

Abstract
Background
Exercise training, as well as catecholaminergic stimulation, increase the incidence of arrhythmic events in patients affected with arrhythmogenic right ventricular cardiomyopathy (ARVC) correlated with plakophilin-2 (PKP2) mutations. Separate data show that reduced abundance of PKP2 leads to dysregulation of intracellular Ca2+ (Ca2+i) homeostasis. Here, we study the relation between exercise and/or catecholaminergic stimulation, Ca2+i homeostasis and arrhythmogenesis in PKP2-deficient murine hearts.
Methods
Experiments were carried out in a murine model of PKP2 deficiency (PKP2cKO). For training, mice underwent 75 minutes of treadmill running once per day, 5 days each week for six weeks. We used multiple methods including imaging, high-resolution mass spectrometry and electrocardiography, as well as pharmacological approaches to study the functional properties of cells/hearts in vitro and in vivo.
Results
In myocytes from PKP2cKO animals, training increased sarcoplasmic reticulum Ca2+ load, increased the frequency and amplitude of spontaneous ryanodine receptor (RyR2)-mediated Ca2+ release events (“sparks”), and changed the time course of sarcomeric shortening. Phosphoproteomics analysis revealed that training led to hyper-phosphorylation of phospholamban (Pln) in residues 16 and 17, suggesting a catecholaminergic component. Isoproterenol-induced increase in Ca2+i transient amplitude showed a differential response to beta-adrenergic blockade that depended on the purported ability of the blockers to reach intracellular receptors. Additional experiments showed significant reduction of ISO-induced Ca2+i sparks and ventricular arrhythmias in PKP2cKO hearts exposed to an experimental blocker of RyR2 channels.
Conclusions
Exercise disproportionately affects Ca2+i homeostasis in PKP2-deficient hearts, in a manner facilitated by stimulation of intracellular beta-ARs and hyper-phosphorylation of Pln. These cellular changes create a pro-arrhythmogenic state that can be mitigated by RyR2 blockade. Our data unveil an arrhythmogenic mechanism for exercise-induced, or catecholaminergic life-threatening arrhythmias in the seting of PKP2 deficit. We suggest that membrane-permeable beta-blockers are potentially more efficient for ARVC patients, highlight the potential for RyR2 channel blockers as treatment for the control of heart rhythm in the population at risk and propose that PKP2-dependent, and Pln-dependent ARVC-related arrhythmias have a common mechanism.
Keywords: arrhythmogenic right ventricular cardiomyopathy, plakophilin 2, exercise, beta1 adrenergic receptors, phospholamban
INTRODUCTION
Desmosomes are intercellular structures that maintain mechanical continuity between cells. Plakophilin-2 (PKP2) is an essential component of the desmosome, and of the connexome as a whole.1,2 Mutations in PKP2 lead to arrhythmogenic right ventricular cardiomyopathy (ARVC), a disease characterized by high propensity to life-threatening arrhythmias in young individuals and progressive fibrofatty infiltration of the ventricular walls at the expense of muscle mass.2,3 The arrhythmogenic and cardiomyopathic components do not progress together and sudden cardiac arrest in young individuals often occurs during the concealed stage of the disease.4–6 Understanding arrhythmia mechanisms remains a necessary step to improve risk assessment, prevention and therapy.
Exercise negatively impacts ARVC progression and arrhythmia incidence.7–9 The mechanism remains unclear, though a catecholaminergic component has been proposed.10,11 There is evidence that sympathetic input can be particularly arrhythmogenic in ARVC patients. In fact, an isoproterenol (ISO) bolus can unmask a concealed arrhythmia risk in patients suspect of ARVC,12,13 and sympathectomy is an effective method of treatment in some patients.14 Yet, the relation between catecholaminergic input, exercise, and arrhythmias in the setting of PKP2 deficiency remains understudied.
PKP2 deficiency can disrupt the equilibrium of intracellular calcium (Ca2+i), facilitating triggered activity and arrhythmogenesis.15,16 Here we test the hypotheses that: a) Ca2+i dysregulation in PKP2-deficient ventricular myocytes is accentuated by exercise (treadmill running) and/or by beta1-adrenergic receptor (beta1-AR) stimulation, b) the effect is related to changes in the phosphorylation state of key molecular components that control Ca2+i homeostasis and c) the catecholaminergic arrhythmias can be prevented by RyR2 blockade. Our results unveil an arrhythmogenic mechanism for exercise-induced, or catecholaminergic life-threatening arrhythmias in the seting of PKP2 deficit. We propose that membrane-permeable beta-blockers are potentially more effective in the prevention of exercise/catecholaminergic-related arrhythmias in ARVC, highlight the potential for RyR2 channel blockers as treatment for the control of heart rhythm in these patients and suggest that PKP2-dependent, and Pln-dependent ARVC-related arrhythmias have a common mechanism.
METHODS
Experiments were performed in 3-6 month old mice expressing a cardiac-specific, tamoxifen (TAM)-activated deletion of PKP2 (PKP2cKO), and age- and gender-matched controls.15 Cre-negative, flox-positive, TAM-injected littermates were used as controls. Procedures conformed with the Guide for Care and Use of Laboratory Animals of the National Institutes of Health and were approved by the NYU-IACUC committee. Unless specified, hearts were studied 21 days post-TAM, a point at which an arrhythmogenic cardiomyopathy of right ventricular predominance is manifest.15 For training, mice underwent a 75-minutes treadmill running per day, 5 days a week for six weeks (three weeks before the first TAM injection and three weeks afterwards; Figure S1). Details of the training protocol and its effects on murine cardiac function are in Data supplement and in.17
Procedures for cardiomyocyte dissociation, Ca2+ imaging, measurements of sarcomere shortening, tissue homogenisation, peptide preparation, TMT labelling, phosphopeptide enrichment, off-line peptide fractionation, mass spectrometry, and electrocardiography followed those published and are detailed in Data supplement. For mass spectrometry and for cellular work in hearts of mice 21 days post-TAM, hearts were collected and LV dissected out. Only LV was used for analysis given the extensive fibrosis seen in the RV free wall at this stage.15
Isoproterenol (ISO) and two beta-blockers, propranolol (1 μM) and sotalol (25 μM), were used. Ventricular myocytes were pre-incubated for 5 minutes with antagonists before ISO incubation and Ca2+ transients examined accordingly, following published protocols.18 The RyR2 blocker ent-(+)-verticilide (ent-vert)19 was used in isolated cells (25 μM) and in anesthetized mice (30 mg/kg). Preparation and methods of administration in Data supplement.
Data availability
Mass spectrometry proteomics and phosphoproteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository with dataset identifier PXD025956 (accessible through https://www.ebi.ac.uk/pride/archive) and project name “Investigation of changes in the proteome and the phosphoproteome of the heart induced by exercise, either in control or in PKP2-deficient hearts in relation to ARVC”. Additional information can be made available from the corresponding authors upon reasonable request.
Statistical analysis
Numerical results reported as mean and standard deviation (SD). Pooled data originating from separate animals were tested using the workflow designed by Sikkel et al20 to determine the validity of the assumption of independence vis a vis data clustering. When clustering was identified, hierarchical analysis was employed. All data sets were tested for normal distribution (Shapiro-Wilk and Kolmogorov-Smirnov tests), and significance determined by parametric or non-parametric methods, as appropriate. All comparisons were evaluated for effect size (Cohen’s d). Statistical significance was corroborated by linear mixed-effect analysis (GraphPad Prism). All statistical data for non-omics experiments are reported in Table S1. Specific methods were tailored to individual datasets and are specified in the figure legends. Data were analyzed using GraphPad Prism v8.0. Sarcomere shortening data were analyzed using IonWizard (IonOptix, Westwood, MA, USA).
RESULTS
Exercise leads to increased SR load and spontaneous Ca2+ release events in PKP2cKO myocytes
Previous studies indicate that loss of PKP2 leads to dysregulation of Ca2+i and increased RyR2-mediated Ca2+ release.15,16 Separate studies have shown that exercise is a negative factor in ARVC progression.7–9 We tested the hypothesis that Ca2+i dysregulation in PKP2cKO myocytes is accentuated by exercise.
Unless specified, myocytes were obtained 21 days post-TAM, a time at which a phenotype of arrhythmogenic cardiomyopathy of right ventricular predominance is observed.15 Isolated myocytes were loaded with the Ca2+ indicator Fluo-8 AM (see16 and methods) and confocal line-scan images obtained to detect spontaneous Ca2+ release events (sparks). Acquisition was preceded by a train of electrical pulses (1 Hz for 10 seconds). Fig.1A shows representative line scans obtained from myocytes isolated from mice either sedentary control (left), sedentary PKP2cKO (middle), or PKP2cKO that completed six-weeks of training (right).17 We confirmed that PKP2 loss leads to increased frequency and amplitude of Ca2+ sparks (Fig.1B–C, comparing control sedentary to PKP2cKO sedentary; also).15. More importantly, we found a further increase in both parameters in myocytes from trained PKP2cKO mice (Fig.1B–C; comparing PKP2cKO sedentary vs PKP2cKO trained). Yet, contrary to what happens in PKP2cKO myocytes, training in control animals did not lead to an increase in the frequency or amplitude of Ca2+ sparks (Fig. 1B–C, green bar outline and green data points; compare control sedentary vs control trained). Additional data are provided in Figure S2. Of note, PKP2cKO myocytes obtained a week prior (14 days post-TAM), during the concealed stage of the disease and therefore at a point where separate dissection of LV and RV was feasible (see16) showed exercise-induced increase in spark frequency in RV but not LV myocytes (Figure S3).
Figure 1. Ca2+ sparks in cardiac myocytes from control and plakophilin-2 conditional knockout (PKP2cKO) hearts.
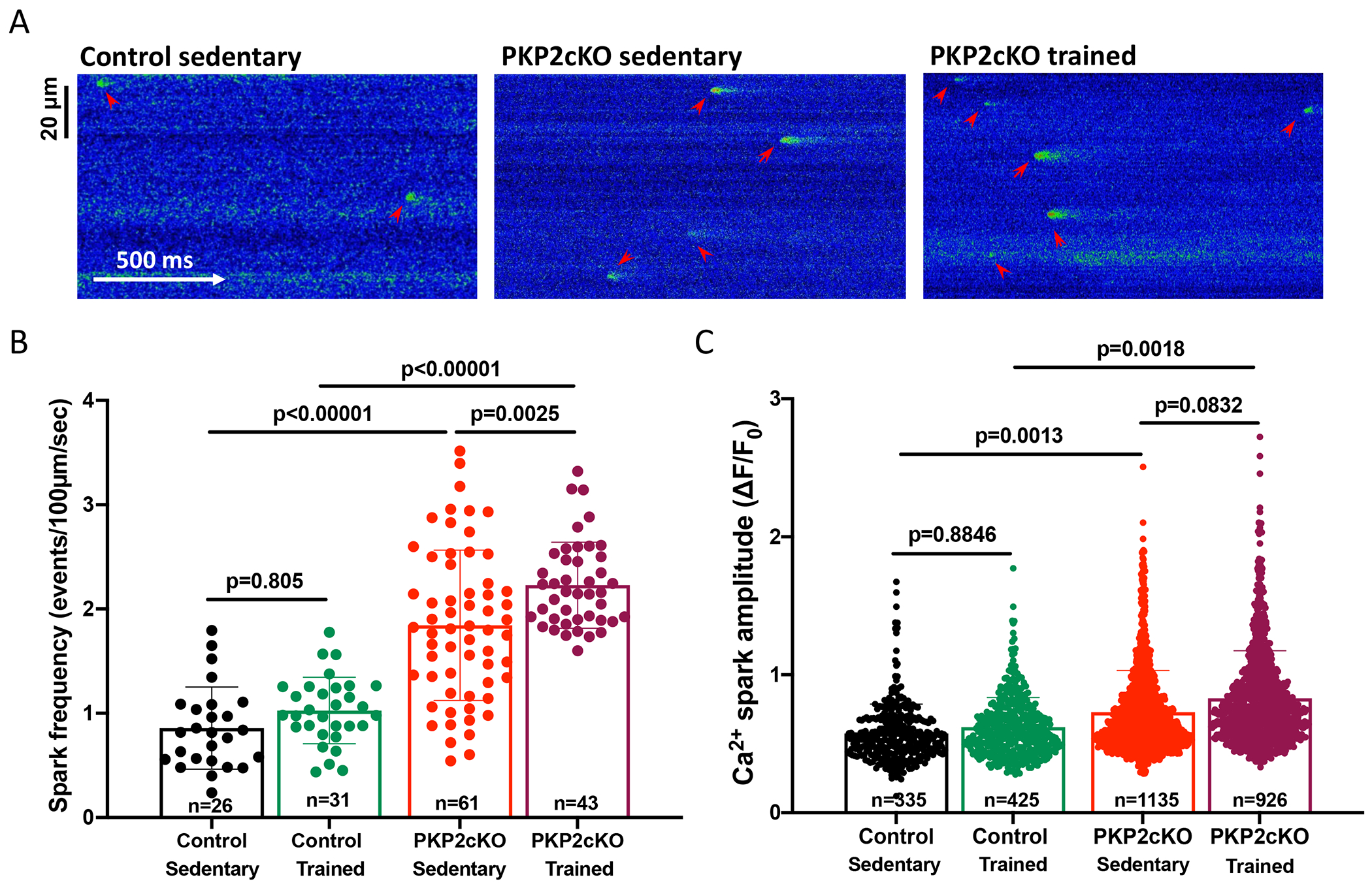
(A) Confocal line-scan images of Ca2+ sparks (green; emphasized by red arrowheads) recorded from cardiac myocytes isolated from sedentary control, sedentary PKP2cKO and trained PKP2cKO mice 21 days post-tamoxifen injection. (B) Mean frequency of Ca2+ sparks, reported as average number of events per second in a 100 μm line. (C) Average peak amplitude (ΔF/F0). Data are presented as mean ± SD. Black bars and symbols: sedentary control; green bars and symbols: trained control; red bars and symbols: sedentary PKP2cKO; purple bars and symbols: trained PKP2cKO mice. Number of cells studied noted in the corresponding bars, 3–6 mice per group. Test for clustering versus validity of assumption of independence between data obtained from more than one mouse and one cell within a group, as in Sikkel et al (20). Significance of clustering is reported in Table S1. For spark frequency, significance was assessed by two-way ANOVA followed by Tukey’s multiple comparison’s test. For spark amplitude, hierarchical statistics on animal and cell level were performed; data were logarithmically transformed before statistical analysis. Statistical significance was corroborated by linear mixed-effects analysis (Table S1). Effect size (Cohen’s d) and determination of normal distribution (Shapiro-Wilk and Kolgomorov-Smirnov tests) of both datasets also reported in Table S1.
Increased spark frequency/amplitude may result, at least in part, from increased abundance (“load”) of Ca2+ within the sarcoplasmic reticulum (SR). SR load was measured as the peak amplitude of a Ca2+i transient elicited by a caffeine pulse (details in “Methods” and16). Fig.2A shows exemplary time-space plots and traces of caffeine-induced Ca2+i transients from sedentary control, sedentary PKP2cKO, and trained PKP2cKO myocytes. As in the case of Ca2+ sparks (Figure 1), training leads to increased SR Ca2+ content in PKP2cKO myocytes (purple bar outline and symbols in Fig.2B) but not in control cells (green bar outline and symbols in Fig.2B). No differences were observed in kinetics of rise or relaxation of the caffeine-induced Ca2+i transient (Figure S4). Overall, the data indicate a genotype-specific response to exercise that can lead to a pro-arrhythmogenic state after loss of PKP2.
Figure 2. Ca2+ content in sarcoplasmic reticulum of control and plakophilin-2 conditional knockout (PKP2cKO) cardiac myocytes.
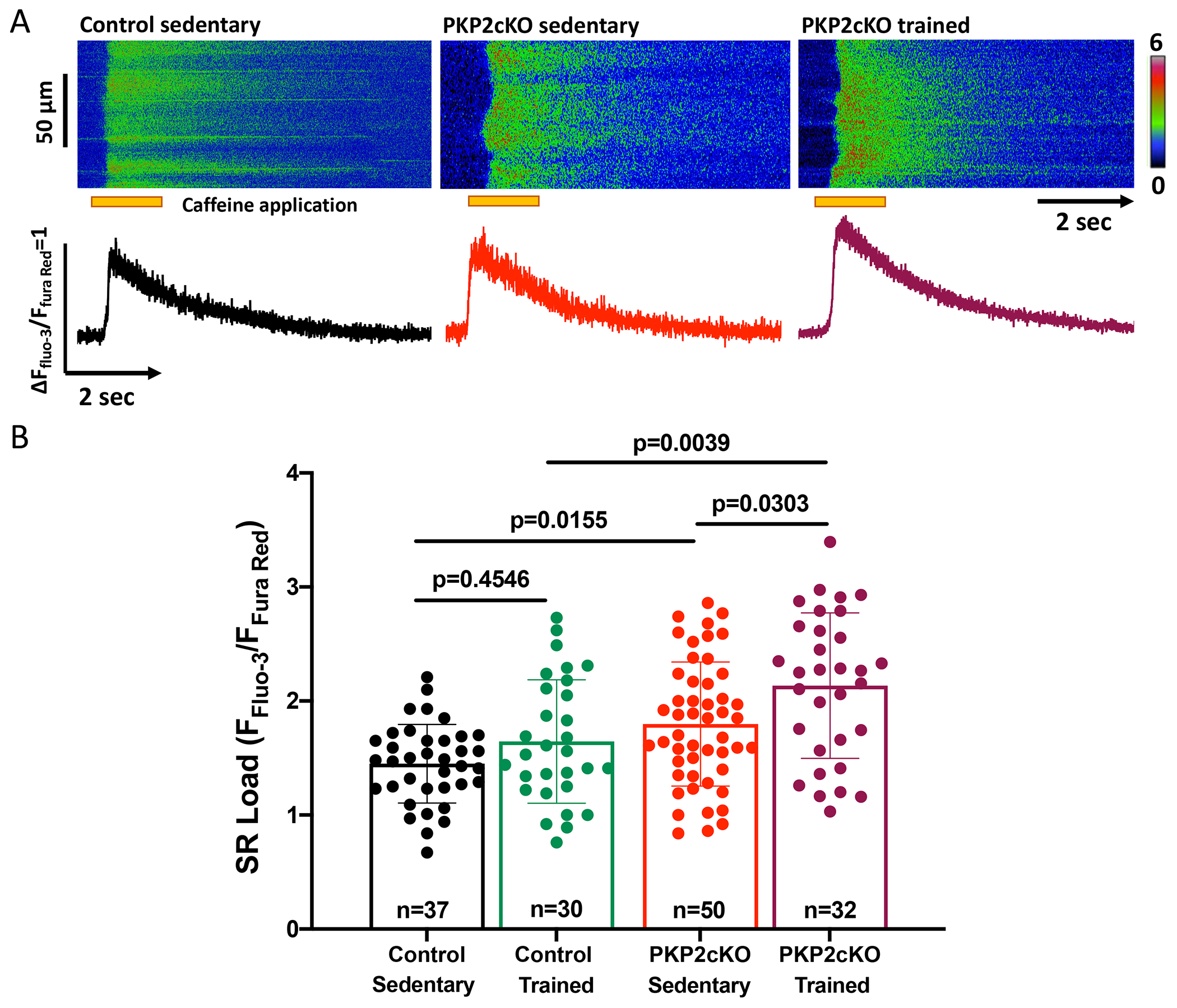
(A) Upper panel; Confocal line-scan images (1.43 ms/line) recorded from cardiac myocytes isolated from sedentary control, sedentary PKP2cKO or trained PKP2cKO mice 21 days post-tamoxifen injection. The pulse of caffeine (10 mmol/L) is indicated by the orange bar at the bottom of the image. Intracellular calcium changes detected by a ratiometric method (FFluo-3/FFura Red; see also Methods). Bottom panel: Time course of calcium transients displayed in the top. (B): Cumulative data, presented as mean ± SD. Black bars and symbols: sedentary control; green bars and symbols: trained control; red bars and symbols: sedentary PKP2cKO; purple bars and symbols: trained PKP2cKO mice. Number of cells studied is noted in the corresponding bars, 3-6 mice per group. Test for clustering versus validity of assumption of independence between data obtained from more than one mouse within a group, as in Sikkel et al (20). Results determined no need for hierarchical statistics (significance of clustering in Table S1). Significance was assessed by two-way ANOVA followed by Tukey’s multiple comparison’s test. Statistical significance was corroborated by linear mixed-effects analysis (Table S1). Effect size (Cohen’s d) and determination of normal distribution (Shapiro-Wilk and Kolgomorov-Smirnov tests) also reported in Table S1.
Exercise induces a differential proteome and phosphoproteome in control and in PKP2cKO hearts.
We speculated that the observed phenotypes may result, at least partially, from changes in abundance or phosphorylation state of proteins relevant to Ca2+i homeostasis. We therefore performed an unbiased investigation of changes in the cardiac proteome and phosphoproteome induced by training, either in control or PKP2cKO mice. The workflow is illustrated in Figure 3A. For both control and PKP2cKO, results from sedentary animals were compared to those from the trained group. Specific comparisons were “Control trained vs Control sedentary,” and “PKP2cKO trained vs PKP2cKO sedentary”. In both cases, the variable was “training.” LV tissue was subjected to quantitative proteome and phosphoproteome measurements applying tandem mass tag (TMT) labeling and high-resolution mass spectrometry (see Methods). Peptides were enriched with titanium dioxide chromatography and samples pre-fractionated (12 fractions) for phosphoproteomics. Number of proteins and phosphorylated peptides per heart sample are shown in Figure 3B. Technical reproducibility of TMT sets of Control and PKP2cKO measurements was high, with average Pearson correlation coefficients of biological replicates of 0.95 for both proteome and phosphoproteome (Figures S4 & S5). From these data we quantified 4,378 proteins in control (Table S2) and 4,353 proteins in PKP2cKO animals (Table S3). Principal component analysis (PCA) of the data classified the samples according to the exercise variable (Figure S6).
Figure 3. Workflow and data summary of proteomics and phosphoproteomics experiments.
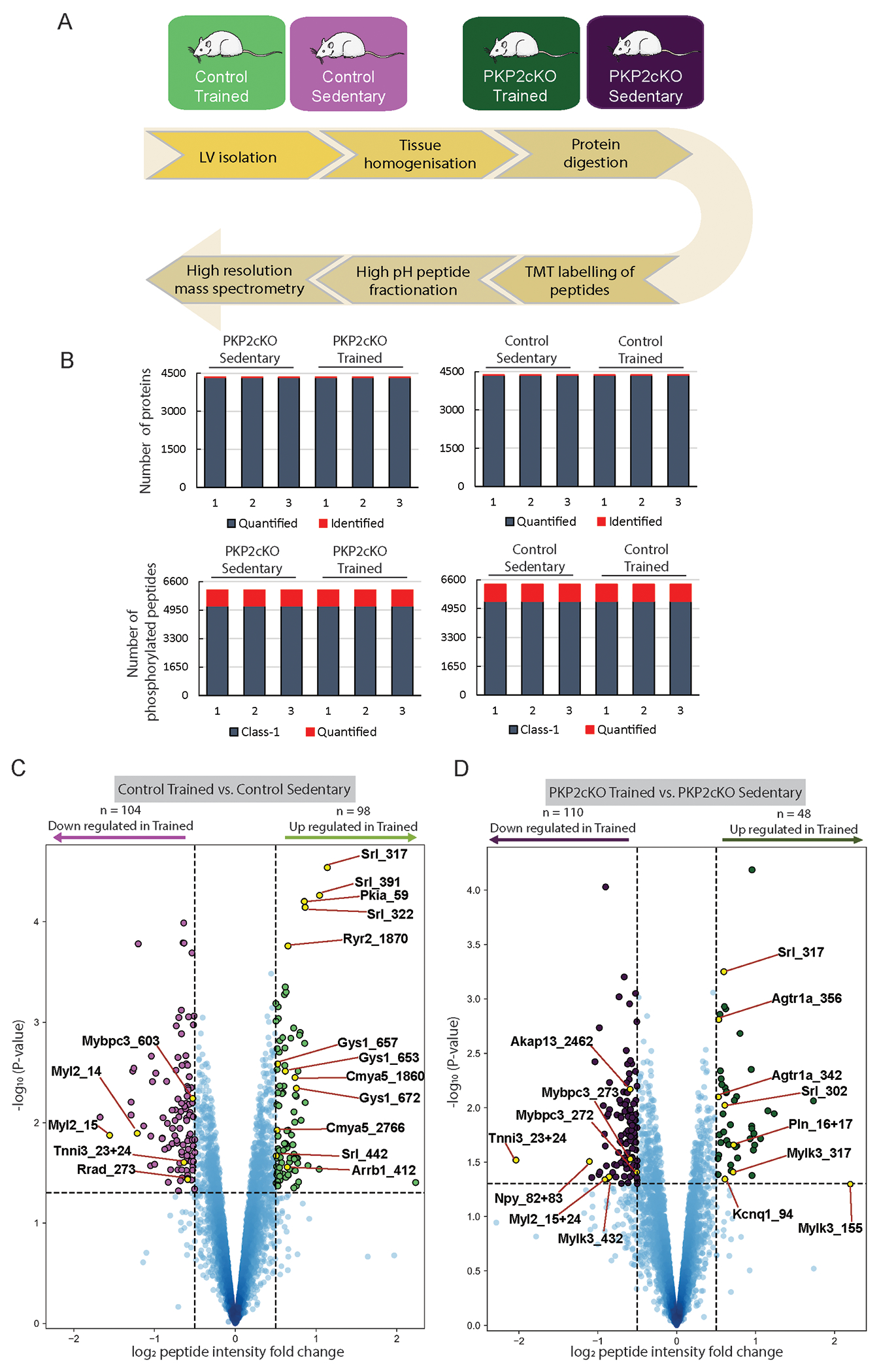
(A) Control and PKP2cKO mice were either kept sedentary, or trained (treadmill running 1 hour a day; six times a week for six weeks; green). Details of training protocol in Online Methods. Left ventricular tissue was isolated, proteins extracted and subjected to quantitative proteome and phosphoproteome measurements by LC-MS/MS analysis. (B) Bar graphs summarize the number of proteins (top) and phosphorylated peptides (bottom) measured for each heart sample. For proteome samples red indicates the number of identified proteins and blue the number of proteins quantified across samples. For phosphoproteome, red indicates the number of phosphorylated peptides quantified across samples and blue indicates that the phosphorylation site localization is deemed a class 1 site (i.e., localization probability greater than 0.75). (C-D) Volcano plot analysis showing phosphorylated peptides that are either downregulated (purple) or upregulated (green) in response to exercise for control (C) or PKP2cKO animals (D). Phosphorylated peptides were considered regulated at a combined cut-off of p-value < 0.05 and a numerical log2 fold change greater than 0.50. A few selected phosphorylation sites are indicated in the plot. Each point in the volcano plots in panels 3C and 3D represent a phosphorylated peptide. For each of these peptides, the fold change of the abundance of the phosphorylated peptide between trained and sedentary animals were calculated as the difference between logarithmized mean intensities measured in trained animals (n=3) and those measured in sedentary animals (n=3).
Nine proteins were upregulated and 32 downregulated by exercise in control (Control trained vs Control sedentary), whereas 10 and 26 were upregulated and downregulated, respectively, in PKP2cKO hearts (PKP2cKO.trained vs PKP2cKO.sedentary; Figure S7; Tables S2–S3). Thus, training itself had a very limited effect on the cardiac proteome. We observed a trend toward increased abundance of Pln, though below statistical significance.
Training, on the other hand, had a large impact on phosphorylation-mediated signaling. We quantified 6,330 phosphorylation events in controls (Table S4) and 6,118 phosphorylation events in PKP2cKO animals (Table S5). From these, 98 events were upregulated and 104 downregulated in controls (Control trained vs Control sedentary; Figure 3C), whereas 48 and 110 sites were upregulated and downregulated, respectively, in PKP2cKO hearts (PKP2cKO.trained vs PKP2cKO.sedentary (Figure 3D).
Kinase Substrate Enrichment Analysis (KSEA) estimates differential kinase activity based on the collective phosphorylation changes of their identified substrates. In Figure 4A, specific kinases are noted (left panel) and the abundance and phosphorylation state of the kinase itself is displayed (right panel). For the latter, the color of the inner circle depicts a differential in protein abundance (log2Fc; proteome) and the colors in the outer circle, depict the measured differential in the phosphorylation state of the residues specified by the small numbers (log2FC; Phospho-proteome). Tones of red or blue indicate increased or decreased abundance, respectively. The analysis showed prominent differential kinase activities for calcium/calmodulin-dependent protein kinase II delta (Camk2d) and protein kinase C (PKC), with magnitude and sign depending on PKP2 expression (Figure 4A and Table S6), whereas the activity of protein kinase A (PKA), a major kinase downstream of the beta-adrenoreceptors (beta-ARs), was similarly reduced in both groups (Control and PKP2cKO). Furthermore, from a total of ~500 phosphosites reported by Lundby et al to be regulated consequent to exposure to beta1-AR agonists,21 ~200 were identified in our experiments. Yet very few were actually upregulated by exercise, either in control (3 sites) or in the PKP2cKO group (1 paired site; Figure S8A–B). A similar pattern was observed when considering the proteins, regardless of the specific phosphorylation site (Figure S8C–D). The one paired phosphorylation paired site prominently regulated in PKP2cKO animals and previously known as downstream target of beta-ARs was serine 16 and threonine 17 of phospholamban (PlnS16,T17), which was upregulated in PKP2cKO upon training but not in the control group. Phosphorylation of Pln is known as critical to the regulation of Ca2+i homeostasis.22 Thus, we focused our attention on proteins key to excitation-contraction coupling in cardiomyocytes, including those that participate in Ca2+i buffering during the cardiac cycle and whose function is regulated by their phosphorylation state.
Figure 4. Phosphorylation mediated signaling changes in response to exercise.
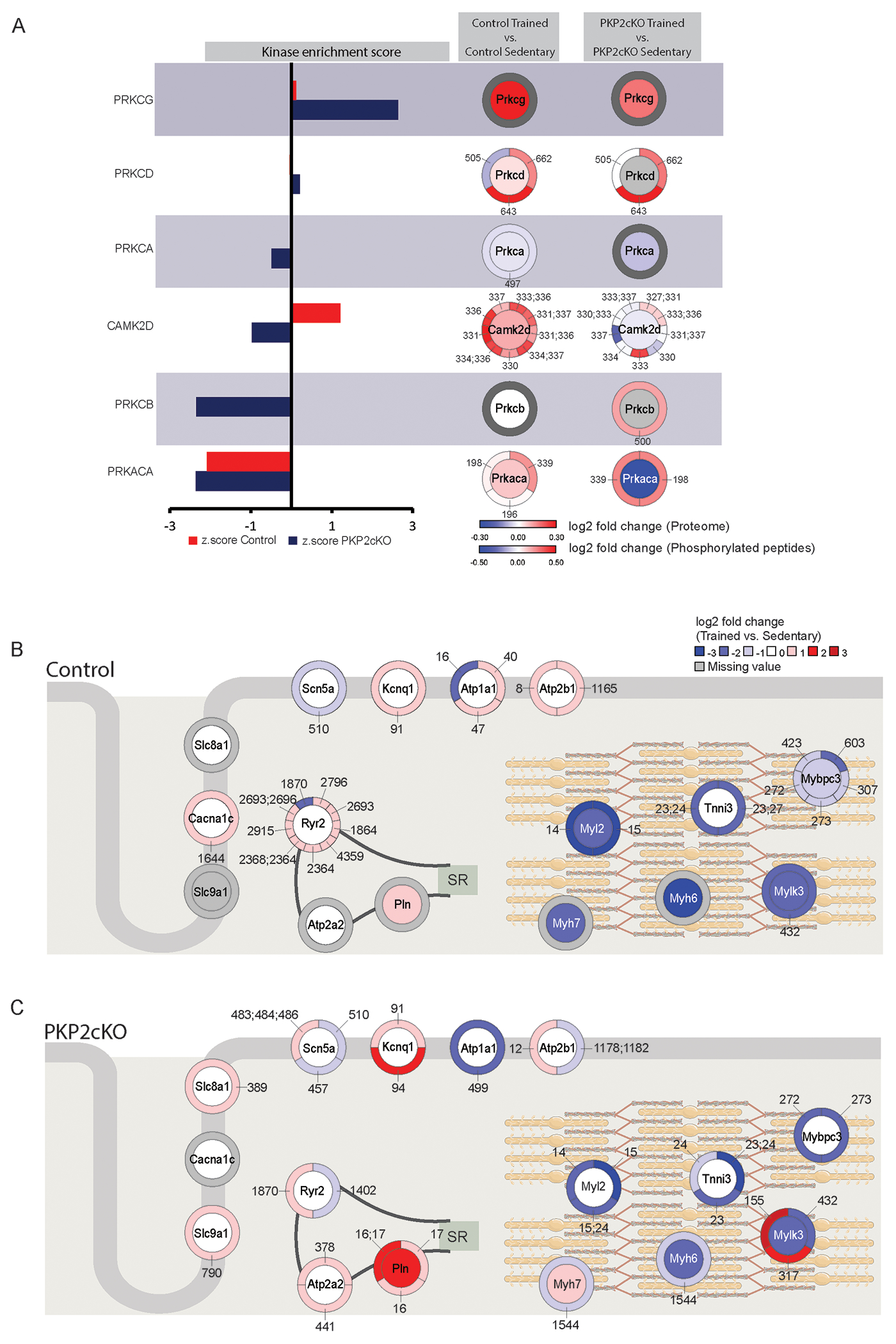
(A) KSEA results that show kinases with either up (positive z-score) or downregulated (negative z-score) activity in control (red) or PKP2cKO (blue) hearts upon exercise. Next to each kinase, data from proteome and phosphoprotome is visualized for control and PKP2cKO groups. The inner circle represents changes in protein abundance and the outer circle, changes in phosphorylation state of sites specified (i.e., small numbers note the position of the phosphosite in the protein sequence). Colors are coded as per the scales in the bottom-right of the panel. (B-C): Diagrammatic representation including a heat map that summarizes differential abundance (inner circle) and phosphorylation state (outer circle) of a selected set of proteins residing in the dyad (left) or the sarcomeric complex (right). B): control; (C): PKP2cKO. Both cases reflect differential proteome/phosphoproteome data obtained by comparing trained versus sedentary animals. Tones of red indicate increased abundance in exercised animals and tones of blue, decrease. Only absolute log2FC values higher than 0.3 are represented; white inner circle: protein differential remained below threshold. Small numbers pointing to segments of the outer circles indicate the position of the phospho-site in the sequence of the protein.
Figure 4B–C present heat maps that summarize the differential abundance (inner circle) and phosphorylation state (outer circle) of a selected set of proteins that contribute to excitation (left) and contraction (right) of the myocyte via the Ca2+-mediated coupling of these two functions (see also Tables S2–S5). Panels 4B and 4C show the heatmaps from control (Control trained-Control sedentary) and PKP2cKO (PKP2cKO trained vs PKP2cKO sedentary) hearts, respectively. A general pattern became apparent, as phosphorylation sites pertaining to proteins prominently (though not exclusively) located in the dyad were colored mostly in tones of red, such as Slc8a1 (coding for the sodium-calcium exchanger, NCX), Atp2a2 (coding for SR Ca2+-ATPase 2a (SERCA2a)), RyR2 and Pln, whereas proteins localized to the contractile apparatus showed the opposite trend. Looking specifically at changes for which a functional association has been established, we noted drastic reduction in phosphorylation of residues 23 and 24 of troponin I3 (Tnni323,24), residues 14 and 15 of myosin light chain 2 (Myl214,15) and residues 272 and 273 of cardiac myosin binding protein C3 (Mybpc3272,273). These changes were found, though to a different extent, in both control and PKP2cKO groups. Yet, a drastic increase in the abundance of phosphorylated Pln in two functionally critical sites (PlnS16, T17) was noted only in the PKP2cKO group.
Time course of sarcomeric shortening and Ca2+i transients in trained PKP2cKO myocytes
The changes noted above were expected to differentially impact sarcomeric shortening kinetics of exercised control or PKP2cKO myocytes. Specifically, reduced phosphorylation of Tnni323,24 and Myl214,15 would increase calcium sensitivity of myofilament components, prolonging overall time course of contraction of trained myocytes. Increased Pln phosphorylation on the other hand would facilitate Ca2+i reuptake into the SR and as such, accelerate relaxation. Figure 5 shows the results. Sarcomeric shortening was measured in single, isolated myocytes by tracking the position of z lines during pacing (details in Methods). As shown in Figure 5A, the time point at which the cell reached maximum shortening divided the time course into two components: from onset to peak (“time to peak” or “TTP”) and from peak to 90% relaxation (“time to rest” or “TTR”). Myocytes from control trained animals had a prolongation of both TTP and TTR (panels B-C). In PKP2cKO cells, however, TTP was also prolonged with exercise, but relaxation was abbreviated, consistent with an increased rate of Ca2+i reuptake (panels B-C). The time constant of relaxation yielded similar results, that is, a slower relaxation in trained controls and a faster relaxation in trained PKP2cKO myocytes (panel D). Same trend was found in the time course of relaxation of paced Ca2+i transients in PKP2cKO myocytes (panel E). Calcium transient amplitude and peak amplitude of sarcomeric shortening were higher in PKP2cKO trained myocytes, consistent with data previously reported (see Figure S9; also17). In summary, exercise in control animals prolonged the entire duration of sarcomere shortening and relaxation, consistent with the observed reduced phosphorylation in both Tnni323,24 and Myl214,15. Exercise in PKP2cKO myocytes on the other hand, accelerated the relaxation process, as expected from an increase in the phosphorylated state of PlnS16,T17. Additional quantitative immunofluorescence experiments confirmed that the signal intensity for PlnS16,T17 was higher, and the signal for Tnni323,24 and Myl215 was lower, in PKP2cKO myocytes of trained animals than in myocytes obtained from PKP2cKO sedentary hearts (Figure S10A–C).
Figure 5. Time course of sarcomere shortening and Ca2+ transients in trained control and plakophilin-2 conditional knockout (PKP2cKO) cardiac myocytes.
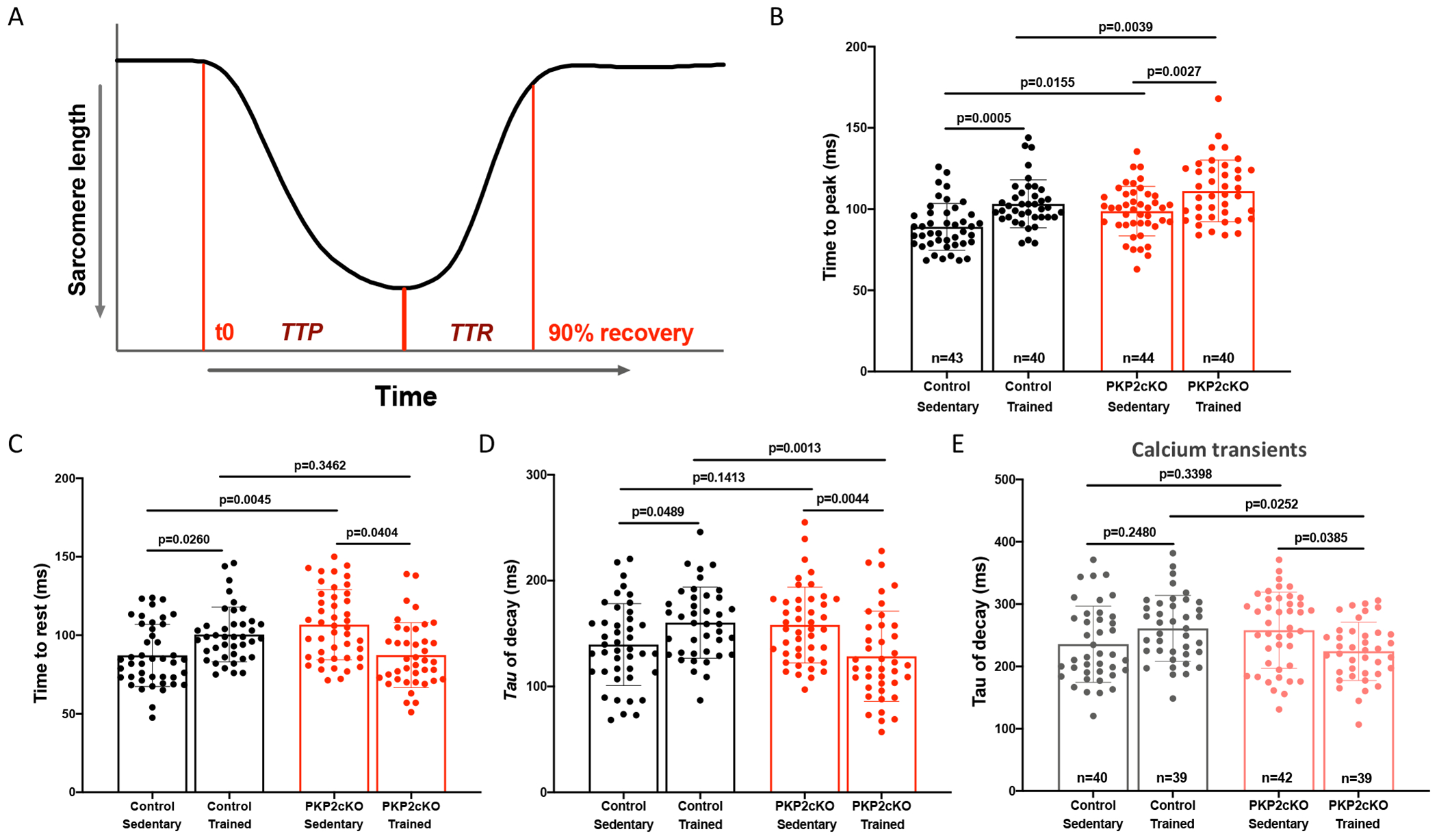
(A) Time course of sarcomere shortening during 1Hz field stimulation. Point of maximum shortening divides two components: Onset to peak (“time to peak” or “TTP”) and peak to 90% relaxation (“time to rest” or “TTR”). (B) Time to peak (ms), (C) time to rest (ms) and (D) time constant of decay (ms) reported as average of 15 consecutive contractions. (E) Time constant of Ca2+ transient decay (ms) obtained by confocal line-scans (1.43 ms/line) during 1Hz field stimulation. Black bars: control myocytes; red bars: PKP2cKO myocytes 21 days post-TAM. Data presented as mean ± SD. Number of cells noted in the corresponding bars; 4-5 mice per group. Test for clustering versus validity of assumption of independence between data obtained from more than one mouse within a group, as in Sikkel et al (20). Results determined no need for hierarchical statistics (significance of clustering in supplemental Table 1). Significance was assessed by two-way ANOVA followed by Tukey’s multiple comparison’s test. Statistical significance was corroborated by linear mixed-effects analysis (Table S1). Effect size (Cohen’s d) and determination of normal distribution (Shapiro-Wilk and Kolgomorov-Smirnov tests) are also reported in Table S1.
Exercise-induced redistribution of functional beta1-ARs in PKP2cKO myocytes
The functional results converged with the phosphoproteome analysis indicating reduced phosphorylation in sarcomeric molecules known to be regulated by beta1-ARs21 and yet, hyperphosphorylation of a protein prominently localized to the SR (Pln) in PKP2cKO hearts. We speculated that this discrepancy may reflect a shift in the distribution of beta receptors to specific subcellular territories. To test this hypothesis, we utilized the experimental protocol introduced by Wang et al18. Specifically, we examined the amplitude and time course of Ca2+i transients elicited by isoproterenol (ISO), a beta-AR agonist to both sarcolemmal and intracellular receptors, in the presence of either propranolol (a blocker of both sarcolemmal and intracellular receptors) or sotalol, a beta-blocker that does not cross the sarcolemma. Data are shown in Figure 6. Panels A-D depict Ca2+i transient amplitude in cells isolated from control sedentary (panel A), control trained (panel B), PKP2cKO sedentary (C) or PKP2cKO trained hearts (D). For each panel, bars reflect transient amplitude without ISO (black bar outline and symbols), with ISO (red), or in combination treatment (either propranolol+ISO, or sotalol+ISO; brown; see Methods). Propranolol completely blocked the ISO-induced increase in transient amplitude in all four groups (ISO vs ISO+Propranolol; Figure 6A–D), and sotalol significantly dampened the ISO response in both control groups and in sedentary PKP2cKO myocytes (ISO vs ISO+sotalol; Figures 6A–C). Yet, sotalol failed to block the ISO response in trained PKP2cKO myocytes (ISO vs ISO+Sotalol; Figure 6D). Quantitative immunofluorescence experiments confirmed that the signal intensity for PlnS16,T17 from isolated myocytes in the presence of ISO was significantly reduced by pre-treatment with propranolol and with sotalol (Figure S10D–E). Moreover, consistent with the fact that repolarizing currents in murine myocytes are not susceptible to block by sotalol,23 Ca2+i transient amplitude and time course of either control or PKP2cKO myocytes were not affected by sotalol exposure (Figure S11). Overall, the data support the notion that the response to ISO in the trained PKP2cKO group is predominantly mediated by intracellular (sotalol-inaccessible) receptors.
Figure 6. Adrenergic regulation of Ca2+ transients in cardiac myocytes from control and plakophilin-2 conditional knockout (PKP2cKO) mice.
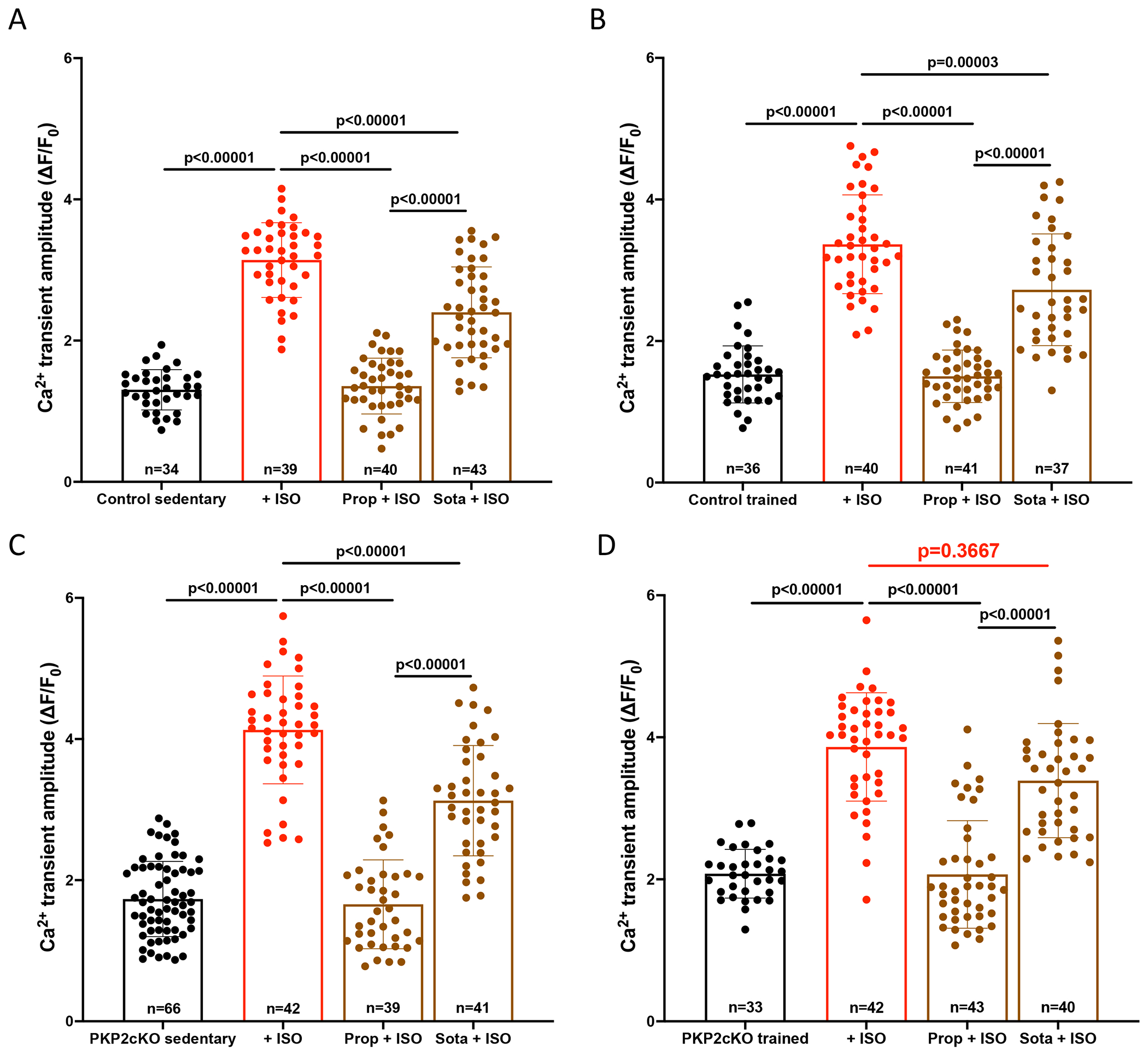
(A-D) Compiled data: Average Ca2+ transient amplitude (relative amplitude; ∆F/F0) obtained by confocal line-scans (1.43 ms/line) during 1Hz field stimulation. Cardiac myocytes from sedentary control (A), trained control (B), sedentary PKP2cKO (C) and trained PKP2cKO (D) mice were used and imaged at baseline or in the presence of 100 nM isoproterenol (ISO), alone or in conjunction with 1 μM propranolol (prop) or 25 μM sotalol (Sota). Cardiac myocytes were pre-incubated for 5 minutes with propranolol or sotalol before ISO incubation (10 minutes). Black bars represent data at baseline, red bars upon ISO, brown bars after pre-incubation with beta blockers. Data presented as mean ± SD. Number of cells noted in the corresponding bars; 4-6 mice per group. Test for clustering versus validity of assumption of independence between data obtained from more than one mouse within a group, as in Sikkel et al (20). Significance of clustering reported in Table S1. Hierarchical statistics used for sedentary PKP2cKO data. Significance was assessed by one-way ANOVA followed by a Bonferroni test for sedentary control and for trained control data. Significance was assessed by Kruskal Wallis followed by Dunn’s multiple comparison’s test for trained PKP2cKO data. Statistical significance was corroborated by a linear mixed-effects analysis (Table S1). Effect size (Cohen’s d) and determination of normal distribution (Shapiro-Wilk and Kolgomorov-Smirnov tests) also reported in Table S1.
Increased SR load after stimulation of the beta1-AR pathway in PKP2cKO myocytes
Separate from exercise, an adrenergic surge unrelated to exercise (e.g., an emotional event; ISO infusion) can trigger arrhythmogenic events in patients with PKP2 mutations.12,24 We examined the effect of ISO on SR load and spontaneous Ca2+i release in cells from sedentary animals. Results are shown in Figure 7. As shown in Figure 7A (exemplary traces) and in the cumulative data (Figure 7B), SR load was higher in PKP2cKO cells (red symbols and bar contour) than in control (black symbols and bar contour), consistent with previous reports,16 and the amplitude of the caffeine-elicited Ca2+i transient was higher in ISO-treated than in untreated cells.
Figure 7. SR Ca2+ content and frequency of Ca2+ sparks in response to ISO; susceptibility to RyR2 block.
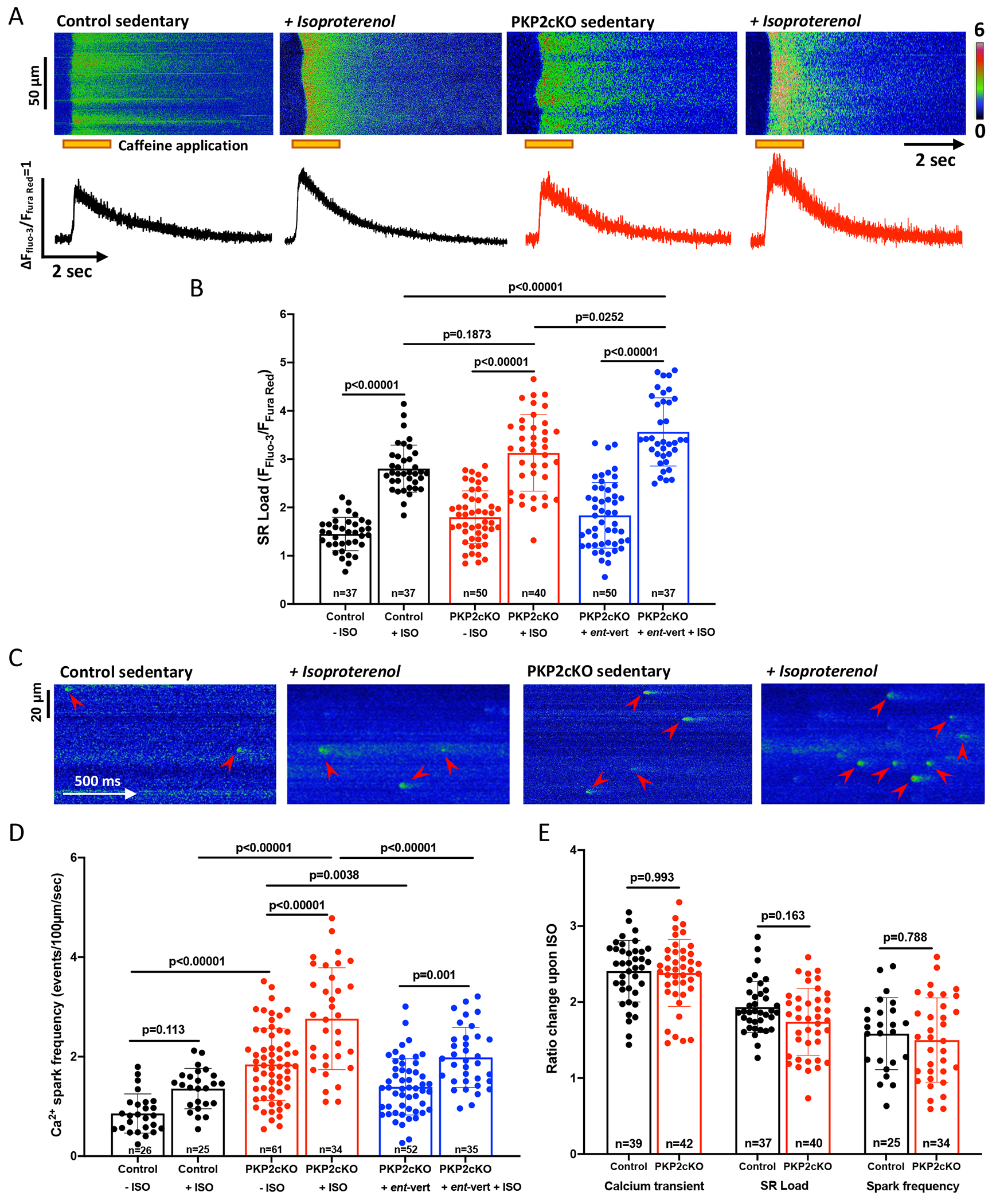
(A) Upper panel; Confocal line-scan images (1.43 ms/line) recorded from cardiac myocytes isolated from sedentary control and sedentary PKP2cKO mice 21 days post-tamoxifen injection, at baseline or exposed to 100 nM isoproterenol. The pulse of caffeine (10 mmol/L) is indicated by the orange bar at the bottom of the image. Intracellular Ca2+ changes were detected by a ratiometric method (FFluo-3/FFura Red; see also Methods). Bottom panel; Time course of change in fluorescence elicited by caffeine pulse. Cumulative data in (B), black bars and symbols represent data from control myocytes, red bars and symbols, PKP2cKO myocytes, blue bars and symbols, PKP2cKO myocytes pre-treated with ent-verticilide. Data represented as mean ± SD. Number of cells noted in the corresponding bars; 5-6 mice per group. (C) Confocal line-scan images of Ca2+ sparks (green; emphasized by red arrowheads) recorded from sedentary control and sedentary PKP2cKO myocytes 21 days post-TAM, at baseline or exposed to 100 nM ISO. Cumulative data are shown in (D), colors of bars and symbols same as in panel B. Number of cells studied are noted in the corresponding bars, 4-6 mice per group. (E) Ratio of change in Ca2+ transient amplitude, SR load and frequency of Ca2+ sparks upon ISO in sedentary control (black bars) and sedentary PKP2cKO (red bars) myocytes, normalized to values obtained in the absence of ISO. Number of cells noted in the corresponding bars, 4-6 mice per group. For all statistical comparisons reported in this figure, test for clustering versus validity of assumption of independence between data obtained from more than one mouse within a group, as in Sikkel et al (20). Significance of clustering reported in Table S1. Results indicated no need for hierarchical statistics. Significance was assessed by Two-way ANOVA followed by Tukey’s multiple comparison’s test (values reported in the Figure and in Table S1) and corroborated by linear mixed-effects analysis (Table S1). Effect size (Cohen’s d) and determination of normal distribution (Shapiro-Wilk and Kolgomorov-Smirnov tests) also reported in Table S1. Control data (no ISO) for spark frequency and SR load also shown in Figures 1 and 2, respectively.
In previous studies, we have reported that loss of PKP2 expression leads to increased rate of diastolic RyR2 channel openings and hence spontaneous SR Ca2+ release. This “Ca2+ leak” from the SR into the cytoplasm would reduce SR Ca2+ content. Thus, we repeated the experiments in the presence of the selective RyR2 inhibitor ent-vert.19 As expected, (Figure 7B, blue contour and symbols), SR load in the presence of ISO was further increased when cells were pretreated with ent-vert (bar labelled Ent-vert+ISO). Under these conditions, SR load in PKP2cKO myocytes after ISO was significantly larger than control.
Ca2+ sparks in beta-AR stimulated PKP2cKO cells; effect of ent-vert
We examined whether the increased SR load associate with increased frequency of spontaneous Ca2+ release events. Figure 7C shows examples of Ca2+ sparks in control and PKP2cKO myocytes exposed to ISO; cumulative data are shown in Fig.7D. ISO led to increased frequency of spontaneous Ca2+ release events in control and PKP2cKO cells, though the spark frequency reached by PKP2cKO cells was significantly higher than in control (Figure 7D). As expected, ent-vert dampened the effects. Additional parameters are presented in Figure S12. Of note, an ISO-induced increase in spark frequency and amplitude was observed in both RV and LV myocytes at 14 days post-TAM, though the frequency of sparks was highest in the myocytes dissociated from the RV of PKP2cKO hearts (Figure S13). Also of note, when measured relative to the values obtained in the absence of ISO, the increases observed in PKP2cKO myocytes were similar to the ones in control cells (Fig.7E), suggesting that the increased magnitude of the response in PKP2cKO cell may be consequent to the increased initial (pre-ISO) value.
Ent-vert reduces the incidence of ISO-induced ectopic ventricular beats in PKP2-deficient hearts
The in vitro efficacy of ent-vert led us to test its efficacy in preventing ISO-induced arrhythmias in whole animals. PKP2cKO mice (21 dpi) were anesthetized and injected (i.p.) with a bolus of ISO (3 mg/kg), a manipulation previously shown to provoke ventricular extrasystolic beats in PKP2cKO mice.15 Two groups were studied: one pre-treated for 30 minutes with ent-vert (30 mg/kg), and one pre-treated with vehicle. The number of premature ventricular contractions (PVCs) within the first 30 minutes after ISO injection were recorded. As shown in Figure 8A, 5 out of 10 mice treated with vehicle (dimethyl sulfoxide; DMSO) presented more than 100 PVCs during 30 minutes of recording (average of 412.6 ± 275.22). In contrast, ent-vert blunted the arrhythmic response; in fact, none of the mice developed more than 100 PVCs in 30 minutes of recording. Taken together, there was an average of over 200 extrasystoles recorded from the control group, and ~20 in the treated group (Figure 8B; exemplary traces in Figure 8C). These results strongly support the hypothesis that Ca2+i dysregulation due to Pln hyperphosphorylation plays a key role for the pro-arrhythmic effect of chronic exercise in hearts deficient in PKP2.
Figure 8. Isoproterenol-induced arrhythmias in PKP2cKO hearts upon treatment with ent-verticilide.
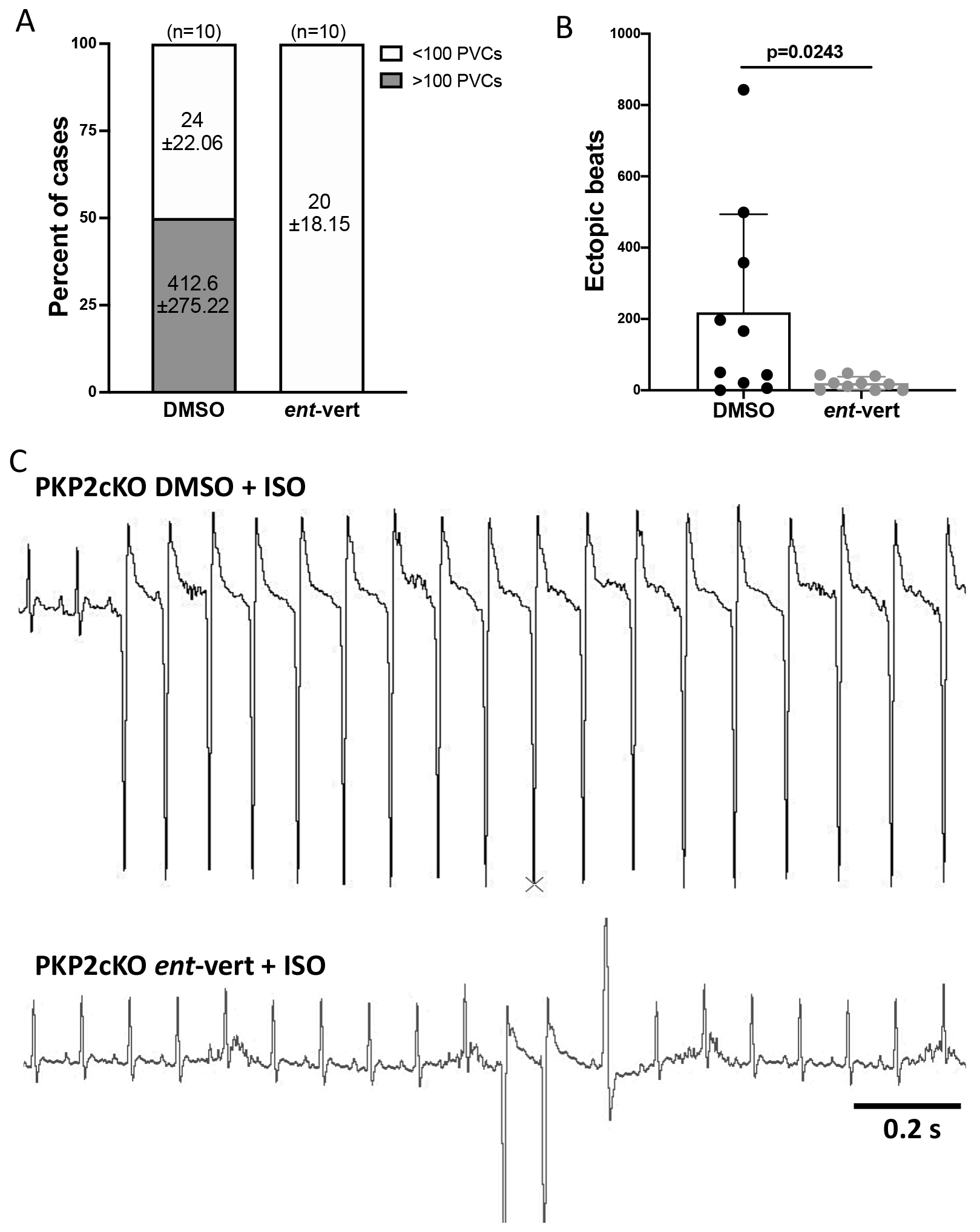
(A) Incidence of isoproterenol-induced (ISO) PVCs during 30 minutes of recording in anesthetized PKP2cKO mice treated with DMSO or ent-verticilide. Data reported as percent of total animals studied per condition; number of animals at top of each bar. Numbers inside bars indicate mean ± SD of ventricular extrasystoles. (B) Cumulative data of ectopic beats in DMSO and ent-verticilide treated mice. Data presented as mean ± SD. Significance as per Mann Whitney test. Effect size (Cohen’s d) and determination of normal distribution (Shapiro-Wilk and Kolgomorov-Smirnov tests) reported in Table S1. (C) Top panel: Example of ISO-induced ventricular tachycardia in a DMSO treated PKP2cKO mouse. Bottom panel: Example of ISO-induced ventricular extrasystoles in an ent-verticilide treated PKP2cKO mouse. Scale bar = 200 ms.
DISCUSSION
Exercise training is considered an independent risk factor for arrhythmias consequent to PKP2 deficiency.7–9 Separate work shows that reduced PKP2 abundance leads to Ca2+i dysregulation.15,16 We therefore explored the relation between exercise training and Ca2+i using a previously characterized model of PKP2 deficiency. Our results support the notion that exercise disproportionately affects Ca2+i homeostasis in PKP2-deficient hearts in a manner facilitated by stimulation of intracellular beta-ARs, creating a pro-arrhythmogenic state that can be mitigated by an RyR2 blocker. We provide mechanistic insight into the PKP2 cardiac endophenotype, with possible implications to the understanding of exercise-induced and catecolaminergic-dependent arrhythmias in individuals with PKP2 deficiency.
We used high resolution mass-spectrometry to obtain an unbiased view of the molecular landscape of hearts from trained and sedentary animals. We measured more than 5,000 class-1 phosphorylation sites from all of our samples, with high reproducibility between samples and minimal inputation (Figure S6). Samples were collected and processed as separate pairs (control trained vs control sedentary in one pair; PKP2cKO trained vs PKP2cKO sedentary in a separate pair) thus allowing comparisons within each pair with minimal normalization (Figure S6). For both cases, the variable was “training.” We decided to stay within the fringes of those comparisons to avoid further data manipulation. The high quality of the data provided us with the statistical power to make quantitative comparisons and interpret the differential phosphoproteomes acquired.
In a recent study, Wang et al18 showed the presence of junctional SR (jSR)-associated intracellular beta1-ARs, and demonstrated that the function of intracellular vs sarcolemmal receptors could be assessed by the response to ISO stimulation in the presence of blockers that are either membrane-impermeable (sotalol) or membrane-permeable (propranolol). Using this protocol we found that propranolol blocks, but sotalol is unable to block, the ISO-induced increase in Ca2+i transient amplitude in trained PKP2cKO myocytes. These results, together with our phosphoproteome data, allow us to propose that in cardiomyocytes from trained PKP2cKO animals, the dominant population of beta1ARs are intracellular and in proximity to sites populated by Pln, such as the jSR. Future experiments, utilizing genetic modifications to reduce the abundance of the catecholamine transporter Oct3 (organic cation transporter 3), will further assess the latter hypothesis. Complementary to that, the data suggest that there is a significant reduction in the abundance of functional sarcolemmal beta1-ARs. Whether this is related to the process of exercise-induced beta1-AR “desensitization,” as previously described,25–27 remains to be determined. Of note, sotalol is considered a Class III antiarrhythmic given its blocking effect on IKr, a potassium current present in human cardiomyocytes.28 This current, however, is not functionally present in murine ventricular myocytes.23 Thus, it is highly unlikely that the effect of sotalol observed in this study is consequent to potassium channel blockade.
The increased phosphorylation state of Pln detected in our studies contrasted with the reduced phosphorylation of Tnni323,24 and Myl214,15. Tnni3 is a major regulator of the Ca2+ buffering capacity of the myocyte via its inhibitory interaction with troponin C,29,30 an action facilitated by Tnni3 phosphorylation at residues 23, 24. Reduced Tnni23,24 phosphorylation would increase Ca2+ sensitivity of the myofilaments and decrease their rate of Ca2+ dissociation.31–33 Myl214,15 also contributes to Ca2+ sensitivity of the myofilaments.34 Overall, reduced phosphorylation of Myl214,15 and Tnni23,24 are consistent with the prolonged time to peak and total duration of sarcomere shortening (Figure 5) in myocytes from trained control and trained PKP2cKO animals, while relaxation in the specific case of PKP2cKO trained animals was likely facilitated by PlnS16,T17-dependent increase in Ca2+i reuptake into the SR.
While increased PlnS16,T17 and reduced Myl214,15 and Tnni23,24 were prominent changes in our differential phosphoproteome, other differences were also noted. Interestingly, upregulation of phosphosites was more common in components of the SR (including the dyad), whereas sarcomeric molecules showed a tendency toward downregulation. This dichotomy was surprising, given previous evidence that the effect of intracellular beta1-AR activation can reach the sarcomere.18,35 On the other hand, changes in the jSR nanostructure in PKP2cKO myocytes, which have been previously noted,15 could affect the precise location of intracellular receptors in relation to Pln vis a vis its distance from the troponin complex. Future experiments, including a crossing of PKP2cKO mice with those deficient in Oct3 (and thus unable to internalize catecholamines18) will dissect in detail the subcellular redistribution of beta1-ARs observed in our study.
We analyzed the occurrence of phosphorylation events in the context of previous characterization of events downstream of beta1-AR stimulation.21 It should be noted though that in our case, mice were subjected to a more chronic adrenergic stimulation via exercise training, whereas in the previous study, Lundby et al reported acute effects. The prolonged time course may have allowed for molecular remodeling of receptors and targets, thus explaining the differences between datasets.
It is worth noting that, from the statistical perspective, the calculated effect size (Cohen’s d model) was not large in many cases (see Table S1). Yet, the meaning of effect size from the biological perspective is difficult to establish, as intracellular signaling often acts as ampification cascades and as such, a small effect when comparing two variables that are close in a pathway can translate into large effects when looking at the final outcomes downstream.
The importance of RyR2 activity in PKP2-related arrhythmogenesis has been previously documented.15,16 Though founded on different mechanisms, our results are reminiscent of the phenotype that characterizes catecholaminergic polymorphic ventricular tachycardia (CPVT;36), a condition most commonly resulting from mutations in the RyR2 gene.37 The studies of Watanabe et al first demonstrated the efficacy of flecainide to block RyR2 channels and to prevent arrhythmias in experimental models of CPVT38 and then in patients.39,40 Following on those observations, we showed that flecainide can decrease the incidence of arrhythmic events in PKP2-deficient mice.15 The latter was consistent with preliminary observations in humans,41 and lent support to a clinical trial currently in progress to assess flecainide efficacy to treat arrhythmias in patients with ARVC (NCT 03685149). More recently, Batiste et al19 reported that the unnatural enantiomer of verticilide (ent-vert) is a potent inhibitor of RyR2 activity. Our data show that ent-vert suppresses the increased spark frequency in ISO-treated PKP2cKO myocytes, and the ISO-induced arrhythmic activity in PKP2cKO mice. These results open the possibility that existing or future RyR blockers may prove effective to treat or prevent arrhythmias in ARVC patients and to permit increased physical activity.
In addition to RyR2 blockers, our results showed that a membrane-impermeable beta-blocker (sotalol) has no benefit in controlling ISO-induced increase in Ca2+i transient amplitude observed in myocytes from trained PKP2cKO animals. The latter may have translational implications, as beta-blocking drugs are commonly administered to patients with ARVC and the selection of the right beta-blocker directly affects therapeutical effectiveness. These results may help explain the reason for the occasional failure of sotalol in controlling arrhythmias in ARVC patients.
For the most part, our studies are limited to the time point at which the mice present an arrhythmogenic cardiomyopathy of right ventricular predominance (21 days post-TAM; see15). However, when looking at a preceding time point, during the concealed stage of the disease (i.e., 14 days post-TAM;15), we found that the largest frequency of Ca2+ sparks was observed in the trained, and in the ISO-exposed, RV myocytes (Figure S3 and Figure S8). This is consistent with previous observations showing that RV myocytes are first at manifesting the effects of PKP2 deficiency.16 It is important to note that electrical heterogeneity can facilitate ventricular arrhythmias,42,43 and that it is during this concealed period of likely electrical right-left heterogeneity that life-threatening arrhythmias in ARVC patients commonly occur.44
Our studies are limited to one experimental model and for one particular gene (PKP2). It is interesting, however, that our unbiased search for modified phosphopeptides led us to a protein that when mutated is also causative of ARVC (Pln). The latter may be more than a coincidence. Rather, an indication that desmosome-dependent and Pln-dependent ARVC phenotypes converge into a common mechanism. While direct mutations on serine 16 or threonine 17 have not been described in ARVC patients, the phosphorylation state of those residues directly impact on the function of the protein to control SERCA activity and intracellular calcium homeostasis, 22,45 and may be altered by mutations in their proximity (e.g., PlnR14del46–48). How the two pathways (desmosome-initiated and Pln-initiated) converge to yield a similar overall cardiomyopathic phenotype, remains unclear.
We conducted our studies using an experimental animal model of PKP2 deficiency. It is not our intent to “recapitulate ARVC.” In fact, to our knowledge, no animal or cell model has done so. The reasons are simple, as mouse models, even those involving expression of ARVC-relevant mutations, do not reproduce the actual genetic defect, the complete phenotype, and/or the right environmental conditions. Cells in a dish, even those derived from human samples, are even farther from recapitulating the disease, given their immaturity and their minimalist environment. But all of these models, the one used here included, teach us about phenotypes that relate to our gene of interest. In our case, what we do is not to directly study ARVC but to characterize, in adult hearts, the endophenotype of a gene that when mutated, causes the disease. Lessons learned from understanding the endophenotype can then be used to speculate about mechanisms behind the human disease. So far, we and others have identified a number of similarities that have, in fact, guided potential therapeutic approaches.15,49–50 Yet, translation to the case of humans with ARVC needs to be done with caution.
Supplementary Material
CLINICAL PERSPECTIVE.
What is new?
We have used an animal model of plakophilin-2 deficiency to investigate possible molecular mechanisms underlying the increased susceptibility to exercise-induced life-threatening arrhythmias in patients with arrhythmogenic right ventricular cardiomyopathy (ARVC).
We found that cardiac myocytes deficient in Plakophilin-2 are particularly susceptible to exercise-induced alterations in intracellular calcium homeostasis.
Hyperphosphorylation of phospholamban resulting from activation of beta-adrenergic receptors emerges as a key mechanistic event possibly linking desmosomal-initiated ARVC to cases that result from mutations in the PLN gene itself.
What are the clinical implications?
We propose a mechanism causative of life-threatening arrhythmias associated to exercise in patients with ARVC.
We suggest that membrane-permeable beta-blockers are potentially more efficient for ARVC patients.
Our results highlight the potential for RyR2 channel blockers as treatment for the control of heart rhythm in the population at risk.
SOURCES OF FUNDING
Work supported by NIH grants RO1-HL134328, RO1-HL136179 and RO1-HL145911 (MD), a Transatlantic Network of Excellence from the Leducq Foundation (MD, AL), a Transformational Project award from the American Heart Association 18TPA34230006 (MC), the Wilton W. Webster Fellowship in Pediatric Electrophysiology from Heart Rhythm Society, and an American Heart Association Postdoctoral Fellowship (CvO). The mass spectrometry measurements were performed at The Novo Nordisk Foundation Center for Protein Research, which is funded in part by a generous donation from the Novo Nordisk Foundation (NNF14CC0001). The project was supported by The Danish Council for independent Research (DFF-0134-00054B) and the Novo Nordisk Foundation (NNF18OC0052844) to AL. The Vanderbilt group was supported in part by NIH grants NHLBI R35 HL144980 (BCK), NHLBI R01 HL151223 (JNJ, BCK), NHLBI F32 HL140874 (DJB), NHLBI F31 HL151125 (ANS), the Leducq Foundation grant 18CVD05 (BCK), the American Heart Association grant 19SFRN34830019 (BCK), PhRMA Foundation Postdoctoral Award (DJB).
Non-standard Abbreviations and Acronyms
- ARVC
arrhythmogenic right ventricular cardiomyopathy
- beta-ARs
beta-adrenergic receptors
- Ca2+
calcium
- Ca2+i
intracellular calcium
- Camk2d
calcium/calmodulin-dependent protein kinase II delta
- CPVT
catecholaminergic polymorphic ventricular tachycardia
- DMSO
dimethyl sulfoxide
- ent-vert
ent-verticilide
- ISO
isoproterenol
- jSR
junctional sarcoplasmic reticulum
- KSEA
Kinase Substrate Enrichment Analysis
- LV
left ventricle
- Mybpc3
cardiac myosin binding protein C3
- Myl2
myosin light chain 2
- NCX
sodium-calcium exchanger
- NIH
National Institutes of Health
- OCT3
organic cation transporter 3
- PCA
Principal component analysis
- PKA
protein kinase A
- PKC
protein kinase C
- PVCs
premature ventricular contractions
- Pln
phospholamban
- PlnS16,T17
phospholamban, serine 16 and threonine 17
- PKP2
plakophilin-2
- PKP2cKO
cardiomyocyte-specific, tamoxifen-activated, PKP2 knockout
- RyR2
ryanodine receptor 2
- RV
right ventricle
- SD
standard deviation
- SERCA
sarco/endoplasmic reticulum Ca2+-ATPase
- SR
sarcoplasmic reticulum
- TAM
tamoxifen
- TMT
tandem mass tag
- Tnni3
troponin I3
- TTP
time to peak
- TTR
time to rest
Footnotes
DISCLOSURES
Authors have no conflict of interests to disclose.
REFERENCES
- 1.Agullo-Pascual E, Cerrone M, Delmar M. Arrhythmogenic cardiomyopathy and Brugada syndrome: Diseases of the connexome. In: FEBS Letters. 2014;17:588:1322–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Austin KM, Trembley MA, Chandler SF, Sanders SP, Saffitz JE, Abrams DJ, Pu WT . Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2019;16:519–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corrado D, Link MS, Calkins H. Arrhythmogenic Right Ventricular Cardiomyopathy. N Engl J Med. 2017;376:1489–1490. [DOI] [PubMed] [Google Scholar]
- 4.Corrado D, Link MS, Calkins H. Arrhythmogenic Right Ventricular Cardiomyopathy. N Engl J Med. 2017;376:61–72. [DOI] [PubMed] [Google Scholar]
- 5.Groeneweg JA, Bhonsale A, James CA, te Riele AS, Dooijes D, Tichnell C, Murray B, Wiesfeld ACP, Sawant AC, Kassamali B, et al. , Calkins H. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ Cardiovasc Genet. 2015;8:437–446. [DOI] [PubMed] [Google Scholar]
- 6.Bhonsale A, Groeneweg JA, James CA, Dooijes D, Tichnell C, Jongbloed JDH, Murray B, te Riele ASJM, van den Berg MP, Bikker H, et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur Heart J. 2015;36:847–855. [DOI] [PubMed] [Google Scholar]
- 7.James CA, Bhonsale A, Tichnell C, Murray B, Russell SD, Tandri H, Tedford RJ, Judge DP, Calkins H. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62:1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sawant AC, Bhonsale A, te Riele ASJM, Tichnell C, Murray B, Russell SD, Tandri H, Tedford RJ, Judge DP, Calkins H, et al. Exercise has a disproportionate role in the pathogenesis of arrhythmogenic right ventricular dysplasia/cardiomyopathy in patients without desmosomal mutations. J Am Heart Assoc. 2014;3:e001471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang W, Tichnell C, Murray BA, Agafonova J, Cadrin-Tourigny J, Chelko S, Tandri H, Calkins H, James CA. Exercise restriction is protective for genotype-positive family members of arrhythmogenic right ventricular cardiomyopathy patients. Europace. 2020;22:1270–1278. [DOI] [PubMed] [Google Scholar]
- 10.Tester DJ, Ackerman JP, Giudicessi JR, Ackerman NC, Cerrone M, Delmar M, Ackerman MJ. Plakophilin-2 Truncation Variants in Patients Clinically Diagnosed With Catecholaminergic Polymorphic Ventricular Tachycardia and Decedents With Exercise-Associated Autopsy Negative Sudden Unexplained Death in the Young. JACC Clin Electrophysiol. 2019;5:120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y, Li C, Shi L, Chen X, Cui C, Huang J, Chen B, Hall DD, Pan Z, Lu M, et al. Integrin β1D Deficiency-Mediated RyR2 Dysfunction Contributes to Catecholamine-Sensitive Ventricular Tachycardia in Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation. 2020;141:1477–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denis A, Sacher F, Derval N, Martin R, Lim HS, Pambrun T, Massoullie G, Duchateau J, Cochet H, Pillois X, et al. Arrhythmogenic response to isoproterenol testing vs. exercise testing in arrhythmogenic right ventricular cardiomyopathy patients. Europace. 2018;20:f30–f36. [DOI] [PubMed] [Google Scholar]
- 13.Denis A, Sacher F, Derval N, Lim HS, Cochet H, Shah AJ, Daly M, Pillois X, Ramoul K, Komatsu Y, et al. Diagnostic value of isoproterenol testing in arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol. 2014;7:590–597. [DOI] [PubMed] [Google Scholar]
- 14.Assis FR, Krishnan A, Zhou X, James CA, Murray B, Tichnell C, Berger R, Calkins H, Tandri H, Mandal K. Cardiac sympathectomy for refractory ventricular tachycardia in arrhythmogenic right ventricular cardiomyopathy. Hear Rhythm. 2019;16:1003–1010. [DOI] [PubMed] [Google Scholar]
- 15.Cerrone M, Montnach J, Lin X, Zhao Y-T, Zhang M, Agullo-Pascual E, Leo-Macias A, Alvarado FJ, Dolgalev I, Karathanos TV, et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat Commun. 2017;8:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim J-C, Pérez-Hernández Duran M, Alvarado FJ, Maurya SR, Montnach J, Yin Y, Zhang M, Lin X, Vasquez C, Heguy A, et al. Disruption of Ca2+i Homeostasis and Cx43 Hemichannel Function in the Right Ventricle Precedes Overt Arrhythmogenic Cardiomyopathy in PKP2-Deficient Mice. Circulation. 2019;140:1015–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cerrone M, Marron-Linares GM, van Opbergen CJM, Costa S, Bourfiss M, Perez-Hernandez M, Schlamp F, Sanchis F, Malkani K, Drenkova K, et al. Role of PKP2 expression on the exercise-mediated differential transcriptome of adult cardiac ventricular myocytes. Eur Heart J. 2021;19;ehab772. [Google Scholar]
- 18.Wang Y, Shi Q, Li M, Zhao M, Reddy Gopireddy R, Teoh J-P, Xu B, Zhu C, Ireton KE, Srinivasan S, et al. Intracellular β(1)-Adrenergic Receptors and Organic Cation Transporter 3 Mediate Phospholamban Phosphorylation to Enhance Cardiac Contractility. Circ Res. 2021;128:246–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Batiste SM, Blackwell DJ, Kim K, Kryshtal DO, Gomez-Hurtado N, Rebbeck RT, Cornea RL, Johnston JN, Knollmann BC. Unnatural verticilide enantiomer inhibits type 2 ryanodine receptor-mediated calcium leak and is antiarrhythmic. Proc Natl Acad Sci U S A. 2019;116:4810–4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sikkel MB, Francis DP, Howard J, Gordon F, Rowlands C, Peters NS, Lyon AR, Harding SE, MacLeod KT. Hierarchical statistical techniques are necessary to draw reliable conclusions from analysis of isolated cardiomyocyte studies. Cardiovasc Res. 2017;113:1743–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lundby A, Andersen MN, Steffensen AB, Horn H, Kelstrup CD, Francavilla C, Jensen LJ, Schmitt N, Thomsen MB, Olsen JV. In vivo phosphoproteomics analysis reveals the cardiac targets of β-adrenergic receptor signaling. Sci Signal. 2013;6:rs11. [DOI] [PubMed] [Google Scholar]
- 22.Kranias EG, Hajjar RJ. The Phospholamban Journey 4 Decades After Setting Out for Ithaka. Circ Res. 2017;120:781–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nerbonne JM, Nichols CG, Schwarz TL, Escande D. Genetic manipulation of cardiac K(+) channel function in mice: what have we learned, and where do we go from here? Circ Res. 2001;89:944–956. [DOI] [PubMed] [Google Scholar]
- 24.Fornes P, Ratel S, Lecomte D. Pathology of arrhythmogenic right ventricular cardiomyopathy/dysplasia--an autopsy study of 20 forensic cases. J Forensic Sci. 1998;43:777–783. [PubMed] [Google Scholar]
- 25.Hart E, Dawson E, Rasmussen P, George K, Secher NH, Whyte G, Shave R. Beta-adrenergic receptor desensitization in man: insight into post-exercise attenuation of cardiac function. J Physiol. 2006;577:717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hammer KP, Mustroph J, Stauber T, Birchmeier W, Wagner S, Maier LS. Beneficial effect of voluntary physical exercise in Plakophilin2 transgenic mice. PLoS One. 2021;16:e0252649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Welsh RC, Warburton DER, Humen DP, Taylor DA, McGavock J, Haykowsky MJ. Prolonged strenuous exercise alters the cardiovascular response to dobutamine stimulation in male athletes. J Physiol. 2005;569:325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–53. [DOI] [PubMed] [Google Scholar]
- 29.Gomes AV, Harada K, Potter JD. A mutation in the N-terminus of troponin I that is associated with hypertrophic cardiomyopathy affects the Ca(2+)-sensitivity, phosphorylation kinetics and proteolytic susceptibility of troponin. J Mol Cell Cardiol. 2005;39:754–765. [DOI] [PubMed] [Google Scholar]
- 30.Smith GL, Eisner DA. Calcium Buffering in the Heart in Health and Disease. Circulation. 2019;139:2358–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salhi HE, Hassel NC, Siddiqui JK, Brundage EA, Ziolo MT, Janssen PML, Davis JP, Biesiadecki BJ. Myofilament Calcium Sensitivity: Mechanistic Insight into TnI Ser-23/24 and Ser-150 Phosphorylation Integration. Front Physiol. 2016;7:567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Biesiadecki BJ, Kobayashi T, Walker JS, Solaro RJ, de Tombe PP. The troponin C G159D mutation blunts myofilament desensitization induced by troponin I Ser23/24 phosphorylation. Circ Res. 2007;100:1486–1493. [DOI] [PubMed] [Google Scholar]
- 33.Du C-K, Morimoto S, Nishii K, Minakami R, Ohta M, Tadano N, Lu Q-W, Wang Y-Y, Zhan D-Y, Mochizuki M, et al. Knock-in mouse model of dilated cardiomyopathy caused by troponin mutation. Circ Res. 2007;101:185–194. [DOI] [PubMed] [Google Scholar]
- 34.Sheikh F, Lyon RC, Chen J. Functions of myosin light chain-2 (MYL2) in cardiac muscle and disease. Gene. 2015;569:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Surdo NC, Berrera M, Koschinski A, Brescia M, Machado MR, Carr C, Wright P, Gorelik J, Morotti S, Grandi E, et al. FRET biosensor uncovers cAMP nano-domains at β-adrenergic targets that dictate precise tuning of cardiac contractility. Nat Commun. 2017;8:15031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas NL, Maxwell C, Mukherjee S, Williams AJ. Ryanodine receptor mutations in arrhythmia: The continuing mystery of channel dysfunction. FEBS Lett. 2010;584:2153–2160. [DOI] [PubMed] [Google Scholar]
- 37.Priori SG, Chen SRW. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ Res. 2011;108:871–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, Duff HJ, Roden DM, Wilde AAM, Knollmann BC. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med. 2009;15:380–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van der Werf C, Kannankeril PJ, Sacher F, Krahn AD, Viskin S, Leenhardt A, Shimizu W, Sumitomo N, Fish FA, Bhuiyan ZA, et al. Flecainide Therapy Reduces Exercise-Induced Ventricular Arrhythmias in Patients With Catecholaminergic Polymorphic Ventricular Tachycardia. J Am Coll Cardiol. 2011;57:2244–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watanabe H, van der Werf C, Roses-Noguer F, Adler A, Sumitomo N, Veltmann C, Rosso R, Bhuiyan ZA, Bikker H, Kannankeril PJ, et al. Effects of flecainide on exercise-induced ventricular arrhythmias and recurrences in genotype-negative patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2013;10:542–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ermakov S, Gerstenfeld EP, Svetlichnaya Y, Scheinman MM. Use of flecainide in combination antiarrhythmic therapy in patients with arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm. 2017;14:564–569. [DOI] [PubMed] [Google Scholar]
- 42.van Veen TAB, Stein M, Royer A, Le Quang K, Charpentier F, Colledge WH, Huang CL-H, Wilders R, Grace AA, Escande D, et al. Impaired impulse propagation in Scn5a-knockout mice: combined contribution of excitability, connexin expression, and tissue architecture in relation to aging. Circulation. 2005;112:1927–1935. [DOI] [PubMed] [Google Scholar]
- 43.Smith RM, Velamakanni SS, Tolkacheva EG. Interventricular heterogeneity as a substrate for arrhythmogenesis of decoupled mitochondria during ischemia in the whole heart. Am J Physiol Heart Circ Physiol. 2012;303:H224–233. [DOI] [PubMed] [Google Scholar]
- 44.Andrews CM, Srinivasan NT, Rosmini S, Bulluck H, Orini M, Jenkins S, Pantazis A, McKenna WJ, Moon JC, Lambiase PD, et al. Electrical and Structural Substrate of Arrhythmogenic Right Ventricular Cardiomyopathy Determined Using Noninvasive Electrocardiographic Imaging and Late Gadolinium Magnetic Resonance Imaging. Circ Arrhythm Electrophysiol. 2017;10:e005105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sato D, Uchinoumi H, Bers DM. Increasing SERCA function promotes initiation of calcium sparks and breakup of calcium waves. J Physiol. 2021;599:3267–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Rijsingen IAW, van der Zwaag PA, Groeneweg JA, Nannenberg EA, Jongbloed JDH, Zwinderman AH, Pinto YM, Dit Deprez RHL, Post JG, Tan HL, et al. Outcome in phospholamban R14del carriers: results of a large multicentre cohort study. Circ Cardiovasc Genet. 2014;7:455–465. [DOI] [PubMed] [Google Scholar]
- 47.Raad N, Bittihn P, Cacheux M, Jeong D, Ilkan Z, Ceholski D, Kohlbrenner E, Zhang L, Cai C-L, Kranias EG, et al. Arrhythmia Mechanism and Dynamics in a Humanized Mouse Model of Inherited Cardiomyopathy Due to Phospholamban R14del Mutation. Circulation. 2021;10;144:441–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Opbergen CJM, Delmar M, van Veen TAB. Potential new mechanisms of pro-arrhythmia in arrhythmogenic cardiomyopathy: focus on calcium sensitive pathways. Netherlands Heart J. 2017;25:157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chelko SP, Keceli G, Carpi A, Doti N, Agrimi J, Asimaki A, Beti CB, Miyamoto M, Amat-Codina N, Bedja D, et al. Exercise triggers CAPN1-mediated AIF truncation, inducing myocyte cell death in arrhythmogenic cardiomyopathy. Sci Transl Med. 2021;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chelko SP, Asimaki A, Lowenthal J, Bueno-Beti C, Bedja D, Scalco A, Amat-Alarcon N, Andersen P, Judge DP, Tung L, et al. Therapeutic Modulation of the Immune Response in Arrhythmogenic Cardiomyopathy. Circulation. 2019;140:1491–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cerrone M, van Opbergen CJM, Malkani K, Irrera N, Zhang M, Van Veen TAB, Cronstein B, Delmar M. Blockade of the Adenosine 2A Receptor Mitigates the Cardiomyopathy Induced by Loss of Plakophilin-2 Expression. Front Physiol. 2018;9:1750. [DOI] [PMC free article] [PubMed] [Google Scholar]; 51. Sanchis-Gomar F, Pareja-Galeano H, Martinez-Bello VE . PPARgamma agonist pioglitazone does not enhance performance in mice. Drug Test Anal. 2014;6:922–929. [DOI] [PubMed] [Google Scholar]
- 52.Davies KJ, Packer L, Brooks GA. Biochemical adaptation of mitochondria, muscle, and whole-animal respiration to endurance training. Arch Biochem Biophys. 1981;209:539–554. [DOI] [PubMed] [Google Scholar]
- 53.Derbré F, Gomez-Cabrera MC, Nascimento AL, Sanchis-Gomar F, Martinez-Bello VE, Tresguerres JA, Fuentes T, Gratas-Delamarche A, Monsalve M, Viña J. Age associated low mitochondrial biogenesis may be explained by lack of response of PGC-1α to exercise training. Age (Dordr). 2012;34:669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Picht E, Zima AV, Blatter LA, Bers DM. SparkMaster: automated calcium spark analysis with ImageJ. Am J Physiol Cell Physiol. 2007;293:C1073–1081. [DOI] [PubMed] [Google Scholar]
- 55.Nilbratt M, Porras O, Marutle A, Hovatta O, Nordberg A. Neurotrophic factors promote cholinergic differentiation in human embryonic stem cell-derived neurons. J Cell Mol Med. 2010;14:1476–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Linscheid N, Logantha SJRJ, Poulsen PC, Zhang S, Schrölkamp M, Egerod KL, Thompson JJ, Kitmitto A, Galli G, Humphries MJ, et al. Quantitative proteomics and single-nucleus transcriptomics of the sinus node elucidates the foundation of cardiac pacemaking. Nat Commun. 2019;10:2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zecha J, Satpathy S, Kanashova T, Avanessian SC, Kane MH, Clauser KR, Mertins P, Carr SA, Kuster B. TMT Labeling for the Masses: A Robust and Cost-efficient, In-solution Labeling Approach. Mol Cell Proteomics. 2019;18:1468–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bekker-Jensen DB, Kelstrup CD, Batth TS, Larsen SC, Haldrup C, Bramsen JB, Sørensen KD, Høyer S, Ørntoft TF, Andersen CL, et al. An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes. Cell Syst. 2017;4:587–599.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tyanova S, Temu T, Cox J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc. 2016;11:2301–2319. [DOI] [PubMed] [Google Scholar]
- 60.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. [DOI] [PubMed] [Google Scholar]
- 61.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods. 2016;13:731–740. [DOI] [PubMed] [Google Scholar]
- 62.Kammers K, Cole RN, Tiengwe C, Ruczinski I. Detecting Significant Changes in Protein Abundance. EuPA Open Proteomics. 2015;7:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wiredja DD, Koyutürk M, Chance MR. The KSEA App: a web-based tool for kinase activity inference from quantitative phosphoproteomics. Bioinformatics. 2017;33:3489–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Horn H, Schoof EM, Kim J, Robin X, Miller ML, Diella F, Palma A, Cesareni G, Jensen LJ, et al. KinomeXplorer: an integrated platform for kinome biology studies. Nat Methods. 2014;11:603–604. [DOI] [PubMed] [Google Scholar]
- 65.Cohen J Statistical Power Analysis for the Behavioral Sciences. Routledge. 1988;ISBN 978-1-134-74270-7. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mass spectrometry proteomics and phosphoproteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository with dataset identifier PXD025956 (accessible through https://www.ebi.ac.uk/pride/archive) and project name “Investigation of changes in the proteome and the phosphoproteome of the heart induced by exercise, either in control or in PKP2-deficient hearts in relation to ARVC”. Additional information can be made available from the corresponding authors upon reasonable request.