

Abstract
The germinal center (GC) reaction is a coordinated and dynamic ensemble of cells and processes that mediate the maturation and selection of high-affinity GC B cells (GCBs) from lower affinity precursors and ultimately results in plasma (PC) and memory cell fates that exit the GC. It is of great interest to identify intrinsic and extrinsic factors that control the selection process. The transcription factor IRF4, induced upon BCR and CD40 signaling, is essential for the acquisition of PC and GCB cell fates. We hypothesized that beyond this early requirement, IRF4 continuously operates at later phases of the B cell response. We show that IRF4 is expressed in GCBs above levels seen in resting cells and plays a role in efficient selection of high-affinity GCBs. Halving Irf4 gene copy number in an antigen-specific murine B cell model, we found that antigen presentation, isotype switching, GC formation and zonation, somatic hypermutation rates, and proliferation were comparable to cells with a full Irf4 allelic complement. In contrast, Irf4 haploinsufficient GCBs exhibited impaired generation of high affinity cells. Mechanistically, we demonstrate sub-optimal Blimp-1 regulation amongst high affinity Irf4 haploinsufficient GCBs. Furthermore, in cotransfer settings, we observed a marked disadvantage of Irf4 haploinsufficient cells for GC entry, evidential of ineffective recruitment of T cell help. We propose that, analogous to its role in early GC entry, IRF4 continues to function in the late phase of the antibody response to promote productive Tfh interactions and to activate optimal Blimp-1 expression during GC selection and affinity maturation.
Introduction
T-dependent antibody responses generate differentiated plasma cell (PC), Germinal Center B cell (GCB), and memory B cell progeny from activated antigen specific clonal precursor B cells1. Critical cell intrinsic and extrinsic mechanisms allocate PC and GCB fates to control the quantity and composition, respectively, of antibody to mediate immune protection. IRF4 has emerged as a critical cell fate determinant of PC and GCB2–4. Specifically, the PC gene program promotes antibody secretion and thus antibody quantity. Alternatively, the GCB gene program promotes somatic hypermutation (SHM) of the antibody VDJ heavy chain and VJ light chain genes to generate structural variants of which a few are subsequently selected for, termed affinity maturation. Selected clones differentiate into PC and/or memory B cells and exit the germinal center (GC); clones with high affinity mutations are enriched in the PC compartment. Entry, selection, and exit of GCBs are coordinated by T follicular helper cells (Tfh)5.
Upon antigen recognition BCR signaling initiates mitogenesis, reprogrammed bioenergetics, and chemokine-dependent migration to the B-T zone border6. Subsequent antigen processing and presentation generates peptide-MHCII (pMHC) complexes that are presented to likewise antigen-activated cognate Tfh cells7. In fact, both BCR signal strength and pMHC density have been shown to limit B cell clonal entry into the GC8,9. These events are integrated during prolonged B-T interactions that involve adhesion10, stabilization11, and costimulation12. Productive cell conjugates lead to CD40:CD40L and ICOS:ICOSL signaling13, B cell proliferation and GCB differentiation.
The GC is classically composed of two zones – the light and the dark zones (LZ/DZ), defined by chemokines and guidance cues for zonal recirculation1,14. Interestingly, recent studies suggest greater complexity and have identified multiple distinct GCB populations within the DZ, including actively proliferating cells (gray zone, GZ/DZp), cells undergoing AID-dependent SHM (DZd), as well as others in the LZ15–17. Upon cessation of SHM, proliferation, and pMHC degradation18, clones migrate to the LZ to test mutated BCRs19–21. Competition for antigen on Follicular Dendritic Cells (FDCs), BCR signaling, and subsequent pMHC presentation to Tfh cells all play critical roles in selection of the fittest mutated clones. Interestingly, we note that this sequence of events parallel that of B-T conjugate interactions that initiate the GC response. If pMHC density is above a certain threshold, strong CD40 signals22,23 lead to sustained Foxo124–26, c-Myc27–29, AP4 transcription factor expression30 and DZ re-entry, proliferation, differentiation, and exit as high affinity PC or as memory B cells. In contrast, lower pMHC density causes DZ re-entry and additional mutagenesis before retesting in the LZ, termed cyclic re-entry21,31,32.
The IRF4 transcription factor, an immediate-early gene downstream of BCR signaling33, is cell autonomously essential for PC and GCB differentiation2–4,34. Mechanistic studies highlight the role of IRF4 levels in this process; low levels directly induce Bcl6 upregulation, GCB differentiation, and inhibition of PC fate whereas high levels directly induce Prdm1 (Blimp-1) upregulation, PC differentiation, and suppression of GCB fate2,4,35. Interestingly, GCBs with conditionally deleted Irf4 exhibit somewhat lower SHM, suggesting that IRF4 plays roles in GCBs beyond regulating Bcl6 and GCB fate3. Furthermore, tight regulation of IRF4 expression is important for normal GCB function, as elevated IRF4 protein results in precocious PC differentiation and decreased generation of high affinity cells36.
Given the mechanistic parallels between GC entry and GCB selection, and prior mechanistic studies that demonstrate the importance of IRF4 levels during priming, we hypothesized that IRF4 activity is limiting for efficient GC selection. Herein, we show that GCBs express a basal level of IRF4 and that IRF4 is required for efficient selection of high affinity GCBs in part by upregulating ideal levels of Blimp-1. Furthermore, Irf4 haploinsufficient cells are competitively disadvantaged for Tfh help and GC entry. We propose that efficient affinity maturation depends on optimal levels of IRF4 and Blimp-1 expression, driven by successful recruitment of Tfh help during competitive Tfh cell interactions.
Materials and Methods
Mice, immunizations and injections
In all experiments recipient mice were WT CD45.1+/+. Our colony of SWHEL mice37 were bred with Irf4−/− mice38 to generate desired genotypes. CD45.1+CD45.2+ Irf4+/+ cells were generated by breeding CD45.1+/+ with CD45.2+/+ SWHEL mice. Irf4+/+ or Irf4+/− SWHEL mice were bred to Prdm1:YFP mice39 and were CD45.2+/+. Matched genders of donor cells and host mice were used in the experiments and no effect of gender was observed. Mice were housed in specific pathogen-free conditions and were used and maintained in accordance with the Institutional Animal Care and Use Committee guidelines of UC Davis.
In all adoptive transfer experiments, numbers of SWHEL B cells from the spleen of donor mice were quantified by flow cytometry and a total spleen mixture containing 5×104 SWHEL cells were adoptively transferred with 2×108 sheep red blood cells (SRBC; Lampire Biological Laboratories) conjugated to either HELWT or HEL2X protein in PBS, i.v., as previously described37. For experiments analyzing the day 1 response, 5×105 SWHEL cells were adoptively transferred i.v., into mice immunized with 75 μg of HEL-IEα in Imject Alum at a 1:1 ratio with PBS (Thermo Fisher).
For S-phase determination, mice were injected with either 1mg of EdU diluted in PBS i.v., 4 hours prior or 2 mg of BrdU diluted in PBS i.v., 2 hours prior to splenic harvest.
HEL2X-IEα production
The HEL2X-IEα fusion gene was subcloned into a pRMHa3 copper-inducible expression vector, and this plasmid was used to stably transfect D. melanogaster S2 cells. Transfectants were identified by induction with copper sulfate and the presence of HEL2X-IEα in supernatants by western blotting using anti-HEL polyclonal antibodies (Rockland Immunochemicals). Secreted HEL-IEα was affinity purified on a nickel column (Bio-Rad NGC Quest 10 Chromatography System).
Flow cytometry
RBC-depleted splenic cell suspensions were prepared and washed in isotonic buffer40 containing sodium azide and 1% FBS (TC buffer) before extracellular staining in PBS containing sodium azide, 1% FBS FC buffer, Brilliant Stain Buffer (BD) and rat serum (2% v/v; Jackson ImmunoResearch). Detection of total SWHEL cells and high affinity HEL3X-binding cells was conducted as previously described by incubating cells with non-conjugated HEL antigens prior to detection with a fluorchrome-conjugated HyHel9 antibody37. For some experiments in the single transfer setting, host CD45.1+/+ cells were depleted using an immuno-magnetic bead approach consisting of CD45.1-biotin antibody (Biolegend), Streptavidin magnetic beads (Miltenyi), and MACS LS columns (Miltenyi) prior to HEL3X incubation. Detection of intracellular markers (such as IRF4) was performed by fixing and permeabilizing cells with Fix/Perm staining kit (Invitrogen). Flow cytometry was conducted on a BD Fortessa or BD Symphony and analysis performed using FlowJo software (Tree Star, Inc.). Detection of EdU was performed using the Click-iT EdU Plus kit from ThermoFisher (C10636) as per the manufacturer’s instructions. Detection of BrdU was performed using a BrdU Monoclonal Antibody from ThermoFisher in conjunction with a BrdU Permeabilization Buffer Plus kit and protocol from BD. To assess absolute cell number, AccuCount Blank Particles (Spherotech) were used.
Flow Cytometry Antibodies and Clones
B220 (RA3–6B2), Bcl6 (K112–91), CD4 (RM4–5), CD45.1 (A20), CD45.2 (104), CD86 (GL1), Fas/CD95 (Jo2), IgG1 (A85–1), Ly108 (13G3), PDL1/CD274 (M1H5), and Streptavidin were from BD. CCR6/CD196 (29–2L17), CD3 (145–2C11), CD38 (90), CD4 (GK1.5), CD40 (3/23), CD8 (53–6.7), CD80 (16–10A1), GL7 (GL7), ICOSL (HK5.3), IRF4 (3E4), MHCII:I-A/I-E (M5/114.15.2), PDL2/CD273 (TY25), and SLAM/CD150 (TC15–12F12.2) were from BioLegend. CXCR4 (2B11/CXCR4), ICAM (YN1/1.7.4), and YAe/Ea 52–68 (eBioY-Ae) were from eBioscience. BrdU (MoBU-1) and Active Caspase-3 (C92–605) were from Thermo Fisher. HyHel9, specific for HEL (UCSF Monoclonal Antibody Core), was conjugated in-house using an Alexa Fluor 647 Antibody Labeling Kit (Thermo Fisher).
VDJ Sequencing
Post-immunization, GC-resident HELWT-specific or HEL3X-specific donor B cells were individually sorted into a 96-well plate containing digest buffer (100 μl 10x Taq PCR reaction buffer (no MgCl2), 50 μl Proteinase K (10 mg/ml in H2O), 10 μl 10 mM EDTA, 10 μl Tween 20 (10% solution), in final volume 1 ml H2O,37 using a Astrios EQ high speed cell sorter (Beckman Coulter). The VDJ region of single sorted SWHEL B cells was amplified by PCR and Sanger-sequenced at the UC Davis DNA Technologies and Expression Analysis Core on an ABI Prism 3730 Genetic Analyzer with ABP Prism 3730 Data collection software (v3.0) and ABI Prism DNA Sequencing Analysis Software (v5.2). Chromatogram analysis was performed on Chromas software (v2.6.4, Technelysium Pty Ltd), and multiple alignment and mutation analysis of sequences was conducted using UGENE software (v1.28, Unipro) and Excel (Microsoft).
Cell preparation for scRNA-seq
For multi cell sorting prior to scRNA-sequencing, cells were stained as described in the flow cytometry section. Two CD45.1 mice were cotransferred with a 1:3 mixture of CD45.1+CD45.2+ Irf4+/+ and CD45.2+ Irf4+/− SWHEL cells and immunized with HEL2X-SRBC. On day 10 of the response, CD45.1 expression was distinguished using CD45.1-biotin and TotalSeq-A0951 PE Streptavidin (Biolegend) and equivalent numbers of splenic CD45.1+CD45.2+ Irf4+/+ and CD45.2+ Irf4+/− FashiCD38lo HEL-specific donor B cells were bulk sorted into a single polypropylene 5mL tube containing PBS using an Astrios EQ high speed cell sorter (Beckman Coulter). Viability was confirmed using a Countess (ThermoFisher) prior to loading onto the 10X Genomics Chromium fluidic system using 10X Chromium Single Cell 3’ GEM, Library & Gel Bead Kit v3 for single cell RNA extraction and processing. Sequencing was performed on a NovaSeq instrument (Illumina).
Informatics for scRNA-seq
Raw fastq files were processed with the 10x Genomics software Cell Ranger v. 3.1.041 to perform alignment, filtering, barcode demultiplexing, and counting of UMI (unique molecular indices), using the 10X Genomics supplied mm10–3.0.0 genome and annotation. The CD45.2 (Irf4+/−) and CD45.1/CD45.2 (Irf4+/+) identity of the cells in the population was determined based on the frequency of the CITE-seq-encoded anti-CD45.1 biotin-streptavidin-PE-barcode (See Supplementary Fig. 2E). The two genotypes were separated by defining the breakpoint between distributions. Preprocessing, clustering, and visualization of single-cell gene counts was conducted using the R package Seurat, version 3.1.442, in R version 3.6.1 (https://www.r-project.org/). Sequence counts from both experiments were pooled, log transformed, normalized, and batch corrected, then clustering was performed and t-SNE dimension-reduction coordinates were calculated on the top 100 principal components of the normalized, batch-corrected data. Identification of cluster markers was conducted using Wilcoxon rank sum tests as implemented in the Seurat function FindAllMarkers, and differential expression between groups was similarly conducted using Wilcoxon rank sum tests as implemented in the Seurat function FindMarkers. Enrichment analyses were conducted by using Fisher’s exact test to compare the proportion of genes in each custom set that was expressed in more than 50% of cells of a given cluster to what would be expected under random chance. Signature gene lists reported in Victora et al.43 Supplementary Table 4e (derived from GSE38696) were used for cluster enrichment of LZ_up, DZ_up, LMP1_up, CD40_up, Myc_up, and Mitosis. Memory signature enrichment analysis used GSE89897 from Laidlaw et al.44. The scRNA-seq data has been deposited at Gene Expression Omnibus with the GSE185188 identifier.
In vitro stimulation of B cells
Spleens of CD45.1+ CD45.2+ Irf4+/+ or CD45.2+/+ Irf4+/− mice were processed and proportion of B cells determined by flow cytometry. Equivalent numbers of Irf4+/+ or Irf4+/− B cells were then added to a 96-well plate and B cells were cultured in RPMI medium (10% vol/vol FCS, 2 mM glutamine, 100 IU/mL penicillin, 0.1 mg/mL streptomycin, 20 mM HEPES buffer (all from Invitrogen), 1 mM Sodium Pyruvate, 1 mM nonessential amino acids (Gibco), and 2 μM β-mercaptoethanol (Invitrogen)) and stimulated with IgD dextran (0.2 μg/ml, Fina Biosolutions, LLC).
In vitro HEL2X-IEα presentation
After B cell enrichment by MACs (Miltenyi), equal numbers of Irf4+/+ or Irf4−/− cells were co-cultured and number-matched to ensure each genotype had 37,500 SWHEL cells seeing the antigen. Irf4+/+ cells were labeled with CFSE to differentiate from Irf4-/−. Cells were stimulated with an 11x titration series of HEL2X-IEα, from 5 μg to 0.04 μg (and a 0 μg control) or HEL (0.2 μg) and incubated in 96 well flat bottom plates for 24 hours.
Statistical Analysis
Statistical analyses of parametric unpaired t-tests, paired t-tests, and Mann-Whitney U tests were performed using Prism software.
Results
Development of Irf4+/− SWHEL B cell system.
Expression levels of IRF4 regulate activated antigen specific B cell fate choices. Given the requirement for IRF4 during antigen specific priming and GC formation as well as the observation of diminished mutation frequency in Irf4-conditionally deleted polyclonal GCBs3, we hypothesized that IRF4 haploinsufficiency in responding B cells would compromise affinity maturation in the GC. We utilized a previously published VDJ knock-in mouse model (SWHEL) in which B cells express the VDJ from the HyHEL10 mAb, specific for hen egg lysozyme (HEL). In this system, small numbers of CD45.2+ SWHEL cells are adoptively transferred into CD45.1 mice, which upon immunization with HEL conjugated to SRBC (HEL-SRBC), antigen-specific B cell responses can be tracked and functionally interrogated37. SWHEL B cells are identified by flow cytometry using the CD45 congenic system as well as binding the HELWT antigen. In these experiments, we used an engineered form of HEL (HEL2X) composed of 2 amino acid substitutions, R73E and D101R, that decrease its affinity for the HyHEL10 mAb by ~240 fold compared to wild type HEL45. SWHEL mice were bred to mice bearing the Irf4 null allele to generate SWHEL Irf4+/+ and Irf4+/− mice that are CD45.2+. Advantageously, this monoclonal system enables the analysis of specific gene mutations on B cell fate and function during the antibody response without the confounder of cell competition in polyclonal systems.
Irf4+/− SWHEL B cells form normal GCs.
Either Irf4+/+ or Irf4+/− SWHEL B cells were adoptively transferred into CD45.1+ congenic hosts and immunized with HEL2X-SRBC (Fig. 1A). SWHEL B cells activated and expanded well regardless of IRF4 gene copy number and sustained the response at intermediate (day 10) and late (day 15) timepoints, however, there were fewer Irf4+/− SWHEL cells at day 5 which corresponded to a decrement of PC frequencies as determined by downregulation of B22045 (Fig. 1B, Supplementary Fig. 1A). This observation was corroborated using a BAC Prdm1:YFP reporter approach (see below and Fig. 6A). In addition, class switch recombination (CSR) to IgG1 was equally unaffected (Fig. 1C). Furthermore, GCB frequencies and numbers were unaffected as a function of Irf4 gene copy number, regardless of whether they were quantitated by Fas+CD38-, Fas+GL7+ (Fig. 1D, Supplementary Fig. 1B), or by expression of the GC master transcription factor, Bcl6 (Fig. 1E). Due to the important roles of the DZ, LZ, and GZ in affinity maturation, we analyzed whether halving Irf4 gene copy number affected these cell states. Irf4+/− SWHEL GCBs exhibited comparable DZ, GZ, and LZ frequencies to wild type SWHEL GCBs (Fig. 1F). Additionally, the proportions of DZ, GZ, and LZ antigen-specific cells in S phase, as measured by EdU incorporation, were indistinguishable (Fig. 1G). Finally, the expression of levels of key receptors that mediate GC entry and biology appear unchanged between the two genotypes (Supplementary Fig. 1C). These results indicate that to all appearances, Irf4 hemizygosity does not affect the ability of antigen specific B cells to differentiate into GCBs nor maintain the GC cell state over time.
Figure 1: Irf4+/− SWHEL GCB differentiation and function are normal.
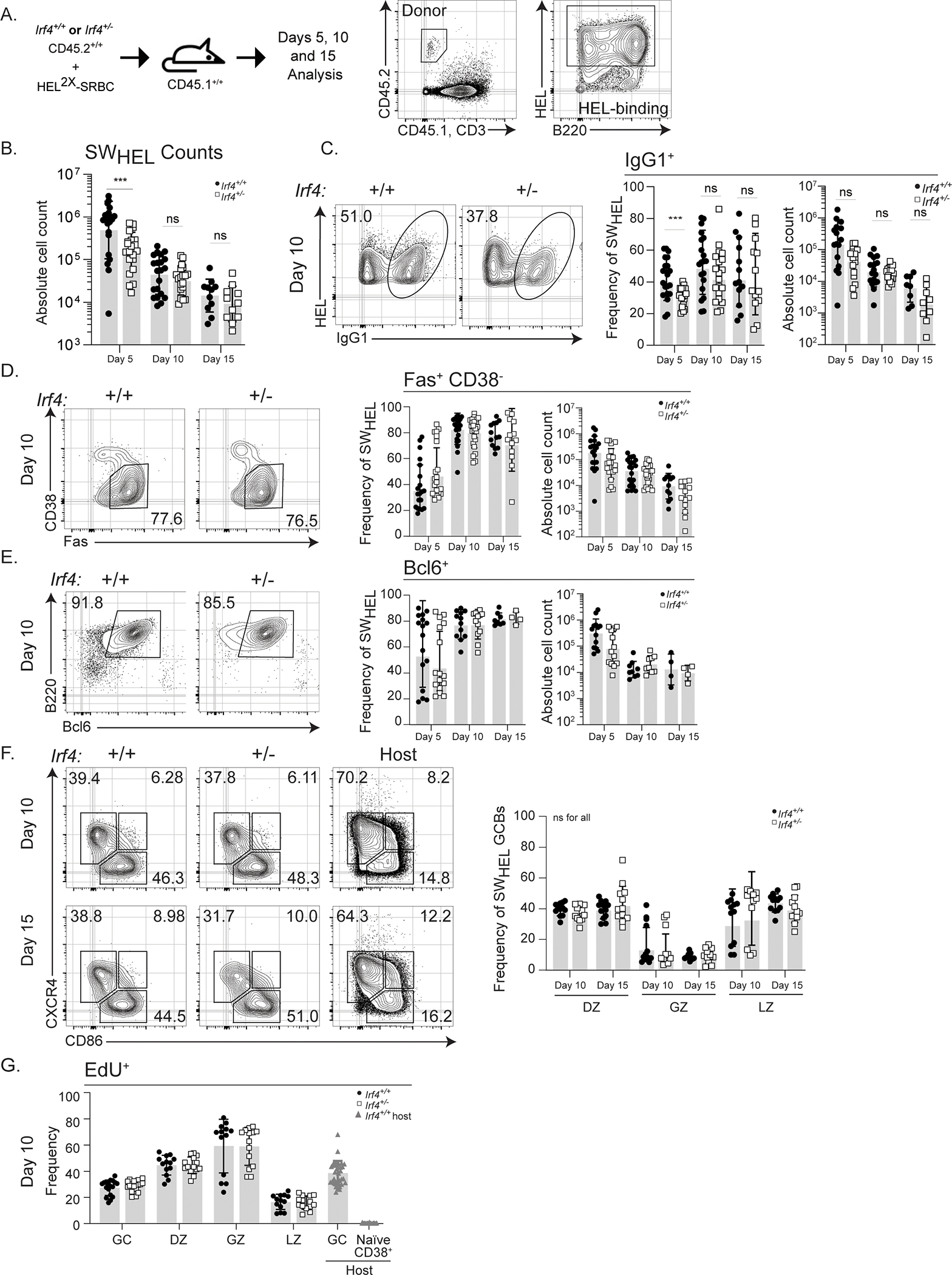
A) Schematic of experimental design and SWHEL gating strategy. 50,000 CD45.2+ Irf4+/+ or Irf4+/− SWHEL B cells were mixed with HEL2X-SRBC and adoptively transferred i.v. into CD45.1+ hosts. Spleens were harvested at days 5, 10, or 15 for analysis. Antigen-specific responding donor B cells were identified as CD45.2+CD45.1-CD3-B220+HELWT+. Flow plots represent sequential gating strategy. B) Absolute numbers of Irf4+/+ or Irf4+/− SWHEL B cells, days 5, 10, and 15 post immunization. C-E) Representative flow cytometry plots, frequency, and absolute cell counts of C) IgG1-switched, D) CD38loFas, E) BCL6+, Irf4+/+ or Irf4+/−SWHEL B cells at the indicated time points after immunization. F) Representative flow cytometry plots and frequency of CD38loFas+ DZ (CXCR4+CD86-), GZ (CXCR4+CD86+), or LZ (CXCR4-CD86+) Irf4+/+ or Irf4+/−SWHEL GCBs at the indicated times after immunization. G) Frequency of EdU incorporation after a 4-hour pulse in Irf4+/+ or Irf4+/− SWHEL GCBs within the DZ, GZ, and LZ sub-compartments gated as in F). B-G) Bar graphs represent mean ± SD with dots representing individual mice. Solid circle, Irf4+/+ and open square, Irf4+/− SWHEL cells. Experiments in B–F are from 16 mice in 4 experiments performed while G is from 18 mice in 4 experiments performed; contour plots are concatenated files from all mice of a given group in a given experiment. Unpaired t tests were performed to determine significance. *** p ≤ 0.001; ns, not significant (p > 0.05)
Figure 6: Irf4 haploinsufficiency results in suboptimal Blimp-1 expression in high affinity cells.
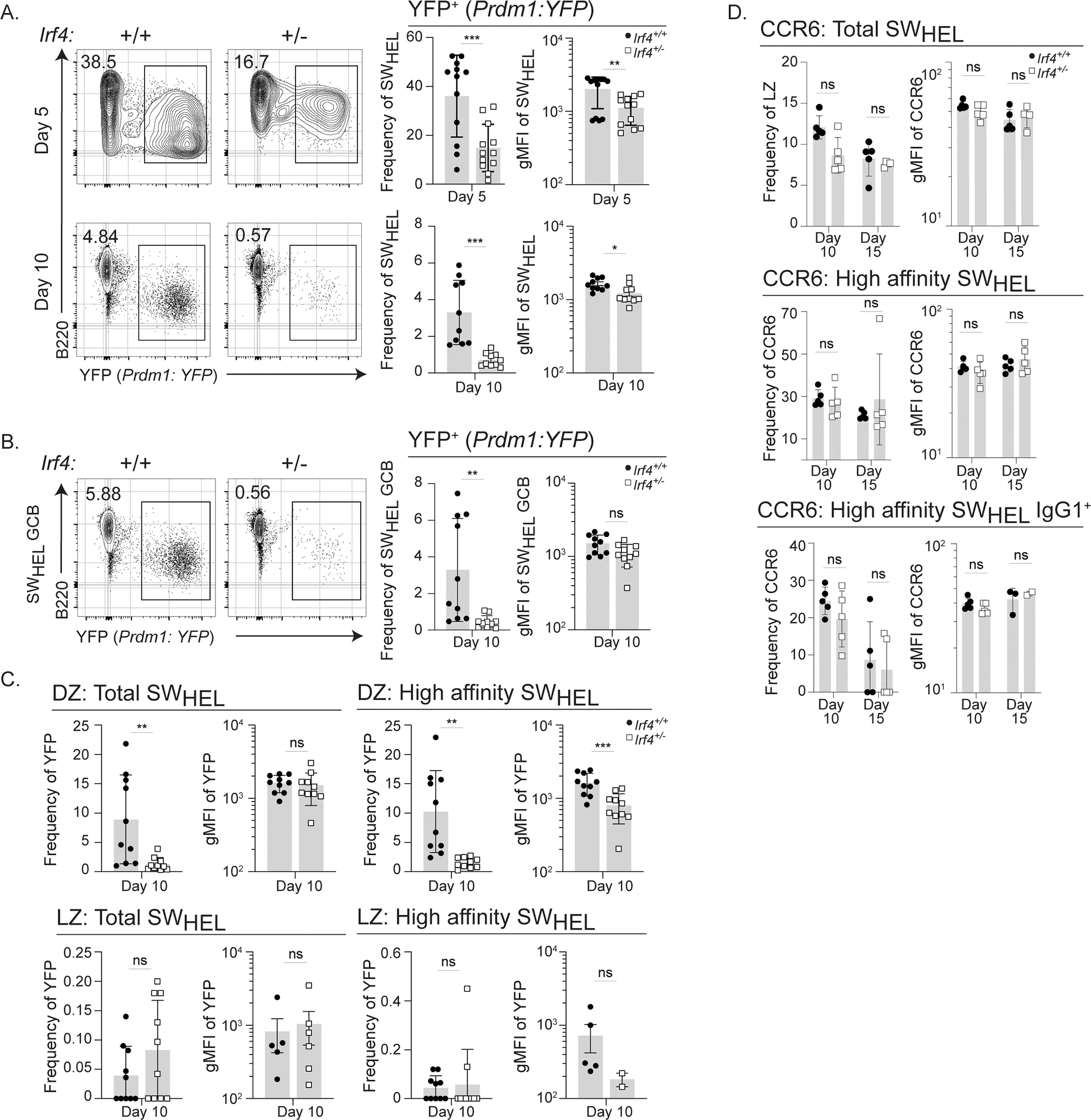
A-D) Cells from Irf4+/+ or Irf4+/− Prdm1:YFP SWHEL mice were transferred into recipient mice and immunized as in Fig. 1A prior to harvesting splenocytes at days 5 or 10 for analysis. Donor-specific responding B cells were identified as CD45.2+CD45.1-CD3-B220+HELWT+. A-B) Representative flow cytometry plots, frequency, and gMFI of YFP expression on A) total SWHEL or B) total SWHEL GCBs, identified by CD38loFas+. C-D) Frequency and gMFI of YFP expression in C) total SWHEL GCB DZ or LZ subsets or high affinity SWHEL GCB DZ or LZ subsets (gated as in Fig. 1F). D) Cells from Irf4+/+ or Irf4+/− SWHEL mice were transferred into recipient mice and immunized as in Fig. 1A prior to harvesting splenocytes at days 10 or 15 for analysis. Total SWHEL, high affinity SWHEL, or IgG1+ high affinity SWHEL cells were gated for LZ GCBs as in Fig. 1F and the frequency and gMFI of CCR6 is depicted. Bar graphs represent mean ± SD or mean ± SEM (gMFI graphs) with dots representing individual mice. Experiments in A are from 17 mice in 4 experiments performed, experiments in B-C are from 10 mice in two experiments performed, experiment in D is from 10 mice in 1 experiment performed; contour plots are concatenated files from all mice of a given group in a given experiment. Unpaired t tests were performed to determine significance. ** p ≤ 0.01, *** p ≤ 0.001, ns, not significant (p > 0.05).
GCBs express low levels of IRF4.
These initial findings were surprising and prompted us to confirm a reduction in IRF4 levels in Irf4+/− SWHEL cells and GCBs. For this quantitative analysis, we co-transferred equal numbers of CD45.1+CD45.2+ Irf4+/+ and CD45.2+ Irf4+/− SWHEL B cells into CD45.1+ hosts (Fig. 2A). Flow cytometric analysis of IRF4 protein expression at days 5 and 10 revealed a consistent 10–20% reduction in IRF4 gMFI in total SWHEL cells as well as Bcl6-expressing GCB Irf4+/− HELWT-specific B cells compared to that of Irf4+/+ (Fig. 2B). Importantly, IRF4 expression in host resting B cells was lower than activated Irf4+/− SWHEL cells, demonstrating that the Irf4+/− SWHEL cells experienced BCR-dependent IRF4 upregulation (Fig. 2B). Polyclonal host GCBs, which are Irf4+/+, also expressed higher levels of IRF4 compared to the Irf4+/− SWHEL GCBs. Further, BCR stimulation in vitro using an anti-IgD dextran agonistic reagent, resulted in roughly two-fold less induced IRF4 protein expression in Irf4+/− compared to Irf4+/+ cells (Supplementary Fig. 1D). Together, these results demonstrate that Irf4+/− SWHEL GCBs undergo BCR-dependent IRF4 induction that is reduced in an Irf4 gene copy-dependent manner.
Figure 2: IRF4 expression in hemizygous cells and in GCBs.
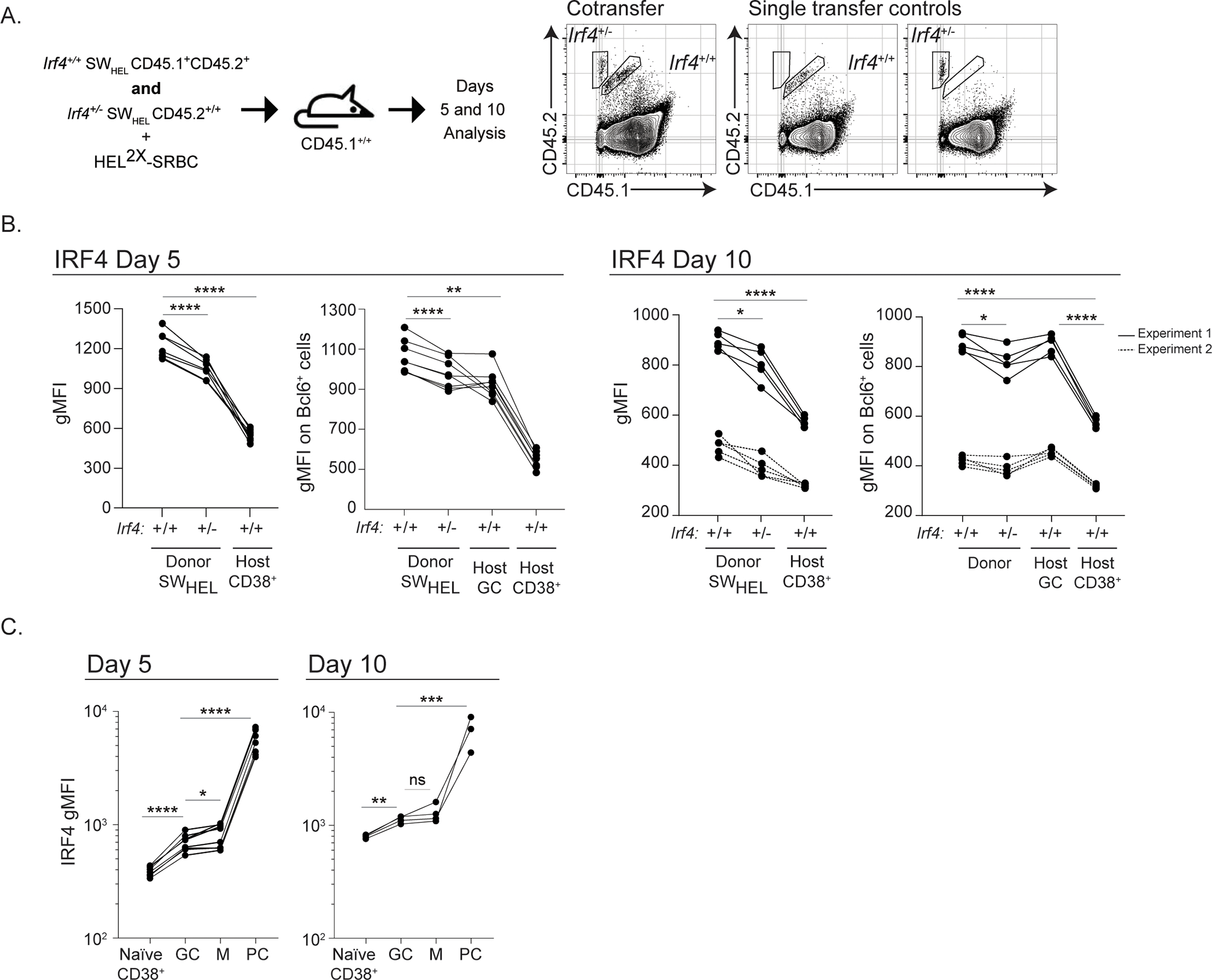
A) Schematic of experimental design and donor gating strategy. Irf4+/+ (CD45.1+CD45.2+) and Irf4+/− (CD45.2+) SWHEL cells were transferred at a ratio of 1:1 (total 50,000 cells), mixed with HEL2X-SRBC, and adoptively transferred i.v. into CD45.1+ hosts. Spleens were harvested at days 5 or 10 for analysis. Representative gating strategy is shown to identify Irf4+/+ and Irf4+/− SWHEL responders by allotypic markers. Identification of B220+HELWT+ cells is performed as in Fig. 1A. B) IRF4 geometric mean fluorescence intensity (gMFI) of total SWHEL cells (left) or BCL6+ GCB SWHEL cells (host CD38+ are not Bcl6+) (right) at the indicated time points after immunization. Lines linking dots denote measurements from a single mouse. At day 10, solid line indicates gMFI values from experiment 1, dotted line indicates gMFI values from experiment 2. C) IRF4 gMFI in Irf4+/+ total SWHEL B cell subsets at the indicated time points after immunization. Naïve host CD38+ B cells (CD45.2-CD45.1+B220+CD38+Fas-), GC: SWHEL GCBs (CD45.2+CD45.1-B220+HEL+CD38loFas+), M: SWHEL memory B cells (CD45.2+CD45.1-B220+HEL+CD38hi), PC: SWHEL PCs (CD45.2+CD45.1-B220loHEL+CD38+). Lines linking dots denote measurements from a single mouse. Experiments in B (Day 5) and C are from 5 mice in 1 experiment performed, while experiments in B (Day 10) are from 8 mice in 2 experiments performed. Paired t tests were performed to determine significance. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001, ns, not significant (p > 0.05)
In these experiments, we noticed that basal IRF4 expression in Irf4+/+ SWHEL GCBs was higher than that of CD38+ resting B cells. Because this contradicted earlier results measured using immunohistochemistry46, we quantified IRF4 expression as a function of resting CD38+ B cells, GCB, memory, and PC cell states by flow cytometry (Fig. 2C). As expected, PCs expressed the highest levels of IRF4 protein, ≥10-fold higher than that of resting B cells. GCBs express twice as much IRF4 protein compared to resting B cells. Together, these data indicate that GCBs express IRF4; this new basal level is greater than that of resting B cells but less than that of PCs.
Irf4 haploinsufficiency impairs affinity maturation.
To test the functionality of affinity maturation in Irf4+/− GCBs, we analyzed the generation of high affinity cells using a system developed by the Brink lab37. Specifically, immunization with low affinity HEL drives the formation of GCBs exhibiting a canonical SHM pattern that can be enumerated by flow cytometry by virtue of binding to sub-saturating amounts of HEL3X. HEL3X contains an additional amino acid substitution (R21Q) compared to HEL2X, which results in a ~40 fold diminished affinity compared to HEL2X to the parental unmutated SWHEL BCR45,47. Using this system, analysis of Irf4+/+ SWHEL cells at day 10 revealed that approximately 30% of IgG1-switched GCBs bound HEL3X (Fig. 3A). In contrast, analysis of similarly treated Irf4+/− SWHEL cells resulted in roughly 10% of IgG1-switched GCBs binding HEL3X (Fig. 3A). We confirmed specificity of HEL3X binding using a separate cohort of mice that were immunized with HELWT and known to not develop47 GCBs capable of binding subsaturating amounts of HEL3X. The reduction in HEL3X-binding cells was not specific to IgG1-switched B cells because analysis of total B220+ SWHEL GCBs that contain other isotypes revealed a similar decrement (Fig. 3B). We asked whether this reduction was due to reduced BCR abundance; however, based on HELWT- or anti-BCR staining we saw no difference in SWHEL BCR gMFI on GCBs at day 10 or 15 (Supplementary Fig. 1E). At day 15, proportions of HEL3X-binding GCBs increase. Interestingly, we observed comparable HEL3X-binding frequencies between Irf4+/+ and Irf4+/− -switched or total SWHEL GCBs at this later time point. Despite compromised early affinity maturation, the ratio of LZ:DZ populations among high affinity cells was equivalent in Irf4+/+ and Irf4+/− at both days 10 and 15 (Fig. 3C). Further, the defect in efficient affinity maturation at day 10 was not due to cell proliferative capacity; comparable proportions of Irf4+/+ and Irf4+/− high affinity cells were in S-phase in all GC compartments (Fig. 3D). Together, these results demonstrate delayed affinity maturation efficiency in Irf4 haploinsufficient GCBs.
Figure 3: Irf4 haploinsufficiency impairs affinity maturation.
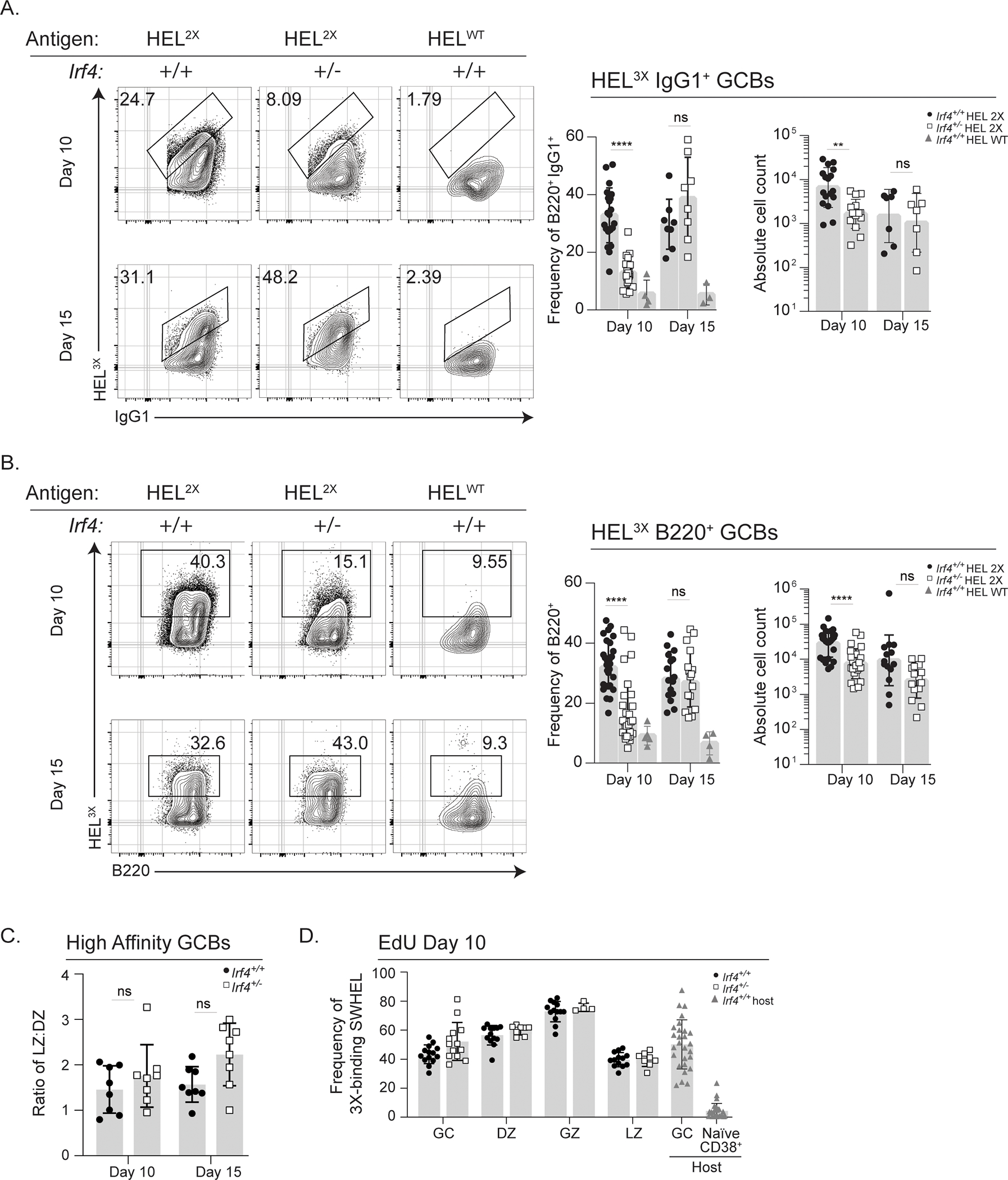
Irf4+/+ or Irf4+/− SWHEL B cells were transferred into recipient mice and immunized as in Fig. 1A prior to harvesting splenocytes at days 10 or 15 for analysis. A-D) Donor-specific SWHEL GCBs were identified as CD45.2+CD45.1-CD3-B220+CD38loFashi or CD45.2+CD45.1-CD3-B220+CD38loFashiIgG1+ and high affinity cells as those that bound subsaturating amounts of HEL3X. A-B) Mice immunized with either HEL2X-SRBC or HELWT-SRBC and representative flow cytometry plots, frequency, and absolute cell counts of A) IgG1+, and B) B220+ high affinity SWHEL B cells were assessed. C) Ratio of high affinity Irf4+/+ or Irf4+/− in the LZ and DZ compartments, identified as in Fig. 1F. D) Frequency of EdU incorporation after a 4-hour pulse in high affinity SWHEL GCB subsets. A-D) Bar graphs represent mean ± SD with dots representing individual mice. Experiments in A–C are from 15 mice in 3 experiments performed, and experiment D is from 14 mice in 3 experiments performed; contour plots are concatenated files from all mice of a given group in a given experiment. Unpaired t tests were performed to determine significance. ** p ≤ 0.01, **** p ≤ 0.0001, ns, not significant (p > 0.05)
Irf4 hemizygous GCBs exhibit comparable rates of SHM.
The Aicda gene, which encodes the AID protein, has been shown to be regulated by IRF4 activity2,3. To determine whether attenuated affinity maturation in Irf4+/− GCBs is due to issues with AID-induced SHM, single SWHEL GCBs were sorted on days 10 and 15 and the heavy chain VDJ region was sequenced. For both genotypes, the number of DNA mutations increases over time, with an average of 3 mutations/cell on day 10, to 5 mutations/cell on day 15, indicating comparable rates of SHM (Fig. 4A). In both Irf4+/+ and Irf4+/− GCBs, mutations are dispersed across the entirety of the VDJ region and the vast majority of mutations are within AID hotspots (Supplementary Fig. 2A and not shown), confirming equivalent AID activity. Furthermore, the DNA mutations were not silent because similar proportions of amino acid substitutions per cell were observed (Fig. 4B). Despite these findings, the proportions of cells with the canonical high affinity mutations47, Y53D and/or Y58F, are reduced in Irf4+/− GCBs on day 10 (Fig. 4C) and are consistent with diminished HEL3X-binding frequencies (Fig. 3A–B). In contrast, by day 15, proportions of high affinity mutations are similar between genotypes, consistent with HEL3X-binding frequencies (Fig. 3A–B). In addition to sorting and sequencing from total SWHEL, we sorted and sequenced from the HEL3X-binding populations to enrich for somatically mutated cells and observed the same trends (Fig. 4D–F). We note that, in some samples, the enrichment was somewhat inefficient, likely due to the position of the gate during the sort. Together, we observe impaired generation of high affinity mutations in Irf4+/− SWHEL GCBs at early time points despite efficient rates of SHM.
Figure 4: Irf4 hemizygous GCBs exhibit comparable rates of SHM.
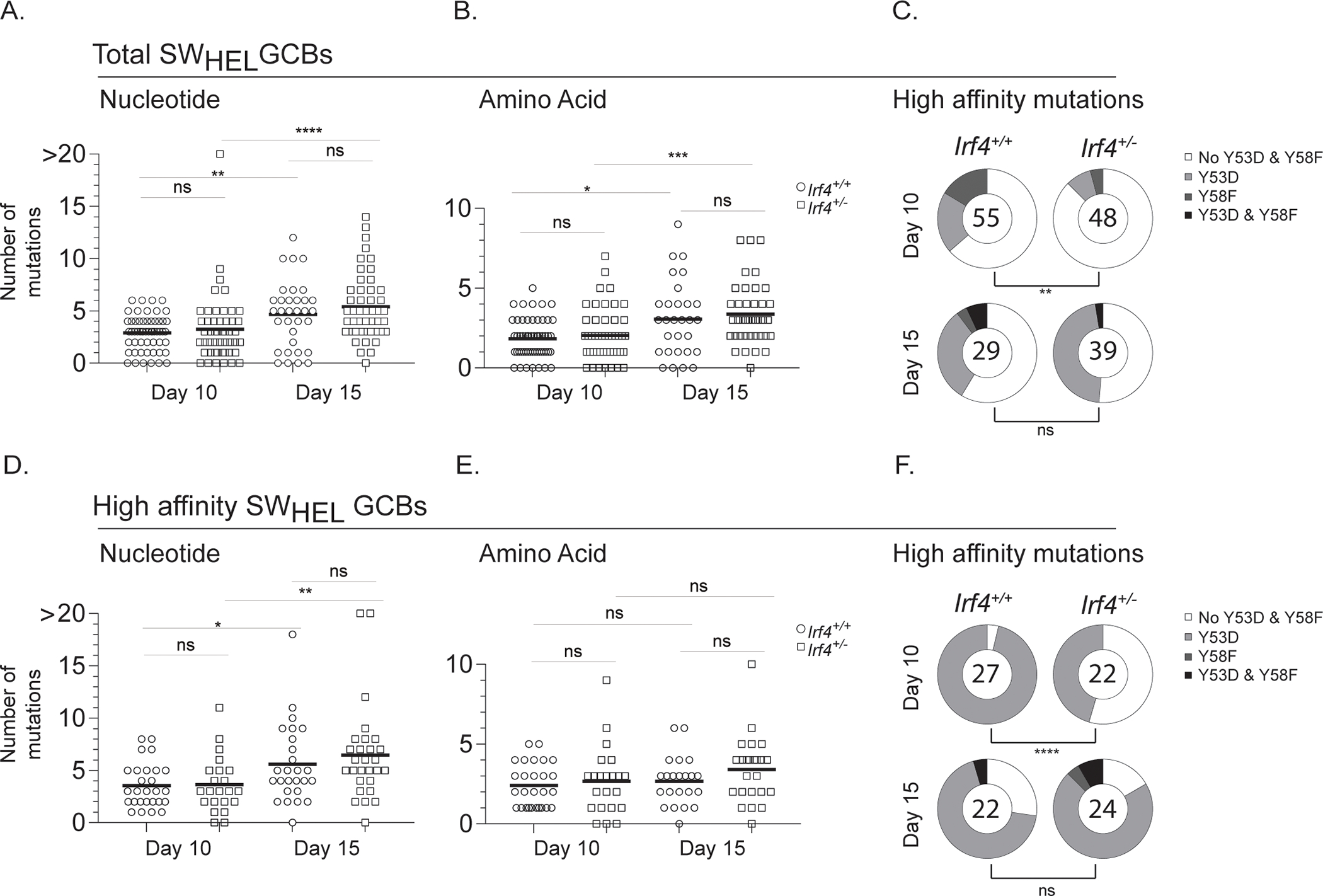
A-F) Cells were transferred into 3 recipient mice per group and immunized as in Fig. 1A prior to harvesting splenocytes at days 10 or 15 for sorting. Individual SWHEL GCB clones were sorted by FACS on days 10 and 15 post immunization and the heavy chain VDJ was sequenced. A-C) Total SWHEL donor-specific responding GCBs were identified as CD45.2+CD45.1-CD3-B220+HELWT+CD38loFas+. A) Number of DNA mutations per clone, B) Number of amino acid mutations per clone, and C) Proportion of clones with high affinity mutations Y53D and Y58F, number in pie represents total number of clones sequenced. D-F) Analysis as in A-C for GCBs that bind subsaturating amounts of HEL3X (high affinity cells). A,B,D,E) Each dot represents an individual clone, with the median indicated as a black line. Mann-Whitney U tests were performed to determine significance. C,F) Fisher’s exact tests were performed to determine significance; analysis based on total high affinity mutations. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001, ns, not significant (p > 0.05).
GC subpopulations are intact despite Irf4 haploinsufficiency.
Cumulatively, the impaired affinity maturation of Irf4+/− SWHEL cells cannot be accounted by GCB phenotype, rates of SHM, or apparent proliferative abilities. Recent observations suggest deeper divisions in GCB cell subset identity beyond the foundational LZ/DZ paradigm15–17, raising the possibility that Irf4+/− SWHEL B cell state transitions contribute to impaired affinity maturation. To address this, we performed droplet-based scRNA-seq of Irf4+/+ and Irf4+/− SWHEL GCBs. To enable a direct comparison of cells within the same GC microenvironments, we co-transferred cells of each genotype into the same host in a 3:1 ratio of Irf4+/− to Irf4+/+ SWHEL B cells; the results of the cotransfer are described in more detail in a following section. Equal numbers of Irf4+/+ and Irf4+/− SWHEL GCBs from 2 mice were separately sorted and processed into two scRNA-seq runs. Viable cells were processed on the 10x Genomics Chromium platform and RNA-seq libraries were generated for a total of 8360 cells comprised of GCBs of both genotypes. We utilized CITE-seq, Cellular Indexing of Transcriptomes and Epitopes by Sequencing, to resolve the SWHEL cells of the respective genotypes that differed in expression of CD45.1 (see methods)48. An average of 139,000 reads/cell and 900 genes/cell were identified from both experiments. Importantly, sequence counts of the CITE-seq barcode yielded a bi-modal distribution clearly separating cells from each genotype (Supplementary Fig. 2E). Sequence counts from both experiments were pooled, log transformed, normalized, and batch corrected, then clustering was performed and t-SNE dimension-reduction coordinates were calculated on the top 100 principal components of the normalized, batch-corrected data (Fig. 5A). While 10 cell clusters were initially identified, three were removed because they represented T, myeloid/dendritic cell, and non-MHCII expressing lineages (data not shown); the genes distinguishing each cluster are depicted in Supplementary Figure 3A. Within the remaining 7 clusters, there was comparable representation of cells from each genotype except for cluster 4, which comprised of 15% Irf4+/− cells (Fig. 5B). Cells were also projected as a function of the cell cycle and consistent with the EdU metabolic labelling analysis, we observed equal representation of cells of each genotype in each cell cycle phase (Supplementary Fig. 3B). Gene set enrichment analysis was used to assign cell state identity of each cluster. Specifically, Fisher’s exact test was used to compare the proportion of genes in each custom set that was expressed in more than 50% of cells of a given cluster to what would be expected under random chance (Fig. 5C). As with recently published analyses, this resulted in the resolution of multiple GCB subsets that corresponded to LZ, DZ, GZ, and LZ-like populations. Differential gene expression within each cluster was next used to determine whether the cell state was differentially programmed by Irf4 haploinsufficiency. This analysis, at the sequencing depth attained, retrieved few significant differentially expressed genes not known to be relevant for GC biology and only in clusters 0,1,2, and 4 (Supplementary Table I). Together, this analysis suggests that impaired affinity maturation of Irf4 haploinsufficient cells is not attributable to disadvantaged representation of GCB cell subcompartments, except perhaps a pre-memory compartment (cluster 4).
Figure 5: GC subpopulations are intact despite Irf4 haploinsufficiency.
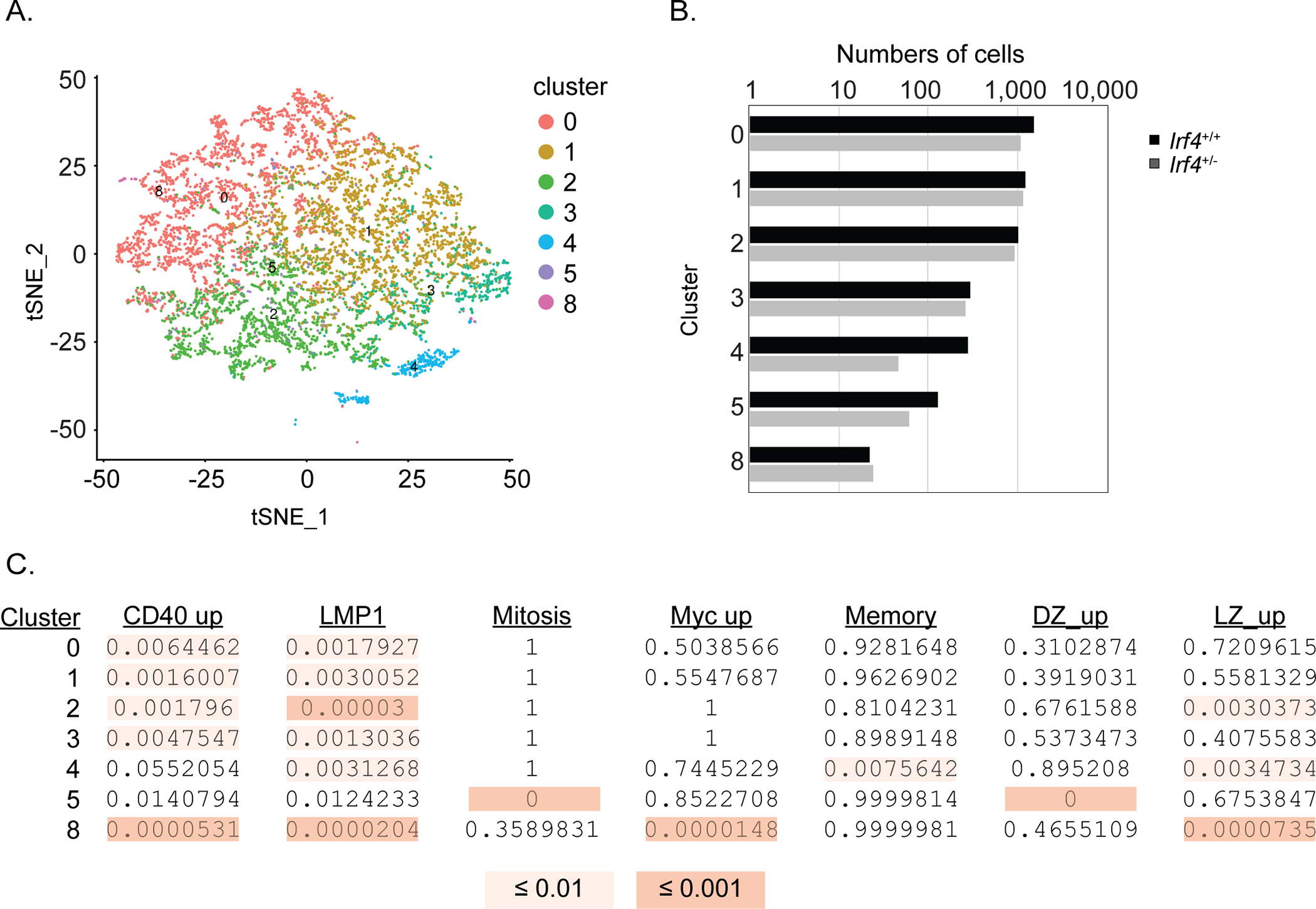
Irf4+/+ or Irf4+/− SWHEL B cells were co-transferred into recipient mice at a 1:3 ratio and immunized as in Fig. 2A prior to sorting GCBs at day 10 (identified as in Fig. 1D) and droplet-based scRNA-seq analysis of 8360 cells in two separate adoptive transfers. CITE-seq using a barcoded Streptavidin-PE reagent bound to anti-CD45.1 biotin was used to discriminate the Irf4+/+ and Irf4+/− genotypes among sequenced cells (see Methods). A) t-SNE dimension-reduction coordinates were calculated on the top 100 principal components of the normalized, batch-corrected data. B) Number of cells in each cluster that are Irf4+/+ (black bars) or Irf4+/− (gray bars). C) Enrichment of indicated gene signatures within each cluster calculated using Fisher’s exact test.
Irf4 haploinsufficiency results in suboptimal Blimp-1 expression in high affinity cells
To determine the basis for impaired affinity maturation in Irf4+/− GCBs, we sought to determine whether the high affinity cells exhibited diminished proportions of presumptive pre-PC49,50. To identify pre-PC cells, we utilized Prdm1:YFP mice39 that encode a YFP-modified BAC allele of genomic Prdm1 (the gene encoding the PC fate determinant, Blimp-1) and generated SWHEL Blimp-1YFP reporter mice with either half or the full complement of Irf4 gene copies. As previously shown, expression of YFP indicates activation of Blimp-1 expression and marks cells with a PC and pre-PC phenotype. At the peak of the extrafollicular PC response, day 5, Irf4+/− SWHEL cells generated 2–3 fold fewer YFP-expressing PCs compared to Irf4+/+ SWHEL cells (Fig. 6A) as previously predicted. By day 10, the frequency of YFP+ cells markedly contracted (Fig. 6A). Interestingly, the intensity of YFP expression, as measured by geometric mean fluorescence intensity (gMFI), was decreased amongst Irf4+/− PCs compared to Irf4+/+ counterparts at day 5 (Fig. 6A). To specifically quantify newly selected YFP+ pre-PCs49,50, we gated on SWHEL GCBs and in the zonal compartments (Fig. 6B–C). Similar to what was observed in total SWHEL B cells at days 5 and 10, we observed a marked decrease in Irf4+/− YFP+ SWHEL GCBs (Fig. 6B). As previously shown, the YFP+ GCBs of both genotypes were enriched in the DZ compartment and rare in the LZ (Fig. 6C). Strikingly, we observed a decrease in YFP intensity among Irf4 haploinsufficient high affinity HEL3X-binding SWHEL DZ GCBs (although not in total SWHEL DZ GCBs). Together, these results confirm predictions regarding IRF4 cell concentrations and the generation of PCs, as well as revealed a dose response relationship between IRF4 amounts and Blimp-1 levels. Furthermore, the results raise the possibility that suboptimal expression of Blimp-1 attenuates the generation of high affinity GCBs during affinity maturation.
Similarly, as precocious generation of presumptive pre-memory cells could dampen affinity maturation dynamics, we determined the proportions of CCR6-expressing LZ GCBs51. This analysis revealed comparable frequencies amongst total and high affinity HEL3X-binding SWHEL LZ GCBs (Fig. 6D). Thus, Irf4 haploinsufficiency does not alter the generation of GCB memory precursor cells.
Irf4+/− SWHEL cells fail to recruit optimal T cell help when in a competitive environment.
Our cumulative findings demonstrate impaired affinity maturation and Blimp-1 expression levels among presumptive positively-selected HEL3X-binding DZ GCBs despite comparable rates and targeting of AID-dependent SHM. We reasoned that a competition experiment between Irf4+/+ and Irf4+/− SWHEL cells would illuminate which check point(s) is critical for efficient affinity maturation when haploinsufficient for Irf4. For this analysis, we performed a co-transfer of equal numbers of Irf4+/+ and Irf4+/− SWHEL B cells; Irf4+/− cells were CD45.2, Irf4+/+ cells were CD45.1/CD45.2 and the host mouse was CD45.1 (Fig. 2A) and determined whether Irf4+/− day 10 GCBs might be disadvantaged upon HEL2X-SRBC immunization. Strikingly, we found that Irf4+/− GCBs were ~10 fold outcompeted (Fig. 7A). Moreover, adjustment of the Irf4+/− to Irf4+/+ SWHEL cell ratio to 3:1 also resulted in outcompeting Irf4+/+ GCBs (Supplementary Fig. 2B). To determine the timing of Irf4 haploinsufficient cell failure, we measured their frequencies in a 2:1 Irf4+/−:Irf4+/+ transfer over time (Fig. 7B). Importantly, we observed a strong depletion of responding Irf4+/− SWHEL cells and their differentiated progeny by day 5 of the response that was most pronounced for the PC fate, demonstrating the dominance of two copies of Irf4 compared to one in the response (Fig. 7C). At day 5, we estimated cell proliferation by metabolic labeling with the BrdU thymidine nucleoside analog and cell death by detection of cleaved activated Caspase 3 using a specific mAb. The analysis demonstrated comparable incorporation of BrdU and frequencies of cells undergoing Caspase 3 cleavage (Fig. 7D). Interestingly, at later time points the proportions of Irf4+/− GCBs declined at a slower rate (Fig. 7B) without a noticeable effect on a particular zonal subset (data not shown). Impaired generation of HEL3X-binding cells was also observed in this setting (Supplementary Fig. 2C). Thus, we conclude that a large fraction of Irf4 haploinsufficient cells is disadvantaged for T cell help; however, those that successfully compete proliferate and survive with near-comparable frequencies. Interestingly, this experiment highlights the dependency of PC for the full complement of Irf4 alleles.
Figure 7: Irf4+/− SWHEL cells fail to recruit optimal T cell help when in a competitive environment.
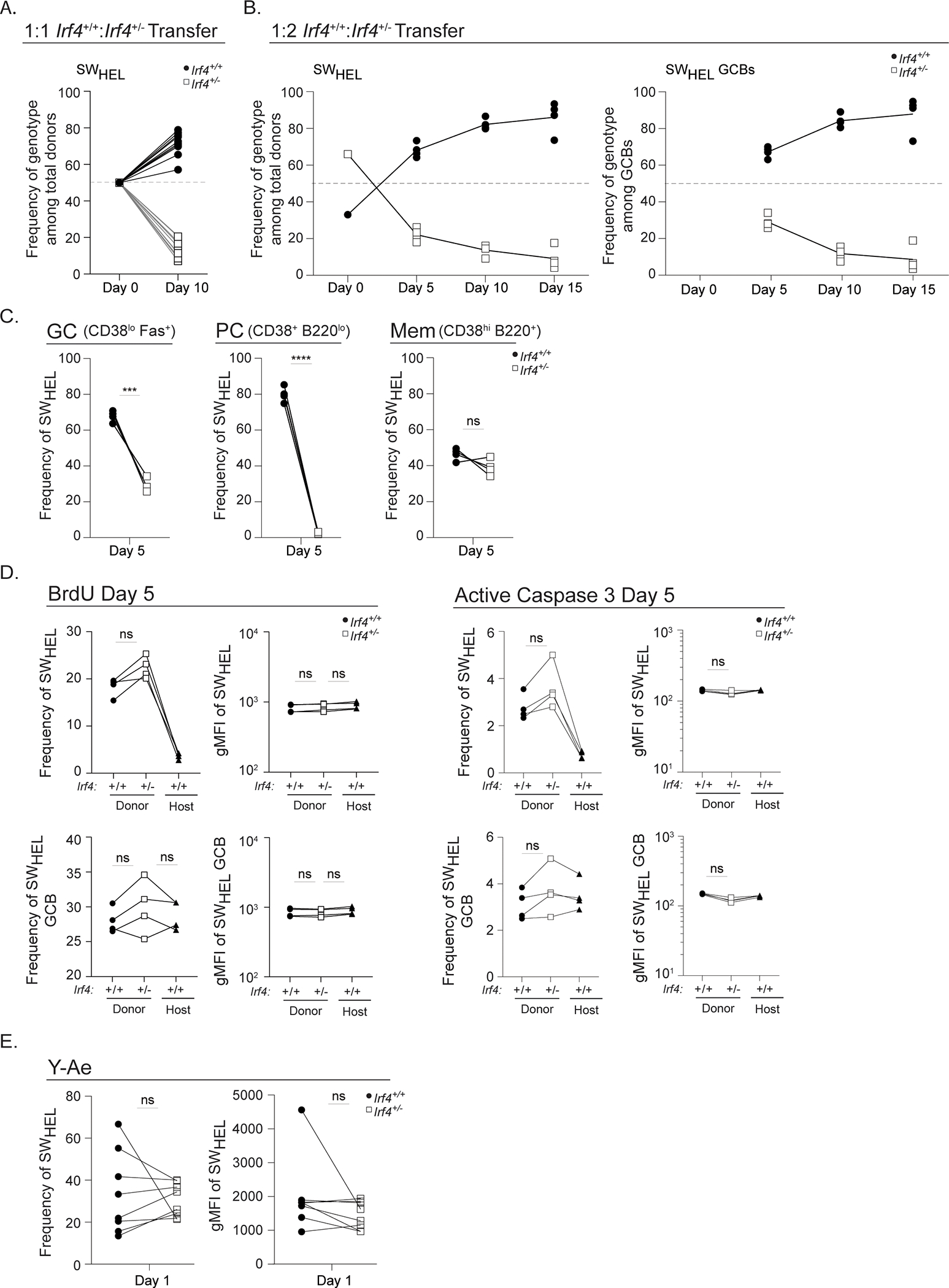
Irf4+/+ or Irf4+/− SWHEL B cells were co-transferred into recipient mice and immunized as in Fig. 2A prior to analysis at the indicated time points. Donor-specific responding Irf4+/+ cells were identified as CD45.2+CD45.1+CD3-B220+HELWT+ and donor- specific responding Irf4+/− cells were identified as CD45.2+CD45.1-CD3-B220+HELWT+. A) Frequency of Irf4+/+ and Irf4+/− total SWHEL cells at day 10, 1:1 initial cotransfer ratio. B) Frequency of Irf4+/+ and Irf4+/− total SWHEL cells or total SWHEL GCBs at indicated time points, 1:2 initial cotransfer ratio. C-D) Flow cytometry analyses of 1:2 Irf4+/+:Irf4+/− cotransfer at day 5. C) Proportion of GCB, PC, or memory populations (identified as in Fig. 2C legend) among total SWHEL donors. D) Frequency and gMFI of BrdU incorporation or active Caspase 3 among total SWHEL or total SWHEL GCB cells at day 5. E) Flow cytometry analyses of 1:1 Irf4+/+:Irf4−/− cotransfer and HEL-IEα s.c. immunization (day 1.5 post transfer and 24 hours post immunization). Frequency and gMFI of Y-Ae on total SWHEL B cells. Lines linking dots denote measurements from a single mouse. Experiment in A is from 10 mice in 2 experiments performed, and experiments in B-D are from 5 mice in one experiment performed. Experiment in E is from 8 mice in 2 experiments performed. Paired t tests were performed to determine significance. *** p ≤ 0.001, **** p ≤ 0.0001, ns, not significant (p > 0.05).
Clonal selection of specified GCBs is T cell dependent9, suggesting that Irf4 haploinsufficient cells compete poorly for T cell help. It has been shown that dendritic cells require IRF4 for efficient antigen presentation52, raising the possibility that Irf4+/− B cells may be presenting less pMHC during this T cell checkpoint. To test this possibility, we developed a fusion protein antigen, HEL-IEα, comprised of HEL2X, IEα46–74, OVA327–339, and a 6XHIS tag. The terminal tag enabled protein purification from supernatants of engineered D. melanogaster S2 cells. The pOVA component is irrelevant for these experiments. Thus, recognition and internalization of HEL-IEα by SWHEL cells would yield pMHC complexes, comprised of IEα:IAb, detectable by the Y-Ae mAb53. Irf4+/+ and Irf4+/− SWHEL cells were transferred into congenic hosts in a 1:1 ratio, immunized with HEL-IEα s.c., and their expression of IAb and Y-Ae were quantified one day later. Interestingly, cells of both genotypes expressed comparable MHCII and Y-Ae density and frequency (Fig. 7E), suggesting that Irf4 haploinsufficiency does not compromise antigen presentation in the B cell lineage. In fact, similar in vitro experiments suggest that complete Irf4 deficiency does not control antigen presentation efficiency by B cells (Supplementary Fig. 2D).
Discussion
The prevailing model for GCB selection upon entry into the LZ from the DZ involves competition for antigen displayed on FDC and subsequent processing and presentation of pMHC complexes for Tfh cells1. Limiting numbers of Tfh19,21,54, their ability to sense pMHC density21, and a subsequent CD40 / ICOSL feed forward loop13 promotes the fittest GCBs for DZ re-entry, mitosis, and differentiation. A subset of these cells have also induced the Myc and IRF4 transcription factors27,28. Here we reveal an Irf4 haploinsufficiency in selection of high affinity cells and their ability to upregulate optimal Blimp-1 expression. In contrast, GCB attributes of proliferation (Fig. 1G, 3D), phenotype (Fig. 1D, S1B), zonal compartmentalization (Fig. 1F, 3C), subset distribution observed in scRNA-seq analysis (Fig. 5), class switch recombination (Fig. 1C) and rates of SHM (Fig. 4) are unaffected by Irf4 gene copy reduction. Interestingly, when in competition, Irf4 heterozygous cells are substantially disadvantaged at the Tfh-mediated GCB entry checkpoint (Fig. 7B), suggesting that (1) diminished recruitment of Tfh signals and (2) subsequently dampened upregulation of high levels of Blimp-1 expression (Fig. 6C) underlies impaired affinity maturation of Irf4 hemizygous GCBs. We note that these two key findings precisely position IRF4 activity at the known IRF4 steps of receipt of signals from selecting T cells and execution of the PC gene program.
These data extend previous observations and reveal new biology of IRF4 in GCBs. First, Cγ1-Cre mediated deletion of Irf4 during the polyclonal response to NP-KLH revealed a trend towards fewer W33L mutations events3, suggesting impaired affinity maturation. Second, ERT2-Cre mediated deletion of Irf4 using Tamoxifen treatment during peak GCB responses showed normal GC functionality23 consistent with the data herein that IRF4 is dispensable for most GC biology. Interestingly, this study also showed that Irf4+/− GCBs are impaired in acquiring a pre-PC phenotype although affinity maturation was not addressed. This observation is consistent with the impaired Blimp-1 expression in Irf4 haploinsufficient cells presented in this work (Fig. 6). Third, augmented IRF4 expression caused by inactivation of the Cbl ubiquitin ligases in GCBs results in precocious PC differentiation accompanied by reduced acquisition of high affinity conferring somatic mutations36. These results, together with the observation that positively selected cells upregulate IRF4, suggest a model whereby levels of induced IRF4 expression are functionally limiting for dictating the fate of cells that have acquired high affinity-conferring somatic mutations and have successfully recruited Tfh selecting signals. In contrast to previous findings that reported negligible expression of IRF4 in GCBs46, we found that IRF4 levels in GCBs were above that seen in resting CD38+ B cells (Fig. 2C), suggesting that GCBs express an augmented basal level of IRF4 expression. The role of this new basal level is unclear given the lack of a GC phenotype in our system and upon tamoxifen-induced deletion23; however, we suggest that this basal level may counter the degradation rate imposed by Cbl activity in order to facilitate the acquisition of an IRF4 high cell state during positive selection and pre-PC differentiation.
We observed Irf4 haploinsufficiency for the generation of somatically mutated high affinity B cells during the onset of selection which was less pronounced at later time points, demonstrating that a two-fold reduction in Irf4 gene copies impairs affinity maturation (Fig. 3A, B). Sequence analysis revealed that the acquisition of the canonical Y53D and Y58F mutations were initially decreased amongst HEL-binding GCBs but not so at the later time point, despite comparable somatic mutation rates throughout (Fig. 4C, F). A possibility we entertained was that cells from the later time point were enriched for superadded high-affinity conferring somatic mutations that might function as a second site suppressor to enhance IRF4 inducibility by more intense BCR signaling. However, the sequence analysis did not support this hypothesis, suggesting that other pathways seemingly function to augment affinity maturation at later time points. Furthermore, we did not observe enhanced frequencies of CCR6 expressing pre-memory cells amongst Irf4+/− high affinity GCBs51 (Fig. 6D), excluding the possibility that Irf4+/− cells were pre-disposed to a memory cell fate trajectory, preventing their accumulation due to premature exit from the cell cycle. In contrast, we found a pronounced Irf4 haploinsufficient role for PCs and putative high affinity pre-PCs which manifests as a reduction in frequencies and YFP intensities, the latter as estimated with a BAC-encoded fluorescent protein reporter system (Fig. 6A–C). This suggests arrested and incomplete transition to a progenitor PC state that is perhaps accompanied with decelerated cell cycle progression55. In addition, this implies that the process of selection is coupled to PC fate decisions. Perhaps the lack of a difference in Blimp-1 YFP intensity amongst total GCB DZ cells reflects the proposition that not all such cells exhibit PC potential50. A deficit in high affinity pre-PCs is supported by our other observations that extrafollicular PCs depend on the full complement of Irf4 alleles, either when alone or in competition. Importantly, these observations also demonstrate, for the first time, that IRF4 not only activates Blimp-1 expression but also sets its levels. Together we propose that Irf4+/− cells are inherently capable of acquiring high affinity conferring somatic mutations yet are defective in leveraging selection signals into productive PC fate outcomes. Future experiments utilizing Blimp-1 hemizygous SWHEL cells may resolve and/or extend this interpretation. Finally, we speculate that the apparent normalization of high affinity cell frequencies at later time points (Fig. 3A,B) continues to reflect an Irf4 haploinsufficient deficit and reflects an accumulation of cells unable to exit, rather than accumulating and exiting. This will be resolved when the rate of the latter can be reliably measured.
The defect in upregulating Blimp-1 to high levels may constitute part of a two-step (or more) defect of Irf4 hemizygous cells. Specifically, when Irf4 wild type and heterozygous cells are in competition, the latter are disadvantaged at stages thought to be dependent on the receipt of T cell help. Interestingly, MHCII levels8 (Fig. S1C), pMHC density9 (Fig. 7E, S2D), ICAM density10 (Fig. S1C), and proliferation (Fig. 1G, 7D), parameters known to compromise this selection checkpoint, are not impaired, suggesting that other, remaining to be identified factors, compromise Irf4 haploinsufficent cells. The disadvantaged state is propagated, albeit to a lesser extent after GC commitment, leading us to propose that this feature of Irf4+/− cells functions during selection of high affinity cells and lies upstream of the defect in upregulating optimal Blimp-1 levels. As Blimp-1 is a target gene of IRF4, it is likely that part of the problem in selection is in the execution of the Irf4 gene regulatory network. However, selection of high affinity GCBs also depends upon successful competition with limiting numbers of selective Tfh. Thus, whether Irf4 haploinsufficient cells are coincidentally defective in sensing T cell signals upon limited interactions with Tfh remains to be investigated. Our findings raise the possibility that stochastic and/or regulated dampening of IRF4 expression plays a role in maintaining GCB clonal diversity56,57. Given the identification of IRF4 loss of function mutations in people58, our work uncovers a B cell specific haploinsufficient effect of Irf4 gene copy loss during antibody responses.
Supplementary Material
Key Points.
IRF4 is limiting for efficient production of high affinity GCBs
Irf4 haploinsufficiency causes sub-optimal regulation of Blimp-1 expression in GCBs
Irf4 haploinsufficient cells are competitively disadvantaged for T cell help
Acknowledgements
We are grateful to the Sciammas lab for valuable scientific and critical input for this project. Technical assistance for bulk and single cell sorting was provided by Ms. Bridget McLaughlin and Mr. Jonathan Van Dyke of The University of California Davis Flow Cytometry Shared Resource Laboratory. The scRNA-seq processing pipeline was carried out by the DNA Technologies and Expression Analysis Core at the UC Davis Genome Center, and its analysis by the UC Davis Bioinformatics Core. We are grateful for the award from the 10x Genomics & UC Davis GC Cores Single-Cell Pilot Study Program.
This project was supported by NIAID grants AI110513 and AI113145 (RS). This project was supported by the University of California Davis Flow Cytometry Shared Resource Laboratory with funding from the NCI P30 CA093373 (Cancer Center), and S10 OD018223 (Astrios Cell Sorter), and S10 RR 026825 (Fortessa Cytometer) grants. Support for the DNA Technologies and Expression Analysis Core at the UC Davis Genome Center by NIH Shared Instrumentation Grant 1S10OD010786-01.
References
- (1).Cyster JG, and Allen CDC. 2019. B Cell Responses: Cell Interaction Dynamics and Decisions. Cell. 177: 524–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, and Singh H. 2006. Graded Expression of Interferon Regulatory Factor-4 Coordinates Isotype Switching with Plasma Cell Differentiation. Immunity. 25: 225–236. [DOI] [PubMed] [Google Scholar]
- (3).Klein U, Casola S, Cattoretti G, Shen Q, Lia M, Mo T, Ludwig T, Rajewsky K, and Dalla-Favera R. 2006. Transcription Factor IRF4 Controls Plasma Cell Differentiation and Class-Switch Recombination. Nat Immunol. 7: 773–782. [DOI] [PubMed] [Google Scholar]
- (4).Ochiai K, Maienschein-Cline M, Simonetti G, Chen J, Rosenthal R, Brink R, Chong AS, Klein U, Dinner AR, Singh H, and Sciammas R. 2013. Transcriptional Regulation of Germinal Center B and Plasma Cell Fates by Dynamical Control of IRF4. Immunity. 38: 918–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Qi H 2016. T Follicular Helper Cells in Space-Time. Nat Rev Immunol. 16: 612–625. [DOI] [PubMed] [Google Scholar]
- (6).Kurosaki T, Shinohara H, and Baba Y. 2010. B Cell Signaling and Fate Decision. Annu Rev Immunol. 28: 21–55. [DOI] [PubMed] [Google Scholar]
- (7).Garside P, Ingulli E, Merica RR, Johnson JG, Noelle RJ, and Jenkins MK. 1998. Visualization of Specific B and T Lymphocyte Interactions in the Lymph Node. Science. 281: 96–99. [DOI] [PubMed] [Google Scholar]
- (8).Yeh C-H, Nojima T, Kuraoka M, and Kelsoe G. 2018. Germinal Center Entry Not Selection of B Cells Is Controlled by Peptide-MHCII Complex Density. Nat Commun. 9: 928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Schwickert TA, Victora GD, Fooksman DR, Kamphorst AO, Mugnier MR, Gitlin AD, Dustin ML, and Nussenzweig MC. 2011. A Dynamic T Cell-Limited Checkpoint Regulates Affinity-Dependent B Cell Entry into the Germinal Center. J. Exp. Med. 208: 1243–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zaretsky I, Atrakchi O, Mazor RD, Stoler-Barak L, Biram A, Feigelson SW, Gitlin AD, Engelhardt B, and Shulman Z. 2017. ICAMs Support B Cell Interactions with T Follicular Helper Cells and Promote Clonal Selection. J Exp Med. 214: 3435–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Qi H, Cannons JL, Klauschen F, Schwartzberg PL, and Germain RN. 2008. SAP-Controlled T-B Cell Interactions Underlie Germinal Centre Formation. Nature. 455: 764–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Watanabe M, Fujihara C, Radtke AJ, Chiang YJ, Bhatia S, Germain RN, and Hodes RJ. 2017. Co-Stimulatory Function in Primary Germinal Center Responses: CD40 and B7 Are Required on Distinct Antigen-Presenting Cells. J Exp Med. 214: 2795–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Liu D, Xu H, Shih C, Wan Z, Ma X, Ma W, Luo D, and Qi H. 2015. T-B-Cell Entanglement and ICOSL-Driven Feed-Forward Regulation of Germinal Centre Reaction. Nature. 517: 214–218. [DOI] [PubMed] [Google Scholar]
- (14).Victora GD, and Nussenzweig MC. 2012. Germinal Centers. Annu Rev Immunol. 30: 429–457. [DOI] [PubMed] [Google Scholar]
- (15).Kennedy DE, Okoreeh MK, Maienschein-Cline M, Ai J, Veselits M, McLean KC, Dhungana Y, Wang H, Peng J, Chi H, Mandal M, and Clark MR. 2020. Novel Specialized Cell State and Spatial Compartments within the Germinal Center. Nat Immunol. 21: 660–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Holmes AB, Corinaldesi C, Shen Q, Kumar R, Compagno N, Wang Z, Nitzan M, Grunstein E, Pasqualucci L, Dalla-Favera R, and Basso K. 2020. Single-Cell Analysis of Germinal-Center B Cells Informs on Lymphoma Cell of Origin and Outcome. J Exp Med. 217: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Laidlaw BJ, Duan L, Xu Y, Vazquez SE, and Cyster JG. 2020. The Transcription Factor Hhex Cooperates with the Corepressor Tle3 to Promote Memory B Cell Development. Nat Immunol. 21: 1082–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Bannard O, McGowan SJ, Ersching J, Ishido S, Victora GD, Shin J-S, and Cyster JG. 2016. Ubiquitin-Mediated Fluctuations in MHC Class II Facilitate Efficient Germinal Center B Cell Responses. J Exp Med. 213: 993–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Allen CDC, Okada T, Tang HL, and Cyster JG. 2007. Imaging of Germinal Center Selection Events during Affinity Maturation. Science. 315: 528–531. [DOI] [PubMed] [Google Scholar]
- (20).Schwickert TA, Lindquist RL, Shakhar G, Livshits G, Skokos D, Kosco-Vilbois MH, Dustin ML, and Nussenzweig MC. 2007. In Vivo Imaging of Germinal Centres Reveals a Dynamic Open Structure. Nature. 446: 83–87. [DOI] [PubMed] [Google Scholar]
- (21).Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, and Nussenzweig MC. 2010. Germinal Center Dynamics Revealed by Multiphoton Microscopy with a Photoactivatable Fluorescent Reporter. Cell. 143: 592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Luo W, Weisel F, and Shlomchik MJ. 2018. B Cell Receptor and CD40 Signaling Are Rewired for Synergistic Induction of the C-Myc Transcription Factor in Germinal Center B Cells. Immunity. 48: 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ise W, Fujii K, Shiroguchi K, Ito A, Kometani K, Takeda K, Kawakami E, Yamashita K, Suzuki K, Okada T, and Kurosaki T. 2018. T Follicular Helper Cell-Germinal Center B Cell Interaction Strength Regulates Entry into Plasma Cell or Recycling Germinal Center Cell Fate. Immunity. 48: 702–715. [DOI] [PubMed] [Google Scholar]
- (24).Sander S, Chu VT, Yasuda T, Franklin A, Graf R, Calado DP, Li S, Imami K, Selbach M, Di Virgilio M, Bullinger L, and Rajewsky K. 2015. PI3 Kinase and FOXO1 Transcription Factor Activity Differentially Control B Cells in the Germinal Center Light and Dark Zones. Immunity. 43: 1075–1086. [DOI] [PubMed] [Google Scholar]
- (25).Dominguez-Sola D, Kung J, Holmes AB, Wells VA, Mo T, Basso K, and Dalla-Favera R. 2015. The FOXO1 Transcription Factor Instructs the Germinal Center Dark Zone Program. Immunity. 43: 1064–1074. [DOI] [PubMed] [Google Scholar]
- (26).Inoue T, Shinnakasu R, Ise W, Kawai C, Egawa T, and Kurosaki T. 2017. The Transcription Factor Foxo1 Controls Germinal Center B Cell Proliferation in Response to T Cell Help. J Exp Med. 214: 1181–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Dominguez-Sola D, Victora GD, Ying CY, Phan RT, Saito M, Nussenzweig MC, and Dalla-Favera R. 2015. The Proto-Oncogene MYC Is Required for Selection in the Germinal Center and Cyclic Reentry. Nat Immunol. 13: 1083–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Calado DP, Sasaki Y, Godinho SA, Pellerin A, Köchert K, Sleckman BP, de Alborán IM, Janz M, Rodig S, and Rajewsky K. 2012. The Cell-Cycle Regulator c-Myc Is Essential for the Formation and Maintenance of Germinal Centers. Nat Immunol. 13: 1092–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Finkin S, Hartweger H, Oliveira TY, Kara EE, and Nussenzweig MC. 2019. Protein Amounts of the MYC Transcription Factor Determine Germinal Center B Cell Division Capacity. Immunity. 51: 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Chou C, Verbaro DJ, Tonc E, Holmgren M, Cella M, Colonna M, Bhattacharya D, and Egawa T. 2016. The Transcription Factor AP4 Mediates Resolution of Chronic Viral Infection through Amplification of Germinal Center B Cell Responses. Immunity. 45: 570–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kepler TB, and Perelson AS. 1993. Cyclic Re-Entry of Germinal Center B Cells and the Efficiency of Affinity Maturation. Immunol Today. 14: 412–415. [DOI] [PubMed] [Google Scholar]
- (32).Bannard O, Horton RM, Allen CDC, An J, Nagasawa T, and Cyster JG. Germinal Center Centroblasts Transition to a Centrocyte Phenotype According to a Timed Program and Depend on the Dark Zone for Effective Selection. Immunity 2013, 39 (5), 912–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Matsuyama T, Grossman A, Mittrücker HW, Siderovski DP, Kiefer F, Kawakami T, Richardson CD, Taniguchi T, Yoshinaga SK, and Mak TW. 1995. Molecular Cloning of LSIRF, a Lymphoid-Specific Member of the Interferon Regulatory Factor Family That Binds the Interferon-Stimulated Response Element (ISRE). Nucleic Acids Res. 23: 2127–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Cook SL, Franke MC, Sievert EP, and Sciammas R. 2020. A Synchronous IRF4-Dependent Gene Regulatory Network in B and Helper T Cells Orchestrating the Antibody Response. Trends Immunol. 41: 614–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Sciammas R, Li Y, Warmflash A, Song Y, Dinner AR, and Singh H. 2011. An Incoherent Regulatory Network Architecture That Orchestrates B Cell Diversification in Response to Antigen Signaling. Mol Syst Biol. 7: 495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Li X, Gadzinsky A, Gong L, Tong H, Calderon V, Li Y, Kitamura D, Klein U, Langdon WY, Hou F, Zou Y-R, and Gu H. 2018. Cbl Ubiquitin Ligases Control B Cell Exit from the Germinal-Center Reaction. Immunity. 48: 530–541. [DOI] [PubMed] [Google Scholar]
- (37).Brink R, Paus D, Bourne K, Hermes JR, Gardam S, Phan TG, and Chan TD. 2015. The SW(HEL) System for High-Resolution Analysis of in Vivo Antigen-Specific T-Dependent B Cell Responses. Methods Mol Biol. 1291: 103–123. [DOI] [PubMed] [Google Scholar]
- (38).Mittrücker HW, Matsuyama T, Grossman A, Kündig TM, Potter J, Shahinian A, Wakeham A, Patterson B, Ohashi PS, and Mak TW. 1997. Requirement for the Transcription Factor LSIRF/IRF4 for Mature B and T Lymphocyte Function. Science. 275: 540–543. [DOI] [PubMed] [Google Scholar]
- (39).Fooksman DR, Schwickert TA, Victora GD, Dustin ML, Nussenzweig MC, and Skokos D. 2010. Development and Migration of Plasma Cells in the Mouse Lymph Node. Immunity. 33: 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Yenson V, and Baumgarth N. 2021. Purification and Immune Phenotyping of B-1 Cells from Body Cavities of Mice. Methods Mol Biol. 2270: 27–45. [DOI] [PubMed] [Google Scholar]
- (41).Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, Gregory MT, Shuga J, Montesclaros L, Underwood JG, Masquelier DA, Nishimura SY, Schnall-Levin M, Wyatt PW, Hindson CM, Bharadwaj R, Wong A, Ness KD, Beppu LW, Deeg HJ, McFarland C, Loeb KR, Valente WJ, Ericson NG, Stevens EA, Radich JP, Mikkelsen TS, Hindson BJ, and Bielas JH. 2017. Massively Parallel Digital Transcriptional Profiling of Single Cells. Nat Commun. 8: 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R. 2019. Comprehensive Integration of Single-Cell Data. Cell. 177: 1888–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Victora GD, Dominguez-Sola D, Holmes AB, Deroubaix S, Dalla-Favera R, and Nussenzweig MC. 2012. Identification of Human Germinal Center Light and Dark Zone Cells and Their Relationship to Human B-Cell Lymphomas. Blood. 120: 2240–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Laidlaw BJ, Schmidt TH, Green JA, Allen CDC, Okada T, and Cyster JG. 2017. The Eph-Related Tyrosine Kinase Ligand Ephrin-B1 Marks Germinal Center and Memory Precursor B Cells. J Exp Med. 214: 639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Paus D, Phan TG, Chan TD, Gardam S, Basten A, and Brink R. 2006. Antigen Recognition Strength Regulates the Choice between Extrafollicular Plasma Cell and Germinal Center B Cell Differentiation. J. Exp. Med. 203: 1081–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Falini B, Fizzotti M, Pucciarini A, Bigerna B, Marafioti T, Gambacorta M, Pacini R, Alunni C, Natali-Tanci L, Ugolini B, Sebastiani C, Cattoretti G, Pileri S, Dalla-Favera R, and Stein H. 2000. A Monoclonal Antibody (MUM1p) Detects Expression of the MUM1/IRF4 Protein in a Subset of Germinal Center B Cells, Plasma Cells, and Activated T Cells. Blood. 95: 2084–2092. [PubMed] [Google Scholar]
- (47).Phan TG, Paus D, Chan TD, Turner ML, Nutt SL, Basten A, and Brink R. 2006. High Affinity Germinal Center B Cells Are Actively Selected into the Plasma Cell Compartment. J Exp Med. 203: 2419–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, and Smibert P. 2017. Simultaneous Epitope and Transcriptome Measurement in Single Cells. Nat Methods. 14: 865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Kräutler NJ, Suan D, Butt D, Bourne K, Hermes JR, Chan TD, Sundling C, Kaplan W, Schofield P, Jackson J, Basten A, Christ D, and Brink R. 2017. Differentiation of Germinal Center B Cells into Plasma Cells Is Initiated by High-Affinity Antigen and Completed by Tfh Cells. J Exp Med. 214: 1259–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Radtke D, and Bannard O. 2018. Expression of the Plasma Cell Transcriptional Regulator Blimp-1 by Dark Zone Germinal Center B Cells During Periods of Proliferation. Front Immunol. 9: 3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Suan D, Kräutler NJ, Maag JLV, Butt D, Bourne K, Hermes JR, Avery DT, Young C, Statham A, Elliott M, Dinger ME, Basten A, Tangye SG, and Brink R. 2017. CCR6 Defines Memory B Cell Precursors in Mouse and Human Germinal Centers, Revealing Light-Zone Location and Predominant Low Antigen Affinity. Immunity. 47: 1142–1153. [DOI] [PubMed] [Google Scholar]
- (52).Vander Lugt B, Khan AA, Hackney JA, Agrawal S, Lesch J, Zhou M, Lee WP, Park S, Xu M, DeVoss J, Spooner CJ, Chalouni C, Delamarre L, Mellman I, and Singh H. 2014. Transcriptional Programming of Dendritic Cells for Enhanced MHC Class II Antigen Presentation. Nat. Immunol. 15: 161–167. [DOI] [PubMed] [Google Scholar]
- (53).Murphy DB, Rath S, Pizzo E, Rudensky AY, George A, Larson JK, and Janeway CA. 1992. Monoclonal Antibody Detection of a Major Self Peptide. MHC Class II Complex. J Immunol. 148: 3483–3491. [PubMed] [Google Scholar]
- (54).Meyer-Hermann ME, Maini PK, and Iber D. 2006. An Analysis of B Cell Selection Mechanisms in Germinal Centers. Math Med Biol. 23: 255–277. [DOI] [PubMed] [Google Scholar]
- (55).Gitlin AD, Shulman Z, and Nussenzweig MC. 2014. Clonal Selection in the Germinal Centre by Regulated Proliferation and Hypermutation. Nature. 509: 637–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Tas JMJ, Mesin L, Pasqual G, Targ S, Jacobsen JT, Mano YM, Chen CS, Weill J-C, Reynaud C-A, Browne EP, Meyer-Hermann M, and Victora GD. 2016. Visualizing Antibody Affinity Maturation in Germinal Centers. Science. 351: 1048–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Kuraoka M, Schmidt AG, Nojima T, Feng F, Watanabe A, Kitamura D, Harrison SC, Kepler TB, and Kelsoe G. 2016. Complex Antigens Drive Permissive Clonal Selection in Germinal Centers. Immunity. 44: 542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Guérin A, Kerner G, Marr N, Markle JG, Fenollar F, Wong N, Boughorbel S, Avery DT, Ma CS, Bougarn S, Bouaziz M, Béziat V, Della Mina E, Oleaga-Quintas C, Lazarov T, Worley L, Nguyen T, Patin E, Deswarte C, Martinez-Barricarte R, Boucherit S, Ayral X, Edouard S, Boisson-Dupuis S, Rattina V, Bigio B, Vogt G, Geissmann F, Quintana-Murci L, Chaussabel D, Tangye SG, Raoult D, Abel L, Bustamante J, and Casanova JL. 2018. IRF4 Haploinsufficiency in a Family with Whipple’s Disease. Elife. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.