

Abstract
Substance use disorders (SUDs) are moderately to highly heritable and are in part cross-transmitted genetically, as observed in twin and family studies. We performed exome-focused genotyping to examine the cross-transmission of four SUDs: alcohol use disorder (AUD, n=4,487); nicotine use disorder (NUD, n=4,394); cannabis use disorder (CUD, n=954); and nonmedical prescription opioid use disorder (NMPOUD, n=346) within a large nationally representative sample (N=36,309), the National Epidemiologic Survey on Alcohol and Related Conditions-III (NESARC-III). All diagnoses were based on in-person structured psychiatric interview (AUDADIS-5). SUD cases were compared alone and together to 3,959 “super controls” who had neither a SUD nor a psychiatric disorder using an exome-focused array assaying 363,496 SNPs, yielding a representative view of within-disorder and cross-disorder genetic influences on SUDs. The 29 top susceptibility genes for one or more SUDs overlapped highly with genes previously implicated by GWAS of SUD. Polygenic scores (PGS) were computed within the European ancestry (EA) component of the sample (N=12,505) using summary statistics from each of four clinically distinct SUDs compared to the 3,959 “super controls” but then used for two distinctly different purposes: to predict SUD severity (mild, moderate, or severe) and to predict each of the other 3 SUDs. Our findings based on PGS highlight shared and unshared genetic contributions to the pathogenesis of SUDs, confirming the strong cross-inheritance of AUD and NUD as well as the distinctiveness of inheritance of opioid use disorder.
Introduction
Access and use of psychoactive substances are associated with risk of substance use disorders (SUDs), which themselves precipitate other risky behaviors. SUDs are both endemic and epidemically emergent as global public health priorities [1]. SUDs are often comorbid, worsening their impact and complicating prevention and treatment, but suggesting shared innate and environmental etiologies. Comorbidity of SUDs is associated with more severe disease course and worse prognosis. Comorbidity undermines the effectiveness of treatment and maintenance services, impairs productivity and return to functionality, and complexifies and increases costs of medical care. Four common SUDs that are frequently comorbid, namely alcohol use disorder (AUD), nicotine use disorder (NUD), cannabis use disorder (CUD) and nonmedical prescription opioid use disorder (NMPOUD), are the focus of this study.
Pathways to comorbidity include the gateway drug hypothesis [2], as well as the shared impact of environmental exposures and pleiotropy of genetic variants to increase, or decrease, risk of different SUDs. SUDs that have been investigated in twin studies are mostly moderately to highly heritable, heritabilities ranging from 39%-72% [3, 4], indicating that alleles shared between blood relatives moderate risk. In addition, it has long been known, both from genetic studies measuring cross-transmission of SUDs in monozygotic and dizygotic twins and from studies comparing siblings, that vulnerabilities to several SUDs are both cross-transmitted and influenced by genetic variants whose effects are limited to one or a few types of SUD [5].
Recently, new light has been shed on the cross-transmission of SUDs and other phenotypes by genome wide association studies (GWAS) that identify constellations of genetic variants predicting risk [6-13]. These genetic variants have been combined into polygenic scores predicting relatively small proportions of genetic risk (heritability) [14] but on the other hand implicating common genetic variants in different psychiatric diseases [15]. A polygenic score catalog https://www.pgscatalog.org/ includes several phenotypes directly related to SUDs (e.g. alcohol dependence) as well as phenotypes that are etiologically relevant (e.g. depression, risky behavior; https://www.pgscatalog.org/, December, 2020 release). GWAS in large samples has yielded a small but partly well-replicated set of loci influencing SUDs. Replication via GWAS of CHRNA5 (for NUD) and the ADH gene cluster (for AUD) has tended to validate the power of GWAS for gene identification in SUDs. Like other complex phenotypes, few new causal genetic variants have been identified to date for SUDs, and the proportion of phenotypic variance explained by those identified variants is modest; however, genes identification has led to new insights into pathways to vulnerability and cross-inheritance of vulnerability, and with the promise of identifying more causal loci accounting for higher proportions of genetic variance in the future.
The National Epidemiologic Survey on Alcohol and Related Conditions-III (NESARC-III) is a sample of the US general population ages 18 years and older [16, 17], providing a unique opportunity to measure gene effects on individual SUDs and cross-transmission of SUDs in a nationally representative framework, the entire sample being ascertained, psychiatrically interviewed and diagnosed in consistent fashion. GWAS of SUD performed using the Kaiser Permanente study [18] and Million Veterans study [19] were based on clinicians’ diagnoses in the electronic health record (EHR). GWAS of SUD-related phenotypes in UK Biobank [20] and 23andMe [9] did not utilize assessment of SUDs by DSM or ICD criteria. Each of these large GWAS had power to detect loci of relatively small effect, and these GWAS studies implicated several of the same genes; however, these studies tested individual and polygenic locus effects (as integrated into polygenic scores (PGS)) and ability of a PGS to predict cross-transmission in particular contexts. The Kaiser Permanente study was based on a HMO located in California; the Million Veterans Program, veterans; the UK Biobank, volunteers; 23andMe, customers. The deCode sample was nationally representative for the country of Iceland [21]. The NESARC-III dataset, representative for the United States, contains sociodemographic characteristics, family history of various disorders, and DSM-5 diagnoses via structured psychiatric interview for substance use, mood, anxiety, and personality disorders.
The diagnosis of substance use disorder underwent substantial revision in the Diagnostic and Statistical Manual of Mental Disorders–Fifth Edition (DSM-5). The DSM-5 definition adds a craving criterion and eliminates the “legal problems” criterion. It removes the abuse category, uses a diagnostic threshold of ≥2 criteria, and categorizes severity based on criteria counts, i.e., mild (2 to 3), moderate (4 to 5), and severe (6+). The focus of the present investigation was the detection and quantitation of shared inheritance between different SUDs and testing SUD Polygenic scores against the three DSM categories of severity for each SUD, in a nationally representative sample. Secondly, we test the ability of this study, performed in a nationally representative, psychiatrically interviewed population, to replicate genes identified in previous large SUD GWAS.
Materials and methods
Sample and data.
The National Epidemiologic Survey on Alcohol and Related Conditions-III (NESARC-III) was a nationally representative cross-sectional survey of the U.S. general population sponsored by the National Institute on Alcohol Abuse and Alcoholism (NIAAA) and conducted 2012-2013. The research protocol was approved by the Institutional Review Boards of the National Institutes of Health and Westat, Inc., and written informed consent was obtained from all participants. As detailed elsewhere [17], the NESARC-III target population was the U.S. noninstitutionalized civilian population, 18 years and older, including residents of selected group quarters (e.g., group homes, workers’ dormitories). Multistage probability sampling was used to randomly select respondents. Primary sampling units (PSUs) were individual counties or groups of contiguous counties, secondary sampling units (SSU) comprised groups of Census-defined blocks, and tertiary sampling units were households within SSUs. Finally, eligible adults within sampled households were randomly selected. Hispanics, Blacks, and Asians were oversampled and resulted in a total of 36,309 respondents. Of the 36,309 respondents, 23,860 provided saliva samples for the genetic study. The individual-level genetic data (22,848 samples) with phenotypic variables (n = 4,320) for NESARC-III (family history, adverse childhood experiences, substance use, mood, anxiety, personality and posttraumatic stress disorders) are available in dbGaP (accession: phs001590.v2.p1).
Clinical Assessment.
All participants were assessed using the Alcohol Use Disorder and Associated Disabilities Interview Schedule 5 (AUDADIS-5), a structured psychiatric interview. The AUDADIS-5 was designed to measure DSM-5 criteria for AUDs, nicotine use disorder (NUD), specific drug use disorders (DUDs), and selected mood, anxiety, trauma related, eating, and personality disorders. Lifetime DSM-5 AUD/NUD/CUD/NMPOUD diagnoses required at least 2 of the 11 corresponding criteria in the 12 months preceding the interview, or a diagnosis before the past 12 months required clustering of at least 2 criteria within a 1-year period [22-24].
Subjects who were genetically inferred as being of European ancestry as described later, and in 4 non-independent SUD categories were analyzed: AUD (n=4,487), NUD (n=4,394), CUD (n=954), NMPOUD (n=346). In all, 6,525 subjects had a SUD, and with comorbidity. The SUD groups were compared alone and together with 3,959 healthy controls who had neither a SUD nor a related psychiatric disorder. As a replication cohort, cases of AUD (n=1,120), NUD (n=1,145), CUD (n=349), NMPOUD (n=71) were identified among African Americans (AA). The SUD groups in AA were subsequently compared alone and together with 1,889 healthy controls in AA from this survey. The overall study design was shown in Supplementary Fig. 1.
Genotyping and quality control.
Of the 23,860 people provided saliva samples for the genetic study, DNA was extracted from saliva and genotyped with an Affymetrix Axiom® Exome Array consisting of 319,283 SNPs and 103,404 custom-selected SNPs. The custom SNPs were selected based on addiction-associated genes, and genomic and transcriptomic findings from five GWAS of alcohol and other psychiatric disorders, and from animal models of addictions. After filtering out poor quality SNPs using the standard Affymetrix SNPs polisher algorithm, 396,581 SNPs were potentially available. Several additional quality control criteria were applied to samples and genetic variants prior to genetic association analysis. Individuals were excluded for the following reasons: a genotyping call rate of <97% (n = 188) or genetically related samples (n=824). SNPs were removed if call rate was <97%, for deviation from Hardy-Weinberg equilibrium (P<1x10−7), or if monomorphic or represented by a single heterozygote. A total of 22,848 samples and 260,779 SNPs were passed for subsequent analyses. Among 260,779 loci, 121,484 SNPs had a MAF of ≥ 0.05. A total of 21,840 SNPs had MAF 0.01-0.05, and the remaining 117,455 SNPs had MAF < 0.01. Sex was determined using 3 SNPs located on the X chromosome and 3 SNPs located on the Y chromosome from the 96 SNP RUID™ DNA QC panel (now commercially available through Fluidigm as SNPtrace™). Because sex indicated in RUID™ DNA QC is more accurate than imputed sex from PLINK data, genetically determined sex from RUID™ DNA QC was used for downstream analyses.
Population stratification.
To identify and correct for population stratification, we included 2,211 reference samples representing 66 populations including indigenous peoples. The reference sources were a) HapMap-3 (924 individuals from 11 populations) [https://www.sanger.ac.uk/data/hapmap-3], b) Human Genome Diversity Project (940 individuals in 51 populations from sub-Saharan Africa, North Africa, Europe, the Middle East, South/Central Asia, East Asia, Oceania, the Americas) [https://www.hagsc.org/hgdp/files.html], and c) 1000 Genome Project (347 individuals from four America populations, including MXL, PUR, CLM and PEL populations) [https://www.internationalgenome.org/home]. First, we applied PLINK [25, 26] to generate genome-wide Identity by State (IBS) estimates between NESARC-III and the reference samples at 10,612 common SNPs. We then produced multi-dimensional scaling (MDS) plots for examination of genetically inferred ancestry of all NESARC-III participants against the reference populations. We also used the Admixture program to quantitate the genetic ancestry of each NESARC-III participant. Via these procedures, 12,505 individuals of predominantly European ancestry (MDS1≤−0.035; MDS2≤−0.01; MDS3[−0.02,0.01]; MDS4[−0.031,0.018]) and 4,418 individuals of predominantly African ancestry (MDS1≥0.06; MDS2≤0; MDS3[−0.02,0.02]; MDS4[−0.01,0.02]) were identified.
Association analysis.
We used Rvtests [27] to test association between phenotypes and common variants (MAF≥0.01) under a score test model. For rare variants, we ran a gene-based analysis (at least 2 rare variants in a single gene) under optimal sequencing kernel association test (SKAT-O model) using Rvtests software with a MAF cutoff of ≤ 5%. We ran an exome wide association analysis controlling for age, sex, and the first 4 multi-dimensional scaling (MDS) scores. Healthy super-controls were participants with no history of SUD nor a related psychiatric disorder (e.g., mood disorders, anxiety disorders, PTSD, personality disorders, and eating disorders).
The GCTA-fastBAT (a fast and flexible gene- or set-Based Association Test) software [28] was used to identify gene-level and genetic pathways associations using GWAS summary data for common variants (MAF≥0.01).
Meta-analysis.
Meta-analysis for genetic association was accomplished using rareMETAL [29], a computationally efficient tool for meta-analysis of both common and rare variants at the gene-level. The rareMETAL program utilizes summary statistics and LD matrices generated by Rvtests.
Expression quantitative trait loci (eQTL) and methylation QTL analysis.
We used summary-data-based Mendelian randomization (SMR) [30] to identify variants pleiotropically associated with both a SUD and level of gene expression (mQTL or eQTL) from 2 different tissues (blood and fetal brain) in EA. SMR was developed to detect pleiotropic associations between complex trait and expression levels of genes using summary level data from GWAS and Methylation QTL (mQTL) or expression quantitative trait loci (eQTL). SMR can ascertain whether the effect of a variant on a phenotype may be mediated by expression level of the corresponding gene and is therefore commonly used to prioritize genes implicated by GWAS.
Multi-trait analysis of GWAS (MTAG).
SUDs are both clinically and genetically correlated. To parse genetic correlations between pairs of SUDs in the presence of clinical comorbidity, we used multi-trait analysis of GWAS (MTAG) [31], a method for joint analysis of summary statistics from multiple GWAS with overlapping samples. This analysis detected loci contributing to individual SUDs and up to 4 different SUDs.
Multivariate association analysis on 4 SUDs (TATES).
We carried out multivariate genetic association analysis on all four SUDs together, through TATES [32], to identify relational pleiotropic effects on multiple SUDs. TATES (Trait-based Association Test uses Extended Simes procedure) combines P-values obtained from univariate associations of single traits while accounting for cross-trait correlations. TATES provides a more comprehensive view of the genetic architecture of complex traits and boosts statistical power to detect underlying causal variants.
Polygenic scores.
Polygenic score (PGS) is a single aggregated estimate of an individual’s genetic propensity to a phenotype based on a series of genetic markers. PGS of each sample was calculated as a sum of effects of risk variants across a genome-wide scale weighted by corresponding genotype effect sizes (i.e., OR) using PRSice-2 [33]. Common genetic variants (MAF ≥ 0.01) were pruned using r2-clumping in PLINK, with a cutoff of r2=0.2 within 250kb windows. Pruned SNPs were also filtered at P value of < 0.05 in case-control association analysis.
For each disorder we also used external GWAS summary statistics (except for CUD for which we used only the NESARC-III summary statistic) [13, 34, 35] as a base association result to calculate externally derived PGS of each SUD for NESARC-III EA samples.
Results
GWAS for SUDs in NESARC-III.
GWAS was performed as a foundation for evaluation of cross-inheritances of the 4 SUDs. The NESARC-III sample was diverse in ancestry (Fig. 1). The ancestry vector space defined by multi-dimensional scaling was anchored using 2,211 individuals from 66 reference populations, also revealing that population admixture was common in the NESARC-III sample (Fig. 1). To reduce the potential impact of ancestry stratification in estimating individual locus effects and computing PGS, we focused on 12,505 individuals of predominantly European Ancestry (EA) and 4,418 individuals -African American (AA)- of predominantly African descent. The characteristics of NESARC-III cases and control as well as psychiatric comorbidity for cases in EA are shown in Supplementary Table 1. Cases and controls within the EA and AA groups used in the analyses were also plotted with regards to their multidimensional scaling scores (Supplementary Fig. 2). Clinically, SUDs were highly comorbid (Fig. 2). Jaccard indices (similarity coefficients) between SUDs ranged from 0.06 (AUD x NMPOUD and NUD x NMPOUD) to a high of 0.38 (AUD x NUD) (all P<10−4).
Fig. 1.
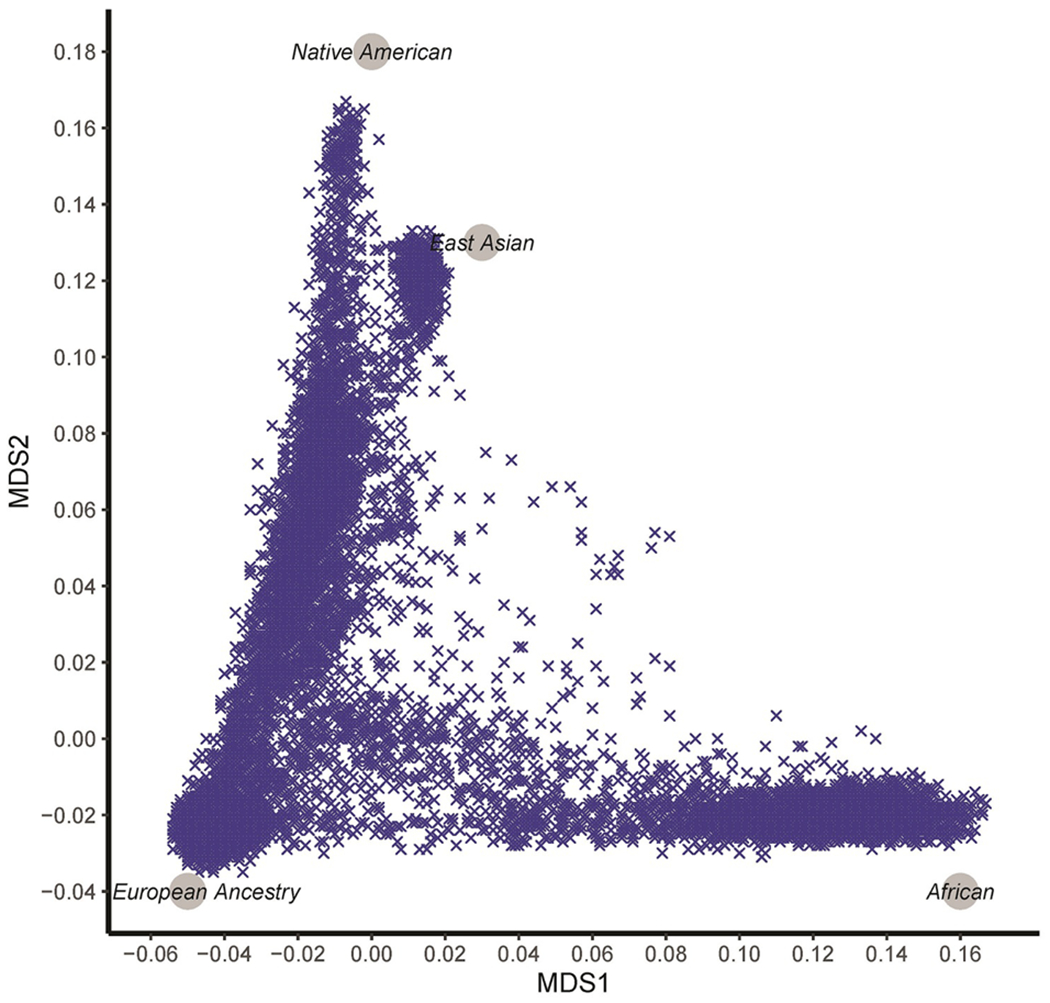
Multidimensional scaling (MDS) of ancestry using the top two MDS scores for 22,848 NESARC-III samples from a nationally representative survey: NESARC-III. Four ancestral continental population clusters, established via genotyping reference obtained from 1000 Genomes and the Human Genome Diversity Project, are shown in grey circles.
Fig. 2.
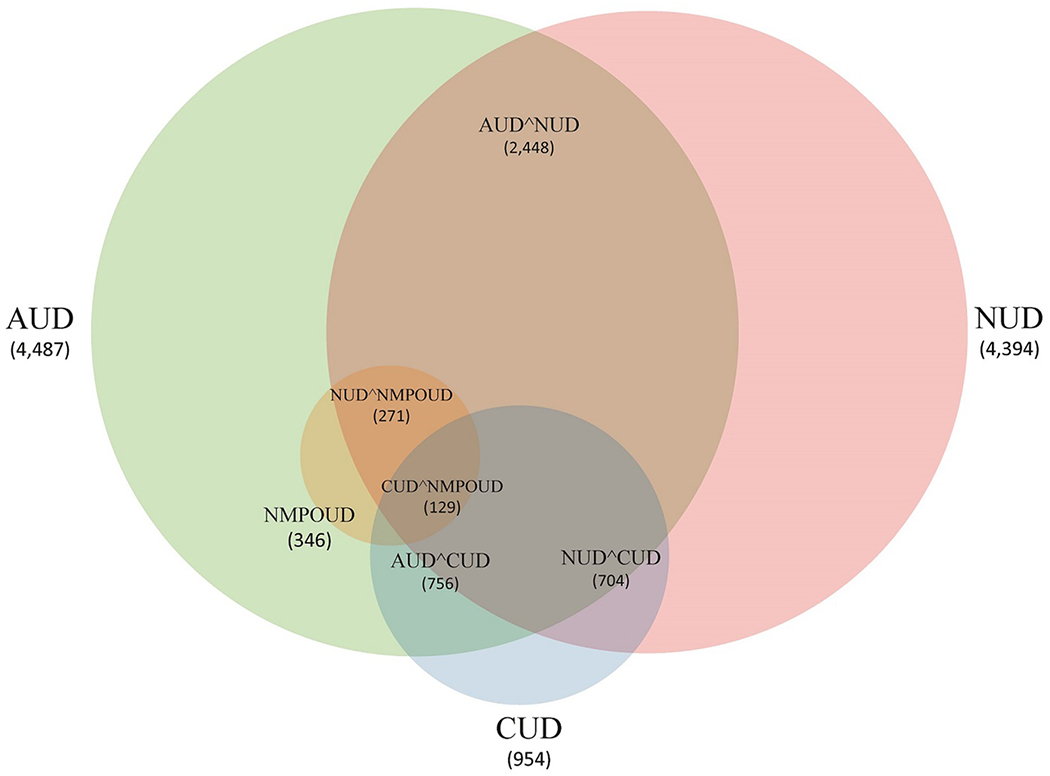
Clinical overlap (comorbidity) of four substance use disorders (SUDs) in EA in NESARC-III, a nationally representative, psychiatrically interviewed sample: AUD (n=4,487), NUD (n=4,394), CUD (n=954) and NMPOUD (n=346). Not shown in the figure for clarity of display: AUD and NMPOUD clinically overlapped in 273 cases. Also, all four SUDs were present in 101 cases.
The quantile-quantile plots for single-SNP case/control associations of common variants (MAF≥0.01) in EA revealed minimal inflation of p values (Supplementary Fig. 3). Lambda values ranged from 1.01 to 1.03 as follows: AUD:1.02, NUD:1.03, CUD:1.03, NMPOUD:1.01, and any SUD:1.01. Therefore, neither polygenicity of SUDs nor residual uncorrected population stratification significantly inflated p values.
In this study, in a relatively small sample for GWAS, we were primarily interested in cross-disorder genetic transmission as predicted by PGS. However, as a foundation for that PGS analysis, we performed within-sample GWAS and we evaluated generalizability of these data by replication in other GWAS samples and by candidate gene replications because of the “addiction gene” focused design of the array we used. Several well-known candidate gene associations to SUDs [3, 36] were replicated in our EA cohort at a nominal level (P<0.05) (Supplementary Table 2). For AUD, replicated genes were ADH1B, P= 8.0x10−4; OPRM1, P=1.8x10−3 and ALDH16A1, P=7.4x10−3. For NUD, replicated genes were CHRNA3, P=3.0x10−3; CHRNA5, P=3.1x10−3, OPRM1, P=1.0x10−2, and ALDH16A1, P=3.5x10−2. CYP2A6, a multi-locus, multi-allelic, gene, was marginally significant at P=0.08. For CUD, the replicated genes were COMT, P=2.4x10−3, OPRM1, P=1.3x10−2 and ADH1B, P=1.9x10−2. For NMPOUD, the replicated genes included COMT, P=1.2x10−2 and OPRM1, P=0.052 was marginally significant. For the grand analysis of all four SUDs, the replicated genes were OPRM1, P= 1.6x10−3, CHRNA3, P= 5.2x10−3, ALDH16A1, P= 6.6x10−3, ADH1B, P= 7.4x10−3, CHRNA5, P= 2.9x10−2 and COMT, P= 4.4x10−2. We also list significance levels (P<0.05) for 24 candidate genes previously implicated in addictions [37], although not via GWAS (Supplementary Table 3).
As illustrated in Supplementary Fig. 4, exome-wide association in EA of single common SNPs in 4 SUDs and in any SUD detected 53 SNPs, including 21 that were genetically independent (r2<0.2), that exceeded significance threshold (P<5x10−5) for at least 1 SUD (Table 1). A total of 143,324 SNPs with MAF>0.01 were tested, which would correspond to a Bonferroni P value threshold of 3.5x10−7 if all these SNPs were genetically independent, which they were not. We concluded that using this Bonferroni threshold would be overly conservative to investigate replication, obscuring a high rate of replication of previous SUD GWAS hits in this national sample. All genes reported here at the nominal P<5x10−5 cutoff were previously implicated in large SUD GWAS [8, 9, 13, 19, 38, 39] as susceptibility loci (P<0.05 for at least one SNP). Among the 53 SNPs, 21 were implicated in AUD, 14 in NUD, 7 in CUD, 3 in NMPOUD, 8 in the grand analysis of four SUDs, and 2 SNPs (rs3756772 in FRK and rs4888599, an intergenic SNP) were associated with 2 different SUDs in NESARC-III data. Among these SNPs, rs72501734 in ULBP3 showed modest replication for association to NUD in African Americans (AA, P=0.03). Interestingly, 3 genes (SPTLC1, ULBP3, ZMYM4) were also implicated by subsequent gene-based analysis of uncommon variants (P<10−4) (Table 2). In addition, among the 53 SNPs, five (NUD: rs72501734, rs7918769, rs9935059; CUD: rs4933836, rs2294669) were also implicated as top SNPs in genetic pathways of the corresponding disorder (P<0.05), those pathway analyses having been performed using GWAS summary-level data in the GCTA-fastBAT program.
Table 1.
Single-point SNPs associated with SUDs in EA (P<5x10−5) in NESARC-III.
| Disorder | Chr | Pos | SNPa | Geneb | Function | Ref | Alt | N | AFc | Effectd | SE | Pathway Pvaluee | Pvalue |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AUD | 2 | 163678535 | rs4574131 | KCNH7 | Intron | T | G | 8446 | 0.676 | 0.16 | 0.04 | 0.15 | 6.39x10−6 |
| AUD | 2 | 163689483 | rs10930073 | KCNH7 | Intron | C | T | 8446 | 0.674 | 0.16 | 0.04 | NA | 8.83x10−6 |
| AUD | 2 | 163703739 | rs12476629 | Intergenic | Intergenic | T | G | 8446 | 0.677 | 0.16 | 0.04 | NA | 8.88x10−6 |
| AUD | 2 | 163682494 | rs12465289 | KCNH7 | Intron | G | T | 8446 | 0.674 | 0.16 | 0.04 | NA | 1.05x10−5 |
| AUD | 2 | 163696863 | rs16847467 | Intergenic | Intergenic | A | G | 8446 | 0.676 | 0.15 | 0.04 | NA | 1.15x10−5 |
| AUD | 2 | 163691715 | rs10180917 | KCNH7 | Intron | T | A | 8446 | 0.674 | 0.15 | 0.04 | NA | 1.36x10−5 |
| AUD | 2 | 163692689 | rs10171918 | KCNH7 | Intron | G | A | 8446 | 0.675 | 0.15 | 0.04 | NA | 1.56x10−5 |
| AUD | 2 | 163687672 | rs12476040 | KCNH7 | Intron | T | C | 8446 | 0.676 | 0.15 | 0.04 | NA | 1.59x10−5 |
| AUD | 2 | 163692347 | rs10169170 | KCNH7 | Intron | G | A | 8446 | 0.671 | 0.15 | 0.04 | NA | 1.64x10−5 |
| AUD | 2 | 163669642 | rs3912909 | KCNH7 | Intron | G | T | 8446 | 0.677 | 0.15 | 0.04 | NA | 1.71x10−5 |
| AUD | 2 | 163690219 | rs11898091 | KCNH7 | Intron | C | T | 8446 | 0.675 | 0.15 | 0.04 | NA | 1.93x10−5 |
| AUD | 2 | 163694878 | rs4667768 | KCNH7 | Intron | C | G | 8446 | 0.671 | 0.15 | 0.04 | NA | 2.12x10−5 |
| AUD | 2 | 163670203 | rs3912911 | KCNH7 | Intron | C | T | 8446 | 0.674 | 0.15 | 0.04 | NA | 2.25x10−5 |
| AUD | 2 | 163681163 | rs12611890 | KCNH7 | Intron | G | A | 8446 | 0.676 | 0.15 | 0.04 | NA | 2.34x10−5 |
| AUD | 2 | 163689167 | rs1389092 | KCNH7 | Intron | G | T | 8446 | 0.676 | 0.15 | 0.04 | NA | 2.34x10−5 |
| AUD | 2 | 163669750 | rs3912910 | KCNH7 | Intron | G | A | 8446 | 0.674 | 0.15 | 0.04 | NA | 2.36x10−5 |
| AUD | 2 | 163684757 | rs9646728 | KCNH7 | Intron | A | G | 8446 | 0.671 | 0.15 | 0.04 | NA | 2.48x10−5 |
| AUD | 2 | 163680351 | rs4667759 | KCNH7 | Intron | C | T | 8446 | 0.676 | 0.15 | 0.04 | NA | 2.51x10−5 |
| AUD | 2 | 144248718 | rs35789697 | ARHGAP15 | Intron | G | A | 8446 | 0.368 | −0.14 | 0.03 | 0.54 | 3.25x10−5 |
| AUD | 2 | 163666127 | rs7601793 | KCNH7 | Intron | G | A | 8446 | 0.669 | 0.15 | 0.04 | NA | 3.25x10−5 |
| AUD | 2 | 163694024 | rs10221628 | KCNH7 | Intron | C | G | 8446 | 0.673 | 0.15 | 0.04 | NA | 3.70x10−5 |
| NUD | 6 | 150390149 | rs72501734 | ULBP3 | Stop_Gain | G | C | 8353 | 0.012 | 0.65 | 0.15 | 5.27x10−3 | 1.10x10−5 |
| NUD | 16 | 77154226 | rs4888599 | Intergenic | Intergenic | C | G | 8353 | 0.456 | −0.14 | 0.03 | NA | 1.26x10−5 |
| NUD | 13 | 67697374 | rs7330064 | PCDH9 | Intron | C | G | 8353 | 0.936 | −0.29 | 0.07 | NA | 2.55x10−5 |
| NUD | 1 | 35847032 | rs34924462 | ZMYM4 | Nonsynonymous | G | A | 8353 | 0.034 | −0.37 | 0.09 | NA | 2.59x10−5 |
| NUD | 12 | 10383316 | rs10772246 | Intergenic | Intergenic | A | G | 8353 | 0.959 | −0.38 | 0.09 | NA | 2.62x10−5 |
| NUD | 10 | 84098252 | rs7918769 | NRG3 | Intron | T | A | 8353 | 0.191 | −0.17 | 0.04 | 4.89x10−3 | 3.34x10−5 |
| NUD | 10 | 84098752 | rs3897738 | NRG3 | Intron | C | T | 8353 | 0.191 | −0.17 | 0.04 | NA | 3.58x10−5 |
| NUD | 10 | 84094771 | rs3862550 | NRG3 | Intron | T | C | 8353 | 0.191 | −0.17 | 0.04 | NA | 3.66x10−5 |
| NUD | 16 | 22237118 | rs9935059 | EEF2K | Nonsynonymous | A | G | 8353 | 0.075 | −0.25 | 0.06 | 2.55x10−4 | 3.69x10−5 |
| NUD | 16 | 22237273 | rs17841292 | EEF2K | Nonsynonymous | C | G | 8353 | 0.073 | −0.25 | 0.06 | NA | 3.96x10−5 |
| NUD | 10 | 84110564 | rs7917348 | NRG3 | Intron | A | G | 8353 | 0.191 | −0.17 | 0.04 | NA | 4.38x10−5 |
| NUD | 10 | 84087761 | rs7476649 | NRG3 | Intron | C | T | 8353 | 0.130 | −0.19 | 0.05 | NA | 4.84x10−5 |
| NUD | 10 | 84113099 | rs11193893 | NRG3 | Intron | A | G | 8353 | 0.191 | −0.16 | 0.04 | NA | 4.87x10−5 |
| NUD | 10 | 84117730 | rs12248513 | NRG3 | Intron | G | A | 8353 | 0.190 | −0.16 | 0.04 | NA | 4.99x10−5 |
| CUD | 6 | 116325142 | rs3756772 | FRK | Nonsynonymous | C | T | 4913 | 0.420 | 0.25 | 0.06 | NA | 1.41x10−5 |
| CUD | 6 | 32814942 | rs1057149 | TAP1 | Nonsynonymous | C | T | 4913 | 0.025 | 0.77 | 0.18 | 0.15 | 1.99x10−5 |
| CUD | 6 | 32816772 | rs41550019 | TAP1 | Nonsynonymous | C | A | 4913 | 0.025 | 0.76 | 0.18 | NA | 2.02x10−5 |
| CUD | 10 | 84137873 | rs4933836 | NRG3 | Intron | C | T | 4913 | 0.581 | 0.24 | 0.06 | 7.25x10−3 | 2.14x10−5 |
| CUD | 10 | 84168912 | rs10787129 | NRG3 | Intron | T | A | 4913 | 0.582 | 0.23 | 0.06 | NA | 2.93x10−5 |
| CUD | 6 | 625673 | rs2294669 | EXOC2 | Intron | C | G | 4913 | 0.715 | 0.25 | 0.06 | 3.40x10−2 | 4.51x10−5 |
| CUD | 15 | 58862825 | rs71478677 | Intergenic | Intergenic | C | T | 4913 | 0.015 | 0.89 | 0.22 | NA | 4.66x10−5 |
| NMPOUD | 16 | 79682751 | rs1424233 | Intergenic | Intergenic | T | C | 4305 | 0.515 | −0.35 | 0.08 | NA | 1.89x10−5 |
| NMPOUD | 16 | 53769677 | rs6499640 | FTO | Intron | G | A | 4305 | 0.608 | −0.35 | 0.08 | NA | 2.58x10−5 |
| NMPOUD | 9 | 94821861 | rs12237598 | SPTLC1 | Intron | A | G | 4305 | 0.019 | 1.20 | 0.30 | 0.07 | 4.96x10−5 |
| Any SUD | 16 | 77154226 | rs4888599 | Intergenic | Intergenic | C | G | 10484 | 0.454 | −0.13 | 0.03 | NA | 6.74x10−6 |
| Any SUD | 12 | 11183108 | rs12318612 | TAS2R31 | Nonsynonymous | G | C | 10484 | 0.269 | 0.15 | 0.03 | 0.74 | 8.78x10−6 |
| Any SUD | 15 | 36139024 | rs2646782 | LOC100507 466 |
Intron | A | G | 10484 | 0.551 | 0.13 | 0.03 | NA | 1.19x10−5 |
| Any SUD | 19 | 58861808 | rs145144275 | A1BG | Nonsynonymous | A | G | 10484 | 0.010 | −0.63 | 0.15 | 0.48 | 2.86x10−5 |
| Any SUD | 7 | 115335567 | rs78431260 | Intergenic | Intergenic | C | T | 10484 | 0.050 | −0.28 | 0.07 | NA | 3.07x10−5 |
| Any SUD | 15 | 22816713 | rs8035524 | Intergenic | Intergenic | A | C | 10484 | 0.351 | −0.13 | 0.03 | NA | 3.56x10−5 |
| Any SUD | 7 | 115336270 | rs74960260 | Intergenic | Intergenic | T | C | 10484 | 0.050 | −0.28 | 0.07 | NA | 3.87x10−5 |
| Any SUD | 6 | 116325142 | rs3756772 | FRK | Nonsynonymous | C | T | 10484 | 0.427 | 0.12 | 0.03 | NA | 4.58x10−5 |
32 SNPs among 53 SNPs were previously reported in large GWAS as susceptibility loci with directional coherence for alcohol/nicotine/drug related phenotypes (P<0.05) as shown in Supplementary Table 12.
All genes (not always with the same SNP) were previously reported in large GWAS for alcohol/nicotine/drug related phenotypes (P<0.05).
AF: Allele freq for ALT allele.
Positive value indicates ALT allele is the risk allele.
P value for Pathway indicates when the SNP is a top SNP in a corresponding pathway.
Table 2.
Gene-based association signals (P<10−4) via uncommon variants in NESARC-III.
| Disorder | Genea | Range | N | Num of Poly variants | Q | rho | Pvalue(EA) | Pvalue(AA) | Pvalue (Burden Meta) |
|---|---|---|---|---|---|---|---|---|---|
| AUD | FER1L6 | 8:124864226-125132302 | 8446 | 12 | 214122 | 0 | 8.26x10−6 | 0.11 | 6.13x10−3 |
| NUD | FER1L6 | 8:124864226-125132302 | 8353 | 11 | 442609 | 0 | 2.17x10−10 | 4.19x10−4 | 7.58x10−3 |
| NUD | OMD | 9:95176526-95186836 | 8353 | 4 | 261850 | 0 | 1.49x10−8 | 8.66x10−2 | 0.11 |
| NUD | TUFT1 | 1:151512780-151556059,1:151512780-151556059 | 8353 | 2 | 120504 | 0.1 | 6.63x10−8 | 1.00 | 0.81 |
| NUD | SEMA3G | 3:52467267-52479043 | 8353 | 2 | 124432 | 0.3 | 3.36x10−6 | 0.45 | 2.60x10−2 |
| NUD | ARHGAP5 | 14:32546494-32628934,14:32546494-32628934 | 8353 | 2 | 148595 | 0.1 | 1.27x10−5 | 0.26 | 2.27x10−5 |
| NUD | ULBP3 | 6:150385742-150390202 | 8353 | 2 | 156889 | 0 | 1.31x10−5 | 0.31 | 6.16x10−2 |
| NUD | ZMYM4 | 1:35734567-35887545 | 8353 | 2 | 133676 | 0 | 3.59x10−5 | 0.55 | 1.14x10−2 |
| NUD | CENPP | 9:95087740-95377437 | 8353 | 15 | 272548 | 0 | 4.94x10−5 | 0.40 | 0.97 |
| NUD | LTBP2 | 14:74964885-75079034 | 8353 | 4 | 257167 | 1 | 9.59x10−5 | 0.18 | 4.08x10−4 |
| CUD | FER1L6 | 8:124864226-125132302 | 4913 | 12 | 103940 | 0 | 2.37x10−7 | 9.89x10−3 | 0.62 |
| CUD | LIPC | 15:58724174-58861073 | 4913 | 42 | 3.76E+06 | 0.1 | 4.04x10−5 | 1.00 | 0.68 |
| CUD | GPR124 | 8:37654400-37701504 | 4913 | 3 | 85829.4 | 0.5 | 9.21x10−5 | 0.77 | 3.93x10−4 |
| NMPOUD | TUBAL3 | 10:5435060-5446793,10:5435060-5446793 | 4305 | 3 | 46331.6 | 1 | 4.01x10−6 | 0.49 | 4.71x10−3 |
| NMPOUD | FER1L6 | 8:124864226-125132302 | 4305 | 12 | 32204.6 | 0 | 1.72x10−5 | 0.32 | 0.95 |
| NMPOUD | CAPRIN2 | 12:30862485-30907448,12:30862485-30907448,12:30862485-30907448,12:30862485-30907448,12:30862485-30907448 | 4305 | 3 | 13924.2 | 0 | 2.49x10−5 | 7.93x10−2 | 0.70 |
| NMPOUD | OMD | 9:95176526-95186836 | 4305 | 4 | 20258.2 | 0.1 | 5.39x10−5 | 0.13 | 8.27x10−3 |
| NMPOUD | SPTLC1 | 9:94793426-94877690,9:94841345-94877690 | 4305 | 2 | 23036.1 | 0 | 5.86x10−5 | 0.87 | 2.68x10−2 |
| NMPOUD | LPCAT2 | 16:55542912-55620582 | 4305 | 7 | 19723.8 | 0.1 | 6.74x10−5 | 0.62 | 1.43x10−3 |
| NMPOUD | C19orf71 | 19:3539154-3544028 | 4305 | 2 | 19822.4 | 0.7 | 9.16x10−5 | 0.28 | 1.23x10−3 |
| Any SUD | FER1L6 | 8:124864226-125132302 | 10484 | 11 | 357823 | 0 | 5.66x10−8 | 1.03x10−2 | 6.87x10−4 |
| Any SUD | TUFT1 | 1:151512780-151556059,1:151512780-151556059 | 10484 | 2 | 104955 | 0.1 | 5.04x10−6 | 0.24 | 0.69 |
| Any SUD | LENG1 | 19:54659378-54663446 | 10484 | 2 | 144494 | 0.8 | 6.51x10−6 | 1.11x10−2 | 2.09x10−6 |
| Any SUD | OMD | 9:95176526-95186836 | 10484 | 4 | 166409 | 0 | 5.34x10−5 | 0.13 | 0.25 |
All genes were previously reported in large GWAS as susceptibility loci for alcohol/nicotine/drug related phenotypes (P<0.05).
A total of 67 additional SNPs generated subthreshold association signals to at least 1 SUD in EA with single-point P values between 5x10−5 and 10−4 (Supplementary Table 4). Among these, 2 for NUD (rs2646782 and rs9571714) were replicated with directional coherence in AA (p<0.05). Furthermore, 4 of these SNPs (rs740160 in ARPC1A, rs283690 in RXRG, rs7179938 in LIPC, rs9935059 in EEF2K) were top SNPs in genetic pathways associated with the corresponding SUD (p<0.05).
Gene-based analysis of uncommon variants (MAF≤0.05).
In EA, gene-level testing for effects of uncommon alleles (MAF≤0.05) implicated 24 genes (P<10−4) associated with SUDs (Table 2 and Supplementary Fig. 5). Similar to single-point analysis and again because the focus of this study was cross-transmission of SUDs rather than locus discovery, we used a nominal threshold (P<10−4) to better test congruence between this national sample and previous SUD GWAS. Although not all genes reported here met the Bonferroni threshold (3.9x10−6) for gene-level testing (12,664 genes), all genes were previously implicated in large SUD GWAS [8, 9, 13, 19, 38, 39] at various significance levels (P<0.05).
Among these, 4 gene signals were replicated in AA at the p<0.05 level: 1 in NUD (FER1L6), 1 in CUD (FER1L6), and 2 in the grand analysis of SUD (FER1L6 and LENG1). In EA, we further identified 52 susceptibility genes (6 in AUD, 14 in NUD, 12 in CUD, 13 in NMPOUD and 7 in the grand analysis of SUD) with subthreshold gene-level P values between 10−4 and 5x10−4 Two of these (NUD: IGSF5 and CUD: BARHL2) were replicated in AA (P<0.05) (Supplementary Table 5).
Genetic pathway and set-based analysis of SNPs with common alleles
GCTA-fastBAT (a fast and flexible set-Based Association Test) was used for gene-based association analysis of common variants (MAF≥0.01) in GWAS summary data. A total of 36 gene signals (P<5x10−4) were identified by gene-based association in EA, and 4 genes were replicated in AA (Supplementary Table 6): 2 in AUD (SMIM17 and ZNF835), 1 in NUD (MXRA7), 1 in CUD (EXOC2), and 1 in a grand analysis of four SUDs (MXRA7). GCTA-fastBAT was also used to perform association analysis of genetic pathways in EA and AA separately, followed by meta-analysis of pathway association analysis in the combined sample using Fisher’s method. A total of 9 genetic pathways yielded P<10−3 in EA (Supplementary Table 7).
Relationship of SUD-associated SNPs to RNA eQTLs and mQTLs
To prioritize genes for follow-up functional studies, we used Summary-data-based Mendelian Randomization (SMR) to identify 20 SNPs (Supplementary Table 8) associated with both SUDs and mQTL/eQTLs (single SNP association P<1x10−4; P_SMR<10−3). These results may indicate that the effects of these SNPs on risk of SUD are mediated by ability of the SNPs to cis-modulate expression of the corresponding genes. There were 4 such genes in AUD (CRYGC, ARPC1A, FRK and ZKSCAN5), 2 in NUD (EEF2K and FRK), 7 in CUD (FRK, TAP1, EXOC2, LIPC and 3 intergenic variants), 3 in NMPOUD (FTO, SPTLC1 and 1 intergenic variant), and 4 in the grand analysis of SUD (TAS2R31, FRK, CRYGC, and 1 intergenic SNP).
Furthermore, we performed a co-localization analysis using the LocusFocus program [40] to test whether the genetic variants (single-point P<5x10−5) were colocalized with eQTLs in GTEx. This analysis yielded supportive evidence for seven genes (ARHGAP15, FRK, FTO, KCNH7, PCDH9, SPTLC1, ZMYM4) with relatively strong colocalization (Simple Sum P<10−3) of association signals with eQTL in various tissues (Supplementary Table 9).
Shared inheritance of substance use disorders.
In the case-control analysis of SUDs, one gene, FER1L6, was associated with all 4 SUDs and the grand analysis of all four SUDs via gene-based association of rare alleles. In the single SNP or gene-based analysis on rare alleles, four genes (NRG3, FRK, OMD, TUFT1) were associated with at least 2 SUDs. Furthermore, the grand analysis of four SUDs implicated 4 independent gene signals (Supplementary Fig. 6).
To boost power to detect gene effects on SUDs, and to identify genetic correlations between SUDs, we used multi-trait analysis of GWAS (MTAG), a method for joint analysis of summary statistics from multiple GWAS with overlapping samples. We jointly analyzed association results for 4 SUDs to identify susceptibility loci specific to individual SUDs, and pleiotropic for more than one SUD. Two SNPs (rs35789697, rs35942385) representing 1 gene (ARHGAP15) were implicated in susceptibility to all 4 SUDs (P<10−4) in EA after controlling for within-subject comorbidity (Supplementary Fig. 6 and Supplementary Table 10).
Finally, we carried out a multivariate genetic association analysis on all 4 SUDs through the TATES program [32], to identify relational pleiotropic effects on multiple SUDs. Multivariate analysis identified 25 SNPs (P<10−4) within 5 genes (NRG3, FRK, KCNH7, PCDH9 and TAP1) or intergenic regions conferring susceptibility to multiple SUDs (Supplementary Fig. 6 and Supplementary Table 11).
Prediction of mild, moderate and severe SUD by four SUD polygenic scores (PGS).
Polygenic scores (PGS) were calculated for all NESARC-III European ancestry samples (N=12,505) using summary statistics obtained from substance-specific GWAS, in which each of the 4 DSM-5 SUDs was compared to 3,959 “super controls” only. Each SUD PGS was then divided into deciles with each decile representing 1250 or 1251 subjects. Each substance-specific PGS was examined to assess its ability to predict severity (mild, moderate or severe) of the corresponding SUD, as well as to predict the other three SUDs. Notably, the GWAS from which the substance-specific SUD PGS was derived was qualitative for the presence or absence of the binary trait, and did not specify the severity of symptoms. Therefore, the relative proportions of mild, moderate and severe SUD in the different PGS deciles represent independent observations. Additionally, samples that were not originally used as cases or controls in the baseline GWAS analysis, such as samples with other SUD or related psychiatric diseases, were included in the PGS derivation.
Within disorder, and for example when using AUD PGS to predict the severity of AUD, the genetic risk and severity of each SUD escalated in higher PGS deciles, especially in the top decile (Fig. 3-6). For example, in the top decile for AUD PGS to AUD, 24.9%, 19.8% and 42.8% of subjects had mild, moderate, and severe AUD, respectively, while the corresponding percentages of the severity levels of AUD cases ranged between 0.2%-0.4% in the bottom decile (Fig. 3a). A similar pattern was observed for NUD PGS in relation to NUD (Fig. 4b). Meanwhile, 89.2% of CUD cases (n=851) and 100% NMPOUD cases (n=346) clustered in the top decile for CUD PGS to CUD and NMPOUD PGS to NMPOUD, respectively, perhaps partially due to the relatively small number of cases (Fig. 5c and Fig. 6d). Moreover, AUD and NUD PGS tended to predict comorbidity of these two SUDs (dual diagnosis) (Fig. 3b and Fig. 4a). Most prominently, the AUD PGS and NUD PGS revealed strong cross inheritances of these disorders (Fig. 3b and Fig. 4a). Conversely, neither AUD PGS nor NUD PGS strongly predicted CUD/NMPOUD (Fig. 3c-d and Fig. 4c-d), and likewise, neither did CUD PGS nor NMPOUD PGS strongly predict AUD or NUD (Fig. 5a-b and Fig. 6a-b). Future studies are needed to extend this analysis to other populations, particularly clinically ascertained SUD patients.
Fig. 3.
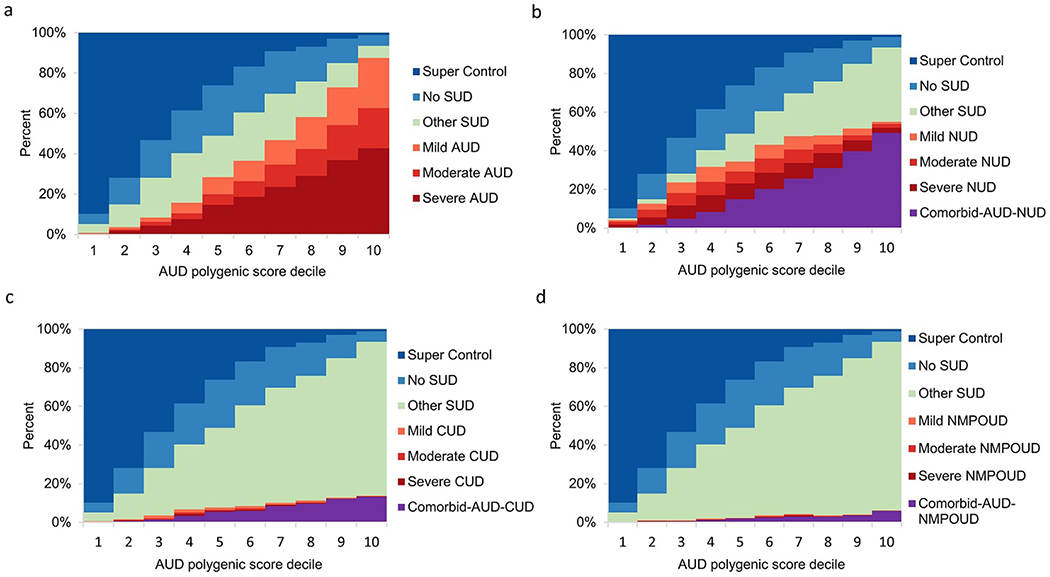
Relationship of AUD polygenic score to severity of four SUDs. Within NESARC-III EA, 12,505 participants were stratified according to decile of the AUD PGS distribution, each decile representing 1,250 or 1,251 individuals. AUD PGS was computed using summary statistics obtained from the AUD GWAS that compared AUD cases to the 3,959 “super controls” and in the absence of severity information. For each SUD, effect of AUD PGS on severity of that SUD, comorbidity between AUD and that SUD, other SUD, non-SUD but with related psychiatric diseases and super controls is shown.
a Ability of AUD PGS to predict AUD severity (mild, moderate or severe).
b Ability of AUD PGS to predict NUD severity (mild, moderate or severe) and NUD comorbid with AUD.
c Ability of AUD PGS to predict CUD severity (mild, moderate or severe) and CUD comorbid with AUD.
d Ability of AUD PGS to predict NMPOUD severity (mild, moderate or severe) and NMPOUD comorbid with AUD.
Fig. 6.
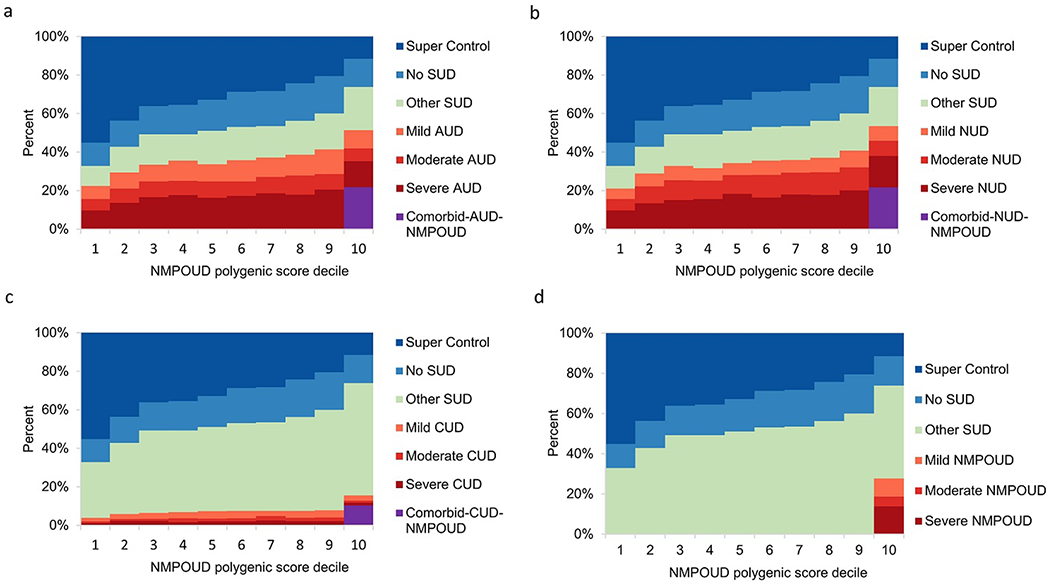
Relationship of NMPOUD polygenic score to severity of four SUDs. Within NESARC-III EA, 12,505 participants were stratified according to decile of the NMPOUD PGS distribution. NMPOUD PGS was computed using summary statistics obtained from the NMPOUD GWAS that compared NMPOUD cases to the 3,959 “super controls” and in the absence of severity information.
a Ability of NMPOUD PGS to predict AUD severity (mild, moderate or severe) and AUD comorbid with NMPOUD.
b Ability of NMPOUD PGS to predict NUD severity (mild, moderate or severe) and NUD comorbid with NMPOUD.
c Ability of NMPOUD PGS to predict CUD severity (mild, moderate or severe) and CUD comorbid with NMPOUD.
d Ability of NMPOUD PGS to predict NMPOUD severity (mild, moderate or severe).
Fig. 4.
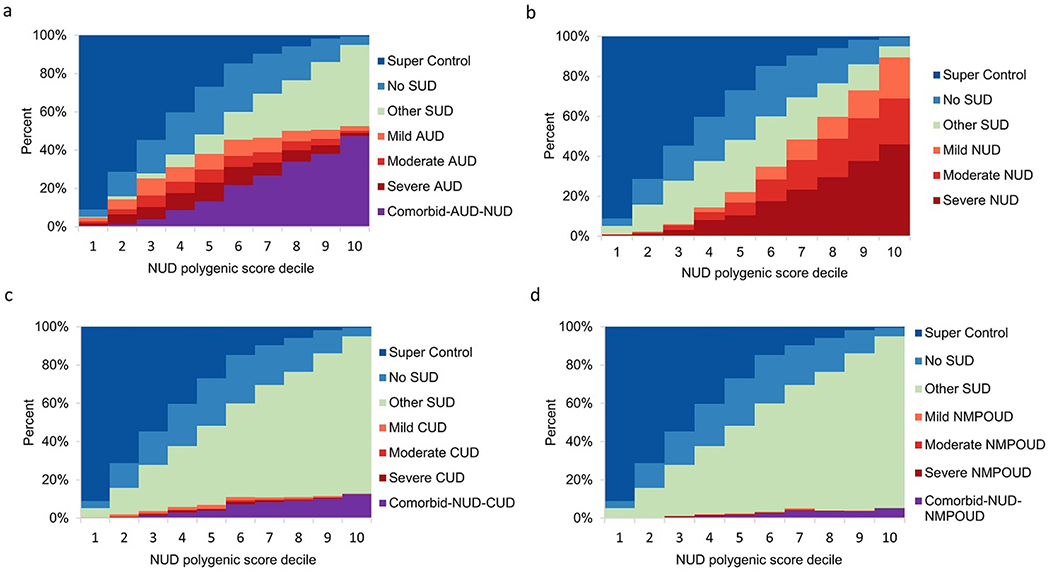
Relationship of NUD polygenic score to severity of four SUDs. Within NESARC-III EA, 12,505 participants were stratified according to decile of the NUD PGS distribution. NUD PGS was computed using summary statistics obtained from the NUD GWAS that compared NUD cases to the 3,959 “super controls” and in the absence of severity information.
a Ability of NUD PGS to predict AUD severity (mild, moderate or severe) and AUD comorbid with NUD.
b Ability of NUD PGS to predict NUD severity (mild, moderate or severe).
c Ability of NUD PGS to predict CUD severity (mild, moderate or severe) and CUD comorbid with NUD.
d Ability of NUD PGS to predict NMPOUD severity (mild, moderate or severe) and NMPOUD comorbid with NUD.
Fig. 5.
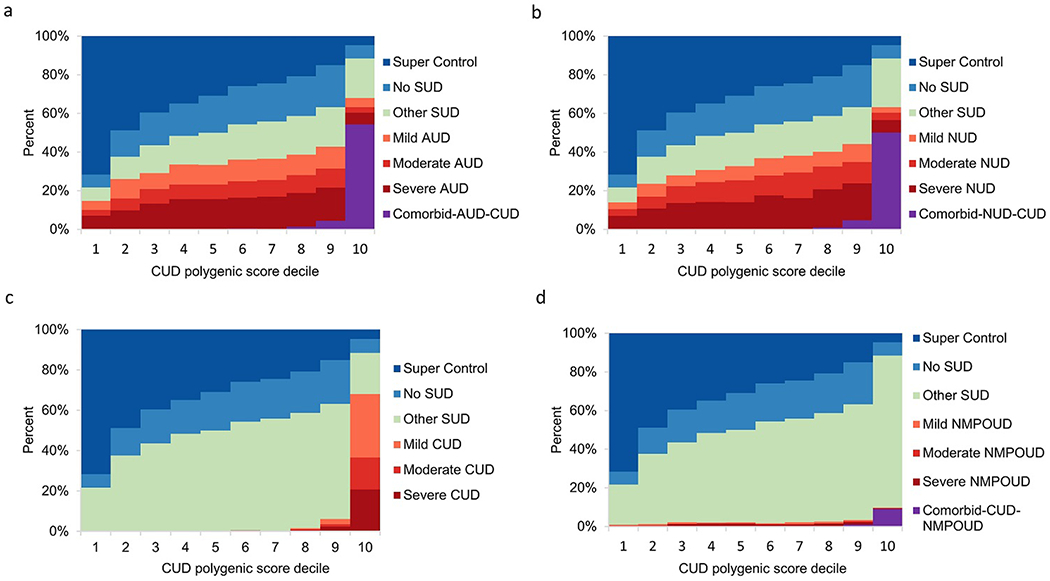
Relationship of CUD polygenic score to severity of four SUDs. Within NESARC-III EA, 12,505 participants were stratified according to decile of the CUD PGS distribution. CUD PGS was computed using summary statistics obtained from the CUD GWAS that compared CUD cases to the 3,959 “super controls” and in the absence of severity information.
a Ability of CUD PGS to predict AUD severity (mild, moderate or severe) and AUD comorbid with CUD.
b Ability of CUD PGS to predict NUD severity (mild, moderate or severe) and NUD comorbid with CUD.
c Ability of CUD PGS to predict CUD severity (mild, moderate or severe).
d Ability of CUD PGS to predict NMPOUD severity (mild, moderate or severe) and NMPOUD comorbid with CUD.
Phenotypic correlation and genetic correlation.
The genetic correlation between phenotypes is the proportion of phenotypic variance attributable to shared genetic components [41]. The NESARC-III study of deeply phenotyped individuals presents a rare opportunity to compare phenotypic and genetic correlations among pairs of SUDs simultaneously in a national sample.
Genetic correlations were calculated as pairwise correlations between polygenic scores (PGS) of 2 SUDs, where PGS scores of NESARC-III samples for each SUD were formulated on the basis of external large GWAS (non-NESARC-III, except for CUD for which we used NESARC-III CUD GWAS results) summary statistics. Here, we used externally derived PGS to exclude the effects of comorbid SUD cases in binary trait GWAS, which is different from the use of internally derived PGS to predict SUD severity that was described earlier. Phenotypic correlations were derived as the phi correlation coefficient (rφ) between a pair of SUDs. Confirming our within-study observations, we noted that AUD and NUD were most strongly genetically and phenotypically correlated. On the other hand, NMPOUD showed the weakest genetic and phenotypic correlations with other SUDs, among all pairs of SUDs (Supplementary Fig. 7).
Discussion
Increasingly, GWAS studies of the same SUDs are implicating the same genes, although with important exceptions due to differences in sampling frameworks and phenotyping. In a nationally representative, psychiatrically interviewed sample, we identified 21 independent SNPs (P<5X10−5) and 17 unique gene-based loci (P<10−4) that were associated with at least 1 SUD. Overall, a total of 29 unique genes were identified as top susceptibility loci to one or more SUDs. For gene identification, large GWAS samples are key. However, the NESARC-III nationally representative sample, with a total of 23,860 human subjects with genetic data, was well-suited for capturing polygenic effects at less than genome-wide significance. All of top nominally significant genes in NESARC-III were previously implicated in SUD GWAS [8, 9, 13, 19, 38, 39], although not always with the same SNP. Unlike prior GWAS for which these genes were identified based on single-SNP analysis of common alleles, some genes were implicated in our study based on tests of different single SNPs in the same gene, or gene-based analysis on rare variants located in coding regions (Supplementary Table 12). Three genes (ULBP3, FER1L6 and LENG1) were further replicated in the NESARC-III AA population. Additionally, we observed that 3 genes (SPTLC1, ULBP3 and ZMYM4) were implicated in SUDs based on both common and uncommon variants, whereas previous GWAS and candidate gene discoveries in SUDs have mainly been based on common variants only. These findings provided evidence that both uncommon and common alleles, sometimes at the same gene, contribute to risk of SUDs. These results support the impact of previously implicated genes in an epidemiologically representative sample, thus setting the stage for analysis of cross-transmission via PGS. In addition, we compared NESARC-III GWAS on AUD with genome-wide meta-analysis of problematic alcohol use in 435,563 individuals [42]. Among 29 independent variants that were genome-wide significant in problematic alcohol use study, 3 SNPs (rs1229984 in ADH1B, rs62250713 in CADM2 and rs8008020 in C14orf2) were replicated with directional coherence in NESARC-III; 11 SNPs were not replicated; the remaining 15 SNPs were not typed in NESARC-III array data. It bears mentioning that although NSENRC-III is a nationally representative sample, the current investigation is largely based on individuals of European ancestry. Due to the limitation of smaller sample size of other ancestry groups, the generalizability of these findings to other ancestry populations (such as African American, Hispanic American, East Asian and Native American) is limited. The fact that only 3 genes among our top hits were replicated in AA with nominal threshold (P<0.05) is related to the observation that PGS derived in one population tend not to be as highly predictive in others [43] although this idea was not tested here for the 4 SUDs.
Due to the depth of psychiatric assessment in NESARC-III we were able to identify a large sample of so-called “super-controls” who had neither a SUD nor another psychiatric disorder. Importantly, all phenotyping, sample collection, genotyping and analyses were performed simultaneously, super-controls and cases with various SUDs only being identified post-hoc. Among the 3,959 super controls, only 499 (12.6%) were lifetime abstainers from alcohol and nicotine. Many SUD GWAS have used exposed users (drinkers and smokers) as controls, assuming lifetime abstainers do not have the opportunity to develop SUD. Nevertheless, the exposure may progress at a slower pace for later onset SUD, and SUD vulnerability itself may be mediated by propensity for exposure (gene x environment correlation). As shown in both the Australia and Virginia twin studies, genes that influence choice to use or age at first use independently alter future risks associated with use [2, 44]. This may be regarded as an example of inverse causation, where “riskiness” of early use reflects, in substantial part, genetic predisposition to early use rather than the effect of early use itself [2, 44]. More importantly, the drinker/smoker may have a second SUD that is highly genetically correlated with the first disorder (e.g., NUD/AUD). Non-SUD users with related psychiatric diseases are also likely to share risk alleles with users who have SUDs. In addition, another common concern for control groups is whether they are more female or younger in composition. Females are less likely to develop SUDs, either because of genetic or cultural factors or both, and younger controls may manifest SUDs later in life. Among our super controls, 59.3% were female. Although prevalences of SUDs were lower in females than males, many other psychiatric diseases were more common in females, including major depressive disorder, persistent depression, anxiety disorders and PTSD. Therefore, our female super-controls were less likely to carry risk genes for those psychiatric diseases, and as could be important if some of these genes pleiotropically increase risk of SUDs. Finally, 65.6% of our super-controls were 45 years of age or older, and 89.0% of super-controls passed beyond mean age at AUD onset (i.e., 26.2 years) [23].
Prior to the advent of GWAS, shared versus unshared inheritance among SUDs was primarily studied in twin and sibling pairs, where cross-inheritance was measured [5] and to some extent also in families where other types of relative pairs were compared for cross-transmission of phenotypes. Consistent with our within-study PGS decile prediction model of SNP-based cross-inheritance of AUD and NUD, and as we replicated with PGS externally derived from outside GWAS, twin and sibling based cross-inheritance studies also found strongest cross-inheritance of AUD and NUD [5]. AUD and NUD are not only frequently comorbid, but are cross-inherited. In contrast, twin studies have revealed that opioid use disorder has the lowest cross-inheritance with other SUDs and the highest substance-specific inheritance [5]. In line with those findings, the three genetic correlations that we calculated as PGS correlations using external GWAS summary between NMPOUD and other SUDs were the lowest.
Genes involved in vulnerability to SUDs include substance-specific genes and genes that act on common genetic pathways involved in multiple, sometimes comorbid substance use disorders [2, 3, 45, 46]. We implicated 5 susceptibility genes (NRG3, FRK, FER1L6, OMD, TUFT1) associated with at least 2 SUDs. The finding of genes shared by SUDs could be due to the confound that the same person has more than 1 SUD. Furthermore, to explore the effects of SUD comorbidity, we adopted the multi-traits method (MTAG program) and discovered 1 gene (ARHGAP15) involved in all 4 SUDs that was among the AUD top hits. Multivariate analysis identified 5 additional genes that increase susceptibility to multiple SUDs.
Beyond identifying SNPs and gene-level association signals that individually account for a small proportion of phenotypic variance, we estimated combined contributions of multiple loci to phenotypic variance, and genetic correlations between pairs of SUDs. Unlike most studies of genetic correlations, which apply summary GWAS statistics obtained from different projects, our analyses on genetic correlation were performed using individual-level data within a single project. To our knowledge, this is the first study to calculate genetic correlation between two disorders using samples from a single project. In addition, we used internally derived PGS scores to predict different phenotypes, including other SUDs and three different levels of SUD severity, as defined by DSM5, and as such the results produced on cross-disorder polygenic effects and within-disorder prediction of severity were not tautological. Of note, the analyses of genetic correlations in previous studies had different limitations, namely sampling frameworks and depth of phenotype. Sampling frameworks can alter effect sizes of alleles and combinations of alleles, and quantitative estimates of cross-transmission. We observed that genetic correlations, extracted independently from phenotypic correlations, strongly predicted phenotypic correlations (comorbidity). This observation points to the importance of genes responsible for innate vulnerability, and common to multiple SUDs, rather than to a gateway drug, or other cause-effect actions of one drug use on another, an important mechanism in the genesis of SUDs [47]. These findings highlight the need to identify genes involved in shared pathways to addiction to different substances; for example, targeting genes that may alter neurofunctional domains such as negative emotionality, executive function, and stimulus salience/reward that can impact vulnerability to different SUDs [48, 49].
In conclusion, exome-focused genotyping analysis of four common SUDs in a nationally representative sample implicates both common and rare genetic variants in the pathogenesis of SUDs and tends to replicate GWAS of SUD performed in other sampling frameworks and using other SUD phenotypes. This study highlights that SNP-based genetic correlations predict comorbidity between pairs of SUDs. More importantly, consistent with previous cross-transmission data from twin and some GWAS studies, AUD and NUD were strongly cross-inherited. Cross-inheritance of different SUDs directly suggests the usefulness of measuring brain phenotypes, for example, differences in executive cognition, reward or emotionality, that are pan-diagnostic in relevance. The results also suggest the need to identify pathways of gene action that are modifiable and beneficial as an integral part of the prevention and treatment of specific SUDs. In this regard, NMPOUD was not highly genetically correlated with other SUDs, and thus, it would be important to focus on differences in the neurobiology of NMPOUD as well as each of the other SUDs, better targeting mechanisms of vulnerability that are unshared.
Supplementary Material
Acknowledgments
We thank the study participants and their families. The National Epidemiologic Survey on Alcohol and Related Conditions-III (NESARC-III) is funded by the National Institute on Alcohol Abuse and Alcoholism (NIAAA) with supplemental support from the National Institute on Drug Abuse. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Footnotes
Conflict of interest
The authors declare no competing interests.
Data availability
The individual-level genetic data (22,848 samples) with phenotypic variables (n=4,320) for NESARC-III (family history, adverse childhood experiences, substance use, mood, anxiety, personality and posttraumatic stress disorders) are available in dbGaP (accession: phs001590.v2.p1). [https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001590.v2.p1]
References
- 1.Whiteford HA, Degenhardt L, Rehm J, Baxter AJ, Ferrari AJ, Erskine HE, et al. Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. Lancet. 2013;382(9904):1575–1586. [DOI] [PubMed] [Google Scholar]
- 2.Kendler KS, Prescott CA, Myers J, Neale MC. The structure of genetic and environmental risk factors for common psychiatric and substance use disorders in men and women. Arch Gen Psychiatry. 2003;60(9):929–937. [DOI] [PubMed] [Google Scholar]
- 3.Ducci F, Goldman D. The genetic basis of addictive disorders. Psychiatr Clin North Am. 2012;35(2):495–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldman D, Oroszi G, Ducci F. The genetics of addictions: uncovering the genes. Nat Rev Genet. 2005;6(7):521–532. [DOI] [PubMed] [Google Scholar]
- 5.Goldman D, Bergen A. General and specific inheritance of substance abuse and alcoholism. Arch Gen Psychiatry. 1998;55(11):964–965. [DOI] [PubMed] [Google Scholar]
- 6.Cheng Z, Zhou H, Sherva R, Farrer LA, Kranzler HR, Gelernter J. Genome-wide Association Study Identifies a Regulatory Variant of RGMA Associated With Opioid Dependence in European Americans. Biol Psychiatry. 2018;84(10):762–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li D, Zhao H, Kranzler HR, Li MD, Jensen KP, Zayats T et al. Genome-wide association study of copy number variations (CNVs) with opioid dependence. Neuropsychopharmacology. 2015;40(4):1016–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu M, Jiang Y, Wedow R, Li Y, Brazel DM, Chen F, et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet. 2019;51(2):237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanchez-Roige S, Palmer AA, Fontanillas P, Elson SL, Adams MJ, Howard DM, et al. Genome-Wide Association Study Meta-Analysis of the Alcohol Use Disorders Identification Test (AUDIT) in Two Population-Based Cohorts. Am J Psychiatry. 2019; 176(2): 107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sherva R, Wang Q, Kranzler H, Zhao H, Koesterer R, Herman A, et al. Genome-wide Association Study of Cannabis Dependence Severity, Novel Risk Variants, and Shared Genetic Risks. JAMA Psychiatry. 2016;73(5):472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stringer S, Minică CC, Verweij KJ, Mbarek H, Bernard M, Derringer J, et al. Genome-wide association study of lifetime cannabis use based on a large meta-analytic sample of 32 330 subjects from the International Cannabis Consortium. Transl Psychiatry. 2016;6(3):e769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tobacco Genetics C. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet. 2010;42(5):441–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walters RK, Polimanti R, Johnson EC, McClintick JN, Adams MJ, Adkins AE, et al. Transancestral GWAS of alcohol dependence reveals common genetic underpinnings with psychiatric disorders. Nat Neurosci. 2018;21(12):1656–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldman D Polygenic Risk Scores in Psychiatry. Biol Psychiatry. 2017;82(10):698–699. [DOI] [PubMed] [Google Scholar]
- 15.Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, et al. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am J Hum Genet. 2017; 101(1):5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grant BF, Goldstein RB, Smith SM, Jung J, Zhang H, Chou SP, et al. The Alcohol Use Disorder and Associated Disabilities Interview Schedule-5 (AUDADIS-5): reliability of substance use and psychiatric disorder modules in a general population sample. Drug Alcohol Depend. 2015;148:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grant BF, Chu A, Sigman R, Amsbary M, Kali J, Sugawara Y, et al. Source and Accuracy Statement: National Epidemiologic Survey on Alcohol and Related Conditions-III (NESARC-III) [https://www.niaaa.nih.gov/sites/default/files/NESARC_Final_Report_FINAL_1_8_15.pdf]. National Institute on Alcohol Abuse and Alcoholism. 2014. [Google Scholar]
- 18.Jorgenson E, Thai KK, Hoffmann TJ, Sakoda LC, Kvale MN, Banda Y, et al. Genetic contributors to variation in alcohol consumption vary by race/ethnicity in a large multi-ethnic genome-wide association study. Mol Psychiatry. 2017;22(9):1359–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kranzler HR, Zhou H, Kember RL, Vickers Smith R, Justice AC, Damrauer S, et al. Genome-wide association study of alcohol consumption and use disorder in 274,424 individuals from multiple populations. Nat Commun. 2019;10(1):1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clarke TK, Adams MJ, Davies G, Howard DM, Hall LS, Padmanabhan S, et al. Genome-wide association study of alcohol consumption and genetic overlap with other health-related traits in UK Biobank (N=112 117). Mol Psychiatry. 2017;22(10): 1376–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thorgeirsson TE, Geller F, Sulem P, Rafnar T, Wiste A, Magnusson KP, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452(7187):638–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chou SP, Goldstein RB, Smith SM, Huang B, Ruan WJ, Zhang H, et al. The Epidemiology of DSM-5 Nicotine Use Disorder: Results From the National Epidemiologic Survey on Alcohol and Related Conditions-III. J Clin Psychiatry. 2016;77(10):1404–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grant BF, Chou SP, Saha TD, Pickering RP, Kerridge BT, Ruan WJ, et al. Prevalence of 12-Month Alcohol Use, High-Risk Drinking, and DSM-IV Alcohol Use Disorder in the United States, 2001-2002 to 2012-2013: Results From the National Epidemiologic Survey on Alcohol and Related Conditions. JAMA Psychiatry. 2017;74(9):911–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grant BF, Saha TD, Ruan WJ, Goldstein RB, Chou SP, Jung J, et al. Epidemiology of DSM-5 Drug Use Disorder: Results From the National Epidemiologic Survey on Alcohol and Related Conditions-III. JAMA Psychiatry. 2016;73(1):39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhan X, Hu Y, Li B, Abecasis GR, Liu DJ. RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics. 2016;32(9):1423–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88(1):76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feng S, Liu D, Zhan X, Wing MK, Abecasis GR. RAREMETAL: fast and powerful meta-analysis for rare variants. Bioinformatics. 2014;30(19):2828–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48(5):481–487. [DOI] [PubMed] [Google Scholar]
- 31.Turley P, Walters RK, Maghzian O, Okbay A, Lee JJ, Fontana MA, et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat Genet. 2018;50(2):229–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Sluis S, Posthuma D, Dolan CV. TATES: efficient multivariate genotype-phenotype analysis for genome-wide association studies. PLoS Genet. 2013;9(1):e1003235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Euesden J, Lewis CM, O’Reilly PF. PRSice: Polygenic Risk Score software. Bioinformatics. 2015;31(9):1466–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song W, Kossowsky J, Torous J, Chen CY, Huang H, Mukamal KJ, et al. Genome-wide association analysis of opioid use disorder: A novel approach using clinical data. Drug Alcohol Depend. 2020;217:108276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watanabe K, Stringer S, Frei O, Umicevic Mirkov M, de Leeuw C, Polderman TJC, et al. A global overview of pleiotropy and genetic architecture in complex traits. Nat Genet. 2019;51(9):1339–1348. [DOI] [PubMed] [Google Scholar]
- 36.Yu C, McClellan J. Genetics of Substance Use Disorders. Child Adolesc Psychiatr Clin N Am. 2016;25(3):377–385. [DOI] [PubMed] [Google Scholar]
- 37.Hodgkinson CA, Yuan Q, Xu K, Shen PH, Heinz E, Lobos EA, et al. Addictions biology: haplotype-based analysis for 130 candidate genes on a single array. Alcohol Alcohol. 2008;43(5):505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou H, Rentsch CT, Cheng Z, Kember RL, Nunez YZ, Sherva RM, et al. Association of OPRM1 Functional Coding Variant With Opioid Use Disorder: A Genome-Wide Association Study. JAMA Psychiatry. 2020;77(10):1072–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Demontis D, Rajagopal VM, Thorgeirsson TE, Als TD, Grove J, Leppala K, et al. Genome-wide association study implicates CHRNA2 in cannabis use disorder. Nat Neurosci. 2019;22(7):1066–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Panjwani N, Wang F, Mastromatteo S, Bao A, Wang C, He G, et al. LocusFocus: Web-based colocalization for the annotation and functional follow-up of GWAS. PLoS Comput Biol. 2020;16(10):e1008336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47(11):1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou H, Sealock JM, Sanchez-Roige S, Clarke TK, Levey DF, Cheng Z, et al. Genome-wide meta-analysis of problematic alcohol use in 435,563 individuals yields insights into biology and relationships with other traits. Nat Neurosci. 2020;23(7):809–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duncan L, Shen H, Gelaye B, Meijsen J, Ressler K, Feldman M, et al. Analysis of polygenic risk score usage and performance in diverse human populations. Nat Commun. 2019;10(1):3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heath AC, Martin NG. Genetic models for the natural history of smoking: evidence for a genetic influence on smoking persistence. Addict Behav. 1993;18(1):19–34. [DOI] [PubMed] [Google Scholar]
- 45.Han C, McGue MK, Iacono WG. Lifetime tobacco, alcohol and other substance use in adolescent Minnesota twins: univariate and multivariate behavioral genetic analyses. Addiction. 1999;94(7):981–993. [DOI] [PubMed] [Google Scholar]
- 46.Hicks BM, Krueger RF, Iacono WG, McGue M, Patrick CJ. Family transmission and heritability of externalizing disorders: a twin-family study. Arch Gen Psychiatry. 2004;61(9):922–928. [DOI] [PubMed] [Google Scholar]
- 47.Kandel DB, Kandel ER. A molecular basis for nicotine as a gateway drug. N Engl J Med. 2014;371(21):2038–2039. [DOI] [PubMed] [Google Scholar]
- 48.Kwako LE, Momenan R, Litten RZ, Koob GF, Goldman D. Addictions Neuroclinical Assessment: A Neuroscience-Based Framework for Addictive Disorders. Biol Psychiatry. 2016;80(3):179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kwako LE, Schwandt ML, Ramchandani VA, Diazgranados N, Koob GF, Volkow ND, et al. Neurofunctional Domains Derived From Deep Behavioral Phenotyping in Alcohol Use Disorder. Am J Psychiatry. 2019;176(9):744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The individual-level genetic data (22,848 samples) with phenotypic variables (n=4,320) for NESARC-III (family history, adverse childhood experiences, substance use, mood, anxiety, personality and posttraumatic stress disorders) are available in dbGaP (accession: phs001590.v2.p1). [https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001590.v2.p1]