

Abstract
The presence of endotoxin in blood can lead to acute kidney injury (AKI) and septic shock. Resolvins, the endogenous lipid mediators derived from docosahexaenoic acid, have been reported to exhibit potent anti-inflammatory action. Using a mouse model of lipopolysaccharide (LPS)-induced AKI, we investigated the effects of aspirin-triggered resolvin D1 (AT-RvD1) on inflammatory kidney injury. Administration of AT-RvD1 1 h after LPS challenge protected the mice from kidney injury as indicated by the measurements of blood urea nitrogen, serum creatinine, and morphological alterations associated with tubular damage. The protective effects were evidenced by decreased neutrophil infiltration in the kidney indicating reduction in inflammation. AT-RvD1 treatment restored kidney cell junction protein claudin-4 expression, which was otherwise reduced after LPS challenge. AT-RvD1 treatment inhibited endotoxin-induced NF-κB activation and suppressed LPS-induced ICAM-1 and VCAM-1 expression in the kidney. Moreover, AT-RvD1 treatment markedly decreased LPS-induced IL-6 level in the kidney and blocked IL-6-mediated signaling including STAT3 and ERK phosphorylation. Our findings demonstrate that AT-RvD1 is a potent anti-inflammatory mediator in LPS-induced kidney injury, and AT-RvD1 has therapeutic potential against AKI during endotoxemia.
Keywords: Acute kidney injury, Endotoxemia, Inflammation, Aspirin-triggered resolvin D1, Sepsis
Introduction
The incidence of sepsis keep rising and the mortality associated with severe sepsis remains very high (Angus et al., 2001; Doi et al., 2009). Sepsis causes multi-organ failure including acute kidney injury (AKI) (Mehta et al., 2007). It has been reported that 45% to 70% of all AKI is associated with sepsis and patients with sepsis-related AKI have extremely high mortality (Bagshaw et al., 2005; Russell et al., 2000; Silvester et al., 2001). AKI is defined as a rapid reduction in renal function with severe damage to renal tubules (Jiang et al., 2012; Zhang et al., 2012). Endotoxin (lipopolysaccharide, LPS) is a component of the outer membrane of Gram-negative bacteria and is involved in the pathogenesis of sepsis-induced AKI (Doi et al., 2009). LPS induces systemic inflammation that mimics many early clinical features of sepsis. LPS exposure also causes kidney injury and microcirculatory failure (Cunningham et al., 2004; Doi et al., 2009; Knotek et al., 2001; Tiwari et al., 2005). So far, there has been no effective treatment for sepsis-induced AKI. Therefore, novel therapeutic interventions are urgently needed to tackle this devastating disease.
The systemic inflammation has been known to mediate organ dysfunction during sepsis (Ricci and Ronco, 2009). Excessive inflammation plays a major role in the initiation of kidney damage and deterioration of kidney function (Grigoryev et al., 2008; Lu et al., 2007; Thurman, 2007). Pro-inflammatory cytokines are major mediators of sepsis-induced AKI (Burne-Taney et al., 2003; Donnahoo et al., 1999; Gabay, 2006; Mitazaki et al., 2013; Takada et al., 1997; Thurman, 2007). Pro-inflammatory mediators are known to regulate acute inflammation by activating the signal transducer and activator of transcription (STAT3) (Mitazaki et al., 2009, 2013; Tanabe et al., 2010) and Nuclear factor (NF)-κB (Deng et al., 2011; Liu and Malik, 2006) pathways. Controlling the production and downstream signaling of pro-inflammatory mediators could be an effective approach to treat inflammatory kidney injury (Thurman, 2007). However, the regulation of inflammatory signaling in sepsis-induced AKI remains largely unknown.
Omega-3 polyunsaturated fatty acids, namely, eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), exhibit therapeutic potential against inflammatory disorders (Bento et al., 2011; Calder, 2002; Ishida et al., 2010; Kremer, 2000; Teitelbaum and Allan Walker, 2001). Resolvins are a group of endogenous lipid mediators derived from EPA and DHA. Resolvins not only possess potent anti-inflammatory action, they also promote resolution of inflammation (Serhan et al., 2002, 2008). Resolvin D1 (RvD1), a mediator derived from DHA, has been shown to protect against organ injury caused by ischemia-reperfusion (Duffield et al., 2006; Kasuga et al., 2008) and LPS-induced acute lung injury (Wang et al., 2011). In the presence of aspirin, aspirin-acetylated cyclooxygenase-2 (COX-2) generates (17(R)-hydroxy docosahexaenoic acid, which can be converted to 17R-RvD1, also known as aspirin-triggered RvD1 (AT-RvD1), by 5-lipoxygenase (LOX) (Bento et al., 2011; Serhan et al., 2002; Spite and Serhan, 2010; Sun et al., 2007). AT-RvD1 has been reported to be more efficient in inhibiting leukocyte infiltration in mouse peritonitis than RvD1. AT-RvD1 is also more resistant to enzymatic inactivation than RvD1 (Eickmeier et al., 2013; Sun et al., 2007), indicating that AT-RvD1 could be a more potent and long lasting anti-inflammatory mediator than RvD1. In the present study, we investigated the reno-protective effects of AT-RvD1 using an endotoxin-induced AKI model in mice.
Materials and methods
Reagents
LPS (Escherichia coli, 0111:B4) and fetal bovine serum (FBS) were purchased from Sigma (St. Louis, MO, USA). 17(R)-Resolvin D1 was obtained from Cayman (Ann Arbor, MI, USA). LPS and AT-RvD1 were dissolved in HyClone Dulbecco’s phosphate-buffered saline (PBS) from Thermo Scientific (West Palm Beach, FL, USA). Serum Creatinine and Blood Urea Nitrogen (BUN) Detection Kit were obtained from Arbor Assays (Ann Arbor, Michigan, USA). Mouse interleukin-6 (IL-6) enzyme-linked immunosorbent assay (ELISA) kits were purchased from BioLegend (San Diego, CA, USA). FITC-conjugated anti-mouse Ly-6G (Gr-1) antibody was purchased from eBioscience (San Diego, CA, USA). Goat anti-ICAM-1, VCAM-1 and Claudin-4 antibodies were obtained from Santa Cruz (Dallas, Texas, USA). Phospho-IκBα, phospho-STAT3, phospho-Erk1/2 antibody, IκBα, STAT3, Erk1/2 and β-actin antibodies were purchased from Cell Signaling Technology (Boston, MA, USA). Excell Plus ready to use histological tissue fixative was ordered from American MasterTech (Lodi, CA, USA).
Acute kidney injury model
C57BL/6 male mice (13 weeks old) were purchased from the Jackson Laboratory. Mice were housed in cages with access to food and water in a temperature-controlled room with a 12-hour dark/light cycle. All experiments and animal care procedures were approved by the Institutional Animal Care and Use Committee of the University of Texas Health Science Center at Tyler. For LPS-induced AKI, mice were given either LPS (5 mg/kg in 300 μl PBS) or vehicle (300 μl PBS) by intraperitoneal (IP) injection. One hour after LPS challenge, mice were given either AT-RvD1 (1 μg/mouse dissolved in 300 μl 3% vol/vol ethanol in PBS) or vehicle (300 μl 3% vol/vol ethanol in PBS) by IP injection. Experiments were terminated 24 h after LPS challenge. Mouse kidney tissue and blood were then collected.
Renal function and histology studies
Serum creatinine and blood urea nitrogen (BUN) levels were tested with a colorimetric detection kit from Arbor Assays. Kidney tissues were fixed in Excell Plus and embedded in paraffin. Sections of 4 μm were stained with hematoxylin and eosin (H&E) and periodic acid-Schiff (PAS) reagent. Histologic changes in kidney cortex and outer medulla were analyzed. Kidney injury was defined as tubular damage score. For each mouse, at least 10 high-power (×400) fields were examined.The histological tubular necrosis was scored as follows: 0, no damage; 1, 0–25% damage; 2, 26–50% damage; 3, 50–75% damage; 4, N75% damage.
ELISA, immunofluorescence and immunoblotting assays
IL-6 levels in kidney tissues were measured with an ELISA assay kit from BioLegend. For immunofluorescence assay, kidney tissue cyrosections were fixed in pre-chilled methanol for 5 min, washed three times in PBS, and then blocked with 0.5% mouse BD Fc Block (San Jose, California, USA) in 3% FBS at room temperature for 30 min. FITC-conjugated anti-mouse Ly-6G (Gr-1) antibody dilution (100 μl; 1:50) was applied per tissue on the slides and incubated for 1 h at room temperature and kept in dark. After three washes in PBS, the slides were mounted with ProLong Antifade Kit (Life technology, Grand Island, NY, USA). Images were captured using Nikon Elipse TE-2000-U microscope with a 40×/1.30 NA objective and NIS Elements software (Nikon, Melville, NY, USA). Kidney tissues were subjected to immunoblotting assay as described previously (Wu et al., 2012).
Statistical analysis
Data are expressed as mean ± SD. Multiple comparisons were examined by one-way ANOVA, followed by individual comparisons with the Turkey post hoc test. Student’s t test was used to test two groups. Statistical significance was assigned to P values smaller than 0.05.
Results
Aspirin-triggered resolvin D1 protects mice from LPS-induced acute kidney injury
Serum creatinine and blood urea nitrogen (BUN) levels have been used as major indices of renal function. Their levels are often elevated when kidney function is compromised (Jeong et al., 2013). We examined the effects of Aspirin-triggered resolvin D1 (AT-RvD1) on LPS-induced acute kidney injury in mice. A single dose of AT-RvD1 or vehicle control was administered 1 h after LPS challenge. Twenty-four hours after the LPS challenge, the mice exhibited a significant increase in both serum BUN (27.63 ± 4.29 mg/dL for PBS group vs. 57.01 ± 9.47 mg/dL for LPS group; Fig. 1A) and serum creatinine (0.500 ± 0.081 mg/dL for PBS group vs. 0.845 ± 0.142 mg/dL for LPS group; Fig. 1B), indicating that renal function was hampered. AT-RvD1 treatment significantly attenuated the rise of BUN (35.21 ± 5.27 mg/dL for LPS/AT-RvD1 group; Fig. 1A) and serum creatinine (0.598 ± 0.067 mg/dL for LPS/AT-RvD1 group; Fig. 1B).
Fig. 1.
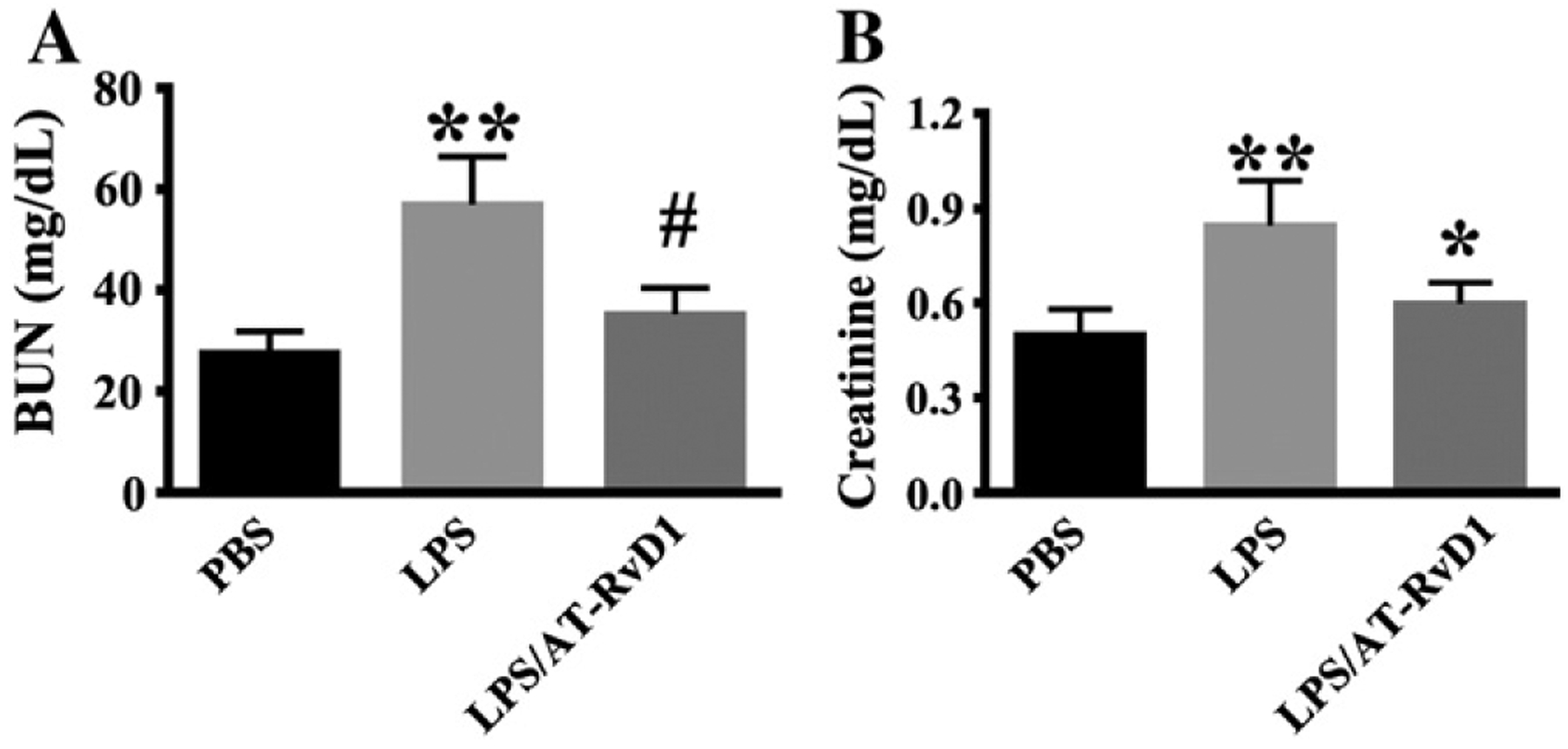
AT-RvD1 protects against renal dysfunction during LPS-induced kidney injury. Mice were divided into three groups: PBS control (PBS); 5 mg/Kg LPS (LPS); 5 mg/kg LPS/1 μg AT-RvD1 (LPS/AT-RvD1). Mice were treated with AT-RvD1 (1 μg) or PBS at 1 h after LPS challenge. (A) Blood urea nitrogen levels in each group at 24 h after LPS (PBS: n = 9 mice; LPS: n = 10 mice; LPS/AT-RvD1: n = 7 mice). (B) Serum creatinine levels in each group at 24 h after LPS challenge (PBS: n = 5 mice; LPS: n = 10 mice; LPS/AT-RvD1: n = 8 mice). Data are presented as mean ± SD. **P < 0.0001 vs PBS group; #P < 0.0001 vs LPS group; *P < 0.001 vs LPS group.
AT-RvD1 treatment diminishes LPS-induced renal tubular damage
We then conducted histological studies with hematoxylin and eosin (H&E) and periodic acid-Schiff (PAS) staining to examine kidney damage. Compared with mouse kidneys from control group, LPS challenge caused tissue damage in the renal cortex and outer medulla (Fig. 2A, LPS), as showed by tubular epithelial cells sloughing, loss of brush border, tubular dilation and distortion. AT-RvD1 treatment 1 h after LPS challenge markedly reduced kidney tubular damage (Fig. 2A, LPS/ATRvD1). The measurement of tubular damage score was performed as described in the Material and Methods section. The data confirmed that AT-RvD1 treatment significantly reduced tubular injury in LPS-challenged mice (Fig. 2B).
Fig. 2.
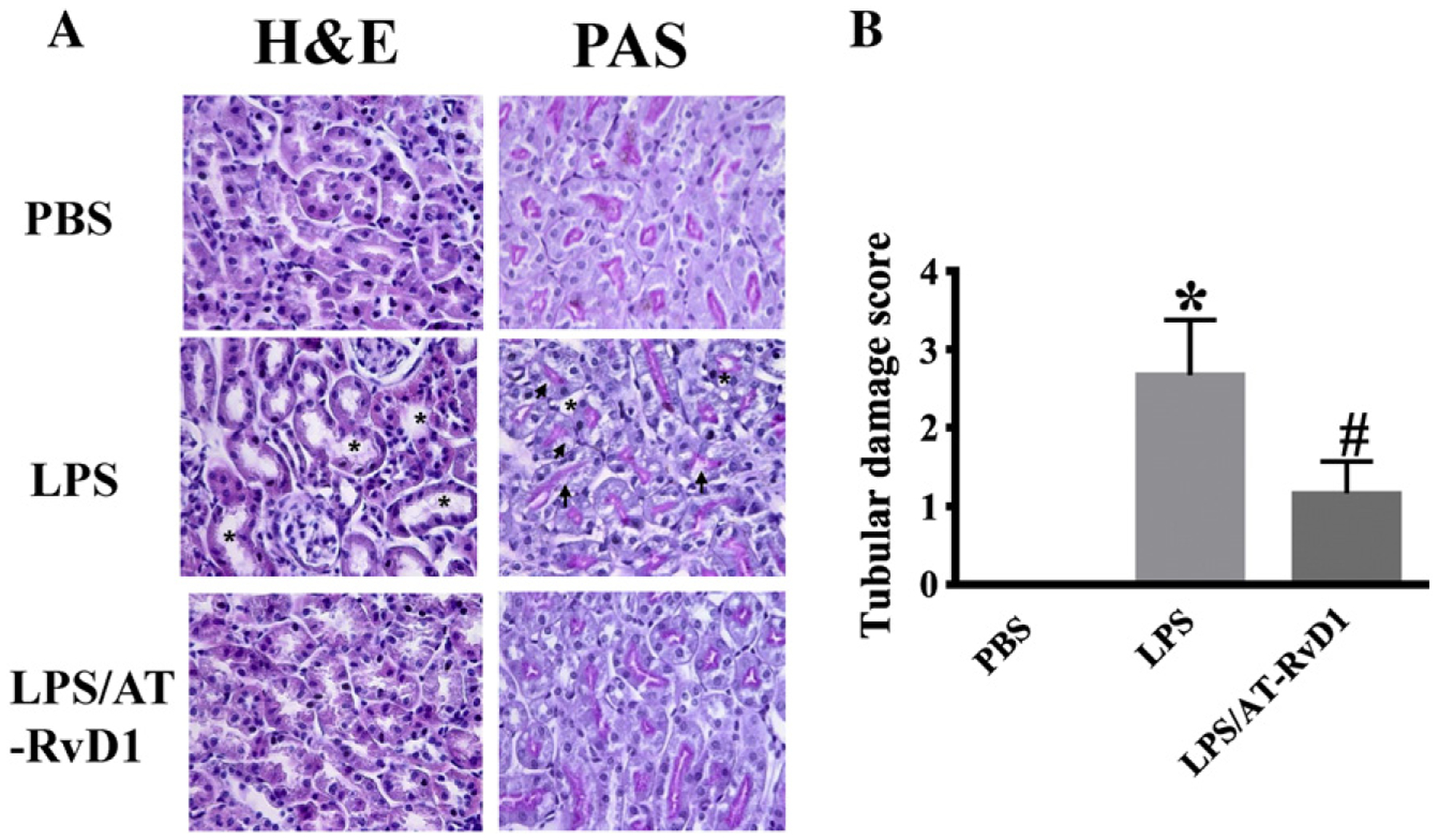
AT-RvD1 protects against LPS-induced renal tubular damage. Kidney tissues were collected at 24 h after LPS challenge for H&E and PAS staining (PBS: n = 6 mice; LPS: n = 9 mice; LPS/AT-RvD1: n = 6 mice). (A) Representative H&E staining (A) and PAS staining (B) of kidney cortex and outer medulla tissues. Compared with WT mice, LPS-induced renal tubular injury with tubular dilation or cast formation (*) and vacuolization of renal tubular cells (arrows). (C) Pathological scores of tubular damage in LPS and LPS/AT-RvD1 groups. Data are presented as mean ± SD. *P < 0.0001 vs PBS group, #P < 0.001 vs LPS group.
AT-RvD1 inhibits neutrophil infiltration in LPS-induced acute kidney injury
Previous studies have shown that neutrophil infiltration is associated with an early inflammatory response during the first 24 h of kidney injury (Liano and Pascual, 1996). Here, we assessed neutrophil infiltration using FITC-conjugated antibody for GR-1, which is highly expressed by neutrophils (Fig. 3). AT-RvD1 treatment at 1 h after LPS challenge resulted in reduced GR-1-positive cells (Duffield et al., 2005), suggesting that neutrophil infiltration in the kidney is inhibited.
Fig. 3.
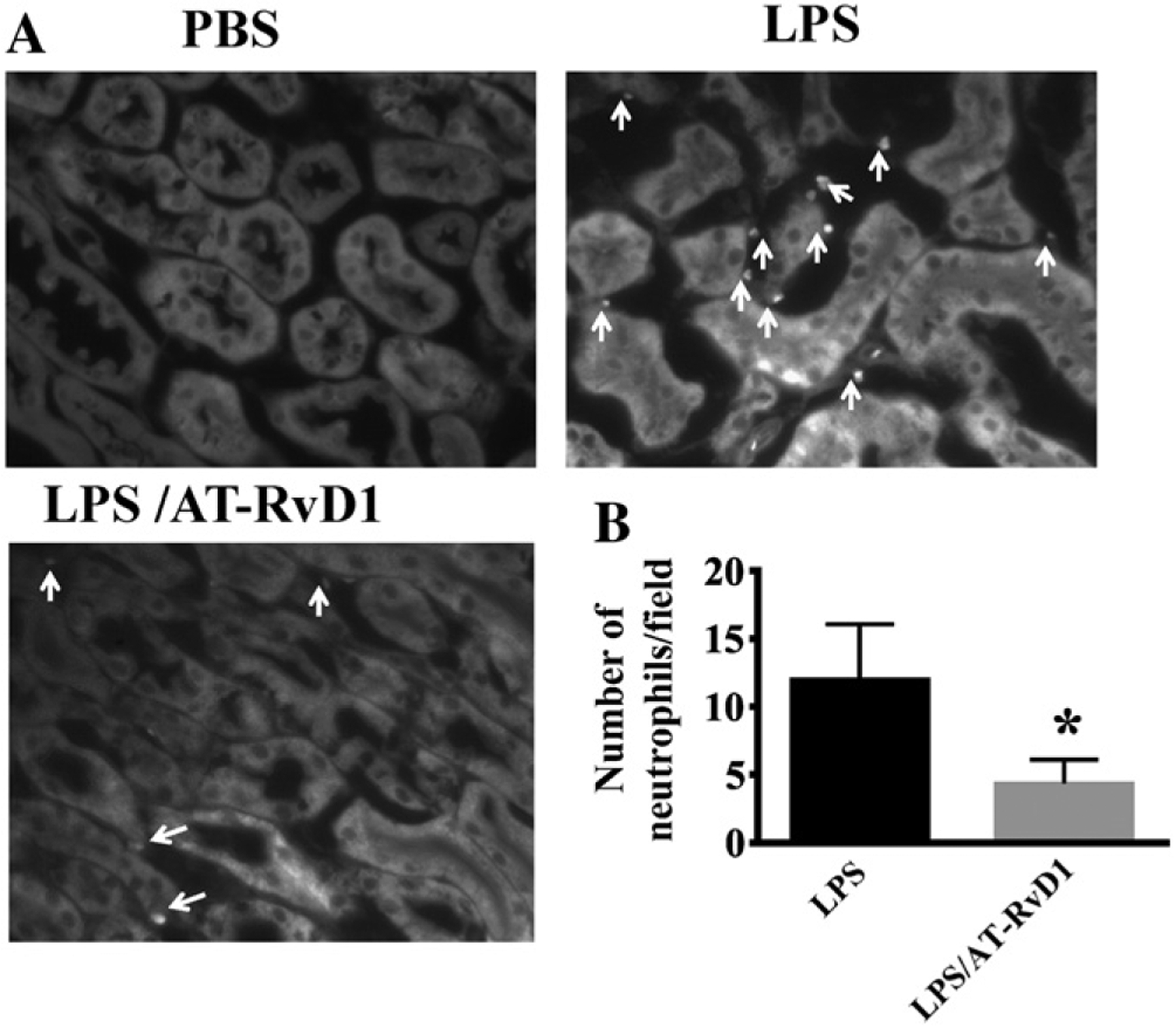
AT-RvD1 inhibits neutrophil infiltration in LPS-induced acute kidney injury. Kidney sections were stained with FITC-anti-GR1 antibody specific for neutrophils. (A) The outer medullary region of sections was photographed at ×200. White arrows indicate neutrophils. (B) Neutrophil count/field (LPS: n = 8 mice; LPS/AT-RvD1: n = 6 mice). *P < 0.001 vs LPS group.
AT-RvD1 regulates the cell junction protein claudin-4 expression in LPS-induced acute kidney injury
Claudin-4 is one of the most important components of the tight junctions. Claudin-4 has also been found to regulate paracellular permeability in the distal nephron (Hou et al., 2010; Le Moellic et al., 2005). We determined the effect of AT-RvD1 treatment on claudin-4 expression in LPS-induced AKI by immunoblotting assay. As shown in Fig. 4, LPS challenge significantly decreased claudin-4 expression. AT-RvD1 treatment after LPS challenge, restored claudin-4 expression to baseline levels comparable to that observed in the kidney tissues of the control mice. The data suggest that AT-RvD1 is able to inhibit LPS-induced disruption of barrier function in the kidney.
Fig. 4.
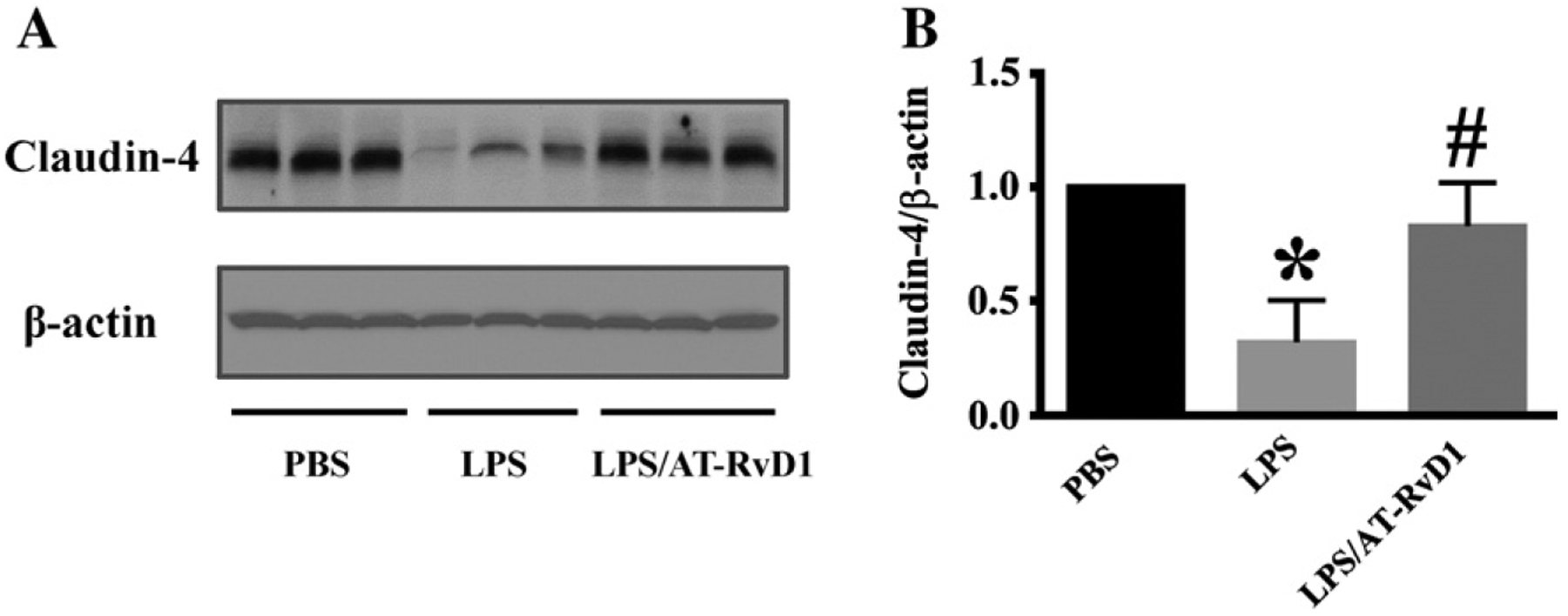
AT-RvD1 regulates the cell junction protein claudin-4 expression in acute kidney injury. Kidney tissues were subjected to immunoblotting assay (PBS: n = 8 mice; LPS: n = 10 mice; LPS/AT-RvD1: n = 9 mice). (A) Claudin-4 expression in kidney tissues. Blots were re-probed for actin to normalize total protein content. (B) Data are expressed as the relative ratio to actin (right panel), and presented as mean ± SD. *P < 0.05 vs PBS group; #P < 0.05 vs LPS group.
AT-RvD1 decreases ICAM-1, VCAM-1 expression and inhibits IL-6 expression in LPS-induced kidney injury
Up-regulated ICAM-1 and VCAM-1 expression mediates early and persistent inflammatory responses (Hopkins et al., 2004). Here, we examined the expression of ICAM-1 and VCAM-1 in the kidney tissues by immunoblotting assays. ICAM-1 and VCAM-1 were increased (8- to 10-fold) in kidney tissues from LPS-treated mice when compared with that of kidney tissues from the control mice treated with PBS (Fig. 5A,B). AT-RvD1 treatment significantly inhibited LPS-induced increase of ICAM-1 and VCAM-1 expression (Fig. 5A, B). The results indicate that AT-RvD1 can inhibit kidney inflammatory responses by down-regulating ICAM-1 and VCAM-1 expression.
Fig. 5.
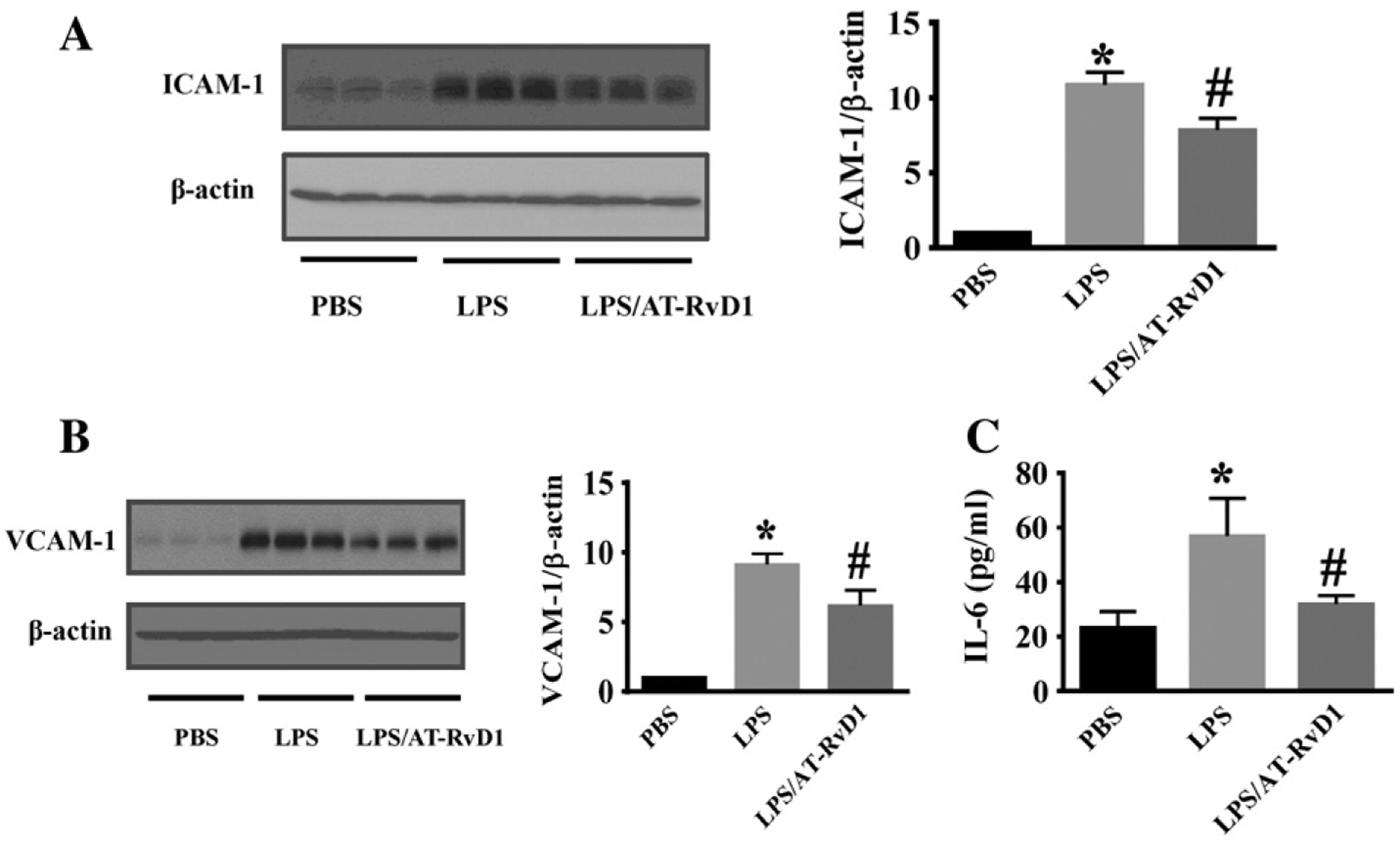
AT-RvD1 inhibits LPS-induced kidney inflammation. Kidney tissues were subjected to immunoblotting assay to assess ICAM-1 (A) and VCAM-1 (B) expression (PBS: n = 8 mice; LPS: n = 10 mice; LPS/AT-RvD1: n = 9 mice). Immunoblots were re-probed for actin to normalize total protein content. Data are expressed as the relative ratio to actin and presented as mean ± SD. *P < 0.0001, vs PBS group; #P < 0.05, vs LPS group. (C) Kidney IL-6 levels were determined by ELISA (n = 5 mice/group). Data are presented as mean ± SD. *P < 0.001 vs PBS group; #P < 0.05 vs LPS group.
IL-6 is a pro-inflammatory cytokine that is up-regulated during tissue injury and inflammation (Naugler and Karin, 2008). We examined kidney IL-6 levels in LPS-induced AKI. Compared with the control group, IL-6 level in the kidney of LPS challenged mice was significantly increased, and AT-RvD1 treatment decreased IL-6 level in the kidney (Fig. 5C).
AT-RvD1 inhibits NF-κB activation in LPS-induced acute kidney injury
NF-κB is a transcription factor that regulates the expression of a wide variety of pro-inflammatory mediators (Deng et al., 2011; Liu and Malik, 2006). NF-κB is present in the cytosol in an inactive state through its binding to the inhibitory IκB proteins. The phosphorylation of IκBα leads to proteasome-mediated degradation and results in the release and nuclear translocation of active NF-κB (Deng et al., 2011; Liu and Malik, 2006). Our data showed that LPS challenge led to NF-κB activation in mouse kidney tissues as indicated by significantly increased phosphorylation of IκBα relative to actin and total IκBα (Fig. 6A). AT-RvD1 treatment reduced IκBα phosphorylation (Fig. 6A). Our data indicate that LPS-induced NF-κB activation was inhibited by AT-RvD1 treatment (Fig. 6B,C).
Fig. 6.
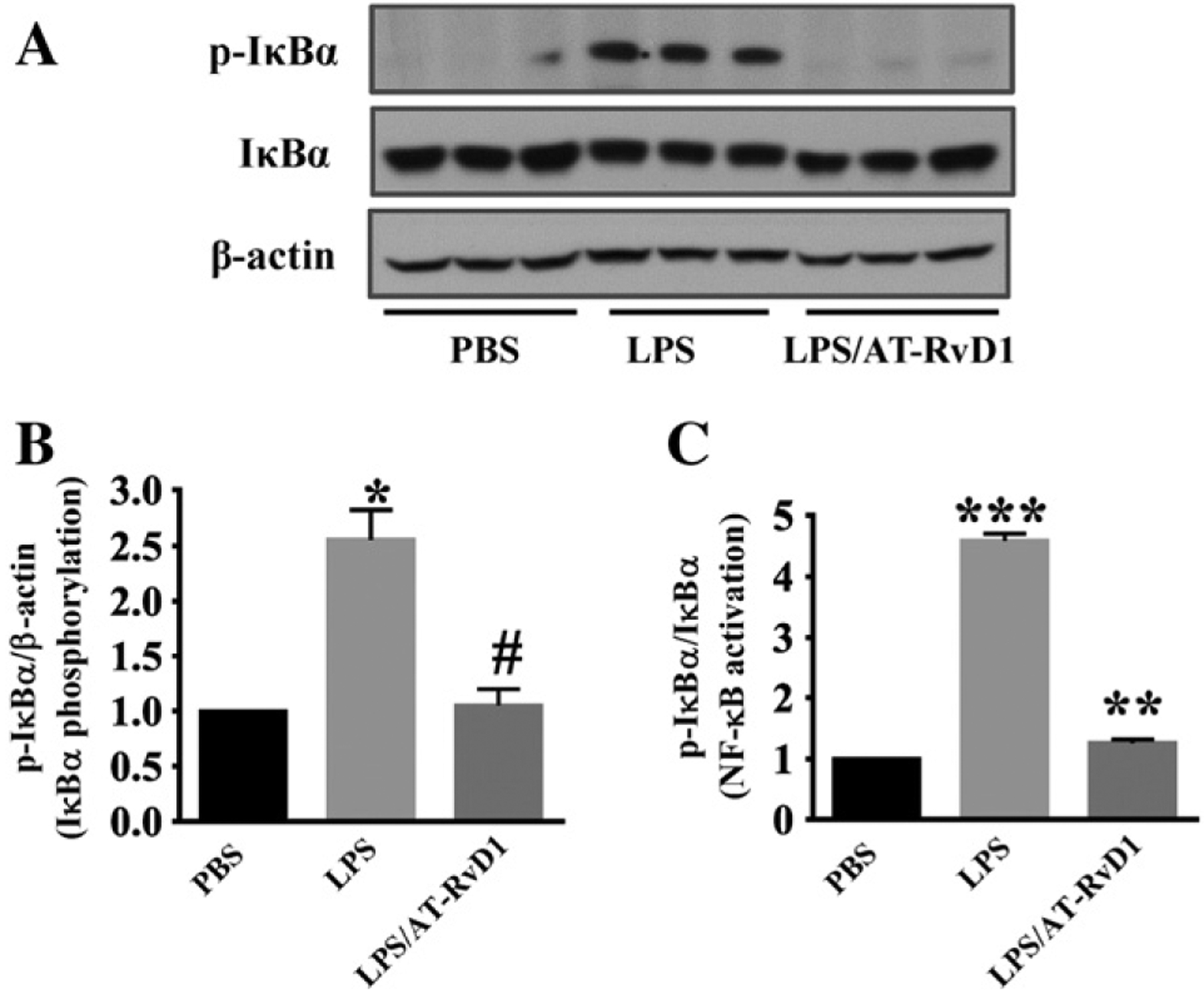
AT-RvD1 inhibits NF-κB activation in LPS-induced acute kidney injury. Immunoblotting assays will conducted to examine phosphorylated IκBα, total IκBα and actin levels in kidney tissues (n = 6 mice/group). (A) Representative blots of phosphorylated IκBα, IκBα and actin. (B, C) Densitometry analysis is presented as relative ratios of phosphorylated IκBα to actin and total IκBα. Data are presented as mean ± SD. *P < 0.05 vs PBS group; #P < 0.05 vs LPS group. ***P < 0.0001 vs PBS group; **P < 0.001 vs LPS group.
AT-RvD1 inhibits IL-6-mediated inflammatory signaling in LPS-induced acute kidney injury
STAT3 activation is a critical signaling mechanism in IL-6-mediated inflammatory signaling. We examined STAT3 activation in LPS-induced AKI. Compared with the control group, STAT3 phosphorylation was significantly increased in the kidney after LPS challenge (Fig. 7A). AT-RvD1 treatment inhibited LPS-induced STAT3 phosphorylation (Fig. 7A), which is consistent with the decreased IL-6 level in the kidney after AT-RvD1 treatment. ERK is activated in response to pro-inflammatory cytokines (Meloche and Pouyssegur, 2007). We determined whether AT-RvD1 treatment was able to inhibit ERK activation in the kidney and found that ERK phosphorylation was remarkably increased in kidney tissues after LPS challenge (Fig. 7B). AT-RvD1 treatment significantly decreased LPS-induced ERK phosphorylation (Fig. 7B). The data suggest that AT-RvD1 can suppress LPS-induced kidney inflammation by modulating STAT3/ERK signaling.
Fig. 7.
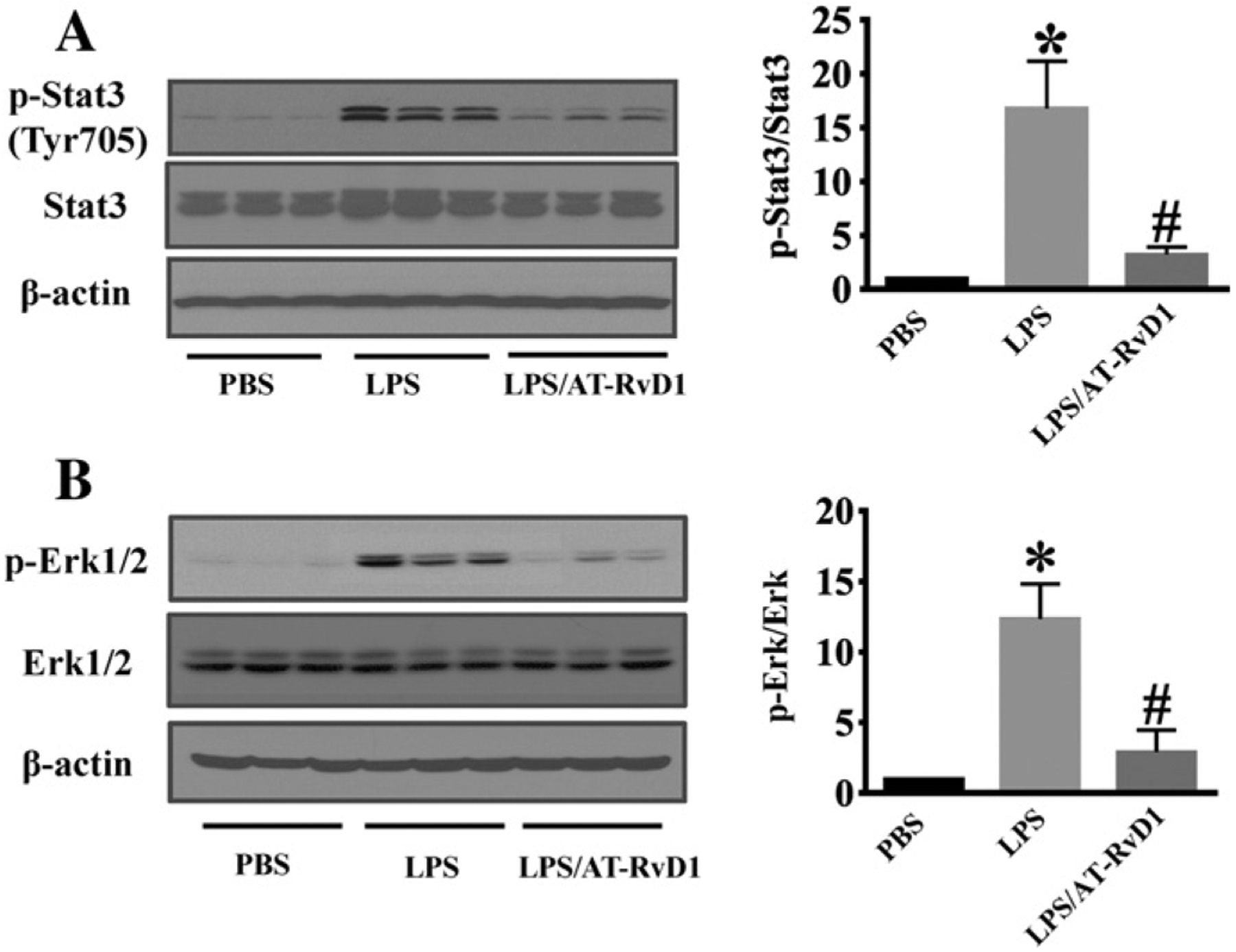
AT-RvD1 inhibits inflammatory signaling during LPS-acute kidney injury. Kidney tissues was collected for immunoblotting assays (n = 6 mice/group). (A) Representative blots of LPS-induced STAT3 phosphorylation. (B) Representative blots of LPS-induced ERK phosphorylation. Densitometry analysis is presented as the relative ratio of each protein to non-phosphorylated proteins. Data are presented as mean ± SD. *P < 0.05 vs PBS group; #P < 0.05 vs LPS group.
Discussion
AT-RvD1 exhibits potent anti-inflammatory action and has been reported to inhibit inflammatory injury in various animal models (Eickmeier et al., 2013). In the present study, we investigated the effects of AT-RvD1 on endotoxin-induced AKI in mice. AT-RvD1 treatment inhibited the production of pro-inflammatory mediators, and less tubular damage was observed in AT-RvD1-treated mice. In our studies, AT-RvD1 was administrated at 1 h after the induction of AKI by LPS. Using this post-treatment regime, we were able to improve renal function and attenuate AKI caused by LPS challenge in mice (Coldewey et al., 2013). Our studies support the therapeutic potential of AT-RvD1 in LPS-induced AKI.
The protective effects of AT-RvD1 are linked to its inhibition of LPS-induced kidney inflammation. AT-RvD1 treatment diminished PMN infiltration and down-regulated inflammatory responses in the kidney, which was associated with reduced production of pro-inflammatory mediators in the injured tissue. AT-RvD1 treatment suppressed LPS-induced ICAM-1 and VCAM-1 up-regulation and inhibited the disruption of cell–cell junction protein expression to support endothelial and epithelial barrier function. Our findings indicate that AT-RvD1 attenuates inflammatory kidney injury through several specific protective mechanisms.
Claudin-4, a cell–cell junction protein involved in barrier function (Amasheh et al., 2011), is expressed in the distal convoluted tubule and collecting duct (Balkovetz, 2006; Eadon et al., 2012; Kiuchi-Saishin et al., 2002). Claudin-4 is a major component of tight junction (Le Moellic et al., 2005). Claudin-4 has also been reported to participate in paracellular anion transport in the distal nephron (Hou et al., 2010; Le Moellic et al., 2005). However, the regulation of Claudin-4 expression during kidney injury is not clear. Our data showed that Claudin-4 protein expression in kidney tissue was markedly decreased after LPS challenge. AT-RvD1 inhibited Claudin-4 down-regulation, which may promote the integrity of tight junction during AKI. Several studies also support the role of Claudin-4 in controlling paracellular ion permeability through the tight junctions (Van Itallie et al., 2001). Further studies are needed to determine whether AT-RvD1 is a direct modulator of Claudin-4 function at the tight junction. Mechanistic investigation are needed to explore the regulation of Claudin-4 expression in LPS-induced AKI.
Adhesion molecules such as ICAM-1 and VCAM-1 mediate neutrophil/endothelial cells interactions and play an important role in LPS-induced inflammation (Kim et al., 2009; Smith et al., 1991). The role of ICAM-1 and VCAM-1 in mediating neutrophil infiltration during AKI is also well established (Kim et al., 2009; Smith et al., 1991). LPS increased the expression of ICAM-1, which indicates an early inflammatory response. The down-regulation of ICAM-1 expression decreases the infiltration of leukocytes into injured tissues (Kim et al., 2009). LPS-induced ICAM-1, and VCAM-1 up-regulation was markedly attenuated by AT-RvD1 treatment, indicating that AT-RvD1 can modulate early inflammatory responses in the kidney.
Mortality in sepsis has been reported to correlate with the up-regulation of IL-6 (Hotchkiss et al., 1999; Wang et al., 2009). The role of IL-6 as a pro-inflammatory cytokine in kidney injury has been well established. Our studies demonstrated that LPS-induced increase of IL-6 level in the kidney tissue was suppressed after AT-RvD1 treatment. AT-RvD1 may inhibit inflammatory response by modulating IL-6-mediated signaling. Signal transducer and activator of transcription (STAT) proteins play a crucial role in IL-6-mediated inflammation and immunity (Fang et al., 2013). STAT3 is a major downstream signaling target of IL-6 (Yuan et al., 2011). Our results suggest that AT-RvD1 can modulate inflammatory signaling through STAT3 in LPS-induced AKI. ERK, another target of inflammatory signaling, is involved in several pathways leading to tissue damage during AKI (Wang et al., 2013). We showed that LPS-induced ERK phosphorylation was markedly decreased after AT-RvD1 treatment. The inhibition of ERK activation by AT-RvD1 may contribute to its protection against LPS-induced kidney injury. NF-κB pathway has been known to play an important role in mediating inflammatory responses in sepsis (Liu and Malik, 2006). NF-κB activation requires the phosphorylation of the inhibitory IκB proteins. Our data suggest that AT-RvD1 can inhibit LPS-induced NF-κB activation by down-regulating IκB phosphorylation.
In summary, our data suggest that the DHA-derived lipid mediator AT-RvD1 can inhibit kidney inflammation, NF-κB activation and STAT3 and ERK signaling, which leads to attenuation of LPS-induced inflammatory kidney injury. Therefore, AT-RvD1 may serve as a novel therapeutic agent in the treatment of LPS-induced AKI and other inflammatory kidney diseases.
Footnotes
Disclosure
All the authors declared no competing interests.
References
- Amasheh S, Fromm M, Gunzel D, 2011. Claudins of intestine and nephron—a correlation of molecular tight junction structure and barrier function. Acta Physiol 201, 133–140. [DOI] [PubMed] [Google Scholar]
- Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR, 2001. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit. Care Med 29, 1303–1310. [DOI] [PubMed] [Google Scholar]
- Bagshaw SM, Laupland KB, Doig CJ, Mortis G, Fick GH, Mucenski M, Godinez-Luna T, Svenson LW, Rosenal T, 2005. Prognosis for long-term survival and renal recovery in critically ill patients with severe acute renal failure: a population-based study. Crit. Care 9, R700–R709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkovetz DF, 2006. Claudins at the gate: determinants of renal epithelial tight junction paracellular permeability. Am. J. Physiol. Ren. Physiol 290, F572–F579. [DOI] [PubMed] [Google Scholar]
- Bento AF, Claudino RF, Dutra RC, Marcon R, Calixto JB, 2011. Omega-3 fatty acidderived mediators 17(R)-hydroxy docosahexaenoic acid, aspirin-triggered resolvin D1 and resolvin D2 prevent experimental colitis in mice. J. Immunol 187, 1957–1969. [DOI] [PubMed] [Google Scholar]
- Burne-Taney MJ, Kofler J, Yokota N, Weisfeldt M, Traystman RJ, Rabb H, 2003. Acute renal failure after whole body ischemia is characterized by inflammation and T cell-mediated injury. Am. J. Physiol. Ren. Physiol 285, F87–F94. [DOI] [PubMed] [Google Scholar]
- Calder PC, 2002. Dietary modification of inflammation with lipids. Proc. Nutr. Soc 61,345–358. [DOI] [PubMed] [Google Scholar]
- Coldewey SM, Khan AI, Kapoor A, Collino M, Rogazzo M, Brines M, Cerami A, Hall P, Sheaff M, Kieswich JE, Yaqoob MM, Patel NS, Thiemermann C, 2013. Erythropoietin attenuates acute kidney dysfunction in murine experimental sepsis by activation of the beta-common receptor. Kidney Int 84, 482–490. [DOI] [PubMed] [Google Scholar]
- Cunningham PN, Wang Y, Guo R, He G, Quigg RJ, 2004. Role of Toll-like receptor 4 in endotoxin-induced acute renal failure. J. Immunol 172, 2629–2635. [DOI] [PubMed] [Google Scholar]
- Deng Y, Edin ML, Theken KN, Schuck RN, Flake GP, Kannon MA, DeGraff LM, Lih FB, Foley J, Bradbury JA, Graves JP, Tomer KB, Falck JR, Zeldin DC, Lee CR, 2011. Endothelial CYP epoxygenase overexpression and soluble epoxide hydrolase disruption attenuate acute vascular inflammatory responses in mice. FASEB J 25, 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi K, Leelahavanichkul A, Yuen PS, Star RA, 2009. Animal models of sepsis and sepsis-induced kidney injury. J. Clin. Invest 119, 2868–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnahoo KK, Meng X, Ayala A, Cain MP, Harken AH, Meldrum DR, 1999. Early kidney TNF-alpha expression mediates neutrophil infiltration and injury after renal ischemia-reperfusion. Am. J. Physiol 277, R922–R929. [DOI] [PubMed] [Google Scholar]
- Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP, 2005. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Invest 115, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffield JS, Hong S, Vaidya VS, Lu Y, Fredman G, Serhan CN, Bonventre JV, 2006. Resolvin D series and protectin D1 mitigate acute kidney injury. J. Immunol 177, 5902–5911. [DOI] [PubMed] [Google Scholar]
- Eadon MT, Hack BK, Xu C, Ko B, Toback FG, Cunningham PN, 2012. Endotoxemia alters tight junction gene and protein expression in the kidney. Am. J. Physiol. Ren. Physiol 303, F821–F830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eickmeier O, Seki H, Haworth O, Hilberath JN, Gao F, Uddin M, Croze RH, Carlo T, Pfeffer MA, Levy BD, 2013. Aspirin-triggered resolvin D1 reduces mucosal inflammation and promotes resolution in a murine model of acute lung injury. Mucosal Immunol 6, 256–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Z, Tang Y, Fang J, Zhou Z, Xing Z, Guo Z, Guo X, Wang W, Jiao W, Xu Z, Liu Z, 2013. Simvastatin inhibits renal cancer cell growth and metastasis via AKT/mTOR, ERK and JAK2/STAT3 pathway. PLoS One 8, e62823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabay C, 2006. Interleukin-6 and chronic inflammation. Arthritis Res. Ther 8 (Suppl. 2), S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryev DN, Liu M, Hassoun HT, Cheadle C, Barnes KC, Rabb H, 2008. The local and systemic inflammatory transcriptome after acute kidney injury. J. Am. Soc. Nephrol 19, 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins AM, Baird AW, Nusrat A, 2004. ICAM-1: targeted docking for exogenous as well as endogenous ligands. Adv. Drug Deliv. Rev 56, 763–778. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, Buchman TG, Karl IE, 1999. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit. Care Med 27, 1230–1251. [DOI] [PubMed] [Google Scholar]
- Hou J, Renigunta A, Yang J, Waldegger S, 2010. Claudin-4 forms paracellular chloride channel in the kidney and requires claudin-8 for tight junction localization. Proc. Natl. Acad. Sci. U. S. A 107, 18010–18015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida T, Yoshida M, Arita M, Nishitani Y, Nishiumi S, Masuda A, Mizuno S, Takagawa T, Morita Y, Kutsumi H, Inokuchi H, Serhan CN, Blumberg RS, Azuma T, 2010. Resolvin E1, an endogenous lipid mediator derived from eicosapentaenoic acid, prevents dextran sulfate sodium-induced colitis. Inflamm. Bowel Dis 16, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong M, Kim YW, Min JR, Kwon M, Han BS, Kim JG, Jeong SH, 2013. Change in kidney damage biomarkers after 13 weeks of exposing rats to the complex of Paecilomyces sinclairii and its host Bombyx mori larvae. Food Chem. Toxicol 59C, 177–186. [DOI] [PubMed] [Google Scholar]
- Jiang M, Wei Q, Dong G, Komatsu M, Su Y, Dong Z, 2012. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int 82, 1271–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasuga K, Yang R, Porter TF, Agrawal N, Petasis NA, Irimia D, Toner M, Serhan CN, 2008. Rapid appearance of resolvin precursors in inflammatory exudates: novel mechanisms in resolution. J. Immunol 181, 8677–8687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Jung YJ, Lee AS, Lee S, Kang KP, Lee TH, Lee SY, Jang KY, Moon WS,Choi KS, Yoon KH, Sung MJ, Park SK, Kim W, 2009. COMP-angiopoietin-1 decreases lipopolysaccharide-induced acute kidney injury. Kidney Int 76, 1180–1191. [DOI] [PubMed] [Google Scholar]
- Kiuchi-Saishin Y, Gotoh S, Furuse M, Takasuga A, Tano Y, Tsukita S, 2002. Differential expression patterns of claudins, tight junction membrane proteins, in mouse nephron segments. J. Am. Soc. Nephrol 13, 875–886. [DOI] [PubMed] [Google Scholar]
- Knotek M, Rogachev B, Wang W, Ecder T, Melnikov V, Gengaro PE, Esson M, Edelstein CL, Dinarello CA, Schrier RW, 2001. Endotoxemic renal failure in mice: role of tumor necrosis factor independent of inducible nitric oxide synthase. Kidney Int 59, 2243–2249. [DOI] [PubMed] [Google Scholar]
- Kremer JM, 2000. n-3 fatty acid supplements in rheumatoid arthritis. Am. J. Clin. Nutr 71, 349S–351S. [DOI] [PubMed] [Google Scholar]
- Le Moellic C, Boulkroun S, Gonzalez-Nunez D, Dublineau I, Cluzeaud F, Fay M, Blot-Chabaud M, Farman N, 2005. Aldosterone and tight junctions: modulation of claudin-4 phosphorylation in renal collecting duct cells. Am. J. Physiol. Cell Physiol 289, C1513–C1521. [DOI] [PubMed] [Google Scholar]
- Liano F, Pascual J, 1996. Epidemiology of acute renal failure: a prospective, multicenter, community-based study. Madrid Acute Renal Failure Study Group. Kidney Int 50, 811–818. [DOI] [PubMed] [Google Scholar]
- Liu SF, Malik AB, 2006. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol 290, L622–L645. [DOI] [PubMed] [Google Scholar]
- Lu CY, Hartono J, Senitko M, Chen J, 2007. The inflammatory response to ischemic acute kidney injury: a result of the ‘right stuff’ in the ‘wrong place’? Curr. Opin. Nephrol. Hypertens 16, 83–89. [DOI] [PubMed] [Google Scholar]
- Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, Levin A, Acute Kidney Injury N, 2007. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit. Care 11, R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meloche S, Pouyssegur J, 2007. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 26, 3227–3239. [DOI] [PubMed] [Google Scholar]
- Mitazaki S, Kato N, Suto M, Hiraiwa K, Abe S, 2009. Interleukin-6 deficiency accelerates cisplatin-induced acute renal failure but not systemic injury. Toxicology 265, 115–121. [DOI] [PubMed] [Google Scholar]
- Mitazaki S, Hashimoto M, Matsuhashi Y, Honma S, Suto M, Kato N, Nakagawasai O, Tan-No K, Hiraiwa K, Yoshida M, Abe S, 2013. Interleukin-6 modulates oxidative stress produced during the development of cisplatin nephrotoxicity. Life Sci 92, 694–700. [DOI] [PubMed] [Google Scholar]
- Naugler WE, Karin M, 2008. The wolf in sheep’s clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol. Med 14, 109–119. [DOI] [PubMed] [Google Scholar]
- Ricci Z, Ronco C, 2009. Pathogenesis of acute kidney injury during sepsis. Curr. Drug Targets 10, 1179–1183. [DOI] [PubMed] [Google Scholar]
- Russell JA, Singer J, Bernard GR, Wheeler A, Fulkerson W, Hudson L, Schein R, Summer W, Wright P, Walley KR, 2000. Changing pattern of organ dysfunction in early human sepsis is related to mortality. Crit. Care Med 28, 3405–3411. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, Moussignac RL, 2002. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med 196, 1025–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Chiang N, Van Dyke TE, 2008. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol 8, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvester W, Bellomo R, Cole L, 2001. Epidemiology, management, and outcome of severe acute renal failure of critical illness in Australia. Crit. Care Med 29, 1910–1915. [DOI] [PubMed] [Google Scholar]
- Smith CW, Entman ML, Lane CL, Beaudet AL, Ty TI, Youker K, Hawkins HK, Anderson DC, 1991. Adherence of neutrophils to canine cardiac myocytes in vitro is dependent on intercellular adhesion molecule-1. J. Clin. Invest 88, 1216–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spite M, Serhan CN, 2010. Novel lipid mediators promote resolution of acute inflammation: impact of aspirin and statins. Circ. Res 107, 1170–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YP, Oh SF, Uddin J, Yang R, Gotlinger K, Campbell E, Colgan SP, Petasis NA, Serhan CN, 2007. Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J. Biol. Chem 282, 9323–9334. [DOI] [PubMed] [Google Scholar]
- Takada M, Nadeau KC, Shaw GD, Marquette KA, Tilney NL, 1997. The cytokine-adhesion molecule cascade in ischemia/reperfusion injury of the rat kidney. Inhibition by a soluble P-selectin ligand. J. Clin. Invest 99, 2682–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe K, Matsushima-Nishiwaki R, Yamaguchi S, Iida H, Dohi S, Kozawa O, 2010. Mechanisms of tumor necrosis factor-alpha-induced interleukin-6 synthesis in glioma cells. J. Neuroinflammation 7, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teitelbaum JE, Allan Walker W, 2001. Review: the role of omega 3 fatty acids in intestinal inflammation. J. Nutr. Biochem 12, 21–32. [DOI] [PubMed] [Google Scholar]
- Thurman JM, 2007. Triggers of inflammation after renal ischemia/reperfusion. Clin. Immunol 123, 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari MM, Brock RW, Megyesi JK, Kaushal GP, Mayeux PR, 2005. Disruption of renal peritubular blood flow in lipopolysaccharide-induced renal failure: role of nitric oxide and caspases. Am. J. Physiol. Ren. Physiol 289, F1324–F1332. [DOI] [PubMed] [Google Scholar]
- Van Itallie C, Rahner C, Anderson JM, 2001. Regulated expression of claudin-4 decreases paracellular conductance through a selective decrease in sodium permeability. J. Clin. Invest 107, 1319–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Bansal S, Falk S, Ljubanovic D, Schrier R, 2009. Ghrelin protects mice against endotoxemia-induced acute kidney injury. Am. J. Physiol. Ren. Physiol 297, F1032–F1037. [DOI] [PubMed] [Google Scholar]
- Wang B, Gong X, Wan JY, Zhang L, Zhang Z, Li HZ, Min S, 2011. Resolvin D1 protects mice from LPS-induced acute lung injury. Pulm. Pharmacol. Ther 24, 434–441. [DOI] [PubMed] [Google Scholar]
- Wang S, Wei Q, Dong G, Dong Z, 2013. ERK-mediated suppression of cilia in cisplatin-induced tubular cell apoptosis and acute kidney injury. Biochim. Biophys. Acta 1832, 1582–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Liu MC, Liang M, Fu J, 2012. Sirt1 protects against thrombomodulin down-regulation and lung coagulation after particulate matter exposure. Blood 119, 2422–2429. [DOI] [PubMed] [Google Scholar]
- Yuan K, Huang C, Fox J, Gaid M, Weaver A, Li G, Singh BB, Gao H, Wu M, 2011. Elevated inflammatory response in caveolin-1-deficient mice with Pseudomonas aeruginosa infection is mediated by STAT3 protein and nuclear factor kappaB (NF-kappaB). J. Biol. Chem 286, 21814–21825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MZ, Yao B, Yang S, Jiang L, Wang S, Fan X, Yin H, Wong K, Miyazawa T, Chen J, Chang I, Singh A, Harris RC, 2012. CSF-1 signaling mediates recovery from acute kidney injury. J. Clin. Invest 122, 4519–4532. [DOI] [PMC free article] [PubMed] [Google Scholar]