In mammals, USF2 is ubiquitously expressed with a molecular weight of 44 kDa. It can form homo- and heterodimers with USF1 and recognize in vitro a CACGTG core sequence termed E box [Sirito et al., 1994]. Through binding to the E boxes of target genes, USF1 and USF2 have been shown to be involved as regulators of a high number of genes in numerous cellular processes [Rippe et al., 1997; Vallet et al., 1997; Qian et al., 1999; Kingsley-Kallesen et al., 2001; Nicolas et al., 2001; Bidder et al., 2002; Zhu et al., 2005; Chen et al., 2006; Corre and Galibert, 2005]. Studies from Valount and co-workers demonstrate the contribution of USF1 and USF2 to hepatic glucose responses in mice possessing homologous disruption of these two transcription factors genes [Vallet et al., 1997, 1998]. Although USFs have not been found to be altered by glucose concentrations in liver cells, studies from ours and others demonstrated the regulation of USFs by high glucose in mesangial cells as well as in vascular smooth muscle cells [Bidder et al., 2002; Wang et al., 2004; Weigert et al., 2004b; Zhu et al., 2005]. USFs have been shown to regulate TGF-β1 [Weigert et al., 2004a; Zhu et al., 2005] and TGF-β2 [Scholtz et al., 1996; Kingsley-Kallesen et al., 1999] gene expression, which are the key genes implicated in the pathogenesis of diabetic nephropathy. In addition, we demonstrated that glucose stimulated USF2 protein accumulation in rat glomerular mesangial cells and enhanced protein-DNA interactions on the TSP1 gene promoter, resulting in increased TSP1 protein levels and TSP1-dependent TGF-β activation [Wang et al., 2004], suggesting a role of USF2 in the development of diabetic renal complications.
In the present study, we determined the molecular mechanisms of glucose-mediated increase in USF2 expression. We first demonstrated the transcriptional mechanism of high glucose mediated increases in USF2 expression in primary rat mesangial cells. Our data showed that elevated glucose levels stimulated accumulation of USF2 protein levels is not due to increased protein stability-a posttranslational mechanism, since similar USF2 protein degradation rate was observed under either normal or high glucose exposure in mesangial cells after the cycloheximide treatment to inhibit de novo protein synthesis. Thus, increased USF2 synthesis may be a mechanism contributing to high glucose-mediated accumulation of USF2 protein levels. This mechanism is supported by glucose-mediated increase in USF2 mRNA levels. USF2 mRNA stability is not altered by high glucose exposure, which excludes the possibility of a post-transcriptional regulation. In addition, high glucose exposure stimulates the transcriptional activity of the full-length mouse USF2 promoter. Taken together, these results support the transcriptional regulation of USF2 by glucose.
We further characterized the transcriptional regulation of the USF2 expression in response to high glucose treatment. We first employed a series of 5’-deletion constructs and identified a glucose-responsive region in the USF2 promoter from −1,742 to −1,620. There is no obvious carbohydrate response element (ChoRE, consisting of two E-box motif separated by five base pairs) in this region. Within this region, there are binding sites for HSF, c-Myb and CREB (cAMP-response element-binding protein). We mutated these sites in the context of the −1,742/+1 pGL3/Basic construct. Our results demonstrated that mutation of binding sites for HSF or c-Myb had no effect on glucose-mediated USF2 promoter activity. However, mutation of CRE site abolished glucose-induced USF2 promoter activity. This CRE site has high similarity between mouse, rat and human USF2 promoter, suggesting that this sequence has important regulatory function. The chromatin immunoprecipitation assay demonstrated the binding of CREB to the USF2 promoter. This protein-DNA association is enhanced by high glucose treatment. To further confirm that CREB is involved in glucose-induced USF2 transcription, we performed the transactivation studies using eukaryotic expression vector. The experiment revealed that overexpression of a dominant-negative CREB mutant abolished glucose-induced USF2 promoter activity. Finally, siRNA-mediated knock-down of CREB inhibited glucose-induced USF2 expression. Based on these observations, CREB appears to be an important transcriptional activator of the USF2 promoter in responsive to glucose treatment in mesangial cells.
In summary, we have demonstrated that high glucose levels upregulate USF2 expression in mesangial cells at the transcriptional level. A glucose-responsive region in the USF2 promoter (−1,742 to −1,620 bp) is further identified, which is bound by CREB and mediates glucose-induced USF2 expression in mesangial cells.
ACKNOWLEDGMENTS
This work was supported by a grant from American Heart Association 04351332N (to S.W.). We thank Dr. Dennis Bruemmer (University of Kentucky) for providing the dominant—negative CREB construct.
Grant sponsor: American Heart Association Grant number: 04351332N.
REFERENCES
- Ahn S, Olive M, Aggarwal S, Krylov D, Ginty DD, Vinson C. 1998. A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Mol Cell Biol 18:967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidder M, Shao JS, Charlton-Kachigian N, Loewy AP, Semenkovich CF, Towler DA. 2002. Osteopontin transcription in aortic vascular smooth muscle cells is controlled by glucose-regulated upstream stimulatory factor and activator protein-1 activities. J Biol Chem 277: 44485–44496. [DOI] [PubMed] [Google Scholar]
- Bloomgarden ZT. 2005. Diabetic nephropathy. Diabetes Care 28:745–751. [DOI] [PubMed] [Google Scholar]
- Chen L, Shen YH, Wang X, Wang J, Gan Y, Chen N, Wang J, LeMaire SA, Coselli JS, Wang XL. 2006. Human prolyl-4-hydroxylase alpha(I) transcription is mediated by upstream stimulatory factors. J Biol Chem 281: 10849–10855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corre S, Galibert MD. 2005. Upstream stimulating factors: Highly versatile stress-responsive transcription factors. Pigment Cell Res 18:337–348. [DOI] [PubMed] [Google Scholar]
- Ishimura E, Sterzel RB, Morii H, Kashgarian M. 1992. Extracellular matrix protein: Gene expression and synthesis in cultured rat mesangial cells. Nippon Jinzo Gakkai Shi 34:9–17. [PubMed] [Google Scholar]
- Kingsley-Kallesen ML, Kelly D, Rizzino A. 1999. Transcriptional regulation of the transforming growth factor-beta2 promoter by cAMP-responsive element-binding protein (CREB) and activating transcription factor-1 (ATF-1) is modulated by protein kinases and the coactivators p300 and CREB-binding protein. J Biol Chem 274:34020–34028. [DOI] [PubMed] [Google Scholar]
- Kingsley-Kallesen M, Luster TA, Rizzino A. 2001. Transcriptional regulation of the transforming growth factor-beta2 gene in glioblastoma cells. In Vitro Cell Dev Biol Anim 37:684–690. [DOI] [PubMed] [Google Scholar]
- Nicolas G, Bennoun M, Devaux I, Beaumont C, Grand-champ B, Kahn A, Vaulont S. 2001. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci USA 98:8780–8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poczatek MH, Hugo C, Darley-Usmar V, Murphy-Ullrich JE. 2000. Glucose stimulation of transforming growth factor-beta bioactivity in mesangial cells is mediated by thrombospondin-1. Am J Pathol 157:1353–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Kaytor EN, Towle HC, Olson LK. 1999. Upstream stimulatory factor regulates Pdx-1 gene expression in differentiated pancreatic beta-cells. Biochem J 341(Pt 2): 315–322. [PMC free article] [PubMed] [Google Scholar]
- Rippe RA, Umezawa A, Kimball JP, Breindl M, Brenner DA. 1997. Binding of upstream stimulatory factor to an E-box in the 3-flanking region stimulates alpha1(I) collagen gene transcription. J Biol Chem 272:1753–1760. [DOI] [PubMed] [Google Scholar]
- Sawadogo M, Roeder RG. 1985. Interaction of a gene-specific transcription factor with the adenovirus major late promoter upstream of the TATA box region. Cell 43:165–175. [DOI] [PubMed] [Google Scholar]
- Scholtz B, Kingsley-Kallesen M, Rizzino A. 1996. Transcription of the transforming growth factor-beta2 gene is dependent on an E-box located between an essential cAMP response element/activating transcription factor motif and the TATA box of the gene. J Biol Chem 271: 32375–32380. [DOI] [PubMed] [Google Scholar]
- Sirito M, Lin Q, Maity T, Sawadogo M. 1994. Ubiquitous expression of the 43- and 44-kDa forms of transcription factor USF in mammalian cells. Nucleic Acids Res 22: 427–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skyler JS. 1996. Diabetic complications. The importance of glucose control. Endocrinol Metab Clin North Am 25: 243–254. [DOI] [PubMed] [Google Scholar]
- Susztak K, Sharma K, Schiffer M, McCue P, Ciccone E, Bottinger EP. 2003. Genomic strategies for diabetic nephropathy. J Am Soc Nephrol 14:S271–S278. [DOI] [PubMed] [Google Scholar]
- Vallet VS, Henrion AA, Bucchini D, Casado M, Raymond-jean M, Kahn A, Vaulont S. 1997. Glucose-dependent liver gene expression in upstream stimulatory factor 2−/−mice. J Biol Chem 272:21944–21949. [DOI] [PubMed] [Google Scholar]
- Vallet VS, Casado M, Henrion AA, Bucchini D, Raymond-jean M, Kahn A, Vaulont S. 1998. Differential roles of upstream stimulatory factors 1 and 2 in the transcriptional response of liver genes to glucose. J Biol Chem 273:20175–20179. [DOI] [PubMed] [Google Scholar]
- Wang S, Shiva S, Poczatek MH, Darley-Usmar V, Murphy-Ullrich JE. 2002. Nitric oxide and cGMP-dependent protein kinase regulation of glucose-mediated thrombospondin 1-dependent transforming growth factor-beta activation in mesangial cells. J Biol Chem 277:9880–9888. [DOI] [PubMed] [Google Scholar]
- Wang S, Skorczewski J, Feng X, Mei L, Murphy-Ullrich JE. 2004. Glucose upregulates thrombospondin 1 gene transcription and transforming growth factor-beta activity through antagonism of cGMP-dependent protein kinase repression via upstream stimulatory factor 2. J Biol Chem 279:34311–34322 [Epub 2004 June 7]. [DOI] [PubMed] [Google Scholar]
- Watson PA, Vinson C, Nesterova A, Reusch JE. 2002. Content and activity of cAMP response element-binding protein regulate platelet-derived growth factor receptor-alpha content in vascular smooth muscles. Endocrinology 143:2922–2929. [DOI] [PubMed] [Google Scholar]
- Weigert C, Brodbeck K, Sawadogo M, Haring HU, Schleicher ED. 2004a. Upstream stimulatory factor (USF) proteins induce human TGF-beta1 gene activation via the glucose-response element-1013/−1002 in mesangial cells: Upregulation of USF activity by the hexosamine biosynthetic pathway. J Biol Chem 279:15908–15915. [DOI] [PubMed] [Google Scholar]
- Weigert C, Brodbeck K, Sawadogo M, Haring HU, Schleicher ED. 2004b. USF proteins induce human TGF-beta1 gene activation via the glucose response element −1013/−1002 in mesangial cells—Upregulation of USF activity by the hexosamine biosynthetic pathway. J Biol Chem 2:2. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Casado M, Vaulont S, Sharma K. 2005. Role of upstream stimulatory factors in regulation of renal transforming growth factor-beta1. Diabetes 54:1976–1984. [DOI] [PubMed] [Google Scholar]